

Abstract
Background
Coxsackievirus B (CVB) is the most common cause of viral myocarditis. It targets cardiomyocytes through coxsackie and adenovirus receptor, which is highly expressed in the fetal heart. We hypothesized CVB3 can precipitate congenital heart defects when fetal infection occurs during critical window of gestation.
Methods and Results
We infected C57Bl/6 pregnant mice with CVB3 during time points in early gestation (embryonic day [E] 5, E7, E9, and E11). We used different viral titers to examine possible dose‐response relationship and assessed viral loads in various fetal organs. Provided viral exposure occurred between E7 and E9, we observed characteristic features of ventricular septal defect (33.6%), abnormal myocardial architecture resembling noncompaction (23.5%), and double‐outlet right ventricle (4.4%) among 209 viable fetuses examined. We observed a direct relationship between viral titers and severity of congenital heart defects, with apparent predominance among female fetuses. Infected dams remained healthy; we did not observe any maternal heart or placental injury suggestive of direct viral effects on developing heart as likely cause of congenital heart defects. We examined signaling pathways in CVB3‐exposed hearts using RNA sequencing, Kyoto Encyclopedia of Genes and Genomes enrichment analysis, and immunohistochemistry. Signaling proteins of the Hippo, tight junction, transforming growth factor‐β1, and extracellular matrix proteins were the most highly enriched in CVB3‐infected fetuses with ventricular septal defects. Moreover, cardiomyocyte proliferation was 50% lower in fetuses with ventricular septal defects compared with uninfected controls.
Conclusions
We conclude prenatal CVB3 infection induces congenital heart defects. Alterations in myocardial proliferate capacity and consequent changes in cardiac architecture and trabeculation appear to account for most of observed phenotypes.
Keywords: congenital heart defect, Coxsackie type B, infection, pregnancy, virus
Subject Categories: Pregnancy, Pediatrics, Cardiomyopathy
Nonstandard Abbreviations and Acronyms
- CAR
coxsackievirus and adenovirus receptor
- CVB
coxsackievirus B
- DORV
double‐outlet right ventricle
- E
embryonic day
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- TGF
transforming growth factor
- VP1
viral capsid protein
Clinical Perspective
What Is New?
Our study found that maternal coxsackievirus B (CVB) 3 infection during pregnancy caused various congenital heart defects, including ventricular septal defects, double‐outlet right ventricle, and ventricular noncompaction, as well as fetal demise in a murine model.
Infection during the critical E7 through E9 period of gestation resulted in the highest incidence of congenital heart defects in fetuses, with infection at E9 in particular effecting the greatest impact.
CVB3 suppresses cardiomyocyte proliferation through stimulation of the transforming growth factor‐β1 pathway.
What Are the Clinical Implications?
Although CVB3 is a well‐known cause of viral myocarditis, evidence about its role in the pathogenesis of congenital heart disease has been scarce; CVB3 infection during pregnancy represents a previously unknown cause behind congenital heart defects and may account for a significant number of cases without a clear causative agent.
Further studies assessing the role of maternal CVB3 infection in babies with congenital heart defects are needed to corroborate our findings in humans.
Epidemiological measures aimed at reducing and preventing CVB3 infection in women of reproductive age may reduce the incidence of congenital heart disease.
Congenital heart defects (CHDs) are the most common anomalies in newborns, affecting 1 in 100 newborns annually. 1 Although many lesions are now treatable with excellent outcomes, still many experience a shortened lifespan, whereas others endure lifelong residual cardiovascular problems, 2 many of whom continue to need invasive treatments and not an insignificant number eventually requiring heart transplantation. 3 Pedigree analysis of families with multiple affected individuals as well as other detailed genetic studies have provided insights into the genetic basis of several CHDs, and potential heterogeneous pathogenic causes 4 ; however, for a significant portion of CHDs, the cause remains largely elusive.
Since the Rubella epidemic of 1964 in United States, it has been recognized that viruses could play a role in cause of many birth defects, including CHDs. 5 , 6 Some of these studies suggest a strong association between viral infections and fetal congenital anomalies of the nervous and cardiovascular systems. 7 Overall, prenatal infections are presumed to account for 2% to 3% of all congenital anomalies, most often by infections with “TORCH” organisms: toxoplasmosis, rubella, cytomegalovirus, herpes, and “other.” 8 The latter category of “other” pathogens is continually expanding: for example, a high incidence of congenital defects has been reported in infants born coincident with seasonal influenza epidemics and Zika virus infection during early pregnancy. 9 , 10
Coxsackievirus B (CVB) infects 10 million US citizens annually, allowing for a potentially large exposure burden. Six serotypes (B1–B6) are recognized, with serotypes B2, B3, and B4 considered endemic in the United States, and serotypes B1 and B5 exhibiting epidemic patterns. 11 Coxsackievirus and adenovirus receptor (CAR; encoded by CXADR in humans) is believed to be the primary mediator of CVB infection, serving as a point for attachment and internalization for all CVB serotypes. 12 CXADR is expressed in the human placenta during the first trimester, 13 and transplacental transfer of CVB is well documented in humans, 14 suggesting the potential for exposure of the developing fetus to viral infections. Evidence of CVB infection in late gestation mouse placenta and fetus is seen within 2 to 3 days of maternal infection, with a short duration of fetal viremia (2–4 days). 15 That the developing heart can be a target for virus‐induced pathological features is also suggested from studies showing that following perinatal maternal infection with CVB, viral RNA is detected in the neonatal heart for up to 5 days 16 and is associated with damage in neonatal cardiomyocytes. 17 Studying serologic responses, Brown and Evans (1967) were the first to suggest a possible clinical association of early gestation maternal CVB infection with various congenital heart and brain defects, later posited by others as well. 18 , 19 , 20 , 21 No further studies, however, have provided direct evidence that CVB causes CHD or a plausible mechanism to account for the clinical observations.
We have hypothesized that infection by “cardiotropic” viruses (ie, viruses, such as CVB, that enter the cardiomyocyte via specific receptors in the fetal heart) can induce CHD by interfering with normal cardiomyocyte proliferation, provided infection occurs during a critical window (first trimester) of cardiogenesis. To test our hypothesis, we used a mouse model to examine fetal exposure to CVB in early gestation. We also examined the influence of sex in this mechanism because prior studies have suggested sex differences in susceptibility to infection by CVB, as well as in the incidence of certain CHDs. 22 , 23
Methods
The authors declare that all supporting data are available within the article (and its online supplementary files), and any additional information is available from the corresponding author on reasonable request.
Propagation of Coxsackievirus and Infectivity Assays
LLC‐MK2 derivative cells (American Type Culture Collection CCL‐7.1) were maintained in minimum essential medium (Corning‐Mediatech, 10‐009‐CV) containing 10% fetal bovine serum (Millipore, TMS‐013‐B) and 2 mmol/L (1×) GlutaMAX‐I (Gibco, 35050‐061) at 37°C in a 5% CO2 incubator. CVB3, Nancy (American Type Culture Collection, VR‐30) was propagated in monolayers of LLC‐MK2, derivative cells. When cytopathic effect reached 95% to 100%, virus stocks were prepared by subjecting cells and media to 3 freeze‐thaw cycles, clarifying the media by centrifugation (2800g, 10 minutes) and aliquoting the supernatant. Aliquots were quick frozen on dry ice and stored at −80°C. Virus concentration was determined using an end point dilution assay on LLC‐MK2, derivative cells and expressed as a 50% tissue culture infective dose per milliliter, as calculated by the Spearman and Kärber algorithm. 24 To determine the presence of infectious virus, fetal tissues were homogenized in serum‐free minimal essential media containing penicillin, streptomycin, and amphotericin B. The suspension was frozen on dry ice, quickly thawed at 37°C, and then clarified by centrifugation at 2000g for 10 minutes. A portion of the supernatant was used to inoculate LLC‐MK2 derivative cells seeded the day prior in 24‐well plates. Cultures were observed daily for the presence of cytopathic effect for at least 1 week. Live cells remaining on the plates following the observation period were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet.
Experimental Animals
C57Bl6/J mice were purchased from The Jackson Laboratory and allowed to acclimate for 1 week before any procedures. Animal protocols were approved by the Institutional Animal Care and Use Committee at Washington University School of Medicine (protocol No. 20170070). All procedures conformed to the guidelines from the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Matings were set up overnight, and the following morning, considered embryonic day (E) 1, the females were examined for the presence of a copulation plug. Weights were monitored throughout the gestation period, and weight gain was used as an indicator of the progression of pregnancy and maternal health after infection. Pregnant females were randomly assigned into either a control group (n=7) or groups inoculated by intraperitoneal injection with the virus (n=42) at the selected gestational day. We used a matrix with 4 time points of CVB3 infection (E5, E7, E9, and E11) and 3 viral inoculation doses (1 × 106, 2.5 × 106, and 5 × 106 50% tissue culture infective dose). Dams were observed daily following inoculation for signs of illness (ie, lethargy and rough coat). Fetoplacental units from all groups were collected by laparotomy under general anesthesia at the desired gestational stage. Dams were euthanized by exsanguination and cardiectomy under inhaled 5% isoflurane anesthesia. Placental and fetal tissues were either flash frozen, embedded in optimal cutting temperature compound (Tissue‐Tek, 4583), and then frozen or fixed in 4% paraformaldehyde and embedded in paraffin for immunohistochemical analysis. The fetuses were collected at E15 to E17, as normal cardiac formation would be complete and, hence, any abnormalities identified would not be residua of incomplete development.
Histological Analysis and Immunohistochemistry
Transverse serial 7‐µm sections of the mouse fetus and placenta were stained with hematoxylin and eosin to assess heart morphological features. Maternal heart sections were stained with Masson trichrome stain to detect cardiac injury. Sections for immunohistochemistry were deparaffinized and rehydrated, and then antigen retrieval was performed using the citrate‐based solution (H‐3300) and high temperature‐pressure protocol of Vector Laboratories (Burlingame, CA). Samples were blocked for 1 hour at room temperature with 10% fetal bovine serum and then incubated overnight at 4°C with primary antibodies PHH3 (Millipore 06‐570) to identify mitotic cells, MF20 (Developmental Studies Hybridoma Bank, MF20) to identify myocytes, and Mouse Anti‐Enterovirus Clone 5‐D8/1 (Dako M7064) to identify CVB VP1 (viral capsid protein) region. For immunofluorescence detection, sections were incubated with Alexa Fluor–conjugated secondary antibodies (Invitrogen, goat anti‐rabbit IgG No. A11011 and goat anti‐mouse IgG No. A11001) for 2 hours at room temperature. Slides were mounted with Vectashield mounting media with 4′,6‐diamidino‐2‐phenylindole (Vector Labs, H‐1200) and imaged using a Zeiss fluorescent microscope (Axiovert with Apotome 2 apparatus). Apoptosis in the experimental groups was assessed in a blinded manner by terminal deoxynucleotidyl transferase dUTP nick end labeling staining using the In Situ Cell Death Detection Kit, TMR red (Roche 12156792910), according to manufacturer’s instructions.
Placental Histological Features
Placental sections obtained from infected and uninfected groups were processed for histological analysis. Hematoxylin and eosin staining was performed on sections, as per standard protocol. Placental sections (n=3–4) of the decidua, junctional zone, and labyrinth zone from each group were examined in a blinded manner (C.J. and I.M.) for evidence of pathological features.
Fetal Heart Culture
Fetal hearts were collected at E11 from uninfected and CVB3 infected (E9) dams and cultured on Millicell EZ 8‐well slides (Millipore Sigma PEZGS0816) in high‐glucose DMEM (American Type Culture Collection 30‐2002) supplemented with 10% fetal bovine serum (Millipore TMS‐013‐B), 1% penicillin‐streptomycin (Gibco 15140‐122), and 1% amphotericin B (Corning 30‐003‐CF). Cells were grown for 3 days, to achieve an age comparable to E14 gestation, and fixed with paraformaldehyde for immunohistochemistry.
Mouse Fetal Sex Determination
Genomic DNA was extracted from frozen tissue using Tissue Direct polymerase chain reaction (PCR) kit (Lamda Biotech D300‐100), amplified using Taq Polymerase 2× PCR premix (Intact Genomics, 3249) and SX primers (SX_forward, 5ʹ‐GATGATTTGAGTGGAAATGTGAGGTA‐3ʹ; SX_reverse, 5ʹ‐CTTATGTTTATAGGCATGCACCATGTA‐3ʹ), 25 and the PCR products were analyzed on a 2% agarose gel.
Determination of Cell Proliferation
Cell proliferation in fetal hearts was determined by mitotic index, which was expressed as the percentage of pHH3 stained nuclei in MF20‐positive cardiomyocytes/total number of nuclei in MF20‐positive cardiomyocytes. Cell counting was calculated in 4 biological replicates using ImageJ image processing software (US National Institutes of Health). 26 Individual data points together with the average measurements are reported as the mean±SD.
RNA Extraction
RNA extraction was done using QIAamp Viral RNA mini kit (Qiagen 52904) for reverse transcription–PCR and Arcturus PicoPure RNA isolation kit (Applied Biosystems 12204‐01) for quantitative PCR and sequencing using manual guidelines. cDNA synthesis was done using SuperScript III First‐Stand (Invitrogen 18080‐051) and RT All‐in‐One master mix (Lamda Biotech G208‐100). For sequence analysis, RNA was extracted from fetal tissues embedded in optimal cutting temperature; tissues were sectioned at 10 µm onto polyethylene naphthalate membrane slides (Leica, 11505158). Fetal heart tissue was collected using a Leica LMD7000 Laser Microdissection System from CVB3‐infected fetuses, showing a ventricular septal defect (VSD) and uninfected control fetuses.
PCR Analysis
End point PCR was done using EV primers (forward, CGGCCCCTGAATGCGGCTAATCC; reverse, TTGTCACCATAAGCAGCCA). 27 PowerUp SYBR Green Mastermix (Applied Biosystems A25741) was used for quantitative PCR to quantify CVB (forward, CCCCGGACTGAGTATCAATA; reverse, GCAGTTAGGATTAGCCGCAT), 28 Transforming growth factor (TGF)‐β1 (forward, CAACAATTCCTGGCGTTACCTTGG; reverse, GAAAGCCCTGTATTCCGTCTCCTT), GAPDH (forward, GCATGGCCTTCCGTGTTC; reverse, GATGTCATCATACTTGGCAGGTTT), 29 BMP2 (bone morphogenetic protein 2) (forward, TGTGGGCCCTCATAAAGAAGCAGA; reverse, AGATCCCTGCTTCTCAAAGGCACT), 30 Mothers against decapentaplegic homolog 1 (Smad1; forward, CGCTCCACGGCACAGTTAAG; reverse, GCCAGTTGATTTGCGAACAGAA), Mothers against decapentaplegic homolog 5 (Smad5; forward, TGCAGCTTGACCGTCCTTACC; reverse, GCAGACCTACAGTGCAGCCATC), and Mothers against decapentaplegic homolog 9 (Smad9; forward, CGATCATTCCATGAAGCTGACAA; reverse, TGGGCAAGCCAAACCGATA). 31
RNA Sequencing
RNA was extracted from the heart tissue and submitted to the Genome Technology Access Center (Department of Genetics at Washington University in St. Louis) to perform RNA sequencing using next‐generation sequencing for transcriptome profiling. Total RNA integrity was determined using an Agilent Bioanalyzer. Library preparation was performed with 10 ng of total RNA with a Bioanalyzer RNA integrity number score >8.0. Double‐stranded cDNA was prepared using the SMARTer Ultra Low RNA kit for Illumina Sequencing (Takara‐Clontech), per manufacturer’s protocol. cDNA was fragmented using a Covaris E220 sonicator using peak incident power 18, duty factor 20%, and cycles per burst 50 for 120 seconds. cDNA was blunt ended, had an A base added to the 3’ ends, and then had Illumina sequencing adapters ligated to the ends. Ligated fragments were then amplified for 12 to 15 cycles using primers incorporating unique dual index tags. Fragments were sequenced on an Illumina HiSeq 3000 with single‐end 50 base‐pair reads. Basecalls and demultiplexing were performed with Illumina’s bcl2fastq software and a custom python demultiplexing program with a maximum of one mismatch in the indexing read. RNA‐sequencing reads were then aligned to the Ensembl release 76 top‐level assembly with STAR version 2.0.4b. 32 Gene counts were derived from the number of uniquely aligned unambiguous reads by Subread:featureCount version 1.4.5. 33 Isoform expression of known Ensembl transcripts was estimated with Sailfish version 0.6.13. 34 Sequencing performance was assessed for the total number of aligned reads, total number of uniquely aligned reads, and features detected. The ribosomal fraction, known junction saturation, and read distribution over known gene models were quantified with RSeQC version 2.3. 35
All gene counts were then imported into the R/Bioconductor package EdgeR, 36 and trimmed mean of M values normalization size factors were calculated to adjust for samples for differences in library size. Genes or transcripts not expressed in any sample were excluded from further analysis. The trimmed mean of M values size factors and the matrix of counts were then imported into the R/Bioconductor package Limma. 37 Weighted likelihoods based on the observed mean‐variance relationship were then derived for every gene with Voom. 38 The performance of all genes was assessed with plots of the residual SD of every gene to their average log count with a robustly fitted trend line of the residuals. Differential expression analysis was then performed to analyze for differences between conditions, and the results were filtered for only those genes with Benjamini‐Hochberg false‐discovery rate adjusted P≤0.05.
The Kyoto Encyclopedia of Genes and Genomes (KEGG) database was used to identify biological pathways linked to differentially expressed genes enriched in CVB3‐infected fetal hearts. For each contrast extracted with Limma, global perturbations in known Gene Ontology terms and KEGG pathways were detected using the R/Bioconductor package GAGE 39 to test for changes in expression of the reported log 2 fold changes reported by Limma in each term versus the background log 2 fold changes of all genes found outside the respective term. The R/Bioconductor packages heatmap3 40 and Pathview 41 were used to display heat maps or annotated KEGG graphs across groups of samples for each Gene Ontology term or KEGG pathway, respectively, with a Benjamini‐Hochberg false‐discovery rate adjusted P≤0.05.
Data have been deposited to Gene Expression Omnibus (accession No. GSE157522).
Statistical Analysis
Two‐sample t test was used to compare the differences between control and test groups when the outcomes are continuous, and χ2 test was used to compare the differences between control and test groups when the outcomes are categorical. P≤0.05 was considered statistically significant. For multiple biological replicates, SD was calculated. To quantify amount of virus in tissue samples, standard curve equation was used along with its coefficient of determination (R 2). Differences in observed phenotypes between sexes were compared using a logistic regression, with outcomes reported as odds ratio (95% CI). Statistical analysis was performed using SPSS 27.0 (IBM Corporation, Armonk, NY).
Results
CVB3 Passes From Infected Pregnant Dam Into the Fetuses
To demonstrate passage of CVB3 from the infected mother to the developing fetus, initially we performed infectivity assay and end point PCR on fetal lysates (Figure 1A and 1B). To confirm the presence of virus within the fetus (and to localize the viral particles), we made multiple attempts at RNA in situ hybridization without success. We, therefore, used quantitative PCR not only to confirm the presence of the virus but also to quantify the viral load within the fetus. Using a standard curve based on our CVB3 stock (Figure 1D), we measured CVB3 levels in placenta (Figure 1E) and various fetal organs (Figure 1F) at different time points (E11, E14, and E17) following inoculation at E9. These data show that high levels of virus persist for at least 6 days within the placenta. CVB3 quantification in different fetal organs was done with specific emphasis on those that express CAR. Because dissection of individual organs was not possible at E11, we elected to divide the fetus into head (brain), midtorso (heart), and lower torso (liver and intestine) segments for analysis at that gestational age. At E14, we examined heart, brain, midtorso (predominantly liver), and lower torso. At E17, we could dissect out all individual organs. We found the maximum amount of CVB3 in the fetal heart at E14, followed by fetal brain at E17 (Figure 1F).
Figure 1. Vertical transmission of coxsackievirus B (CVB) 3 from infected pregnant dams to the fetus.
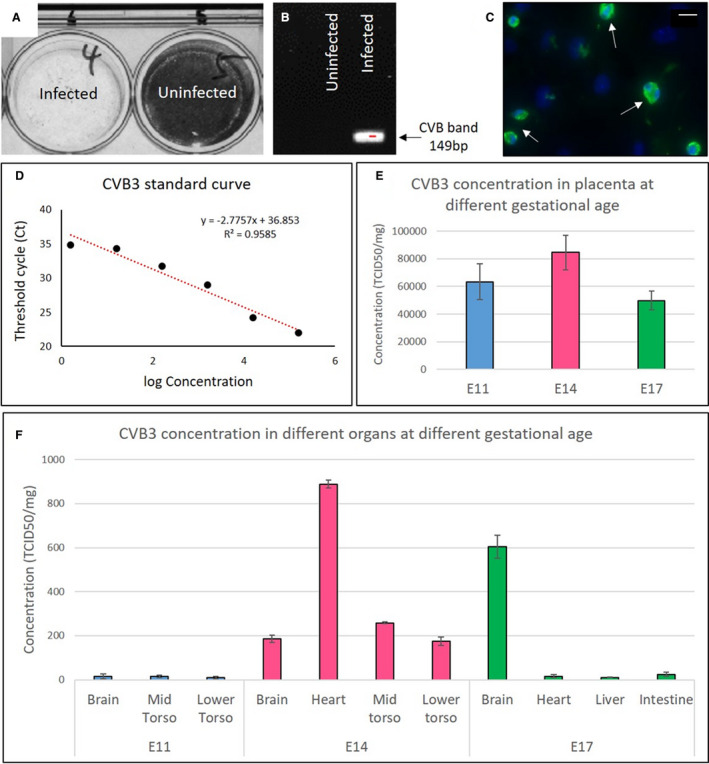
A, The presence of infectious virus in fetal lysates from infected and uninfected dams (embryonic day [E] 9) was analyzed using a cell culture infectivity assay; the lack of stained cells indicates the presence of virus. B, Reverse transcription–polymerase chain reaction (PCR) with enterovirus (EV)–specific primers, showing amplification of the 149‐bp (determined by agarose gel electrophoresis) virus band only in the tissue lysate of a fetus from an infected dam (at E9). C, Immunohistochemistry using anti‐EV antibody on cultured fetal heart (~E14) explanted at E11 from a dam infected at E9 (white arrows). Virus quantification by quantitative PCR. D, Standard curve based on the concentration of CVB3 stock. E, Virus present in placenta at E11, E14, and E17 after infection at E9 (n=6 for each gestational age). F, Virus present in fetal organs at E11, E14, and E17 after infection at E9 (n=6 for each gestational age). To quantify the amount of virus in tissue samples, the standard curve equation was used along with its coefficient of determination (R 2). Bar=20 µm. TCID50 indicates 50% tissue culture infective dose.
We also performed immunohistochemistry to detect VP1 on both fetal tissue sections and explanted hearts (at E11) that were cultured for 3 days (~E14). Although the anti‐VP1 antibody did not reliably detect the virus in tissue sections of the fetal hearts, with in vitro culture we could easily detect VP1, confirming the continued presence of the virus in the hearts of fetuses from CVB3 infected dams (Figure 1C).
Maternal CVB3 Infection Leads to Abnormal Heart Development in Fetal Mice
At E17, approximately one third of the fetuses from CVB3 infected dams were either dead or resorbed (37.2%; n=124/333; P=0.001) and could not be processed for assessment of heart morphological features (Figure 2A and 2F). Of the 209 infected fetuses assessed, 78 (37.3%; P=0.00002) had one or multiple CHDs: 66 (31.6%; P=0.0005) exhibited VSD (Figure 2C and 2F; Figure S1a‐c for more representative sections), 48 (23%; P=0.00002) exhibited alteration in myocardial architecture grossly resembling noncompaction of the ventricular myocardium (Figure 2D and 2F; Figure S1d‐f for more representative sections), and 9 (4.3%; P=0.2) exhibited double‐outlet right ventricle (DORV) phenotype (Figure 2E and 2F). Of all the VSDs seen, most were perimembranous in nature and only 6% were muscular VSDs (Figure S1a). The viral loads determined in the fetuses showed resorbed fetuses had the highest viral loads per milligram tissue, followed by fetuses with multiple CHDs, and the lowest load was seen in those with isolated VSD or noncompaction (Figure 2 table). Immune response was examined in fetal heart section using cluster of differentiation 45 antibody, which showed no difference between infected and control fetuses (data not shown). Part of resorbed fetus and lower torso of fully formed fetuses were used to assess viral load. No other major heart pathological feature was noted.
Figure 2. Coxsackievirus B (CVB) 3 infection of pregnant dams leads to fetal cardiac defects and death.
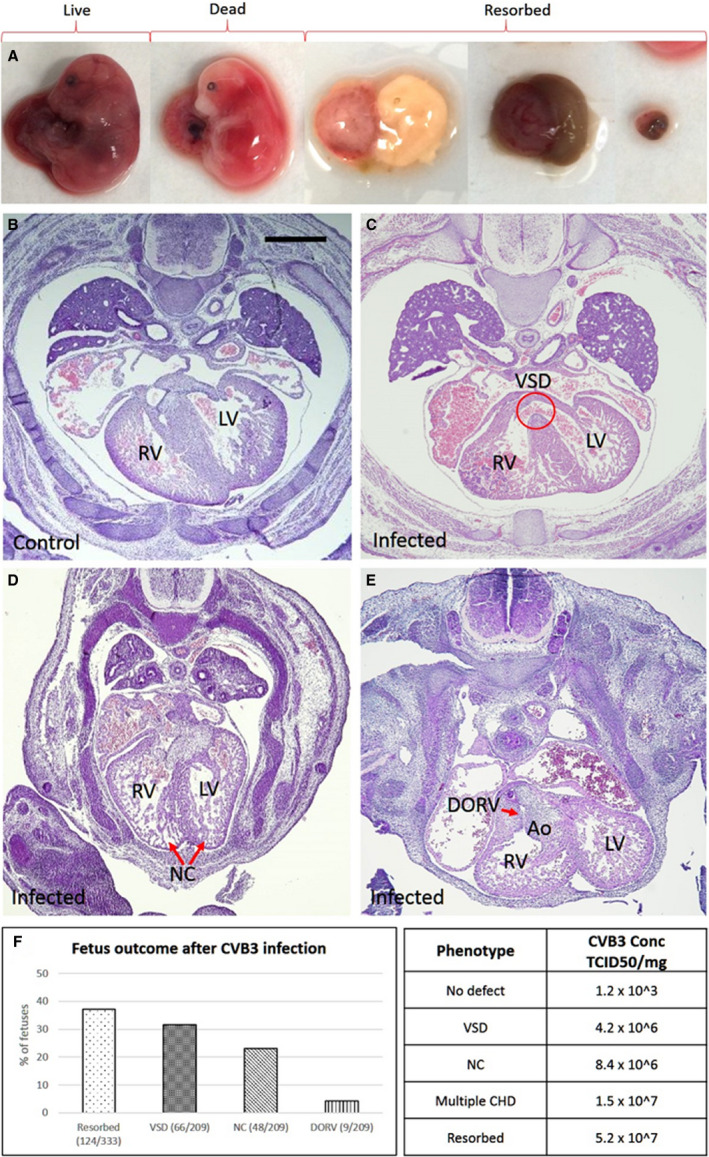
A, Representative images of embryonic day (E) 17 fetuses from CVB3‐infected dams, exhibiting specific phenotypes (left to right): viable, fully formed dead, and 3 images showing fetuses at various stages of resorption. B–E, Hematoxylin and eosin–stained heart sections of E17 fetuses from uninfected (B) and infected dams (C–E), and the various cardiac defects are indicated. F, Graph showing the percentage of fetuses from CVB3‐infected dams with the indicated cardiac defects. The χ2 test was used to analyze statistical significance between control and test groups by calculating P values. Table showing the amount of virus present in resorbed fetuses and fetuses with congenital heart defect (CHD), determined by quantitative polymerase chain reaction using CVB3 quantification standard curve (Figure 1d). Part of resorbed fetus and lower torso of fully formed fetuses were used to assess viral load. Bar=100 µm. Ao indicates aorta; DORV, double‐outflow RV; LV, left ventricle; NC, noncompaction; RV, right ventricle; TCID50, 50% tissue culture infective dose; and VSD, ventricular septum defect.
E7 to E9 Is the Critical Window for CVB3 Induction of CHDs in Fetal Mice
Because expression of CAR, the receptor for CVB, is essential between E10 and E11 for cardiomyocyte development, 42 we determined the time in gestation when CVB infection had the greatest impact on the incidences of the induced CHD phenotype, based on the timeline of mouse cardiac development (Figure 3A). The highest proportion of fetuses exhibiting VSDs occurred after infection at E9, regardless of the viral dose, with even low doses of CVB3 infection leading to abnormal heart development (Figure 3B; P=0.00001). The same pattern was observed for fetuses with noncompaction and DORV, with more incidences observed after infection at E9 for all CVB3 doses (Figure S2). Similarly, although fetal resorption occurred following CVB infection at each gestational stage, the most cases followed infection at E9 (Figure 3C; P=0.00001).
Figure 3. The effect of viral dose and gestational age at infection on the incidence of ventricular septal defect (VSD) and fetal demise.
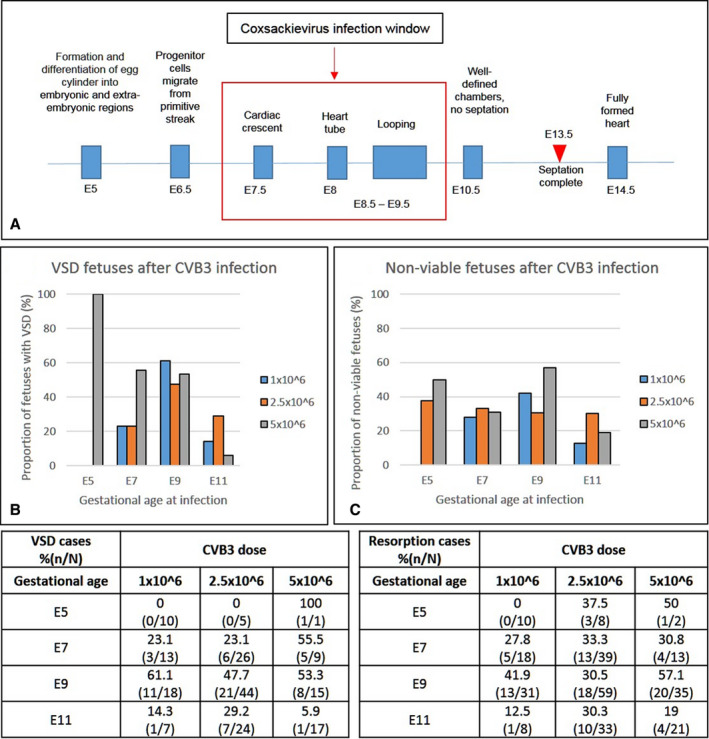
A, Schematic representation of the timeline of heart development in the mouse and critical window for coxsackievirus B (CVB) 3 infection. B and C, Dams were infected at various stages of gestation (embryonic day [E] 5, E7, E9, or E11) with different doses of virus (1.0, 2.5, and 5.0 × 106 50% tissue culture infective dose). Graphs and their corresponding tables show the percentage of fetuses with VSDs (B) and nonviable (dead or resorbed) fetuses (C) along with the total examined in each experimental group. The χ2 test was used to analyze statistical significance between control and test groups by calculating P values.
CHD Is Attributable to Direct Effect of CVB Infection on the Fetuses
To confirm the CHDs observed were a direct effect of CVB infection of the fetuses as opposed to poor health of the moms or placental pathological features, we evaluated maternal health and examined placental histological features. Infected dams did not show outward signs of illness, such as lethargy or rough coat following CVB3 infection. We compared maternal weight gain before and after infection between infected dams and uninfected control dams and found equivalent weight gain between the 2 groups (data not shown). Masson trichrome staining of maternal heart tissue from control and infected dams also showed no evidence of myocarditis or cardiac injury (Figure 4A through 4F). Placentas from control and E9 infected fetuses collected at E11 and E17 were analyzed by hematoxylin and eosin staining and examined blindly by placental biology experts (C.J. and I.M.). No placental damage, insufficiency, or vascular injury was identified (Figure 4G through 4L). These results suggest the observed CHDs are likely not simply a result of poor maternal health or placental insufficiency.
Figure 4. Cardiac defects in fetuses are not secondary to poor maternal health.
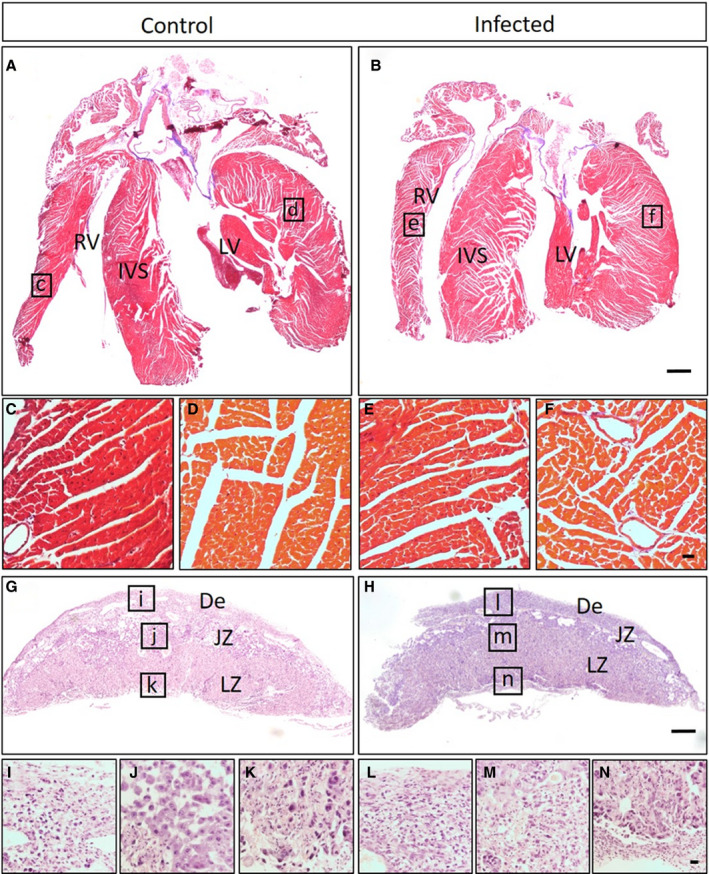
A–F, Masson trichrome–stained heart sections of uninfected control (n=5) (A) and infected dam (n=5) (B). Higher magnifications of control right ventricle (RV) (C), control left ventricle (LV) (D), infected RV (E), and infected LV (F) show no evidence of cardiac injury. G–N, Hematoxylin and eosin–stained placental sections of embryonic day 17 fetuses from uninfected (n=10) (G) and infected dams (n=10) (H). Higher magnifications of control (I, J, and K) and infected (L, M, and N) tissue show no evidence of placental damage in different parts of placenta. Bar=1000 µm. De indicates decidua; IVS, interventricular septum; JZ, junctional zone; and LZ, labyrinth zone.
CVB3 Stimulates the TGF‐β1 Signaling Pathway and Suppresses Cardiomyocyte Proliferation
We found the maximum amount of CVB3 at E14 (Figure 1F); therefore, we used this time point for KEGG enrichment analysis of differentially expressed transcripts in VSD‐exhibiting CVB3‐infected and uninfected fetal hearts. Transcriptome analysis detected 13,118 protein‐coding genes in the fetal heart, and differentially expressed genes were mapped to determine which were upregulated or downregulated because of CVB3 infection (Gene Expression Omnibus accession No. GSE157522) (Figure S3). The top 5 pathways most significantly enriched in CVB3‐infected hearts (log fold change >1.9; P<0.05) were those in the Hippo signaling, tight junction, TGF‐β1, cell cycle, and extracellular matrix receptor pathways (Figure 5A). Numbers of genes upregulated in these individual pathways were as follows: 4 of 118 genes in Hippo signaling, 3 of 119 genes in tight junction, 7 of 61 genes in TGF‐β1, and 10 of 61 genes in extracellular matrix receptor pathways.
Figure 5. The effect of coxsackievirus B (CVB) 3 infection on different signaling pathways and cell proliferation.
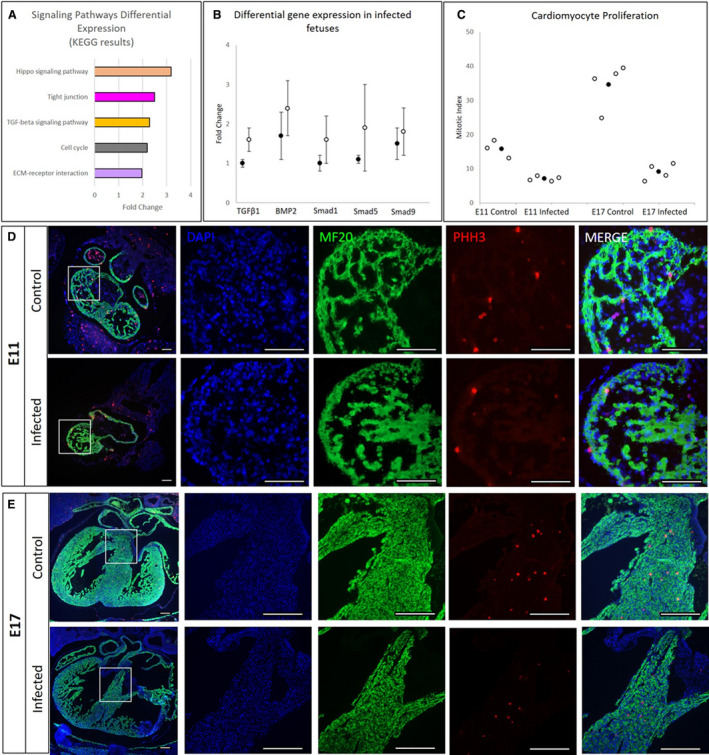
A, Top 5 differentially regulated Kyoto Encyclopedia of Genes and Genomes (KEGG) biological pathways in heart tissue from embryonic day (E) 14 fetuses infected with CVB3 at E9. B, Comparison of RNA‐sequencing (black dot, n=4) and quantitative reverse transcription–polymerase chain reaction (white dot, n=4) data for upregulated genes within the pathways (transforming growth factor [TGF]‐β1, BMP2 [bone morphogenetic protein 2], Smad1, Smad5, and Smad9), normalized to Gapdh expression in heart tissue from E14 fetuses infected with CVB3 at E9. C, pHH3‐stained cells (individual data points as white dots and mean as black dot±SD), indicating lower cell proliferation in the CVB3‐infected fetal hearts (E11 and E17) compared with uninfected controls (n=4 for each gestational age). D and E, Immunofluorescence images showing 4′,6‐diamidino‐2‐phenylindole (DAPI)–stained DNA (blue), MF20‐stained cardiomyocytes (green), and pHH3‐stained proliferating cells (red) in the E11 control and infected fetuses (D) and E17 control and infected fetuses (E) (magnified images of the white box in the panels on the left). The number of stained cells was determined using ImageJ software. The images are representative of 4 biological replicates. Bar=100 µm. ECM indicates extracellular matrix.
Because increased TGF‐β1 expression has been reported in several CVB3 infection‐related models, 43 and because the expression of TGF‐β1 transcript has been found in regions actively undergoing cardiac septation in murine embryos, 44 we examined whether the expression of other members of the TGF‐β superfamily, known to be associated with cardiogenesis, was altered. Quantitative PCR was used to validate differentially expressed genes, and the findings were consistent with data obtained by RNA sequencing. In particular, we found upregulation of genes and transcripts encoding TGF‐β1, BMP2, and downstream mediators SMAD1/5/9 in fetal hearts with VSDs (Figure 5B).
We assessed cell proliferation and apoptosis at E11, focusing on a developmental time point before septal completion (E13.5) (Figure 3A). We also examined E17 fetal heart sections to determine if the effects of CVB3 infection on proliferation were temporary or persistent. In heart sections stained with an antibody against phosphorylated histone H3, a marker for mitosis, cell proliferation in fetuses infected at E9 and collected at E11 and E17 was significantly lower than that in control fetuses with a mitotic index of 7.8% versus 15.8% at E11 and 9.1% versus 36.6% at E17 (P=0.0006 and P=0.0002, respectively) (Figure 5C through 5E). Cell apoptosis levels, assessed by terminal deoxynucleotidyl transferase dUTP nick end labeling staining, were similar in CVB3‐infected and control fetuses (data not shown).
Female Mouse Fetuses Appear More Susceptible to CVB3‐Induced CHDs Than Males
Because sex differences have been reported in the prevalence of congenital anomalies and specific cardiac defects, 45 we examined whether there were differences based on sex with CVB3‐induced CHDs. We found more female fetuses developing CHDs than males, although not reaching statistical significance (Figure 6). A 1.5‐fold higher (P=0.1) VSD incidence among females was observed compared with males. Females appeared also more susceptible to DORV and noncompaction after CVB3 infection than males, with a 3.4‐fold (P=0.1) and a 1.4‐fold (P=0.2) increase, respectively. Furthermore, 1.2‐fold (P=0.4) more female fetuses were resorbed compared with males following in utero CVB3 infection, suggestive of perhaps greater susceptibility of the female fetuses to infection.
Figure 6. Difference in incidence of observed defects following coxsackievirus B (CVB) 3 infection based on sex.
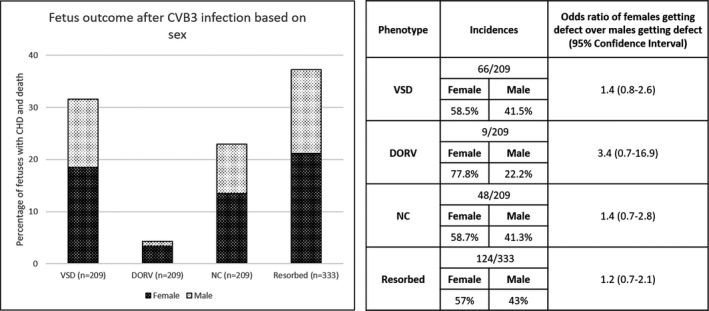
The percentage of fetuses from CVB3‐infected dams exhibiting the indicated abnormal phenotype. Differences in observed phenotypes between sexes were compared using a logistic regression, with outcomes reported as odds ratio (95% CI). CHD indicates congenital heart defect; DORV, double‐outflow right ventricle; NC, noncompaction; and VSD, ventricular septal defect.
Discussion
Viral infections in early pregnancy are recognized as important factors that can affect cardiovascular development ever since an association between intrauterine rubella infection and CHD was first reported in humans >45 years ago. 46 Similarly, suggestive, although inconclusive, evidence had been reported associating coxsackievirus infection during pregnancy and CHDs. 47 In the current study, we show conclusively prenatal CVB3 infection can induce CHDs, confirming prior suspicions based on epidemiologic studies (Figure 7I). Moreover, we find viral load to directly relate to both severity and incidence of fetal pathological features, showing highest viral loads in the resorbed fetuses, followed by fetuses with multiple CHDs, and lowest virus levels in fetuses with isolated CHDs. The major heart defect observed in our study, occurring in ~30% of fetal mice exposed to CVB in utero, was perimembranous VSD, one of the most prevalent CHDs in humans. Although not the focus of our study, it is of interest the second fetal organ where we identified abundant levels of CVB3 was the brain. Some studies have suggested the co‐occurrence of central nervous system pathological conditions with CHDs, although often these are attributed to secondary effects of altered in utero circulation from the primary CHD. Last, we identified a critical window during mouse development (between E7–E9) when prenatal CVB3 infection has the greatest impact on inducing heart defects (Figure 7I).
Figure 7. Mouse model of coxsackievirus B (CVB) 3 infection during pregnancy and proposed mechanism for ventricular septal defect (VSD) formation after infection.
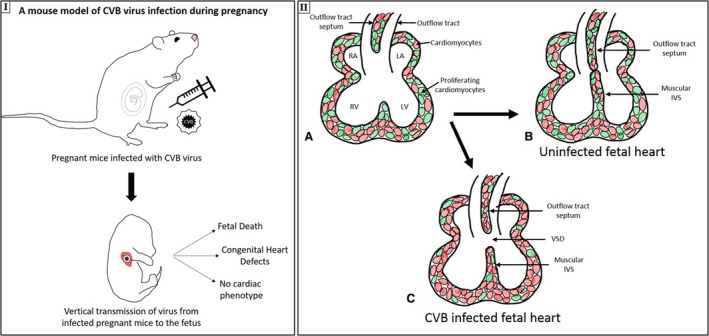
I, Mouse model of CVB virus infection during pregnancy, leading to vertical transmission of virus from infected pregnant mice to the fetus, resulting in 3 outcomes: fetal death, congenital heart defect, and no cardiac phenotype. IIa–c, The proposed mechanism of VSD generation. Normal intraventricular septum (IVS) formation (a), with the proliferation of the muscular IVS and the outflow tract septum, resulting in a fully formed septum (b), is shown (proliferating cardiomyocytes are shown in green). c, In CVB‐infected fetuses, cell proliferation is suppressed, which leads to incomplete septum formation and VSD. LA indicates left atrium; LV, left ventricle; RA, right atrium; and RV, right ventricle.
Prior reports using late gestation fetal mice suggested concentrations of CVB3 in fetal tissues peak at approximately 3 days following maternal infection. 48 We observed a similar pattern and therefore speculate maternal CVB3 infections between E7 and E9 in our experiments resulted in peak viral concentrations in the fetal heart around E10 to E12, when septation of the primitive cardiac tube is completing. Exposure of the fetal heart shortly before or during the period of septation could explain the temporal and quantitative relationship between CVB3 infection and the incidence of VSD that we observed in fetal mice. Because we did not observe any obvious placental injury and because the infected moms remained healthy throughout pregnancy, we suspect the heart defects resulted directly from infection of the fetus rather than secondary to placental insufficiency or poor maternal health. Although not every maternal heart was analyzed by Masson trichrome staining, there are reports stating that C57BL/6 mice are less susceptible to CVB3‐induced myocarditis compared with mice from other genetic backgrounds. 49 Our viral quantification studies suggest the placenta does perform a superb job at filtering much of the viral wave. Some viruses, however, clearly manage to get across the placenta and persist for some time in the fetus, with preferential proliferation in certain tissues (eg, heart and brain).
It is of interest we also observed abnormal trabeculations and myocardial architecture resembling noncompaction in CVB3‐infected fetal hearts. Noncompaction of the left ventricle, diagnosed in some children with CHD, 50 can be isolated or associated with other congenital cardiac malformations. Although the cause of noncompaction is presently unknown, noncompaction of the left ventricle is attributed to arise from the arrest of the normal myocardial maturation process during ontogenesis, with familial or genetic triggers. 51 Despite some controversy about diagnosis of noncompaction, most agree noncompaction is a genetically and phenotypically heterogeneous disease that can affect both ventricles, and has been associated with several genetic variants as well as mutations in cytoskeletal and sarcomeric proteins. 52 Although our data showing that ≈25% of CVB3‐infected fetal hearts develop abnormalities of trabeculation somewhat similar to noncompaction, perhaps attributable to CVB3‐mediated arrested myocardial development, additional studies are required to correlate our findings to those observed in the clinical setting as well as potential mechanisms involved.
We examined signaling pathways to obtain insights into the molecular mechanisms responsible for the observed phenotypes in our mice, focusing on differentially regulated genes as a result of CVB3 infection. Enrichment of genes encoding for components of the TGF‐β1 signaling pathway in fetuses with CVB3‐induced heart defects, like elevated expression of BMP2 and its downstream signaling transcription factors, Smad 1, 5, and 9, may be related to observed VSD as well as altered trabeculation patterns in our mice. Increase in TGF‐β1 expression is associated with cardiomyopathies in general but also with CVB3‐induced dilated cardiomyopathy. 43 , 53 A critical role for BMP signaling in cardiac development has been extensively studied; deletion of Bmp2 from the endocardial lineage results in perimembranous VSDs in mouse embryos. 54 Moreover, upregulation of BMP2 in the cardiac fields phosphorylates SMAD1/5/9, and enhanced SMAD signaling suppresses cell proliferation. 55 We found CVB3 suppressed cardiomyocyte proliferation in the fetal heart, suggesting it is one of the possible reasons behind observed phenotypes in our mice, 56 and the resultant septation (VSD) and alignment defects (DORV) (Figure 7II).
KEGG pathway enrichment analysis also showed that genes in the Hippo signaling pathways, known to regulate cardiovascular development and function, were enriched in CVB3‐infected fetal hearts compared with uninfected controls. The Hippo signaling pathway is conserved across species and has a critical role in regulating embryonic organ (including heart) size and cardiomyocyte cell proliferation and apoptosis. 57 Our observations of altered fetal cardiomyocyte proliferative capacity are therefore consistent with upregulation of Hippo in our model. We also found expression of tight junction signaling pathways to be enriched in fetal hearts exposed to CVB3. Of note, the receptor for CVB3, CAR, is highly expressed in the developing murine heart, and CAR knockouts result in embryonic lethality from cardiac defects if expression is repressed during a critical developmental window (E10 and E12). 42 This critical window coincides with the time at which fetal hearts in our model would be exposed to the highest levels of CVB3 following maternal infection at E7 to E9. Although our transcriptomic data showed equal expression of CAR and zonula occludens‐1 in CVB3‐infected and uninfected heart tissue (data not shown), we plan to pursue further analysis of these pathway components. With regard to all transcriptomic studies, it is critical that changes at the protein level may still occur without seeing a change in transcript levels. In totality, however, our RNA‐sequencing data do suggest CHD observed in our model is likely related to alterations in proliferation capacity of fetal cardiomyocytes from CVB3 infection.
Sex hormones are thought to affect the susceptibility or response to CVB3 infection as well as CVB3‐induced myocarditis in adult mice. In the preceding studies, males show more susceptibility to effects of infection than females. 58 Moreover, testosterone has been shown to alter cardiac remodeling during CVB3‐induced myocarditis. 59 We observed some semblance of sex‐influenced differences as well, albeit the reverse of preceding observations; however, despite the rather large number of fetuses examined, our observed ratios did not reach statistical significance. Clinicians and epidemiologists have reported unequal sex distribution of various cardiac defects in clinical cohorts, 23 , 35 although frequently with conflicting results: equal male/female ratios for VSD, but higher DORV incidence in males than females, 60 versus in results from a large Dutch study of 4110 adults with CHD, the incidence of VSD was higher among females (53%) than males. 61 Female fetuses are often overrepresented in stillbirth and recurrent miscarriage cases. 62 We are interested to further dissect the influence of sex in our model in future studies as likely these subtle differences reflect as of yet not understood but important mechanisms.
In conclusion, although birth malformations caused by maternal viral infections, such as congenital rubella syndrome and those by the Zika virus, have been well documented, 10 we present novel data that maternal infection with a common enterovirus (CVB) can also result in development of structural heart defects. We found differential regulation between the uninfected and CVB‐infected fetal hearts of signaling pathways associated with cardiac development. On the basis of these findings, we posit BMP/SMAD2 signaling‐led suppressed cardiomyocyte proliferation as one possible mechanism of CHD pathogenesis associated with CVB infection. We believe our findings could have simple but important implications from the perspective of possible public health measures to reduce the incidence of CHD if clinical observations corroborate our findings.
Sources of Funding
The Genome Technology Access Center is partially supported by National Cancer Institute Cancer Center Support Grant P30 CA91842 to the Siteman Cancer Center and by Institute of Clinical and Translational Science/Clinical and Translational Science Award (ICTS/CTSA) Grant UL1TR002345 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH. Work done by Dr Mysorekar was funded by NIH/Eunice Kennedy Shriver National Institute of Child Health and Human Development R01HD091218.
Disclosures
None.
Supporting information
Figures S1–S3
Acknowledgments
We thank Connor Mullen, Alma Muller, Brian Dailey, and Daniel Perry for data collection and histology support and Horacio Carvajal for clinical perspective and SPSS analysis. We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis.
Author contributions: Dr Eghtesady devised the project and the main conceptual ideas and supervised the project. Dr Sharma, L.S. Goessling, Dr Brar, and Dr Eghtesady conceived and planned the experiments. Dr Sharma and L.S. Goessling performed the experiments. Dr Sharma analyzed the data and wrote the manuscript (with input and feedback from Dr Eghtesady, Dr Brar, L.S. Goessling, and Dr Mysorekar). Drs Joshi and Mysorekar performed placental histopathological analysis.
(J Am Heart Assoc.2021;10:e017995. DOI: 10.1161/JAHA.120.017995.)
Supplementary material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.017995
For Sources of Funding and Disclosures, see page 14.
Preprint posted on BioRxiv, May 22, 2020. DOI: 10.1101/2020.05.19.104844.
References
- 1. Triedman JK, Newburger JW. Trends in congenital heart disease. Circulation. 2016;133:2716–2733. [DOI] [PubMed] [Google Scholar]
- 2. Spector LG, Menk JS, Knight JH, McCracken C, Thomas AS, Vinocur JM, Oster ME, St Louis JD, Moller JH, Kochilas L. Trends in long‐term mortality after congenital heart surgery. J Am Coll Cardiol. 2018;71:2434–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Triedman JK, Newburger JW. Trends in congenital heart disease: the next decade. Circulation. 2016;133:2716–2733. [DOI] [PubMed] [Google Scholar]
- 4. Vaughan CJ, Basson CT. Molecular determinants of atrial and ventricular septal defects and patent ductus arteriosus. Am J Med Gen Seminars Med Gen. 2000;97:304–309. [DOI] [PubMed] [Google Scholar]
- 5. Yazigi A, De Pecoulas AE, Vauloup‐Fellous C, Grangeot‐Keros L, Ayoubi JM, Picone O. Fetal and neonatal abnormalities due to congenital rubella syndrome: a review of literature. J Maternal‐Fetal Neon Med. 2017;30:274–278. [DOI] [PubMed] [Google Scholar]
- 6. Kleinman DS, Poole BD, Beckman GM, Hammersly MF, Rubella MTA. Some comments on the 1964–65 epidemic in California. Calif Med. 1968;109:279–285. [PMC free article] [PubMed] [Google Scholar]
- 7. Racicot K, Mor G. Risks associated with viral infections during pregnancy. J Clin Inves. 2017;127:1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stegmann BJ, Carey JC, TORCH Infections . Toxoplasmosis, Other (syphilis, varicella‐zoster, parvovirus B19), Rubella, Cytomegalovirus (CMV), and Herpes infections. Curr Women"s Health Rep. 2002;2:253–258. [PubMed] [Google Scholar]
- 9. Liang Q, Gong W, Zheng D, Zhong R, Wen Y, Wang X. The influence of maternal exposure history to virus and medicine during pregnancy on congenital heart defects of fetus. Environ Sci Pollut Res Int. 2017;24:5628–5632. [DOI] [PubMed] [Google Scholar]
- 10. Liang B, Guida JP, Costa M.L., Mysorekar IU. Host and viral mechanisms of congenital Zika syndrome. Virulence. 2019;10:768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moore M, Kaplan MH, McPhee J, Bregman DJ, Klein SW. Epidemiologic, clinical, and laboratory features of coxsackie B1–B5 infections in the United States, 1970–79. Public Health Rep. 1984;99:515–522. [PMC free article] [PubMed] [Google Scholar]
- 12. Coyne CB, Bergelson JM. CAR: a virus receptor within the tight junction. Adv Drug Deliv Rev. 2005;57:869–882. [DOI] [PubMed] [Google Scholar]
- 13. Koi H, Zhang J, Makrigiannakis A, Getsios S, MacCalman CD, Kopf GS, Strauss Iii JF, Parry S. Differential expression of the coxsackievirus and adenovirus receptor regulates adenovirus infection of the placenta. Biol Reprod. 2001;64:1001–1009. [DOI] [PubMed] [Google Scholar]
- 14. Bendig JWA, Franklin OM, Hebden AK, Backhouse PJ, Clewley JP, Goldman AP, Piggott N. Coxsackievirus B3 sequences in the blood of a neonate with congenital myocarditis, plus serological evidence of maternal infection. J Med Virol. 2003;70:606–609. [DOI] [PubMed] [Google Scholar]
- 15. Modlin JF, Crumpacker CS. Coxsackievirus B infection in pregnant mice and transplacental infection of the fetus. Infect Immun. 1982;37:222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jaidane H, Halouani A, Jmii H, Elmastour F, Mokni M, Aouni M. Coxsackievirus B4 vertical transmission in a murine model. Virol J. 2017;14:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Neumann DA, Lane JR, LaFond‐Walker A, Allen GS, Wulff SM, Herskowitz A, Rose NR. Heart‐specific autoantibodies can be eluted from the hearts of Coxsackievirus B3‐infected mice. Clin Exp Immunol. 1991;86:405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown GC, Evans TN. Serologic evidence of Coxsackievirus etiology of congenital heart disease. J Am Med Assoc. 1967;199:183–187. [PubMed] [Google Scholar]
- 19. Euscher E, Davis J, Holzman I, Nuovo GJ. Coxsackie virus infection of the placenta associated with neurodevelopmental delays in the newborn. Obstet Gynecol. 2001;98:1019–1026. [DOI] [PubMed] [Google Scholar]
- 20. Lansdown ABG. Coxsackievirus B3 infection in pregnancy and its influence on foetal heart development. Br J Exp Pathol. 1977;58:378–385. [PMC free article] [PubMed] [Google Scholar]
- 21. Sharma V, Goessling LS, Brar AK, Eghtesady P. In utero infection with Coxsackievirus‐B is associated with congenital pulmonary atresia. medRxiv. 2020;2020:18.20070813. [Google Scholar]
- 22. Klein SL, Huber S. Sex differences in susceptibility to viral infection. In: Klein S, Roberts C eds. Sex Hormones Immunity Infection. Berlin, Heidelberg: Springer; 2010. DOI: 10.1007/978-3-642-02155-8_4. [DOI] [Google Scholar]
- 23. Tennant PW, Samarasekera SD, Pless‐Mulloli T, Rankin J. Sex differences in the prevalence of congenital anomalies: a population‐based study. Birth Def Res Part A Clin Mol Teratol. 2011;91:894–901. [DOI] [PubMed] [Google Scholar]
- 24. Hierholzer JC, Killington RA. Virus isolation and quantitation. Virol Methods Manual. 1996;25–46. [Google Scholar]
- 25. McFarlane L, Truong V, Palmer JS, Wilhelm D. Novel PCR assay for determining the genetic sex of mice. Sex Dev. 2013;7:207–211. [DOI] [PubMed] [Google Scholar]
- 26. Lee LH, Yang H, Bigras G. Current breast cancer proliferative markers correlate variably based on decoupled duration of cell cycle phases. Sci Rep. 2015;4. DOI: 10.1038/srep05122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Halonen P, Rocha E, Hierholzer J, Holloway B, Hyypia T, Hurskainen P, Pallansch M. Detection of enteroviruses and rhinoviruses in clinical specimens by PCR and liquid‐phase hybridization. J Clin Microbiol. 1995;33:648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hwang JY, Lee KM, Kim YH, Shim HM, Bae YK, Hwang JH, Park H. Pregnancy loss following coxsackievirus B3 infection in mice during early gestation due to high expression of coxsackievirus‐adenovirus receptor (CAR) in uterus and embryo. Exp Anim. 2014;63:63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chung ACK, Huang XR, Zhou L, Heuchel R, Lai KN, Lan HY. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol Dial Transplant. 2009;24:1443–1454. [DOI] [PubMed] [Google Scholar]
- 30. Wang Y, Wu B, Chamberlain AA, Lui W, Koirala P, Susztak K, Klein D, Taylor V, Zhou B. Endocardial to myocardial notch‐wnt‐bmp axis regulates early heart valve development. PLoS ONE. 2013;8:e60244. DOI: 10.1371/journal.pone.0060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsukamoto S, Mizuta T, Fujimoto M, Ohte S, Osawa K, Miyamoto A, Yoneyama K, Murata E, Machiya A, Jimi E, et al. Smad9 is a new type of transcriptional regulator in bone morphogenetic protein signaling. Sci Rep. 2015;4. DOI: 10.1038/srep07596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dobin A, Davis CA, Schlesigner F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. [DOI] [PubMed] [Google Scholar]
- 34. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias‐aware quantification of transcript expression. Nat Methods. 2017;14:417–419. DOI: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang L, Wang S, Li W. RSeQC: quality control of RNA‐seq experiments. Bioinformatics. 2012;28:2184–2218. [DOI] [PubMed] [Google Scholar]
- 36. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. DOI: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Liu R, Holik AZ, Su S, Jansz N, Chen K, Leong HS, Blewitt ME, Asselin‐Labat M‐L, Smyth GK, Ritchie ME. Why weight? Modelling sample and observational level variability improves power in RNA‐seq analyses. Nucleic Acids Res. 2015;43:e97. DOI: 10.1093/nar/gkv412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luo W, Friedman M, Shedden K, Hankenson K, Woolf P. GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics. 2009;10:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao S, Guo Y, Sheng Q, Shyr Y. Advanced heat map and clustering analysis using heatmap3. Bio Res Int. 2014;2014:1–6. DOI: 10.1155/2014/986048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luo W, Brouwer C. Pathview: an R/Bioconductor package for pathway‐based data integration and visualization. Bioinformatics. 2013;29:1830–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen JW, Zhou B, Yu QC, Shin SJ, Jiao K, Schneider MD, Baldwin HS, Bergelson JM. Cardiomyocyte‐specific deletion of the coxsackievirus and adenovirus receptor results in hyperplasia of the embryonic left ventricle and abnormalities of sinuatrial valves. Circ Res. 2006;98:923–930. [DOI] [PubMed] [Google Scholar]
- 43. Chen P, Xie Y, Shen E, Li GG, Yu Y, Zhang CB, Yang Y, Zou Y, Ge J, Chen R, et al. Astragaloside IV attenuates myocardial fibrosis by inhibiting TGF‐β1 signaling in coxsackievirus B3‐induced cardiomyopathy. Eur J Pharmacol. 2011;658:168–174. [DOI] [PubMed] [Google Scholar]
- 44. Millan FA, Denhez F, Kondaiah P, Akhurst RJ. Embryonic gene expression patterns of TGF beta 1, beta 2 and beta 3 suggest different developmental functions in vivo. Development. 1991;111:131–143. [DOI] [PubMed] [Google Scholar]
- 45. Verheugt CL, Uiterwaal CSPM, Van Der Velde ET, Meijboom FJ, Pieper PG, Vliegen HW, Van Dijk APJ, Bouma BJ, Grobbee DE, Mulder BJM. Gender and outcome in adult congenital heart disease. Circulation. 2008;118:26–32. [DOI] [PubMed] [Google Scholar]
- 46. Liang Q, Gong W, Zheng D, Zhong R, Wen Y, Wang X. The influence of maternal exposure history to virus and medicine during pregnancy on congenital heart defects of fetus. Environ Sci Pollut Res. 2017;24:5628–5632. [DOI] [PubMed] [Google Scholar]
- 47. Watson WJ, Awadallah S, Jo JM. Intrauterine infection with Coxsackievirus: is it a cause of congenital cardiac malformations? Infec Dis Obst Gynecol. 1995;3:79–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Soike K. Coxsackie B‐3 virus infection in the pregnant mouse. J Infect Dis. 1967;117:203–208. [DOI] [PubMed] [Google Scholar]
- 49. Opavsky MA, Martino T, Rabinovitch M, Penninger J, Richardson C, Petric M, Trinidad C, Butcher L, Chan J,Lie PP. Enhanced ERK‐1/2 activation in mice susceptible to coxsackievirus‐induced myocarditits. J Clin Invest. 2002;109:1561–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tian T, Yang Y, Zhou L, Luo F, Li Y, Fan P, Dong X, Liu Y, Cui J, Zhou X. Left ventricular non‐compaction: a cardiomyopathy with acceptable prognosis in children. Heart Lung Circ. 2018;27:28–32. [DOI] [PubMed] [Google Scholar]
- 51. Zhang W, Chen H, Qu X, Chang CP, Shou W. Molecular mechanism of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC). Am J Med Genet C Semin Med Genet. 2013;163C:144–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Miszalski‐Jamka K, Jefferies JL, Mazur W, Glowacki J, Hu J, Lazar M, Gibbs RA, Liczko J, Klys J, Venner E, et al. Novel genetic triggers and genotype–phenotype correlations in patients with left ventricular noncompaction. Circ Cardiovasc Genet. 2017;10:e001763. DOI: 10.1161/circgenetics.117.001763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Khan R, Sheppard R. Fibrosis in heart disease: understanding the role of transforming growth factor‐β1 in cardiomyopathy, valvular disease and arrhythmia. Immunology. 2006;118:10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Saxon JG, Baer DR, Barton JA, Hawkins T, Wu B, Trusk TC, Harris SE, Zhou B, Mishina Y, Sugi Y. BMP2 expression in the endocardial lineage is required for AV endocardial cushion maturation and remodeling. Dev Biol. 2017;430:113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Prall OWJ, Menon MK, Solloway MJ, Watanabe Y, Zaffran S, Bajolle F, Biben C, McBride JJ, Robertson BR, Chaulet H, et al. An Nkx2‐5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128:947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Anderson RH, Webb S, Brown NA, Lamers W, Moorman A. Development of the heart: (2) septation of the atriums and ventricles. Heart. 2003;89:949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Heallen T, Zhang M, Wang J, Bonilla‐Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lyden D, Olsewski J, Huber SA. Influence of sex hormones on coxsackie virus group B, type 3 induced myocarditis in Balb/c mice. Eur Heart J. 1987;8:389–391. [Google Scholar]
- 59. Coronado MJ, Brandt JE, Kim E, Bucek A, Bedja D, Abston ED, Shin J, Gabrielson KL, Mitzner W, Fairweather D. Testosterone and interleukin‐1β increase cardiac remodeling during coxsackievirus B3 myocarditis via serpin A 3n. Am J Physiol Heart Circ Physiol. 2012;302:H1726–H1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miller‐Hance WC, Tacy TA. Gender differences in pediatric cardiac surgery: the cardiologist's perspective. J Thorac Cardiovasc Surg. 2004;128:7–10. [DOI] [PubMed] [Google Scholar]
- 61. Engelfriet P, Mulder BJM. Gender differences in adult congenital heart disease. Netherlands Heart J. 2009;17:414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hadar E, Melamed N, Sharon‐Weiner M, Hazan S, Rabinerson D, Glezerman M, Yogev Y. The association between stillbirth and fetal gender. J Maternal‐Fetal Neonatal Med. 2012;25:158–161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S3