

Abstract
Congenital heart disease is the most common congenital defect observed in newborns. Within the spectrum of congenital heart disease are left‐sided obstructive lesions (LSOLs), which include hypoplastic left heart syndrome, aortic stenosis, bicuspid aortic valve, coarctation of the aorta, and interrupted aortic arch. These defects can arise in isolation or as a component of a defined syndrome; however, nonsyndromic defects are often observed in multiple family members and associated with high sibling recurrence risk. This clear evidence for a heritable basis has driven a lengthy search for disease‐causing variants that has uncovered both rare and common variants in genes that, when perturbed in cardiac development, can result in LSOLs. Despite advancements in genetic sequencing platforms and broadening use of exome sequencing, the currently accepted LSOL‐associated genes explain only 10% to 20% of patients. Further, the combinatorial effects of common and rare variants as a cause of LSOLs are emerging. In this review, we highlight the genes and variants associated with the different LSOLs and discuss the strengths and weaknesses of the present genetic associations. Furthermore, we discuss the research avenues needed to bridge the gaps in our current understanding of the genetic basis of nonsyndromic congenital heart disease.
Keywords: aortic stenosis, bicuspid aortic valve, coarctation of the aorta, congenital heart disease, hypoplastic left heart syndrome, interrupted aortic arch
Subject Categories: Congenital Heart Disease, Genetics
Nonstandard Abbreviations and Acronyms
- AD
autosomal dominant
- AS
aortic stenosis
- AV
aortic valve
- BAV
bicuspid aortic valve
- CHD
congenital heart disease
- CoA
coarctation of the aorta
- DNV
de novo variant
- ES
exome sequencing
- GS
genome sequencing
- HLHS
hypoplastic left heart syndrome
- IAA
interrupted aortic arch
- iPSC‐CM
induced pluripotent stem cell derived cardiomyocytes
- LOF
loss of function
- LSOL
left‐sided obstructive lesion
- NS‐CHD
non‐syndromic CHD
- S‐CHD
syndromic CHD
- SVAS
supravalvular aortic stenosis
- TAA
thoracic aortic aneurysm
Congenital heart disease (CHD) is a spectrum of structural malformations of the heart that impair efficient blood flow. It is the most common class of congenital abnormality, with a global prevalence of ≈1 in 100 live births and ≈1.35 million infants born with CHD each year. 1 , 2 Advances in diagnosis, surgical intervention, and perioperative management have significantly reduced mortality. Between 1987–1990 and 2002–2005, there was a 59% and 16% decrease in childhood and adult mortality respectively. 3 As a result of these advancements, the number of adults living with CHD now exceeds the number of children. 3 As more adults with CHD are able to live to have children of their own, understanding the genetics of CHD is critical to ensure early diagnosis and treatment of affected children.
Burden of Left‐Sided Obstructive Lesions
In a structurally normal heart, deoxygenated blood enters the right heart and travels through the pulmonary artery to the lungs. After gas exchange, oxygenated blood enters the left heart and is pumped through the aorta to the systemic circulation. Within the landscape of CHD, the class of left‐sided obstructive lesions (LSOLs) includes structural and stenotic lesions that block left ventricular filling, output, and systemic blood flow. Among these are hypoplastic left heart syndrome (HLHS), aortic stenosis (AS), bicuspid aortic valve (BAV), coarctation of the aorta (CoA), and interrupted aortic arch (IAA). HLHS, a lethal univentricular defect, is characterized by an underdeveloped left ventricle and ascending aorta. The estimated population prevalence of HLHS is 2.60 (95% CI, 2.46–2.75) per 10 000 live births. 4 AS is a valvular defect that obstructs left ventricular output and prevalence per 1000 births is 0.46 (95% CI, 0.25–0.73). 5 AS is further subclassified as supravalvular, valvular, and subvalvular depending on the location of the stenotic lesion. In a classical tricuspid aortic valve, valvular AS can arise if the leaflets are dysplastic and narrow the aortic orifice but more commonly, congenital AS is due to cusp fusion. 6 BAV is the most common subtype of valvular AS and is the most prevalent form of CHD overall with a population prevalence of 1% to 2%. 7 BAV is frequently asymptomatic and undiagnosed until adulthood when AV stenosis, regurgitation, or other aortopathies develop. Though rare, cases of unicuspid and quadricuspid AVs have also been observed. 6 , 8 CoA is characterized by a narrowing of the aorta with an estimated prevalence of 5.57 (95% CI, 5.26–5.79) per 10 000 live births. 4 IAA is a rare defect with population prevalence 0.62 (95% CI, 0.55–0.70) per 10 000 live births and is characterized by the complete absence of part of the aortic arch. 4
Heritability of Left‐Sided Obstructive Lesions
Overall, the etiology of CHD is heterogenous and can roughly be divided into CHD in the setting of extracardiac abnormalities, so‐called syndromic CHD (S‐CHD), and nonsyndromic CHD (NS‐CHD) where congenital abnormalities are isolated to the heart. Syndromic causes of LSOLs include a number of well‐defined genetic syndromes resulting primarily from aneuploidies or variation in the typical diploid arrangement of portions of the chromosomes known as copy number variants (CNVs). 9 Down syndrome results from an extra copy of chromosome 21 and is associated with CHD in 40% to 50% of cases. 9 Turner syndrome is caused by the partial or total loss of the X chromosome in females and CHD occurs in 20% to 40% of these patients. 9 DiGeorge syndrome is caused by microdeletion at the 22q11.2 locus, resulting in the loss of over 40 genes. 9 Other LSOL‐associated syndromes can be caused by single‐gene variation, including Cantu syndrome (ABCC9), Kabuki syndrome (KMT2D), and Rubinstein‐Taybi (CBP and EP300). 9
NS‐CHD has clear evidence for a heritable basis of disease and is the focus of this review. In contrast to S‐CHD, NS‐CHD typically have normal chromosomal number and arrangement without CNV. The incidence of CHD in a sibling of an affected child is 4% to 22% in comparison to the population prevalence of ≈1% among live births. 10 , 11 Nonsyndromic LSOLs are associated with a 20% incidence of CHD in first‐degree relatives, with BAV being the most commonly detected lesion. 12 , 13 Given this high sibling recurrence risk, echocardiographic screening of first‐degree relatives is recommended. 14 , 15 , 16 The risk of CHD varies by the proband's defect but is higher than the general population with prevalence in first‐degree relatives of 19% for HLHS, 9% for CoA, and 1% for transposition of the great arteries. 16 Pedigree analysis of HLHS in 3 generations of 38 families found that over 55% of HLHS probands had more than 1 member affected and the recurrence risk of HLHS in siblings was 8%, which increased to 21% with an affected parent. 11 In the case of BAV, 9.4% of first‐ and second‐degree relatives of a BAV proband were also affected. 17 Family studies demonstrate a heritable factor for CoA; however, ascertaining the genetics of CoA is difficult because only 16.2% of patients with CoA have isolated disease, whereas the majority have concomitant cardiac defects. 18 , 19 The high heritability of these lesions is strongly suggestive of an underlying genetic cause for LSOLs. 20
Traditionally these inherited lesions are believed to be caused by the interaction of unidentified environmental and genetic factors. 21 However, the early presentation and familial clustering of CHD is reminiscent of early onset monogenic diseases such as severe autosomal recessive disorders and genetically more complex diseases such as Alzheimer disease, dyslipidemias, and autoimmune conditions. 22 , 23 In addition, a compound heterozygous inheritance pattern, a type of autosomal recessive inheritance where each copy of an allele harbors a different variant, has been identified in several LSOLs. 24 , 25 Finally, a number of cardiovascular diseases have been linked to autosomal dominant (AD), monogenic causes, including cardiomyopathies, channelopathies, and connective tissue diseases. 26 Forms of CHD associated with a high sibling recurrence risk and a multigenerational family history are consistent with an AD mode of inheritance with variable penetrance, such as in BAV, 27 , 28 , 29 and HLHS. 11 Sporadic cases of CHD may be because of either recessively inherited variants or de novo AD variants. Exome sequencing (ES) of 1365 trios, 68 affected sibling pairs, and 458 singleton probands with S‐CHD and NS‐CHD revealed that de novo loss of function (LOF) variants were enriched in syndromic CHD, whereas inherited rare pathogenic variants were enriched in NS‐CHD. 30 Incomplete penetrance of these rare inherited variants may explain the phenotypic spectrum observed in familial CHD and point toward a mono‐ or oligogenic cause.
Transcription Factors Are Implicated in Left‐Sided Obstructive Lesions
The heart is the first fetal organ to develop and arises from 2 cell lineages in the anterior lateral plate mesoderm. Gastrulation brings the cardiogenic mesodermal cells to form the cardiac crescent, and shortly after, the cells migrate to the midline to form a beating heart tube. Rightward looping occurs thereafter and positions the heart chambers, inflow, and outflow tracts, which are established by subsequent septal division. This process is governed by dynamic interaction between cell signaling cascades and transcription factors, namely NKX2‐5, GATA, and NOTCH, among others. 31 Given the tight genetic control throughout cardiogenesis, any number of alterations in cardiogenic transcription factors or signal transduction could underlie NS‐CHD phenotypes.
The Genetic Landscape of Left‐Sided Obstructive Lesions
The genetic causes of nonsyndromic LSOLs are heterogeneous with overlapping genetic substrate (Figure 1). Many of the genes implicated are associated with myocyte contractility (MYH6) or the transcriptional regulators of cardiac development (Figure 2). Identification of putative genetic loci have provided a foundation for understanding the genetics underlying heart development and CHD; however, there are limitations to these studies that are summarized next.
Figure 1. Venn diagram showing overlap of genes associated with LSOLs based on disease subtype.
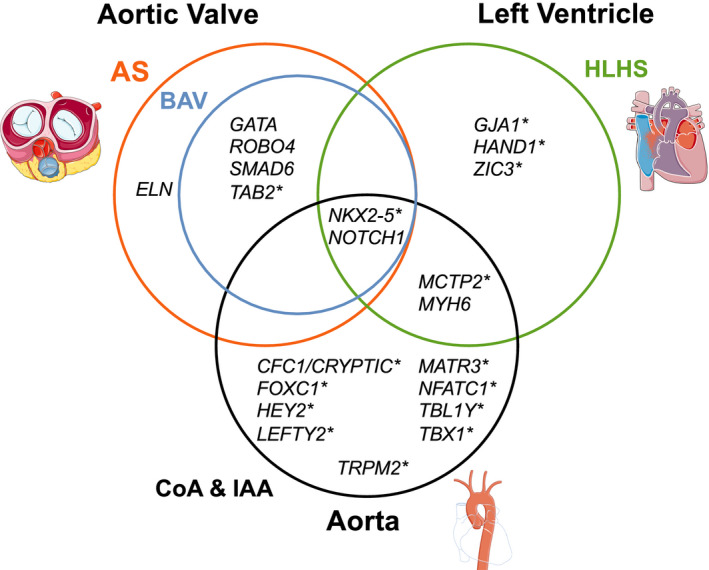
Asterisk indicates genes without robust evidence of association. AS indicates aortic stenosis; BAV, bicuspid aortic valve; CoA, coarctation of the aorta; HLHS, hyperplastic left heart syndrome; IAA, interrupted aortic arch; and LSOL, left‐sided obstructive lesion. This figure was created using images modified from Servier Medical Art Commons, licensed under a Creative Commons Attribution 3.0 Unported License (http://smart.servier.com).
Figure 2. Genetic landscape of left‐sided obstructive lesions.
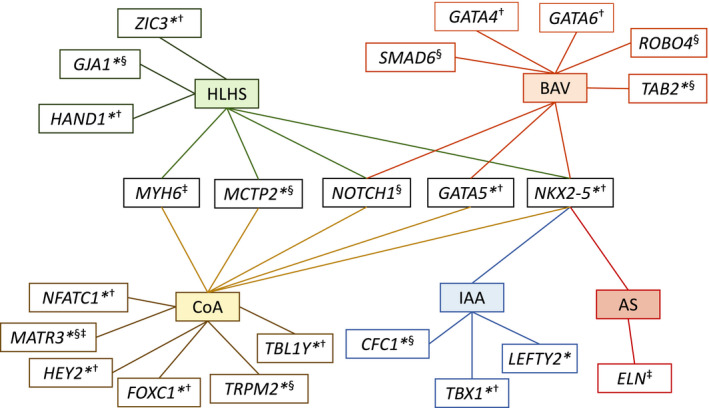
Asterisk indicates genes without robust evidence of association. Genes code for †transcription factors, ‡structural or contractile proteins, §cell signaling components. AS indicates aortic stenosis; BAV, bicuspid aortic valve; CoA, coarctation of the aorta; HLHS, hyperplastic left heart syndrome; IAA, interrupted aortic arch; and LSOL, left‐sided obstructive lesion.
Hypoplastic Left Heart Syndrome
NOTCH1‐Encoded Notch Receptor 1
NOTCH1 encodes the NOTCH1 receptor, 1 of 4 type 1 transmembrane receptors that interact with Jagged and Delta‐like receptors on neighboring cells. When activated, the NOTCH receptors undergo a series of cleavage events that release the NOTCH intracellular domain, which interacts with transcriptional repressor RBP‐J to target genes in the HESR, CHF, and Hrt families. Despite the observation that Notch1 +/− mice show no overt phenotype, several NOTCH1 variants have been identified in patients with HLHS. 32 For instance, exome sequencing (ES) of 4 patients with HLHS identified 1 with a heterozygous protein‐truncating variant C4662A, inherited through the patient's unaffected father. 33 Of note, the patient's paternal aunt was known to have tricuspid atresia, suggesting that this pathogenic variant may have variable expressivity. In another study, ES of 49 HLHS‐affected families found likely pathogenic NOTCH1 variants in 6% of HLHS probands and variants of unknown significance in 16% of the cohort. 34 Following this, a primary genetic association study in 1085 individuals with left ventricular outflow tract (LVOT) obstruction identified 2 rare intronic variants (g.chr9:139427582C>T and g.chr9:139435649C>T) with strong linkage disequilibrium, though the effects of these intronic variants remain unknown. 34 In addition to large genetic association studies that have implicated noncoding variation, family studies have identified variants in NOTCH1 associated with likely autosomal recessive CHD. In 1 family, genome sequencing (GS) of an HLHS proband and 4 family members, 2 of whom had bicuspid aortic or pulmonary valves, identified compound heterozygous NOTCH1 variants in the proband (P1964L and P1256L). 24 The P1256L variant was inherited from the patient's unaffected father, whereas the P1964L was inherited from the patient's mother who had BAV. The proband's induced pluripotent stem cell‐derived cardiomyocytes (iPSC‐CMs) exhibited reduced Notch signaling, impaired myofilament organization, and altered nitrous oxide signaling while parent cells were unaffected. Independent HLHS‐iPSC studies also observe impaired Notch signaling and differentiation, even in patient‐derived cells without known NOTCH1 variants. 35 NOTCH1 variants associated with HLHS are repeatedly observed in unbiased genetic studies, though the current assessment by ClinVar is not currently supportive of a clear monogenic cause of disease based on the absence of many of these variants, and 1 is currently listed as benign. Animal and iPSC models have begun to delineate the role of NOTCH1 in HLHS, but variants are still uncommon in the HLHS population and may represent a pathogenic substrate rather than a causative agent.
MYH6‐Encoded Myosin 6
MYH6 encodes the minor ventricular myosin heavy chain isoform expressed during fetal development and the alpha‐heavy chain subunit of cardiac myosin (MYH6). 36 Myh6 ablation in mice is embryonic lethal and associated with gross cardiac defects, and recent identification of variants in HLHS patients suggests a role for MYH6 in human CHD. 37 Unbiased genetic screens provide the strongest evidence for this association with HLHS, and suggest MYH6 variants may predict poorer prognosis. GS in 5 patients with HLHS and reduced right ventricular ejection fraction post‐Fontan operation identified 2 patients with rare inherited compound heterozygous variants (I704N and T1379M; D588A and E1207K) affecting the tail and head domains of MYH6. 38 Heterozygous family members were unaffected, and interestingly, MYH6 variants were not identified in 21 patients with normal post‐Fontan ejection fraction. Further, 1 group performed GS in a family with recurrent HLHS identified a rare inherited MYH6‐R443P variant in both affected and unaffected individuals. 25 Case‐control association testing in 190 unrelated patients with HLHS (who had previously undergone ES or GS) identified damaging variants in 20 patients (10.5%) versus 2.9% of controls. Of those with MYH6 variants, 95% were heterozygous carriers. 25 Just as the previous study associated MYH6 variants with reduced post‐Fontan ejection fraction, transplant‐free survival was significantly lower in MHY6 variant carriers in the case‐control cohort. 25 , 38 This suggests that beyond being a potential cause of HLHS, knowledge of MYH6 variant status could inform the provider's choice between univentricular repair, biventricular repair, or transplant. Finally, a group derived iPSC‐CMs from 16 patients with HLHS, 8 of whom were MYH6 variant carriers. MYH6 variant iPSC‐CMs had 350% increased MYH7 expression in atrial and ventricular tissue, along with inefficient cardiomyocyte differentiation and dysmorphic sarcomeres, all of which may represent compensatory responses to MYH6 haploinsufficiency. 25 Taken together, MYH6 variants in HLHS are well characterized by GS, ES, and iPSC models; however, ClinVar considers many of these variants of conflicting or uncertain significance. Given that independent studies have associated MYH6 variants with a poorer post‐surgery prognosis, it is possible that they are a disease‐modifying factor rather than a disease‐causing substrate.
GJA1‐Encoded Gap Junction Alpha‐1 Protein
GJA1 encodes the connexin43 (CX43) subunit of gap junction channels abundantly expressed in the heart. During development, these gap junctions are critical to facilitating cell‐cell adhesion and communication via electrical and molecular signals. 39 GJA1 knockout is embryonic lethal in mice and causes severe conotruncal defects and LVOT obstruction; however, the role in human LSOLs is debated. 40 GJA1 variants were identified in an early study via denaturing gradient gel electrophoresis in 8 of 14 patients with HLHS post‐transplant. 41 Each of these patients had 2 silent polymorphisms and missense variants R362Q and R376Q that, in vitro, abolish phosphorylation of the CX43 regulation domain. 41 Interestingly, this sequence is that of the GJA1 pseudogene and the authors suggested that illicit recombination and loss of regulatory phosphorylation contributes to the HLHS phenotype. The GJA1 pseudogene is implicated in nonsyndromic deafness and has been identified as a regulator of tumor growth; it has not been identified in HLHS probands in more recent studies and was likely an artifact from poor variant resolution by gel electrophoresis. 41 , 42 , 43 An independent group evaluated 300 patients with CHD for GJA1 variants, including only 4 HLHS probands, and found no nonsynonymous variants in any patient with CHD. 44 Surprisingly, knock‐in mice with variants blocking critical CK1 (S325A/S328Y/S330A) and PKC (S368A) phosphorylation sites shown in vitro to alter cell‐cell communication exhibit no CHD. 44 Though animal models suggest an important role for GJA1 in LVOT development, modern genetic screens, mouse models, and current ClinVar assessments do not support the original pseudogene hypothesis or GJA1 variants as a genetic cause of HLHS.
NKX2‐5‐Encoded NK2 Homeobox 5
The NKX2‐5 homeobox transcription factor is expressed in the early first heart field, the second heart field pharyngeal mesoderm, and the pharyngeal endoderm. 45 Nkx2‐5−/− mice develop a linear heart tube but fail to initiate looping, and NKX2‐5 haploinsufficiency in humans is associated with a spectrum of CHD. 46 , 47 The approach to NKX2‐5 variant identification in HLHS has been candidate‐gene based because of established associations with ventricular septal defect, atrial septal defect, and tetralogy of Fallot but in patients with HLHS, NKX2‐5 variants are rarely observed. The first study describing an association between genetic variants in NKX2‐5 and HLHS identified a T178M variant in a patient with HLHS and a family history of atrial septal defect and atrioventricular conduction block. 48 Though a later study found a heterozygous R25C variant in 1 of 80 patients with HLHS, several groups have since sequenced NKX2‐5 in HLHS cohorts and repeatedly fail to identify any NKX2‐5 variant associated with sporadic or familial HLHS. 48 , 49 , 50 , 51 Despite this, iPSC‐CMs from patients with HLHS exhibit downregulated NKX2‐5 expression and increased H3K27 methylation at the NKX2‐5 promoter relative to iPSC‐CMs from patients with biventricular CHD. 52 Though mouse models illustrate the importance of NKX2‐5 in cardiogenesis, the few variants identified through targeted sequencing are conflicting in the ClinVar database, and current evidence does not support NKX2‐5 variants as a significant monogenic cause of HLHS.
HAND1‐Encoded Heart and Neural Crest Derivatives‐Expressed Protein 1
The HAND1 gene encodes a basic helix‐loop‐helix (bHLH) transcription factor (HAND1) that is required for cardiac morphogenesis and regulates the balance between cardioblast proliferation and differentiation. 53 , 54 HAND1 overexpression in mice leads to elongation of the LVOT whereas LOF causes abnormal looping and embryonic lethality. 54 , 55 The critical role and expression pattern of HAND1 in left‐heart development makes it a candidate gene in the etiology of HLHS; however, the existence and role of HAND1 variants in the development of HLHS is controversial. Drawing on the outflow tract defects observed in mice, 1 group sequenced HAND1 in formalin‐fixed left ventricles and identified the HAND1 frameshift variant A126fs (A126Pfs13X), in 24/31 patients with HLHS. 56 Formalin fixation can cause sequence artifacts due to cytosine deamination, crosslinking, and DNA fragmentation, and recent studies suggest that the A126fs variant was artefactual. 50 , 57 , 58 Direct HAND1 sequencing in fresh frozen tissue from 24 HLHS hearts identified no HAND1 variants. 50 This was corroborated by another group that failed to find HAND1 variants in ventricular samples from 14 patients with HLHS. 33 Furthermore, recent ES studies also fail to find evidence of HAND1 variants in HLHS, as do current ClinVar assessments. 30 , 59 , 60 , 61 Although current understanding does not support any specific HAND1 variant in the etiology of HLHS, it is possible that undiscovered alterations in HAND1 regulatory elements may yet play an undiscovered role.
Aortic Stenosis
Supravalvular Aortic Stenosis
ELN‐Encoded Elastin
The ELN gene, located at 7q11.23, encodes the elastin protein, a significant component of the extracellular matrix of the aorta. Large deletions of the ELN region cause Williams syndrome whereas smaller deletions affecting between 30 and 100 kb of the gene are associated with isolated supravalvular aortic stenosis (SVAS). 62 , 63 , 64 , 65 , 66 , 67 SVAS has an AD pattern of inheritance and >200 ELN variants, including microdeletions, intronic splice site, nonsense, and missense variants, are estimated to explain up to 35% of isolated SVAS. 66 , 68 , 69 , 70 , 71 , 72 , 73 , 74 Eln−/− mice died by P4.5 and recapitulate the aortic phenotype of SVAS whereas Eln+/− mice only recapitulate the hypertension, arterial stiffness, and compensatory overexpression of elastic lamellae and smooth muscle. 75 , 76 Overall, ELN haploinsufficiency is a well‐established cause of inherited SVAS in a significant proportion of the patient population, but single gene variants underlying SVAS in the remaining patients have not been well described.
Subvalvar Aortic Stenosis
Although subvalvar, or subaortic, stenosis is typically believed to be an acquired condition because of its progressive nature and association with LVOT hypertrophy and scarring, there are a few case reports of familial occurrence. 77 , 78 , 79 , 80 Although a genetic predisposition has been observed in Newfoundland dogs, no recent genetic studies have identified subvalvar aortic stenosis associated genes in humans. 81
Bicuspid Aortic Valve
ROBO4‐Encoded Roundabout Homolog 4
ROBO4 is expressed in the endothelial and intimal cells of the aorta and regulates vascular permeability. 82 Zebrafish homozygous for a small deletion in exon 6 demonstrate perturbed outflow tract function, whereas mice with homozygous deletion of exons 1 to 3 demonstrate incompletely penetrant AV thickening, BAV, AS, regurgitation, and ascending aortic aneurysm. 82 These animal models were generated after discovery of ROBO4 variants through ES in patients with BAV, and the consistent phenotypes of animal models support a causative role of ROBO4 variants in a small percentage of BAV. Initially, ES in 5 affected individuals of a family with a history of BAV and ascending aortic aneurysm identified a heterozygous splice site variant at exon 13 (c.2056+1G>T). Further, ES in a mother with atrial septal defect/AS and her son with BAV/atrial septal defect/AS identified the rare heterozygous variant R64C. 82 After identification of these initial variants in family‐based studies, candidate ROBO4 sequencing in 441 probands with BAV/ascending aortic aneurysm identified 7 with heterozygous ROBO4 variants (A95T, T232M, H411G, R568X, R64C, V247A, Y280S, G534Efs*49, N622H, A749L, and N510V) including an independent individual with the R64C variant. 82 Several of these variants are considered likely pathogenic in ClinVar; however, larger independent ES and functional studies are needed to more clearly define the role and prevalence of ROBO4 variants in BAV.
GATA Factors
The GATA family of zinc‐finger transcription factors includes GATA4, ‐5, and ‐6; each of which are required for cardiogenesis. GATA5 variants are well characterized through candidate sequencing studies, and unlike Gata4 and Gata6, Gata5 knockout mice are not embryonic lethal but exhibit mild left ventricle hypertrophy and 25% frequency of BAV. 83 Targeted GATA5 sequencing in 3 independent BAV cohorts identified several heterozygous variants, including L233P, S19Y, Y143H, G166S, Y16D, T252P, and a Q3R variant that appeared in 2 studies. 84 , 85 , 86 Although GATA5 variants may be better characterized in the literature, a potential role for GATA4 variants is supported by a genome‐wide association study in a cohort of 466 patients with BAV that identified 2 novel variants, including an intergenic variant (rs6601627) identified in 8.3% of BAV cases relative to 4.2% in controls, as well as a near‐significant S337G variant. 87 In addition, GATA4 sequencing in a family with AD BAV found a heterozygous E147X variant that caused loss of transcriptional activity in vitro. 88 Finally, Gata6+/− mice exhibit BAV in 56% of males and 27% of females, consistent with the male predilection of human BAV. 89 GATA6 sequencing in 152 patients with BAV identified a novel heterozygous E38X variant in 1 proband with a family history of BAV, and this variant caused a loss of transcriptional activity in vitro. 90 Current evidence suggests these rare variants play some causal role in between 0.4% and 8.3% of patients with BAV, but despite aforementioned genetic screens and promising animal models, neither GATA4 nor GATA6 variants appear in ClinVar, and only GATA5‐Y16D is considered pathogenic.
NOTCH1‐Encoded Notch Receptor 1
The Notch signaling cascade coordinates cell migration and differentiation in the conotruncal cushions that give rise to the aortic and pulmonary valves. 91 , 92 A potential role for NOTCH1 variants in BAV was first discovered through linkage analysis of a family with AV disease that revealed association with the 9q34‐35 locus. 91 Direct sequencing of NOTCH1 in this and another CHD‐affected family revealed that the R1108X and H1505del nonsense variants segregated with CHD in the respective families. 91 NOTCH1 variants have also been identified in sporadic BAV. One study found NOTCH1 variants predicted to be pathogenic in silico, T596M and P1797H, in 2/48 patients with sporadic BAV. 93 Given that BAV is associated with an elevated risk of thoracic aortic aneurysm (TAA), NOTCH1 was sequenced in 48 patients with BAV/TAA and 4 nonsynonymous NOTCH1 variants in 5 probands (R1350L, P1377S, A1343V, and P1390T) were identified in both familial and sporadic cases. 94 Two of these variants, P1390T and A1343V, were absent from healthy controls and affect highly conserved residues. Whereas genetic variants in NOTCH1 are emerging in their association with BAV and TAA, dysregulated Notch signaling in aortic endothelial cells may be present more broadly in BAV. 92 NOTCH1 LOF variants would directly cause this dysregulation via haploinsufficiency, but the cause of attenuated Notch signaling in patients without NOTCH1 variants warrants further investigation. Current ClinVar assessments consider the aforementioned variants benign or conflicting with the exception of R1107X, which is considered pathogenic. Further functional and animal studies are needed to better define the role not only of variants in NOTCH1 but of other genes along the Notch signaling pathway.
SMAD6‐Encoded Mothers Against Decapentaplegic Homolog 6
SMAD6 negatively regulates bone morphogenic proteins in response to elevated transforming growth factor‐β signaling. 95 In mice, knockout of the SMAD6 analog Madh6 causes septation defects and cardiac valve hyperplasia, a finding that has driven SMAD6 sequencing in patients with BAV. 95 In 1 study, targeted sequencing of unrelated probands found the P415L and C484F variants in patients with BAV/AS and BAV/CoA respectively, and both show inefficient bone morphogenic protein inhibition in vitro. 96 In another, pathogenic SMAD6 variants were observed in 2.5% of a large BAV/TAA cohort, including 2 frameshift deletions (K242NfsX300 and Gly166VfsX23), 1 in‐frame deletion (G26_S27del), 2 nonsense variants (Y279X and Y288X), and 6 missense variants clustered in the MH1 and MH2 functional domains (V239M, P257L, G271W, G406C, H408Q, and R443H). 97 The MH1 domain binds DNA and the MH2 domain interacts with transforming growth factor‐β and bone morphogenic protein signaling cascades both of which are critical to SMAD6 function. 97 Finally, SMAD6 resequencing in 473 patients with TAA, 65 of whom also had BAV, identified variants in 1.5% of the overall cohort and only in patients with concomitant BAV. 98 Of note, half of the SMAD6 variant‐positive patients with BAV/TAA had a family history of cardiovascular anomalies. 98 The P415L, C484F, and P257L variants are considered pathogenic in ClinVar but have not been validated by other independent studies at the present time. Other variants are either unlisted or of uncertain significance. In vivo interrogation of these variants, particularly those affecting the MH1 and MH2 domains, is still needed to fully understand the role of SMAD6 in the development of BAV.
NKX2‐5‐Encoded NK2 Homeobox 5
As with HLHS, the search for NKX2‐5 variants in BAV has been driven by associations of variants with other forms of CHD and animal models that underscore the importance of NKX2‐5 signaling in heart development. In the C57BI/6 mouse strain, 11% of Nkx2‐5+/− mice have BAV relative to the 1.4% in wild‐type mice; however, this elevated frequency is not recapitulated in other strains and likely represents background genetic effect. 99 NKX2‐5 sequencing of 142 BAV probands and relatives identified the K192X variant in a family with AD BAV, which in vitro caused loss of transcriptional activity. 100 A second study, though limited by cohort size, failed to identify deleterious NKX2‐5 variants in 19 patients with BAV. 101 ClinVar does not consider any NKX2‐5 variant associated with BAV pathogenic and overall, the gene has modest association as a monogenic cause of BAV.
Other Candidate Genes
Recently identified variants have been proposed but their causal role warrants further study. A rare MAT2A‐E344A variant has been associated with BAV/TAA. 102 NRF2F candidate sequencing identified an inherited heterozygous variant in the NRF2F transcription factor (C96X) in a family with nonsyndromic‐BAV that caused complete loss of transcriptional activity in vitro. 103 Other variants identified in at least 2 patients through targeted sequencing include those in AXIN1/2 (R841Q; A684V), MCTP2 (T545M; L847F), NFATC1 (P77L; V210M), and TBX5 (S372L; V263M), each of which are conflicting, uncertain, or unlisted in ClinVar. 104 Further study of the functional consequences and heritability of each are needed to validate their role in the etiology of BAV.
Interrupted Aortic Arch
TBX1‐Encoded T‐Box Transcription Factor 1
IAA is a rare form of CHD and can be divided into subtypes A, B, and C based on the location of interruption. IAA type B interrupts the aorta between the left carotid and left subclavian arteries and is both the most common and genetically homogenous form of IAA and links to the 22q11 locus. 105 22q11 deletion is most commonly associated with DiGeorge syndrome and is rare in isolated CHD; only 1 in a cohort of 628 patients with nonsyndromic conotruncal defects had 22q11 deletion. 106 No single gene is a definitive cause of the cardiovascular abnormalities associated with DiGeorge syndrome, though TBX1 has been proposed as a likely candidate, as TBX1 variants alone can recapitulate the cardiovascular and craniofacial defects, and Tbx1+/− mice develop abnormal aortic arch phenotypes. 107 , 108 , 109 , 110 , 111 Truly pathogenic TBX1 variants in isolated IAA are uncommon; 1 study sequenced TBX1 in 105 patients identified a 466‐476dup10 duplication in a proband with IAA. 109 Further, another group failed to identify TBX1 variants in 41 patients with conotruncal defects, and ClinVar does not consider TBX1 variants pathogenic in IAA. 108
CRKL‐Encoded Crk‐Like Protein
Like TBX1, CRKL is a candidate genetic susceptibility locus for the cardiac defects observed in children with DiGeorge syndrome. CRKL resides within the typical 3 Mb deletion that is characteristic of DiGeorge syndrome, and knockout in mice is embryonic lethal with severe outflow tract defects. 112 , 113 Interestingly, though Crkl+/ − mice are typically normal, compound heterozygous Crkl+/;Tbx1+/− mice develop cardiac defects including IAA and other outflow tract defects, suggesting dose dependent interaction between TBX1 and CRKL may underlie CHD in a subset of patients. 112 To date, there are no studies identifying CRKL genetic variants in patients with nonsyndromic LSOLs. Further investigation is needed to assess if CRKL is involved in cardiac defects apart from DiGeorge syndrome.
Other Identified Genes
Mouse studies have also identified genes in which LOF causes IAA and aortic arch defects. For example, Foxc2 LOF causes the IAA phenotype in mice, Pitx2 LOF causes a spectrum of conotruncal defects, and Tgfβ1 LOF causes fourth pharyngeal arch artery hypoplasia. 111 , 114 , 115 Although promising animal models exist, the difficulty of assembling a large nonsyndromic cohort remains a barrier to defining the genetics of IAA.
Coarctation of the Aorta
MYH6‐Encoded Myosin 6
Genome‐wide association study of 120 Icelanders with CoA, both with and without other CHD, was paired with GS data from 15 220 Icelanders, 39 of whom had CoA. 116 The genome‐wide association study implicated the MYH6‐encoding 14q11 locus, and GS identified the R721Y variant in 20% of the 39 chip‐typed individuals with CoA. 116 This variant is rare outside of the Icelandic population and is not present in 6503 exomes from the NHBLI Exome Sequencing Project nor in ClinVar and appears only once in the 126 216 exomes and 15 136 genomes in the Genome Aggregation Database. 116 This variant also associates with BAV and other forms of CHD, but further functional studies and animal models, as well as further interrogation outside of the founder population of Iceland, are needed to establish the pathogenicity of this variant. 116
HEY2‐Encoded Hairy/Enhancer‐of‐Split Related With YRPW Motif Protein 2 and NOTCH Signaling
The zebrafish gridlock variant in hey2 causes CoA and treatment with vascular endothelial growth factor pathway activators during angioblast cell fate determination and migration rescue the CoA phenotype. 117 Hey2 −/− mice do not have CoA but do develop lethal postnatal cardiac hypertrophy; however, human HEY2 variants have not been associated with CoA. 118 HEY2 is a NOTCH1 target gene, and a large candidate‐gene study of NOTCH1 variants in families with CHD identified the E1262‐G1301del and Y1843X variants in 2 probands with CoA and other cardiac defects. 119 The Y1843X variant was identified in 2 family members with AS and aortic valve insufficiency, whereas the E1262‐G1301del was found in both affected and unaffected family members. 119 Neither variant is listed in ClinVar and an association of NOTCH1 variants with CoA cannot be made because of the presence of other concomitant cardiac defects in variant positive individuals.
Other Candidate Loci
CNV analyses have identified a handful of CoA associated loci, including in TRPM2, FOXC1 binding sites, and along the X chromosome. 120 , 121 Beyond CNVs, a translocation in MATR3 was identified in a proband with CoA and Noonan‐like syndrome, but heterozygous Matr3 ablation in mice causes diverse and incompletely penetrant cardiac defects. 122 MCTP2 knockdown in Xenopus embryos causes outflow tract defects, and MCTP2 duplications, deletions, and missense variants (A60T, G203D, and Y235C) have been found in patients with CoA both with and without other cardiac defects. 123 Finally, 1 study associated TBL1Y missense variants (D69H and R176W) with CoA, but cardiac defects associated with TBL1Y LOF have not been identified in animal models and have not been assessed in ClinVar. 120 As a whole, genetic studies identifying single gene variants directly associated with CoA are not currently present.
Exome and Genome‐Based Studies
Many inherited NS‐CHD‐associated genes were first identified through linkage analysis and targeted sequencing of large CHD‐affected families. Initial findings led to candidate gene sequencing in larger CHD cohorts that typically return a handful of variants; however, it is rare for any gene to account for a significant proportion of affected individuals (Table 1). This low yield of known genetic variants is compounded by variable expression of disease. Many variants are shared among different LSOLs as well as other forms of CHD including tetralogy of Fallot and other right‐sided or conotruncal defects. In one example, a paternally inherited NOTCH1 variant was found in a patient with HLHS whose paternal aunt had a hypoplastic right ventricle. 33 Although the aunt was not available for sequencing to confirm presence of the putative variant, this report illustrates the variable expressivity that underlies the genetics of CHD. 33 The incidence of both right‐ and left‐sided CHD in the same family also suggests these defects may also share common genetic and/or development etiology. In another example, the frequent association of BAV with CoA and AS invites the possibility that these LSOLs exist on a spectrum of disease severity.
Table 1.
Genes Implicated in Nonsyndromic Left‐Sided Obstructive Congenital Heart Lesions
| Gene | Chr. | Protein | Mode of Inheritance | Genetic Yield | Online Mendelian Inheritance in Man | Reference |
|---|---|---|---|---|---|---|
| HLHS | ||||||
| GJA1 | 6q22.31 | Gap junction alpha‐1 protein | AR | Rare | 121014 | 41, 44, 124 |
| HAND1 | 5q33.2 | Heart and neural crest derivatives‐expressed protein 1 | Rare | 241550 | 56, 125, 126 | |
| MCTP2 | 15q26.2 | Multiple C2 and transmembrane domain‐containing protein 2 | Rare | 616297 | 128 | |
| MYH6 | 14q11.2 | Myosin‐6 | AR, AD, de novo, CH | ≈11% | 160710 | 25, 38 |
| NKX2‐5 | 5q35.1 | Homeobox protein Nkx‐2.5 | AD | Rare | 600584 | 47, 51, 129 |
| NOTCH1 | 9q34.3 | Neurogenic locus notch homolog protein 1 | AD | ≈6% to 22% | 190198 | 24, 33, 130, 131 |
| ZIC3 | Xq26.3 | Zinc finger protein ZIC3 | XR | Unknown | 300265 | 132 |
| Aortic stenosis | ||||||
| ELN | 7q11.23 | Elastin | AD | ≈11% to 35% | 185500 | 63, 64, 133 |
| NKX2‐5 | 5q35.1 | Homeobox protein Nkx‐2.5 | Unknown | 600584 | 47 | |
| Bicuspid aortic valve | ||||||
| GATA4 | 8p23.1 | Transcription factor GATA‐4 | AD | ≈0.4% to 8% | 600576 | 87, 88 |
| GATA5 | 20q13.33 | Transcription factor GATA‐5 | AD | ≈3% to 4% | 611496 | 83, 84 |
| GATA6 | 18q11.2 | Transcription factor GATA‐6 | AD | Rare | 600001 | 90, 134 |
| NKX2.5 | 5q35.1 | Homeobox protein Nkx‐2.5 | AD | Rare | 600584 | 100 |
| NOTCH1 | 9q34.3 | Neurogenic locus notch homolog protein 1 | AD | ≈4% to 10% | 190198 | 24, 93, 135 |
| ROBO4 | 11q24.2 | Roundabout homolog 4 | AD | ≈2% | 607528 | 82 |
| SMAD6 | 15q22.31 | Mothers against decapentaplegic homolog 6 | ≈2% to 3% | 602931 | 97, 98 | |
| TAB2 | 6q25.1 | TAK1‐binding protein 2 | AD | Rare | 605101 | 136, 137 |
| Interrupted aortic arch | ||||||
| CFC1 | 2q21.1 | Cryptic protein | Unknown | 605194 | 138 | |
| LEFTY2 | 1q42.12 | Left‐right determination factor 2 | Unknown | 601877 | 139 | |
| NKX2.5 | 5q35.1 | Homeobox protein Nkx‐2.5 | AD | Unknown | 600584 | 51 |
| TBX1 | 22q11.21 | T‐box transcription factor TBX‐1 | Rare | 602054 | 107, 109, 110 | |
| Coarctation of the aorta | ||||||
| FOXC1 | 6p25/3 | Forkhead box protein C1 | Rare* | 601090 | 121 | |
| GATA5 | 20q13.33 | Transcription factor GATA‐5 | Unknown | 611496 | 83 | |
| HEY2 | 6q22.31 | Hairy/enhancer‐of‐split related with YRPW motif protein 2 | Unknown | 604674 | 117, 118 | |
| MATR3 | 5q31.2 | Matrin‐3 | Unknown | 164015 | 122 | |
| MCTP2 | 15q26.2 | Multiple C2 and transmembrane domain‐containing protein 2 | AD | Rare* | 616297 | 128 |
| MYH6 | 14q11.2 | Myosin‐6 | AD, de novo | ≈0% to 20% | 160710 | 116 |
| NFATC1 | 18q23 | Nuclear factor of activated T‐cells, cytoplasmic 1 | Unknown | 600489 | 104 | |
| NKX2.5 | 5q35.1 | Homeobox protein Nkx‐2.5 | AD | Unknown | 600584 | 47 |
| NOTCH1 | 9q34.3 | Neurogenic locus notch homolog protein 1 | AD | Rare* | 190198 | 119 |
| SMAD6 | 15q22.31 | Mothers against decapentaplegic homolog 6 | Unknown | 602931 | 98 | |
| TBL1Y | Yp11.2 | F‐box‐like/WD repeat‐containing protein TBL1Y | Rare* | 400033 | 140 | |
| TRPM2 | 21q22.3 | Transient receptor potential cation channel subfamily M member 2 | Rare* | 603749 | 120 | |
Asterisk indicates a higher percentage was reported in a small study, but robust, independent evaluation is required for an association to be established. Unknown indicates the variant was associated with concomitant defects and a disease‐specific yield could not be established. AD indicates autosomal dominant; AR, autosomal recessive; CH, compound heterozygous; and XR, X‐linked recessive.
It is likely that this variable expressivity is due to a myriad of factors, both intrinsic and extrinsic to the developing heart. In HLHS, for example, it is still unclear whether ventricular hypoplasia arises as the result of intrinsic defect in the left ventricular myocardium, whether it is secondary to hemodynamic changes caused by valvular defects or if both mechanisms are at play. 141 To this end, it is necessary to understand not only what genetic variants are associated with a particular lesion but also the cell population and tissue location that is perturbed developmentally. In addition, the developmental stage at the time of the extrinsic/environment stressor exposure also plays a key role in modifying the phenotypic expression of pathologic genetic variation. 141 For example, retinoic acid is involved in first and second heart‐field development, myocardial proliferation, and coronary artery angiogenesis. Vitamin A deficiency, or toxicity, at any stage of cardiac development will lead to altered gene expression with downstream developmental consequences. 142
Taken together, epigenetic and environmental influences play a role in CHD development and may further modify a pathologic genetic substrate. This complex interplay has been elegantly reviewed elsewhere. 143 , 144 Briefly, hypermethylation of cardiac transcription factors like NKX2‐5 and HAND1 have been associated with tetralogy of Fallot, and monozygotic twins discordant for CHD have different DNA methylation at transcription factor binding sites. 145 , 146 Expression‐altering histone modifications and miRNA dysregulation are also implicated in several defects. 143 , 147 These epigenetic changes may mediate the effects of environmental risk factors for CHD, including vitamin A exposure, thalidomide, maternal diabetes mellitus, and rubella infection, among others. 144 , 148 The precise mechanisms for these effects are poorly understood and represent the complex interaction between genetics, epigenetics, and the environment. Here, we discuss how an unbiased, systematic study of the heritable component of CHD through ES, GS, and transcriptomics is critical in defining the complex genetic mechanisms underlying CHD.
Overall Yield of NS‐CHD Genes by ES Is Low
A recent multi‐institutional study of the exomes of 2871 individuals with CHD, including 2645 trios, found significant association with several of the genes mentioned in this review, namely NOTCH1, SMAD6, ELN, and GATA6. In this large cohort, recessive genotypes contributed to 0.9% of CHD cases, de novo variants (DNVs) alone contributed to 3.1% of isolated CHD and inherited and DNVs together contributed to 10.1% of CHD. 149 Notably, this study included probands with neurodevelopmental defects and extracardiac abnormalities, and it is likely that cohorts stratified based on defect type and the presence of extracardiac syndromes would reduce the heterogeneity of returned candidate genes. In the case of laterality defects, ES of 323 unrelated probands identified 28 candidate variants in known heterotaxy related genes, nearly all of which were inherited from unaffected parents, potentially the result of parental mosaicism. 150 Gene‐based aggregation analyses significantly associated PXDNL and BMS1, and in total, monogenic candidate variants were identified in 7.1% of the heterotaxy cohort. 150 In nonsyndromic‐LSOLs specifically, a series of large ES studies have made strides in identifying putative pathogenic variants and many genes are shared between cohorts (DNAH5, ACVR1, KMT2D, NOTCH1, POGZ, ROCK2, JARID2). 30 , 59 , 60 , 61 Even in these large studies, the yield of variant‐positive ES is low, ranging between 7.8% and 23.5%. 59 , 60 , 61
Common and Noncoding Genetic Variation
There is evidence that common variants in genes expressed in cardiac development such as in ERBB4, BMP4, and ISL1 may confer risk for LSOLs and might be otherwise missed in conventional searches for rare nonsynonymous variants. 151 , 152 , 153 This points toward the complex genetic architecture underlying CHD and a need to better understand how common, rare, inherited, and de novo variants interact to produce a CHD phenotype. ES analyses interrogating the differences between recessive and de novo CHD identified an enrichment of cilia‐related gene variants in recessive CHD but an enrichment in chromatin‐modifying genes in de novo CHD genotypes. 154 Similar findings were observed in an independent ES study that identified an excess of DNVs in genes involved in H3K4 and H3K27 methylation and H2BK120 ubiquitination. 155 These chromatin‐modifying variants are also observed in patients with extracardiac and neurodevelopmental delays, suggesting these DNVs may play a greater role in syndromic CHD, a finding supported by previous ES analyses suggesting a higher burden of DNVs in S‐CHD relative to NS‐CHD. 30 , 155
Genome Studies are Needed to Close the Gap in Our Understanding of NS‐CHD Heritability
Although informative in identifying coding variants, ES alone fails to capture the full genetic landscape of inherited CHD, which is reflected in its overall yield of around 10%. GS has been a powerful tool in the accurate diagnosis of pediatric disease, particularly in critically ill neonates. A 2018 study performed rapid trio‐based GS in critically ill neonates admitted to the neonatal intensive care unit had a diagnostic yield of 45% and was actionable within 2 to 5 days of sample collection. 156 Moreover, the Undiagnosed Diseases Network regularly leverages the power of ES and GS to identify a genetic cause of disease in over 40% of cases and identifies intronic and CNVs otherwise undetected by ES. 157 , 158 Given that LSOLs are associated with a spectrum of morbidity and mortality that ranges from HLHS, which is uniformly fatal if not corrected within the first few days of life, to BAV, which can remain undetected until adulthood when AS or aortic aneurysm develop and reveal the underlying congenital defect, GS could serve to significantly improve long‐term quality of life through early intervention. GS will soon be widely used for all newborns, sick and ostensibly healthy. If the genetics underlying LSOL, or CHD in general, can be fully delineated, it opens the door for identifying at‐risk infants by genetic testing.
Future Directions: Overcoming the Challenges of Genome Sequencing in LSOL
Implementation of GS is not without practical challenges, the most important of which is the need to distinguish truly pathogenic variants from background genetic noise. This is most challenging in noncoding areas of the genome that are not amenable to classic transgenic tools. Allele‐specific expression analysis may provide a methodology for identifying candidate noncoding genetic variation through a combination of RNAseq and GS to identify variants predicted to cause changes in expression, including those in noncoding regions. This technique has identified pathogenic noncoding variation in several complex genetic diseases and similar methodologies have been applied to CHD. 159 , 160 , 161 One study linked RNAseq and ES in a cohort of 144 patients with surgically repaired CHD and observed significant expression differences pointing toward new candidate genes. 162 More recent studies have extended this strategy to combine GS and transcriptome sequencing in a cohort of 13 sudden cardiac death and sudden unexplained death in infancy victims with previously negative exome studies. 163 Of the 23 candidate variants identified in cardiac gene regulatory regions, the most significant was a NEXN‐promoter variant associated with decreased NEXN expression and cardiac hypertrophy. 163 Finally, the largest GS in the CHD population sequenced 763 CHD trios with previously variant‐negative ES studies. 164 Transcriptomic profiling through effect neural network analysis and ATAC‐seq identified an enrichment in damaging DNVs in noncoding cardiac regulatory regions in CHD trios relative to controls. 164 Overall, the group estimated that noncoding DNVs contribute to between 17% and 45% of CHD cases, underscoring the need to fully appreciate the role of the noncoding genome in the etiology of CHD. 164 Further stratification based on disease phenotype is needed to understand the precise underpinnings of each defect.
CONCLUSIONS
Inheritance studies suggest that LSOLs have a significant heritable component, and though progress has been made in identifying genes and variants associated with LSOLs, each fails to explain more than a small fraction of the patient population. A change in the approach to ascertaining the genetic underpinnings of CHD is needed. GS, paired with transcriptomics and allele‐specific expression analysis, has the potential to detect variation in regulatory regions that cause haploinsufficiency. Streamlining this comprehensive, unbiased approach will pave the way for the development of better diagnostic algorithms and unlock potential therapeutic avenues.
RESOURCES
A number of genetics data repositories and resources are available to interested clinicians and researchers to aid in the study of LSOL genetics or interpretation of genetic variant pathogenicity (Table 2). 165 , 166 , 167 , 168 , 169 , 170 The Genome Aggregation Database provides information on sequence variation in the general population, whereas Online Mendelian Inheritance in Man serves as an encyclopedia of genetic disease in humans. 165 , 166 ClinVar relies on user submission of variants and phenotypes and interprets pathogenicity accordingly. 167 Similarly, ClinGen is a data sharing portal to speed identification of clinically relevant variants. 168 The Human Gene Mutation Database accomplishes a similar aim by curating a list of published pathogenic genetic lesions. 169 The Pediatric Cardiac Genomics Consortium aims to identify and characterize CHD causing variants and makes limited data available to researchers on an application basis. 170 Finally, the most recent scientific statement from the American Heart Association on the genetic basis for congenital heart disease provides a comprehensive overview of genetics underlying syndromic and nonsyndromic heart lesions. 9 Given that many of these resources rely on user submission of data, there are inevitable information gaps. Further, our understanding of the pathogenicity of noncoding variation is lacking, and the recent advances in sequencing and analysis technology will help bridge this gap.
Table 2.
Resources for Studying Congenital Heart Disease Genetics
| Resource | URL | Reference |
|---|---|---|
| ClinGen | https://clinicalgenome.org/ | 168 |
| ClinVar | https://www.ncbi.nlm.nih.gov/clinvar/ | 167 |
| Genome Aggregation Database | https://gnomad.broadinstitute.org/ | 165 |
| Human Gene Mutation Database | http://www.hgmd.cf.ac.uk/ac/index.php | 169 |
| Online Mendelian Inheritance in Man | https://www.omim.org/about | 166 |
| Pediatric Cardiac Genomics Consortium | https://benchtobassinet.com/ | 170 |
Sources of Funding
This work is supported by the Duke Children's Health & Discovery Initiative, Duke University School of Medicine, and the Duke University School of Medicine Medical Scientist Training Program.
Disclosures
None.
(J Am Heart Assoc.2021;10:e019006. DOI: 10.1161/JAHA.120.019006.)
For Sources of Funding and Disclosures, see page 12.
REFERENCES
- 1. van der Linde D, Konings EEM, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJM, Roos‐Hesselink JW. Birth prevalence of congenital heart disease worldwide: a systematic review and meta‐analysis. J Am Coll Cardiol. 2011;58:2241–2247. [DOI] [PubMed] [Google Scholar]
- 2. van der Bom T, Zomer AC, Zwinderman AH, Meijboom FJ, Bouma BJ, Mulder BJ. The changing epidemiology of congenital heart disease. Nat Rev Cardiol. 2011;8:50–60. [DOI] [PubMed] [Google Scholar]
- 3. Khairy P, Ionescu‐Ittu R, Mackie AS, Abrahamowicz M, Pilote L, Marelli AJ. Changing mortality in congenital heart disease. J Am Coll Cardiol. 2010;56:1149–1157. [DOI] [PubMed] [Google Scholar]
- 4. Mai CT, Isenburg JL, Canfield MA, Meyer RE, Correa A, Alverson CJ, Lupo PJ, Riehle‐Colarusso T, Cho SJ, Aggarwal D, et al. National population‐based estimates for major birth defects, 2010–2014. Birth Defects Res. 2019;111:1420–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marelli AJ, Ionescu‐Ittu R, Mackie AS, Guo L, Dendukuri N, Kaouache M. Lifetime prevalence of congenital heart disease in the general population from 2000 to 2010. Circulation. 2014;130:749–756. [DOI] [PubMed] [Google Scholar]
- 6. Singh GK. Congenital aortic valve stenosis. Children (Basel). 2019;6:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Movahed MR, Hepner AD, Ahmadi‐Kashani M. Echocardiographic prevalence of bicuspid aortic valve in the population. Heart Lung Circ. 2006;15:297–299. [DOI] [PubMed] [Google Scholar]
- 8. Edwards JE. Pathology of left ventricular outflow tract obstruction. Circulation. 1965;31:586–599. [DOI] [PubMed] [Google Scholar]
- 9. Pierpont ME, Brueckner M, Chung WK, Garg V, Lacro RV, McGuire AL, Mital S, Priest JR, Pu WT, Roberts A, et al. Genetic basis for congenital heart disease: revisited: a scientific statement from the American Heart Association. Circulation. 2018;138:e653–e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brodwall K, Greve G, Leirgul E, Tell GS, Vollset SE, Oyen N. Recurrence of congenital heart defects among siblings‐a nationwide study. Am J Med Genet A. 2017;173:1575–1585. [DOI] [PubMed] [Google Scholar]
- 11. Hinton RB, Martin LJ, Tabangin ME, Mazwi ML, Cripe LH, Benson DW. Hypoplastic left heart syndrome is heritable. J Am Coll Cardiol. 2007;50:1590–1595. [DOI] [PubMed] [Google Scholar]
- 12. Kerstjens‐Frederikse WS, Du Marchie Sarvaas GJ, Ruiter JS, Van Den Akker PC, Temmerman AM, Van Melle JP, Hofstra RM, Berger RM. Left ventricular outflow tract obstruction: should cardiac screening be offered to first‐degree relatives? Heart. 2011;97:1228–1232. [DOI] [PubMed] [Google Scholar]
- 13. McBride KL, Pignatelli R, Lewin M, Ho T, Fernbach S, Menesses A, Lam W, Leal SM, Kaplan N, Schliekelman P, et al. Inheritance analysis of congenital left ventricular outflow tract obstruction malformations: segregation, multiplex relative risk, and heritability. Am J Med Genet A. 2005;134a:180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lewin MB, McBride KL, Pignatelli R, Fernbach S, Combes A, Menesses A, Lam W, Bezold LI, Kaplan N, Towbin JA, et al. Echocardiographic evaluation of asymptomatic parental and sibling cardiovascular anomalies associated with congenital left ventricular outflow tract lesions. Pediatrics. 2004;114:691–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hales AR, Mahle WT. Echocardiography screening of siblings of children with bicuspid aortic valve. Pediatrics. 2014;133:e1212–e1217. [DOI] [PubMed] [Google Scholar]
- 16. Loffredo CA, Chokkalingam A, Sill AM, Boughman JA, Clark EB, Scheel J, Brenner JI. Prevalence of congenital cardiovascular malformations among relatives of infants with hypoplastic left heart, coarctation of the aorta, and d‐transposition of the great arteries. Am J Med Genet A. 2004;124a:225–230. [DOI] [PubMed] [Google Scholar]
- 17. Cripe L, Andelfinger G, Martin LJ, Shooner K, Benson DW. Bicuspid aortic valve is heritable. J Am Coll Cardiol. 2004;44:138–143. [DOI] [PubMed] [Google Scholar]
- 18. Teo LL, Cannell T, Babu‐Narayan SV, Hughes M, Mohiaddin RH. Prevalence of associated cardiovascular abnormalities in 500 patients with aortic coarctation referred for cardiovascular magnetic resonance imaging to a tertiary center. Pediatr Cardiol. 2011;32:1120–1127. [DOI] [PubMed] [Google Scholar]
- 19. Boon AR, Roberts DF. A family study of coarctation of the aorta. J Med Genet. 1976;13:420–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sanchez‐Cascos A. The recurrence risk in congenital heart disease. Eur J Cardiol. 1978;7:197–210. [PubMed] [Google Scholar]
- 21. Nora J. Multifactorial inheritance hypothesis for the etiology of congenital heart diseases: the genetic‐environmental interaction. Circulation. 1968;38:604–617. [DOI] [PubMed] [Google Scholar]
- 22. Ropers HH. New perspectives for the elucidation of genetic disorders. Am J Hum Genet. 2007;81:199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Peltonen L, Perola M, Naukkarinen J, Palotie A. Lessons from studying monogenic disease for common disease. Hum Mol Genet. 2006;15 Spec No 1:R67–R74. [DOI] [PubMed] [Google Scholar]
- 24. Theis JL, Hrstka SC, Evans JM, O'Byrne MM, de Andrade M, O'Leary PW, Nelson TJ, Olson TM. Compound heterozygous notch1 mutations underlie impaired cardiogenesis in a patient with hypoplastic left heart syndrome. Hum Genet. 2015;134:1003–1011. [DOI] [PubMed] [Google Scholar]
- 25. Tomita‐Mitchell A, Stamm KD, Mahnke DK, Kim MS, Hidestrand PM, Liang HL, Goetsch MA, Hidestrand M, Simpson P, Pelech AN, et al. Impact of myh6 variants in hypoplastic left heart syndrome. Physiol Genomics. 2016;48:912–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McBride KL, Garg V. Impact of Mendelian inheritance in cardiovascular disease. Ann N Y Acad Sci. 2010;1214:122–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mordi I, Tzemos N. Bicuspid aortic valve disease: a comprehensive review. Cardiol Res Pract. 2012;2012:196037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Giusti B, Sticchi E, De Cario R, Magi A, Nistri S, Pepe G. Genetic bases of bicuspid aortic valve: the contribution of traditional and high‐throughput sequencing approaches on research and diagnosis. Front Physiol. 2017;8:612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huntington K, Hunter AG, Chan KL. A prospective study to assess the frequency of familial clustering of congenital bicuspid aortic valve. J Am Coll Cardiol. 1997;30:1809–1812. [DOI] [PubMed] [Google Scholar]
- 30. Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Turki SH, Thienpont B, McRae J, Fitzgerald TW, Singh T, Swaminathan GJ, et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016;48:1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zaffran S, Frasch M. Early signals in cardiac development. Circ Res. 2002;91:457–469. [DOI] [PubMed] [Google Scholar]
- 32. Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994;8:707–719. [DOI] [PubMed] [Google Scholar]
- 33. Durbin MD, Cadar AG, Williams CH, Guo Y, Bichell DP, Su YR, Hong CC. Hypoplastic left heart syndrome sequencing reveals a novel NOTCH1 mutation in a family with single ventricle defects. Pediatr Cardiol. 2017;38:1232–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Helle E, Cordova‐Palomera A, Ojala T, Saha P, Potiny P, Gustafsson S, Ingelsson E, Bamshad M, Nickerson D, Chong JX, et al. Loss of function, missense, and intronic variants in notch1 confer different risks for left ventricular outflow tract obstructive heart defects in two european cohorts. Genet Epidemiol. 2019;43:215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yang C, Xu Y, Yu M, Lee D, Alharti S, Hellen N, Ahmad Shaik N, Banaganapalli B, Sheikh Ali Mohamoud H, Elango R, et al. Induced pluripotent stem cell modelling of HLHS underlines the contribution of dysfunctional NOTCH signalling to impaired cardiogenesis. Hum Mol Genet. 2017;26:3031–3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. England J, Loughna S. Heavy and light roles: myosin in the morphogenesis of the heart. Cell Mol Life Sci. 2013;70:1221–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jones WK, Grupp IL, Doetschman T, Grupp G, Osinska H, Hewett TE, Boivin G, Gulick J, Ng WA, Robbins J. Ablation of the murine alpha myosin heavy chain gene leads to dosage effects and functional deficits in the heart. J Clin Invest. 1996;98:1906–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Theis JL, Zimmermann MT, Evans JM, Eckloff BW, Wieben ED, Qureshi MY, O'Leary PW, Olson TM. Recessive MYH6 mutations in hypoplastic left heart with reduced ejection fraction. Circ Cardiovasc Genet. 2015;8:564–571. [DOI] [PubMed] [Google Scholar]
- 39. Britz‐Cunningham SH, Shah MM, Zuppan CW, Fletcher WH. Mutations of the Connexin43 gap‐junction gene in patients with heart malformations and defects of laterality. N Engl J Med. 1995;332:1323–1329. [DOI] [PubMed] [Google Scholar]
- 40. Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–1834. [DOI] [PubMed] [Google Scholar]
- 41. Dasgupta C, Martinez AM, Zuppan CW, Shah MM, Bailey LL, Fletcher WH. Identification of connexin43 (alpha1) gap junction gene mutations in patients with hypoplastic left heart syndrome by denaturing gradient gel electrophoresis (DGGE). Mutat Res. 2001;479:173–186. [DOI] [PubMed] [Google Scholar]
- 42. Bier A, Oviedo‐Landaverde I, Zhao J, Mamane Y, Kandouz M, Batist G. Connexin43 pseudogene in breast cancer cells offers a novel therapeutic target. Mol Cancer Ther. 2009;8:786–793. [DOI] [PubMed] [Google Scholar]
- 43. Hong HM, Yang JJ, Shieh JC, Lin ML, Li ML, Li SY. Novel mutations in the connexin43 (GJA1) and GJA1 pseudogene may contribute to nonsyndromic hearing loss. Hum Genet. 2010;127:545–551. [DOI] [PubMed] [Google Scholar]
- 44. Huang GY, Xie LJ, Linask KL, Zhang C, Zhao XQ, Yang Y, Zhou GM, Wu YJ, Marquez‐Rosado L, McElhinney DB, et al. Evaluating the role of connexin43 in congenital heart disease: screening for mutations in patients with outflow tract anomalies and the analysis of knock‐in mouse models. J Cardiovasc Dis Res. 2011;2:206–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang L, Nomura‐Kitabayashi A, Sultana N, Cai W, Cai X, Moon AM, Cai CL. Mesodermal Nkx2.5 is necessary and sufficient for early second heart field development. Dev Biol. 2014;390:68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lyons I, Parsons LM, Hartley L, Li R, Andrews JE, Robb L, Harvey RP. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2‐5. Genes Dev. 1995;9:1654–1666. [DOI] [PubMed] [Google Scholar]
- 47. Schott J‐J, Benson DW, Basson CT, Pease W, Silberbach GM, Moak JP, Maron BJ, Seidman CE, Seidman JG. Congenital heart disease caused by mutations in the transcription factor NKX2‐5. Science. 1998;281:108–111. [DOI] [PubMed] [Google Scholar]
- 48. Elliott DA, Kirk EP, Yeoh T, Chandar S, McKenzie F, Taylor P, Grossfeld P, Fatkin D, Jones O, Hayes P, et al. Cardiac homeobox gene NKX2‐5 mutations and congenital heart disease: associations with atrial septal defect and hypoplastic left heart syndrome. J Am Coll Cardiol. 2003;41:2072–2076. [DOI] [PubMed] [Google Scholar]
- 49. Stallmeyer B, Fenge H, Nowak‐Göttl U, Schulze‐Bahr E. Mutational spectrum in the cardiac transcription factor gene NKX2.5 (CSX) associated with congenital heart disease. Clin Genet. 2010;78:533–540. [DOI] [PubMed] [Google Scholar]
- 50. Esposito G, Butler TL, Blue GM, Cole AD, Sholler GF, Kirk EP, Grossfeld P, Perryman BM, Harvey RP, Winlaw DS. Somatic mutations in NKX2‐5, GATA4, and HAND1 are not a common cause of tetralogy of Fallot or hypoplastic left heart. Am J Med Genet A. 2011;155a:2416–2421. [DOI] [PubMed] [Google Scholar]
- 51. McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E. Nkx2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol. 2003;42:1650–1655. [DOI] [PubMed] [Google Scholar]
- 52. Kobayashi J, Yoshida M, Tarui S, Hirata M, Nagai Y, Kasahara S, Naruse K, Ito H, Sano S, Oh H. Directed differentiation of patient‐specific induced pluripotent stem cells identifies the transcriptional repression and epigenetic modification of NKX2‐5, HAND1, and NOTCH1 in hypoplastic left heart syndrome. PLoS One. 2014;9:e102796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Riley P, Anson‐Cartwright L, Cross JC. The Hand1 bHLH transcription factor is essential for placentation and cardiac morphogenesis. Nat Genet. 1998;18:271–275. [DOI] [PubMed] [Google Scholar]
- 54. Risebro CA, Smart N, Dupays L, Breckenridge R, Mohun TJ, Riley PR. Hand1 regulates cardiomyocyte proliferation versus differentiation in the developing heart. Development. 2006;133:4595–4606. [DOI] [PubMed] [Google Scholar]
- 55. Firulli AB, McFadden DG, Lin Q, Srivastava D, Olson EN. Heart and extra‐embryonic mesodermal defects in mouse embryos lacking the bHLH transcription factor HAND1. Nat Genet. 1998;18:266–270. [DOI] [PubMed] [Google Scholar]
- 56. Reamon‐Buettner SM, Ciribilli Y, Inga A, Borlak J. A loss‐of‐function mutation in the binding domain of HAND1 predicts hypoplasia of the human hearts. Hum Mol Genet. 2008;17:1397–1405. [DOI] [PubMed] [Google Scholar]
- 57. Williams C, Pontén F, Moberg C, Söderkvist P, Uhlén M, Pontén J, Sitbon G, Lundeberg J. A high frequency of sequence alterations is due to formalin fixation of archival specimens. Am J Pathol. 1999;155:1467–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Do H, Dobrovic A. Sequence artifacts in DNA from formalin‐fixed tissues: causes and strategies for minimization. Clin Chem. 2015;61:64–71. [DOI] [PubMed] [Google Scholar]
- 59. Sevim Bayrak C, Zhang P, Tristani‐Firouzi M, Gelb BD, Itan Y. De novo variants in exomes of congenital heart disease patients identify risk genes and pathways. Genome Med. 2020;12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Preuss C, Capredon M, Wunnemann F, Chetaille P, Prince A, Godard B, Leclerc S, Sobreira N, Ling H, Awadalla P, et al. Family based whole exome sequencing reveals the multifaceted role of notch signaling in congenital heart disease. PLoS Genet. 2016;12:e1006335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Li AH, Hanchard NA, Furthner D, Fernbach S, Azamian M, Nicosia A, Rosenfeld J, Muzny D, D'Alessandro LCA, Morris S, et al. Whole exome sequencing in 342 congenital cardiac left sided lesion cases reveals extensive genetic heterogeneity and complex inheritance patterns. Genome Med. 2017;9:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pierquin G, Vliers A, Dodinval P. [Supravalvular aortic stenosis. Autosomal dominant form of congenital cardiopathy]. J Genet Hum. 1988;36:485–489. [PubMed] [Google Scholar]
- 63. Olson TM, Michels VV, Urban Z, Csiszar K, Christiano AM, Driscoll DJ, Feldt RH, Boyd CD, Thibodeau SN. A 30 kb deletion within the elastin gene results in familial supravalvular aortic stenosis. Hum Mol Genet. 1995;4:1677–1679. [DOI] [PubMed] [Google Scholar]
- 64. Olson TM, Michels VV, Lindor NM, Pastores GM, Weber JL, Schaid DJ, Driscoll DJ, Feldt RH, Thibodeau SN. Autosomal dominant supravalvular aortic stenosis: localization to chromosome 7. Hum Mol Genet. 1993;2:869–873. [DOI] [PubMed] [Google Scholar]
- 65. Curran ME, Atkinson DL, Ewart AK, Morris CA, Leppert MF, Keating MT. The elastin gene is disrupted by a translocation associated with supravalvular aortic stenosis. Cell. 1993;73:159–168. [DOI] [PubMed] [Google Scholar]
- 66. Tassabehji M, Metcalfe K, Donnai D, Hurst J, Reardon W, Burch M, Read AP. Elastin: genomic structure and point mutations in patients with supravalvular aortic stenosis. Hum Mol Genet. 1997;6:1029–1036. [DOI] [PubMed] [Google Scholar]
- 67. Ewart AK, Morris CA, Ensing GJ, Loker J, Moore C, Leppert M, Keating M. A human vascular disorder, supravalvular aortic stenosis, maps to chromosome 7. Proc Natl Acad Sci USA. 1993;90:3226–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li DY, Toland AE, Boak BB, Atkinson DL, Ensing GJ, Morris CA, Keating MT. Elastin point mutations cause an obstructive vascular disease, supravalvular aortic stenosis. Hum Mol Genet. 1997;6:1021–1028. [DOI] [PubMed] [Google Scholar]
- 69. Jelsig AM, Urban Z, Hucthagowder V, Nissen H, Ousager LB. Novel eln mutation in a family with supravalvular aortic stenosis and intracranial aneurysm. Eur J Med Genet. 2017;60:110–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sugiyama K, Horigome H, Lin L, Murakami T, Shiono J, Yamashiro Y, Matsuura H, Yoda H, Yanagisawa H. Novel ELN mutation in a japanese family with a severe form of supravalvular aortic stenosis. Mol Genet Genomic Med. 2019;7:e986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Park S, Seo EJ, Yoo HW, Kim Y. Novel mutations in the human elastin gene (ELN) causing isolated supravalvular aortic stenosis. Int J Mol Med. 2006;18:329–332. [PubMed] [Google Scholar]
- 72. Urbán Z, Michels VV, Thibodeau SN, Donis‐Keller H, Csiszár K, Boyd CD. Supravalvular aortic stenosis: a splice site mutation within the elastin gene results in reduced expression of two aberrantly spliced transcripts. Hum Genet. 1999;104:135–142. [DOI] [PubMed] [Google Scholar]
- 73. Hayano S, Okuno Y, Tsutsumi M, Inagaki H, Fukasawa Y, Kurahashi H, Kojima S, Takahashi Y, Kato T. Frequent intragenic microdeletions of elastin in familial supravalvular aortic stenosis. Int J Cardiol. 2019;274:290–295. [DOI] [PubMed] [Google Scholar]
- 74. Metcalfe K, Rucka AK, Smoot L, Hofstadler G, Tuzler G, McKeown P, Siu V, Rauch A, Dean J, Dennis N, et al. Elastin: mutational spectrum in supravalvular aortic stenosis. Eur J Hum Genet. 2000;8:955–963. [DOI] [PubMed] [Google Scholar]
- 75. Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998;393:276–280. [DOI] [PubMed] [Google Scholar]
- 76. Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, Stenzel P, Boak B, Keating MT. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Invest. 1998;102:1783–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Abdallah H, Toomey K, O'Riordan AC, Davidson A, Marks LA. Familial occurrence of discrete subaortic membrane. Pediatr Cardiol. 1994;15:198–200. [DOI] [PubMed] [Google Scholar]
- 78. Petsas AA, Anastassiades LC, Constantinou EC, Antonopoulos AG. Familial discrete subaortic stenosis. Clin Cardiol. 1998;21:63–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fatimi SH, Ahmad U, Javed MA, Shamim S, Ahmad R. Familial membranous subaortic stenosis: review of familial inheritance patterns and a case report. J Thorac Cardiovasc Surg. 2006;132:1484–1486. [DOI] [PubMed] [Google Scholar]
- 80. Piacentini G, Marino B, Digilio MC. Familial recurrence of discrete membranous subaortic stenosis. J Thorac Cardiovasc Surg. 2007;134:818–819; author reply 819. [DOI] [PubMed] [Google Scholar]
- 81. Pyle RL, Patterson DF, Chacko S. The genetics and pathology of discrete subaortic stenosis in the newfoundland dog. Am Heart J. 1976;92:324–334. [DOI] [PubMed] [Google Scholar]
- 82. Gould RA, Aziz H, Woods CE, Seman‐Senderos MA, Sparks E, Preuss C, Wünnemann F, Bedja D, Moats CR, McClymont SA, et al. ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nat Genet. 2019;51:42–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Laforest B, Andelfinger G, Nemer M. Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin Invest. 2011;121:2876–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bonachea EM, Chang SW, Zender G, LaHaye S, Fitzgerald‐Butt S, McBride KL, Garg V. Rare GATA5 sequence variants identified in individuals with bicuspid aortic valve. Pediatr Res. 2014;76:211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Padang R, Bagnall RD, Richmond DR, Bannon PG, Semsarian C. Rare non‐synonymous variations in the transcriptional activation domains of GATA5 in bicuspid aortic valve disease. J Mol Cell Cardiol. 2012;53:277–281. [DOI] [PubMed] [Google Scholar]
- 86. Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu L, Liu H, Li RG, Xu YJ, Wang Q, et al. GATA5 loss‐of‐function mutations associated with congenital bicuspid aortic valve. Int J Mol Med. 2014;33:1219–1226. [DOI] [PubMed] [Google Scholar]
- 87. Yang B, Zhou W, Jiao J, Nielsen JB, Mathis MR, Heydarpour M, Lettre G, Folkersen L, Prakash S, Schurmann C, et al. Protein‐altering and regulatory genetic variants near GATA4 implicated in bicuspid aortic valve. Nat Commun. 2017;8:15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Li RG, Xu YJ, Wang J, Liu XY, Yuan F, Huang RT, Xue S, Li L, Liu H, Li YJ, et al. Gata4 loss‐of‐function mutation and the congenitally bicuspid aortic valve. Am J Cardiol. 2018;121:469–474. [DOI] [PubMed] [Google Scholar]
- 89. Xin M, Davis CA, Molkentin JD, Lien CL, Duncan SA, Richardson JA, Olson EN. A threshold of GATA4 and GATA6 expression is required for cardiovascular development. Proc Natl Acad Sci USA. 2006;103:11189–11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xu YJ, Di RM, Qiao Q, Li XM, Huang RT, Xue S, Liu XY, Wang J, Yang YQ. GATA6 loss‐of‐function mutation contributes to congenital bicuspid aortic valve. Gene. 2018;663:115–120. [DOI] [PubMed] [Google Scholar]
- 91. Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. [DOI] [PubMed] [Google Scholar]
- 92. Kostina AS, Uspensky V, Irtyuga OB, Ignatieva EV, Freylikhman O, Gavriliuk ND, Moiseeva OM, Zhuk S, Tomilin A, Kostareva A, et al. Notch‐dependent EMT is attenuated in patients with aortic aneurysm and bicuspid aortic valve. Biochim Biophys Acta. 2016;1862:733–740. [DOI] [PubMed] [Google Scholar]
- 93. Mohamed SA, Aherrahrou Z, Liptau H, Erasmi AW, Hagemann C, Wrobel S, Borzym K, Schunkert H, Sievers HH, Erdmann J. Novel missense mutations (p. T596m and p.P1797h) in NOTCH1 in patients with bicuspid aortic valve. Biochem Biophys Res Commun. 2006;345:1460–1465. [DOI] [PubMed] [Google Scholar]
- 94. McKellar SH, Tester DJ, Yagubyan M, Majumdar R, Ackerman MJ, Sundt TM. Novel NOTCH1 mutations in patients with bicuspid aortic valve disease and thoracic aortic aneurysms. J Thorac Cardiovasc Surg. 2007;134:290–296. [DOI] [PubMed] [Google Scholar]
- 95. Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, Fairchild‐Huntress V, Dixon KL, Dunmore JH, Gimbrone MA, et al. A role for smad6 in development and homeostasis of the cardiovascular system. Nat Genet. 2000;24:171–174. [DOI] [PubMed] [Google Scholar]
- 96. Tan HL, Glen E, Töpf A, Hall D, O'Sullivan JJ, Sneddon L, Wren C, Avery P, Lewis RJ, ten Dijke P, et al. Nonsynonymous variants in the smad6 gene predispose to congenital cardiovascular malformation. Hum Mutat. 2012;33:720–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gillis E, Kumar AA, Luyckx I, Preuss C, Cannaerts E, van de Beek G, Wieschendorf B, Alaerts M, Bolar N, Vandeweyer G, et al. Candidate gene resequencing in a large bicuspid aortic valve‐associated thoracic aortic aneurysm cohort: Front Physiol. 2017;8:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Luyckx I, MacCarrick G, Kempers M, Meester J, Geryl C, Rombouts O, Peeters N, Claes C, Boeckx N, Sakalihasan N, et al. Confirmation of the role of pathogenic smad6 variants in bicuspid aortic valve‐related aortopathy. Eur J Hum Genet. 2019;27:1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Biben C, Weber R, Kesteven S, Stanley E, McDonald L, Elliott DA, Barnett L, Köentgen F, Robb L, Feneley M, et al. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2‐5. Circ Res. 2000;87:888–895. [DOI] [PubMed] [Google Scholar]
- 100. Qu XK, Qiu XB, Yuan F, Wang J, Zhao CM, Liu XY, Zhang XL, Li RG, Xu YJ, Hou XM, et al. A novel NKX2.5 loss‐of‐function mutation associated with congenital bicuspid aortic valve. Am J Cardiol. 2014;114:1891–1895. [DOI] [PubMed] [Google Scholar]
- 101. Majumdar R, Yagubyan M, Sarkar G, Bolander ME, Sundt TM. Bicuspid aortic valve and ascending aortic aneurysm are not associated with germline or somatic homeobox NKX2‐5 gene polymorphism in 19 patients. J Thorac Cardiovasc Surg. 2006;131:1301–1305. [DOI] [PubMed] [Google Scholar]
- 102. Guo DC, Gong L, Regalado ES, Santos‐Cortez RL, Zhao R, Cai B, Veeraraghavan S, Prakash SK, Johnson RJ, Muilenburg A, et al. MAT2A mutations predispose individuals to thoracic aortic aneurysms. Am J Hum Genet. 2015;96:170–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wang J, Abhinav P, Xu YJ, Li RG, Zhang M, Qiu XB, Di RM, Qiao Q, Li XM, Huang RT, et al. NR2F2 loss‐of‐function mutation is responsible for congenital bicuspid aortic valve. Int J Mol Med. 2019;43:1839–1846. [DOI] [PubMed] [Google Scholar]
- 104. Bonachea EM, Zender G, White P, Corsmeier D, Newsom D, Fitzgerald‐Butt S, Garg V, McBride KL. Use of a targeted, combinatorial next‐generation sequencing approach for the study of bicuspid aortic valve. BMC Med Genomics. 2014;7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Lewin MB, Lindsay EA, Jurecic V, Goytia V, Towbin JA, Baldini A. A genetic etiology for interruption of the aortic arch type B. Am J Cardiol. 1997;80:493–497. [DOI] [PubMed] [Google Scholar]
- 106. Marino B, Digilio MC, Toscano A, Anaclerio S, Giannotti A, Feltri C, de Ioris MA, Angioni A, Dallapiccola B. Anatomic patterns of conotruncal defects associated with deletion 22q11. Genet Med. 2001;3:45–48. [DOI] [PubMed] [Google Scholar]
- 107. Yagi H, Furutani Y, Hamada H, Sasaki T, Asakawa S, Minoshima S, Ichida F, Joo K, Kimura M, Imamura S, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet. 2003;362:1366–1373. [DOI] [PubMed] [Google Scholar]
- 108. Conti E, Grifone N, Sarkozy A, Tandoi C, Marino B, Digilio MC, Mingarelli R, Pizzuti A, Dallapiccola B. Digeorge subtypes of nonsyndromic conotruncal defects: evidence against a major role of TBX1 gene. Eur J Hum Genet. 2003;11:349‐351. [DOI] [PubMed] [Google Scholar]
- 109. Gong W, Gottlieb S, Collins J, Blescia A, Dietz H, Goldmuntz E, McDonald‐McGinn DM, Zackai EH, Emanuel BS, Driscoll DA, et al. Mutation analysis of TBX1 in non‐deleted patients with features of DGS/VCFS or isolated cardiovascular defects. J Med Genet. 2001;38:E45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Lindsay EA, Vitelli F, Su H, Morishima M, Huynh T, Pramparo T, Jurecic V, Ogunrinu G, Sutherland HF, Scambler PJ, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. [DOI] [PubMed] [Google Scholar]
- 111. Merscher S, Funke B, Epstein JA, Heyer J, Puech A, Lu MM, Xavier RJ, Demay MB, Russell RG, Factor S, et al. TBX1 is responsible for cardiovascular defects in velo‐cardio‐facial/DiGeorge syndrome. Cell. 2001;104:619–629. [DOI] [PubMed] [Google Scholar]
- 112. Guris DL, Duester G, Papaioannou VE, Imamoto A. Dose‐dependent interaction of Tbx1 and Crkl and locally aberrant RA signaling in a model of del22q11 syndrome. Dev Cell. 2006;10:81–92. [DOI] [PubMed] [Google Scholar]
- 113. Racedo SE, McDonald‐McGinn DM, Chung JH, Goldmuntz E, Zackai E, Emanuel BS, Zhou B, Funke B, Morrow BE. Mouse and human CRKL is dosage sensitive for cardiac outflow tract formation. Am J Hum Genet. 2015;96:235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Liu C, Liu W, Palie J, Lu MF, Brown NA, Martin JF. Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development. 2002;129:5081–5091. [DOI] [PubMed] [Google Scholar]
- 115. Uddin MKM, Kimura W, Amin MB, Nakamura K, Islam MJ, Yamagishi H, Miura N. The loss of Foxc2 expression in the outflow tract links the interrupted arch in the conditional Foxc2 knockout mouse. In: Nakanishi T, Markwald RR, Baldwin HS, Keller BB, Srivastava D, Yamagishi H, eds. Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology. Tokyo: Springer; 2016:211–213. Copyright 2016, The Author(s). [Google Scholar]
- 116. Bjornsson T, Thorolfsdottir RB, Sveinbjornsson G, Sulem P, Norddahl GL, Helgadottir A, Gretarsdottir S, Magnusdottir A, Danielsen R, Sigurdsson EL, et al. A rare missense mutation in myh6 associates with non‐syndromic coarctation of the aorta. Eur Heart J. 2018;39:3243–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Peterson RT, Shaw SY, Peterson TA, Milan DJ, Zhong TP, Schreiber SL, MacRae CA, Fishman MC. Chemical suppression of a genetic mutation in a zebrafish model of aortic coarctation. Nat Biotechnol. 2004;22:595–599. [DOI] [PubMed] [Google Scholar]
- 118. Gessler M, Knobeloch KP, Helisch A, Amann K, Schumacher N, Rohde E, Fischer A, Leimeister C. Mouse gridlock: no aortic coarctation or deficiency, but fatal cardiac defects in Hey2 ‐/‐ mice. Curr Biol. 2002;12:1601–1604. [DOI] [PubMed] [Google Scholar]
- 119. Kerstjens‐Frederikse WS, van de Laar IM, Vos YJ, Verhagen JM, Berger RM, Lichtenbelt KD, Klein Wassink‐Ruiter JS, van der Zwaag PA, du Marchie Sarvaas GJ, Bergman KA, et al. Cardiovascular malformations caused by NOTCH1 mutations do not keep left: data on 428 probands with left‐sided CHD and their families. Genet Med. 2016;18:914–923. [DOI] [PubMed] [Google Scholar]
- 120. Moosmann J, Uebe S, Dittrich S, Rüffer A, Ekici AB, Toka O. Novel loci for non‐syndromic coarctation of the aorta in sporadic and familial cases. PLoS One. 2015;10:e0126873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sanchez‐Castro M, Eldjouzi H, Charpentier E, Busson PF, Hauet Q, Lindenbaum P, Delasalle‐Guyomarch B, Baudry A, Pichon O, Pascal C, et al. Search for rare copy‐number variants in congenital heart defects identifies novel candidate genes and a potential role for FOXC1 in patients with coarctation of the aorta. Circ Cardiovasc Genet. 2016;9:86–94. [DOI] [PubMed] [Google Scholar]
- 122. Quintero‐Rivera F, Xi QJ, Keppler‐Noreuil KM, Lee JH, Higgins AW, Anchan RM, Roberts AE, Seong IS, Fan X, Lage K, et al. MATR3 disruption in human and mouse associated with bicuspid aortic valve, aortic coarctation and patent ductus arteriosus. Hum Mol Genet. 2015;24:2375–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Shin OH, Han W, Wang Y, Südhof TC. Evolutionarily conserved multiple C2 domain proteins with two transmembrane regions (MCTPs) and unusual Ca2+ binding properties. J Biol Chem. 2005;280:1641–1651. [DOI] [PubMed] [Google Scholar]
- 124. Lodi FR, Palma LF, de Victo NC, Alonso LG, de Moraes LOC. Expression of connexin‐43 in the cardiac muscle of children diagnosed with hypoplastic left heart syndrome: a Western blot and confocal laser scanning microscopy study. Cardiol Young. 2020;30:238–242. [DOI] [PubMed] [Google Scholar]
- 125. Reamon‐Buettner SM, Ciribilli Y, Traverso I, Kuhls B, Inga A, Borlak J. A functional genetic study identifies hand1 mutations in septation defects of the human heart. Hum Mol Genet. 2009;18:3567–3578. [DOI] [PubMed] [Google Scholar]
- 126. Vincentz JW, Barnes RM, Firulli AB. Hand factors as regulators of cardiac morphogenesis and implications for congenital heart defects. Birth Defects Res A Clin Mol Teratol. 2011;91:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Firulli BA, Toolan KP, Harkin J, Millar H, Pineda S, Firulli AB. The HAND1 frameshift A126FS mutation does not cause hypoplastic left heart syndrome in mice. Cardiovasc Res. 2017;113:1732–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Lalani SR, Ware SM, Wang X, Zapata G, Tian Q, Franco LM, Jiang Z, Bucasas K, Scott DA, Campeau PM, et al. MCTP2 is a dosage‐sensitive gene required for cardiac outflow tract development. Hum Mol Genet. 2013;22:4339–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Stallmeyer B, Fenge H, Nowak‐Gottl U, Schulze‐Bahr E. Mutational spectrum in the cardiac transcription factor gene NKX2.5 (CSX) associated with congenital heart disease. Clin Genet. 2010;78:533–540. [DOI] [PubMed] [Google Scholar]
- 130. Iascone M, Ciccone R, Galletti L, Marchetti D, Seddio F, Lincesso AR, Pezzoli L, Vetro A, Barachetti D, Boni L, et al. Identification of de novo mutations and rare variants in hypoplastic left heart syndrome. Clin Genet. 2012;81:542–554. [DOI] [PubMed] [Google Scholar]
- 131. McBride KL, Riley MF, Zender GA, Fitzgerald‐Butt SM, Towbin JA, Belmont JW, Cole SE. NOTCH1 mutations in individuals with left ventricular outflow tract malformations reduce ligand‐induced signaling. Hum Mol Genet. 2008;17:2886–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Ware SM, Peng J, Zhu L, Fernbach S, Colicos S, Casey B, Towbin J, Belmont JW. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am J Hum Genet. 2004;74:93–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kumar A, Olson TM, Thibodeau SN, Michels VV, Schaid DJ, Wallace MR. Confirmation of linkage of supravalvular aortic stenosis to the elastin gene on chromosome 7q. Am J Cardiol. 1994;74:1281–1283. [DOI] [PubMed] [Google Scholar]
- 134. Gharibeh L, Komati H, Bossé Y, Boodhwani M, Heydarpour M, Fortier M, Hassanzadeh R, Ngu J, Mathieu P, Body S, et al. GATA6 regulates aortic valve remodeling, and its haploinsufficiency leads to right‐left type bicuspid aortic valve. Circulation. 2018;138:1025–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Garg V. Notch signaling in aortic valve development and disease. In: Nakanishi T, Markwald RR, Baldwin HS, Keller BB, Srivastava D, Yamagishi H, eds. Etiology and Morphogenesis of Congenital Heart Disease: From Gene Function and Cellular Interaction to Morphology. Tokyo: Springer; 2016:371–376. Copyright 2016, The Author(s). [PubMed] [Google Scholar]
- 136. Weiss K, Applegate C, Wang T, Batista DA. Familial TAB2 microdeletion and congenital heart defects including unusual valve dysplasia and tetralogy of Fallot. Am J Med Genet A. 2015;167A:2702–2706. [DOI] [PubMed] [Google Scholar]
- 137. Thienpont B, Zhang L, Postma AV, Breckpot J, Tranchevent LC, Van Loo P, Møllgård K, Tommerup N, Bache I, Tümer Z, et al. Haploinsufficiency of TAB2 causes congenital heart defects in humans. Am J Hum Genet. 2010;86:839–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Goldmuntz E, Bamford R, Karkera JD, dela Cruz J, Roessler E, Muenke M. CFC1 mutations in patients with transposition of the great arteries and double‐outlet right ventricle. Am J Hum Genet. 2002;70:776–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Kosaki K, Bassi MT, Kosaki R, Lewin M, Belmont J, Schauer G, Casey B. Characterization and mutation analysis of human LEFTY A and LEFTY B, homologues of murine genes implicated in left‐right axis development. Am J Hum Genet. 1999;64:712–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tagariello A, Breuer C, Birkner Y, Schmidt S, Koch AM, Cesnjevar R, Ruffer A, Dittrich S, Schneider H, Winterpacht A, et al. Functional null mutations in the gonosomal homologue gene TBL1Y are associated with non‐syndromic coarctation of the aorta. Curr Mol Med. 2012;12:199–205. [DOI] [PubMed] [Google Scholar]
- 141. Grossfeld P, Nie S, Lin L, Wang L, Anderson RH. Hypoplastic left heart syndrome: a new paradigm for an old disease? J Cardiovasc Dev Dis. 2019;6:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Nakajima Y. Retinoic acid signaling in heart development. Genesis. 2019;57:e23300. [DOI] [PubMed] [Google Scholar]
- 143. Jarrell DK, Lennon ML, Jacot JG. Epigenetics and mechanobiology in heart development and congenital heart disease. Diseases. 2019;7:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Kalisch‐Smith JI, Ved N, Sparrow DB. Environmental risk factors for congenital heart disease. Cold Spring Harb Perspect Biol. 2020;12:a037234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Sheng W, Qian Y, Wang H, Ma X, Zhang P, Diao L, An Q, Chen L, Ma D, Huang G. DNA methylation status of NKX2‐5, GATA4 and HAND1 in patients with tetralogy of Fallot. BMC Med Genomics. 2013;6:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Lyu G, Zhang C, Ling T, Liu R, Zong L, Guan Y, Huang X, Sun L, Zhang L, Li C, et al. Genome and epigenome analysis of monozygotic twins discordant for congenital heart disease. BMC Genom. 2018;19:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Blakeslee WW, Demos‐Davies KM, Lemon DD, Lutter KM, Cavasin MA, Payne S, Nunley K, Long CS, McKinsey TA, Miyamoto SD. Histone deacetylase adaptation in single ventricle heart disease and a young animal model of right ventricular hypertrophy. Pediatr Res. 2017;82:642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Moreau JLM, Kesteven S, Martin E, Lau KS, Yam MX, O'Reilly VC, Del Monte‐Nieto G, Baldini A, Feneley MP, Moon AM, et al. Gene‐environment interaction impacts on heart development and embryo survival. Development. 2019;146:dev172957. [DOI] [PubMed] [Google Scholar]
- 149. Jin SC, Homsy J, Zaidi S, Lu Q, Morton S, DePalma SR, Zeng X, Qi H, Chang W, Sierant MC, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017;49:1593–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Li AH, Hanchard NA, Azamian M, D'Alessandro LCA, Coban‐Akdemir Z, Lopez KN, Hall NJ, Dickerson H, Nicosia A, Fernbach S, et al. Genetic architecture of laterality defects revealed by whole exome sequencing. Eur J Hum Genet. 2019;27:563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. McBride KL, Zender GA, Fitzgerald‐Butt SM, Seagraves NJ, Fernbach SD, Zapata G, Lewin M, Towbin JA, Belmont JW. Association of common variants in ERBB4 with congenital left ventricular outflow tract obstruction defects. Birth Defects Res A Clin Mol Teratol. 2011;91:162–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Stevens KN, Hakonarson H, Kim CE, Doevendans PA, Koeleman BP, Mital S, Raue J, Glessner JT, Coles JG, Moreno V, et al. Common variation in isl1 confers genetic susceptibility for human congenital heart disease. PLoS One. 2010;5:e10855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Qian B, Mo R, Da M, Peng W, Hu Y, Mo X. Common variations in BMP4 confer genetic susceptibility to sporadic congenital heart disease in a han chinese population. Pediatr Cardiol. 2014;35:1442–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Watkins WS, Hernandez EJ, Wesolowski S, Bisgrove BW, Sunderland RT, Lin E, Lemmon G, Demarest BL, Miller TA, Bernstein D, et al. De novo and recessive forms of congenital heart disease have distinct genetic and phenotypic landscapes. Nat Commun. 2019;10:4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano‐Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. De novo mutations in histone‐modifying genes in congenital heart disease. Nature. 2013;498:220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Farnaes L, Hildreth A, Sweeney NM, Clark MM, Chowdhury S, Nahas S, Cakici JA, Benson W, Kaplan RH, Kronick R, et al. Rapid whole‐genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med. 2018;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Gilissen C, Hehir‐Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, Kwint M, Janssen IM, Hoischen A, Schenck A, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–347. [DOI] [PubMed] [Google Scholar]
- 158. Shashi V, Schoch K, Spillmann R, Cope H, Tan QK, Walley N, Pena L, McConkie‐Rosell A, Jiang YH, Stong N, et al. A comprehensive iterative approach is highly effective in diagnosing individuals who are exome negative. Genet Med. 2019;21:161–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Jung H, Lee D, Lee J, Park D, Kim YJ, Park WY, Hong D, Park PJ, Lee E. Intron retention is a widespread mechanism of tumor‐suppressor inactivation. Nat Genet. 2015;47:1242–1248. [DOI] [PubMed] [Google Scholar]
- 160. Li Y, Grupe A, Rowland C, Nowotny P, Kauwe JS, Smemo S, Hinrichs A, Tacey K, Toombs TA, Kwok S, et al. DAPK1 variants are associated with Alzheimer's disease and allele‐specific expression. Hum Mol Genet. 2006;15:2560–2568. [DOI] [PubMed] [Google Scholar]
- 161. de la Chapelle A. Genetic predisposition to human disease: allele‐specific expression and low‐penetrance regulatory loci. Oncogene. 2009;28:3345–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. McKean DM, Homsy J, Wakimoto H, Patel N, Gorham J, DePalma SR, Ware JS, Zaidi S, Ma W, Lifton RP, et al. Loss of RNA expression and allele‐specific expression associated with congenital heart disease. Nat Commun. 2016;7:12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Andersen JD, Jacobsen SB, Trudsø LC, Kampmann ML, Banner J, Morling N. Whole genome and transcriptome sequencing of post‐mortem cardiac tissues from sudden cardiac death victims identifies a gene regulatory variant in NEXN. Int J Legal Med. 2019;133:1699–1709. [DOI] [PubMed] [Google Scholar]
- 164. Richter F, Morton SU, Kim SW, Kitaygorodsky A, Wasson LK, Chen KM, Zhou J, Qi H, Patel N, DePalma SR, et al. Genomic analyses implicate noncoding de novo variants in congenital heart disease. Nat Genet. 2020;52:769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Omim HA. OMIM.Org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–D798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, et al. ClinGen—the clinical genome resource. N Engl J Med. 2015;372:2235–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, et al. The Human Gene Mutation Database (HGMD(®)): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139:1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Gelb B, Brueckner M, Chung W, Goldmuntz E, Kaltman J, Kaski JP, Kim R, Kline J, Mercer‐Rosa L, Porter G, et al. The congenital heart disease genetic network study: rationale, design, and early results. Circ Res. 2013;112:698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]