

SUMMARY
Immunomodulatory drugs can inhibit bacterial growth, yet their mechanism of action, spectrum, and clinical relevance remain unknown. Methotrexate (MTX), a first-line rheumatoid arthritis (RA) treatment, inhibits mammalian dihydrofolate reductase (DHFR) but whether it directly impacts gut bacteria is unclear. We show that MTX broadly alters the human gut microbiota. Drug sensitivity varied across strains but the mechanism of action against DHFR appears conserved between mammalian and bacterial cells. RA patient microbiotas were sensitive to MTX and changes in gut bacterial taxa and gene family abundance were distinct between responders and non-responders. Transplantation of post-treatment samples into germ-free mice given an inflammatory trigger led to reduced immune activation relative to pre-treatment controls, enabling identification of MTX-modulated bacterial taxa associated with intestinal and splenic immune cells. Thus, conservation in cellular pathways across domains of life can result in broad off-target drug effects on the human gut microbiota with consequences for immune function.
Keywords: rheumatoid arthritis, human gut microbiome, off-target effects, purine and pyrimidine biosynthesis, immune activation, methotrexate
Graphical Abstract
eTOC Blurb
Nayak et al show that methotrexate, an anti-inflammatory drug designed to target human cells, has off-target effects on the growth, transcription, and metabolic activity of diverse human gut bacteria with downstream consequences for host immunity.
INTRODUCTION
Despite >60 years of use, the mechanism of action of MTX in rheumatic diseases remains unclear. At the higher doses used to treat cancer (Treon and Chabner, 1996) MTX inhibits the mammalian enzyme dihydrofolate reductase (DHFR) (Bleyer, 1978), necessary to convert folic acid into tetrahydrofolate (THF) and enable the production of purines and pyrimidines for cell cycle progression (Bleyer, 1978). The lower doses used in RA coupled to the frequent co-administration of folic acid (Cronstein, 1996) implies that MTX acts via other mechanisms in RA patients. A leading hypothesis is that MTX inhibits 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) transformylase, resulting in an increase in extracellular levels of the anti-inflammatory mediator adenosine (Cronstein, 1996; Cronstein and Sitkovsky, 2017). Alternatively, MTX may decrease the proinflammatory cytokine TNFα (Gerards et al., 2003) or increase the anti-inflammatory cytokine IL-10 (Seitz et al., 2001), perhaps by affecting DHFR in lymphocytes. Yet the potential for MTX to act by inhibiting gut bacterial growth remains unexplored, despite the broad impacts of the gut microbiome on both intestinal and systemic immunity (Atarashi et al., 2013; Hooper et al., 2012) and emerging evidence that the microbiome is associated with RA (Chen et al., 2016; Pianta et al., 2017; Scher et al., 2013; Zhang et al., 2015).
The major target of MTX, DHFR, is conserved across all domains of life. MTX can directly bind DHFR from multiple bacteria (Bolin et al., 1982) and DHFR overexpression rescues the growth of a sensitive E. coli strain (Kopytek et al., 2000). More recently, a high-throughput screen of human gut bacterial isolates identified multiple MTX sensitive strains (Maier et al., 2018), experiments in rats and mice revealed shifts in the gut microbiota in response to intraperitoneal MTX (Letertre et al., 2020; Zhou et al., 2018), and clinical studies suggest that some of the differences between the microbiomes of RA patients relative to healthy controls (Scher et al., 2013) are reversed during treatment (Zhang et al., 2015). However, it remains unclear whether the observed changes to the gut microbiota are a direct result of MTX or reflective of concomitant treatment with other medications and dietary supplements, improvement of the disease process, reduction of inflammation, and/or other confounding factors that may be present in observational studies. Additional gaps in knowledge include the relevance of MTX to complex human gut microbial communities and RA patients; the robustness of MTX-induced changes in the gut microbiota to different doses, delivery routes, and co-therapies; the effects of MTX on the transcriptome and metabolome of bacteria; and the downstream consequences for host immunological phenotypes.
Here, we begin to address these questions by employing metagenomics, gnotobiotic mice, culturing of isolates and complex communities, transcriptomics, metabolomics, and longitudinal analysis of human patients. These data provide an in-depth view of the impact of MTX on the structure and function of the human gut microbiome. Our results demonstrate that drugs intended to target host pathways can have biologically relevant off-target effects on the gut microbiome and provide a strong foundation for addressing long-standing questions about inter-individual variations in drug response for rheumatic and perhaps other disease areas.
RESULTS
Methotrexate has a broad impact on the human gut microbiota in gnotobiotic mice
To control for the potential confounding effects of disease or prior treatment with immunomodulatory drugs, we colonized germ-free mice with a stool sample collected from a healthy human male (Donor 1; Tables S1A and S1B) prior to the oral administration of vehicle, low-dose MTX (1 mg/kg), or high-dose MTX (50 mg/kg), selected to span doses used to treat arthritis (Koyama et al., 2017) and cancer (Chabner and Young, 1973) in murine models (see Methods). Daily stool samples and endpoint samples from the cecum and colon were analyzed using 16S rRNA gene sequencing (16S-seq; Tables S2A and S2B) and quantitative PCR (qPCR), and daily weights were assessed (Table S3A). Total colonization (Figure 1A) and microbial richness (Figure 1B) were comparable between groups. MTX significantly altered gut microbial community structure after 1 day (ANOSIM, R=0.60, p=0.006) and this effect persisted to the final day of treatment (ANOSIM, R=0.75, p=0.006) (Figure 1C). There was no significant difference between groups prior to treatment (ANOSIM, R=0.23 p=0.109). High-dose MTX significantly decreased the Bacteroidetes phylum compared to vehicle controls (Day 4 DESeq padj=0.001, Figure 1D). Analyses of longitudinal trends with a generalized linear mixed-effects model, confirmed that Bacteroidetes were decreased (slope=−0.066, p=0.007). No other phyla were significantly altered using either analysis. High-dose MTX altered 14 bacterial genera and 81 amplicon sequence variants (ASVs) spanning multiple phyla including Actinobacteria, Firmicutes, and Proteobacteria (Figure 1E; Table S2C). Low-dose MTX had a more modest effect that was significant when taking into account longitudinal trends [PERMANOVA: R2=0.22, p=0.001, low-dose; R2=0.38, p=0.001, high-dose]. Low-dose MTX significantly altered 26 ASVs (Figure 1E; Table S2C), including 19 that were also altered in a consistent direction in the high-dose group (Figure 1F). Endpoint samples from the cecum and colon revealed significant differences in community structure (Figures 1G and 1H) and ASVs (Table S2C) that were comparable to stool samples (Figure 1I).
Figure 1. MTX alters the gut microbiotas of humanized mice within days.
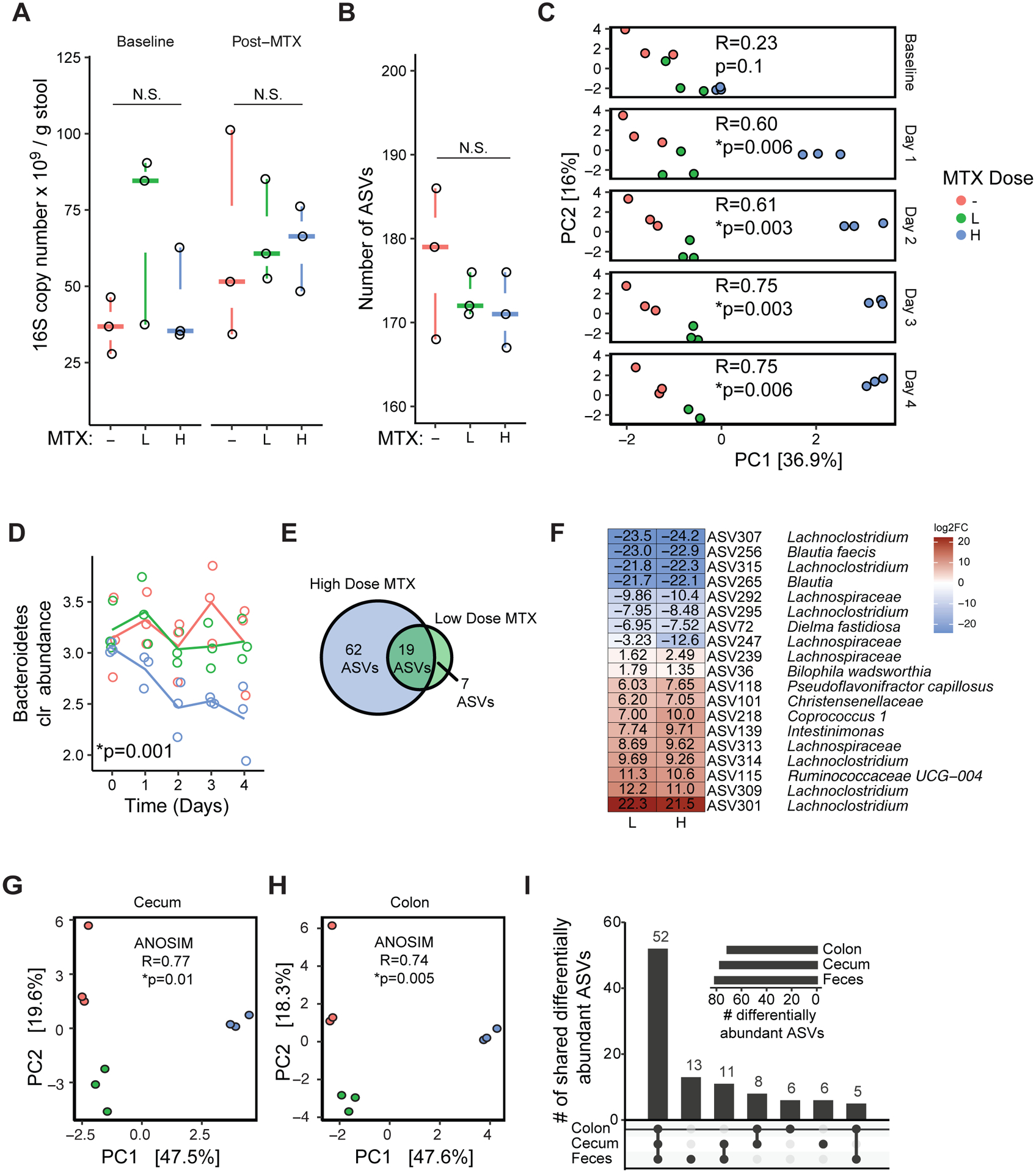
(A) qPCR was used to assess bacterial colonization level (16S copy number per gram stool) at baseline and after 4 days of MTX treatment. Treatments: “-”, vehicle; “L”, low-dose MTX (1 mg/kg); “H” high-dose MTX (50 mg/kg). n=3 mice/group, 1 cage/group.
(B) Number of amplicon sequence variants (ASVs) detected using 16S rRNA gene sequencing. Statistical results of Kruskal-Wallis test reported.
(C) Principal components analysis (PCA) of Euclidean distances using clr-transformed values from longitudinal stool samples. ANOSIM results are shown. Shifts in panel C were confirmed by PERMANOVA also (R2=0.68, p=0.001; Day 4).
(D) Bacteroidetes significantly decreased with high-dose MTX treatment relative to vehicle controls (DESeq padj=0.001, Day 4). Lines depict the mean for each group.
(E) Differentially abundant ASVs for each dose relative to vehicle (DESeq padj<0.01, Day 4).
(F) All of the ASVs differentially abundant in response to both doses showed consistent directionality (p=1.02 × 10−7, Hypergeometric test). Colors indicate log2 fold changes (log2FC) (Day 4, relative to vehicle).
(G-H) Principal components analysis (PCA) of Euclidean distances using clr-transformed values endpoint cecal (G) and colon contents (H). ANOSIM results are shown for each graph.
(I) UpSet plot of differentially abundant ASVs when comparing high-dose MTX vs. vehicle in endpoint cecum, colon and fecal samples (Day 4).
See also Figures S1, S2, and S3.
The impact of MTX (50 mg/kg) on the gut microbiota was robust to the donor sample used, co-housing, route of delivery, and co-administration of folic acid (Table S2C). First, we replicated our original experimental design using individually housed mice colonized with stool from a treatment naïve RA patient (Donor 2, Tables S1A and S1B). Microbial richness was comparable between groups (p=0.4, Wilcoxon rank sum) (Figure S1A). MTX significantly altered gut microbial community structure (Figures S1B–D), with changes to the abundance of 8 genera (Table S2C) and 23 ASVs (Figure S1E). No phylum level changes were detected. Next, we used a different treatment naïve RA patient (Donor 3) to colonize germ-free mice prior to the oral or intraperitoneal (IP) administration of MTX (route) and prior to oral MTX with or without an equimolar level of folic acid (rescue). Neither route nor rescue resulted in a significant difference in α- or β-diversity between groups (Figures S2A–D). A combined analysis of both treatment groups revealed significant shifts in the gut microbiota [Day 0 vs. 2; ANOSIM R=0.89, p=0.007 (route); ANOSIM R=0.99 p=0.002 (rescue)]. Multiple phyla were consistently affected in both experiments (Figures S2E and S2F), along with shifts at the genus and ASV levels (Table S2C; Figure S2G). Notably, we observed consistent shifts in 41 ASVs that overlapped between route and rescue (Figure S2G).
We next compared the results of these experiments to identify common trends. Among ASVs that were differentially abundant with MTX treatment, members of the Bacteroidetes phylum were enriched in 4 out of 5 treatment groups relative to their number in the community (Table S2D). Other phyla did not exhibit such enrichment. To identify taxa that were similarly affected by MTX across donors and multiple experiments, we combined data from these gnotobiotic mouse experiments using multi-factor design models. As before, α-diversity was not affected by MTX (Figure S3A, p>0.05, ANOVA). Community composition was significantly affected by donor (Figure S3B) and MTX treatment (Figure S3C). Four phyla (Figure S3D), 31 genera (Table S2C), and 61 ASVs (padj<0.05, DESeq; Figure S3E) were differentially abundant.
Methotrexate perturbs the growth of human gut bacteria at physiological concentrations
We assembled a panel of 45 bacterial isolates from 6 phyla, 42 of which are commonly found in the human gut microbiota (Table S4). Each isolate was incubated for 48 hours with vehicle controls or MTX (1.7–900 μg/ml, 2–2,000 μM). Minimal inhibitory concentration (MIC) ranged across the full gradient, with 11 isolates resistant to the maximum concentration tested (Figure 2A). We did not detect any significant correlation between growth parameters in vehicle controls and MTX sensitivity (|rho|<0.19, p>0.28, Data File S1). Bacteroidetes tended to be sensitive to MTX relative to the other phyla (Figure 2B). We also observed multiple cases where nearest neighbors had discrepant phenotypes; for example, Parabacteriodes merdae vs. Parabacteroides distasonis and Lactococcus lactis vs. Clostridium innocuum (Figure 2A).
Figure 2. MTX directly inhibits the growth of human gut bacterial isolates.
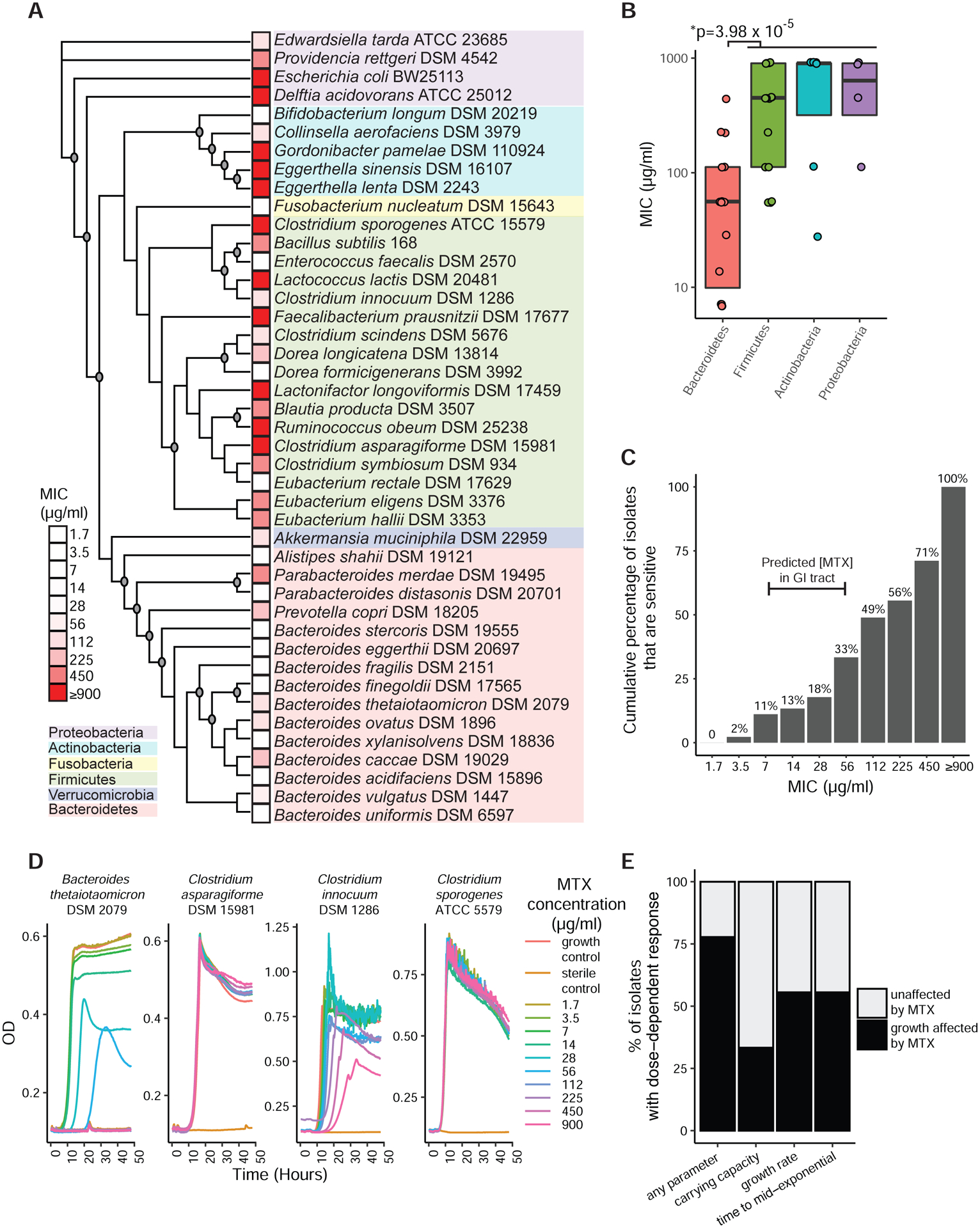
(A) A diverse panel of 45 isolates were incubated with varying concentrations of MTX and the minimal inhibitory concentration (MIC) was measured. A maximum likelihood phylogenetic tree using full-length 16S rRNA gene sequences for each organism was constructed, with bootstrap values greater than 70/100 iterations indicated by circles at branch points. The tree shows 43 of the isolates (2 additional E. lenta strains were tested but only one of each species was included in the tree).
(B) The MICs of various isolates spanning 4 major phyla. Bacteroidetes were compared to other phyla (Wilcoxon rank-sum test). Boxplot top and bottom hinges correspond to the first and third quartiles, respectively, and horizontal lines denote the median.
(C) The cumulative percentage of 45 tested human gut bacterial isolates that have MICs at or lower than a given concentration of MTX.
(D) Growth curves of 4 representative isolates incubated with 10 concentrations of MTX. Each isolate was tested in duplicate and averages are shown.
(E) Carrying capacity, growth rate, and lag phase parameters were affected in a dose-dependent manner by MTX among a significant proportion of bacteria.
Multiple observations support the physiological relevance of MTX for gut bacterial growth and physiology. The estimated concentration of MTX in the proximal gastrointestinal (GI) tract (22–220 μM or 10–100 μg/ml) would be sufficient to inhibit 11–33% (5–15) of the tested isolates (Figure 2C). Furthermore, growth curves for a subset of strains revealed that 78% of the tested isolates had a least one growth parameter affected in a dose-dependent manner at sub-MIC concentrations (Figures 2D and 2E). In total, 36/45 (80%) isolates exhibited either full growth inhibition or alterations in growth curve parameters upon exposure to MTX at concentrations that are predicted to be found in the human gut.
Methotrexate impacts conserved pathways necessary for cell growth
We used RNA sequencing (RNA-seq) to identify differentially expressed transcripts in the presence or absence of MTX (100 μg/ml). We selected 4 isolates (1 Bacteroides and 3 Clostridia) with varying sensitivity to the growth-inhibitory effects of MTX (Table S4). As expected, MTX induced a profound shift in gene expression in the most sensitive strain, B. theta, affecting 83% of genes in the transcriptome (Figure 3A). In contrast, the three Clostridia tested had markedly distinct transcriptional responses that did not correlate to their sensitivity to MTX. Clostridium sporogenes and C. symbiosum exhibited a defined shift (21 and 55 genes, respectively) whereas C. asparagiforme had a robust transcriptional response (468 genes; Figure 3A; Table S5A). Many of these changes were detected at more stringent p-adjusted cutoffs (Table S5B) and with an alternative analysis method (see Methods).
Figure 3. Impact of MTX on human gut bacterial transcriptomes and metabolomes.
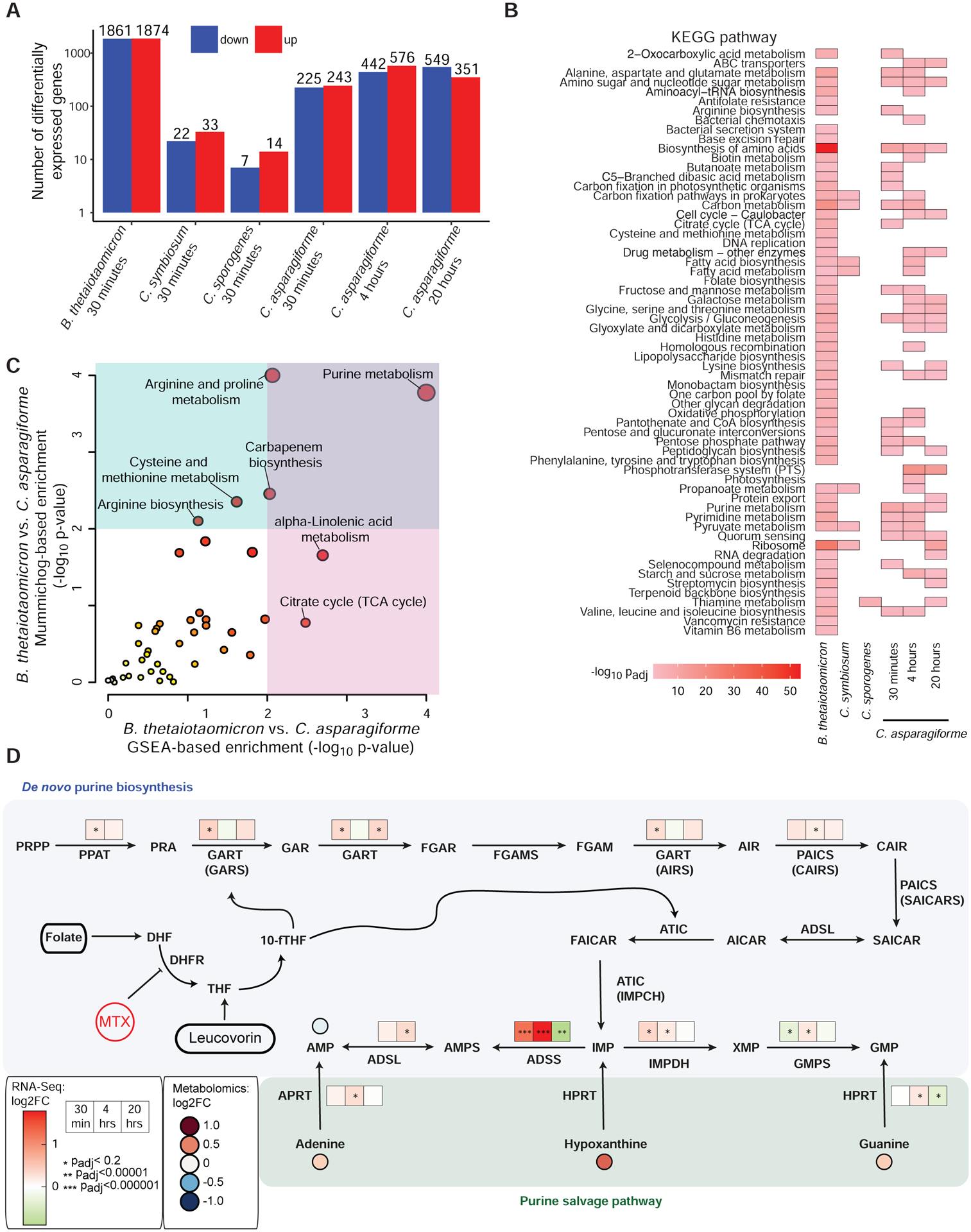
(A) A variable number of transcripts, as determined by RNA-seq, were differentially expressed (padj<0.2, DESeq) upon 30 minutes of MTX 100 μg/ml (compared to vehicle control) in 4 bacterial isolates with varying sensitivity to the growth inhibitory effects of the drug (n=3 per treatment). Clostridium asparagiforme was also treated for 4 and 20 hours.
(B) A heatmap of KEGG pathway enrichments (padj<0.05, BH corrected) of differentially expressed genes (padj<0.2, DESeq) in bacterial isolates treated with 100 μg/ml of MTX vs. vehicle control (n= 3 per treatment / isolate). Colors represent log10 adjusted p-value.
(C) Pathways enriched among metabolite features exhibiting a significant interaction between bacterial isolate and MTX treatment in the supernatants of B. theta and C. asparagiforme cultures exposed to MTX (100 μg/ml for 24 hours) as assessed by mummichog and gene set enrichment analysis (GSEA) (n=2 replicates / isolate for DMSO vs. n=4 replicates/isolate for MTX). Red circles are significant, and circle size indicates number of metabolite features.
(D) Multiple enzymes involved in purine metabolism (Pedley and Benkovic, 2017) were affected at some point during the time course study that was performed on C. asparagiforme. A 3-box heatmap of log2 fold change values at 30 minutes, 4 hours, and 20 hours is shown above each enzyme, with asterisks indicating padj values. Asterisks indicate padj: *<0.2, **<0.00001, ***<0.000001. Four metabolites were detected with high confidence by untargeted metabolomics at 24 hours of treatment (n=2 DMSO and n=6 for MTX): AMP, adenine, hypoxanthine and guanine. Levels of AMP were decreased relative to vehicle control, and the rest were increased, but these differences were not statistically significant (Wilcoxon rank sum). DHF, dihydrofolate; DHFR, dihydrofolate reductase; THF, tetrahydrofolate; 10-fTHF, 10-formyl-tetrahydrofolate; PPRP, phosphoribosylpyrophosphate; PPAT, PPRP amidotransferase; PRA, 5-phosphoribosylamine; GART, trifunctional enzyme consisting of phosphoribosylglycinamide synthetase (GARS), phosphoribosylglycinamide formyltransferase (GAR Tfase), and phosphoribosylaminoimidazole synthetase (AIRS); GAR, glycineamide ribonucleotide; FGAR, N-formylglycinamide ribonucleotide; FGAMS, phosphoribosyl formylglycinamidine synthase; FGAM, N-formylglycinamidine ribonucleotide; AIR, aminoimidazole ribonucleotide; PAICS, bifunctional enzyme consisting of phosphoribosyl aminoimidazole carboxylase (CAIRS) and phosphoribosyl aminoimidazole succinocarboxamide synthetase (SAICARS); CAIR, 5’-phosphoribosyl-4-carboxy-5-aminoimidazole; SAICAR, N-succinocarboxyamide-5-aminoimidazole ribonucleotide; ADSL, adenylosuccinate lyase; AICAR, aminoimidazole-4-carboxamide ribonucleotide; ATIC, bifunctional enzyme consisting of 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase (AICAR Tfase) and IMP cyclohydrolase (IMPCH); FAICAR, 5-formamidoimidazole-4-carboxamide ribotide; IMP, inosine 5’-monophosphate; ADSS, adenylosuccinate synthase; AMPS, adenylosuccinate; AMP, adenosine monophosphate; APRT, adenine phosphoribosyltransferase; HPRT, hypoxanthine-guanine phosphoribosyltransferase; IMPDH, IMP dehydrogenase; XMP, xanthosine monophosphate; GMPS, GMP synthase; GMP, guanosine 5’-monophosphate.
Next, we searched for differentially expressed metabolic pathways and modules. Both purine and pyrimidine metabolism were significantly affected in C. asparagiforme and B. theta, among other pathways (Figure 3B; Tables S5C and S5D). While B. theta exhibited multiple pathway enrichments (57 pathways enriched among transcripts with padj<0.2), purine and pyrimidine metabolism were among the top 10 pathways (9th and 5th, respectively) when ranked by p-value (p<0.05 with Benjamini-Hochberg (BH) adjustment), and these enrichments were largely insensitive to the padj threshold used in our analyses (Table S5E). C. asparagiforme exhibited enrichment of 23 pathways, and purine and pyrimidine metabolism were 2nd and 11th among this list (Table S5D). Additionally, pathways contributing to protein synthesis, which is also known to be targeted by MTX in patients (Cronstein, 1996), were enriched in both B. theta and C. asparagiforme, such as “biosynthesis of amino acids” and more specific pathways such as “valine, leucine, and isoleucine biosynthesis” and “alanine, aspartate and glutamate metabolism” (Figure 3B; Tables S5C and S5D).
To assess the generalizability of these sub-MIC MTX responses to distinct growth phases, we performed a time course experiment on the drug resistant but transcriptionally responsive C. asparagiforme, comparing transcriptional profiles in response to MTX at mid- and late-exponential as well as stationary phase. Consistent with our original analyses, purine and pyrimidine metabolism continued to be among the pathways that were affected at later timepoints (Figure 3B; Table S5D). Of the 41 transcripts that were differentially expressed at all three timepoints (padj<0.2, DESeq), 21 consistently changed in the same direction (5 upregulated, 16 downregulated), whereas 20 demonstrated more complicated dynamics (Table S5F). There were no significant differences in DHFR or AICAR transformylase expression; however, multiple genes encoding enzymes upstream and downstream of these enzymes were differentially expressed. Genes involved in de novo purine biosynthesis as well as the salvage pathway for purine synthesis showed differential expression (Figure 3D). For example, adenylosuccinate synthase (ADSS), which is involved in converting inosine monophosphate (IMP) into adenylosuccinate (AMPS) (Pedley and Benkovic, 2017), was significantly upregulated along with other members of this pathway.
To gain further insights into the metabolic consequences of MTX for gut bacteria, we subjected B. theta and C. asparagiforme to untargeted metabolomics. We found 418 metabolic features that changed similarly in the two isolates and 220 that differed by species (padj<0.01, two-factor ANOVA with BH correction; Table S6A), and both sets showed multiple significant pathway enrichments (Table S6B) with purine metabolism being among the most significantly enriched as assessed by two different enrichment algorithms, mummichog and GSEA (Chong et al., 2019) (Figure 3C; Table S6B). These results suggest the metabolomic response to MTX involves differential impacts on purine metabolism in these isolates. Multiple enriched pathways were consistent with our RNA-seq data (Figure 3B), including purine metabolism, cysteine and methionine metabolism, arginine biosynthesis, and citrate (TCA) cycle.
To provide functional validation of the observed transcriptomic and metabolomic changes, we quantified the ability of folic acid and leucovorin (see Figure 3D) to rescue MTX-induced growth inhibition of four isolates of varying MTX sensitivity (Table S4). B. theta growth was improved in a dose-dependent manner in response to folic acid in the presence and absence of MTX (Figures 4A–C). In contrast, dose-dependent increases in growth for B. vulgatus and C. innocuum were only evident in the presence of MTX (Figures 4A–C). Surprisingly, MTX resistant C. symbiosum was sensitized to MTX in the presence of folic acid at high doses of MTX (Figures 4A–C). In contrast, leucovorin improved growth of 3 of the 4 tested isolates (Figure 4D and 4E) but did not have any significant dose-dependent effects on growth in the presence of MTX (Figure 4F). Taken together, these findings support the hypothesis that MTX acts on bacterial DHFR (Figure 3D).
Figure 4. Folic acid and leucovorin partially rescue the effects of MTX.
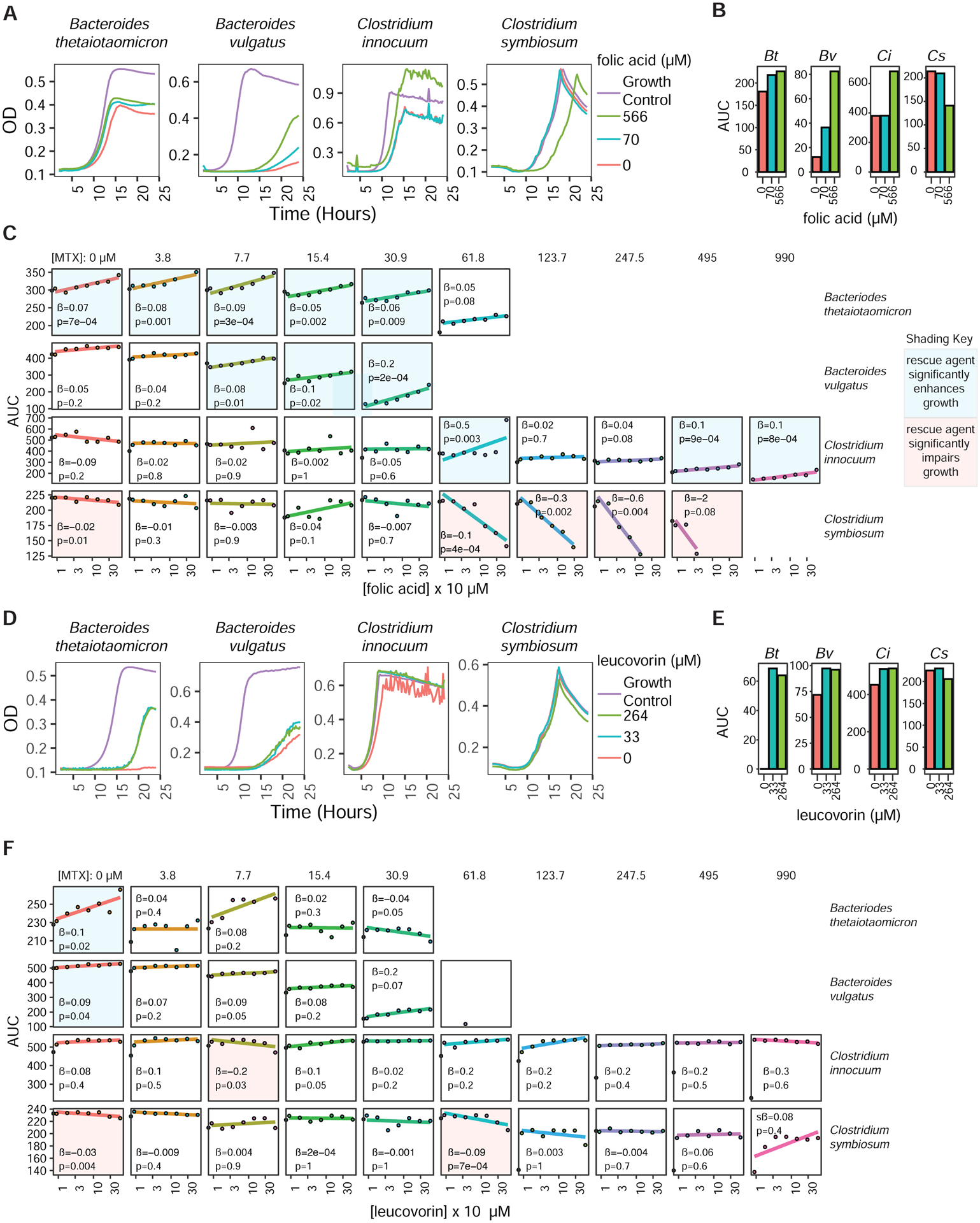
(A–B) Growth curves (A) and area under the growth curve (AUC) (B) of 4 isolates incubated with MTX (61 μM, 28 μg/ml) and varying concentrations of folic acid. Growth control curves are shown for comparison.
(C) Slope estimates (β) and p-values from linear regressions estimating the effects of folic acid on AUC for 4 isolates incubated with 9 concentrations of MTX and 7 concentrations of folic acid.
(D–E) Growth curves (D) and area under the growth curve (AUC) (E) of 4 isolates incubated with MTX (61 μM, 28 μg/ml) and varying concentrations of leucovorin. Growth control curves are shown for comparison.
(F) Slope estimates (β) and p-values from linear regressions estimating the effects of leucovorin on AUC for 4 isolates incubated with 9 concentrations of MTX and 7 concentrations of leucovorin.
(C,F) Panel backgrounds are colored blue or red depending on whether the rescue agent significantly (p<0.05; linear regression) enhances or impairs growth, respectively.
Clinical relevance of the interaction between methotrexate and the human gut microbiome
To test the impact of MTX in the context of a complex human gut microbial community we incubated stool samples obtained from 30 MTX-naïve patients (Table S1A) with MTX (100 μg/ml) or vehicle for 48 hours. MTX significantly impaired the growth of these complex microbial communities (Figure 5A), resulting in a significant decrease in carrying capacity (Figure S4A) and a significant delay in the lag phase (Figure S4B). 16S-seq of endpoint samples revealed a significant decrease in richness (Figure S4C) and shift in community structure (Figures 5B–E) in response to MTX. Consistent with our studies in mice and on isolates, Bacteroidetes decreased (Figure 5F) and Actinobacteria increased (Figure 5G). We also detected significant shifts in the abundance of 17 genera and 20 ASVs (Table S2C, Figure 5H). This included multiple MTX-modulated ASVs seen in our in vivo experiments: B. thetaiotaomicron (ASV23), B. uniformis (ASV14), B. vulgatus (ASV26), C. aerofaciens (ASV44), and B. ovatus (ASV496), which changed in a direction similar to what was seen in vivo.
Figure 5. MTX alters the human gut microbiota ex vivo and in treatment responsive patients.
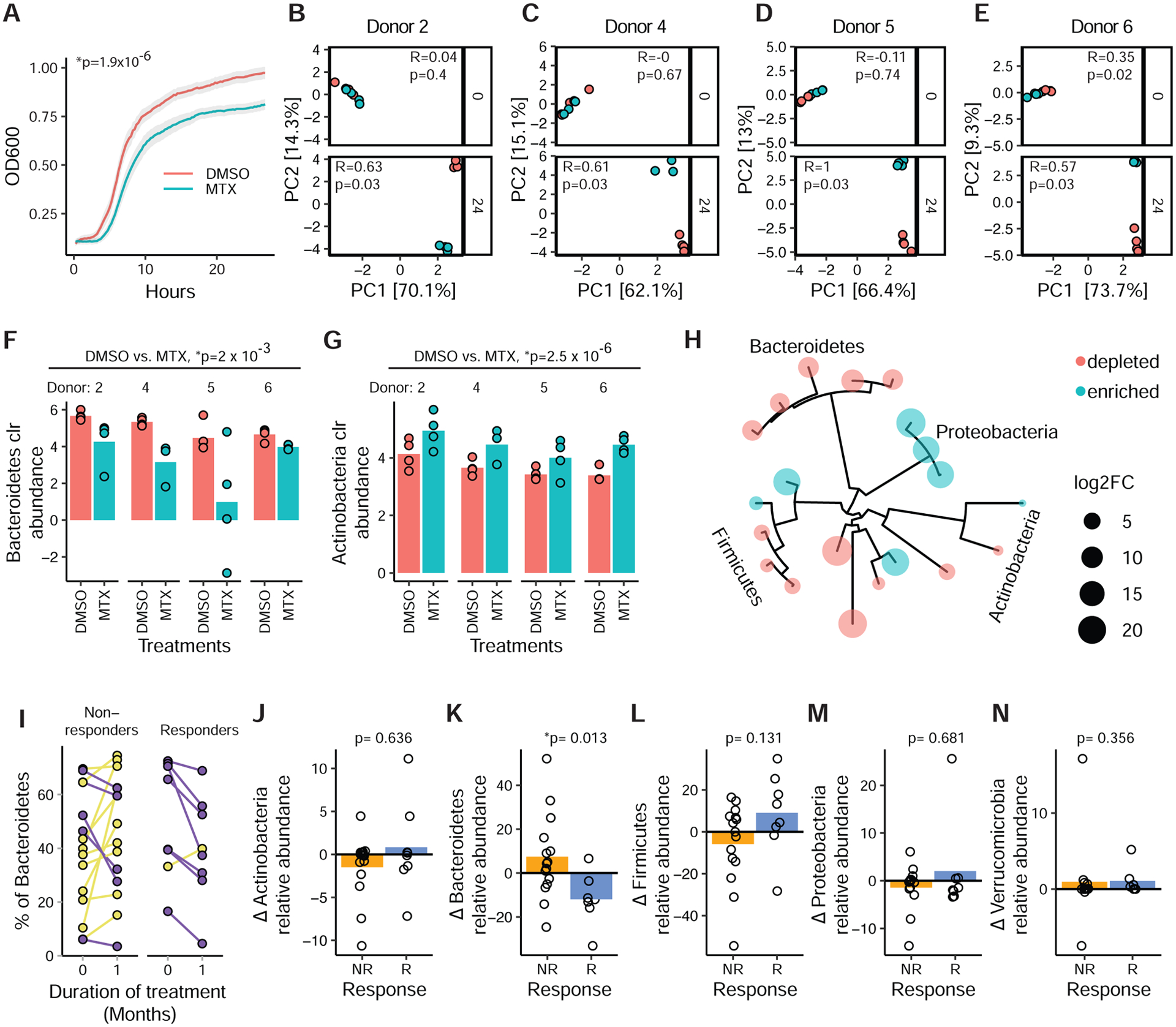
(A) Growth of fecal suspensions from treatment-naive RA patients (n=30, 4 replicates/treatment) in the presence of MTX 100 μg/ml or DMSO. The average growth curves of 30 patients are shown. Shaded areas represent ±SEM. Carrying capacity was reduced (p=1.9×10−6, paired Student’s t-test).
(B–E) PCA of Euclidean distances using clr-transformed values from ex vivo patient samples (MTX-R) treated with MTX vs. vehicle control at 0 and 24 hours. ANOSIM testing was performed comparing MTX to vehicle control at the different time points.
(F–G) Normalized abundances (clr) of Bacteroidetes (F) and Actinobacteria (G) phylum levels in 4 ex vivo microbial communities treated with MTX (100 μg/ml) or DMSO for 24 hours (n=4 replicates/treatment per patient) (padj<0.01, DESeq comparing DMSO vs. MTX).
(H) Phylogenetic tree of 20 ASVs that are differentially abundant (padj<0.01, DESeq) with MTX treatment ex vivo in 4 patient samples.
(I) Fecal samples from 23 RA patients were subjected to 16S sequencing before treatment with MTX and 1 month after treatment. Relative abundance of Bacteroidetes is shown for each individual (blue, decreased; yellow, increased).
(J–N) Change in relative abundance of 5 major phyla with MTX treatment in responders compared to non-responders (Wilcoxon rank-sum).
See also Figure S4.
Next, we asked whether these findings extended to RA patients by 16S-seq (n=23) and metagenomic sequencing (n=17) on longitudinal stool samples collected at baseline and 1 month after treatment initiation (Tables S1A and S2A). There were no significant differences in richness (p=0.4, Wilcoxon rank sum), community structure (Figure S4D), or the abundance of bacterial phyla, genera, or ASVs (padj>0.05, DESeq) in pre- vs. post MTX samples across the entire cohort. There were also no significant differences in bacterial taxa abundance (ranging from phylum to species) in our metagenomic data (q-value<0.25, MaAsLin2). In contrast, analysis of gene abundance revealed a significant shift in the gut microbiome following treatment initiation: 6,409 gene families were differentially abundant over time at a nominal p-value<0.05, and 96 passed multiple testing correction (MaAsLin2 q-value<0.25, Table S7A). 334 (pnominal<0.05) and 5 (q<0.25) KEGG orthologs (KOs) were differentially abundant (Table S7A). These included gene families involved in pyrimidine synthesis (e.g., thymidylate synthase, uracil phosphoribosyltransferase) and protein synthesis (e.g., 50S ribosomal protein L22). We also detected KO enrichments for ABC transporters (padj<0.2, Table S7B).
Finally, we sought to determine if any of the differences in the gut microbiome following treatment initiation were distinctive between MTX responders (MTX-R) and non-responders (MTX-NR). We used a binary classification of drug response defined as a decrease in DAS28 (Wells et al., 2009) of ≥1.8 and continued use of oral MTX without addition of other disease-modifying anti-rheumatic drugs or biologics. There were no significant differences in baseline disease activity parameters between MTX-R (n=8) and MTX-NR (n=15; Table S1C). Baseline differences in the gut microbiome between these two patient populations are the focus of another manuscript (Artacho et al., 2020). When examining changes over time, MTX-R exhibited a significant decrease in Bacteroidetes relative to MTX-NR, without any significant differences in the other phyla (Figures 5I–N). Lower-level taxa (genus and ASV) did not significantly differ between responder groups (pnominal>0.05, Wilcoxon rank sum), nor did taxonomic abundances (ranging from phylum to species) as assessed by shotgun sequencing (q>0.25, MaAsLin2). We detected a significant shift in 2 gene families in MTX-R, whereas MTX-NR had 508 gene families and 5 KOs with significant differences in abundance (q<0.25, MaAsLin2; Table S7A; Figure 5O). This included gene families involved in purine and pyrimidine metabolism (e.g., thymidylate synthase and adenylosuccinate lyase; Table S7A; Figure 5P), with enrichments for KOs involved in biosynthesis of secondary metabolites, 2-oxocarboxylic acid metabolism, biosynthesis of amino acids, thiamine synthesis, and folate biosynthesis among others (padj<0.2, Table S7B).
The post-treatment microbiome decreases host immune activation
We tested the functional impact of MTX-altered microbiota on mucosal and peripheral T cell populations in gnotobiotic mice with or without an inflammatory trigger. Germ-free mice were colonized with pre- and post-treatment stool samples from the 3 MTX-Rs with the largest decrease in Bacteroidetes (Figure S5A). Each colonization group was split into unchallenged and challenged groups, using dextran sodium sulfate (DSS) as an inflammatory trigger. In unchallenged mice, 3 immunocyte populations (including B220+ and Foxp3+ CD4+ cells) were significantly lower in the post-MTX relative to pre-MTX recipients and 1 population was significantly higher (Figure 6; range: 0.48–1.3 fold change relative to pre-MTX recipients). In challenged mice, we observed immune infiltration and histologic colitis with DSS treatment in both groups (Figure S5B). There were no significant differences in body weight change, colitis score, or colon length between recipient groups (Figures S5C–E). In challenged mice, 7 immunocyte populations (including B220+, CD44+ CD69+ CD4 +, and Gr+ CD11b+ cells), were significantly lower and 1 was significantly higher in the post-MTX recipients (Figure 6; range: 0.64 – 1.33).
Figure 6. MTX-altered microbiotas dampen host immune activation.
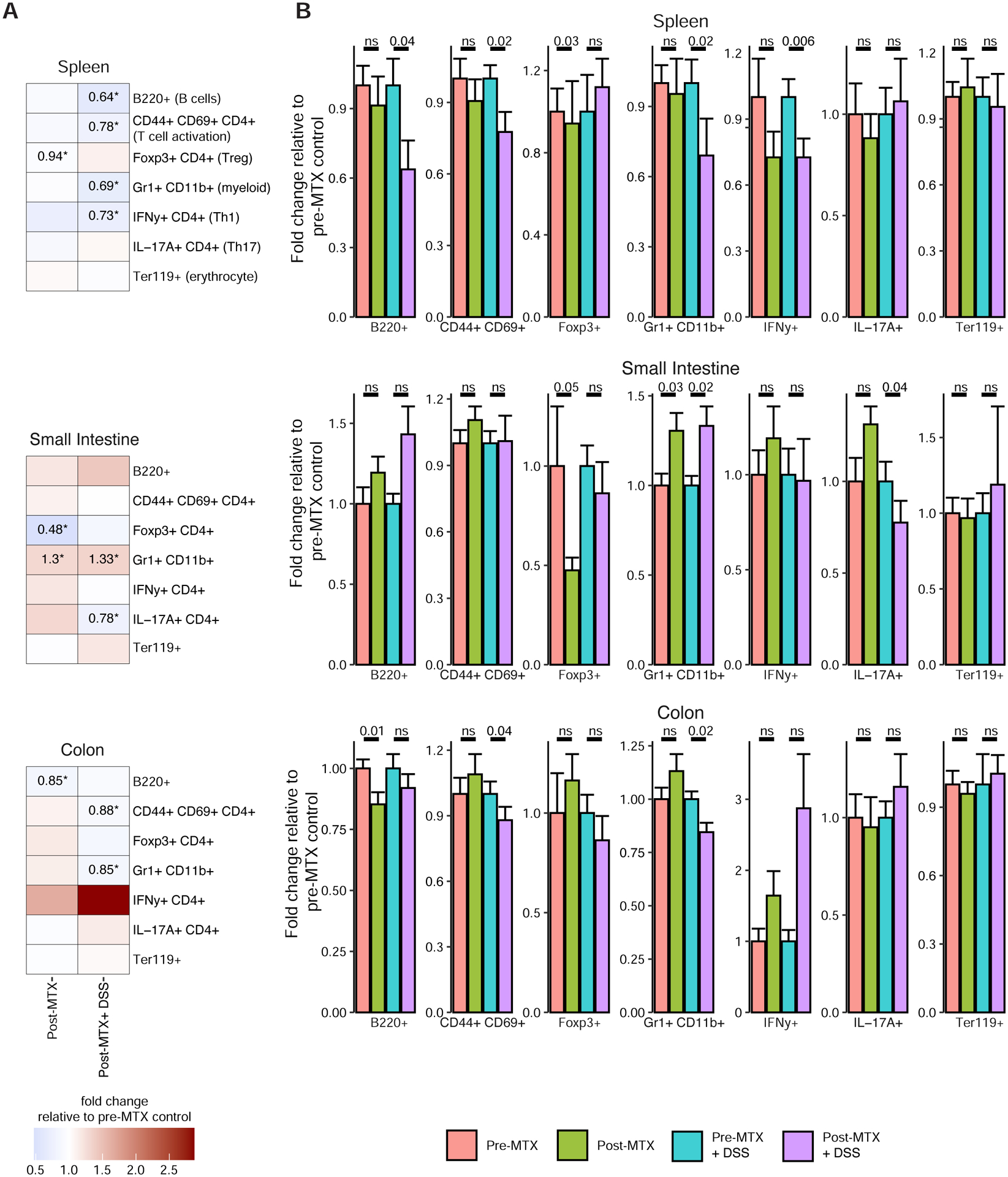
(A) Heatmap showing the relative percentage of cell populations in post-MTX recipient mice under unchallenged and challenged conditions (normalized to the pre-MTX recipient group for each condition) (n=20–22 mice/donor sample, 2–5 cages/treatment group). Populations that significantly differed (p<0.05; linear mixed effects model) are indicated by an asterisk and the fold change percentage in post-MTX recipient mice is indicated (i.e., post-MTX levels divided by pre-MTX levels).
(B) Immune cell populations in mice in 3 donor experiments. Each bar indicates the mean fold change percentage (relative to pre-MTX recipient mice for each condition). P-values from linear mixed effects models are reported.
See also Figures S5 and S6.
To identify candidate bacterial effectors of the observed immune responses to colonization, we performed a 16S-seq on the gut microbiota of recipient mice following microbiota transplantation. Microbial community structure was distinct between donor groups (Figure S5F), and we observed shifts in relative abundance following colonization (Figure S5G). There were significant differences between the pre- and post-MTX recipient groups for each pair of donor samples (Figures S5H–J). One phylum (Proteobacteria), 26 genera, and 41 ASVs were differentially abundant in a combined analysis of the recipients (padj<0.05, DESeq; Figure S5K; Table S2C). These included ASVs that were also differentially abundant in mice or ex vivo microbial communities treated with MTX (Table S2C): Dielma fastidiosa (ASV72), Prevotella copri (ASV914), Eisenbergiella tayi (ASV224), Hungatella effluvia/hathewayi (ASV230), Dorea longicatena (ASV284), and Phascolarctobacterium faecium (ASV97).
We next asked if MTX-modulated ASVs are associated with immune cell phenotypes. Of 23 ASVs that were shown to be modulated by MTX in a prior experiment (see Figure S1 and Table S1B, Experiment 2), 12 were present in at least 10 of 20 mice in which immunocyte populations were also measured (Table S1B, Experiment 5). This enabled us to ask whether these MTX-modulated bacteria were associated with immune phenotypes. We correlated abundances of these 12 ASVs with immune cell populations that showed differential levels in the spleen, small intestine, and colon (Figure 7A; Table S3B). In the spleen, there were 18 significant positive correlations and 1 negative correlation (p<0.05, Spearman’s correlation among 20 mice using day 14 abundances, Figures 7A–D). No significant correlations were detected between MTX-dependent ASVs and small intestinal immune cell populations. 6 ASV-immune correlations were found in the colon, all of which were negative (Figures 7A and 7E).
Figure 7. Gut microbial taxa altered by MTX are associated with immune cell populations.
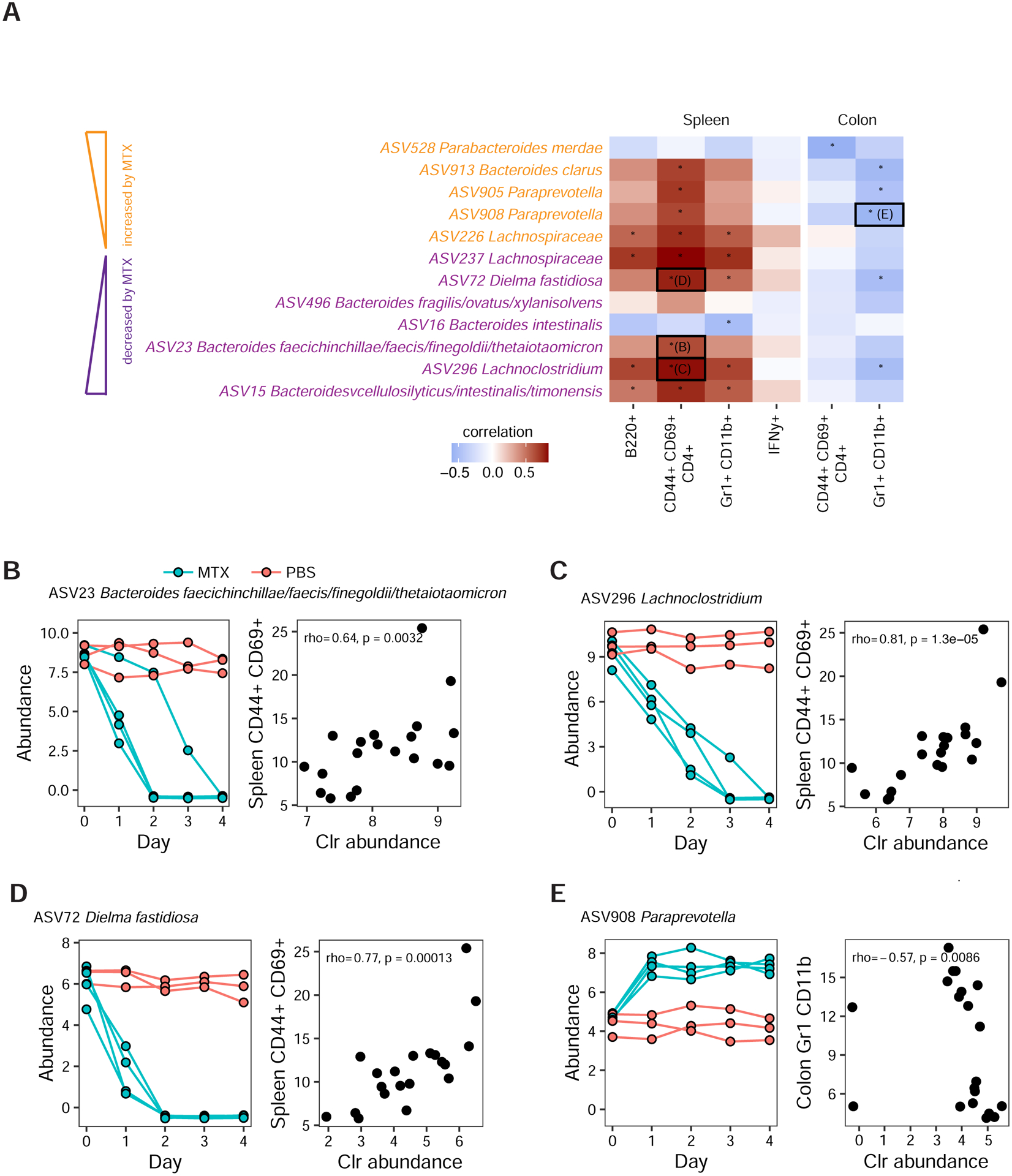
(A) Of 12 ASVs that were modulated by MTX in prior experiments (see Figure S1D), 11 ASVs exhibit 25 significant correlations with immune markers in the spleen and colon of Donor 2 pre-MTX and post-MTX recipient mice (mice from Experiment 5, Table S1B). Four representative ASV-immunocyte correlations are boxed and underlying data are shown in panels B-E. Asterisks denote significant correlations. ASVs that were previously shown to be downregulated or upregulated by MTX are colored purple or yellow, respectively.
(B–E) The left and right panels depict data from two separate experiments. The left panel depicts ASV abundances from individually housed mice treated with high-dose MTX (Table S1B, Experiment 2). The right panel depicts correlations between ASV abundance and immune cell percentages in mice that were transplanted with pre-MTX or post-MTX stool samples from patients treated with MTX (Table S1B, Experiment 5). Mice in both of these experiments were colonized by the same RA patient donor (Donor 2).
(B) ASV23 (Bacteroides faecichinchillae/faecis/finegoldii/thetaiotaomicron) abundance is reduced by MTX (left panel) and is positively associated with T cell activation markers (CD44+ CD69+ CD4+) in the spleen (right panel).
(C) ASV296 (Lachnoclostridium) abundance is reduced by MTX (left panel) and is positively associated with T cell activation markers (CD44+ CD69+ CD4+) in the spleen (right panel).
(D) Dielma fastidiosa (ASV72) abundance is decreased by MTX (left panel) and this ASV is positively correlated with T cell activation markers in the spleen (right panel).
(E) ASV908 (Paraprevotella) abundance is increased by MTX (left panel) and this ASV is negatively correlated with myeloid cell markers in the colon (right panel).
See also Figures S5 and S6.
DISCUSSION
Our results demonstrate that the off-target effects of MTX on the human gut microbiota are far broader than previously appreciated (Bolin et al., 1982; Maier et al., 2018; Wood et al., 1961). While there was considerable variability between mouse experiments, common trends in the microbiota’s response to MTX were observed across multiple donors, including healthy control and RA patients, and from the phylum to the ASV level. We also observed microbiota shifts across oral and intraperitoneal dosing, consistent with prior reports of MTX enterohepatic circulation (Grim et al., 2003; Steinberg et al., 1982). Folic acid, which is commonly co-administered with MTX in RA, did not markedly rescue the gut microbiota, perhaps due to the absorption of this supplement in the proximal GI tract (Visentin et al., 2014). Similar studies in patient cohorts are needed to test the translational relevance of these findings and to evaluate additional factors that could shape the sensitivity of the gut microbiota to MTX and other immunomodulatory drugs, including inter-individual variations in drug disposition (Hoekstra et al., 2004), genetic risk factors (Weyand et al., 1995), and baseline microbial community structure or function (Zhang et al., 2015).
While the effects of MTX were detectable across multiple bacterial phyla, just a single phylum (the Bacteroidetes) was consistently affected in mice, bacterial isolates, mixed communities, and non-responsive patients. The underlying determinants of variability in MTX sensitivity at the cellular, community, and ecosystem level remain to be investigated, are likely multifactorial, and may involve one or more of the following: drug influx and efflux (Kopytek et al., 2000), drug metabolism (Valerino et al., 1972), compensatory pathways (i.e., de novo synthesis of folic acid) (Wood et al., 1961), and redundancies in the cellular pathways to produce reduced folates (Myllykallio et al., 2003), as well as microbe-microbe or host-microbe interaction. Consistent with the importance of these higher-order interactions, we were able to detect ASVs with discrepant sensitivities in vitro and in vivo (e.g., Alistipes shahii). From a translational perspective, the observation of reproducible findings at the phylum level may be a simple and useful biomarker of therapeutic efficacy, which would complement ongoing attempts to integrate data on the pre-treatment microbiome with more established risk factors like host genetics and smoking status (Halilova et al., 2012).
Our in-depth analysis of select Firmicutes and Bacteroidetes isolates is consistent with the hypothesis that MTX acts by inhibiting bacterial DHFR with broad downstream consequences for purine and pyrimidine biosynthesis among other cellular pathways, including amino acid biosynthesis and replication. These results are consistent with data from human cells (Allegra et al., 1987; Genestier et al., 2000), wherein DHFR inhibition leads to changes in the expression of purine and pyrimidine pathways, which rely on folate as a co-factor for multiple key reactions. Rescue experiments with folic acid and leucovorin further support this hypothesis; however, the existence of partial and opposite effects underscore both the broader effects of MTX beyond targeting of bacterial DHFR and the complexities of interactions between metabolites (e.g., folate) and anti-metabolite drugs (e.g., MTX). Even in the absence of growth inhibition, there were marked changes in transcriptional and metabolic activity, consistent with our prior work on other non-antibiotic drugs (Maurice et al., 2013). These results emphasize the importance of considering the broader impacts of drugs on gut microbial physiology and metabolic activity, even in the absence of marked changes in community structure or colonization level. More work is needed to dissect the components of these transcriptional and metabolic changes that are specific to MTX and the degree to which they contribute to drug tolerance or metabolism.
Microbiome transplantations into germ-free mice given an inflammatory stimulus provided initial evidence that MTX-induced changes in the gut microbiota lead to changes in immune cell levels. This includes a reduction in multiple cell populations in the periphery, including activated T cells, IFN-γ+ T cells, myeloid cells, and B cells. We also found reductions in activated T cells, Th17 cells and myeloid cells in the intestinal mucosa. Th1, Th17 and B cells have been previously implicated in RA pathogenesis (Imboden, 2009) and prior studies suggest that MTX decreases IL-17 levels in RA patients (Yue et al., 2010), consistent with our observed decreases in Th17 cells. We observed reduced ileal Foxp3+ in an unchallenged state, suggesting a tonic reduction of Tregs (a T cell population that reduces inflammation) perhaps as a result of reduced post-MTX microbiota mediated stimulation of the immune system and the reduced need for tolerogenic induction of Tregs (Omenetti and Pizarro, 2015). Taken together, with the majority of changes (10/12 or 83%) exhibiting a reduction in post-MTX recipient mice, these findings suggest that MTX-induced shifts to the microbiota reduce its inflammatory potential. Additional studies are needed in gnotobiotic mouse models of RA (Maeda et al., 2016) coupled to genetic and pharmacological immune perturbations to dissect the host pathways responsible.
The specific members of the gut microbiota that contribute to these effects also remain to be elucidated; however, our computational analyses have identified multiple candidate bacterial species that are consistent with the prior literature. This includes the model Bacteroidetes B. theta (ASV23), which is sensitive to MTX, positively associated with immune activation, and exacerbates a mouse model of colitis (Hickey et al., 2015). Other Bacteroidetes may also be important, given their ability to activate Th1 cells in the context of intestinal parasitic infection (Heimesaat et al., 2006). Of note, Prevotella copri (ASV914), which is a Bacteroidetes member and has been previously associated with RA and Th17 activation (Maeda et al., 2016; Pianta et al., 2017), was decreased with MTX treatment (Table S2C) in gnotobiotic mice and ex vivo communities, as well as post-MTX recipient mice (Figure S5K). Another promising candidate that has previously been associated with RA is the gut Actinobacterium Collinsella aerofaciens (ASV44), which was sensitive to MTX in multiple transplant experiments and aggravates inflammatory arthritis via Th17 (Chen et al., 2016). Other candidates include Dielma fastidiosa (ASV72), ASV908 (Paraprevotella) and ASV296 (Lachnoclostridium) which were directly impacted by MTX and showed associations with immune cell phenotypes.
In conclusion, our results emphasize the importance of taking a broader view of pharmacology that encompasses the unintended consequences of non-antibiotic drugs for our associated microbial communities. These findings demonstrate the utility of integrated studies in vitro, in gnotobiotic mice, ex vivo, and in drug naïve patients to begin to elucidate the causality and mechanism for these complex drug-microbiome-host interactions. MTX-induced changes in microbial community structure were associated with patient response, providing a potential biomarker for accelerating the stable initiation of therapy and a first step towards determining which bacterial taxa contribute to or interfere with treatment outcomes.
STAR METHODS
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Turnbaugh (peter.turnbaugh@ucsf.edu).
MATERIALS AVAILABILITY
This study did not generate new unique reagents.
DATA AND CODE AVAILABILITY
All sequencing data generated in the preparation of this manuscript has been deposited in NCBI’s Sequence Read Archive (SRA), under Bioproject ID PRJNA656577. Representative code used in the analyses of 16S-seq data are described here (Callahan et al., 2016b) and custom code is available at https://github.com/turnbaughlab/2020_Nayak_MTX_Effects.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Gnotobiotic mouse studies
C57BL/6J mice (females, ages 6–16 weeks) were obtained from the UCSF Gnotobiotics core facility (gnotobiotics.ucsf.edu) and housed in gnotobiotic isolators for the duration of each experiment (Class Biologically Clean). Key details for each gnotobiotic experiment can be found in Table S1B. Housing: Mice were co-housed in some experiments, distributed across multiple cages per treatment group, or individually housed (Table S1B). Individually housed mice were housed in Single Mouse Duplexed Cages (Thoren, Cat#15). Colonization: Mice were colonized with stool from human donors, either a healthy male donor or a treatment-naïve (re-MTX) female and male donors with rheumatoid arthritis [as defined by American College of Rheumatology classification criteria, either (Arnett et al., 1988) or (Aletaha et al., 2010)]; samples obtained 1 month after starting MTX were used for “post-MTX” transplants. For colonization with a human microbiome, stool was diluted 1:10 g/mL in reduced PBS or saline and homogenized in an anaerobic chamber using pre-equilibrated reagents and supplies. Insoluble material was separated from supernatant by centrifugation at 50g for 1 minute. Aliquots of supernatant (100–200 μl per mouse) were gavaged into mice. In fecal transplant experiments using pre-MTX and post-MTX samples from RA donors, we did not detect MTX at the limit of quantification (100 nM) using targeted LC-MS, particularly in the post-MTX samples, suggesting that only microbiota (and not drug) was being transferred into recipient mice. Mice were colonized for at least 1–2 weeks before initiation of treatment with MTX (Pharmaceutical-grade, Fresenius Kabi, NDC 63323-122-50, Product # 102250). Mice were colonized for at least 2 weeks prior to dextran sodium sulfate treatment (DSS) treatment. Stool samples were collected on days 3, 7–8, and 14 prior to DSS treatment. Drug treatment: In experiments examining MTX treatment, mice were treated either with saline/PBS, MTX 1 mg/kg or 50 mg/kg daily or folic acid 50 mg/kg. Treatment was carried out either by oral gavage or intra-peritoneal injection. Mice were monitored and weighed daily during treatment (Table S3A). No gross signs of toxicity and minimal weight loss were observed for the short MTX treatment durations used in this study. Stool samples were collected daily during treatment. Duration of treatment with MTX was 4 days in the dose-response experiment and in individually housed mice. For the route and rescue experiments, duration of treatment was reduced to 2 days because in the dose-response experiment, with high-dose MTX, changes in community composition were seen within 1–2 days so subsequent treatments with high-dose MTX were done for 2 days given the large effect size seen with a single day of treatment. According to the UCSF Animal Studies protocols, we try to adhere to the three R’s of animal research (Guhad, 2005), and in this case, we were implementing the “refinement” principle, which advocates that researchers modify “husbandry or experimental procedures to minimize pain and distress, and to enhance the welfare of an animal used in science from the time it is born until its death.” Upon completion of treatment, mice were euthanized, and samples from the following tissues were harvested: I8 segment of the ileum and its contents (unconcentrated), cecal tissue and contents, and distal colon tissue and its contents. In DSS experiments, colon length was measured prior to sectioning of the colon. For DSS treatment (Alfa Aesar, Cat no. 9011-18-1), mice were given 2% DSS (w/v) ad libitum in their drinking water for 6–8 days, and DSS water was replaced on days 3 or 4. DSS was used as an inflammatory trigger to challenge the immune system instead of a murine arthritis model because of several favorable properties of the DSS model combined with the inherent difficulties of murine arthritis models in gnotobiotic mice: (1) DSS is highly penetrant in gnotobiotic mice, (2) DSS has been used to understand host-microbiota effects in inflammatory arthritis (Scher et al., 2013), (3) DSS has a rapid onset, and (4) many murine models of arthritis rely on a genetic abnormality which would need to be rederived in a gnotobiotic setting (Vincent et al., 2012). All mouse experiments were approved by the University of California San Francisco Institutional Animal Care and Use Committee.
NYU Human RA patient samples acquisition
Consecutive patients from the New York University Langone Medical Center’s rheumatology clinics and offices were screened for the presence of RA based on ACR criteria (Aletaha et al., 2010). After informed consent was signed, each patient’s medical history (according to chart review and interview/questionnaire), diet, and medications were determined. A screening musculoskeletal examination and laboratory assessments were also performed or reviewed. All RA patients who met the study criteria were offered enrollment. The criteria for inclusion in the study required that patients meet the American College of Rheumatology/European League Against Rheumatism 2010 classification criteria for RA (Aletaha et al., 2010), including seropositivity for rheumatoid factor and/or anti–citrullinated protein antibodies, and that all subjects be age 18 years or older. New-onset RA was defined as disease duration of a minimum of 6 weeks and up to 6 months since diagnosis, and absence of any treatment with disease-modifying anti-rheumatic drugs, biologic therapy or steroids (ever). The exclusion criteria applied to all groups were as follows: recent (<3 months prior) use of any antibiotic therapy, current extreme diet (e.g., parenteral nutrition or macrobiotic diet), known inflammatory bowel disease, known history of malignancy, current consumption of probiotics, any gastrointestinal tract surgery leaving permanent residua (e.g., gastrectomy, bariatric surgery, colectomy), or significant liver, renal, or peptic ulcer disease. This study was approved by the Institutional Review Board of New York University School of Medicine protocols #09–0658 and as previously published (Scher et al., 2013). All new onset rheumatoid arthritis (NORA) patients (Table S1A) were recruited using established protocols from a previously described study (Scher et al., 2013). Patients received oral MTX at standard of care doses as prescribed by their treating rheumatologists. Stool samples were collected at baseline and 1 month after MTX initiation and metadata were obtained at baseline and 4 months after therapy initiation. Clinical and demographic data was de-identified and recorded in RedCap by the designated study personnel. Clinical responder status (MTX-R) was defined a priori as any NORA patient whose DAS28 score was greater than 2 at baseline and improved by at least 1.8 by month 4 post-treatment. DNA was extracted from human fecal samples (n=23 patients, 46 stool samples) as we have previously described (Scher et al., 2013), using the MoBio Powersoil DNA extraction kit, based on cell membrane disruption by high-speed shaking in the presence of beads. The V4 hypervariable region of bacterial 16S ribosomal RNA (rRNA) was performed using a MiSeq Illumina platform (150-bp read length, paired-end protocol) at the New York University Genome Technology Center as previously described (Manasson et al., 2018). For each sample, fastq files are available in NCBI’s Sequence Read Archive (SRA), accession number PRJNA656577.
METHOD DETAILS
MTX dose selection in animal models
The doses used in this study were selected based upon a careful consideration of the doses used previously in humans and mice. While it is dangerous to overinterpret the relevant doses in mice to any human disease, the current gold standard is based upon allometric scaling (Nair and Jacob, 2016). Based on this simple conversion (animal dose × 0.081 = human dose), our two doses are the equivalent to 81 μg/kg and 4.05 mg/kg in humans. Given the average 60 kg humans, this equates to 4.86 mg/day and 243 mg/day, respectively. According to Mayo Clinic (www.mayoclinic.org/drugs-supplements/methotrexate-oral-route/proper-use/drg-20084837), doses in for cancer patients range from 5–50 mg/day, while doses for rheumatoid arthritis are typically ~7.5 – 20 mg/day (administered once per week). Thus, our low dose is slightly lower than the lower end of the human dosing regimens, whereas our high dose is higher than what is typically used in patients. However, we were cautious not to overinterpret the doses used herein, given the potential differences in MTX metabolism, absorption, clearance, and distribution between humans and mice that could complicate these matters, in addition to microbiome-dependent factors such as the differences in the ability of bacteria to convert MTX to DAMPA (Valerino et al., 1972) and/or other metabolites. Additional studies examining pharmacokinetics in mice and patients are needed to capture the genetic and microbiologic factors that could influence drug disposition. Thus, quantitative comparisons of dose equivalency in humans and mice remain highly speculative at this time.
16S-seq of humanized mouse gut microbiota
Broadly, aliquots of mouse fecal, ileal, cecal and colon samples (Table S2A) were homogenized using a bead-beating (Mini-Beadbeater-24, BioSpec) method followed by DNA extraction and purification. For the dose-response gnotobiotic experiment, bead beating was achieved using Lysing Matrix E 2mL Tube beds (MP Biomedicals) and using the digestion solution and lysis buffer of a Wizard SV 96 Genome DNA kit (Promega). The samples were then centrifuged for 10 minutes at 16,000g and the supernatant was transferred to the binding plate. The DNA was purified according to the manufacturer’s instructions. DNA from the remaining experiments was extracted using ZymoBIOMICS 96 well MagBead ZymoBIOMICS 96 MagBead DNA Kit (Cat#D4302) as per the manufacturer’s protocol.
GoLay-barcoded 515F/806R primers (Caporaso et al., 2012) were used to carry out 16S rRNA gene PCR according to the methods of the Earth Microbiome Project (earthmicrobiome.org). For the dose-response experiment, the following were combined: 2μL of DNA, 25 μL of AmpliTaq Gold 360 Master Mix (Life Technologies), 5 μL of primers (2μM each GoLay-barcoded 515/806R), and 18μL H2O. Amplification was as follows: 10 minutes at 95°C, 25x (30 seconds at 95°C, 30 seconds at 50°C, 30 seconds at 72°C), and 7 minutes at 72°C. For the dose-response gnotobiotic experiment, amplicons were quantified with PicoGreen (Quant-It dsDNA; Life Technologies) and pooled at equimolar concentrations. Libraries were quantified (NEBNext Library Quantification Kit; New England Biolabs) and sequenced with a 600 cycle MiSeq Reagent Kit (251×151; Illumina) with ~10% PhiX. For the remaining sequencing experiments, samples underwent primary PCR for amplification and secondary PCR to add flow cell adaptors and indices as previously described (Gohl et al., 2016). Normalization was achieved using SequalPrep Normalization (Life Tech A10510–0) kits. Pooled libraries were purified and concentrated with MinElute PCR Purification kit (Qiagen #28004), run on 1% gel, size-selected and purified using MinElute Gel Extraction kits (Qiagen, #28604). Pooled libraries were run at the Chan Zuckerberg Biohub using Illumina MiSeq platform.
In vitro bacterial growth studies
The isolates used in this study are shown in Table S4. 42/45 of the tested isolates are commonly found in the human gut microbiota, with the exceptions of Bacteroides acidifaciens (found in mice), Delftia acidovorans (found in soil), and Bacillus subtilis 168 (found in soil). Each of these strains was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) culture collection. A single colony of each isolate was subcultured in Bacto Brain Heart Infusion (BD Biosciences, 37 g/L) supplemented with L-cysteine-HCl (0.05%, w/v), menadione (1 μg/mL), and hemin (5 μg/mL) (referred to hereafter as BHI+) for 48 hours in an anaerobic chamber (Coy Laboratory Products) at 37°C with an atmosphere composed of 2–3% H2, 20% CO2, and the balance N2. This subculture was diluted down to an OD600 of 0.08–0.1, which was then further diluted 100-fold, and then used to inoculate a microtiter plate with 2-fold serial dilutions of MTX concentrations ranging from 0 – 900 μg/ml. Plates were incubated at 37°C with shaking in an Eon Microplate Spectrophotometer (BioTek Instruments, Inc) over a 48 to 72-hour period in the anaerobic chamber. Growth was monitored every 15 minutes at OD600 and corrected for background (no growth control). Data were exported using the Gen5 (v 2.0) software. The minimal inhibitory concentration (MIC) was measured as the lowest concentration of MTX resulting in >90% growth inhibition after 48 hours of incubation. We were unable to get growth curves for 6 of the studied isolates (Table S4) due to technical issues but were still able to assess MIC at 48 hours based on visual inspection and endpoint OD600 measurement. Growth curves from these experiments are shown in Data File S1 (panels A-D). As with any in vitro system, the growth of cells may be dependent on the growth medium used, and for our studies, we used a rich medium (BHI+). It remains to be determined whether the growth and transcriptional responses observed in our in vitro studies are mitigated or exacerbated in the gastrointestinal environment where there may be competition for limited resources.
In vitro bacterial rescue studies with folic acid and leucovorin
As described in our in vitro bacterial growth studies, 48-hour overnight cultures were grown in an anaerobic chamber in liquid BHI+ for the following isolates: Bacteroides vulgatus, Bacteroides thetaiotaomicron, Clostridium innocuum and Clostridium symbiosum. Each subculture was diluted down to an OD600 of 0.08–0.1, which was then further diluted 100-fold, and then used to inoculate a 96-well microtiter plate with 2-fold serial dilutions of MTX (Sigma, Cat#M9929) concentrations ranging from 0 – 450 μg/ml (0 – 990 μM) along the columns (1–10) and 2-fold serial dilutions of folic acid (Fischer Scientific, BP25195) ranging from 0 – 250 μg/ml (0–566 μM) along the rows (A–H). Each plate included media and growth controls. The same setup was used to test the rescue effects of 2-fold serial dilutions of leucovorin (Spectrum Chemicals, Cat#LE117) at concentrations ranging from 0–125 μg/ml (0 – 264 μM) and the following isolates were tested: B. vulgatus, B. thetaiotaomicron, C. innocuum, and C. symbiosum. Solubility limits restricted the max concentrations for each compound. Growth curves from these experiments are shown in Data File S1 (panels F–G).
Tree construction
Full-length ribosomal sequences for each isolate were extracted from the Greengenes (DeSantis et al., 2006) database (May, 2013). Sequences were imported into UGENE (Okonechnikov et al., 2012) (v 1.31.0), and aligned using MUSCLE (Edgar, 2004). Gaps occurring in > 50% of sequences were removed, and a maximum likelihood tree was generated using PhyML (Guindon et al., 2010) with 100 bootstraps and the GTR substitution model. For trees generated from 16S-seq from gnotobiotic mice or ex vivo RA samples, we used the ggtree R package (v 2.0.2)(Yu et al., 2018).
Estimated abundance of bacterial species in human metagenomes
Bacterial abundances were quantified using data from MetaQuery (Nayfach et al., 2015), a web-based application that provides taxonomic abundances from >1,900 publicly available human gut metagenomes. For each isolate, we queried the “metaphlan2” database and recorded the mean abundance value. The version of MetaPhlAn2 used by MetaQuery was 2.2.
Predicted concentration of MTX in the gastrointestinal (GI) tract
The predicted concentration of MTX in the proximal GI tract was estimated by taking the oral dose used for rheumatoid arthritis (25 mg) and dividing it by 250 ml (Dahlgren and Lennernas, 2019; Dressman et al., 1985; Shekhawat and Pokharkar, 2017), giving a concentration of 100 μg/ml or 220 μM. Further refinement of this estimate was done based on variations in the absorption of oral MTX in the proximal intestine (Grim et al., 2003), where it is estimated that 10 – 70% of the dose is not absorbed and instead delivered to the distal GI tract in RA patients. Thus, the estimated range of MTX in the GI tract was calculated to be 10–70 μg/ml with 100 μg/ml being the highest predicted concentration (prior to absorption by the host). When considering the doses used for cancer therapy (50 mg/day of oral MTX), the predicted range is 20–140 μg/ml with 200 μg/ml being the highest dose. MTX can also be given intravenously (IV) or intrathecally for the treatment of cancer (Treon and Chabner, 1996), and is typically given at much higher doses, including as high as 800–1000 mg IV over 24 hours for the treatment of cancers such as lymphoma or acute lymphoblastic leukemia. Assuming that 30% of this is enterohepatically circulated (Grim et al., 2003; Steinberg et al., 1982), then between 240 and 300 mg would enter the GI tract, and the predicted concentration in the GI tract would be between 960 – 1200 μg/ml.
MTX treatment for RNA-seq
The bacterial strains used in RNA-seq are given in Table S4. Genomes were obtained from NCBI’s GenBank Assembly database (see Table S4 for accession numbers). Culture media was composed of BHI+ and allowed to equilibrate in an anaerobic environment prior to use. Briefly, bacteria were cultured in BHI+ at 37 °C in an anaerobic chamber. Cultures for each isolate were grown to mid-exponential (achieving an OD600 ~ 0.5), aliquoted into triplicates, treated for 30 minutes with either DMSO or MTX 100 μg/ml, and then removed from the anaerobic chamber. The mid-exponential time point was selected to facilitate comparisons across isolates, as has been done by others (O’Rourke et al., 2020), since lag and stationary phases would present challenges in term of biomass (lag phase with low biomass) and differing physiology (i.e., sporulation among some isolates and not others). For C. asparagiforme, cultures were incubated for 4 and 20 hours as well and profiled. Cultures were centrifuged at 2000 rpm for 10 minutes at 4°C to facilitate removal of supernatant, and the remaining bacterial pellet was flash-frozen in liquid nitrogen.
Total RNA extraction
Each bacterial pellet was incubated with 1 ml of Tri reagent (Sigma Aldrich, catalog #: T9424) at room temperature for 10 minutes. The cell suspension was transferred into Lysing Matrix E tubes (MP Biomedicals, 116914050), and homogenized in a bead-beater (Mini-Beadbeater-24, BioSpec) for 5 minutes at room temperature. The sample was incubated with 200 μL of chloroform at room temperature for 10 minutes, followed by centrifugation at 16,000 × g for 15 minutes at 4 °C. Next, 500 μL of the upper aqueous phase was transferred into a new tube and 500 μL of 100% ethanol was added. To isolate RNA, we used the PureLink RNA Mini Kit (Life Technologies, catalog #: 12183025). This mixture was transferred onto a PureLink spin column and spun at ≥ 12,000 × g for 30 seconds. The column was washed with 350 μl of wash buffer I as described in the PureLink manual. The column was incubated with 80 μl of PureLink DNase (Life Technologies, catalog #: 12185010) at room temperature for 15 minutes, and washed with 350 μl of wash buffer I. The column was washed with wash buffer II twice as described in the PureLink manual. Total RNA was recovered in 50 μl of RNAase-free water. A second round of off-column DNAse treatment was undertaken. The RNA was incubated with 6 μl of TURBO DNAse (Ambion, ThermoFisher, catalog #: AM2238) at 37°C for 30 minutes. To stop the reaction, 56 μl of lysis buffer from the PureLink kit and 56 μl of 100% ethanol was added to the sample and vortexed. This suspension was transferred onto a PureLink column and washed once with 350 μl of wash buffer I and twice with 500 μl of wash buffer II. The RNA was recovered in 30 μl of RNAse-free water.
rRNA depletion, library generation, and RNA sequencing
Total RNA was subjected to rRNA depletion using Ribo-Zero Bacterial rRNA Depletion (Illumina, catalog #: MRZB12424), following the manufacturer’s protocol. RNA fragmentation, cDNA synthesis, and library preparation proceeded using NEBNext Ultra RNA Library Prep Kit for Illumina (New England BioLabs, catalog #: E7530) and NEBNext Multiplex Oligos for Illumina, Dual Index Primers (New England BioLabs, catalog #: E7600), following the manufacturer’s protocol. All samples were single end sequenced (1×50 bp) using an Illumina HiSeq2500 platform (High Output, v4 chemistry) at UCSF’s Institute for Human Genomics. For each sample, fastq files are available in NCBI’s Sequence Read Archive (SRA), accession number PRJNA656577.
Untargeted metabolomics of in vitro cultures
As described in our in vitro bacterial growth studies, 48-hour overnight cultures were grown in an anaerobic chamber in liquid BHI+ for the following isolates: Bacteroidetes thetaiotaomicron and Clostridium asparagiforme (Table S4). Each subculture was diluted down to an OD600 of 0.08–0.1, which was then further diluted 100-fold, and then used to inoculate a 96-well 2-ml deep well plate with 100 μg/ml MTX (n=6 replicates per isolate) (Sigma, Cat#M9929) or DMSO (n=2 replicates per isolate). After 24 hours, aliquots were removed, centrifuged on a tabletop centrifuge at 4°C for 5 minutes at ~2000 rcf, and supernatant was prepared for extraction and untargeted LC-MS as described.
Untargeted metabolomics sample processing
Extraction was done in 96 well plates. In brief, 60μL of the supernatant from the 96 well culture plate was added to 120μL of pre-cooled methanol/acetonitrile (1:1, v/v) in a non-sterile 96 well plate. The samples were then incubated at −20°C for 1 hour. After incubation, samples were mixed at 4°C at 650 rpm using small block tube shaker. The plates were then centrifuged at 3000 rpm for 10 minutes. After centrifugation, using multi-channel pipette, 80μL of supernatant from each well was transferred to non-sterile 96 well polypropylene plate and then sealed with pre-slit silicone mat. A 1:1:1 mix of LC-MS grade H2O, LC-MS grade acetonitrile and LC-MS grade methanol was used as blank.
UPLC-MS experiments for untargeted metabolomics data acquisition
Samples (5 μl) were separated by hydrophilic interaction chromatography HPLC using a Vanquish UHPLC system (Thermo Fisher Scientific, Waltham, MA) with a Waters (Milford, MA) Acquity UPLC BEH Amide Column (2.1 × 100 mm × 1.7μm particle size) maintained at 65 °C and a 17 minute gradient,at a flow rate of 400 μl/min. Solvent A was 100% HPLC grade water with 0.1% formic acid and 10 mM ammonium formate, Solvent B was 95% HPLC grade acetonitrile and 5% HPLC grade water with 0.1% formic acid and 10 mM ammonium formate. The initial condition was 100 % B, decreasing to 70% B at 7.7 minutes, 40% B at 9.5 minutes, 30% B at 10.25 minutes, and 100% B at 12.750 minutes where it was held until 17 minutes. The eluate was delivered into an Orbitrap Fusion Lumos Tribrid™ mass spectrometer using a H-ESI™ ion source (all Thermo Fisher Scientific). The mass spectrometer was scanned from 75–750 m/z at a resolution of 120,000 and operated in polarity switching mode. The capillary voltage was set at 3.25 kV in positive ion mode, and 2.025 kV in negative ion mode with an RF lens value of 30%, and an AGC target of 1.0E + 6 with a maximum injection time of 50 ms. Product ion MS/MS spectra were acquired in AcquireX mode, with one exclusion blank, then three subsequent sample injections. The parent ion was isolated using the quadrupole with an isolation window of 1.5 Da, a stepped HCD (10,25,40 V) was used for activation, the mass spectrometer was operated at a resolution of 7500, and 3 microscans were acquired for each MS2 spectra.
Metabolomics dataset processing
The raw data were converted into mzML format using ProteoWizard v3.0. Converted HILIC files were then analyzed using MS-DIAL v4.16 for deconvolution, peak detection, alignment, and identification (Parameters file can be provided upon request). MassBank of North America (MoNA) reference database was used for identification. Alignment results were exported and then normalized by TIC of each sample. Statistical analyses were done using R (v4.0.1) as described below.
Ex vivo incubation of RA patient stool samples
All work was carried out in an anaerobic chamber. For each patient, stool was aliquoted into a pre-equilibrated cryovial, diluted in reduced PBS at 10 ml per 1 gram of stool, and vortexed to homogenize the sample. The sample was spun at ~20g for 1 minute on a mini-centrifuge to facilitate settling of sediment, and the sediment-free supernatant was then aliquoted into a new pre-equilibrated cryovial for evaluation of ex vivo growth. Growth was evaluated by inoculating liquid BHI with 1:50 dilution of this fecal slurry, with OD600 readings performed every 15 minutes for 48 hours with a 2-minute shake prior to each reading. Samples were treated with MTX 100 μg/ml or an equal volume of DMSO at time zero. Each patient’s fecal slurry and treatment was evaluated in quadruplicate. Samples from four individuals (Donors 2, 4, 5, and 6) underwent 16S sequencing and analysis at 0 and 24 hours after of treatment with either DMSO or MTX treatments (4 replicates per treatment group) as described in the section titled “16S-seq of humanized mouse gut microbiota.”
Shotgun sequencing of rheumatoid arthritis patient samples
Stool samples from 17 RA patients (Table S1A) underwent DNA extraction using ZymoBIOMICS 96 MagBead DNA Kit (Cat#D4302) as per the manufacturer’s protocol. Sequencing libraries were generated using the Nextera DNA Flex Library Kit (Illumina, #20018705) and Nextera Compatible Unique Dual Indices - Set A primers (Illumina, #20027213). Library concentrations were normalized using PicoGreen (Quant-It dsDNA; Life Technologies) and pooled for sequencing on the NovaSeq using S1 or S2 platform at the Chan Zuckerberg Biohub.
Colon Histology
Approximately 1 cm sections from the distal colon were collected for histology from mice treated with or without DSS in the fecal microbiota transplant (FMT) studies. Samples were fixed in formalin for at least 24 hours and subsequently stored in 70% ethanol. Samples were processed by the UCSF Biorepository and Tissue Biomarker Technology Core. Tissues were embedded in wax and 4 μm cross-sections were H & E stained.
Lamina propria lymphocyte isolation
Lamina propria lymphocytes were isolated with slight modifications of previously described methods (Atarashi et al., 2011; Kubinak et al., 2015; Round et al., 2011). In brief, small intestinal (SI) Peyer’s patches were removed and colons and SI tissue were splayed longitudinally with mucus removed by scraping and stored in complete RPMI (10% fetal bovine serum, 100 units per ml penicillin and streptomycin, β-mercaptoethanol, glutamate, sodium pyruvate, HEPES and non-essential amino acids). Supernatants were removed by filtering through a 100μM filter, and remaining tissue incubated in 1X HBSS (without Ca2+ and Mg2+) containing 5 mM EDTA (Promega) and 1 mM DL-Dithiothreitol (DTT) (Bioplus chemicals) for 45 minutes at 37°C on a shaker. Supernatant was removed by filtering through a 100 μM filter, and remaining tissue was incubated for 45 minutes (colon) or 35 minutes (SI) at 37°C on a shaker in a solution containing 1X HBSS containing 5% (v/v) fetal bovine serum (GIBCO heat inactivated), 1 U/ml Dispase (Sigma), 0.5 mg/ml Collagenase VIII (Sigma), and 20 μg/ml DNaseI (Sigma). The supernatant was filtered over a 40 mm cell strainer into ice-cold sterile 1X PBS. Cells were subjected to a Percoll (VWR) gradient (40%/80% [v/v] gradient) and spun at 2000 rpm for 20 minutes with no brake and no acceleration. Cells at the interface were collected, washed in PBS and prepared for flow cytometry analysis.
Flow cytometry
Since RA pathophysiology is associated with a dysregulated T cell response (Imboden, 2009), we focused on the T cell compartment in the spleen and the intestinal lamina propria of the small intestine and colon using flow cytometry. Lymphocytes were isolated from the colonic and small intestinal lamina propria as described above. Spleen cells were prepped through gentle mashing with a syringe plunger. Spleen cells were treated with 1X RBC Lysis Buffer (Biolegend) to lyse and remove red blood cells. Surface staining for lymphocytes was done in staining buffer (1X HBSS (Corning) supplemented with 10 mM HEPES (Cellgro), 2 mM EDTA (Promega), and 0.5% (v/v) fetal bovine serum (GIBCO heat inactivated) for 20 minutes at 4°C. Cells were then washed twice in supplemented 1X HBSS and enumerated via flow cytometry. The following antibodies were used: anti-CD3 (17A2, Invitrogen, Cat. 11-0032-82), anti-CD4 (GK1.5, Biolegend, Cat. 100428), anti-CD69 (H1.2F3, Biolegend, Cat. 104511-BL), anti-CD11b (M1/70, Biolegend, Cat. 101228-BL), anti-CD44 (IM7, Tonbo biosciences, Cat. 10050–486), anti-Gr1 (clone RB6–8C5, Life Technologies, Cat. 108408), anti-TER119 (APC clone TER-119, Biolegend, Cat. 116212), and anti-B220 (clone RA3–6B2, Invitrogen, Cat. 5014055). For intracellular staining, cells were first stimulated with ionomycin (1000 ng/ml), PMA (50 ng/ml), and Golgi Plug (1 μl/sample) (BD Bioscience) overnight at 37°C. Stimulated cells were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain Kit (Thermo Fisher) CD3+ CD4+ cells were assessed within the live population. Cells were surface stained, washed, and then fixed/permeabilized in 100μl Perm/Fix buffer (BD Bioscience). Cells were washed twice in Perm/Wash buffer (BD Bioscience) and then stained for intracellular cytokines with the following antibodies: anti-IFN-γ (XMG1.2, Millipore, Cat. MABF151), anti-IL-17A (ebio17B7, Invitrogen, Cat. 25-7177-82), anti-Foxp3 (150D, Biolegend, Cat. BDB563902). Cells were washed twice in Perm/Wash buffer and then placed in staining buffer for flow cytometry analysis. Gating cell populations was done using isotype and single stain controls. Representative gating strategies are provided in Figure S6. These data were collected with a BD LSR Fortessa and analyzed with FlowJo software.
QUANTIFICATION AND STATISTICAL ANALYSIS
General
No statistical methods were used to pre-determine sample sizes. All statistical analyses were performed using the R environment. “N.S.” or “ns” indicates “not significant.”
16S rRNA amplicon analysis of mouse and human samples
Reads that were not already demultiplexed were demultiplexed using QIIME (Caporaso et al., 2010) v1.9.1 (split_libraries_fastq.py). For all reads except NYU RA patient samples, QIIME2 (Bolyen et al., 2019)(v 2020.2) was used to trim reads, denoise the data, and create a feature table using the following commands: qiime cutadapt trim-paired, qiime dada2 denoise-paired, qiime feature-table filter-samples. Within QIIME2, taxonomy was assigned using the DADA2 (Callahan et al., 2016a) implementation of the RDP Naïve Bayesian classifier (Wang et al., 2007) using the DADA2-formatted SILVA v128 training set; taxonomic assignments to the species level were chosen by exact match of the ASV to the reference database, and where multiple matches were present, all are reported. For figures and in the text, genus and species assignments are reported when available, and when neither was available, we included the family taxonomic assignment; we also report a unique ASV identifier. This ASV identifier can be used to look up the full taxonomic assignment from kingdom to species and the associated sequence variant in Table S2B. A phylogenetic tree was constructed in QIIME2 using the command: phylogeny align-to-tree-mafft-fasttree. The QIIME2R package was used to import QIIME2 artefacts into R (https://github.com/jbisanz/qiime2R). For RA patient samples from NYU, reads were demultiplexed using QIIME (Caporaso et al., 2010) v1.9.1 (split_libraries_fastq.py) before denoising and processing with DADA2 (Callahan et al., 2016a) v1.1.5 under MRO v3.2.5. Taxonomy was assigned using the DADA2 implementation of the RDP classifier (Wang et al., 2007) using the DADA2-formatted SILVA v128 training set (benjjneb.github.io/dada2/assign.html). Low abundance taxa were filtered out (>10 raw reads) as well as ASVs that could not be assigned taxonomy past the kingdom level. Diversity metrics were generated using Vegan (v2.5–6) and Phyloseq (v1.30.0) (McMurdie and Holmes, 2013), with principal coordinate analysis (PCoA) or principal components analysis (PCA) carried out with Ape (v5.3) or Vegan, respectively. Analyses were carried out on either: (1) centered log2-ratio (clr) normalized taxonomic abundances calculated as Aclr=[log2(A1/ga), log2(A2/ga),… log2(An/ga),], where A is a vector of non-zero read counts and ga is the geometric mean of all values of A, or (2) relative abundance calculated as proportion of reads. ANOSIM and PERMANOVA were used to detect changes in community composition using counts from rarefied data and Bray-Curtis distances. DESeq2 (v1.26.0) (Love et al., 2014) was used to determine differentially abundant taxa on raw count data. Some ASVs were reported to have log2 fold changes that were 20 or higher. We found these large fold changes to be driven by outliers, which can sometimes occur with 16S-seq datasets. We used the shrinkage estimators suggested by the creators of DESeq2 but found that because the calculations were driven by outliers, the log2 fold changes using these estimators were also large. Therefore, we report the results as is, and note that the percentage of differentially abundant ASVs with log2 fold changes > 20 represent a small fraction of all the significant findings (ranging from 0–34% of significant ASVs, with a median of 14%). Significance testing of longitudinal trends was determined using generalized mixed effects models using the cplm package (Sharpton et al., 2017; Zhang, 2013) (v. 0.7–7) on clr normalized values. UpSet plots were created with the UpSetR (v1.4.0) package in R (Conway et al., 2017). For each sample, fastq files are available in NCBI’s Sequence Read Archive (SRA), accession number PRJNA656577.
Summary of 16S rRNA amplicon sequencing from multiple gnotobiotic mouse experiments
To assess phylum-level enrichments among differentially abundant ASVs (Table S2D), a hypergeometric test was used to compare the number in each phylum among differentially abundant ASVs vs. the number present in the microbial community. The phyper function from the stats (v3.5.1) package in R was used. ANOVA was used to compare alpha diversity from multiple gnotobiotic experiments in which mice were treated with MTX. To assess changes at the phylum, genus and ASV level, a multi-factor design was carried out using DESeq2 including experiment (dose-response, individually housed, route, rescue; Table S1B, Experiment 1–4) and MTX treatment (absence, presence) (e.g., ~ experiment + MTX). For this analysis, the low-dose samples from the dose-response experiment were excluded. We used the comparator groups that we used when analyzing each experiment on its own. For the initial dose-response experiment (Table S1B, Experiment 1), high-dose was compared to PBS on day 4. Similarly, for individually housed mice, high-dose MTX was compared to PBS (Table S1B, Experiment 2) on day 4. For the route and rescue (Table S1B, Experiment 3 and 4), day 0 was compared to day 2. These were coded as “absence” or “presence” of MTX. Multi-factor design was also used to assess changes at the phylum, genus, and ASV level on day 14 after colonization when comparing pre-MTX and post-MTX recipient mice microbiota in the FMT experiments (Table S1B, Experiment 5–7) (e.g., ~donor + colonization).
qPCR for 16S copy number determination
qPCR of total 16S rRNA gene copies was carried out in triplicate. Ten μL reactions with 200nM 891F(5’-TGGAGCATGTGGTTTAATTCGA-3’)/1003R(5’-TGCGGGACTTAACCCAACA-3’) primers using a BioRad CFX384 thermocycler with iTaq™ Universal Probes Supermix (BioRad 1725132) and probe 1002P ([Cy5]CACGAGCTGACGACARCCATGCA[BHQ3]) were carried out according to the manufacturer’s instructions and an annealing temperature of 60°C. Absolute quantifications were calculated against a standard curve of 8F/1542R amplified from purified bacterial DNA. Reactions were performed in triplicate and mean values were taken for further analyses. Absolute bacterial abundance was derived by adjustments for dilutions during DNA extraction, normalization, and PCR reaction preparation dividing by the total fecal mass used for DNA extraction in grams. Significance was assessed using two-way ANOVA comparing baseline and post-treatment measurements (copyNumber~Treatment*Day).
In vitro bacterial growth and rescue studies
Growth parameters (carrying capacity, time to mid-exponential and growth rate) were calculated in R using GrowthCurveR (Sprouffske and Wagner, 2016) (v0.2.1). Curves that could not be fit by the software (i.e., generated a warning/error message) were excluded from further analysis. We visually inspected the fits for the remaining curves and removed curves with an area under the curve of <52, or estimated time to mid-exponential > 48 hours which we found to be indicators of poor fit in our dataset. Determination of dose-dependency was done by fitting a linear regression with methotrexate concentration as the independent variable and the estimated growth parameter as the dependent variable using lm from the stats package (v3.5.1). For rescue studies, as above, growth parameters (carrying capacity, time to mid-exponential, growth rate, and area under the curve) were calculated in R using GrowthCurveR (Sprouffske and Wagner, 2016) (v0.2.1). We evaluated 4 isolates with varying sensitivity to MTX (B. thetaiotaomicron, B. vulgatus, C. innocuum and C. symbiosum). C. asparagiforme was excluded because rescue of growth would be difficult to identify in this MTX resistant strain (Figure 2D). We measured growth curves at 9 concentrations of MTX tested against 7 concentrations of folic acid. Determination of dose-dependency at each tested concentration of MTX was done by fitting a linear regression with folic acid or leucovorin concentration as the independent variable and the estimated growth parameter as the dependent variable using lm from the stats package (v3.5.1). We also included an interaction term to assess whether the rescue agent demonstrated an interaction with MTX concentration (e.g., AUC ~ MTX + rescue + MTX:rescue). We used a p-value cutoff of p<0.05 to identify effects that were dose-dependent based on the linear regression.
RNA-seq analysis
Reads were trimmed using Trimmomatic (v0.32) (Bolger et al., 2014), and ribosomal reads were removed using SortMeRNA (v2.1) (Kopylova et al., 2012). Reads were mapped to reference genomes using Bowtie2 (Langmead and Salzberg, 2012) using the following options: -q, --met-file, --end-to-end, --sensitive. HTSeq (v 0.8.0) was used to count the number of transcripts mapping to genes (Anders et al., 2015) using the following options: --type=CDS, --idattr:ID, --stranded=no, --minaqual=10. Differential gene expression was assessed using DESeq2 (Love et al., 2014) using the DESeqDataSetFromHTSeqCount and ddsHTSeq functions and default options. Different padj thresholds ranging from 0.01 to 0.2 were used to determine the number of differentially expressed genes, and irrespective of the threshold used, consistent percentages of each bacterial genome were affected by MTX, thus a padj threshold of 0.2 was used in subsequent analyses. We also used edgeR to identify differential gene expression (Robinson et al., 2010) using the glmQLFit and glmQLFTest and the default settings, and found slightly fewer number of differentially expressed genes per isolate, but similar numbers of up- versus downregulated genes in each isolate. BlastKOALA (Kanehisa et al., 2016) was used to map protein sequences from each organism to KO terms using the species_prokaryote database. KEGG pathway enrichment was carried out using clusterProfiler (Yu et al., 2012) (v3.4.1) using the enrichKEGG function. KO terms for all differentially expressed genes (both up- and downregulated with padj<0.2, DESeq) were provided and the organism parameter was set to “ko”. Heatmaps depicting enrichments were generated using the geom_tile function of ggplot2 R package (v3.3.0).
Analysis of untargeted metabolomics of in vitro cultures
For each sample, metabolite feature areas were normalized to relative total ion counts (TIC). For Figure 3D, multiple metabolite features mapped with high confidence to AMP, guanine, adenine, and hypoxanthine, so the TICs for these were summed in each sample. For statistical analysis, metabolite features that were differentially abundant between the sterile samples with MTX vs. DMSO were excluded from further analyses (p<0.05, Wilcoxon rank sum test). To identify metabolite features that responded differently to MTX in Bacteroides thetaiotaomicron versus Clostridium asparagiforme, we assessed for peaks showing a significant interaction between bacterial isolate and MTX treatment; to do this, we used a two-factor ANOVA with terms for bacterial isolate, drug, and the interaction between the two. We used the BH method to correct for multiple testing using p.adjust in the stats package (v3.5.1) in R. We identified metabolite features showing a significant change with drug (drug padj<0.01) or a significant interaction between bacterial isolate and drug (interaction term, padj<0.01). Lists containing m/z values, retention times, and interaction term p-values was submitted for analysis to the MetaboAnalyst software suite “MS Peaks to Pathways” pipeline. The following parameters were set: molecular weight tolerance = 10 ppm, positive mode, p-value, enforce primary ions checked, version 2.0, mummichog and GSEA checked, P-value cutoff = 0.01, KEGG version current (Oct2019), and Pathway library “eco_kegg.”
Analysis of ex vivo incubation of RA patient stool samples
Growth curves were averaged by treatment and individual, and growth parameters were estimated using the GrowthCurveR package (v 0.2.1). Paired Student’s t-tests were used to determine changes in growth parameters. Of the 30 RA patients whose baseline fecal samples were incubated with MTX in this analysis, 23 were also included in a companion paper (Artacho et al., 2020)(Table S1A). Seven additional patients included here were excluded from the Artacho et al paper because of failure to follow-up and lack of information on clinical response to MTX.
Shotgun sequencing of rheumatoid arthritis patient samples
Demultiplexed reads were filtered/trimmed using fastp (Chen et al., 2018) (v0.20.0; parameters --detect_adapter_for_pe, --cut_front, --cut_tail, --cut_window_size 4, --cut_mean_quality 20, --length_required 60), aligned to the human genome (GCR38) using Bowtie2 (Langmead and Salzberg, 2012). Taxonomic profiling was done using MetaPhlAn2 (v2.7.7) (Truong et al., 2015) and functional profiling was done using HUMAnN2 (v0.11.1) (Franzosa et al., 2018) using default parameters. Of 1,136,322 gene families quantified by HUMAnN2, 168,804 were present in at least 50% of samples, and these were carried forward for further analysis. There were 5,011 KOs identified by HUMAnN2 (using the utility function humann2_regroup_table). MaAsLin2 (v 0.99.12) (Mallick) was used to determine differential abundance of gene families and KOs using default parameters. Patient ID, clinic site of stool collection, and sequencing run were coded as random variables. Fixed variables included Month, Age, Gender, Ethnicity, and Disease Duration. KEGG pathway enrichment was carried out using clusterProfiler (Yu et al., 2012) (v3.4.1) using the enrichKEGG function. KO terms for all differentially expressed genes (both up- and down-regulated with q-value<0.25) were provided and the organism parameter was set to “ko”. For each sample, fastq files are available in NCBI’s Sequence Read Archive (SRA), accession number PRJNA656577. Determination of whether a gene family was associated with purine or pyrimidine synthesis was achieved by using the pubmedR package (v 0.0.2) to search PubMed with the following query “gene family name and (purine or pyrimidine).” Results were manually curated.
DSS Colitis Scoring
Mice were monitored for disease progression and weighed daily. Gross signs of toxicity, including hematochezia and weight loss were observed in this study. Stools were scored as follows: 0 = normal stool consistency, 1 = soft stool, 2 = blood in stool, 3 = bloody anus, 4 = prolapsed anus, 5 = moribund/death. Scoring was done by an observer who was blinded to the transplant group.
Analysis of immune cell markers from transplanted gnotobiotic mice
Quantification of cell populations was done by one of the authors (M.A.) who was blinded to the transplant group. All reported values are reported as percentage of cells or fold change percentage in which values are normalized to the pre-MTX group for each donor and treatment condition. Linear mixed effects modeling was carried out using the lme function within the nlme R package (v3.1), and donor ID was coded as a random variable. Post-MTX group was compared to the pre-MTX group in either the unchallenged or challenged mice. A heatmap of the fold change percentages in each treatment group and site was generated using the geom_tile function in the ggplot2 R package (v3.3.0).
Correlation Analysis
We correlated ASV abundances and immunocyte populations from a gnotobiotic experiment (Table S1B, Experiment 5) in which 20 mice were transplanted with pre- and post-MTX microbiota from Donor 2 (Figure S5H). Only a subset of ASVs and immunocyte populations were assessed for correlation. For ASVs, we chose to test those that were previously shown to be modulated by MTX. Thus, we started with 23 ASVs that were altered by MTX in individually housed mice (Figure S1D) colonized with pre-MTX microbiota from Donor 2 (Table S1B, Experiment 2); 21 of the 23 ASVs were present in the communities of mice in Experiment 5. Of these, 12 were present in at least 10 mice, and these were carried forward for assessing correlations. ASV clr-transformed abundances at day 14 were used, which represented microbial abundances prior to perturbation by DSS. For immunocyte populations, we tested only immune markers in each organ that were differentially affected in challenged mice (Figure 6A): B220, CD44+ CD69+ CD4+, Gr1+ CD11b+, and IFN-γ+ CD4+ in the spleen; IL-17A+ CD4+ and Gr1+ CD11b+ in the small intestine; CD44+ CD69+ CD4+ and Gr1+ CD11b+ and in the colon. Spearman’s rank correlation is reported. We report correlations that were nominally significant with p<0.05.
We did not assess correlations in mice transplanted with microbiota from Donors 4 and 5 (Table S1B, Experiments 6 and 7) because these mice had fewer of the 23 MTX-modulated ASVs (ranging between 10–16 ASVs) and the prevalence of these ASVs were low among recipient mice (ranging from 4–11 mice), making it difficult to calculate correlations. This may be expected given that microbial community structure was distinct between donor groups (Figure S5F).
Supplementary Material
Table S1. Metadata for All Human Samples Included in This Study, Related to Figures 1, 5, 6, and 7.
Table S1A. Donors / Patients Included in Our Study and Assays Performed on Them.
Table S1B. Details of Gnotobiotic Experiments Using Human Donor Samples.
Table S1C. Rheumatoid Arthritis Patient Baseline Characteristics.
Table S2. Metadata for All Sequenced Samples and 16S rRNA Gene Sequencing Analysis Results, Related to Figures 1, 5, 6, and 7.
Table S2A. Sample Information for Samples That Underwent 16S rRNA, RNA-Seq, and Shotgun Sequencing.
Table S2B. Taxonomic Lookup Table.
Table S2C. Differentially Abundant ASVs in Multiple 16S-Seq Experiments.
Table S2D. Phylum-Level Enrichments Among Differentially Abundant ASVs (padj<0.01, DESeq).
Table S3. Mouse Phenotypic Results, Related to Figures 1 and 7.
Table S3A. Weight Change With MTX Treatment.
Table S3B. ASVs That Were Correlated With Immune Markers in Transplanted Mice.
Table S5. RNA Sequencing Results, Related to Figure 3.
Table S5A. Differentially Expressed Genes in 4 Isolates Treated With MTX (padj<0.2).
Table S5B. Number of Differentially Expressed Genes at Different Padj Thresholds.
Table S5C. KEGG Enrichments Among Differentially Expressed Transcripts in Bacteroidetes thetaiotaomicron at 30 Minutes (MTX vs. Vehicle Control).
Table S5D. KEGG Enrichments Among Differentially Expressed Transcripts in Clostridium asparagiforme at 30 Minutes, 4 Hours, and 20 Hours (MTX vs. Vehicle Control).
Table S5E. Padj (DESeq) Thresholds and Purine/Pyrimidine Enrichments in B. thetaiotaomicron and C. asparagiforme.
Table S5F. 41 Differentially Expressed Transcripts at All Three Time Points in C. asparagiforme.
Table S6. KEGG Pathway Enrichments Among Differentially Abundant Metabolite Features, Related to Figure 3.
Table S6A. Metabolomic Features That Significantly Differed (padj<0.01, Two-Factor ANOVA With BH correction).
Table S6B. KEGG Pathway Enrichments Among Differentially Abundant Metabolite Features.
Table S6C. Raw Metabolomics Data.
Table S7. Metagenomics Results, Related to Figure 5.
Table S7A. Metagenomic Analysis of the RA Patient Gut Microbiome.
Table S7B. KEGG Pathway Enrichments Among Differentially Abundant KOs.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3 | Fisher Scientific | 11-0032-82 (Clone: 17A2); RRID:AB_2572431 |
| IL-17a | Fisher Scientific | 25-7177-82 (Clone: ebio17b7); RRID:AB_10732356 |
| CD4 | Biolegend | 100428 (Clone: GK1.5); RRID:AB_493647 |
| IFNy | Fisher Scientific | 45-7311-80 (Clone: XMG1.2); RRID:AB_906239 |
| Foxp3 | Fisher Scientific | BDB563902 (Clone: R16–715); RRID:AB_2630318 |
| Gr1 | Biolegend | 108408 (Clone: RB6–8C5); RRID:AB_313373 |
| CD11b | VWR (Biolegend) | 101228-BL (Clone: M1/70); RRID:AB_893232 |
| TER119 | Biolegend | 116212 (Clone: TER-119); RRID:AB_313713 |
| B220 | Fisher Scientific (Invitrogen) | 5014055 (Clone: RA3–6B2); RRID:AB_11220275 |
| CD44 | VWR (Tonbo) | 10050–486 (Clone: IM-7); RRID:AB_2621572 |
| CD69 |
VWR (Biolegend) | 104511-BL (Clone: H1.2F3); RRID: AB_493565 |
| LIVE/DEAD Fixable Dead Cell Stain Kit | Life technologies | L34957 |
| Bacterial and Virus Strains | ||
| Bacteroides thetaiotaomicron | DSM | 2079 |
| Clostridium asparagiforme | DSM | 15981 |
| Clostridium sporogenes | ATCC | 15579 |
| Clostridium symbiosum | DSM | 934 |
| Akkermansia muciniphila | DSM | 22959 |
| Alistipes shahii | DSM | 19121 |
| Bacillus subtilis 168 | DSM | 402 |
| Bacteroides acidifaciens | DSM | 15896 |
| Bacteroides caccae | DSM | 19029 |
| Bacteroides eggerthii | DSM | 20697 |
| Bacteroides finegoldii | DSM | 17565 |
| Bacteroides fragilis | DSM | 2151 |
| Bacteroides ovatus | DSM | 1896 |
| Bacteroides stercoris | DSM | 19555 |
| Bacteroides uniformis | DSM | 6597 |
| Bacteroides vulgatus | DSM | 1447 |
| Bacteroides xylanisolvens | DSM | 18836 |
| Bifidobacterium longum subsp longum | DSM | 20219 |
| Blautia producta | DSM | 3507 |
| Clostridium innocuum | DSM | 1286 |
| Clostridium scindens | DSM | 5676 |
| Collinsella aerofaciens | DSM | 3979 |
| Delftia acidovorans | ATCC | 25012 |
| Dorea formicigenerans | DSM | 3992 |
| Dorea longicatena | DSM | 13814 |
| Edwardsiella tarda | ATCC | 23685 |
| Eggerthella lenta 1-3-56 | DSM | 110906 |
| Eggerthella lenta | DSM | 2243 |
| Eggerthella sinensis | DSM | 16107 |
| Eggerthella sp. | DSM | 11767 |
| Enterococcus faecalis | DSM | 2570 |
| Escherichia coli BW25113 | DSM | 27469 |
| Eubacterium eligens | DSM | 3376 |
| Eubacterium hallii | DSM | 3353 |
| Eubacterium rectale | DSM | 17629 |
| Faecalibacterium prausnitzii | DSM | 17677 |
| Fusobacterium nucleatum subsp. nucleatum | DSM | 15643 |
| Gordonibacter pamelae | DSM | 110924 |
| Lactococcus lactis | DSM | 20481 |
| Lactonifactor longoviformis | DSM | 17459 |
| Parabacteroides distasonis | DSM | 20701 |
| Parabacteroides merdae | DSM | 19495 |
| Prevotella copri | DSM | 18205 |
| Providencia rettgeri | DSM | 4542 |
| Ruminococcus obeum | DSM | 25238 |
| Biological Samples | ||
| Stool samples from healthy and rheumatoid arthritis patients | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Methotrexate (Pharmaceutical Grade) for mouse experiments | Fresenius Kabi | Product # 102250 |
| Methotrexate for in vitro and ex vivo studies | Sigma Aldrich | M9929–100MG |
| Folic acid | Fisher Scientific | BP25195 |
| Leucovorin | Spectrum Chemicals | LE117–100MG |
| Dextran sodium sulfate | Alfa Aesar | Cat no. 9011-18-1 |
| Critical Commercial Assays | ||
| ZymoBIOMICS 96 MagBead DNA Kit | Zymo | D4302 |
| KAPA HiFi Hot Start PCR kit | Kapa Biosystems | KK2502 |
| SequalPrep Normalization Plate | Life Technologies | A10510–01 |
| Qiagen MinElute Gel Extraction Kit | Qiagen | 28604 |
| Qiagen MinElute PCR Purification Kit | Qiagen | 28004 |
| Nextera DNA Flex Library Kit | Illumina | 20018705 |
| Nextera Compatible Unique Dual Indices - Set A primers | Illumina | 20027213 |
| Deposited Data | ||
| 16S rRNA, shotgun, and RNA-seq data | NCBI SRA | PRJNA656577 |
| Metabolomics data | This paper | Table S6C |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Germ free C57BL/6 | UCSF Gnotobiotics Core | N/A |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and Algorithms | ||
| R (v3.6.2) | Packages for analytical parts: phyloseq, tidyverse, ape, vegan, nlme, stats, DEseq2, edgeR, GrowthCurveR, MaSaLin2, ggtree, clusterprofiler, dada2, UpSetR, pubmedR | https://www.r-project.org/ |
| QIIME (v1.91) | Caporaso et al., 2010 | http://qiime.org/ |
| QIIME2 (v2020.2) | Boylen et al., 2019 | https://qiime2.org/ |
| UniPro UGENE (v33) | Okonechnikov et al., 2012 | http://ugene.net/ |
| Trimmomatic (v0.32) | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| SortMeRNA (v2.1) | Kopylova et al., 201 | https://github.com/biocore/sortmerna |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| BlastKOALA | Kanehisa et al., 2016 | https://www.kegg.jp/blastkoala/ |
| Fastp (v0.20.0) | Chen et al., 2018 | https://github.com/OpenGene/fastp |
| MetaPhlAn2(v2.7.7) | Truong et al., 2015 | https://huttenhower.sph.harvard.edu/metaphlan |
| HUMAnN2 (v0.11.1) | Franzosa et al., 2018 | https://huttenhower.sph.harvard.edu/humann |
| Other | ||
Highlights.
Methotrexate alters the human gut microbiota in cell culture, mice, and patients
Methotrexate impacts the expression and activity of conserved metabolic pathways
Shifts in the gut microbiota are associated with clinical drug response
The post-methotrexate gut microbiota decreases immune activation in mice
ACKNOWLEDGEMENTS
Special thanks to Lindsey Criswell, Kathy Lam, Mary Nakamura, and Katie Pollard for comments on the manuscript. We are indebted to Jessie Turnbaugh, Gregory Ostolaza, and the UCSF Gnotobiotic Core (gnotobiotics.ucsf.edu) for technical assistance as well as the Institute for Human Genetics for help with RNA sequencing and the CZ Biohub for DNA sequencing. We thank Jordan E. Bisanz and Elizabeth N. Bess for their assistance with development of sequencing methods. We thank Dr. Philip B. Smith from the Metabolomics Core Facility at The Pennsylvania State University. This work was funded by the National Institutes of Health: R01HL122593, 1R21CA227232 (P.J.T.); 5T32AR007304–37, TR001871, 1K08AR073930 (R.R.N.); R03AR072182 (J.U.S.); S10OD021750 (A.D.P); and R01AR074500 P.J.T./J.U.S.). Additional support was provided by the Searle Scholars Program (SSP-2016–1352), MINECO (SAF2017–90083-R), and the Rheumatology Research Foundation (AWD00003947). P.J.T. is a Chan Zuckerberg Biohub investigator and was a Nadia’s Gift Foundation Innovator supported, in part, by the Damon Runyon Cancer Research Foundation (DRR-42–16). J.U.S is further supported by The NYU Colton Center for Autoimmunity, The Riley Family Foundation, and The Snyder Family Foundation. This work was supported by the UCSF Breakthrough Program for Rheumatoid Arthritis-related Research (BPRAR; partially funded by the Sandler Foundation), MedImmune, and the Arthritis Foundation Center for Excellence.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
P.J.T is on the scientific advisory board for Kaleido, Pendulum, Seres, and SNIPRbiome; there is no direct overlap between the current study and these consulting duties. R.R.N, J.U.S. and P.J.T are listed as inventors on a patent application (33167/55261P1) related to this work.
REFERENCES
- Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD, et al. (2010). 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum 62, 2569–2581. [DOI] [PubMed] [Google Scholar]
- Allegra CJ, Hoang K, Yeh GC, Drake JC, and Baram J (1987). Evidence for direct inhibition of de novo purine synthesis in human MCF-7 breast cells as a principal mode of metabolic inhibition by methotrexate. J Biol Chem 262, 13520–13526. [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al. (1988). The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31, 315–324. [DOI] [PubMed] [Google Scholar]
- Artacho A, Isaac S, Nayak RR, Flor-Duro A, Alexander M, Koo I, Manasson J, Smith P, Rosenthal P, Homsi Y, et al. (2020). The pre-treatment gut microbiome is associated with lack of response to methotrexate in new onset rheumatoid arthritis. Arthritis Rheum In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleyer WA (1978). The clinical pharmacology of methotrexate: new applications of an old drug. Cancer 41, 36–51. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolin JT, Filman DJ, Matthews DA, Hamlin RC, and Kraut J (1982). Crystal structures of Escherichia coli and Lactobacillus casei dihydrofolate reductase refined at 1.7 A resolution. I. General features and binding of methotrexate. J Biol Chem 257, 13650–13662. [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol 37, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, and Holmes SP (2016a). DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, Sankaran K, Fukuyama JA, McMurdie PJ, and Holmes SP (2016b). Bioconductor workflow for microbiome data analysis: from raw reads to community analyses. F1000Res 5, 1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabner BA, and Young RC (1973). Threshold methotrexate concentration for in vivo inhibition of DNA synthesis in normal and tumorous target tissues. J Clin Invest 52, 1804–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Wright K, Davis JM, Jeraldo P, Marietta EV, Murray J, Nelson H, Matteson EL, and Taneja V (2016). An expansion of rare lineage intestinal microbes characterizes rheumatoid arthritis. Genome Med 8, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong J, Wishart DS, and Xia J (2019). Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr Protoc Bioinformatics 68, e86. [DOI] [PubMed] [Google Scholar]
- Conway JR, Lex A, and Gehlenborg N (2017). UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronstein BN (1996). Molecular therapeutics. Methotrexate and its mechanism of action. Arthritis Rheum 39, 1951–1960. [DOI] [PubMed] [Google Scholar]
- Cronstein BN, and Sitkovsky M (2017). Adenosine and adenosine receptors in the pathogenesis and treatment of rheumatic diseases. Nat Rev Rheumatol 13, 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren D, and Lennernas H (2019). Intestinal permeability and drug absorption: predictive experimental, computational and in vivo approaches. Pharmaceutics 11, 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, and Andersen GL (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72, 5069–5072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressman JB, Amidon GL, and Fleisher D (1985). Absorption potential: estimating the fraction absorbed for orally administered compounds. J Pharm Sci 74, 588–589. [DOI] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, et al. (2018). Species-level functional profiling of metagenomes and metatranscriptomes. Nat Methods 15, 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genestier L, Paillot R, Quemeneur L, Izeradjene K, and Revillard JP (2000). Mechanisms of action of methotrexate. Immunopharmacology 47, 247–257. [DOI] [PubMed] [Google Scholar]
- Gerards AH, de Lathouder S, de Groot ER, Dijkmans BA, and Aarden LA (2003). Inhibition of cytokine production by methotrexate. Studies in healthy volunteers and patients with rheumatoid arthritis. Rheumatology (Oxford) 42, 1189–1196. [DOI] [PubMed] [Google Scholar]
- Gohl DM, Vangay P, Garbe J, MacLean A, Hauge A, Becker A, Gould TJ, Clayton JB, Johnson TJ, Hunter R, et al. (2016). Systematic improvement of amplicon marker gene methods for increased accuracy in microbiome studies. Nat Biotechnol 34, 942–949. [DOI] [PubMed] [Google Scholar]
- Grim J, Chladek J, and Martinkova J (2003). Pharmacokinetics and pharmacodynamics of methotrexate in non-neoplastic diseases. Clin Pharmacokinet 42, 139–151. [DOI] [PubMed] [Google Scholar]
- Guhad F (2005). Introduction to the 3Rs (refinement, reduction and replacement). Contemp Top Lab Anim Sci 44, 58–59. [PubMed] [Google Scholar]
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, and Gascuel O (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59, 307–321. [DOI] [PubMed] [Google Scholar]
- Halilova KI, Brown EE, Morgan SL, Bridges SL Jr., Hwang MH, Arnett DK, and Danila MI (2012). Markers of treatment response to methotrexate in rheumatoid arthritis: where do we stand? Int J Rheumatol 2012, 978396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimesaat MM, Bereswill S, Fischer A, Fuchs D, Struck D, Niebergall J, Jahn HK, Dunay IR, Moter A, Gescher DM, et al. (2006). Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J Immunol 177, 8785–8795. [DOI] [PubMed] [Google Scholar]
- Hickey CA, Kuhn KA, Donermeyer DL, Porter NT, Jin C, Cameron EA, Jung H, Kaiko GE, Wegorzewska M, Malvin NP, et al. (2015). Colitogenic Bacteroides thetaiotaomicron antigens access host immune cells in a sulfatase-dependent manner via outer membrane vesicles. Cell Host Microbe 17, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra M, Haagsma C, Neef C, Proost J, Knuif A, and van de Laar M (2004). Bioavailability of higher dose methotrexate comparing oral and subcutaneous administration in patients with rheumatoid arthritis. J Rheumatol 31, 645–648. [PubMed] [Google Scholar]
- Hooper LV, Littman DR, and Macpherson AJ (2012). Interactions between the microbiota and the immune system. Science 336, 1268–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imboden JB (2009). The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol 4, 417–434. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, and Morishima K (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol 428, 726–731. [DOI] [PubMed] [Google Scholar]
- Kopylova E, Noe L, and Touzet H (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. [DOI] [PubMed] [Google Scholar]
- Kopytek SJ, Dyer JC, Knapp GS, and Hu JC (2000). Resistance to methotrexate due to AcrAB-dependent export from Escherichia coli. Antimicrob Agents Chemother 44, 3210–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama A, Tanaka A, and To H (2017). Daily oral administration of low-dose methotrexate has greater antirheumatic effects in collagen-induced arthritis rats. J Pharm Pharmacol 69, 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O’Connell RM, and Round JL (2015). MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe 17, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letertre MPM, Munjoma N, Wolfer K, Pechlivanis A, McDonald JAK, Hardwick RN, Cherrington NJ, Coen M, Nicholson JK, Hoyles L, et al. (2020). A two-way interaction between methotrexate and the gut microbiota of male Sprague-Dawley rats. J Proteome Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Kurakawa T, Umemoto E, Motooka D, Ito Y, Gotoh K, Hirota K, Matsushita M, Furuta Y, Narazaki M, et al. (2016). Dysbiosis contributes to arthritis development via activation of autoreactive T Cells in the intestine. Arthritis Rheumatol 68, 2646–2661. [DOI] [PubMed] [Google Scholar]
- Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, Brochado AR, Fernandez KC, Dose H, Mori H, et al. (2018). Extensive impact of non-antibiotic drugs on human gut bacteria. Nature 555, 623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick HT,TL; McIver LJ; Rahnavard G; Nguyen LH; Weingart G; Ma S; Ren B; Schwager E; Subramanian A; Paulson JN; Franzosa EA; Corrada, Bravo H; Huttenhower C (2020). Multivariable association in population-scale meta’omic surveys. In Submission. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manasson J, Shen N, Garcia Ferrer HR, Ubeda C, Iraheta I, Heguy A, Von Feldt JM, Espinoza LR, Garcia Kutzbach A, Segal LN, et al. (2018). Gut microbiota perturbations in reactive arthritis and postinfectious spondyloarthritis. Arthritis Rheumatol 70, 242–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice CF, Haiser HJ, and Turnbaugh PJ (2013). Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllykallio H, Leduc D, Filee J, and Liebl U (2003). Life without dihydrofolate reductase FolA. Trends Microbiol 11, 220–223. [DOI] [PubMed] [Google Scholar]
- Nair AB, and Jacob S (2016). A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 7, 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S, Fischbach MA, and Pollard KS (2015). MetaQuery: a web server for rapid annotation and quantitative analysis of specific genes in the human gut microbiome. Bioinformatics 31, 3368–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Rourke A, Beyhan S, Choi Y, Morales P, Chan AP, Espinoza JL, Dupont CL, Meyer KJ, Spoering A, Lewis K, et al. (2020). Mechanism-of-action classification of antibiotics by global transcriptome profiling. Antimicrob Agents Chemother 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonechnikov K, Golosova O, Fursov M, and team U (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. [DOI] [PubMed] [Google Scholar]
- Omenetti S, and Pizarro TT (2015). The Treg/Th17 axis: a dynamic balance regulated by the gut microbiome. Front Immunol 6, 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedley AM, and Benkovic SJ (2017). A new view into the regulation of purine metabolism: the purinosome. Trends Biochem Sci 42, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pianta A, Arvikar S, Strle K, Drouin EE, Wang Q, Costello CE, and Steere AC (2017). Evidence of the immune relevance of Prevotella copri, a gut microbe, in patients with rheumatoid arthritis. Arthritis Rheumatol 69, 964–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, and Mazmanian SK (2011). The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science 332, 974–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher JU, Sczesnak A, Longman RS, Segata N, Ubeda C, Bielski C, Rostron T, Cerundolo V, Pamer EG, Abramson SB, et al. (2013). Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. Elife 2, e01202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz M, Zwicker M, and Wider B (2001). Enhanced in vitro induced production of interleukin 10 by peripheral blood mononuclear cells in rheumatoid arthritis is associated with clinical response to methotrexate treatment. J Rheumatol 28, 496–501. [PubMed] [Google Scholar]
- Sharpton T, Lyalina S, Luong J, Pham J, Deal EM, Armour C, Gaulke C, Sanjabi S, and Pollard KS (2017). Development of inflammatory bowel disease is linked to a longitudinal restructuring of the gut metagenome in mice. mSystems 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhawat PB, and Pokharkar VB (2017). Understanding peroral absorption: regulatory aspects and contemporary approaches to tackling solubility and permeability hurdles. Acta Pharm Sin B 7, 260–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprouffske K, and Wagner A (2016). Growthcurver: an R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinformatics 17, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg SE, Campbell CL, Bleyer WA, and Hillman RS (1982). Enterohepatic circulation of methotrexate in rats in vivo. Cancer Res 42, 1279–1282. [PubMed] [Google Scholar]
- Treon SP, and Chabner BA (1996). Concepts in use of high-dose methotrexate therapy. Clin Chem 42, 1322–1329. [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, and Segata N (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 12, 902–903. [DOI] [PubMed] [Google Scholar]
- Valerino DM, Johns DG, Zaharko DS, and Oliverio VT (1972). Studies of the metabolism of methotrexate by intestinal flora. I. Identification and study of biological properties of the metabolite 4-amino-4-deoxy-N 10 -methylpteroic acid. Biochem Pharmacol 21, 821–831. [DOI] [PubMed] [Google Scholar]
- Vincent TL, Williams RO, Maciewicz R, Silman A, Garside P, and Arthritis Research, U.K.a.m.w.g. (2012). Mapping pathogenesis of arthritis through small animal models. Rheumatology (Oxford) 51, 1931–1941. [DOI] [PubMed] [Google Scholar]
- Visentin M, Diop-Bove N, Zhao R, and Goldman ID (2014). The intestinal absorption of folates. Annu Rev Physiol 76, 251–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, and Cole JR (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells G, Becker JC, Teng J, Dougados M, Schiff M, Smolen J, Aletaha D, and van Riel PL (2009). Validation of the 28-joint Disease Activity Score (DAS28) and European League Against Rheumatism response criteria based on C-reactive protein against disease progression in patients with rheumatoid arthritis, and comparison with the DAS28 based on erythrocyte sedimentation rate. Ann Rheum Dis 68, 954–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand CM, McCarthy TG, and Goronzy JJ (1995). Correlation between disease phenotype and genetic heterogeneity in rheumatoid arthritis. J Clin Invest 95, 2120–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood RC, Ferone R, and Hitchings GH (1961). The relationship of cellular permeability to the degree of inhibition by amethopterin and pyrimethamine in several species of bacteria. Biochem Pharmacol 6, 113–124. [DOI] [PubMed] [Google Scholar]
- Yu G, Lam TT, Zhu H, and Guan Y (2018). Two methods for mapping and visualizing associated data on phylogeny using ggtree. Mol Biol Evol 35, 3041–3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, and He QY (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue C, You X, Zhao L, Wang H, Tang F, Zhang F, Zhang X, and He W (2010). The effects of adalimumab and methotrexate treatment on peripheral Th17 cells and IL-17/IL-6 secretion in rheumatoid arthritis patients. Rheumatol Int 30, 1553–1557. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang D, Jia H, Feng Q, Wang D, Liang D, Wu X, Li J, Tang L, Li Y, et al. (2015). The oral and gut microbiomes are perturbed in rheumatoid arthritis and partly normalized after treatment. Nat Med 21, 895–905. [DOI] [PubMed] [Google Scholar]
- Zhang YW (2013). Likelihood-based and Bayesian methods for Tweedie compound Poisson linear mixed models. Stat Comput 23, 743–757. [Google Scholar]
- Zhou B, Xia X, Wang P, Chen S, Yu C, Huang R, Zhang R, Wang Y, Lu L, Yuan F, et al. (2018). Induction and amelioration of methotrexate-induced gastrointestinal toxicity are related to immune response and gut microbiota. EBioMedicine 33, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Metadata for All Human Samples Included in This Study, Related to Figures 1, 5, 6, and 7.
Table S1A. Donors / Patients Included in Our Study and Assays Performed on Them.
Table S1B. Details of Gnotobiotic Experiments Using Human Donor Samples.
Table S1C. Rheumatoid Arthritis Patient Baseline Characteristics.
Table S2. Metadata for All Sequenced Samples and 16S rRNA Gene Sequencing Analysis Results, Related to Figures 1, 5, 6, and 7.
Table S2A. Sample Information for Samples That Underwent 16S rRNA, RNA-Seq, and Shotgun Sequencing.
Table S2B. Taxonomic Lookup Table.
Table S2C. Differentially Abundant ASVs in Multiple 16S-Seq Experiments.
Table S2D. Phylum-Level Enrichments Among Differentially Abundant ASVs (padj<0.01, DESeq).
Table S3. Mouse Phenotypic Results, Related to Figures 1 and 7.
Table S3A. Weight Change With MTX Treatment.
Table S3B. ASVs That Were Correlated With Immune Markers in Transplanted Mice.
Table S5. RNA Sequencing Results, Related to Figure 3.
Table S5A. Differentially Expressed Genes in 4 Isolates Treated With MTX (padj<0.2).
Table S5B. Number of Differentially Expressed Genes at Different Padj Thresholds.
Table S5C. KEGG Enrichments Among Differentially Expressed Transcripts in Bacteroidetes thetaiotaomicron at 30 Minutes (MTX vs. Vehicle Control).
Table S5D. KEGG Enrichments Among Differentially Expressed Transcripts in Clostridium asparagiforme at 30 Minutes, 4 Hours, and 20 Hours (MTX vs. Vehicle Control).
Table S5E. Padj (DESeq) Thresholds and Purine/Pyrimidine Enrichments in B. thetaiotaomicron and C. asparagiforme.
Table S5F. 41 Differentially Expressed Transcripts at All Three Time Points in C. asparagiforme.
Table S6. KEGG Pathway Enrichments Among Differentially Abundant Metabolite Features, Related to Figure 3.
Table S6A. Metabolomic Features That Significantly Differed (padj<0.01, Two-Factor ANOVA With BH correction).
Table S6B. KEGG Pathway Enrichments Among Differentially Abundant Metabolite Features.
Table S6C. Raw Metabolomics Data.
Table S7. Metagenomics Results, Related to Figure 5.
Table S7A. Metagenomic Analysis of the RA Patient Gut Microbiome.
Table S7B. KEGG Pathway Enrichments Among Differentially Abundant KOs.
Data Availability Statement
All sequencing data generated in the preparation of this manuscript has been deposited in NCBI’s Sequence Read Archive (SRA), under Bioproject ID PRJNA656577. Representative code used in the analyses of 16S-seq data are described here (Callahan et al., 2016b) and custom code is available at https://github.com/turnbaughlab/2020_Nayak_MTX_Effects.