

SUMMARY
Mucoepidermoid carcinoma (MEC) is a life-threatening salivary gland cancer that is driven primarily by a transcriptional coactivator fusion composed of cyclic AMP-regulated transcriptional coactivator 1 (CRTC1) and mastermind-like 2 (MAML2). The mechanisms by which the chimeric CRTC1/MAML2 (C1/M2) oncoprotein rewires gene expression programs that promote tumorigenesis remain poorly understood. Here, we show that C1/M2 induces transcriptional activation of the non-canonical peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) splice variant PGC-1α4, which regulates peroxisome proliferator-activated receptor gamma (PPARγ)-mediated insulin-like growth factor 1 (IGF-1) expression. This mitogenic transcriptional circuitry is consistent across cell lines and primary tumors. C1/M2-positive tumors exhibit IGF-1 pathway activation, and small-molecule drug screens reveal that tumor cells harboring the fusion gene are selectively sensitive to IGF-1 receptor (IGF-1R) inhibition. Furthermore, this dependence on autocrine regulation of IGF-1 transcription renders MEC cells susceptible to PPARγ inhibition with inverse agonists. These results yield insights into the aberrant coregulatory functions of C1/M2 and identify a specific vulnerability that can be exploited for precision therapy.
Graphical Abstract
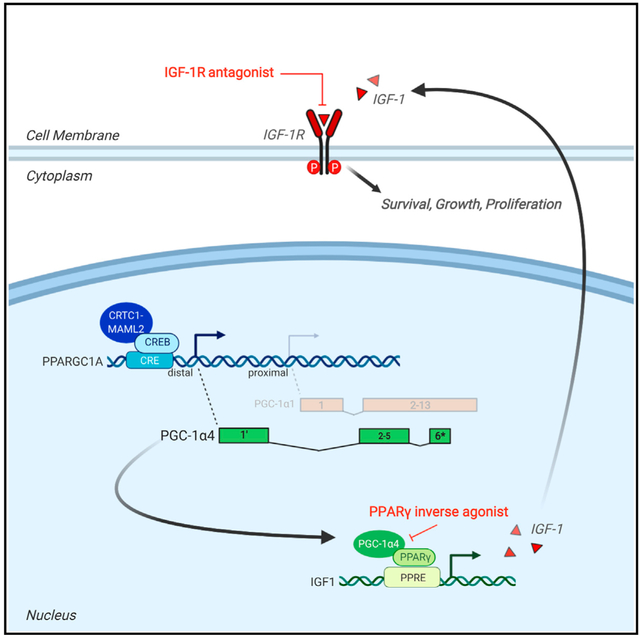
In brief
Musicant et al. demonstrate that the CRTC1-MAML2 gene fusion aberrantly regulates expression of the minor splice variant PGC-1α4, which in turn coactivates PPARγ to induce IGF-1 expression. This synthetic signal circuit establishes pro-growth and pro-survival signaling in mucoepidermoid carcinomas, sensitizing tumors to drugs that disable IGF-1 signaling by targeting PPARγ.
INTRODUCTION
Transcriptional coregulators (coactivators and corepressors) regulate gene expression primarily by acting as bridges between DNA-bound transcription factors and basic transcriptional machinery (Rosenfeld et al., 2006). Chromosomal translocations that create oncogenic fusion genes involving transcriptional coregulators are predicted to cause profound changes to normal developmental, homeostatic, and/or cellular identity programs in cancer (Lee and Young, 2013; Mitelman et al., 2019; Rabbitts, 1994; Tuna et al., 2019).
Mucoepidermoid carcinoma (MEC) is the most common salivary gland malignancy, and patients with advanced recurrent or metastatic tumors often suffer from unresectable, lethal disease marked by a 5-year survival rate of <40% (Bell and Hanna, 2012; El-Naggar et al., 2017; McHugh et al., 2012). Salivary MEC tumors can arise within the major or minor salivary glands and are characterized by significant intra-tumoral cellular heterogeneity fueled by cancer stem cells (CSCs) that give rise to multiple cell types, including epidermoid, mucus, and intermediate cells (Adams et al., 2015; Seethala et al., 2010; Stewart et al., 1945; Volkmann, 1895). The intermediate cells represent a poorly differentiated, proliferative cell type thought to give rise to the terminally differentiated epidermoid and mucus cell populations. Furthermore, the relative proportion of intermediate cells increases in high-grade tumors and correlates with poor prognosis (Batsakis, 1980). Transcriptional programs that control cellular identity and support tumor cell functions can directly influence tumor grade and therefore disease progression. Genomic characterization of salivary MEC tumors implicates either recurrent t(11;19) chromosomal translocation, resulting in fusion of two transcriptional coactivators—cyclic AMP (cAMP)-regulated transcriptional coactivator 1 (CRTC1) and mastermind-like 2 (MAML2)—to generate the oncogenic coactivator fusion CRTC1/MAML2 (C1/M2; fusion-positive MEC), or mutations in the tumor suppressor p53 (fusion-negative MEC) (El-Naggar et al., 1996; Kang et al., 2017; Tonon et al., 2003; Wang et al., 2017). Most MEC cases are fusion positive (50%–85%) (O’Neill, 2009), and these tumors harbor a strikingly low somatic mutational burden, indicating that C1/M2 fusion is the primary oncogenic driver event. Numerous other examples exist of cancers driven primarily by gene fusions in the absence of high mutational burden (Gao et al., 2018; Kadoch and Crabtree, 2013; Missiaglia et al., 2012; Riggi et al., 2014). Although surgical resection is often sufficient to treat patients with low-grade, fusion-positive tumors, some patients expressing C1/M2 develop recurrent, chemoradiation-resistant, high-grade tumors (Chen et al., 2007; Seethala and Chiosea, 2016; Warner et al., 2013). This underscores the critical need to develop targeted therapeutic strategies for this subset of salivary MEC patients.
Molecular properties of the chimeric oncoprotein C1/M2 have been extensively characterized and reveal that the t(11;19) chromosomal translocation fuses the coiled-coil domain of CRTC1, which promotes binding to the transcription factor cAMP-response element binding protein (CREB), with the strong transcriptional activation domain of MAML2 (Coxon et al., 2005; Wu et al., 2005). Consequently, C1/M2 functions as a rogue co-activator of CREB; however, it also displays gain-of-function interactions with and activation of the master transcription factor and proto-oncogene MYC (Amelio et al., 2014). Notably, C1/M2 is localized to the nucleus, where it binds to CREB in a Ser133 phosphorylation-independent manner yet retains the ability to recruit CBP/p300 through the MAML2 transactivation domain, bypassing the need for traditional cAMP signaling inputs (Conkright et al., 2003; Wu et al., 2005). Unfortunately, the absence of ligand binding sites on these transcription factors renders them impractical targets for developing selective inhibitors. Thus, efforts have been directed toward defining the downstream pathways reprogrammed by C1/M2. Although candidate CREB and MYC target genes have been identified (Amelio et al., 2014; Chen et al., 2015), the subordinate transcriptional pathways dysregulated by aberrant activation of these transcription factors remain poorly defined.
Here, we performed transcriptomic profiling of fusion-positive salivary MEC tumors and report that C1/M2 initiates a transcriptional cascade that eventuates with upregulation of the potent growth hormone insulin-like growth factor (IGF) 1 via induction of a peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) alternative splice variant, PGC-1α4. This has functional consequences for the growth, survival, and oncogenic transformation of salivary gland precursors. Elucidating this pathway establishes a molecular basis for the unique capacity of the C1/M2 coactivator fusion to control complex and extensive transcriptional networks beyond CREB and MYC, providing evidence of a role for the PGC-1α4 splice variant in human cancer pathogenesis. Integrating drug screening and mechanistic data exposed multiple biologic nodes of drug sensitivity, revealing a C1/M2-positive, MEC subtype-selective therapeutic vulnerability to peroxisome proliferator-activated receptor gamma (PPARγ) inhibition.
RESULTS
Activation of IGF-1 signaling is a hallmark of C1/M2-positive salivary MEC
IGF-1 receptor (IGF-1R) overexpression and/or ligand-induced activation of downstream signaling occurs in many cancers and is often required by oncogenes to promote transformation and malignancy (Pollak, 2008). Our previous work investigating the pathobiology of C1/M2-positive MEC revealed that several genes involved in anabolic, pro-growth receptor tyrosine kinase (RTK) pathways, including IGF-1R signaling, are induced by the C1/M2 fusion oncogene (Amelio et al., 2014). Here, we confirmed that IGF-1 is upregulated >100-fold upon ectopic induction of C1/M2 expression (Table S1). Given the importance of IGF-1 in tumor cell growth and survival (Pollak, 2008; Yu and Rohan, 2000) and in salivary gland growth and development (Amano and Iseki, 1993; Kerr et al., 1995; Ryan et al., 1992; Werner and Katz, 2004), we hypothesized that IGF-1R signaling plays a key role in salivary MEC tumorigenesis. To validate the clinical relevance of this finding, we first obtained a cohort of 18 human primary salivary MEC samples from the surgical pathology department at University of North Carolina (UNC) Hospitals (Chapel Hill, NC) and assessed C1/M2 status (Figure 1A). In accordance with previous reports (Birkeland et al., 2017), we found that 10 of these samples (56%) were C1/M2 positive. Next, we performed RNA sequencing on all MEC samples (fusion positive and negative) to identify differentially expressed genes (DEGs) relative to six normal salivary gland controls (Table S2). Unbiased and unsupervised hierarchical clustering revealed that C1/M2-positive tumors constitute a gene expression subtype distinct from C1/M2-negative tumors (Figure S1A). This finding is in agreement with recent studies that suggested that C1/M2-positive and C1/M2-negative MEC are two distinct tumor types driven by distinct signaling mechanisms (Kang et al., 2017; Wang et al., 2017). We identified IGF-1 among 3,971 upregulated genes that are significantly differentially expressed (fold change > 2 and padj < 0.05) in C1/M2-positive tumors relative to normal salivary glands (Figure 1B; Table S3). Gene set enrichment analysis (GSEA) of curated gene lists within the Molecular Signatures Database (MSigDB) revealed that IGF signaling pathways rank among the top signatures associated with C1/M2-positive tumors (Figure S1B). Thus, we generated a refined gene list (Musicant_MEC_CRTC1-MAML2_IGF1) by curating IGF-1 pathway-related genes with a fold change > 2 (padj < 0.05) that reflect a gene signature specific to C1/M2-positive MEC tumors (Figures 1C and 1D; Table S4). We then evaluated the ability of our curated Musicant_MEC_CRTC1-MAML2_IGF1 gene set to distinguish C1/M2-positive MEC compared with the established MSigDB GNF2_IGF1 gene set (Figures S1C and S1D). Importantly, we examined gene expression in these MEC cases by qPCR and found that the C1/M2 copy number is significantly correlated (r2 = 0.7251; p < 0.0001) with IGF-1 expression (Figure 1E). These results demonstrate that C1/M2-positive MEC is characterized by increased IGF-1 expression and suggest an important role for IGF-1R signaling in this MEC tumor subtype.
Figure 1. Bioinformatics analyses identify IGF-1 as a hormone associated with C1/M2-fusion-positive salivary MEC.

(A) Quantitative real-time PCR analysis of C1/M2 expression in human salivary MEC tumor samples. C1/M2 copy number per 10 ng input RNA was calculated based on a standard curve. Samples with <500 C1/M2 transcripts per 10 ng RNA were classified as fusion negative (gray bars), whereas those with ≥500 C1/M2 transcripts per 10 ng RNA were classified as fusion positive (blue bars). Data are presented as the mean ± SEM (n = 4).
(B) RNA sequencing was performed on ten C1/M2-positive MEC tumor samples and six normal salivary gland samples. The volcano plot shows genes that are significantly upregulated (red) and downregulated (blue) in C1/M2-positive MEC compared with normal salivary glands (padj < 0.05).
(C) Heatmap of IGF-1-related DEGs between fusion-positive MEC and normal salivary gland samples. Normal (e.g., N #1 and N #2) and tumor (e.g., case #1 and case #7) samples are indicated at the top of the heatmap in black and green, respectively. C1/M2 status is indicated in gray (negative) and blue (positive). Hierarchical clustering was performed using ComplexHeatmap R package version 2.0.0.
(D) Violin plot highlighting the significance of IGF-1 pathway-related genes within our curated Musicant_MEC_CRTC1-MAML2_IGF1 gene set between fusion-positive MEC and normal salivary gland samples (Wilcoxon rank sum test; *p = 0.0075).
(E) Comparison of C1/M2 and IGF-1 CT values in qPCR data from human C1/M2-positive MEC tumor (blue) and normal salivary gland (gray) samples. The fitted regression line demonstrates the correlation between the expression levels of the two genes (r2 = 0.7251, p < 0.0001). 95% confidence intervals are indicated by curved lines on either side of the linear regression.
See also Figure S1.
C1/M2-positive MEC tumor cell lines display selective molecular sensitivities to targeted IGF-1R inhibition
To investigate the role of constitutive IGF-1 expression and downstream signaling in C1/M2-positive MEC cells, we obtained a panel of five MEC (H292, H3118, HMC1, HMC3A, and HMC3B) and three epidermoid (A253, A388, and A431) cell lines. We first confirmed that all MEC cell lines are positive for expression of the C1/M2 fusion transcript and that all epidermoid carcinoma cell lines are fusion negative (Figure 2A). Furthermore, all C1/M2-positive MEC cell lines display robust IGF-1 expression relative to C1/M2-negative cell lines, and this increased IGF-1 expression significantly correlates (r2 = 0.8664; p = 0.0008) with C1/M2 expression in fusion-positive MEC cells (Figures 2B and S2A). Finally, histologic analysis of tumor xenografts confirms that C1/M2-positive salivary MEC cell lines generate significant intra-tumoral cellular heterogeneity, which mimics the mucus, epidermoid, and intermediate cell types characteristic of human salivary MEC, demonstrating that these cell lines are capable of forming representative MEC tumors in vivo (Behboudi et al., 2006; Bell and Hanna, 2012; Warner et al., 2013). In contrast, fusion-negative epidermoid carcinoma cells generate xenograft tumors with typical solid epidermoid morphology (Figures 2C and S2B). Immunohistochemical staining demonstrated that C1/M2-positive tumors are associated with increased IGF-1 production in vivo (Figures 2C and S2B).
Figure 2. A focused small-molecule drug screen identifies sensitivity of C1/M2-positive tumor cells to compounds targeting IGF-1R.
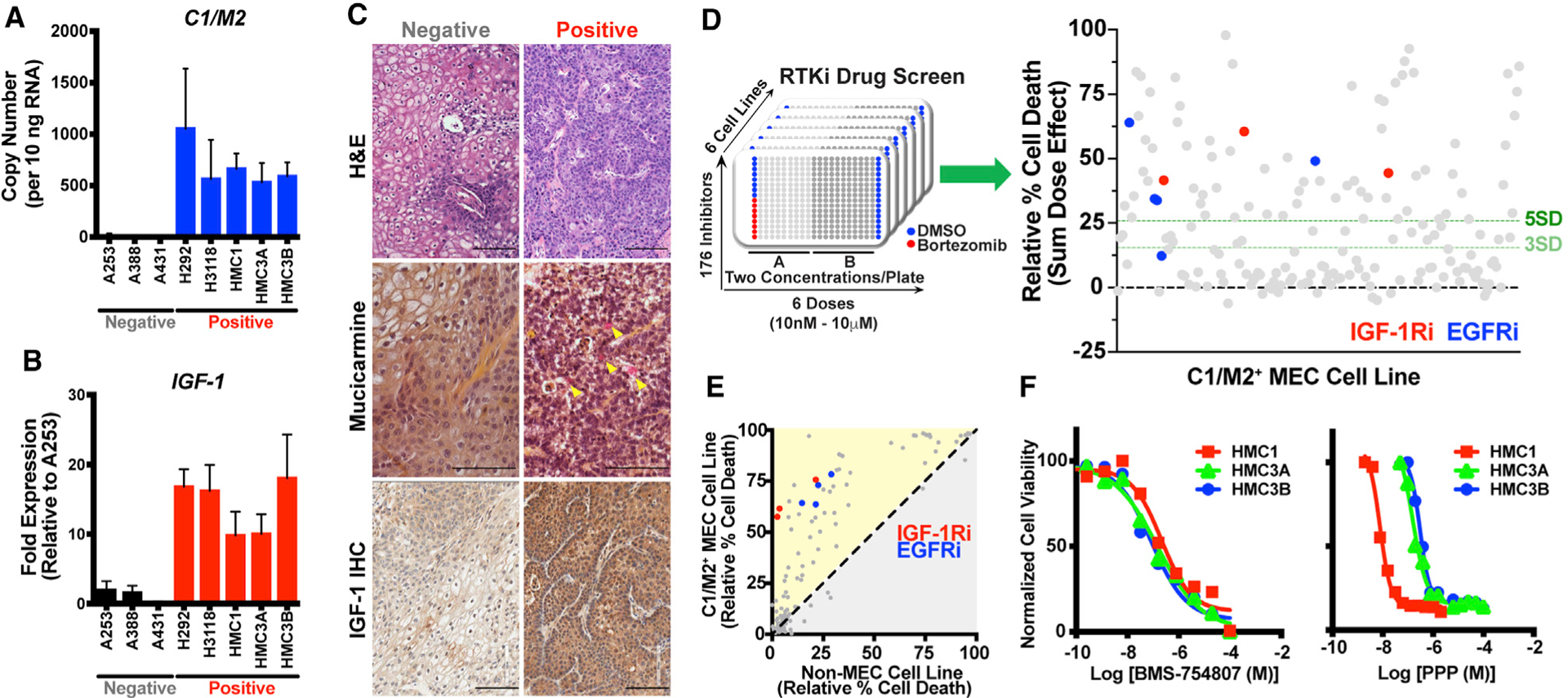
(A) Quantitative real-time PCR analysis of C1/M2 expression in MEC cell lines and control epidermoid carcinoma cell lines. C1/M2 copy number per 10 ng input RNA was calculated based on a standard curve. Cell lines are listed along the x axis, with C1/M2 status indicated below in gray (negative) or red (positive). Data are presented as the mean ± SEM (n = 3 biologic replicates, 4 technical replicates each).
(B) Quantitative real-time PCR analysis of IGF-1 expression in C1/M2-positive and C1/M2-negative cell lines. Cell lines are listed along the x axis, with C1/M2 status indicated below in gray (negative) or red (positive). Relative fold expression is shown normalized to RPL23 mRNA levels. Data are presented as the mean ± SEM (n = 3 biologic replicates, 4 technical replicates each).
(C) Histologic and immunohistochemical analysis of C1/M2-positive (HMC3A) and C1/M2-negative (A253) xenografts. Representative xenograft sections were formalin fixed, embedded, and H&E stained (top). Mucicarmine staining (middle) revealed mucus cells (pink staining, indicated by yellow arrowheads) only in C1/M2-positive xenografts. IHC staining for IGF-1 (bottom) revealed increased IGF-1 expression in C1/M2-positive xenografts. Scale bar, 100 μm.
(D) Drug screen performed in five C1/M2-positive MEC cell lines and one C1/M2-negative epidermoid carcinoma cell line. 176 inhibitors were tested, in duplicate, across all cell lines at six concentrations ranging from 10 nM to 10 μM. DMSO (1%) and bortezomib (1 μM) were used as negative and positive controls, respectively, on each plate. Several IGF-1Ri and EGFR inhibitors emerged as top hits, inducing cell death in C1/M2-positive cell lines with >5 SD above baseline (DMSO).
(E) Relative cell death induced by drug screen inhibitors. IGF-1Ri and EGFR inhibitors more effectively induced cell death in C1/M2-positive MEC cells compared with C1/M2-negative epidermoid carcinoma cells.
(F) Representative dose-response curves showing the viability of three C1/M2-positive cell lines (HMC1, HMC3A, and HMC3B) treated with increasing concentrations of IGF-1Ri (BMS-754807 and PPP). IC50 values were calculated using a non-linear curve fit (log[agonist] versus response, four parameter-variable slope) in GraphPad Prism. The experiment was performed in biologic triplicate (n = 3 technical replicates for each experiment), with one representative experiment shown. See Table 1 for a summary of the IC50 values.
See also Figures S2 and S3.
To test whether C1/M2-positive MEC cells depend on IGF-1 signaling, we performed an unbiased small-molecule drug screen focused on RTK inhibitors (RTKi) at concentrations ranging from 10 nM to 10 μM in all five C1/M2-positive cells lines relative to a fusion-negative control cell line (Figure 2D, left). Calculation of the Z′ factor demonstrated robust assay quality such that the distribution between positive (bortezomib) and negative (DMSO) controls indicates a low likelihood of false-positive hits (Z′ > 0.75) (Figure S3A). Multiple independent inhibitors targeting IGF-1R and epidermal growth factor receptor (EGFR) emerged as significant hits (p ≥ 3 SD) in this screen (Figure 2D, right). EGFR has been explored as a therapeutic target, though with limited clinical success (Chen et al., 2015), and its emergence as a significant hit validated the robustness of our screen results. Compared with the non-MEC fusion-negative control cells, IGF-1R inhibitors (IGF-1Ri) induced selective and robust cell death in all C1/M2-positive cell lines (Figures 2E, S3B, and S3C), indicating the relative importance of IGF-1/IGF-1R signaling in C1/M2-positive MEC. Dose titrations of two separate IGF-1Ri, BMS-754807 and picropodophyllin (PPP), confirmed that the C1/M2-positive cell lines are sensitive to IGF-1R inhibition, with IC50 values for both compounds in the low nanomolar range (Figure 2F; Table 1).
Table 1.
Calculated IC50 of IGF-1Ri and PPARγ inverse agonists on the viability of C1/M2-fusion-positive and C1/M2-fusion-negative cancer cell lines
| Fusion status | Cancer cell line | Tissue | BMS-754807a | PPP | SR-10221 | SR-2595 | T0070907 |
|---|---|---|---|---|---|---|---|
| C1/M2-negative non-MEC | A-253 | salivary gland | 58.9 ± 2.7 | 0.719 ± 0.224 | NDb | ND | ND |
| UM-HMC-1 | salivary gland | 0.213 ± 0.047 | 0.066 ± 0.083 | 114.5 ± 38.4 | 40.1 ± 16.1 | 328.1 ± 36.3 | |
| UM-HMC-3A | salivary gland | 0.148 ± 0.057 | 0.211 ± 0.111 | 72.7 ± 17.4 | 50.2 ± 22.0 | 222.7 ± 12.8 | |
| C1/M2-positive MEC | UM-HMC-3B | salivary gland | 0.088 ± 0.019 | 0.216 + 0.108 | 148.4 + 30.7 | 78.6 ± 34.6 | 329.7 ± 19.0 |
| NCI-H292 | lung | ND | ND | 76.6 ± 2.1 | ND | ND | |
| NCI-H3118 | salivary gland | ND | 0.489 ± 0.075 | 98.5 ± 12.6 | ND | ND |
Cell viability was assessed by ATPlite assay, and IC50 values were calculated in GraphPad Prism using a non-linear curve fit (log[agonist] versus response, four parameter-variable slope).
Reported IC50 values (in micromolars) are reflective of the average of three biologic replicates (mean ± SEM).
ND, not determined.
IGF-1R signaling is critical for C1/M2-positive MEC tumor cell growth and survival
We next wanted to examine the functional role of IGF-1R signaling on human C1/M2-positive MEC cell growth and survival via pharmacologic inhibition and/or genetic knockdown (validated in Figures 3A, S4A, and S4B). Live-cell, kinetic proliferation assays revealed that wild-type HMC3A cells reach 50% confluency after 28 h (GC50 = 28 h), but pharmacologic inhibition of IGF-1R with PPP significantly increases this time to confluency (p < 0.05) relative to control (Figure 3B). To test whether blocking IGF-1R impairs the tumorigenic potential of MEC cells to grow as clonogenic colonies ex vivo, C1/M2-positive cells were plated at low density and observed for colony formation. We found that both PPP and BMS-754807 markedly blunted the clonogenic potential of several independent MEC cell lines relative to vehicle-treated cells (Figures 3C and S4C). Several recent studies have documented the existence of CSC subpopulations within various head and neck cancers, including salivary MEC, and these CSCs are associated with malignant potential (Adams et al., 2015; Curtarelli et al., 2018; Keysar et al., 2016). Because tumor spheroids are uniquely enriched for CSCs, we also quantified the efficacy of PPP on 3D tumor spheroid formation in Matrigel and found that sustained IGF-1Ri treatment (7 days) significantly blocks (p < 0.0001) MEC 3D sphere formation (Figures 3D and 3E). Finally, we investigated the mechanism underlying the observed decrease in proliferation and tumorigenic potential and found that PPP stimulates a significant dose-dependent increase (p < 0.001) in apoptosis compared with vehicle-treated cells (Figure 3F).
Figure 3. Inhibition of IGF-1R blocks MEC cell growth and induces apoptosis.

(A) Left, western blot showing expression of total and phosphorylated (Tyr1135) IGF-1R protein in HMC1 cells treated with BMS-754807 or PPP. The blot is representative of three independent biologic replicates. Right, quantification of phosphorylated (Tyr1135) IGF-1R protein levels. Band intensities were normalized to β-actin and to total IGF-1R and are shown as the fold change in phosphorylated band intensity relative to the untreated condition. Data are presented as the mean ± SEM (n = 3 biologic replicates; one-way ANOVA with Holm-Sidak post hoc test; ns, not significant, *p ≤0.05).
(B) Cell proliferation assay showing relative confluency of HMC3A cells treated with DMSO or PPP. GC50 values for each condition are shown in hours and indicate the time required for cells to reach 50% confluency. ND, not determined. The experiment was performed in biologic triplicate (n = 4 technical replicates per experiment) with one representative experiment shown (mean ± SEM; Benjamini-Hochberg pairwise comparisons;**p ≤0.01, ***p ≤0.001). Data were collected using the InCucyte live-cell imager; see STAR methods for details.
(C) Number of colonies formed by HMC3A cells treated with DMSO or PPP. A colony is defined as a cluster of ≥50 cells. Representative images of wells treated with DMSO and PPP (IC50 concentration) are shown to the left. Data are presented as the mean ± SEM (n = 3 biologic replicates; Student’s t test; ****p ≤0.0001). See Table 1 for a summary of the IC50 values.
(D) Representative images of HMC3A tumorspheres on day 1 (top) and 7 days after treatment with DMSO (vehicle control) or increasing concentrations of PPP.
(E) Tumorsphere formation in HMC3A cells treated with DMSO or increasing concentrations of PPP. The percent change in the tumorsphere area is calculated as [TumorAreaDayX]/[TumorAreaDay1] × 100. >50 individual tumorspheres per condition were tracked for each experiment. Data are presented as the mean ± SEM (n = 3 biologic replicates; Student’s t test; ****p ≤0.0001).
(F) Apoptosis levels (measured as the percentage of cleaved caspase-3/7) in HMC3A cells treated with DMSO or PPP. Data were collected via flow cytometry and normalized to a non-stained control for each condition. Data are presented as the mean ± SEM (n = 3 biologic replicates; Student’s t test; **p ≤ 0.01).
See also Figure S4.
C1/M2 establishes a synthetic PGC-1α4 circuit that regulates IGF-1 expression in MEC tumor cells
To determine the mechanism by which C1/M2 regulates IGF-1 expression in MEC cells, we used our engineered C1/M2-inducible stable cell line (doxycycline-regulated HEK293-CMVTetR TetOC1/M2) (Amelio et al., 2014) (Figure S5A). Overexpression of C1/M2 in this cell line significantly increased IGF-1 expression at both transcript and protein levels (Figures 4A, 4B, S5B, and S5C). C1/M2 coactivates the transcription factor CREB, which in turn binds to specific DNA sequences called cAMP-response elements (CREs). Examination of IGF-1 promoter sequences revealed a non-canonical CRE site, indicating that C1/M2 could possibly drive IGF-1 signaling through direct upregulation of IGF-1 transcription. However, chromatin immunoprecipitation (ChIP) assays failed to identify C1/M2 enrichment at the IGF-1 promoter relative to the control canonical CREB target gene NR4A2 (Figure 4C).
Figure 4. C1/M2 induces expression of the PGC-1α4 alternative splice variant, leading to IGF-1 upregulation.

(A) Quantitative real-time PCR analysis of IGF-1 mRNA levels in C1/M2-inducible cells (doxycycline-regulated HEK293-CMVTetRTetOC1/M2 cells) treated with and without 1 μg/mL of doxycycline. Relative fold expression is shown normalized to RPL23 mRNA levels. Data are presented as the mean ± SEM (n = 3 biologic replicates, 4 technical replicates each; Student’s t test; ***p < 0.001).
(B) AlphaLISA-based analysis of secreted IGF-1 in media from C1/M2-inducible cells (HEK293-CMVTetRTetOC1/M2) with and without 1 μg/mL doxycycline treatment. Data are presented as the mean ± SEM (n = 3 biologic replicates; Student’s t test; **p = 0.003).
(C) Chromatin immunoprecipitation analysis of FLAG-C1/M2 promoter occupancy in C1/M2-inducible cells (HEK293-CMVTetRTetOC1/M2). Data are expressed as promoter occupancy in doxycycline-treated cells normalized to promoter occupancy in vehicle-treated cells. GAPDH and NR4A2 promoters were used as negative and positive controls, respectively. Data are presented as the mean ± SEM (n = 3 biologic replicates; Student’s t test; ***p < 0.001, **p < 0.01).
(D) Luciferase assay measuring activation of the distal and proximal PGC-1α promoters in C1/M2-inducible cells (HEK293-CMVTetRTetOC1/M2) with and without 1 μg/mL doxycycline treatment. Data are presented as the mean ± SEM (n = 3 biologic replicates, 4 technical replicates each; Student’s t test; ***p < 0.001).
(E) Quantitative real-time PCR analysis of PGC-1α4 mRNA expression in HMC3A cells with and without shRNA-mediated C1/M2 knockdown. Relative fold expression is shown normalized to RPL23 mRNA levels and to the mock transduced condition. shNS, non-specific shRNA; shMAML2_#1 and shMAML2_#3, shRNAs targeting C1/M2 and MAML2. Data are presented as the mean ± SEM (n = 3 biologic replicates, 4 technical replicates each; Student’s t test; **p < 0.01).
(F) Temporal analysis of C1/M2, PGC-1α4, and IGF-1 expression kinetics in C1/M2-inducible cells (HEK293-CMVTetRTetOC1/M2) treated with 1 μg/mL of doxycycline for 0–48 h. C1/M2 and PGC-1α4 expression is indicated on the left y axis, whereas IGF-1 expression is indicated on the right y axis. Relative fold expression is shown normalized to RPL23 mRNA levels. Data are presented as the mean ± SEM (n = 3 biologic replicates; Student’s t test; ***p < 0.001).
(G) Quantitative real-time PCR analysis of PGC-1α4 expression in C1/M2-positive and C1/M2-negative cell lines. Cell lines are listed along the x axis, with C1/M2 status indicated below in gray (negative) or red (positive). Relative fold expression is shown normalized to RPL23 mRNA levels. Data are presented as the mean ± SEM (n = 3 biologic replicates, 4 technical replicates each).
(H and I) Quantitative real-time PCR analysis of IGF-1 mRNA levels in HMC3A cells transduced with unique gRNAs targeting the PGC-1α1 isoform only (PGC-1α1 sgRNA) or all PGC-1α isoforms (Pan-PGC-1α sgRNA) (H). Quantitative real-time PCR analysis of PGC-1α4 and IGF-1 mRNA expression in PGC-1α4-inducible cells (doxycycline-regulated HEK293-PGKTetOn3GTRE3GSPGC−1α4 cells) treated with 1 μg/mL of doxycycline for 0–48 h (I). Relative fold expression is shown normalized to RPL23 mRNA levels. Data are presented as the mean ± SEM (n = 3 biologic replicates, 4 technical replicates each; Student’s t test; **p < 0.01, ***p < 0.001, ****p < 0.0001).
See also Figure S5.
We previously showed that CREB regulates stress-inducible expression of a PPARγ coactivator 1α splice variant (PGC-1α4) transcribed from an upstream (~13 kbp) distal promoter of the PGC-1α locus (Bruno et al., 2014), which in turn selectively regulates IGF-1 expression (Ruas et al., 2012). Thus, we performed ChIP assays and identified C1/M2 enrichment specifically at this distal PGC-1α promoter (Figure 4C). Moreover, luciferase reporter assays confirmed that C1/M2 activates the distal CRE-containing PGC-1α promoter, but not the proximal promoter (Figure 4D), suggesting that C1/M2 may indirectly regulate IGF-1 expression by controlling PGC-1α4 expression in MEC cells. We next performed short hairpin RNA (shRNA)-mediated C1/M2 knockdowns and found that PGC-1α4 levels are dramatically reduced similar to control NR4A2, confirming that C1/M2 directly upregulates PGC-1α4 transcription (Figures 4E and S5D). Expression of inducible dominant-negative CREB (A-CREB) in a stable MEC cell line (HMC3A-PGKTetOn3G TRE3GSA-CREB) also blocked PGC-1α4 expression, supporting the role of CREB-dependent regulation of the PGC-1α distal promoter by C1/M2 (Figure S5E). To examine the kinetics of this expression circuit more closely, we performed a time course experiment in C1/M2-inducible stable cells and found that PGC-1α4 transcripts peak at 24 h (~12 h after C1/M2 induction), whereas IGF-1 transcripts begin to rise ~24–36 h but peak 48 h after C1/M2 induction and 24 h after PGC-1α4 upregulation (Figure 4F). Notably, compared with the other PGC-1α transcript isoforms, C1/M2-positive MEC cell lines are all characterized by robust expression of PGC-1α4 relative to C1/M2-negative cell lines (Figures 4G and S5F). To validate that PGC-1α4 is responsible for inducing IGF-1 expression, we engineered MEC cell lines with an inducible CRISPR interference (dCas9-KRAB; CRISPRi) system (Gilbert et al., 2013). Unique gRNAs that repress expression of all PGC-1α isoforms or that selectively repress only PGC-1α1 revealed that the canonical PGC-1α1 variant has no effect on IGF-1 regulation. In contrast, repressing transcription of all isoforms caused a significant decrease (p < 0.0001) in IGF-1 expression (Figures 4H and S5G), further indicating that PGC-1α4 regulates IGF-1. To confirm that this non-canonical PGC-1α4 isoform is responsible for promoting IGF-1 transcription, we generated a PGC-1α4-inducible stable cell line (doxycycline-regulated HEK293-PGKTetOn3G TRE3GSPGC−1α4; validated in Figure S5H) and confirmed that selective overexpression of the PGC-1α4 splice variant is sufficient to upregulate IGF-1 expression, thus acting as the intermediate to C1/M2 (Figure 4I). Finally, shRNA-mediated knockdown of PGC-1α4, as well as other components within this signaling circuit (IGF-1R and C1/M2), significantly blunts proliferation of C1/ M2-positive MEC cells (Figure S5I), confirming that PGC-1α4 is both necessary and sufficient for activating the IGF-1 signaling circuit in fusion-positive MEC.
The PGC-1α4 coregulator activates PPARγ-dependent transcription of IGF-1 in C1/M2-positive MEC tumor cells
PGC-1α is a coactivator that binds to the transcription factor PPARγ to exert its effects on transcription of metabolic target genes (Semple et al., 2006; Sonoda et al., 2008). Strikingly, analysis of the IGF-1 locus identified three canonical PPARγ response element (PPRE) binding motifs (Figure S6A). To test whether PGC-1α4 coactivates PPARγ to modulate IGF-1 transcription, we first confirmed that overexpression of C1/M2 induces transcription of an IGF-1 promoter-driven luciferase reporter comparable to levels of induction observed with a PPRE-driven luciferase reporter (Figures S6B and S6C). Importantly, functional characterization revealed that both the PPRE and the IGF-1 promoter-driven luciferase reporter constructs are responsive to the PPARγ agonist GW1929, indicating that the cloned IGF-1 promoter fragment includes the PPRE motifs that recruit PPARγ (Figure S6D). Therefore, we tested whether the PPARγ inverse agonist SR10221, which functions to recruit transcriptional corepressors to PPARγ that repress target gene transcription, is effective at inhibiting IGF-1 expression. Treatment with SR10221 effectively downregulated PPARγ-mediated transcriptional activity of the IGF-1-luciferase reporter in a dose-dependent manner to below basal levels comparable to those observed with the PPRE-luciferase reporter (Figure 5A). Similarly, shRNA-mediated knockdown of PPARγ resulted in a robust decrease in IGF-1 expression (Figure S6E) and potent induction of apoptosis, as evidenced by significant increases (p < 0.05) in caspase-3/7 activation (Figure S6F). This indicates that the PGC-1α4:PPARγ circuit controls IGF-1 expression and suggests that drugs targeting PPARγ may provide therapeutic benefit against C1/M2-positive salivary MEC.
Figure 5. Inhibition of IGF-1 expression with PPARγ inverse agonists blocks MEC cell growth and induces apoptosis.

(A and B) PPARγ-response element (PPRE)-driven (A, left) and IGF-1 promoter-driven (A, right) luciferase reporter assay showing repression of basal transcriptional activity of endogenously expressed PPARγ in HEK293-CMVTetRTetOC1/M2 cells expressing C1/M2. Representative dose-response curves show cells treated with increasing concentrations of the PPARγ inverse agonist SR10221 for 24 h before measuring luciferase activity. (B) Dose-response curves showing viability of three C1/M2-positive cell lines (HMC1, HMC3A, and HMC3B) treated with increasing concentrations of PPARγ inverse agonists (SR10221, SR2595, and T0070907). IC50 values were calculated using a non-linear curve fit (log[agonist] versus response, four parameter-variable slope) in GraphPad Prism. The experiment was performed in biologic triplicate (n = 3 technical replicates for each experiment), with one representative experiment shown.
(C) Quantitative real-time PCR analysis of IGF-1, PGK1, and PKM2 expression in HMC3A cells treated with vehicle (DMSO) or SR2595. Relative fold expression is shown normalized to RPL23 mRNA levels. Data are presented as the mean ± SEM (n = 3 biologic replicates, 3 technical replicates each; Student’s t test; ***p ≤ 0.001).
(D) Left, western blot showing expression of IGF-1 protein in HMC3A cells treated with increasing concentrations of SR2595. The blot is representative of three independent biologic replicates. Right, quantification of IGF-1 protein levels. Band intensities were normalized to β-actin and are shown as the fold change in band intensity relative to the vehicle-treated condition. Data are presented as the mean ± SEM (n = 3 biologic replicates; Student’s t test; ns, not significant, *p ≤ 0.05).
(E) Cell proliferation assay showing relative confluency of HMC3A cells treated with DMSO or SR10221. The experiment was performed in biologic triplicate (n = 4 technical replicates per experiment) with one representative experiment shown (mean ± SEM; Benjamini-Hochberg pairwise comparisons; ***p ≤ 0.001). Data were collected using the IncuCyte live-cell imager; see STAR methods for details.
(F) Colony formation in HMC1 cells treated with DMSO or SR10221 at 1/2 IC50 concentration or IC50 concentration. A colony is defined as a cluster of ≥50 cells. Data are presented as the mean ± SEM (n = 3 biologic replicates with 3 technical replicates each; Student’s t test; ***p ≤ 0.001).
(G) Tumorsphere formation in HMC3A cells treated with DMSO or 1/2 IC50 SR10221. The percent change in the tumorsphere area is calculated as [TumorAreaDayX]/[TumorAreaDay1] × 100. >50 individual tumorspheres per condition were tracked for each experiment. Data are presented as the mean ± SEM (n = 3 biologic replicates; Student’s t test; **p ≤ 0.01, ***p ≤ 0.001).
(H) Apoptosis levels (measured as the percentage of cleaved caspase-3/7) in HMC3A cells treated with DMSO or SR10221 at IC50 concentration or 23 IC50 concentration. Data were collected via flow cytometry and normalized to a non-stained control for each condition. Data are presented as the mean ± SEM (n = 3 biologic replicates; Student’s t test; ***p ≤ 0.001, ****p ≤ 0.0001).
See Table 1 for a summary of the IC50 values. See also Figure S6.
PPARγ inverse agonists suppress C1/M2-positive MEC tumor growth
To investigate the functional role of PPARγ dependency on MEC cell growth and survival, we tested the effects of several PPARγ inverse agonists in our panel of C1/M2-positive MEC cell lines. First, to determine their potential as anti-MEC agents, we assessed the effect of three inverse agonists (SR10221, SR2595, and T0070907) on cancer cell viability using the full panel of C1/M2-positive MEC cell lines (Figure 5B; Table 1). Dose titration of these compounds revealed that SR10221 potently reduced cancer cell viability at low micromolar concentrations in HMC3A cells (Table 1). Furthermore, treatment with SR10221 or SR2595 decreased expression of IGF-1, as well as the canonical PPARγ target genes PGK1 and PKM2, in C1/M2-positive MEC cells (Figures 5C and 5D). Treatment of C1/M2-positive cell lines with sublethal doses of SR10221 (½ IC50) significantly decreased proliferation, 2D colony formation, and 3D tumor spheroid growth (Figures 5E, 5F, 5G, and S6G). Moreover, this decreased growth and tumorigenic potential coincided with potent induction of apoptosis, as evidenced by significant increases in caspase-3/7 activation (Figure 5H).
To determine the efficacy of PPARγ inverse agonists in vivo, we first tested the potency of SR10221 in subcutaneous HMC3A tumor xenograft models labeled with our LumiFluor bioluminescent reporter (Schaub et al., 2015). Tumor xenografts were allowed to grow to a palpable size (~50 mm3), and animals were randomized into two cohorts. These cohorts then received intra-peritoneal (i.p.) administration of 20 mg/kg of SR10221 or vehicle control once daily for three weeks, which resulted in the significant inhibition (p < 0.0001) of HMC3A tumor xenograft growth (Figures 6A and 6B). Similarly, we tested the in vivo potency of SR2595, because it displays superior pharmacokinetic properties, although we administered SR2595 at 60 mg/kg, because SR10221 was shown to possess ~2- to 3-fold greater potency in vitro (Marciano et al., 2015). Treatment with SR2595 at this concentration was generally well tolerated without body weight loss (Figure 6D) but significantly blocked the growth of C1/M2-positive MEC tumor xenografts (Figures 6C and 6E–6G). Collectively, these results suggest that inhibition of IGF-1 signaling using PPARγ inverse agonists alone or in combination with IGF-1Ri may be a viable therapeutic strategy for the targeted treatment of C1/M2-positive MEC.
Figure 6. PPARγ inverse agonists suppress tumor growth in xenograft models of salivary MEC.

(A) Growth of HMC3A xenograft tumors over time during treatment with either SR10221 or vehicle control. Tumor xenograft volume was quantified by caliper measurements obtained on the indicated days following initiation of drug administration. Decreased tumor volume in the SR10221 cohort correlates directly with suppression of tumor growth in vivo (n = 4, mean ± SEM; Student’s t test; ****p ≤0.0001).
(B) Weight of resected vehicle- and SR10221-treated HMC3A tumor xenografts at the study endpoint (n = 4, mean ± SEM; Student’s t test; **p ≤ 0.01).
(C) Representative in vivo bioluminescent images obtained at the endpoint of Nu/Nu mice bearing subcutaneous HMC3A tumor xenografts and treated with either SR2595 or vehicle control. Images were captured with an open filter and an acquisition time of 5 min (binning = 2; FOV = 25; Fstop = 2; object height = 1.5).
(D) Weights of Nu/Nu mice bearing subcutaneous HMC3A xenograft tumors and treated with SR2595 or vehicle control for up to 24 days (n = 4, mean ± SEM). (E) Comparison of HMC3A xenograft tumor growth following treatment with either SR2595 or vehicle control. Bioluminescence imaging (BLI) signals from tumor xenografts were quantified by region of interest (ROI) analysis of images obtained at the endpoint (day 24 following initiation of drug administration). A decreased BLI signal in the SR2595 cohort correlates directly with suppression of tumor growth in vivo (n = 4, mean ± SEM; Student’s t test; **p ≤ 0.01).
(F) Volume of resected vehicle- and SR2595-treated HMC3A xenograft tumors at the study endpoint (n = 4, mean ± SEM; Student’s t test; *p ≤ 0.05).
(G) Weight of resected vehicle- and SR2595-treated HMC3A xenograft tumors at the study endpoint (n = 4, mean ± SEM; Student’s t test; *p ≤ 0.05).
DISCUSSION
Recurrent chromosomal translocations that generate gene fusions have long been known to have the potential to function as cancer drivers (Rabbitts, 1994, 2009; Rowley, 2001). However, the advent of fusion detection algorithms applied to “omics”-level data not only has enabled the discovery of additional gene fusions composed of splicing factors, signal transduction proteins, transcription factors, and/or transcriptional coregulators (Fernandez-Cuesta et al., 2015; Jang et al., 2020; Kumar-Sinha et al., 2015) but has also aided in elucidating the direct functional consequences of these fusions on cellular processes such as signaling and gene expression (Latysheva et al., 2016; Lee and Young, 2013). These aberrant processes are frequently unique to distinct gene fusions and characteristic of specific cancer subtypes (Fishbein et al., 2017; Gao et al., 2018). C1/M2 represents one such recurrent gene fusion composed of two transcriptional coactivators that is pathognomonic for MEC (O’Neill, 2009; Tonon et al., 2003). Here, we report that the C1/M2 fusion oncogene directs profound reprogramming of transcriptional networks and establishes a synthetic IGF-1 signal circuit through aberrant expression of an alternative PGC-1α splice variant in salivary MEC.
Despite being the most common salivary gland malignancy, MECs are relatively rare compared with other head and neck cancers; consequently, the pathobiology of C1/M2 remains poorly understood. Although it is clear that patients with advanced-stage and/or high-grade tumors display significantly worse overall survival (Chen et al., 2013), the prognostic value of fusion status has been challenged (Seethala and Chiosea, 2016). Most MEC patients are treated with surgical excision, which is sometimes accompanied by adjuvant radiotherapy (Nance et al., 2008). Unfortunately, resistance to chemoradio-therapies and a lack of targeted therapies for recurrent/metastatic disease pose significant challenges to treating patients with aggressive MEC tumors. For example, studies have indicated that paclitaxel, trastuzumab, or RTKi may be viable treatments for fusion-positive MEC; however, preliminary results show only modest responses to these treatments (Coca-Pelaz et al., 2015; McHugh et al., 2012) emphasizing the need for the performance of thorough molecular profiling studies to aid in the development of new targeted therapies.
As a transcriptional coactivator gene fusion, C1/M2 interacts with and activates two master transcription factors, CREB and MYC (Amelio et al., 2014; Tasoulas et al., 2019). Unfortunately, intrinsic disorder and a lack of available binding pockets has made designing small molecules that can effectively target transcription factors a major challenge (Bishop et al., 2019; Chen and Koehler, 2020; Wachtel and Schäfer, 2018). In addition, two recent genomics studies revealed that C1/M2 is the primary oncogenic driver in most salivary MECs and that these fusion-positive cases lack other cooperating mutations that occasionally serve as actionable targets in other cancers (Kang et al., 2017; Wang et al., 2017). In this study, we set out to identify significantly altered and clinically relevant target genes and pathways downstream of C1/M2 that can be exploited to develop novel therapeutic approaches to treat patients with MEC. Using transcriptomic profiling, we show that C1/M2 expression is correlated with increased IGF-1 expression and pathway activation in salivary MEC. Autocrine and paracrine activation of the IGF-1 pathway is known to promote growth and survival of multiple tumor types (Pollak, 2008; Yu and Rohan, 2000). Notably, endocrine IGF-1 signaling is broadly involved in normal tissue growth and development through stimulation of IGF-1R and has been shown to play critical roles in regulating salivary gland growth and development and in stimulating regeneration following injury (Amano and Iseki, 1993; Kerr et al., 1995; Ryan et al., 1992; Werner and Katz, 2004), thus supporting an important role for aberrant IGF-1 expression in salivary tumor development. Through bioinformatics analyses and unbiased small-molecule drug screening, we were successful in pinpointing IGF-1R as an actionable target in salivary MEC, suggesting that treatments directed against IGF-1R may provide therapeutic benefit. However, despite early excitement surrounding the potential utility of these treatments for many cancers (Gualberto and Pollak, 2009), targeting the IGF axis has yielded disappointing results in clinical trials (Chen and Sharon, 2013; Denduluri et al., 2015). Thus, the potential utility of anti-IGF-1R antibodies and small-molecule drug inhibitors, alone or combined with other therapeutic modalities, is being investigated as a viable alternative approach. Therefore, we also sought to explore the underlying mechanism governing IGF-1 activation by C1/M2 in salivary MEC and identified unexpected rewiring of this growth factor signal circuit coordinated by aberrant expression of an alternatively spliced target gene of C1/M2-CREB, PGC-1α4.
PGC-1α has been shown to exert oncogenic activity (Frattini et al., 2018) or tumor-suppressive activity (Torrano et al., 2016), depending on cell type and context-dependent metabolic cues (Mastropasqua et al., 2018; Sancho et al., 2015; Xing et al., 2017). The PGC-1α family of transcriptional coactivators is composed of an expanding list of transcript isoforms generated by multiple promoters and alternative pre-mRNA splicing, and the resulting protein variants are known to display unique functional properties (Martínez-Redondo et al., 2015, 2016). Most current literature focuses on the role of the major PGC-1α transcript, PGC-1α1, in various cancers, including melanoma (Luo et al., 2016, 2020; Vazquez et al., 2013) and prostate cancer (Torrano et al., 2016; Valcarcel-Jimenez et al., 2019; Wallace and Metallo, 2016). However, rare, alternatively spliced transcripts such as PGC-1α4 are emerging as key players in multiple cancer types and thus represent an attractive target for pharmacologic intervention (Kimes et al., 2014; Oltean and Bates, 2014; Wang and Lee, 2018; Zhang and Manley, 2013). To date, the specific role of these PGC-1α variants in cancer has remained elusive. We demonstrate that the C1/M2 fusion selectively induces CREB-dependent expression of PGC-1α4, which we and others have shown is associated with IGF-1 expression in anabolic skeletal muscle (Bruno et al., 2014; Ruas et al., 2012; Tasoulas et al., 2019). We confirm here that C1/M2 directs similar anabolic pro-growth and pro-survival signaling partly by coordinating this autocrine PGC-1α4-IGF-1 signal circuit in fusion-positive salivary MEC.
A model that may explain the transcriptional and pre-mRNA splicing effects of C1/M2 on PGC-1α4 is that C1/M2 acquires a strong transcriptional activation domain from MAML2 and retains the CREB binding domain of CRTC1; however, the splicing domain normally present in full-length CRTC1 is deleted (Amelio et al., 2009; Tasoulas et al., 2019). Importantly, PGC-1α4 retains the activation domain common to other PGC-1α isoforms, which mediates binding to nuclear hormone receptor transcription factors such as PPARγ (Li et al., 2008; Martínez-Redondo et al., 2015; Sonoda et al., 2008). Similar to PGC-1α, dichotomous roles for PPARγ have been described, with evidence pointing to oncogenic activity in some cancers but tumor-suppressor actions in other cancers. This is supported by data using PPARγ agonists or antagonists, respectively (Goldstein et al., 2017; Kardos et al., 2016; Khandekar et al., 2018; Zou et al., 2019). We demonstrate here that PGC-1α4 upregulates IGF-1 in a PPARγ-dependent manner in C1/M2-positive salivary MEC, which sensitizes these tumor cells to treatment with PPARγ inverse agonists. Given the limitations of anti-IGF-1R monotherapy, the identification of PPARγ inverse agonists as potential therapeutics for the treatment of MEC is particularly exciting. However, a limitation of this study is that therapeutic effects achieved with the PPARγ inverse agonists SR10221 and SR2595 were in the low micromolar range, although functional ligand binding and induced PPARγ conformational changes are possible in the nanomolar range (Marciano et al., 2015; Shang et al., 2020). These differences in the inverse agonist concentrations required to achieve corepressor-dependent PPARγ repression may result from higher binding affinity of the PGC-1α4 variant with PPARγ and/or increased levels of competing endogenous ligands (e.g., esterified lipids) in salivary MEC cells. Our bioinformatic analyses revealed that several metabolic pathways with the potential to directly influence endogenous ligand levels, including lipid biosynthesis and modification pathways, are significantly elevated in C1/M2-positive salivary MEC (Table S2). An important caveat to our study is that C1/M2 seems to have wide-ranging effects on multiple genes involved in pro-tumorigenic pathways. Although we demonstrate here that targeting PGC-1α4-IGF-1 signaling using PPARγ inverse agonists effectively reduces MEC cell growth and proliferation, it is likely that the emergence of compensatory mechanisms during prolonged treatment will necessitate the use of combinatorial therapies in clinical studies.
In summary, our study identifies the C1/M2 fusion oncogene as a master regulator of pro-tumorigenic transcriptional networks partly via a PPARγ-dependent PGC-1α4-IGF-1 signaling circuit. Therefore, elucidation of the C1/M2-regulated PGC-1α4-dependent transcriptional program may open new avenues for the identification of other pathways that can be exploited to stratify patients suitable for precision therapies. Our findings establish that targeting this synthetic signaling circuit via IGF-1R inhibition or PPARγ inverse agonism, individually or in combination, is a potential therapeutic option for patients with C1/M2-positive salivary MEC.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Antonio L. Amelio (antonio_amelio@unc.edu).
Materials availability
The plasmids generated in this study have been deposited with Addgene. Cell lines generated in this study are available upon request via a material transfer agreement (MTA).
Data and code availability
The datasets supporting the current study have not been deposited in a public repository because the Institutional Review Board (IRB) protocols supporting this work are secondary analyses IRBs for use of surgical archives without consent, but the datasets are hosted by the UNC Lineberger Bioinformatics Core and are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Clinical samples
All research involving human tumor tissues was reviewed and approved by The University of North Carolina at Chapel Hill Institutional Review Board under IRB protocols 15–1604 and 17–2947. Tissues from these de-identified clinical subjects were identified from chart review and archived FFPE salivary MEC (n = 18; 10 females and 8 males, average age of 49.6 years) or normal salivary gland (n = 6) tissue samples stored at room temperature less than ten years before blocks were sectioned and RNA isolation performed. For all cases, multiple H&E slides were reviewed by a pathologist and sections with tumor were selected for inclusion in the study. Adjacent serial unstained sections were then macrodissected and tumor material submitted for RNA extraction.
Xenograft models
All animal studies were reviewed and approved by The University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee under IACUC protocol 17–202. Male 6–8 week old athymic nude mice (Nu/Nu) were obtained from the Animal Studies Core at the University of North Carolina at Chapel Hill and housed in facilities run by the Division of Comparative Medicine at the University of North Carolina (Chapel Hill, NC, USA). For all xenograft studies, mice were subcutaneously injected with 1×106 UM-HMC-3A cells resuspended in 50% HBSS and 50% Matrigel. When the average tumor size reached a palpable size (~50 mm3), animals were randomized into two groups (Vehicle and SR10221 or SR2595) so that the average tumor size in each group was approximately equal. The vehicle group was intraperitoneally (i.p.) injected daily with 250 uL of a 10% DMSO, 10% Tween-80, 80% PBS solution. Drug-treated animals were i.p. injected daily with a 10% DMSO, 10% Tween-80, 80% PBS solution containing either 20 mg/kg SR10221 (see Methods S1 for synthesis and purification details) or 60 mg/kg SR2595 (Sigma-Aldrich #SML2037) based on an average mouse weight of 25 g. Caliper measurements were collected every 2 days and bioluminescent imaging (BLI) was performed every 5 days throughout the course of the study.
Cell lines
UM-HMC-1 (HMC1), UM-HMC-3A (HMC3A), and UM-HMC-3B (HMC3B) cells (Warner et al., 2013) were kindly provided by Dr. Jacques Nör (University of Michigan, Ann Arbor, MI, USA). NCI-H292 (H292) and NCI-H3118 (H3118) cells (Tonon et al., 2003) were generously provided by Dr. Frederic Kaye (University of Florida, Gainesville, FL, USA). A253, A388, and A431 cells were kindly provided by Dr. Bernard Weissman (University of North Carolina, Chapel Hill, NC, USA). HMC1 (source: male, salivary gland mucoepidermoid carcinoma (MEC)), HMC3A (source: female, hard palate MEC), and HMC3B (source: female, lymph node metastasis of hard palate MEC) parental and stably transduced lines were cultured in DMEM medium (GIBCO #11965–118) supplemented with 10% FBS (Atlanta Biologicals #S11550), 20 ng/mL EGF (Sigma-Aldrich #E9644), 400 ng/mL hydrocortisone (Sigma-Aldrich #H0888), 5 μg/mL insulin (Sigma-Aldrich #I6634), and 1X pen/strep/glutamine (PSG; Life Tech #10378016). H292 (source: female, lung MEC) cells were cultured in RPMI-1640 medium (Life Tech #11875119) supplemented with 10% FBS, 1X GlutaMAX (Life Tech #35050061), 1X NEAA (GIBCO #11140050), 1X NaPyr (Life Tech #11360070), and 1X PSG. HEK293A, HEK293-CMVTetRTetOC1/M2 (Amelio et al., 2014), HEK293-PGKTetOn3GTRE3GSPGC−1α4, Lenti X-293T (viral packaging cell line; Takara #632180), H3118 (source: female, parotid gland MEC), A388 (source: male, squamous cell carcinoma (SCC)), and A431 (source: female, vulvar SCC) cells were cultured in DMEM supplemented with 10% FBS, 1X GlutaMAX, and 1X PSG. A253 (source: male, salivary gland SCC) cells were cultured in McCoy’s 5A medium (Life Tech #16600108) supplemented with 10% FBS, 1X GlutaMAX, and 1X PSG. Cells were passaged using TrypLE (GIBCO #12604013) every 2–3 days or when they reached 90% confluence. All cells were maintained in a 37°C, 5% CO2 atmosphere. All cell lines were confirmed mycoplasma-free by PCR as previously described (Young et al., 2010) and using mycoplasma detection primers (see Table S6).
METHOD DETAILS
Clinical RNA isolation
Formalin-fixed paraffin-embedded (FFPE) tissue samples were sent to the UNC Lineberger Comprehensive Cancer Center Translational Genomics Lab (TGL) for RNA isolation using the Maxwell 16 MDx Instrument (Promega #AS3000) and the Maxwell 16 LEV RNA FFPE Kit (Promega #AS1260) according to the manufacturer’s protocol (Promega #9FB167). Pathology review of a hematoxylin and eosin (H&E) stained slide was used to guide macro-dissection of unstained slides to enrich for tumor RNA. Total RNA quality was measured using a NanoDrop spectrophotometer (Thermo Scientific ND-2000C) and a TapeStation 4200 (Agilent G2991AA). Total RNA concentration was quantified using a Qubit 3.0 fluorometer (Life Technologies Q33216).
RNA-seq
Total RNA sequencing libraries were prepared at TGL using a Bravo Automated Liquid-Handling Platform (Agilent G5562A) and the TruSeq Stranded Total RNA Library Prep Gold Kit (Illumina 20020599) according to the manufacturer’s protocol (Illumina 1000000040499). RNaseq library quality and quantity were measured using a TapeStation 4200 (Agilent G2991AA), pooled at equal molar ratios, and denatured according to the manufacturer’s protocol (Illumina 15050107). Sequencing was performed at the High Throughput Sequencing Facility (HTSF) at UNC Chapel Hill. Two RNA-seq libraries were sequenced per lane on a HiSeq2500 (Illumina SY–401–2501) with 2×50 bp paired-end configuration according to the manufacturer’s protocol (Illumina 15035786).
Bioinformatics
RNaseq data analyses were performed with FASTQ files aligned to the GRCh38 human genome (GRCh38.d1.vd1.fa) using STAR v2.4.2 (Dobin et al., 2013) with the following parameters:–outSAMtype BAM Unsorted,–quantMode TranscriptomeSAM. Transcript abundance for each sample was estimated using Salmon version 0.1.19 (Patro et al., 2017) to quantify the transcriptome defined by Gencode v22. Gene level counts were summed across isoforms and genes with low expression (i.e., samples with fewer than 10 reads) were removed prior to downstream analyses. The R package DESeq2 (version 1.24.0) (Love et al., 2014) was used to test for differentially expressed genes between C1/M2 positive MEC tumors and normal samples or between all MEC tumors and normal samples. The C1/M2-regulated IGF-PI3K gene signature (Musicant_MEC_CRTC1-MAML2_IGF1 curated gene set) includes genes that are differentially expressed between C1/M2 positive MEC samples and normal samples with Benjamini–Hochberg FDR (q-value) < 0.05 and a fold change > 2. The resulting list of genes was refined to include only genes shared with IGF1-related Molecular Signatures Database (Database: MSigDB) curated gene sets. Gene set enrichment analysis was performed on a customized list of gene sets with GSEA software from the Broad Institute (Mootha et al., 2003; Subramanian et al., 2005). The customized gene sets include a list of pathways from MSigDB curated gene sets that are related to IGF1. Hierarchical clustering and heat-map plotting were performed using the ComplexHeatmap R package version 2.0.0 (Gu et al., 2016). An average linkage algorithm with a Euclidean distance function was applied to variance stabilizing transformed (VST) gene expression data for the gene signature.
RTKi drug screen
Compound screening was performed as previously described (Bevill et al., 2019; Lipner et al., 2020). Briefly, drug screening was performed in a 384 well plate format, in duplicate. Cells were seeded in 45 μL of full culture media; A253 (1000 cells/well), H292 (600 cells/well) H3118 (7000 cells/well), HMC1 (500 cells/well), HMC3A (500 cells/well) and HMC3B (700 cells/well). Cells were seeded in 384-well plates using a BioTek microplate dispenser. Cells were allowed to adhere overnight, following which they were treated with 176 individual drugs at 6 doses (10 nM, 100 nM, 300 nM, 1 μM, 3 μM and 10 μM) using a Beckman Coulter Biomek FX Automated Liquid Handling instrument. Each plate also included 1 μM Bortezomib and 1% DMSO as positive and negative controls for growth inhibition, respectively. Furthermore, a full 384 well plate with 1% DMSO treatment was also seeded per cell line and was used to confirm minimal well-location associated intra-plate variability. 72 hr post drug addition, cells were lysed and cell viability was measured using Cell Titer Glo 2.0 (Promega #G9243) according to the manufacturer’s instructions. Luminescence was measured using a PHERAstar FS instrument and growth inhibition was calculated relative to DMSO-treated wells. Z’ scores were calculated as previously described (Zhang et al., 1999). The 1 μM Bortezomib (positive; 0% viability) and 1% DMSO (negative; 100% viability)-treated cells were used to calculate the dynamic range and screen variability/reliability (Z’ score). A Z’ score of > 0.75 was obtained for all cell lines tested. The 3SD/5SD cutoffs were calculated based on the variability observed in the 1% DMSO-negative control plates/wells.
Virus production and transduction
Lentiviral or retroviral expression plasmids were co-transfected with VSV-G envelope plasmid and either δ8.2 gag/pol (lentivirus) or pMD (retrovirus) helper plasmids into Lenti X-293T cells seeded in 10 cm tissue culture dishes. Transfections were performed using 1 mg/mL Polyethyleneimine (PEI) Transfection Reagent (VWR #BT129700). Briefly, 1.5 μg VSV-G, 5 μg δ8.2 or pMD, and 6 μg lentiviral or retroviral plasmid were brought to 500 μL with OptiMEM (LifeTech #1158021) and vortexed briefly. In a separate tube, 25 μL PEI (2 μL PEI/μg DNA) was added to 475 μL OptiMEM and vortexed briefly. Both solutions were incubated at room temperature for 5 min, then combined and incubated for an additional 20 min at room temperature. This mixture was then added dropwise to the seeded Lenti X-293T cells. The next day, cell culture media was replaced with DMEM supplemented with 1x NaPyr, 10 mM HEPES, 1X GlutaMAX, and 1X PSG (no FBS). Two days later, media was collected and filtered through a 0.45 μm PVDF membrane and then viral particles were concentrated via ultracentrifugation (100,000 g for 2 hr at 4°C) into a sucrose cushion. Concentrated virus was resuspended in cold PBS and either stored at −80°C or used immediately for transduction.
To generate stable cells, various cell lines were seeded at 50% confluency in 12-well plates. Concentrated virus and 4 μg/mL polybrene were added directly to cells and then plates were centrifuged at 1200 g for 90 min at 30°C. To generate stable LumiFluor-over-expressing MEC cells, HMC3A cells were transduced with concentrated lentivirus expressing our GpNLuc reporter (Schaub et al., 2015) and selected with puromycin for at least 10 days prior to use in experiments. To generate shRNA-expressing cells, parental HMC3A or HMC3A-PGKGpNLuc cells were transduced with concentrated shRNA-expressing retro- or lentivirus expressing the shRNA of interest. Proliferation assays, quantitative real-time PCR, and western blotting were performed on polyclonal cell populations within 14 days of transduction for all shRNA knockdown experiments. To generate cells that express the PGC-1α4 splice variant, HEK293A cells were first transduced with a concentrated lentivirus that expresses TetOn3G and stable cells selected with puromycin. Subsequently, these HEK293-PGKTetOn3G stables were transduced with a concentrated lentivirus that expresses PGC-1α4 upon tetracycline/doxycycline administration and the resulting HEK293-PGKTetOn3G-TRE3GSPGC−1α4 stable cells were selected with hygromycin for at least 10 days prior to use in experiments. To generate MEC cells with inducible dominant-negative CREB (dnCREB; A-CREB), HMC3A cells were first transduced with a concentrated lentivirus that expresses TetOn3G and stable cells were selected with puromycin. These HMC3A-PGKTetOn3G stables were transduced with a concentrated retrovirus that expresses A-CREB (Ahn et al., 1998) upon tetracycline/doxycycline administration, and the resulting HMC3A-PGKTetOn3G-TetOdnCREB stable cells were selected by fluorescence activated cell sorting (FACS) by gating for tdTomato-positive cells. To generate MEC cells with inducible dCas9 expression, HMC3A cells were first transduced with a concentrated lentivirus that expresses TetOn3G and stable cells were selected with puromycin. These HMC3A-PGKTetOn3G stables were transduced with a concentrated lentivirus encoding the doxycycline induced dCas9-KRAB-IRES-BFP cassette (Addgene #85449) (Fulco et al., 2016). To select for positively transduced cells, 1 μg/mL doxycycline was applied to the cells for 72 hours and then BFP-positive cells were selected by FACS. Finally, the HMC3A-PGKTetOn3G -TRE3GSdCas9-KRAB-IRES-BFP cells were transduced with a lentivirus constitutively expressing the respective PGC-1α sgRNA, a blasticidin resistance cassette, and a 2xNLS-mKate2 fluorescent reporter. Cells were selected with blasticidin for at least 7 days and successful selection was confirmed via nuclear mKate2 fluorescence signal.
Cell viability
HMC1, HMC3A, or HMC3B cells were seeded in 96-well clear-bottom, white-walled plates (Corning #3917) at 15,000 cells/well. The next day, media was replaced with 200 μL fresh media containing either vehicle (DMSO) or increasing concentrations of drug (BMS-754807 (Sigma-Aldrich #BM0003), PPP (Santa Cruz #SC-204008), SR10221 (see Methods S1 for synthesis and purification details), SR2595 (Sigma-Aldrich #SML2037), or T0070907 (Fisher #NC1015539); final concentration 1% DMSO). 72 hr later, the amount of ATP (a proxy for cell viability) in each well was measured using the ATPlite Luminescence Assay System (PerkinElmer #6016949) according to the manufacturer’s instructions. Briefly, plates were removed from the cell culture incubator and equilibrated for 30 min at room temperature in the dark. Then, media was aspirated from each well and 100 μL reconstituted ATPlite 1-step reagent was added to each well. Plates were shaken for 2 min (425 cpm, 3 mm orbit) in a Cytation 5 plate reader (BioTek Instruments, Inc.) and then ATP levels were quantified by measuring total luminescence. IC50 values were calculated in GraphPad Prism (GraphPad, San Diego, CA, USA) using a non-linear curve fit (log[agonist] versus response, four parameter-variable slope). All assays were performed in biologic triplicate with three technical replicates for each condition. Figure panels show one representative experiment (including three technical replicates each). IC50 values presented in Table 1 were calculated using the results from all three biologic replicates.
Cell proliferation assays (Cytation 5)
HMC3A-PGKGpNLuc cells (either non-transduced or transduced with shRNAs targeting PGC-1α, IGF-1R, or MAML2) were seeded in 12-well plates at 200,000 cells/well. The next day, fresh media was applied to each well and then individual wells were imaged at 0, 4, 8, 24, 28, 32, and 48 hr following the media change. Five separate fields of view were imaged for each well at 4X magnification using a fluorescent plate reader (BioTek Cytation 5, BioTek Instruments, Inc.). Between each imaging time point, plates were returned to the cell culture incubator. To determine cell confluence, a primary mask was applied to each fluorescent image using the recommended parameters set out in the BioTek Technical Note “Measuring Confluence Using High Contrast Brightfield” (https://www.biotek.com/resources/technical-notes/measuring-confluence-using-high-contrast-brightfield/). Confluence measurements from multiple fields of view in a single well were used as technical replicates. Each assay was performed in biologic triplicate.
Cell proliferation assays (IncuCyte Zoom)
HMC3A cells were seeded in 48-well plates at 15,000 cells/well. The next day, media was replaced with fresh media containing either vehicle (1% DMSO) or the indicated concentration of PPP (Santa Cruz #SC-204008) or SR10221. Then, plates were imaged at 10X magnification every 2–4 hours on an IncuCyte Zoom (Essen BioScience), which maintained plates at a constant 37°C and 5% CO2 for the duration of the assay. Four separate fields of view were imaged for each well and an image mask was applied to each image to generate confluency measurements. Each assay was performed in biologic triplicate with at least three technical replicates per experiment.
Western blotting
Whole cell lysates were prepared in buffer containing 5% glycerol (Sigma-Aldrich #G5516), 25 mM Tris (pH 7.4; Fisher #BMA51237), 150 mM NaCl (Fisher #BMA51202), 1 mM EDTA (Sigma-Aldrich #E7889), and 1% NP-40 (Fisher #50–147–289) supplemented with protease inhibitors (cOmplete, EDTA-free Protease Inhibitor Cocktail, Roche #04693132001) and phosphatase inhibitors (Phos-STOP, Roche #10917400). Lysates (30–50 mg) were loaded onto mini-10% tris-glycine polyacrylamide gels and proteins were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to a 0.45 μm nitrocellulose membrane (Fisher #0088018) using a Bio-Rad Trans-Blot Turbo system set at 1.3 Amps (constant) and 25 V for 30 min. Membranes were blocked for 1 hr at room temperature in TBS-T + 5% bovine serum albumin (BSA; Sigma #A2153) and then incubated overnight at 4°C with primary antibodies: IGF-1 (0.2 μg/mL; Abcam #ab9572), IGF-1Rβ(1:2500; Cell Signaling #3018S), phosphorylated IGF-1Rβ(Tyr1135; 1:1000; Cell Signaling #3918S), PPARγ (1:1000; Abcam #209350), actin (1:5000; Sigma #A3854), tubulin (1:5000; Sigma #T6074), or Flag (1:5000; Sigma #A8592) diluted in TBS-T + 5% BSA. Following primary antibody incubation, membranes were washed and probed with Horseradish peroxidase (HRP)-conjugated goat anti-mouse (1:5000; Thermo Fisher #31432) or donkey anti-rabbit (1:5000; Thermo Fisher #31458) secondary antibodies diluted in TBS-T supplemented with 5% BSA for 1–2 hr at room temperature. Blots were imaged using Clarity ECL (Bio-Rad #170–5060) and ImageQuant LHS4000 (GE).
2D colony formation
HMC1, HMC3A, and HMC3B cells were seeded in 24-well plates at 800, 600, and 1400 cells/well, respectively. The next day, media was replaced with 500 μL fresh media containing either vehicle (DMSO) or the indicated concentration of drug (BMS-754807 (Sigma-Aldrich #BM0003), PPP (Santa Cruz #SC-204008), SR10221, SR2595 (Sigma-Aldrich #SML2037), or T0070907 (Fisher #NC1015539); final concentration 1% DMSO). Seven days later, media was removed from all wells and cells were fixed in 10% buffered formalin (Fisher #SF994) for 5 min at room temperature. Wells were washed once in ddH2O and then stained in 0.05% crystal violet (Sigma-Aldrich #C6158) for 30 min at room temperature. Wells were then washed an additional 3 times in ddH2O to remove any unbound stain and allowed to dry at room temperature overnight. Once dry, individual wells were imaged at 4X magnification on a BioTek Cytation 5 plate reader (BioTek Instruments, Inc.) and images were stitched using the Gen5 software (v2.09; BioTek Instruments, Inc.) using the default parameters. Colony numbers were quantified manually in ImageJ (Schneider et al., 2012), where a colony is defined as a cluster of at least 50 individual cells.
3D tumor spheroid formation
Individual wells of a 48-well plate were coated with 100 μL of a 25% Matrigel (Fisher #CB4023A) solution diluted in cold 1X DPBS. The plate was spun in a Thermo Scientific Sorvall LYNX 4000 centrifuge at 4°C for 10 min at 2000 g to ensure even distribution across the well surface. The plate was then placed in a 37°C incubator for 1 hr to allow the Matrigel ‘bed’ to completely polymerize. Next, excess DPBS was gently aspirated from the wells and 2000 HMC3A cells resuspended in 100 μL media were added to each well. The plate was centrifuged at 4°C for 5 min at 2000 g to embed the cells into the Matrigel. The plate was returned to the incubator for an additional hour to allow for cell adhesion to the Matrigel. After 1 hr, excess media was carefully aspirated and 50 μL undiluted Matrigel was gently added to each well on top of the cells. The plate was again returned to the incubator for 1 hr to allow for Matrigel polymerization. Finally, 500 μL media (containing either drug or vehicle at a final concentration of 1% DMSO) was added to each well. Every 24 hr, wells were imaged at 4X magnification on a BioTek Cytation 5 plate reader (BioTek Instruments, Inc.) and images were stitched using the Gen5 software (v2.09; BioTek Instruments, Inc.) using the default parameters. Tumorsphere area was manually measured in ImageJ using the ROI area measurement tool. A minimum of 50 individual tumorspheres were quantified for each condition in each experiment.
Caspase 3/7 apoptosis assay
For drug treatment assays, HMC3A cells were seeded in 6-well plates at 350,000 cells/well. The next day, media was replaced with 2 mL fresh media containing either vehicle (DMSO) or the indicated concentration of drug (PPP (Santa Cruz #SC-204008) or SR10221; final concentration 1% DMSO). For shRNA assays, HMC3A cells stably transduced with shNS (control) or shPPARγ were seeded in 6-well plates at 350,000 cells/well. At the indicated time point (either post-treatment (PPP/SR10221) or post-seeding (shRNAs)), cells were trypsinized, spun down, and resuspended in 1 mL 1X DPBS supplemented with 1X GlutaMAX (Life Tech #35050061) and 10% FBS (Atlanta Biologicals #S11550). Apoptotic cells were visualized using the CellEvent Caspase 3/7 Green Flow Cytometry Assay Kit (ThermoFisher #C10427) according to the manufacturer’s instructions. Briefly, 1 μL CellEvent Caspase 3/7 Green Detection Reagent was added per 1 mL of resuspended cells. This mixture was gently vortexed and incubated at 37°C for 25 min. Next, 1μL SYTOX AADvanced Dead Cell Stain was added per 1 mL of resuspended cells. This mixture was gently vortexed and incubated at 37°C for an additional 5 min. Samples were analyzed via flow cytometry on a BD Accuri C6 instrument. An aliquot of unstained cells from each condition was reserved as a control to generate gates.
Chromatin immunoprecipitation (ChIP) assay
HEK293-CMVTetRTetOC1/M2 cells (Amelio et al., 2014) were seeded in 15 cm tissue culture dishes in either plain growth medium or growth medium containing 1 μg/mL doxycycline (Sigma-Aldrich #D9891). After 48 hr, formaldehyde (Fisher #BP531) was added dropwise to each plate to 1% final concentration. Plates were incubated at room temperature for 10 min and then crosslinking was stopped by adding glycine (Fisher #BP381–1) to a final concentration of 120 mM and incubating for an additional 5 min. Cells were then transferred to a tube, pelleted at 4°C, and washed twice with cold 1X DPBS. Finally, washed cells were resuspended in 1X DPBS supplemented with protease inhibitors (cOmplete, EDTA-free Protease Inhibitor Cocktail, Roche #04693132001) to a concentration of 5×106 cells/mL and stored at −80°C in aliquots of 1 mL until lysis. For lysis, individual cell aliquots were pelleted, resuspended in 200 uL SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1) supplemented with protease inhibitors, and rocked at 4°C for at least 20 min. Next, 800 uL ChIP dilution buffer was added and aliquots were immersed in a 100% ethanol ice bath (−11 to −7°C) and sheared (2 Amp for 5 s) using a sonicator (Qsonica #Q700A) equipped with a microtip sonic dismembrator (Model 505, Fisher #4418). A fraction of this sheared sample was reserved as a Pre-IP control. To the rest of the sample, 1 μg anti-Flag M2 antibody (Sigma #F1804) was added and samples were rocked at 4°C overnight. Antibody-chromatin complexes were isolated by rocking with Protein G SureBeads (BioRad #1614821) at 4°C for 4 hr. Beads were washed once each in low salt buffer (0.1% SDS, 1% Triton X-100, 2mM EDTA, 20 mM Tris-HCl, 15 mM NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, 500 mM NaCl), and LiCl buffer (250 mM LiCl, 1% NP-40, 1% Deoxycholate, 1mM EDTA, 10 mM Tris-HCl, pH 8.1) and then twice in TE buffer (1.2 mM EDTA, 10 mM Tris-HCl, pH 8.1). Samples were eluted twice with pre-heated (55°C) elution buffer for 15 min at room temperature with shaking. Cross-links were reversed by adding NaCl to 200 mM and incubating for at least 4 hr at 65°C. Samples were treated for 30 min at 37°C with RNase A at 8 μg/μL final concentration. Finally, samples were treated with Proteinase K solution (10 μL 0.5M EDTA, 10 μL 1M Tris, 1 μL 20 mg/mL Proteinase K (LifeTech #AM2546) for a 500 μL sample) for 1 hr at 50°C and then DNA was purified using the NucleoSpin Gel and PCR Cleanup Kit (Macherey-Nagel #740609.250). Occupancy of target proteins of interest within the promoter regions was assessed by qPCR (Table S6).
Luciferase reporter assay
HEK293-CMVTetRTetOC1/M2 cells were seeded in 24-well plates at 100,000 cells/well. The next day, transfected each well with 500 ng of either PGC-1α proximal promoter or PGC-1α distal promoter driven luciferase reporter using Lipofectamine (Invitrogen #50470) according to the manufacturer’s protocol and using a DNA:lipid ratio of 1:2. The next day, media was replaced with 1 mL fresh media with or without 1 μg/mL doxycycline (Sigma-Aldrich #D9891). After 24 hr, media was removed from all wells and 100 μL 1% Triton X-100 was added to each well. Plates were rocked for 15 min at room temperature. 50 μL of the supernatant from each well was transferred to a white, opaque 96-well plate, and 50 μL Bright-Glo Luciferase Assay Buffer (Promega #E264B) was added to each well. Total luminescence was quantified immediately on a BioTek Cytation 5 plate reader (BioTek Instruments, Inc.). For the dose-response luciferase reporter assays, HEK293 cells were reverse transfected with 3xPPRE promoter or IGF1-P2 promoter driven luciferase reporters using Lipofectamine 2000 Transfection Reagent (Invitrogen #11668019) in 96-well plates at a density of 2×104 cells/well. Forty-eight hours post-transfection, cells were treated with either vehicle (DMSO) or the PPARγ ligands GW1929 (Sigma-Aldrich #5668), SR2595 or SR10221 at the indicated concentrations. 24 hr after treatment, cells were lysed, and luciferase activity was quantified using the Dual-Glo Luciferase Assay System (Promega #E2920). Luminescence was measured using Synergy Neo microplate reader (BioTek). Values were normalized using Renilla expression.
Bioluminescence imaging
Bioluminescent-fluorescent BRET signal was measured non-invasively as previously described (Schaub et al., 2015) with minor modification. Briefly, animals were i.p. injected with 250 μM (1:20 dilution, ~500 μg/kg) Nano-Glo Luciferase Assay Substrate (Promega, #N1120) in sterile PBS. Isoflurane-anesthetized animals were then imaged using an AMI Optical Imaging System (Spectral Instruments Imaging, Inc.) 5 min after injection. Images were captured with open filter and acquisition times of 5 min or less at the indicated settings. Data were analyzed using Aura imaging software (v2.2.0.0).
Histology and immunohistochemistry
All animals showing xenograft tumors > 2 cm3 or other signs of distress were euthanized and subjected to full necropsy. For histological analysis, xenograft tumors were fixed in 10% neutral buffered formalin for approximately 1 week at room temperature. Following fixation, tissues were processed on an ASP6025 automated tissue processor (Leica Biosystems) and embedded in paraffin wax. Blocks were sectioned at 4–6 μm, mounted on glass slides, and FFPE tissue sections were deparaffinized prior to Hematoxylin and eosin (H&E) or mucicarmine staining. Immunohistochemistry was performed on the Discovery Ultra (Ventana Medical Systems) using manufacturers reagents on 4 μm sections. For anti-IGF-1 immunohistochemistry, anti-rabbit IGF-1 (Abcam #ab9572) was prepared using Discovery PSS Diluent (cat. #: 760–212). Antigen retrieval was performed using Ventana’s CC1 (pH 8.5) for 64 min at 90°C. The slides were given a hydrogen peroxide block for 8 min at room temperature and then incubated in the primary antibody diluent (1:100) for 1 hr at room temperature, followed by anti-Rabbit HRP secondary antibody (Vector Labs #MP-7401) for 32 min at room temperature. The slides were then treated with DAB and counterstained with Hematoxylin II for 12 min and then Bluing Reagent for 4 min.
Plasmids
The PGC-1α promoter constructs were generated by cloning the −500 to +52 bp of either the proximal or distal human PGC-1α promoter sequences via 5′ NheI and 3′ HindIII into the pGL4.15 luciferase reporter (Promega, #E6701). The human IGF-1 promoter constructs were generated by cloning either a 2079 bp region encompassing the P1 promoter or a 1547 bp region encompassing the P2 promoter via 5′ BglII and 3′ HindIII into either the pGL4.15 (Promega, #E6701) or the pGL3-Enhancer luciferase reporter (Promega, #E1771). The A-CREB (Ahn et al., 1998) dominant negative CREB cDNA was directionally cloned via 5′ NotI and 3′ MluI restriction enzyme sites into pRetroX-Tight-tdTomato (Amelio et al., 2014). The pTRE3G-MCS_PGK-GpNLuc construct was generated by excising the pTight promoter from the pRetroX-Tight-MCS_PGK-GpNLuc construct (Addgene plasmid #70185) containing our previously described LumiFluor optical reporter (Schaub et al., 2015) and subcloning the TRE3G promoter in its place. Subsequently, a N-terminal FLAG-tagged C1/M2 cDNA was directionally subcloned via 5′ NotI and 3′ MluI restriction enzyme sites into this pTRE3G-MCS-GpNLuc vector. The pLV-TRE3G_PGC1α4-mCherry:T2A:Hygro construct was custom synthesized (Cyagen Bioscience) to contain the human PPARGC1A variant 4 splice isoform (PGC-1α4) (Ruas et al., 2012) downstream of the TetO response element promoter for tet/dox inducible expression. The pFLAG-CMV-2_CRTC1/MAML2 (Tonon et al., 2003) construct was a gift from Frederic Kaye, the pLKO.1 shNS and shMAML2 (Chen et al., 2014) shRNA constructs were gifts from Lizi Wu, the pLMN UltramiR-E shIGF1R (ULTRA-3308043 and ULTRA-3443222) and shPPARGC1A (ULTRA-3197172 and ULTRA-3197171) constructs were from transOMICS Technologies, and the PPRE x3-TK-Luc (Kim et al., 1998) construct was a gift from Bruce Spiegelman. PPARγ knockdown constructs were purchased from Sigma-Aldrich (#SHCLNG-TRCN0000355926, #SHCLNG-TRCN0000001673, and #SHCLNG-TRCN0000001674). All plasmids were amplified and purified from either DH5α (Zymo Research #T3007) or Stbl3 (ThermoFisher Scientific #C737303) E. coli strains.
sgRNA design and cloning
sgRNAs targeting the PGC-1α Proximal or Distal promoters were designed using the CRISPR-ERA resource (http://crispr-era.stanford.edu/).
The PGC-1α proximal promoter sequence was used as the query for generating sgRNAs targeting the PGC-1α1 isoform (GenBank: NM_013261.5):
AGCAAAAAAAAAAAAAAAAAAAAAAAAAGCCCCGTTTGCGCTTTCAAACACTCCCTCAATGAGAAAATGTCTCATAAAAATGCATCATGTGATAAGCTCTTGCTTTAGTCCCAAACTGAGCTTGAGTCCACTTGGAGATCTTAGAATTAAAAAGATTGCAGGGGATTTTGGTTATTATATGGCCAGGGCTCCGTTTAGAGTCTGTGGCATTCAAAGCTGGCTTTAATCACAGCATGATGCTTGAAGCCTCCAAAAGTCTAAGTCTAAGTGTTTCCTTTCTTTCTTTCTTTTCTTTTTCTTTCTTTTTTTTTTTTTTTAAAGCGTTACTTCACTGAAGCAGAGGGCTGCCTTTGAGTGACGTCACGAGTTAGAGCAGCAAGCTGCACAGGGGAAGGGAGGCTGGGTGAGTGACAGCCCAGCCTACTTTTTAATAGCTTTGTCATGTGACTGGGGACTGTAGTAAGACAGGTGCCTTCAGTTCACTCTCAGTAAGGGGCTGGTTGCCTGCA.
The PGC-1α distal promoter sequence was used as the query for generating sgRNAs targeting the PGC-1α4 isoform (JQ772118.1): TAATAACAGGGGACAAAGGGTGAAATAATCCAGTAAAGTTTTGAGCAGCCACTTGACAACGTATTCCAAATAAATGAGAGGAGGAAAACCCTAGCTCTACCAACTGGGGCCATAAAACAGAGTCTTCTACACTCTCTTTAATGTCAATATTAACCAGTTTGCAAATGTTCAGTCAGTTATGGGCATGGATGGATGTCCTGAATGGGTTCCCGGGATAAAGTGTCATCATAGGACAGAAATCACAGGGAGAGTGCACCAAGGAAAAATTACAGTACTGCTATATTTACTTAGTGCCTCTGAACTAGGGTTTTATTTTCCACGGGTTGGAAAGGGAACCACCTGTCTCAATTGCTGATGTCAGAGAGCTCCCTCGAGACACAGGGCTGCTGGAAAGCACATGATACTGTACATATTTGCTCTTACGTTCGTATCTGGCTAAGATTGGGTTTCAGATTTGTGCCCTATTGTGGAGTTCATTTAGTAGTGACTCTGAGATGCCC.
The pgRNA-CKB vector (Addgene #73501) (Mandegar et al., 2016) was used to constitutively express the target guide RNAs. sgRNAs were cloned into the pgRNA-CKB vector using previously published methods (Gilbert et al., 2013). Briefly, the pgRNACKB backbone was digested with Esp3I (NEB #R0734) and dephosphorylated using recombinant Shrimp Alkaline Phosphatase. The required sgRNAs were synthesized (Eton Biosciences, Inc) as oligonucleotides with Esp3I compatible overhangs (Table S6). Oligonucleotides were phosphorylated using T4 Poly Nucleotide Kinase (NEB #M0201) and annealed by adding 50 μM sodium chloride to the phosphorylated oligo reaction, denaturing the oligo mix via boiling (95°C for 10 min), and performing an extended, slow cooling (~0.1°C/sec). 100 ng of the digested/dephosphorylated pgRNA-CKB backbone was ligated overnight with 1 μL of the 0.1 μM phosphorylated/annealed guide RNA using T4 DNA ligase. All sgRNA constructs were sequence verified (Eton Biosciences, Inc) using the primer listed in Table S6. Validation of isoform-specific knockdown revealed that one sgRNA construct (pSico_U6-Pan-PGC1a sgPGC) repressed expression of all PGC-1α isoforms, while another sgRNA construct, (pSico_U6-PGC1a1 sgPGC) selectively repressed expression of the major isoform (PGC-1α1).
PCR and qPCR
For cell lines and fresh-frozen tissues, gene expression was measured by extracting RNA using a Nucleospin RNA kit (Machery-Nagel #740955) according to the manufacturer’s instructions. cDNA was synthesized from 1 μg of RNA using iScript cDNA synthesis kit (Bio-Rad #170–8890). For human tissues, RNA was extracted using the Maxwell 16 MDx Instrument (Promega #AS3000) and the Maxwell 16 LEV RNA FFPE Kit (Promega #AS1260) according to the manufacturer’s protocol (Promega #9FB167). cDNA was made from 1–4 μg of RNA using SuperScript IV Reverse Transcriptase (Invitrogen #18090050) with dNTPs (NEB #N0446S), RNase inhibitor (Applied Biosystems #N808–0119), 25 μM oligo d(T)20 primer (Invitrogen #100023441) and 25 μM MAML2-specific reverse primer (Table S6). C1/M2 copy number was determined by establishing standard curves with 100 to 1×106 copies of FLAG-C1/M2 plasmid. Relative gene expression of target genes was determined using the 2ΔΔCt method and normalized to human RPL23 expression. qPCR was performed using FastStart Universal SYBR Green Master (Rox) Mix (Roche #04913850001) with 1/50 (tissue) or 1/100 (cells) volume of the cDNA iScript reaction, and 0.25 μM of primers (Table S6).
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical tests were executed using GraphPad Prism software or the statistical software R (version 3.1.2). Differences between variables were assessed by 2-tailed Student’s t test, one-way ANOVA with Holm-Sidak post hoc test, or Wilcoxon rank sum tests, where appropriate. For proliferation assays, pairwise comparisons were performed by Benjamini-Hochberg analysis. Data are expressed as mean ± SEM, P values less than 0.05 were considered statistically significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-IGF1 (0.2 μg/mL western blot) (1:300 immunohistochemistry) | Abcam | Cat #ab9572, RRID:AB_308724 |
| Rabbit anti-IGF-1Rβ (1:2500) | Cell Signaling | Cat #3018S, RRID:AB_560943 |
| Rabbit anti-phospho-IGF-1RβTyr1135 (1:1000) | Cell Signaling | Cat #3918S, RRID:AB_10548764 |
| Rabbit anti-PPARγ (1:1000) | Abeam | Cat #209350 |
| Mouse anti-β-actin (1:5000) | Sigma-Aldrich | Cat #A3854, RRID:AB_262011 |
| Mouse anti-α-tubulin (1:5000) | Sigma-Aldrich | Cat #T6074, RRID:AB_477582 |
| Mouse HRP-oonjugated anti-FLAG (1:5000) | Sigma-Aldrich | Cat #A8592, RRID:AB_439702 |
| Goat HRP-conjugated anti-mouse (1:5000) | Fisher | Cat #31432, RRID:AB_228302 |
| Donkey HRP-conjugated anti-rabbit (1:5000) | Fisher | Cat #31458, RRID:AB_228213 |
| Mouse anti-Flag M2 (1.5 μg/mL) | Sigma-Aldrich | Cat #F1804, RRID:AB_262044 |
| Bacterial and virus strains | ||
| DH5α E. coli | Zymo Research | Cat #T3007 |
| Stbl3 E. coli | ThermoFisher Scientific | Cat #C737303 |
| Biological samples | ||
| Healthy adult salivary gland tissue | University of North Carolina at Chapel Hill and UNC Hospitals Surgical Pathology | N/A |
| Salivary mucoepidermoid carcinoma (MEC) tumor tissue | University of North Carolina at Chapel Hill and UNC Hospitals Surgical Pathology | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| SR10221 | This paper | N/A |
| SR2595 | This paper and Sigma-Aldrich | Cat #SML2037 |
| T0070907 | Fisher | Cat #50–136-3114 |
| BMS-754807 | Sigma-Aldrich | Cat #BM0003 |
| PPP | Santa Cruz | Cat #SC-204008 |
| GW1929 | Sigma-Aldrich | Cat #5668 |
| Cell Titer Glo 2.0 | Promega | Cat #G9243 |
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Roche | Cat #04693132001 |
| PhosSTOP phosphatase inhibitor | Roche | Cat #10917400 |
| Matrigel | Fisher | Cat #CB4023A |
| Doxycycline | Sigma-Aldrich | Cat #D9891 |
| Lipofectamine | Invitrogen | Cat #50470 |
| Critical commercial assays | ||
| Nucleospin RNA kit | Macherey-Nagel | Cat #740955 |
| NucleoSpin Gel and PCR Cleanup Kit | Macherey-Nagel | Cat #740609 |
| Maxwell 16 LEV RNA FFPE Kit | Promega | Cat #AS1260 |
| TruSeq Stranded Total RNA Library Prep Gold Kit | Illumina | Cat #20020599 |
| iScript cDNA synthesis kit | Bio-Rad | Cat #170–8890 |
| Superscript IV Reverse Transcriptase | Invitrogen | Cat #18090050 |
| FastStart Universal SYBR Green Master (Rox) Mix | Roche | Cat #04913850001 |
| CellEvent Caspase 3/7 Green Flow Cytometry Assay Kit | Fisher | Cat #C10427 |
| ATPlite Luminescence Assay System | PerkinElmer | Cat #6016949 |
| Dual-Glo Luciferase Assay System | Promega | Cat #E2920 |
| Bright-Glo Luciferase Assay System | Promega | Cat #E264B |
| Nano-Glo Luciferase Assay System | Promega | Cat #N1120 |
| ImmPRESS HRP Horse Anti-Rabbit IgG Polymer Detection Kit | Vector Labs | MP-7401 |
| Deposited data | ||
| Analyzed RNA-seq data | This paper | Available upon request |
| Experimental models: cell lines | ||
| A253 | Gift from Stuart Aaronsen and Bernard Weissman (UNC-CH, Chapel Hill, NC) | RRID: CVCL_1060 |
| A388 | Gift from Stuart Aaronsen and Bernard Weissman (UNC-CH, Chapel Hill, NC) | RRID:CVCL_1063 |
| A431 | Gift from Stuart Aaronsen and Bernard Weissman (UNC-CH, Chapel Hill, NC) | RRID: CVCL_0037 |
| NCI-H292 | Gift from Dr. Frederic Kaye (University of Florida, Gainesville, FL) | RRID: CVCL_0455 |
| NCI-H3118 | Gift from Dr. Frederic Kaye (University of Florida, Gainesville, FL) | RRID: CVCL_A464 |
| UM-HMC-1 | Gift from Dr. Jacques Nör (University of Michigan, Ann Arbor, Ml) | RRID: CVCL_Y473 |
| UM-HMC-3A | Gift from Dr. Jacques Nör (University of Michigan, Ann Arbor, Ml) | RRID: CVCL_Y471 |
| UM-HMC-3B | Gift from Dr. Jacques Nör (University of Michigan, Ann Arbor, Ml) | RRID: CVCL_Y472 |
| HEK293A | ThermoFisher Scientific | Cat #R70507 |
| HEK293-CMVTetRTetOC1/M2 | This paper | N/A |
| HEK293-PGKTetOn3GTRE3GSPGC−1α4 | This paper | N/A |
| HMC3A-PGKTetOn3G TRE3GSdCas9-KRAB-IRES-BFP | This paper | N/A |
| Lenti X-293T | Takara | Cat #632180 |
| Experimental models: organisms/strains | ||
| Mouse: Nu/Nu | Jackson Laboratory | Stock# 002019 |
| Oligonucleotides | ||
| MAML2-specific reverse primer, see Table S6 | This paper | N/A |
| Primers for mycoplasma detection, see Table S6 | Young et al., 2010 | N/A |
| Oligo d(T)20 primer | Invitrogen | Cat #100023441 |
| Primers for qPCR gene expression analysis, see Table S6 | This paper | N/A |
| Primers for chromatin IP, see Table S6 | This paper | N/A |
| Recombinant DNA | ||
| pRetroX-Tight-MCS_PGK-GpNLuc | Schaub et al., 2015 | Addgene plasmid #70185 |
| PGC-1α_Distal_Luciferase | This paper | Addgene plasmid #154260 |
| PGC-1α_Proximal_Luciferase | This paper | Addgene plasmid #154259 |
| pGL4.15 [Luc2p/Hygro] IGF-1 Promoter 1 | This paper | Addgene plasmid #154261 |
| pGL3-Enhancer_IGF-1 Promoter 1 | This paper | Addgene plasmid #154266 |
| pGL4.15 [Luc2p/Hygro] IGF-1 Promoter 2 | This paper | Addgene plasmid #154262 |
| pGL3-Enhancer_IGF-1 Promoter 2 | This paper | Addgene plasmid #154267 |
| pRetroX_Tight_ACREB_PGK-tdTomato | This paper | Addgene plasmid #154264 |
| pTRE3G_FLAG-CRTC1/MAML2_PGK-GpNLuc | This paper | Addgene plasmid #154265 |
| pLV-Tight-PGCv4-mCherry:T2A:Hygro | This paper | Addgene plasmid #154263 |
| pFLAG-CMV-2_CRTC1/MAML2 | Tonon et al., 2003 | N/A |
| pLKO.1_shScramble | Chen et al., 2014 | N/A |
| pLKO.1_shMAML2_1 | Chen etal., 2014 | N/A |
| pLKO.1_shMAML2_3 | Chen etal., 2014 | N/A |
| pLMN UltramiR-E shlGF1R_#1 | transOMICS Technologies | Cat #ULTRA-3308043 |
| pLMN UltramiR-E shlGF1R_#2 | transOMICS Technologies | Cat #ULTRA-3443222 |
| pLMN UltramiR-E shPPARGC1A_#1 | transOMICS Technologies | Cat #ULTRA-3197172 |
| pLMN UltramiR-E shPPARGC1A_#2 | transOMICS Technologies | Cat #ULTRA-3197171 |
| MISSION® shRNA shPPARG_#1 | Sigma-Aldrioh | Cat #SHCLNG-TRCN0000355926 |
| MISSION® shRNA shPPARG_#2 | Sigma-Aldrich | Cat #SHCLNG-TRCN0000001673 |
| MISSION® shRNA shPPARG_#3 | Sigma-Aldrich | Cat #SHCLNG-TRCN0000001674 |
| PPREx3-TK-Luc | Kim et al., 1998 | Addgene plasmid #1015 |
| dCas9-KRAB-IRES-BFP | Fulco et al., 2016 | Addgene plasmid #85449 |
| pgRNA-CKB | Mandegar et al., 2016 | Addgene plasmid #73501 |
| pSico_U6-Pan-PGC1a sgPGC | This paper | Addgene plasmid #165426 |
| pSico_U6-PGC1a1 sgPGC | This paper | Addgene plasmid #165425 |
| Software and algorithms | ||
| STAR (version 2.4.2) | Dobin et al., 2013 | N/A |
| Salmon (version 0.1.19) | Patro et al., 2017. | https://github.com/COMBINE-lab/Salmon |
| DESeq2 (version 1.24.0) | Love et al., 2014 | N/A |
| Gene Set Enrichment Analysis | Mootha et al., 2003; Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| ComplexHeatmap (version 2.0.0) | Gu et al., 2016 | http://www.bioconductor.org/packages/devel/bioc/html/ComplexHeatmap.html |
| GraphPad Prism 6 | N/A | https://www.graphpad.com |
| Gen5 (version 2.09) | BioTek | https://www.biotek.com/products/software-robotics-software/gen5-microplate-reader-and-imager-software/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| R (version 3.1.2) | N/A | https://www.r-project.org/ |
| Other | ||
| Protein G SureBeads | Bio-Rad | Cat #1614821 |
Highlights.
CRTC1-MAML2-fusion-positive salivary mucoepidermoid carcinomas (MECs) express IGF-1
CRTC1-MAML2 coordinates IGF-1 expression via aberrant PGC-1α4 splice variant expression
PGC-1α4 regulates IGF-1 in a PPARγ-dependent manner in CRTC1-MAML2-positive MECs
IGF-1R and PPARγ inhibitors reduce growth and survival of CRTC1-MAML2-positive MECs
ACKNOWLEDGMENTS
The authors thank Gary Johnson and Brian Golitz for technical support in performing the small-molecule drug screen. We also thank Al Baldwin, Gaorav Gupta, Wendell Yarbrough, and members of the Amelio lab for helpful discussions, suggestions, and/or scientific review of this manuscript and Naim Rashid in the UNC Lineberger Biostatistics Core for statistical consultation. The graphical abstract was created with BioRender. The UNC Lineberger Animal Studies, Animal Histopathology, Small Animal Imaging, Flow Cytometry, Office of Genomics Research, Translational Genomics Lab, Translational Pathology Lab, High Throughput Sequencing Facility, Biostatistics, and Bioinformatics Cores are supported in part by the NCI Center Core Support Grant P30-CA016086 to the UNC Lineberger Comprehensive Cancer Center. The UNC Flow Cytometry Core Facility is also supported by the North Carolina Biotech (NCBT) Center Institutional Support Grant 2012-IDG-1006. The Translational Pathology Lab is also supported by NIH U54-CA156733, NIEHS P30-EOS010126, and NCBT 2015-IDG-1007. This work was supported in part by NIH/NIGMS T32-GM007092 and NIH NIDCR F31-DE027282 training grants (to A.M.M.), NIH/NIGMS R01-GM130866 and the American Heart Association 19CDA34660248 (to J.G.), Head and Neck Cancer Fund (to T.G.H. and D.N.H.), UNC Translational Team Science Award (TTSA; to T.G.H. and A.L.A.), University Cancer Research Fund (UCRF; to A.L.A.), and NIH/NCI Howard Temin Pathway to Independence Award in Cancer Research R00-CA157954 (to A.L.A.).
Footnotes
DECLARATION OF INTERESTS
A.L.A. is a GAB member and paid consultant for LG Chem Life Sciences Innovation Center.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108768.
REFERENCES
- Adams A, Warner K, Pearson AT, Zhang Z, Kim HS, Mochizuki D, Basura G, Helman J, Mantesso A, Castilho RM, et al. (2015). ALDH/CD44 identifies uniquely tumorigenic cancer stem cells in salivary gland mucoepidermoid carcinomas. Oncotarget 6, 26633–26650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S, Olive M, Aggarwal S, Krylov D, Ginty DD, and Vinson C (1998). A dominant-negative inhibitor of CREB reveals that it is a general mediator of stimulus-dependent transcription of c-fos. Mol. Cell. Biol 18, 967–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano O, and Iseki S (1993). Expression, localization and developmental regulation of insulin-like growth factor I mRNA in rat submandibular gland. Arch. Oral Biol 38, 671–677. [DOI] [PubMed] [Google Scholar]
- Amelio AL, Caputi M, and Conkright MD (2009). Bipartite functions of the CREB co-activators selectively direct alternative splicing or transcriptional activation. EMBO J. 28, 2733–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio AL, Fallahi M, Schaub FX, Zhang M, Lawani MB, Alperstein AS, Southern MR, Young BM, Wu L, Zajac-Kaye M, et al. (2014). CRTC1/MAML2 gain-of-function interactions with MYC create a gene signature predictive of cancers with CREB-MYC involvement. Proc. Natl. Acad. Sci. USA 111, E3260–E3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batsakis JG (1980). Salivary gland neoplasia: an outcome of modified morphogenesis and cytodifferentiation. Oral Surg. Oral Med. Oral Pathol 49, 229–232. [DOI] [PubMed] [Google Scholar]
- Behboudi A, Enlund F, Winnes M, Andrén Y, Nordkvist A, Leivo I, Flaberg E, Szekely L, Mäkitie A, Grenman R, et al. (2006). Molecular classification of mucoepidermoid carcinomas-prognostic significance of the MECT1-MAML2 fusion oncogene. Genes Chromosomes Cancer 45, 470–481. [DOI] [PubMed] [Google Scholar]
- Bell D, and Hanna EY (2012). Salivary gland cancers: biology and molecular targets for therapy. Curr. Oncol. Rep 14, 166–174. [DOI] [PubMed] [Google Scholar]
- Bevill SM, Olivares-Quintero JF, Sciaky N, Golitz BT, Singh D, Beltran AS, Rashid NU, Stuhlmiller TJ, Hale A, Moorman NJ, et al. (2019). GSK2801, a BAZ2/BRD9 Bromodomain Inhibitor, Synergizes with BET Inhibitors to Induce Apoptosis in Triple-Negative Breast Cancer. Mol. Cancer Res 17, 1503–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkeland AC, Foltin SK, Michmerhuizen NL, Hoesli RC, Rosko AJ, Byrd S, Yanik M, Nör JE, Bradford CR, Prince ME, et al. (2017). Correlation of Crtc1/3-Maml2 fusion status, grade and survival in mucoepidermoid carcinoma. Oral Oncol. 68, 5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop TR, Zhang Y, and Erb MA (2019). Pharmacological Modulation of Transcriptional Coregulators in Cancer. Trends Pharmacol. Sci 40, 388–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno NE, Kelly KA, Hawkins R, Bramah-Lawani M, Amelio AL, Nwachukwu JC, Nettles KW, and Conkright MD (2014). Creb coactivators direct anabolic responses and enhance performance of skeletal muscle. EMBO J. 33, 1027–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, and Koehler AN (2020). Transcription Factor Inhibition: Lessons Learned and Emerging Targets. Trends Mol. Med 26, 508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HX, and Sharon E (2013). IGF-1R as an anti-cancer target—trials and tribulations. Chin. J. Cancer 32, 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen AM, Granchi PJ, Garcia J, Bucci MK, Fu KK, and Eisele DW (2007). Local-regional recurrence after surgery without postoperative irradiation for carcinomas of the major salivary glands: implications for adjuvant therapy. Int. J. Radiat. Oncol. Biol. Phys 67, 982–987. [DOI] [PubMed] [Google Scholar]
- Chen AM, Lau VH, Farwell DG, Luu Q, and Donald PJ (2013). Mucoepidermoid carcinoma of the parotid gland treated by surgery and postoperative radiation therapy: Clinicopathologic correlates of outcome. Laryngoscope 123, 3049–3055. [DOI] [PubMed] [Google Scholar]
- Chen Z, Chen J, Gu Y, Hu C, Li J-L, Lin S, Shen H, Cao C, Gao R, Li J, et al. (2014). Aberrantly activated AREG-EGFR signaling is required for the growth and survival of CRTC1-MAML2 fusion-positive mucoepidermoid carcinoma cells. Oncogene 33, 3869–3877. [DOI] [PubMed] [Google Scholar]
- Chen J, Li J-L, Chen Z, Griffin JD, and Wu L (2015). Gene expression profiling analysis of CRTC1-MAML2 fusion oncogene-induced transcriptional program in human mucoepidermoid carcinoma cells. BMC Cancer 15, 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coca-Pelaz A, Rodrigo JP, Triantafyllou A, Hunt JL, Rinaldo A, Strojan P, Haigentz M Jr., Mendenhall WM, Takes RP, Vander Poorten V, et al. (2015). Salivary mucoepidermoid carcinoma revisited. Eur. Arch. Otorhinolaryngol 272, 799–819. [DOI] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzmán E, Miraglia L, Hogenesch JB, and Montminy M (2003). TORCs: transducers of regulated CREB activity. Mol. Cell 12, 413–423. [DOI] [PubMed] [Google Scholar]
- Coxon A, Rozenblum E, Park Y-S, Joshi N, Tsurutani J, Dennis PA, Kirsch IR, and Kaye FJ (2005). Mect1-Maml2 fusion oncogene linked to the aberrant activation of cyclic AMP/CREB regulated genes. Cancer Res. 65, 7137–7144. [DOI] [PubMed] [Google Scholar]
- Curtarelli RB, Gonçalves JM, Dos Santos LGP, Savi MG, Nör JE, Mezzomo LAM, and Rodríguez Cordeiro MM (2018). Expression of Cancer Stem Cell Biomarkers in Human Head and Neck Carcinomas: a Systematic Review. Stem Cell Rev. Rep 14, 769–784. [DOI] [PubMed] [Google Scholar]
- Denduluri SK, Idowu O, Wang Z, Liao Z, Yan Z, Mohammed MK, Ye J, Wei Q, Wang J, Zhao L, and Luu HH (2015). Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Naggar AK, Lovell M, Killary AM, Clayman GL, and Batsakis JG (1996). A mucoepidermoid carcinoma of minor salivary gland with t(11;19)(q21;p13.1) as the only karyotypic abnormality. Cancer Genet. Cytogenet 87, 29–33. [DOI] [PubMed] [Google Scholar]
- El-Naggar AK, Chan JKC, Grandis JR, and Slootweg PJ (2017). WHO Classification of Head and Neck Tumours (IARC).
- Fernandez-Cuesta L, Sun R, Menon R, George J, Lorenz S, Meza-Zepeda LA, Peifer M, Plenker D, Heuckmann JM, Leenders F, et al. (2015). Identification of novel fusion genes in lung cancer using breakpoint assembly of transcriptome sequencing data. Genome Biol. 16, 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, Lichtenberg TM, Murray BA, Ghayee HK, Else T, et al. ; Cancer Genome Atlas Research Network (2017). Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 31, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini V, Pagnotta SM, Tala, Fan JJ, Russo MV, Lee SB, Garofano L, Zhang J, Shi P, Lewis G, et al. (2018). A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature 553, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, and Engreitz JM (2016). Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Q, Liang W-W, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, Liao W-W, Reynolds SM, Wyczalkowski MA, Yao L, et al. ; Fusion Analysis Working Group; Cancer Genome Atlas Research Network (2018). Driver Fusions and Their Implications in the Development and Treatment of Human Cancers. Cell Rep. 23, 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. (2013). CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JT, Berger AC, Shih J, Duke FF, Furst L, Kwiatkowski DJ, Cherniack AD, Meyerson M, and Strathdee CA (2017). Genomic activation of PPARG reveals a candidate therapeutic axis in bladder cancer. Cancer Res. 77, 6987–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Gualberto A, and Pollak M (2009). Emerging role of insulin-like growth factor receptor inhibitors in oncology: early clinical trial results and future directions. Oncogene 28, 3009–3021. [DOI] [PubMed] [Google Scholar]
- Jang YE, Jang I, Kim S, Cho S, Kim D, Kim K, Kim J, Hwang J, Kim S, Kim J, et al. (2020). ChimerDB 4.0: an updated and expanded database of fusion genes. Nucleic Acids Res. 48 (D1), D817–D824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, and Crabtree GR (2013). Reversible disruption of mSWI/SNF (BAF) complexes by the SS18-SSX oncogenic fusion in synovial sarcoma. Cell 153, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Tan M, Bishop JA, Jones S, Sausen M, Ha PK, and Agrawal N (2017). Whole-Exome Sequencing of Salivary Gland Mucoepidermoid Carcinoma. Clin. Cancer Res 23, 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardos J, Chai S, Mose LE, Selitsky SR, Krishnan B, Saito R, Iglesia MD, Milowsky MI, Parker JS, Kim WY, et al. (2016). Claudin-low bladder tumors are immune infiltrated and actively immune suppressed. JCI Insight 1, e85902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr M, Lee A, Wang PL, Purushotham KR, Chegini N, Yamamoto H, and Humphreys-Beher MG (1995). Detection of insulin and insulin-like growth factors I and II in saliva and potential synthesis in the salivary glands of mice. Effects of type 1 diabetes mellitus. Biochem. Pharmacol 49, 1521–1531. [DOI] [PubMed] [Google Scholar]
- Keysar SB, Le PN, Miller B, Jackson BC, Eagles JR, Nieto C, Kim J, Tang B, Glogowska MJ, Morton JJ, et al. (2016). Regulation of Head and Neck Squamous Cancer Stem Cells by PI3K and SOX2. J. Natl. Cancer Inst 109, djw189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandekar MJ, Banks AS, Laznik-Bogoslavski D, White JP, Choi JH, Kazak L, Lo JC, Cohen P, Wong K-K, Kamenecka TM, et al. (2018). Noncanonical agonist PPARγ ligands modulate the response to DNA damage and sensitize cancer cells to cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 115, 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JB, Wright HM, Wright M, and Spiegelman BM (1998). ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc. Natl. Acad. Sci. USA 95, 4333–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimes PK, Cabanski CR, Wilkerson MD, Zhao N, Johnson AR, Perou CM, Makowski L, Maher CA, Liu Y, Marron JS, and Hayes DN (2014). SigFuge: single gene clustering of RNA-seq reveals differential isoform usage among cancer samples. Nucleic Acids Res. 42, e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar-Sinha C, Kalyana-Sundaram S, and Chinnaiyan AM (2015). Landscape of gene fusions in epithelial cancers: seq and ye shall find. Genome Med. 7, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latysheva NS, Oates ME, Maddox L, Flock T, Gough J, Buljan M, Weatheritt RJ, and Babu MM (2016). Molecular Principles of Gene Fusion Mediated Rewiring of Protein Interaction Networks in Cancer. Mol. Cell 63, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, and Young RA (2013). Transcriptional regulation and its misregulation in disease. Cell 152, 1237–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kovach A, Suino-Powell K, Martynowski D, and Xu HE (2008). Structural and biochemical basis for the binding selectivity of peroxisome proliferator-activated receptor gamma to PGC-1alpha. J. Biol. Chem 283, 19132–19139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipner MB, Peng XL, Jin C, Xu Y, Gao Y, East MP, Rashid NU, Moffitt RA, Herrera Loeza SG, Morrison AB, et al. (2020). Irreversible JNK1-JUN inhibition by JNK-IN-8 sensitizes pancreatic cancer to 5-FU/FOLFOX chemotherapy. JCI Insight 5, vi124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Lim J-H, Lee Y, Granter SR, Thomas A, Vazquez F, Widlund HR, and Puigserver P (2016). A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature 537, 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Balsa E, Perry EA, Liang J, Tavares CD, Vazquez F, Widlund HR, and Puigserver P (2020). H3K27me3-mediated PGC1α gene silencing promotes melanoma invasion through WNT5A and YAP. J. Clin. Invest 130, 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandegar MA, Huebsch N, Frolov EB, Shin E, Truong A, Olvera MP, Chan AH, Miyaoka Y, Holmes K, Spencer CI, et al. (2016). CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 18, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano DP, Kuruvilla DS, Boregowda SV, Asteian A, Hughes TS, Garcia-Ordonez R, Corzo CA, Khan TM, Novick SJ, Park H, et al. (2015). Pharmacological repression of PPARγ promotes osteogenesis. Nat. Commun 6, 7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Redondo V, Pettersson AT, and Ruas JL (2015). The hitchhiker’s guide to PGC-1α isoform structure and biological functions. Diabetologia 58, 1969–1977. [DOI] [PubMed] [Google Scholar]
- Martínez-Redondo V, Jannig PR, Correia JC, Ferreira DMS, Cervenka I, Lindvall JM, Sinha I, Izadi M, Pettersson-Klein AT, Agudelo LZ, et al. (2016). Peroxisome Proliferator-activated Receptor γ Coactivator-1 α Isoforms Selectively Regulate Multiple Splicing Events on Target Genes. J. Biol. Chem 291, 15169–15184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastropasqua F, Girolimetti G, and Shoshan M (2018). PGC1α: Friend or Foe in Cancer? Genes (Basel) 9, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh CH, Roberts DB, El-Naggar AK, Hanna EY, Garden AS, Kies MS, Weber RS, and Kupferman ME (2012). Prognostic factors in mucoepidermoid carcinoma of the salivary glands. Cancer 118, 3928–3936. [DOI] [PubMed] [Google Scholar]
- Missiaglia E, Williamson D, Chisholm J, Wirapati P, Pierron G, Petel F, Concordet J-P, Thway K, Oberlin O, Pritchard-Jones K, et al. (2012). PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol 30, 1670–1677. [DOI] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, and Mertens F, eds. (2019). Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer. https://mitelmandatabase.isb-cgc.org/.
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Nance MA, Seethala RR, Wang Y, Chiosea SI, Myers EN, Johnson JT, and Lai SY (2008). Treatment and survival outcomes based on histologic grading in patients with head and neck mucoepidermoid carcinoma. Cancer 113, 2082–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill ID (2009). t(11;19) translocation and CRTC1-MAML2 fusion oncogene in mucoepidermoid carcinoma. Oral Oncol. 45, 2–9. [DOI] [PubMed] [Google Scholar]
- Oltean S, and Bates DO (2014). Hallmarks of alternative splicing in cancer. Oncogene 33, 5311–5318. [DOI] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak M (2008). Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 8, 915–928. [DOI] [PubMed] [Google Scholar]
- Rabbitts TH (1994). Chromosomal translocations in human cancer. Nature 372, 143–149. [DOI] [PubMed] [Google Scholar]
- Rabbitts TH (2009). Commonality but diversity in cancer gene fusions. Cell 137, 391–395. [DOI] [PubMed] [Google Scholar]
- Riggi N, Knoechel B, Gillespie SM, Rheinbay E, Boulay G, Suvà ML, Rossetti NE, Boonseng WE, Oksuz O, Cook EB, et al. (2014). EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 26, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld MG, Lunyak VV, and Glass CK (2006). Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 20, 1405–1428. [DOI] [PubMed] [Google Scholar]
- Rowley JD (2001). Chromosome translocations: dangerous liaisons revisited. Nat. Rev. Cancer 1, 245–250. [DOI] [PubMed] [Google Scholar]
- Ruas JL, White JP, Rao RR, Kleiner S, Brannan KT, Harrison BC, Greene NP, Wu J, Estall JL, Irving BA, et al. (2012). A PGC-1α isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151, 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan J, Mantle T, McQuaid S, and Costigan DC (1992). Salivary insulin-like growth factor-I originates from local synthesis. J. Endocrinol 135, 85–90. [DOI] [PubMed] [Google Scholar]
- Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, Barneda D, Sellers K, Campos-Olivas R, Graña O, et al. (2015). MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 22, 590–605. [DOI] [PubMed] [Google Scholar]
- Schaub FX, Reza MS, Flaveny CA, Li W, Musicant AM, Hoxha S, Guo M, Cleveland JL, and Amelio AL (2015). Fluorophore-NanoLuc BRET Reporters Enable Sensitive In Vivo Optical Imaging and Flow Cytometry for Monitoring Tumorigenesis. Cancer Res. 75, 5023–5033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seethala RR, and Chiosea SI (2016). MAML2 Status in Mucoepidermoid Carcinoma Can No Longer Be Considered a Prognostic Marker. Am. J. Surg. Pathol 40, 1151–1153. [DOI] [PubMed] [Google Scholar]
- Seethala RR, Dacic S, Cieply K, Kelly LM, and Nikiforova MN (2010). A reappraisal of the MECT1/MAML2 translocation in salivary mucoepidermoid carcinomas. Am. J. Surg. Pathol 34, 1106–1121. [DOI] [PubMed] [Google Scholar]
- Semple RK, Chatterjee VKK, and O’Rahilly S (2006). PPAR gamma and human metabolic disease. J. Clin. Invest 116, 581–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang J, Mosure SA, Zheng J, Brust R, Bass J, Nichols A, Solt LA, Griffin PR, and Kojetin DJ (2020). A molecular switch regulating transcriptional repression and activation of PPARγ. Nat. Commun 11, 956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonoda J, Pei L, and Evans RM (2008). Nuclear receptors: Decoding metabolic disease. FEBS Lett. 582, 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart FW, Foote FW, and Becker WF (1945). Muco-Epidermoid Tumors of Salivary Glands. Ann. Surg 122, 820–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasoulas J, Rodón L, Kaye FJ, Montminy M, and Amelio AL (2019). Adaptive Transcriptional Responses by CRTC Coactivators in Cancer. Trends Cancer 5, 111–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonon G, Modi S, Wu L, Kubo A, Coxon AB, Komiya T, O’Neil K, Stover K, El-Naggar A, Griffin JD, et al. (2003). t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat. Genet 33, 208–213. [DOI] [PubMed] [Google Scholar]
- Torrano V, Valcarcel-Jimenez L, Cortazar AR, Liu X, Urosevic J, Castillo-Martin M, Fernández-Ruiz S, Morciano G, Caro-Maldonado A, Guiu M, et al. (2016). The metabolic co-regulator PGC1α suppresses prostate cancer metastasis. Nat. Cell Biol 18, 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuna M, Amos CI, and Mills GB (2019). Molecular mechanisms and pathobiology of oncogenic fusion transcripts in epithelial tumors. Oncotarget 10, 2095–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcarcel-Jimenez L, Macchia A, Crosas-Molist E, Schaub-Clerigué A, Camacho L, Martín-Martín N, Cicogna P, Viera-Bardón C, Fernández-Ruiz S, Rodriguez-Hernandez I, et al. (2019). PGC1α Suppresses Prostate Cancer Cell Invasion through ERRα Transcriptional Control. Cancer Res. 79, 6153–6165. [DOI] [PubMed] [Google Scholar]
- Vazquez F, Lim J-H, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, and Puigserver P (2013). PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell 23, 287–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann R (1895). Ueber endotheliale Geschwülste, zugleich ein Beitrag zu den Speicheldrüsen- und Gaumentumoren. Deutsche Zeitschrift für Chirurgie 41, 1–180. [Google Scholar]
- Wachtel M, and Schäfer BW (2018). PAX3-FOXO1: Zooming in on an “undruggable” target. Semin. Cancer Biol 50, 115–123. [DOI] [PubMed] [Google Scholar]
- Wallace M, and Metallo CM (2016). PGC1α drives a metabolic block on prostate cancer progression. Nat. Cell Biol 18, 589–590. [DOI] [PubMed] [Google Scholar]
- Wang B-D, and Lee NH (2018). Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers (Basel) 10, 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, McDermott JD, Schrock AB, Elvin JA, Gay L, Karam SD, Raben D, Somerset H, Ali SM, Ross JS, and Bowles DW (2017). Comprehensive genomic profiling of salivary mucoepidermoid carcinomas reveals frequent BAP1, PIK3CA, and other actionable genomic alterations. Ann. Oncol 28, 748–753. [DOI] [PubMed] [Google Scholar]
- Warner KA, Adams A, Bernardi L, Nor C, Finkel KA, Zhang Z, McLean SA, Helman J, Wolf GT, Divi V, et al. (2013). Characterization of tumorigenic cell lines from the recurrence and lymph node metastasis of a human salivary mucoepidermoid carcinoma. Oral Oncol. 49, 1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner H, and Katz J (2004). The emerging role of the insulin-like growth factors in oral biology. J. Dent. Res 83, 832–836. [DOI] [PubMed] [Google Scholar]
- Wu L, Liu J, Gao P, Nakamura M, Cao Y, Shen H, and Griffin JD (2005). Transforming activity of MECT1-MAML2 fusion oncoprotein is mediated by constitutive CREB activation. EMBO J. 24, 2391–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing F, Luan Y, Cai J, Wu S, Mai J, Gu J, Zhang H, Li K, Lin Y, Xiao X, et al. (2017). The Anti-Warburg Effect Elicited by the cAMP-PGC1α Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 18, 468–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L, Sung J, Stacey G, and Masters JR (2010). Detection of Mycoplasma in cell cultures. Nat. Protoc 5, 929–934. [DOI] [PubMed] [Google Scholar]
- Yu H, and Rohan T (2000). Role of the insulin-like growth factor family in cancer development and progression. J. Natl. Cancer Inst 92, 1472–1489. [DOI] [PubMed] [Google Scholar]
- Zhang J, and Manley JL (2013). Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 3, 1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, and Oldenburg KR (1999). A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen 4, 67–73. [DOI] [PubMed] [Google Scholar]
- Zou Y, Watters A, Cheng N, Perry CE, Xu K, Alicea GM, Parris JLD, Baraban E, Ray P, Nayak A, et al. (2019). Polyunsaturated Fatty Acids from Astrocytes Activate PPARγ Signaling in Cancer Cells to Promote Brain Metastasis. Cancer Discov. 9, 1720–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study have not been deposited in a public repository because the Institutional Review Board (IRB) protocols supporting this work are secondary analyses IRBs for use of surgical archives without consent, but the datasets are hosted by the UNC Lineberger Bioinformatics Core and are available from the corresponding author on request.