

Abstract
The TP53 tumor suppressor is the most frequently mutated gene in human cancer. p53 suppresses tumorigenesis by transcriptionally regulating a network of target genes that play roles in various cellular processes. Though originally characterized as a critical regulator for responses to acute DNA damage - activation of apoptosis and cell cycle arrest – recent studies have highlighted new pathways and transcriptional targets downstream of p53 regulating genomic integrity, metabolism, redox biology, stemness, and non-cell autonomous signaling in tumor suppression. Here, we summarize our current understanding of p53-mediated tumor suppression, situating recent findings from mouse models and unbiased screens in the context of previous studies and arguing for the importance of the pleiotropic effects of the p53 transcriptional network in inhibiting cancer.
Keywords: p53, tumor suppression, transcription factor, cancer, mouse models, network
p53: tumor suppressor, stress sensor, and transcription factor
The TP53 gene encodes a transcription factor that is a critical barrier to carcinogenesis. Inactivation of TP53 is the most common mutation in sporadic human cancers, suggesting a strong selection against p53 function during tumorigenesis [1]. The inheritance of a mutant TP53 allele is observed in Li-Fraumeni syndrome, predisposing patients to early onset cancer development, further underscoring the role of p53 in tumor suppression. The importance of p53 as a tumor suppressor is cemented by experimental evidence from p53−/− mice, which develop cancer, mostly thymic lymphomas, with 100% penetrance [2]. Despite this unequivocal characterization of the fundamental role for p53 in tumor suppression, a comprehensive mechanistic understanding of p53 tumor suppressor function is lacking. Understanding the underpinnings of p53 tumor suppressor function is of critical importance to elucidating cancer etiology and developing therapeutic strategies.
p53 is thought to act as a tumor suppressor by serving as a cellular stress sensor (Fig. 1). In unstressed cells, p53 is targeted by the E3 ubiquitin ligase MDM2 for degradation, keeping p53 at low levels. A variety of stress signals, including DNA damage, oncogene expression, and hypoxia, relieve p53 from MDM2 inhibition. p53 binds DNA in a sequence-specific manner and recruits transcriptional machinery components to activate expression of a network of target genes. Transactivation of p53 target genes is compromised by mutations found in human cancers, which occur predominantly in the sequence-specific DNA binding domain. These mutations disable DNA binding capacity by perturbing residues involved in direct contact of DNA or disrupting p53 protein structure, although oncogenic gain-of-function activity has also been reported for some TP53 mutations (reviewed in [3]). The importance of transcriptional activation for p53-mediated tumor suppression is supported by analyses of mice expressing a transactivation-dead mutant, with alterations in TAD1 and TAD2, p5325,26,53,54. In mouse models for a range of cancers, including B- and T-cell lymphomas, lung adenocarcinoma (LUAD), and pancreatic ductal adenocarcinoma (PDAC), this mutant behaves indistinguishably from p53 deletion, suggesting the importance of transcriptional activation for p53-mediated tumor suppression [4–6]. Although we focus in this review primarily on transcriptional programs underlying p53 tumor suppression, roles for p53 independent of transcriptional activation have also been reported (Box 1).
Figure 1. p53 structure and function.
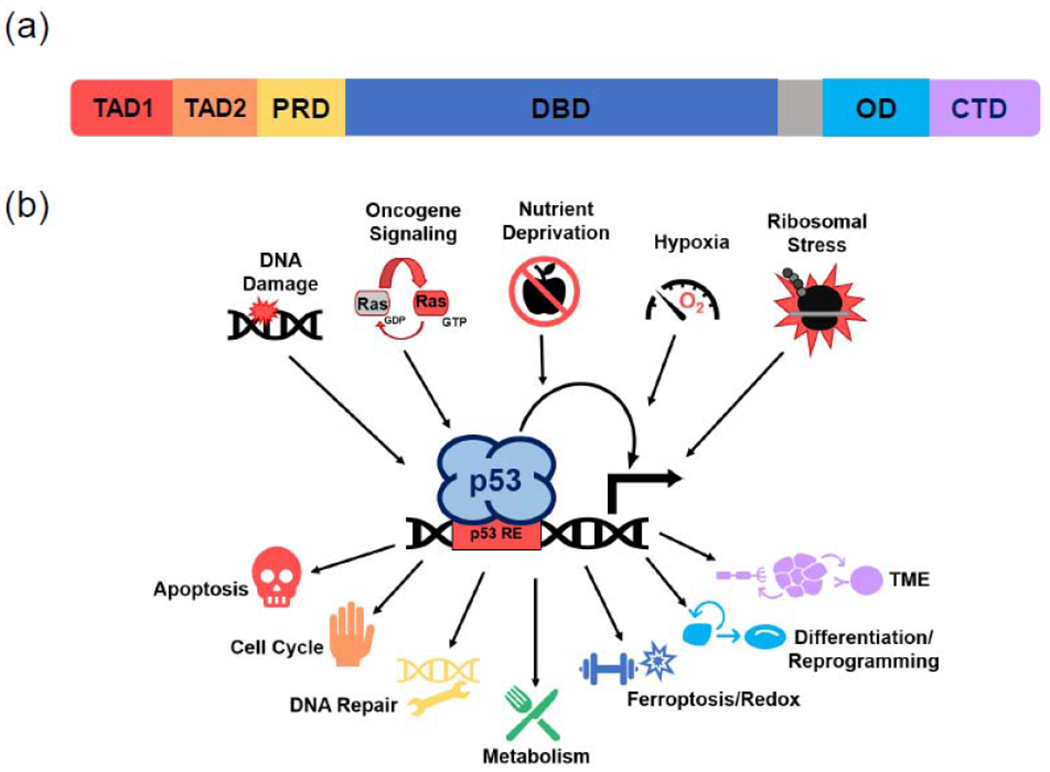
(a) p53 protein includes six major domains essential for transcriptional activation: two amino-terminal transcriptional activation domains (TADs), a proline-rich domain (PRD), a sequence-specific DNA binding domain (DBD), an oligomerization domain (OD), and a C-terminal domain (CTD). (b) p53 is activated by various cellular stresses, driving a transcriptional response that impacts a wide range of cellular processes. A p53 tetramer binds DNA in a sequence-specific fashion at p53 response elements (RE), then activates transcription of target genes involved in different cellular processes.
Box 1. Transactivation-independent p53 functions add another dimension to tumor suppression.
Although most investigation of the mechanisms of p53-mediated tumor suppression has focused on transcriptional activation (transactivation) of p53 target genes, transactivation-independent functions of p53 have also been reported. The best characterized transactivation-independent p53 function is induction of apoptosis at the mitochondria. p53 can bind anti-apoptotic BCL-2 family members to displace them from pro-apoptotic BCL-2 family members or directly activate BAX and BAK to trigger mitochondrial outer matrix permeabilization and apoptosis [75]. p53 has other described transactivation-independent functions both in the cytoplasm and the nucleus that could contribute to tumor suppression. For example, in the cytoplasm p53 can bind and inhibit the rate-limiting enzyme of the pentose phosphate pathway, G6PDH, thus restricting this tumor-supporting metabolic pathway [76]. In the nucleus, p53 can promote genomic integrity through different mechanisms, such as by restraining the mobility of transposons and other classes of repetitive elements and by binding the homologous recombination protein RAD51 to prevent excessive or inaccurate recombination events [75]. Recent GEMM studies have highlighted transactivation-independent roles for p53 in vivo. In one study using mouse models of Wnt-driven intestinal cancer, resulting from either Csnk1a1 deletion or ApcMin/+ mutation in the proximal gut, cancer was suppressed in the presence p53R172H, a DNA binding domain mutant deficient for target gene transactivation, relative to the p53−/− model [77]. This activity, related to suppressing the Wnt pathway through inhibition of TCF4 chromatin binding, suggested p53 transactivation-independent tumor suppression, although further experiments are necessary to clarify whether the activity of this mutant is neomorphic or present in wild-p53 [77]. In another study, analysis of knock-in mice expressing the p53R178E mutant, which is deficient for p53 target gene activation, has revealed that p53R178E behaves like a p53 null allele in both spontaneous and Eμ-Myc-driven mouse tumorigenesis [78]. However, this mutant retains apoptotic activity in vivo both in apoptosis triggered by Mdm2 deficiency in developing embryos and in chemotherapeutic treatment of Eμ-Myc mouse lymphomas [78]. p53R178E can localize to the mitochondria, providing a potential mechanism for p53R178E-triggered transactivation-independent apoptosis [78]. These observations raise the possibility that this transactivation-independent p53 function might have significance in tumor suppression in some settings. Future experiments designed to test the significance of specific transactivation-independent functions of p53 for tumor suppression in vivo will shed more light on the importance of these phenomena for understanding cancer.
The best characterized p53 function is in response to acute DNA damage, when p53 either promotes G1 cell cycle arrest, facilitating DNA repair, or apoptosis, to eliminate damaged cells. Genetic studies in mouse models have suggested, however, that responding to acute DNA damage is dispensable for p53-mediated tumor suppression (Box 2), or perhaps that other responses can compensate when these pathways are compromised. The surprising retention of p53-mediated tumor suppression observed in acute DNA damage response-deficient p5325,26, p533KR, and p21−/−;Puma−/−;Noxa−/− mutant mouse strains (Box 2) emphasizes a need to rethink how the pathways downstream of p53 inhibit tumorigenesis. Recent in vivo studies both highlight new and elaborate on previously described p53 functions, calling for integration and analysis of the expansive catalog of functions ascribed to p53 in tumor suppression. In this review, we will summarize these recent findings, focusing on genetically engineered mouse models (GEMMs), and will propose a model that integrates the many functions of p53 in tumor suppression.
Box 2. Acute DNA damage responses are dispensable for tumor suppression.
Early work on p53 focused on transactivation of target genes like the CDK inhibitor, p21, and the pro-apoptotic BCL-2 family members Bax, Puma and Noxa, which were shown in genetic experiments to be critical for the acute DNA damages responses of G1 cell cycle arrest and apoptosis, respectively [74,79]. These pathways delineated by studying acute DNA damage-induced cell cycle arrest and apoptosis were envisioned to sufficiently explain p53-mediated tumor suppression. Since these studies, however, multiple studies in mouse models suggested that p53-dependent acute DNA damage responses are dispensable for tumor suppression. Using mice expressing temporally-regulatable p53 alleles, two independent studies demonstrated that the presence of p53 during treatment with acute DNA damage is dispensable for tumor suppression, proposing instead that p53 activation by oncogenic signaling is more important for tumor suppression [80,81]. Building on these initial observations, subsequent studies provided deeper mechanistic insight into the issue. The first such study used knock-in mice with inactivating mutations in TAD1 of p53 (p5325,26). Functional analysis revealed that cells expressing p5325,26 fail to undergo cell cycle arrest or apoptosis in response to acute DNA damage, and genome-wide transcriptomics revealed that p5325,26 activates only a subset of target genes activated by wild-type p53. p5325,26 cannot robustly activate canonical targets including p21, Puma, and Noxa, yet it retains the ability to suppress both spontaneous cancer (T-cell lymphoma) and oncogene-driven cancers (KrasG12D-driven lung adenocarcinoma and pancreatic ductal adenocarcinoma, Myc-driven B-cell lymphoma and Patched inactivation-driven medulloblastoma) in GEMMs [4,5], suggesting that the robust transactivation of canonical p53 targets is dispensable for tumor suppression. These findings are supported by p21−/−;Puma−/−;Noxa−/− triple knockout mice and mice expressing p533KR, an acetylation site mutant that cannot activate canonical p53 target genes or acute DNA damage responses, both of which are resistant to the spontaneous cancer development observed in p53−/− mice [13,21]. Furthermore, there is indeed an enhancement of tumorigenesis observed in p21−/−;Puma−/−;Eμ-Myc mice relative to Eμ-Myc counterparts, albeit not even as potent as p53+/−;Eμ-Myc mice [82]. Collectively, these studies have highlighted the need to investigate additional p53-regulated pathways that might compensate for p21, Puma, and Noxa or their associated functions to maintain tumor suppression in their absence. These studies do not strictly rule out a tumor-suppressive role for cell cycle arrest and apoptosis responses activated by other stresses that p53 responds to, such as chronic DNA damage, oncogenic signaling, hypoxia, and nutrient deprivation, amongst others.
Analysis of p5325,26 target genes reveals new players in p53-mediated tumor suppression
The finding that p5325,26 activates only a small subset of p53 target genes yet remains an active tumor suppressor presented an opportunity to interrogate genes downstream of p53 involved in tumor suppression in a focused manner [4]. Gene expression analysis identified 87 genes activated in both oncogene-expressing p53+/+ and p5325,26 murine embryonic fibroblasts (MEFs) [7]. In an unbiased approach to probe tumor suppressor activity in vivo, oncogene-expressing MEFs expressing shRNA and sgRNA libraries targeting these 87 genes were transplanted subcutaneously into mice to seed tumors. Enrichment of sh/sgRNA elements was sought in the resulting tumors to identify tumor suppressors. The top hit identified by both the shRNA and sgRNA screens was the p53 target Zmat3, which encodes an RNA-binding protein, suggesting that Zmat3 is a potent tumor suppressor. The shRNA screen also confirmed the tumor suppressor status of Ptpn14, a p53-inducible gene recently linked with suppression of PDAC as an inhibitor of the Yap oncogene [6]. Follow-up studies confirmed Zmat3 tumor suppressor function in Kras-driven LUAD and hepatocellular carcinoma (HCC) GEMMs [7]. Combined CLIP-seq and RNA-seq analyses revealed that Zmat3 regulates an alternative splicing program by binding RNAs upstream of 3’ splice sites of specific introns and regulating exon skipping, leading to changes in the transcriptome. These findings, along with a recent study that found that ZMAT3 inhibits clonogenicity of cancer cells by controlling the splicing of CD44, suggest a link between p53-mediated tumor suppression and splicing [7,8]. Zmat3 deficiency did not promote tumor growth to the extent of p53 loss, suggesting that Zmat3 is one of multiple tumor suppression effectors downstream of p53. The significance of ZMAT3 to cancer suppression in humans is supported by recent meta-analyses of CRISPR/Cas9 and shRNA screens in human cell lines, which revealed that ZMAT3 depletion significantly promotes proliferation in cells with functional p53 but not with p53-deficiency[7,9]. ZMAT3 and p21 were the two p53 target gene effectors to reach the threshold of significant enrichment in wild-type versus p53-deficient cells set in this “Cancer Dependency Map” (DepMap) meta-analysis, suggesting a centrality of these two target genes to the p53 tumor suppression network [9].
p53 ensures multiple aspects of genomic integrity
The surprising lack of spontaneous tumorigenesis in p21−/−;Puma−/−;Noxa−/− mice spurred efforts to identify p53 target genes that might play redundant or cooperative roles in tumor suppression. Toward this end, functional genetic screens using an shRNA library directed against 166 p53 target genes were conducted in mouse leukemia/lymphoma models in vivo [10]. These screens were performed in sensitized backgrounds, using Eμ-Myc;Puma−/− - or p21−/−;Puma−/− hematopoietic stem/progenitor cells (HSPCs), to better reveal genes that may be involved in tumor suppression [10]. These screens implicated DNA repair genes, most notably Mlh1, as important for suppression of leukemia/lymphoma in vivo, while also confirming Zmat3 as a tumor suppressor in the context of Puma and p21 deletion in HSPCs [10]. While the p53 network is known to comprise target genes involved in various types of DNA repair such as nucleotide-excision repair, base-excision repair, and mismatch repair [11] and activation of DNA repair is a feature of p53 conserved evolutionarily at least through its Caenorhabditis elegans ortholog, cep-1 [12], this study brings renewed attention to p53 activation of DNA repair target genes as being critical for tumor suppression. These findings echoed the observation that p21−/−;Puma−/−;Noxa−/− mice maintain DNA damage repair, suggesting that repair function could compensate for the lack of the classical p53 effectors [13]. Moreover, a role for DNA repair in the absence of p21 suggests that DNA repair need not be coupled to proliferative pause or that other p53 target gene products can dampen proliferation to allow for DNA repair. Further analysis of these target genes and p53-mediated DNA repair in non-hematopoietic tissues will be important for addressing how broadly this p53 function promotes tumor suppression.
The spotlight on DNA repair situates alongside a historic focus of p53 activity in maintaining genomic stability. Genomic instability is one of the hallmarks of cancer kept at bay by p53, and, in the form of aneuploidy, is one of the most striking characteristics of cells derived from p53−/− mice [14]. In vivo studies have sought to establish whether genomic instability contributes to tumorigenesis in the wake of p53 loss or is merely a side effect. p53R172P mice – which retain full genomic stability and partial cell cycle arrest in the absence of apoptotic function – displayed significantly delayed development of spontaneous thymic lymphoma compared to p53−/− mice, suggesting that maintenance of genomic stability could contribute to suppression of early onset lymphomagenesis [15]. Cells from p53R172P;p21−/− mice displayed a total loss of cell cycle control, accompanied by genomic instability, and mice exhibited significantly shorter lymphoma latency, suggesting that p21-coupled cell cycle control and preservation of chromosomal stability play a role in tumor suppression [16]. However, p53R172P;p21−/− mice still displayed significantly prolonged survival compared to p53−/− mice, suggesting that some residual p53 function other than apoptosis, cell cycle arrest, or genomic stability is at play. Two other separation-of-function (see Glossary) p53 mutants, p53ΔP, which lacks the proline-rich domain, and the acetylation mutant p533KR, failed to inhibit chromosomal instability but did not show a commensurate increase in spontaneous tumorigenesis, suggesting that loss of genomic stability is not sufficient for tumorigenesis [17,18]. Thus, studies of p53R172P;p21−/− mice suggest that preserving genomic stability can contribute to p53-mediated tumor suppression, but that its importance is context-dependent.
Collectively, various studies provide insight into the role of p21 in tumor suppression [16]. While p21 deletion alone does not sensitize mice to spontaneous tumorigenesis, even with combined loss of Puma and Noxa, p21 deletion in p53R172P mice reveals a situational role for p21 contributing to suppression of spontaneous tumorigenesis [13,16]. Furthermore, deletion of p21 with Puma in HSPCs reveals a role for p21 in suppressing spontaneous leukemia/lymphoma, and the aforementioned DepMap meta-analyses of human cancer suggest the importance of p21 in p53-mediated proliferation suppression [9,10]. Thus, in some settings, p21 can contribute to tumor suppression. The precise contexts in which it plays a role likely relates to where there may be redundancy between it and other p53 target genes.
p53 regulation of metabolic homeostasis cuts both ways in cancer
Another line of investigation has focused on understanding how the p533KR acetylation mutant, which is defective for acute DNA damage-induced apoptosis, cell cycle arrest, and senescence, can suppress tumorigenesis. This work built on the known role for p53 in dampening the Warburg effect, a shift from oxidative phosphorylation to glycolysis triggered in cancer cells as a strategy to balance increased anabolic needs and energy generation [19]. p53 opposes the Warburg effect by activating target genes involved in oxidative phosphorylation, such as GLS2 and SCO2, and by decreasing the expression of glucose importers, like GLUT1 and GLUT4, that fuel glycolysis [20]. Like wild-type p53, p533KR can inhibit both glucose uptake and glycolysis, suggesting that regulation of metabolism might contribute to the p53 tumor suppression program [21]. Mice expressing the polymorphic variant p53S47 have further supported a role for p53 regulating metabolism in tumor suppression. Like p53−/− mice, p53S47/S47 mice developed spontaneous tumors, albeit primarily HCCs, distinguishing them from p53−/− mice [21]. Cells from p53S47/S47 mice exhibited increased glycolysis and decreased oxygen consumption compared to p53+/+ counterparts, further correlating metabolic regulation and p53-mediated tumor suppression [22,23].
More recent studies have shed further light on mechanisms by which p53 transcriptionally regulates metabolism. Taking advantage of the sometimes reciprocal relationship between the effects of wild-type and mutant p53 on cellular phenotype, one recent study expanded on the observation that mutant p53 upregulates the mevalonate pathway, reporting that wild-type p53 represses the mevalonate pathway by activation of the Abca1 cholesterol transporter gene [24]. In a Myc-driven HCC GEMM, Abca1 displayed tumor suppressor function, linked to its suppression of cholesterol and nonsterol isoprenoids synthesis. p53 deficiency significantly decreased HCC-free survival compared to Abca1 deficiency, suggesting that Abca1 must cooperate with other genes in HCC suppression. In another study aimed at understanding whether the capacity of p53 to regulate metabolism constrains PDAC, metabolomic analysis of cells derived from a PDAC GEMM revealed that p53 reactivation led to an elevated alpha-ketoglutarate (αKG)/succinate ratio – a phenomenon associated with cell fate decisions in some contexts [25], p53-dependent αKG accumulation, through the proposed p53 targets Pcx and Idh1, increased levels of the chromatin modification 5-hydroxymethylcytosine [25]. This epigenetic shift was mediated by the αKG-dependent Tet enzyme family, which is associated with tumor cell differentiation and decreased tumor cell fitness [25]. Restoration of αKG accumulation to p53 null orthotopic tumors in mice resulted in cellular differentiation and tumor suppression similar to p53 restoration, suggesting a role for p53-mediated accumulation of αKG in PDAC suppression. These findings add to the existing body of literature on p53 regulation of metabolism in tumor suppression.
In addition to opposing tumorigenic metabolism, p53 can promote cellular survival under some conditions of nutrient stress [19,26]. p53 target genes implicated in survival pathways include SLC1A3 and SLC7A3, encoding amino acid transporters involved in survival upon glutamine starvation, as well as the acyl-CoA dehydrogenase-encoding Acad11 and the non-coding RNA TRINGS, which permit survival in the face of glucose deprivation [27–30]. The p53 tumor-derived mutant, p53R248W, which is associated with particularly poor patient outcomes, selectively retains the ability of wild-type p53 to survive combined serine and glycine starvation, hinting at a tumor-supporting role for aspects of wild-type p53 function [31]. The p53 target gene Tigar can support the formation of mouse intestinal adenomas by enhancing nucleotide synthesis as well as supporting NADPH production [32,33]. It remains to be determined exactly when a p53 response to metabolic stress is tumor-supporting versus tumor-suppressive (Box 3), although the duration and severity of p53 activation is speculated to have a role, with more severe stress favoring cell death and tumor suppression [26].
Box 3. Is p53 always a tumor suppressor?
p53 seems capable of both negatively and positively regulating many aspects of biology, including anabolic pathways and ROS accumulation. Though p53 has clearly proven itself as important for tumor suppression, it is becoming clearer that p53, as an important hub in cellular stress responses, has some homeostatic functions that can benefit cellular fitness and promote tumor growth. Such a fact should not be surprising given the pleiotropic effects of p53. This observation could explain why p53 loss is typically observed as a late stage hit during tumor evolution of certain cancer types, including carcinomas of the colon, lung, and pancreas [83]. While p53−/− mice have been shown in the DMBA/TPA-induced model of skin cancer to develop more advanced carcinomas than p53+/+ and p53−/− mice, they actually develop fewer benign papillomas compared to p53-proficient mice, suggesting distinct effects of p53 loss depending on tumor stage [84]. If loss of p53 not only has pleiotropic effects that permit tumorigenesis but also has some effects that limit cell survival, the fitness advantage of losing p53 might be dictated by context. Perhaps, in some tissue types, additional mutations or perturbations that overcome the need for p53 to quench oxidative stress or adapt to low-nutrient microenvironments are prerequisites to shifting the effect of losing p53 from negative to positive.
Ferroptosis: a new form of cell death regulated by p53
Further analysis of p533KR revealed that p53 can promote a non-apoptotic, iron-dependent, oxidative form of cell death called ferroptosis. p533KR can promote ferroptosis and suppress cancer, while p534KR, with one additional mutation, loses the ability to support the induction of ferroptosis and suppress xenograft tumor growth, correlating ferroptosis activation with tumor suppression [34]. Similarly, p53S47 lacks both the ability to sensitize cells to ferroptosis and suppress spontaneous tumors in mice, in contrast to the tumor suppression-proficient and ferroptosis-supporting p53P47 polymorphic variant [22]. Various mechanisms to explain how p53 sets the rheostat for ferroptosis sensitivity have been proposed, including reduction of cystine import by direct transcriptional repression of the gene encoding the cystine-glutamate antiporter SLC7A11 as well as transcriptional induction of the genes encoding the metabolic enzymes SAT1, PANK1-3, and GLS2 [22,35–37]. Accordingly, in mouse xenograft models with human cancer cell lines, overexpression of SLC7A11 promotes tumor growth, ostensibly by limiting ferroptosis, while overexpression of SAT1 limits tumor growth [35,36]. However, other studies in cell lines have reported that p53 either suppresses ferroptosis [38,39] or has no effect on the induction of ferroptosis [40]. Disparities between these reports could result from differences in ferroptosis-activating stresses or cell lines used, with expression of the lipoxygenase ALOX12 suggested as a key determinant of the ability of p53 to support ferroptosis in vitro [41]. In the Eμ-Myc-driven B-cell lymphoma mouse model, Alox12 heterozygosity significantly decreased survival and tumors displayed lower levels of the ferroptosis markers Ptgs2 and Chac1 compared to Alox12+/+ mice [41]. Nonetheless, these mice did not die as rapidly as Eμ-Myc;p53+/− mice did, suggesting that activation of ferroptosis is part of a set of processes involved in p53-mediated tumor suppression [41]. Further study is required to answer questions regarding how and when ferroptosis might be triggered during tumorigenesis in physiological settings and in what contexts p53 supports or opposes this non-canonical form of cell death.
Although these recent studies on ferroptosis suggest how oxidative stress can trigger cell death to inhibit tumorigenesis, earlier studies of the relationship between p53 and redox biology focused on the ability of p53 to suppress reactive oxygen species (ROS), which in turn could limit DNA damage and accrual of mutations. In support of an important role for the antioxidant function of p53 in tumor suppression, treatment with the antioxidant N-acetylcysteine significantly impaired spontaneous tumor development in p53−/− mice [42]. p53 can activate a variety of antioxidant target genes, including ALDH4, TP53INP1, TIGAR, and SESN1-3, which limit ROS accumulation and ROS-induced DNA damage [20]. Interrogation of individual antioxidant p53 target genes in in vivo models of tumorigenesis, however, has yielded mixed results. While Trp53inp1 deletion enhances Kras-driven PDAC, suggestive of tumor suppressor function [43], Tigar ablation inhibits the development of intestinal adenomas in mice, suggesting a tumor-supporting role [33]. p53 can dynamically regulate the accumulation of ROS in vitro, initially suppressing and then promoting ROS accumulation after prolonged p53 stabilization, suggesting that p53 might switch between antioxidant function and promotion of ferroptosis [44]. Elucidating the unsolved mechanisms behind this switch between seemingly contradictory activation of both anti- and pro-oxidant p53 transcriptional programs (Box 3) promises to add to our understanding of how p53 regulates carcinogenesis.
p53 inhibits cellular self-renewal and plasticity
Recent in vivo studies of lung homeostasis in GEMMs have provided insight into p53 functions likely involved in tumor suppression. In one study, conditional p53 knockout in club cells of adult mouse lungs revealed a role for p53 in maintaining quiescence in club cells and limiting epithelial cell density in the lungs [45]. This p53 homeostatic function in club cells was attributed in part to the target gene p21. More support for a role for p53 in limiting self-renewal has come from a study of conditional p53 and Rb ablation from neuroendocrine cells in the lung [46]. p53, along with Rb, was found to be part of a program that suppresses self-renewal of neuroendocrine stem cells, thought to be the cell of origin for small cell lung cancer, following naphthalene injury. These studies support previous findings highlighting a role for p53 in dampening self-renewal in neural, mammary, and hematopoietic stem/progenitor cells [47]. Clinical studies correlating wild-type p53 status with the suppression of a stem cell-like transcriptional signature in breast, lung, prostate, and liver cancer patient datasets further support a role for p53 in regulating sternness [48–50].
The role for p53 in limiting self-renewal is only part of a growing understanding of p53 function in stem cell biology, which also comprises restricting cellular plasticity through impeding reprogramming and promoting differentiation. A recent single cell RNA-seq study comparing Kras-driven LUAD with wild-type or p53 null status at various stages of tumorigenesis revealed increased transcriptional heterogeneity with p53 deficiency, supporting a role for p53 in limiting cellular plasticity during tumor evolution [51]. This finding harkens back to studies that found p53 was a major barrier to cellular reprogramming in producing induced pluripotent stem cells, with p21 and mir34 implicated as downstream target genes important for limiting plasticity [52,53]. Roles for the p53 target genes p21, mir34a, and mir145 in promoting differentiation of human embryonic stem cells have also been revealed [54]. Neat1, a p53-inducible non-coding RNA, was also shown to be important for driving differentiation programs and suppressing transdifferentiation of acinar cells into ductal cells, then premalignant lesions, in response to KrasG12D expression in a PDAC GEMM [55]. Thus, the p53 network acts to reinforce the differentiated state.
p53 also inhibits another type of cellular reprogramming known as epithelial-to-mesenchymal transition (EMT). During EMT, the activation of embryonic signaling programs drives the remodeling of cell-cell adhesion complexes, resulting in enhanced motility and invasiveness that have been connected to metastasis. In vitro, p53 restricts the motility and invasiveness of many types of primary and cancer cells and inhibits the EMT transcription factors Snaill, Slug, and Zebl as well as RhoA/ROCK signaling [40,56–58]. In vivo, conditional knockout of p53 was associated with decreased E-cadherin expression, a hallmark of EMT, in a model of DMBA/TPA-induced skin cancer [59]. Studies in a Wnt-driven colorectal carcinoma GEMM revealed a gene set associated with invasion of epithelial cells into the lamina propria unleashed by p53 ablation [60]. This gene set, dubbed the “p53-suppressed invasiveness signature”, is downregulated by p53 and its target gene p21 independently of cell cycle arrest, supporting an in vivo role for p53 in controlling cellular invasion programs. Though not likely involved in primary tumor initiation, loss of p53-mediated suppression of EMT may contribute to tumor progression and metastasis.
Non-cell autonomous tumor suppression: an emerging p53 function
Although p53 clearly impacts various cell-autonomous processes, evidence continues to accumulate suggesting that p53 activity in tumor cells shapes various aspects of the tumor microenvironment (TME) and promotes tumor suppression in a non-cell autonomous fashion. p53 has been reported to exert transcriptional control over both secreted molecules themselves [61–64] and components of the intracellular transport machinery [65], though how p53 modulates the TME non-cell autonomously remains to be completely understood. Most recently, studies of a head and neck cancer GEMM revealed a non-cell autonomous role for p53 in suppressing the pro-tumorigenic transdifferentiation of tumor-associated sensory nerves into adrenergic neurons [61]. Specifically, Rab27-dependent secretion of exosomes containing miRNAs, such as the p53-inducible mir34a, suppresses transdifferentiation of sensory nerves in p53 wild-type tumors [61]. Denervation of xenograft tumors derived from p53 null head and neck cancer cells significantly diminished tumor growth compared to sham surgery, whereas this intervention had no effect on p53 wild-type tumors [61]. In addition to suppressing tumorigenic innervation, p53 also has a well-documented role in inhibiting angiogenesis by activating target genes, such as THBS1 and COL4A1, which lead to decreased tumor growth and enhanced tumor regression in mouse models [66,67].
p53 can also modulate the constitution and functionality of immune cells present in the TME in a non-cell autonomous fashion. Two recent studies have used GEMMs to demonstrate that p53 inhibits cytokine-mediated recruitment of immune cells that promote tumor growth [62,63]. Significant enrichment of immunosuppressive myeloid CD11b+ cells was observed in p53 null tumors relative to wild-type counterparts in two distinct GEMMS, including Kras-driven PDAC and EGFR-driven LUAD, suggesting a role of p53 in maintaining a tumor-suppressive immune response [62]. p53 deletion was found to promote expression of CXCR3/CCR2-associated chemokines and macrophage colony-stimulating factor, resulting in the recruitment of immunosuppressive cells that oppose CD4+ T helper 1 and CD8+ T cell responses, thus promoting tumor growth. This study echoes analysis of a Pten-deficient prostate cancer GEMM in which the presence of wild-type p53 resulted in decreased enrichment of immunosuppressive myeloid Gr-1+CD11b+ cells, pro-tumorigenic macrophages, and Treg cells in the TME compared to p53 null tumors [63]. This difference was attributed to lower expression of pro-tumorigenic chemokines in p53 wild-type tumors compared to p53 null. In another recent study comparing 16 breast cancer GEMMs, p53 suppressed pro-metastatic, neutrophilic inflammation by limiting expression and secretion of Wnt ligands, including WNT1, WNT6 and WNT7A [64]. In an allograft model of medulloblastoma, p53 was shown to be important for MHC-I presentation and recruitment of anti-tumor CD3+ and CD8+ T cells, attributed to activation of the p53 target genes Erap1 and Tap1, which encode proteins involved in peptide transport and processing respectively [65]. These more recent studies expand the non-cell autonomous effects of p53 on the immune system originally uncovered in HCC GEMMs in which p53 activated the senescence-associated secretory phenotype (SASP). The p53-driven SASP induces cytokine and adhesion molecule expression that promotes the presence of anti-tumor neutrophils, macrophages, and natural killer cells in the TME, thus facilitating tumor clearance [68–70]. Given the prominence and promise of immunotherapy and the high rate of p53 mutation in human cancer, studying the effects of p53 on tumor immunology will remain an area of enormous interest.
p53 concurrently modulates diverse cellular processes
While the individual studies reviewed here catalog the various effects of p53 on cellular processes in different settings, it has remained unclear whether p53 plays context-specific roles, regulating different cellular processes in distinct settings, or whether p53 truly regulates many functions in unison in a particular tumor suppression context. This question was recently addressed through assays of a spectrum of cell biological processes in oncogene-expressing primary fibroblasts expressing or lacking p53, a model system in which p53 is critical for inhibiting transformation [40]. This systematic study revealed dysregulation of a surprising number of cellular processes upon p53 loss. Experiments were performed under physiological oxygen tensions (5% compared to the usual 21% oxygen), and some of the p53-regulated phenotypes, such as invasion, were only uncovered in physiological oxygen. Together, the findings of this phenotypic cataloging support the idea that p53 can coordinately regulate many functions ranging from cell death to metabolism to actin arrangement simultaneously in one tumor-suppressive context (Fig. 2). While this comprehensive study also showed some pleiotropy in phenotypes triggered by p53 loss in human HCT116 colorectal cancer cells [40], it remains to be seen whether p53 loss triggers widespread changes in cellular processes in all contexts of p53 tumor suppression.
Figure 2. Two distinct models to explain p53-mediated tumor suppression.
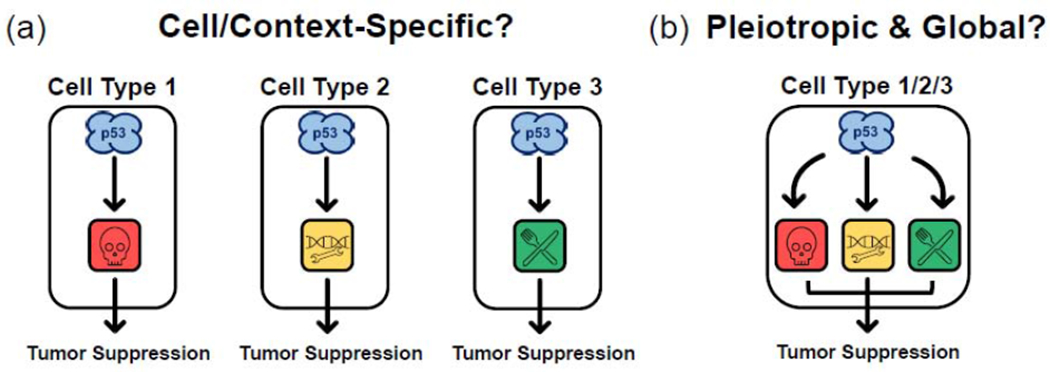
(a) A proposed model for p53-mediated tumor suppression where a cell- or context-specific subset of p53 function is critical. (b) A model attributing p53 tumor suppression to the pleiotropic activity of p53 in modulating many different cellular processes. New evidence detailing the pleiotropy of p53 in a single tumor-suppressive setting supports this model. A limited set of p53-dependent processes are illustrated here to provide an example, rather than to enumerate a conclusive list of important processes.
Accompanying this ability of p53 to regulate numerous cellular processes is its capacity to directly activate a network of hundreds of target genes encompassing effectors that participate in various biological functions (Fig. 3). Unbiased genetic screens and other in vivo approaches have identified specific target genes that are among the tumor suppressive components downstream of p53 yet do not completely recapitulate p53 deficiency when deleted [7,13,16,24,71]. Additional support for the idea that p53-mediated tumor suppression is distributed comes from the relative lack of mutations in p53 target genes in human cancers compared to mutations to TP53 itself [71]. Collectively, these findings support a model for p53-mediated tumor suppression that relies on collective and cooperative activation of the p53 target gene network.
Figure 3. The p53 transcriptional network.

The p53 transcriptional network contains target genes that regulate diverse cellular processes including apoptosis, cell cycle arrest (including senescence), DNA repair, metabolism, ferroptosis/redox levels, cellular differentiation/reprogramming, and regulators of non-cell autonomous functions that impact the tumor microenvironment (TME).
Concluding Remarks
More than 40 years and counting, the study of p53, one of the most important tumor suppressors and signaling hubs in our genome, continues to yield fascinating discoveries and raise intriguing questions (Outstanding Questions Box). Genetic screens and GEMMs have proved useful tools for revealing the importance of numerous p53-mediated target genes and biological functions to tumor suppression that comprise a cooperative tumor suppression network downstream of p53. Careful consideration should be given to which aspects of the p53 network are the most important for continued study. One line of thinking proposes the importance of studying the “core” p53 program [71], comprising the subset of p53 target genes activated by p53 across tissues and activating stresses. While offering a parsimonious answer to why p53 is a potent tumor suppressor in various cancer types, more experimental work is necessary to test the importance of the “core” program for tumor suppression. Another, non-mutually-exclusive approach is to identify the most important nodes in the p53 network by defining those p53 target genes evolutionarily conserved from mice to humans [72], which might explain the conservation of tumor suppression between the species. A recent meta-analysis of transcriptomic and ChIP-seq data sets identified 86 direct, upregulated p53 target genes shared between mouse and human cells [72]. These genes include classic p53 targets (e.g. p21), genes recently demonstrated to have roles in p53-mediated tumor suppression (e.g. Zmat3), and genes involved in diverse cellular processes regulated by p53. Studying p53 in mice comes with the caveat that a particular mechanism of tumor suppression might not be conserved, but focusing study on conserved targets is one way to address this limitation. Finally, offering a contrast to the “core” program model, the relative importance of different target genes downstream of p53 could vary based on context, such as the tissue of origin, mutational landscape, and tumor microenvironment. Indeed, a recent in vivo study revealed tissue-specific transcriptomes and phenotypes resulting from acute p53 activation by genetic ablation of Mdm2, underscoring the possibility that different mechanisms might contribute to p53-mediated tumor suppression in different contexts [73]. Different components of the p53 pathway might also have distinct roles in tumor suppression depending on whether p53 is acting to suppress spontaneous cancer initiation or cancer progression in the context of oncogenic drivers. For example, while Puma deletion does not promote spontaneous tumorigenesis, it has a clear role in the context of suppressing Eμ-Myc-driven lymphomagenesis [74]. Regardless of the approach to finding the most important p53 effectors, the historic focus on target genes activated by acute DNA damage in vitro in cancer cell lines leaves open the possibility that studies in more physiologically relevant settings could reveal new p53 targets. There is a clear need for continued analysis of p53 target genes and their functions if we are to understand the most frequently mutated gene in cancer.
Outstanding Questions Box.
Is there a “core program” of p53 target genes and cellular responses responsible for p53-mediated tumor suppression in a variety of contexts, including in different tissue types and in response to different oncogenic drivers or microenvironmental stresses? Are there also tissue or context-specific tumor suppression programs reflecting the inherent biology of specific tissues, oncogenic drivers, and microenvironmental conditions?
How broadly does p53 exert pleiotropic effects, in which it concurrently governs many diverse cellular processes, in different tumor suppression contexts?
Which p53-activating stress signals are most salient to tumor suppression in vivo?
Will continued study of p53 in vivo in tissue contexts or analysis of understudied p53-activating stresses such as hypoxia and metabolic stress reveal new target genes and cellular programs that contribute to tumor suppression?
How can understanding of p53 target genes and the p53 tumor suppression network be applied to improve cancer therapy?
How can we improve cell culture systems and mouse models to more accurately identify p53 responses most relevant to human cancer?
What are the relative contributions of loss of p53 wild-type function – including both transactivation-dependent and -independent activities – and oncogenic gain-of-function p53 activities to cancer development? How can we test this question in vivo? Does the answer depend on context?
Upon defining p53 target genes contributing to tumor suppression, it will be important to understand if and how these members of the p53 network interact with or compensate for each other. While some pathways affecting different cellular processes would be expected to cooperate in tumor suppression, some p53 functions could be interchangeable and therefore redundant. For example, perhaps the different forms of p53-mediated cell death – ferroptosis, apoptosis, and immune cell-mediated cell death – are somewhat interchangeable in tumor suppression. Redundancy could also be built into the p53 network through multiple target genes regulating the same process. While initial efforts to deconstruct the p53 tumor suppression network have relied on analysis of cooperation between classical effectors like p21, Puma, and Noxa, illuminating the full complement of pathways downstream of p53 will highlight new avenues for deepening our understanding of the intricacies of p53-mediated tumor suppression.
While experimental tools, such as GEMMs and genetic screens, coupled with patient-derived xenograft models and human cancer genome analyses, are certain to reveal further insights into p53-mediated tumor suppression, we should be mindful of how to translate this knowledge to the clinic. Our current understanding of the p53 transcriptional network underscores how this knowledge can be leveraged in cancer therapy by exposing potential vulnerabilities of p53-deficient cancers. For example, identification of the p53 target gene Ptpn14 as an inhibitor of oncogenic Yap signaling and a critical PDAC suppressor revealed that p53-deficient cells are susceptible to verteporfin, a Yap inhibitor [6]. Elucidation of p53 target gene Abca1 as an HCC suppressor through effects on the mevalonate pathway identified atorvastatin as a therapy for p53-deficient murine HCC [24]. The importance of pleiotropy and breadth of the p53 target gene network imply that p53-inspired combinatorial treatments will prove most fruitful for clinical investigation. Given the lack of targeted therapies for p53-deficient cancers and the challenges of restoring p53 to human tumors, harnessing our knowledge of the p53 transcriptional network to identify therapeutic targets remains an area rich for future investigation.
Highlights.
In vivo shRNA and CRISPR/Cas9 screens identify p53 target genes such as Zmat3 and Mlh1 as important for tumor suppression.
Emerging p53 functions potentially important for tumor suppression include activation of DNA damage repair, opposition to metabolic rewiring, stimulation of ferroptosis, inhibition of cellular plasticity, and maintenance of an anti-cancer immune tumor microenvironment.
Through modulation of metabolism and redox biology, p53 can promote cell survival and homeostasis in addition to promoting cell death, raising the possibility that, in some contexts, p53 might support tumor growth.
p53 can concurrently regulate many cellular processes in a given biological setting, suggesting that tumor suppression relies on coordinated effects on cellular biology.
New evidence suggests that p53 suppresses tumorigenesis by modulating a target gene network comprising cooperative and redundant effectors.
Acknowledgements
We thank Elizabeth Valente, Kathryn Hanson, and Edel McCrea for critical reading of the manuscript. We apologize to those whose work we could not cite due to spatial constraints. The authors declare no competing interests.
Glossary
- Club cells
secretory cells found in the bronchioles that can self-renew and differentiate into multiple epithelial cell types.
- Epithelial-to-mesenchymal transition (EMT)
a process by which epithelial cells lose epithelial characteristics, such as intercellular adhesion, and gain mesenchymal characteristics such as increased motility and invasiveness. EMT is thought to play a role in metastasis.
- Ferroptosis
a non-apoptotic, iron-dependent form of programmed cell death characterized by the accumulation of lipid peroxidases.
- Pleiotropy
an effect that occurs when a gene influences two or more distinct phenotypes.
- Senescence-associated secretory phenotype (SASP)
an increase in inflammatory cytokine, growth factor, and protease release accompanying the gene expression changes associated with senescence, a cell fate defined by permanent cell cycle exit.
- Separation-of-function
describes a mutation that impairs some but not all gene functions. In reference to p53, separation-of-function mutations yield impaired activation of some target genes but wild-type level activation of others. Separation-of-function mutants have been important for testing the necessity and sufficiency of different p53-regulated target genes and cellular processes to tumor suppression.
- Tumor microenvironment (TME)
the ecosystem surrounding a tumor, including vasculature, immune cells, fibroblasts, signaling molecules, and extracellular matrix.
- Warburg effect
a metabolic shift from oxidative phosphorylation to glycolysis observed in many cancer cells. Despite the presence of oxygen and the inefficiency of glycolysis in ATP production relative to oxidative phosphorylation, this glycolytic shift helps meet the increased anabolic needs of cancer cells.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kandoth C et al. (2013) Mutational landscape and significance across 12 major cancer types. Nature 502, 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaiser AM and Attardi LD (2018) Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death and Differentiation 25, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y and Lozano G (2017) p53: Multiple Facets of a Rubik’s Cube. Annu Rev Cancer Biol 1, 185–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brady CA et al. (2011) Distinct p53 Transcriptional Programs Dictate Acute DNA Damage Responses and Tumor Suppression. Cell 145, 571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang D et al. (2011) Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proc. Natl. Acad. Sci. U.S.A 108, 17123–17128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mello SS et al. (2017) A p53 Super-tumor Suppressor Reveals a Tumor Suppressive p53-Ptpn14-Yap Axis in Pancreatic Cancer. Cancer Cell 32, 460–473. e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bieging-Rolett KT et al. (2020) Zmat3 Is a Key Splicing Regulator in the p53 Tumor Suppression Program. Mol Cell 80, 452–469. e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muys BR et al. (2020) The p53-induced RNA-binding protein ZMAT3 is a splicing regulator that inhibits the splicing of oncogenic CD44 variants in colorectal carcinoma. Genes Dev DOI: 10.1101/gad.342634.120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giacomelli AO et al. (2018) Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet 50, 1381–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janic A et al. (2018) DNA repair processes are critical mediators of p53-dependent tumor suppression. Nature Medicine 24, 947–953 [DOI] [PubMed] [Google Scholar]
- 11.Sengupta S and Harris CC (2005) p53: traffic cop at the crossroads of DNA repair and recombination. Nature Reviews Molecular Cell Biology 6, 44–55 [DOI] [PubMed] [Google Scholar]
- 12.Hoffman S et al. (2014) C. elegans CEP-1/p53 and BEC-1 Are Involved in DNA Repair. PLoS One 9, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valente LJ et al. (2013) p53 efficiently suppresses tumor development in the complete absence of its cell-cycle inhibitory and proapoptotic effectors p21, Puma, and Noxa. Cell Rep 3, 1339–1345 [DOI] [PubMed] [Google Scholar]
- 14.Eischen CM (2016) Genome Stability Requires p53. Cold Spring Harb Perspect Med 6, a026096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu G et al. (2004) Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nature Genetics 36, 63–68 [DOI] [PubMed] [Google Scholar]
- 16.Barboza JA et al. (2006) p21 delays tumor onset by preservation of chromosomal stability. Proc Natl Acad Sci U S A 103, 19842–19847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adams CJ et al. (2016) The Trp53 delta proline (Trp53ΔP) mouse exhibits increased genome instability and susceptibility to radiation-induced, but not spontaneous, tumor development. Mol Carcinog 55, 1387–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li T et al. (2016) Loss of p53-mediated cell-cycle arrest, senescence and apoptosis promotes genomic instability and premature aging. Oncotarget 7, 11838–11849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruiswijk F et al. (2015) p53 in survival, death and metabolic health: a lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol 16, 393–405 [DOI] [PubMed] [Google Scholar]
- 20.Lacroix M et al. (2020) Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Molecular Metabolism 33, 2–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li T et al. (2012) Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149, 1269–1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jennis M et al. (2016) An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 30, 918–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barnoud T et al. (2019) Tumor cells containing the African-Centric S47 variant of TP53 show increased Warburg metabolism. Oncotarget 10, 1217–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moon S-H et al. (2019) p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 176, 564–580. e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris JP et al. (2019) α-Ketoglutarate links p53 to cell fate during tumour suppression. Nature 573, 595–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Humpton T and Vousden KH (2019) Taking up the reins of power: metabolic functions of p53. J Mol Cell Biol 11, 610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tajan M et al. (2018) A Role for p53 in the Adaptation to Glutamine Starvation through the Expression of SLC1A3. Cell Metab 28, 721–736. e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lowman XH et al. (2019) p53 Promotes Cancer Cell Adaptation to Glutamine Deprivation by Upregulating Slc7a3 to Increase Arginine Uptake. Cell Rep 26, 3051–3060. e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jiang D et al. (2015) Analysis of p53 transactivation domain mutants reveals Acad11 as a metabolic target important for p53 pro-survival function. Cell Rep 10, 1096–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Khan MR et al. (2017) The p53-inducible long noncoding RNA TRINGS protects cancer cells from necrosis under glucose starvation. EMBO J 36, 3483–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humpton TJ et al. (2018) p53-mediated adaptation to serine starvation is retained by a common tumour-derived mutant. Cancer Metab 6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bensaad K et al. (2006) TIGAR, a p53-Inducible Regulator of Glycolysis and Apoptosis. Cell 126, 107–120 [DOI] [PubMed] [Google Scholar]
- 33.Cheung EC et al. (2013) TIGAR Is Required for Efficient Intestinal Regeneration and Tumorigenesis. Dev Cell 25, 463–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S-J et al. (2016) Acetylation Is Crucial for p53-mediated Ferroptosis and Tumor Suppression. Cell Rep 17, 366–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang L et al. (2015) Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ou Y et al. (2016) Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc Natl Acad Sci U S A 113, E6806–E6812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leu JI-J et al. (2019) Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc Natl Acad Sci U S A 116, 8390–8396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie Y et al. (2017) The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Reports 20, 1692–1704 [DOI] [PubMed] [Google Scholar]
- 39.Tarangelo A et al. (2018) p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep 22, 569–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valente LJ et al. (2020) p53 deficiency triggers dysregulation of diverse cellular processes in physiological oxygen. J Cell Biol 219, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chu B et al. (2019) ALOX12 is required for p53-mediated tumor suppression through a distinct ferroptosis pathway. Nat Cell Biol 21, 579–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sablina AA et al. (2005) The antioxidant function of the p53 tumor suppressor. Nat Med 11, 1306–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al Saati T et al. (2013) Oxidative stress induced by inactivation of TP53INP1 cooperates with KrasG12D to initiate and promote pancreatic carcinogenesis in the murine pancreas. Am. J. Pathol 182, 1996–2004 [DOI] [PubMed] [Google Scholar]
- 44.Jiang L et al. (2015) Dynamic roles of p53-mediated metabolic activities in ROS-induced stress responses. Cell Cycle 14, 2881–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McConnell AM et al. (2016) p53 Regulates Progenitor Cell Quiescence and Differentiation in the Airway. Cell Rep 17, 2173–2182 [DOI] [PubMed] [Google Scholar]
- 46.Ouadah Y et al. (2019) Rare Pulmonary Neuroendocrine Cells Are Stem Cells Regulated by Rb, p53, and Notch. Cell 179, 403–416. e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spike BT and Wahl GM (2011) p53, Stem Cells, and Reprogramming: Tumor Suppression beyond Guarding the Genome. Genes Cancer 2, 404–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mizuno H et al. (2010) Inactivation of p53 in breast cancers correlates with stem cell transcriptional signatures. Proc. Natl. Acad. Sci. U.S.A 107, 22745–22750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Markert EK et al. (2011) Molecular classification of prostate cancer using curated expression signatures. Proc Natl Acad Sci U S A 108, 21276–21281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wheeler DA and Roberts LR (2017) Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 169, 1327–1341. e23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marjanovic ND et al. (2020) Emergence of a High-Plasticity Cell State during Lung Cancer Evolution. Cancer Cell 38, 229–246. e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krizhanovsky V and Lowe SW (2009) The promises and perils of p53. Nature 460, 1085–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi YJ et al. (2011) miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nat. Cell Biol 13, 1353–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jain AK et al. (2012) p53 Regulates Cell Cycle and MicroRNAs to Promote Differentiation of Human Embryonic Stem Cells. PLoS Biol 10, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mello SS et al. (2017) Neat1 is a p53-inducible lincRNA essential for transformation suppression. Genes Dev 31, 1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim NH et al. (2011) A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol 195, 417–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang C-J et al. (2011) p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat. Cell Biol 13, 317–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang S-P et al. (2009) p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nat. Cell Biol 11, 694–704 [DOI] [PubMed] [Google Scholar]
- 59.Page A et al. (2016) Protective role of p53 in skin cancer: Carcinogenesis studies in mice lacking epidermal p53. Oncotarget 7, 20902–20918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elyada E et al. (2011) CKIα ablation highlights a critical role for p53 in invasiveness control. Nature 470, 409–413 [DOI] [PubMed] [Google Scholar]
- 61.Amit M et al. (2020) Loss of p53 drives neuron reprogramming in head and neck cancer. Nature 578, 449–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blagih J et al. (2020) Cancer-Specific Loss of p53 Leads to a Modulation of Myeloid and T Cell Responses. Cell Rep 30, 481–496. e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bezzi M et al. (2018) Diverse genetic-driven immune landscapes dictate tumor progression through distinct mechanisms. Nature Medicine 24, 165–175 [DOI] [PubMed] [Google Scholar]
- 64.Wellenstein MD et al. (2019) Loss of p53 triggers Wnt-dependent systemic inflammation to drive breast cancer metastasis. Nature 572, 538–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garancher A et al. (2020) Tumor necrosis factor overcomes immune evasion in p53-mutant medulloblastoma. Nature Neuroscience 23, 842–853 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 66.Teodoro JG et al. (2007) Inhibition of tumor angiogenesis by p53: a new role for the guardian of the genome. J. Mol. Med 85, 1175–1186 [DOI] [PubMed] [Google Scholar]
- 67.Assadian S et al. (2012) p53 Inhibits Angiogenesis by Inducing the Production of Arresten. Cancer Res 72, 1270–1279 [DOI] [PubMed] [Google Scholar]
- 68.Xue W et al. (2007) Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lujambio A et al. (2013) Non-cell-autonomous tumor suppression by p53. Cell 153, 449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iannello A et al. (2013) p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J. Exp. Med 210, 2057–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Andrysik Z et al. (2017) Identification of a core TP53 transcriptional program with highly distributed tumor suppressive activity. Genome Res. 27, 1645–1657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fischer M (2019) Conservation and divergence of the p53 gene regulatory network between mice and humans. Oncogene 38, 4095–4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moyer SM et al. (2020) p53 drives a transcriptional program that elicits a non-cell-autonomous response and alters cell state in vivo. Proc Natl Acad Sci U S A 117, 23663–23673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aubrey BJ et al. (2018) How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 25, 104–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ho T et al. (2019) How the Other Half Lives: What p53 Does When It Is Not Being a Transcription Factor. Int J Mol Sci 21, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jiang P et al. (2011) p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol 13, 310–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kadosh E et al. (2020) The gut microbiome switches mutant p53 from tumour-suppressive to oncogenic. Nature 586, 133–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Timofeev O et al. (2019) Residual apoptotic activity of a tumorigenic p53 mutant improves cancer therapy responses. EMBO J 38, e102096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.el-Deiry WS et al. (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 [DOI] [PubMed] [Google Scholar]
- 80.Christophorou MA et al. (2006) The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 443, 214–217 [DOI] [PubMed] [Google Scholar]
- 81.Hinkal G et al. (2009) Timed Somatic Deletion of p53 in Mice Reveals Age-Associated Differences in Tumor Progression. PLOS ONE 4, e6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Valente LJ et al. (2016) Combined loss of PUMA and p21 accelerates c-MYC-driven lymphoma development considerably less than loss of one allele of p53. Oncogene 35, 3866–3871 [DOI] [PubMed] [Google Scholar]
- 83.Levine AJ et al. (2019) The Roles of Initiating Truncal Mutations in Human Cancers: The Order of Mutations and Tumor Cell Type Matters. Cancer Cell 35, 10–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kemp CJ et al. (1993) Reduction of p53 gene dosage does not increase initiation or promotion but enhances malignant progression of chemically induced skin tumors. Cell 74, 813–822 [DOI] [PubMed] [Google Scholar]