

Abstract
Background
Recent clinical trials have demonstrated the possible pleiotropic effects of SGLT2 (sodium–glucose cotransporter 2) inhibitors in clinical cardiovascular diseases. Atrial electrical and structural remodeling is important as an atrial fibrillation (AF) substrate.
Methods and Results
The present study assessed the effect of canagliflozin (CAN), an SGLT2 inhibitor, on atrial remodeling in a canine AF model. The study included 12 beagle dogs, with 10 receiving continuous rapid atrial pacing and 2 acting as the nonpacing group. The 10 dogs that received continuous rapid atrial pacing for 3 weeks were subdivided as follows: pacing control group (n=5) and pacing+CAN (3 mg/kg per day) group (n=5). The atrial effective refractory period, conduction velocity, and AF inducibility were evaluated weekly through atrial epicardial wires. After the protocol, atrial tissues were sampled for histological examination. The degree of reactive oxygen species expression was evaluated by dihydroethidium staining. The atrial effective refractory period reduction was smaller (P=0.06) and the degree of conduction velocity decrease was smaller in the pacing+CAN group compared with the pacing control group (P=0.009). The AF inducibility gradually increased in the pacing control group, but such an increase was suppressed in the pacing+CAN group (P=0.011). The pacing control group exhibited interstitial fibrosis and enhanced oxidative stress, which were suppressed in the pacing+CAN group.
Conclusions
CAN and possibly other SGLT2 inhibitors might be useful for preventing AF and suppressing the promotion of atrial remodeling as an AF substrate.
Keywords: atrial fibrillation, atrial remodeling, canagliflozin, oxidative stress, sodium–glucose cotransporter 2 inhibitor
Subject Categories: Animal Models of Human Disease, Atrial Fibrillation, Electrophysiology
Nonstandard Abbreviations and Acronyms
- AERP
atrial effective refractory period
- CV
conduction velocity
- DHE
dihydroethidium
- LA
left atrial
- NII
nuclear immunofluorescent intensity
- ROS
reactive oxygen species
- β‐HA
β‐hydroxybutyric acid
Clinical Perspective
What Is New?
This study provides the first experimental documentation of the suppressive effects of canagliflozin on atrial remodeling through the suppression of oxidative stress and fibrosis in a canine atrial fibrillation model.
This antiarrhythmic action may constitute a pleiotropic effect of SGLT2 (sodium–glucose cotransporter 2) inhibitors, although the precise mechanisms remain unclear.
What Are the Clinical Implications?
Other SGLT2 inhibitors may have similar clinical effects on antiatrial remodeling as a result of the same pharmacological class effect.
This research may lead to the development of therapeutic strategies aimed at preventing atrial fibrillation in the general population.
Atrial fibrillation (AF) is a common tachyarrhythmia known to be associated with structural and electrical remodeling of the atria under various pathological conditions. 1 , 2 Several basic studies have demonstrated that inflammatory processes and myocardial hyperoxidative stress might play important roles, especially in the early phase of the process of construction of an arrhythmogenic AF substrate, which is often associated with the promotion of electrical and structural atrial remodeling. 1 , 2 , 3 We previously demonstrated the role of oxidative stress in such a process using a canine AF model. 4 , 5 , 6
Canagliflozin (CAN) is one of the SGLT2 (sodium–glucose cotransporter 2) inhibitors originally designed as clinical antidiabetic medicines. 7 SGLT2 inhibitors reduce the blood sugar level by increasing the amount of sugar loss in the urine through action on the urinary tubules. However, recent studies, such as the EMPA‐REG OUTCOME (Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients–Removing Excess Glucose) trial and DECLARE TIMI 58 (Dapagliflozin Effect on Cardiovascular Events–Thrombolysis in Myocardial Infarction 58) trial, have reported the occurrence of reductions in cardiovascular diseases in populations treated with empagliflozin and dapagliflozin, suggesting the cardiovascular‐protective effect of SGLT2 inhibitors. 8 , 9 Furthermore, the DAPA‐HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) clinical trial indicated a reduction in the risk of worsening heart failure or death from cardiovascular causes was realized among patients who received dapagliflozin, regardless of the presence or absence of diabetes mellitus. 10
As based on the anticardiovascular effects of SGLT2 inhibitors, we hypothesized that CAN would suppress the promotion of an AF substrate. The present study sought to evaluate the suppressive effects of CAN on the development of atrial electrical remodeling, structural remodeling, and oxidative stress states in a canine AF model.
Methods
This study has been approved by the Ethical Review Board of Kitasato University School of Medicine. The authors declare that all supporting data are available within the article.
Initial Surgery
The canine AF model was set up as previously reported. 4 , 5 , 11 Twelve adult female beagle dogs (body weight 9.6±0.6 kg) were anesthetized with dormicum (midazolam 0.2 mg/kg, intravenous) and propofol (propofol 6 mg/kg, intravenous), then given isoflurane (isoflurane 2.5%, inhalation) during the surgery. Two pairs of electrodes were sutured against the left atrial (LA) appendage and right atrial free wall and later used for atrial electrogram monitoring and stimulation. The other ends of the electrode wires were tunneled subcutaneously and exposed at the lower back of the neck. For continuous rapid atrial pacing, a unipolar screw‐in lead (OptiSense model 1999; St. Jude Medical, St. Paul, MN, USA) was inserted through the right external jugular vein and the distal end of the lead was screwed into the endocardial side of the right atrial appendage. The proximal end of the pacing lead was connected to a rapid pulse generator (Activa RC; Medtronic, Minneapolis, MN, USA), which was implanted into a subcutaneous pocket at the upper back of the neck. Atrioventricular block was not performed in this study to mimic the hemodynamic situation of clinical cases of AF. 4 , 5 , 11 All study protocols were performed in accordance with the guidelines specified by the Animal Experimentation and Ethics Committee of the Kitasato University School of Medicine.
Study Protocol
To obtain stable baseline conditions, each dog was allowed to recover after the initial surgery (Day −7) for 1 week without pacing. After the 1‐week recovery period (Day 0), atrial rapid pacing at a rate of 400 beats per minute (bpm) was initiated in 10 of the 12 dogs. The remaining 2 dogs without pacing were assigned to a nonpacing group (sham group). In the present study, this group was set up as the sham group for histopathological evaluation. In the nonpacing (sham) group, similar to in the other groups, initial surgery was performed in the absence of pacing and/or drug administration and the dogs were euthanized after 6 weeks of observation. The 10 dogs receiving atrial rapid pacing were divided into the following 2 subgroups: a pacing control group (n=5), which included dogs without any oral administration of CAN, and a pacing+CAN group (n=5), which included dogs with oral administration of CAN (3 mg/kg per day) starting 3 days before the initiation of rapid pacing (Figure 1). The powder form of CAN in a capsule was orally administered. In these 2 groups, continuous rapid atrial pacing was performed for 3 weeks.
Figure 1. Schematic of the study protocol.
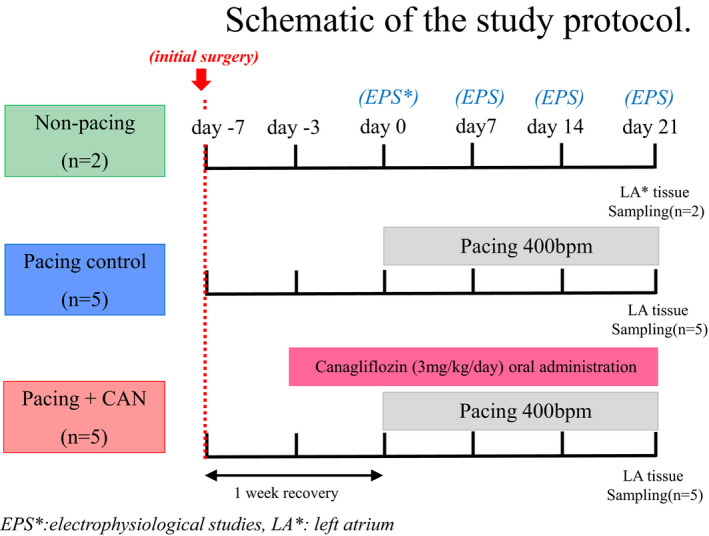
Each dog underwent initial surgery and was allowed to recover for 1 week without pacing before the start of atrial rapid pacing (Day 0). Atrial rapid pacing (400 bpm) was performed for 3 weeks in the pacing control and pacing+CAN groups. In the pacing+CAN group, CAN (3 mg/kg per day) was orally administered from Day −3 until Day 21. Atrial tissue was sampled from each dog at the end of the study period. CAN indicates canagliflozin; EPS, electrophysiological study; and LA, left atrium.
Electrophysiological Studies
During the 3‐week study period, electrophysiological studies were performed every week to evaluate the AF inducibility, atrial effective refractory period (AERP), and atrial conduction velocity (CV) through epicardial wires implanted on the surfaces of both atria in each canine. During the electrophysiological studies, the rapid pacing was temporarily stopped and all measurements were performed under pharmacological block of the autonomic nervous system (infusion of atropine 0.05 mg/kg and propranolol 0.2 mg/kg) to exclude the influence of autonomic nervous tone. 4 , 5 , 11
To evaluate AF inducibility, the incidence of AF induction was evaluated with atrial burst pacing for 3 seconds at the minimal pacing cycle length necessary to achieve 1:1 atrial capture at the LA pacing site. AF induction was delivered 5 times at the LA pacing site at each evaluation time point during the entire study period. 4 , 5 , 11 This pacing was delivered at a level 4 times the diastolic threshold with a pulse width of 2 ms. For this investigation, we defined AF as a spontaneous irregular atrial rhythm lasting longer than 5 seconds. 12 When AF was induced, its duration was measured and AF inducibility was calculated as the ratio (%) of successful AF inductions to the total number of AF induction trials with atrial burst pacing.
At each evaluation time point, the AERP was measured with a basic drive cycle length of 150 ms at the LA site. The pacing energy output was set at 2 times the diastolic threshold during each evaluation. The longest coupling interval of the premature beat that failed to capture the atrium was established as the local AERP. As the AERP data varied among individual dogs owing to the heterogeneity of the canine model itself, the change in the AERP (ΔAERP) was calculated by subtracting the AERP recorded on Day 0. 4 , 5 , 11
The conduction time between the right atrium and left atrium was determined as the time interval between 2 activation times during right atrial appendage pacing with a drive cycle length of 150 ms. As the distance between the right atrial and LA electrodes varied among individual dogs, the delta conduction time was calculated by subtracting the conduction time in Day 0 from each piece of data. The delta CV was evaluated as the reciprocal indirectly by measuring the delta conduction time as described previously, although the delta conduction time was expressed as conduction data in this figure. 4 , 11 , 12
Hemodynamic Evaluation
To estimate the presence or absence of hemodynamic changes caused by CAN administration and/or rapid atrial pacing, hemodynamic parameters—including systemic blood pressure, pulmonary arterial pressure, pulmonary arterial wedge pressure, and cardiac output—were evaluated using a thermodilution catheter at the end of the 3‐week study period in the pacing control and pacing+CAN groups.
Histological Analysis of the Atrial Tissue
At the end of the study period, small portions of the LA free wall were excised for histological and biochemical analyses. The histology of the atrial tissue was evaluated by hematoxylin and eosin and Azan staining. The degree of tissue fibrosis was quantified by measuring the mean % area in digitized images, using the ImageJ program (National Institutes of Health, Bethesda, MD, USA). 4 , 5 The mean value was calculated from the values of 20 randomly selected microscopic windows in each dog. For the analysis of in situ localization of reactive oxygen species (ROS), frozen sections (16 μm) of LA tissues were incubated with fluorophores sensitive to (dihydroethidium [DHE] 10 μmol/L; Sigma‐Aldrich, St. Louis, MO, USA). DHE specifically reacts with intracellular and is converted to the red fluorescent compound ethidium, which then binds irreversibly to double‐stranded DNA and appears as punctuate nuclear staining. The specificity of DHE for was confirmed by preincubation with polyethylene glycol–conjugated superoxide dismutase500 U/mL; Sigma‐Aldrich). Stained sections were observed under a fluorescence microscope (LSM710; Carl Zeiss MicroImaging, Thornwood, NY, USA). To localize the nuclei of cardiomyocytes, 4′,6‐diamidino‐2‐phenylindole staining was performed. The degree of ROS expression on DHE staining was quantified by measuring the mean % area in digitized images, using the ImageJ program (National Institutes of Health). 4 , 5 The relative intensity was calculated by normalizing the immunofluorescence intensity for nuclear staining in cardiomyocytes.
Statistical Analysis
The data are presented as means±SDs or SEs. Statistical differences for the hemodynamic evaluation were analyzed using the Mann–Whitney U test implemented in the JMP statistical software (SAS Institute Inc., Cary, NC, USA). Considering pseudo‐replication and multiple testing, hierarchical statistical techniques 13 based on a generalized linear mixed model (GLMM) framework were employed to detect the statistical significance for the electrophysiological and histological analyses, which were performed using the R packages lmerTest 14 and glmmML in R version 3.5.2 (R Foundation for Statistical Computing, Vienna, Austria) and RStudio. Although the number of subjects was small, the electrophysiological values were measured on the multiple days per subject and the histological values were obtained from 25 cells per subject. The hierarchical statistical analyses were performed using these values linked to the subject ID. The conditions for those hierarchical statistical analyses were described in each figure legend. A P value of less than 0.05 was considered to indicate statistical significance.
Results
Parameters of the Electrophysiological Studies
Figures 2, 3, 4 summarizes the parameters of the electrophysiological studies in the pacing control and pacing+CAN groups. As previously reported, the pacing control group exhibited gradual AERP shortening, a gradual CV decrease, and a gradual AF inducibility increase during the 3‐week rapid atrial pacing protocol. 4 , 11 The AERP shortening was smaller in the pacing+CAN group than in the pacing control group and this difference tended to become significant at Day 21 (ΔAERP for 3 weeks, pacing control versus pacing+CAN; P=0.06 [GLMM]) (Figure 2A and 2B). Additionally, the degree of CV decrease was smaller in the pacing+CAN group than in the pacing control group and the difference became significant at Day 21 (ΔCV for 3 weeks, pacing control versus pacing+CAN; P=0.009 [GLMM]) (Figure 3A and 3B). Moreover, the AF inducibility gradually increased in the pacing control group, whereas such an increase was suppressed in the pacing+CAN group, and the difference became significant at Day 21 (% AF inducibility for 3 weeks, pacing control versus pacing+CAN; P=0.011 [GLMM]) (Figure 4).
Figure 2. Changes in the AERP over the time course in the groups with and without the CAN administration.
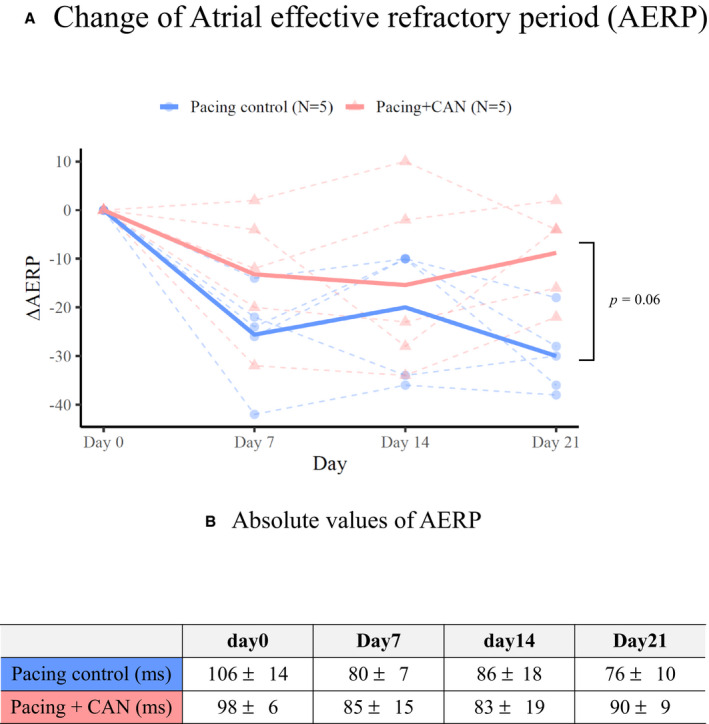
During rapid atrial pacing, gradual AERP shortening were observed in the pacing control group. On the other hand, the AERP shortening was smaller in the pacing+CAN group than in the pacing control group and this difference tended to become significant at Day 21 ([A], ΔAERP for 3 weeks, pacing control vs pacing+CAN; P=0.06 [generalized linear mixed model]). The points‐sets of closed circles and triangles connected by dashed lines indicate the individuals of the control and treatment groups, respectively. Furthermore, (B) indicated absolute values of AERP. In hierarchical statistical analyses, the ΔAERP was modeled with Gaussian distribution and an identity link function. Each model included the date and condition (treated or not) as fixed effects and their interaction and the subject ID as random effects (ie, random intercept mixed model). See text for the details. AERP indicates atrial effective refractory period; AF, atrial fibrillation; and CAN, canagliflozin.
Figure 3. Changes in CV over the time course in the groups with and without the CAN administration.
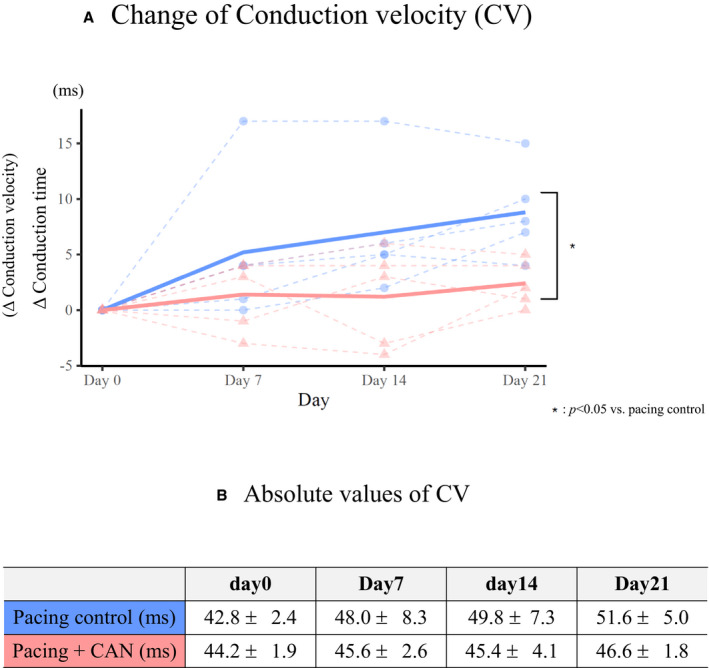
The ΔCV was evaluated as the reciprocal indirectly by measuring the delta conduction time as above although the delta conduction time was expressed as conduction data. The pacing control group exhibited a gradual CV decrease during the 3‐week rapid atrial pacing protocol. In contrast, the degree of CV decrease was smaller in the pacing+CAN group than in the pacing control group and the difference became significant at Day 21 ([A], ΔCV for 3 weeks, pacing control vs pacing+CAN; P=0.009 [generalized linear mixed model]). The points‐sets of closed circles and triangles connected by dashed lines indicate the individuals of the control and treatment groups, respectively. Furthermore, (B) indicated absolute values of CV. In hierarchical statistical analyses, ΔCV was modeled with Gaussian distribution and an identity link function. Each model included the date and condition (treated or not) as fixed effects and their interaction and the subject ID as random effects (ie, random intercept mixed model). See text for the details. AF indicates atrial fibrillation; CAN, canagliflozin; and CV, conduction velocity.
Figure 4. Changes in the inducibility of AF over the time course in the left atrium with and without the CAN administration.
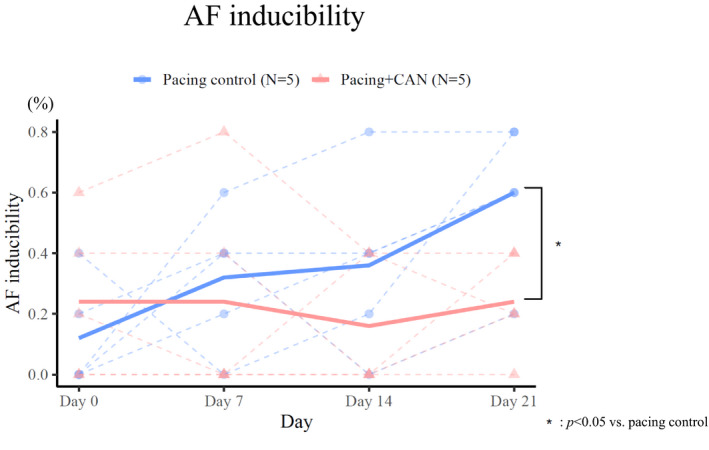
To evaluate AF inducibility, the incidence of AF induction was evaluated with atrial burst pacing for 3 seconds at the minimal pacing cycle length necessary to achieve 1:1 atrial capture at the LA pacing site. When AF was induced, its duration was measured and AF inducibility was calculated as the ratio (%) of successful AF inductions to the total number of AF induction trials with atrial burst pacing. The AF inducibility gradually increased in the pacing control group, while such an increase was suppressed in the pacing+CAN group, and the difference became significant at Day 21 (% AF inducibility for 3 weeks, pacing control vs pacing+CAN; P=0.011 [generalized linear mixed model]). The AF inducibility was modeled with binomial distribution and a logit link function. Each model included the date and condition (treated or not) as fixed effects and their interaction and the subject ID as random effects (ie, random intercept mixed model). See text for the details. AF indicates atrial fibrillation; and CAN, canagliflozin.
Carbohydrate Metabolism Evaluation
Figure 5 shows the time courses of changes in materials that could be affected by CAN administration in the pacing control and pacing+CAN groups. The fasting blood glucose level remained almost entirely unchanged during the protocol and there was no difference between the 2 groups at any evaluation time (Figure 5A). In contrast, the plasma levels of total ketone bodies, acetoacetic acid, and β‐hydroxybutyric acid (β‐HA) immediately increased after the start of rapid atrial pacing (ie, Day 0) in the pacing+CAN group and the levels were significantly higher in the pacing+CAN group than in the pacing control group during the 3‐week protocol (pacing control versus pacing+CAN, total ketone bodies for 3 weeks, P<0.001 [GLMM]; acetoacetic acid for 3 weeks, P<0.01 [GLMM]; β‐HA for 3 weeks, pacing control versus pacing+CAN, P<0.001 [GLMM]) (Figure 5B through 5D).
Figure 5. Carbohydrate metabolism evaluation.
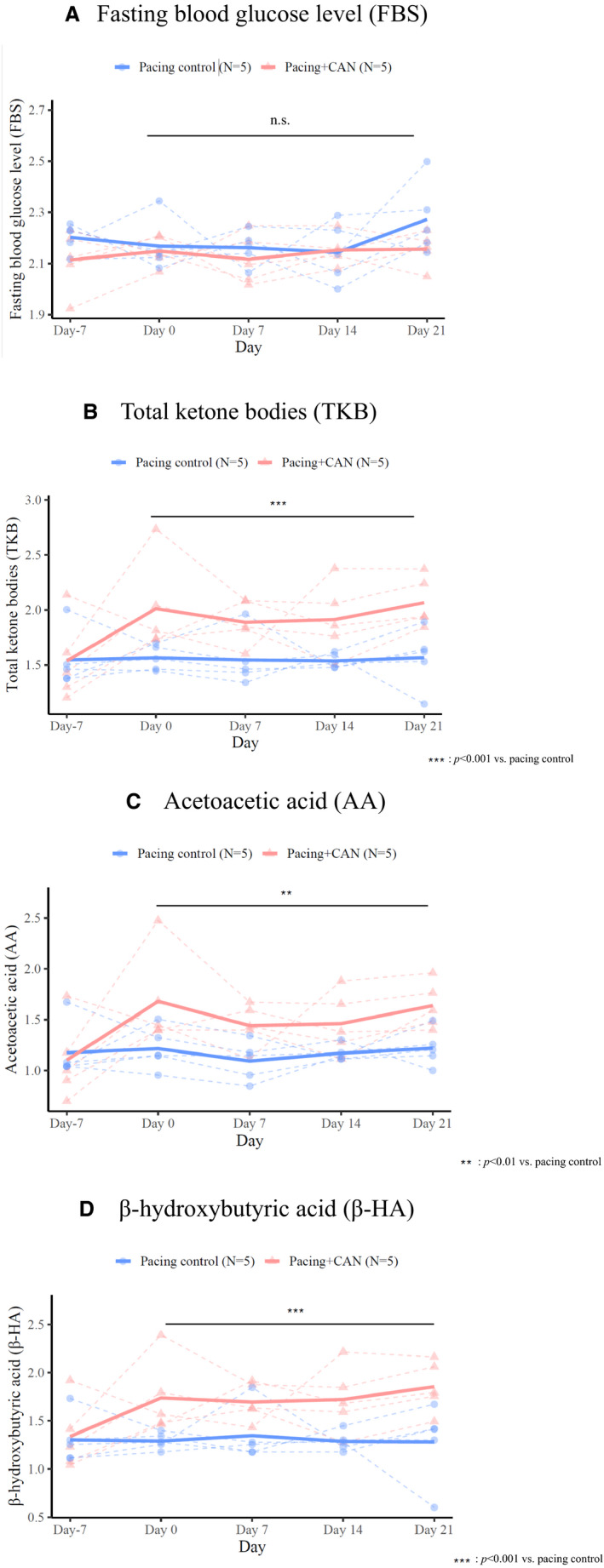
There was no significant difference in the FBS level between the pacing control and pacing+CAN groups (A). In contrast, the levels of plasma ketone bodies, including TKBs, AA, and β‐HA, were higher in the pacing+CAN group than in the pacing control group (B through D). The points‐sets of closed circles and triangles connected by dashed lines indicate the individuals of the control and treatment groups, respectively. In hierarchical statistical analyses, response variables—specifically, log‐transformed FBS, TKBs, AA, and β‐HA—were modeled with Gaussian distribution and an identity link function. Each model included the date and the condition (treated or not) as fixed effects and the subject ID as the random effect. AA indicates acetoacetic acid; CAN, canagliflozin; FBS, fasting blood glucose; TKBs, total ketone bodies; and β‐HA, β‐hydroxybutyric acid.
Hemodynamic Parameters
Table shows the hemodynamic parameters evaluated by using a thermodilution catheter at the end of the 3‐week pacing protocol. There were no significant differences in the hemodynamic parameters between the pacing control and pacing+CAN groups.
Table 1.
Hemodynamic Evaluation
| Pacing Control (N=5) | Pacing+CAN (N=5) | P Value | |
|---|---|---|---|
| Systolic blood pressure, mm Hg | 119±7 | 113±14 | 0.45 |
| Systolic pulmonary arterial pressure, mm Hg | 25±3 | 19±4 | 0.05 |
| Diastolic pulmonary arterial pressure, mm Hg | 11±2 | 9±2 | 0.18 |
| Pulmonary wedge pressure, mm Hg | 14±3 | 9±4 | 0.06 |
| Cardiac output, L/min | 3.2±1.3 | 2.6±1.1 | 0.44 |
CAN indicates canagliflozin.
Histopathology
Figure 6 shows the histological findings. Representative examples of microscopic findings of the LA tissue in the pacing control, pacing+CAN, and nonpacing groups are presented in Figure 6A. The degree of tissue fibrosis was evaluated and the findings are summarized as a bar graph in Figure 6B. Following Azan staining, the pacing control group demonstrated increased histological changes, including the irregularity of cardiomyocytes and interstitial fibrosis, relative to the nonpacing group. On the other hand, the degree of such changes was significantly lower in the pacing+CAN group than in the pacing control group (% area of fibrotic tissue, pacing control versus pacing+CAN: 12.8±1.1 versus 8.1±0.80; P<0.05 [GLMM]).
Figure 6. Histological findings in the left atrial tissue.
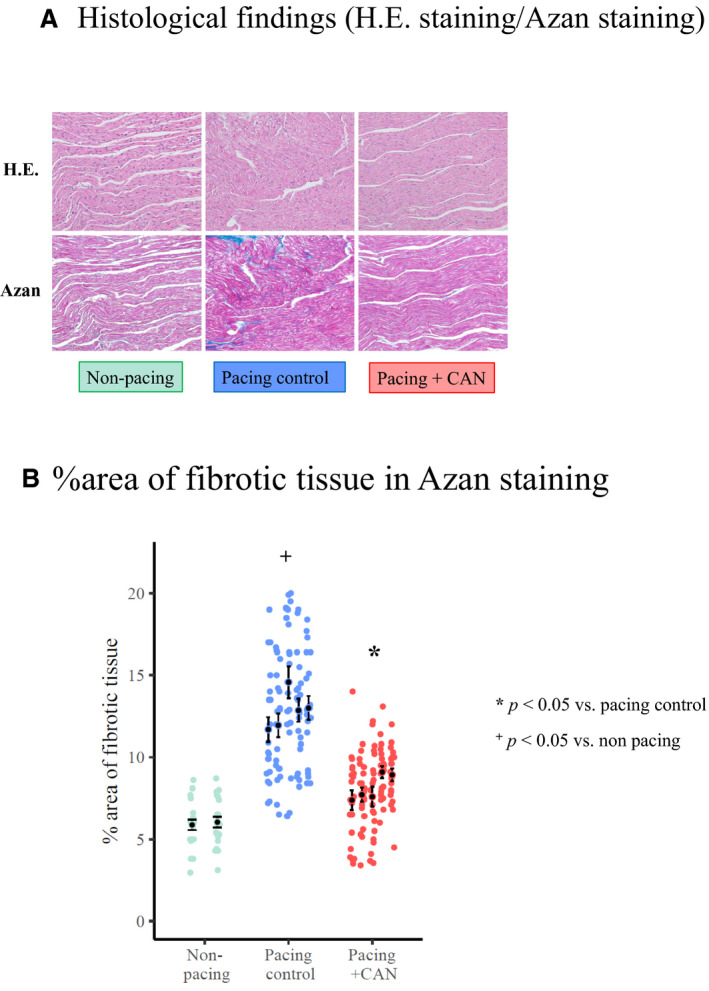
Histological changes, such as the irregularity of cardiomyocytes and interstitial fibrosis, were advanced in the pacing control group when compared with in the nonpacing group. In contrast, the degree of tissue fibrosis was suppressed in the pacing+CAN group relative to in the pacing control group (A). The degree of fibrosis (% area of fibrotic tissue on Azan staining) at the end of the study protocol was higher in the pacing control group than in the nonpacing group (P<0.05) and lower in the pacing+CAN group than in the pacing control group (P<0.05) (B). In hierarchical statistical analysis, the response variable, log‐transformed % area of fibrotic tissue on Azan staining, was modeled with Gaussian distribution and an identity link function. The fixed effect and the random effect were the experimental conditions and subject ID, respectively. CAN indicates canagliflozin; and HE, hematoxylin and eosin.
Expression of ROS
Figure 7 shows the results of DHE staining. Representative examples of immunofluorescent staining of DHE in the pacing control, pacing+CAN, and nonpacing groups are presented in Figure 7A. In the pacing control group, DHE was clearly noted in the myocardial nucleus, which could be localized by a merged image of DHE and 4,6‐diamidino‐2‐phenylindole staining. As this expression was negated by polyethylene glycol–conjugated superoxide dismutase, it was considered to reflect a scenario of enhanced ROS expression. In contrast, in the pacing+CAN group, this expression of DHE was suppressed. The ratio of nuclear immunofluorescent intensity (NII) as an indicator of ROS expression on DHE staining, which was assessed using ImageJ (National Institutes of Health), is presented in Figure 7B. The mean NII was calculated by evaluating the NII values of 25 randomized cells in each group and the ΔNII was calculated by subtracting the mean NII of the nonpacing group. The ΔNII was higher in the pacing control group than in the nonpacing group (ΔNII, pacing control versus nonpacing: 1.9±1.0 versus 0.028±0.89; P<0.05 [GLMM]). In contrast, the ΔNII was lower in the pacing+CAN group than in the pacing control group (pacing control versus pacing+CAN: 1.9±1.0 versus 0.27±1.1; P<0.05 [GLMM]).
Figure 7. Immunofluorescent staining of DHE in the left atrial tissue.
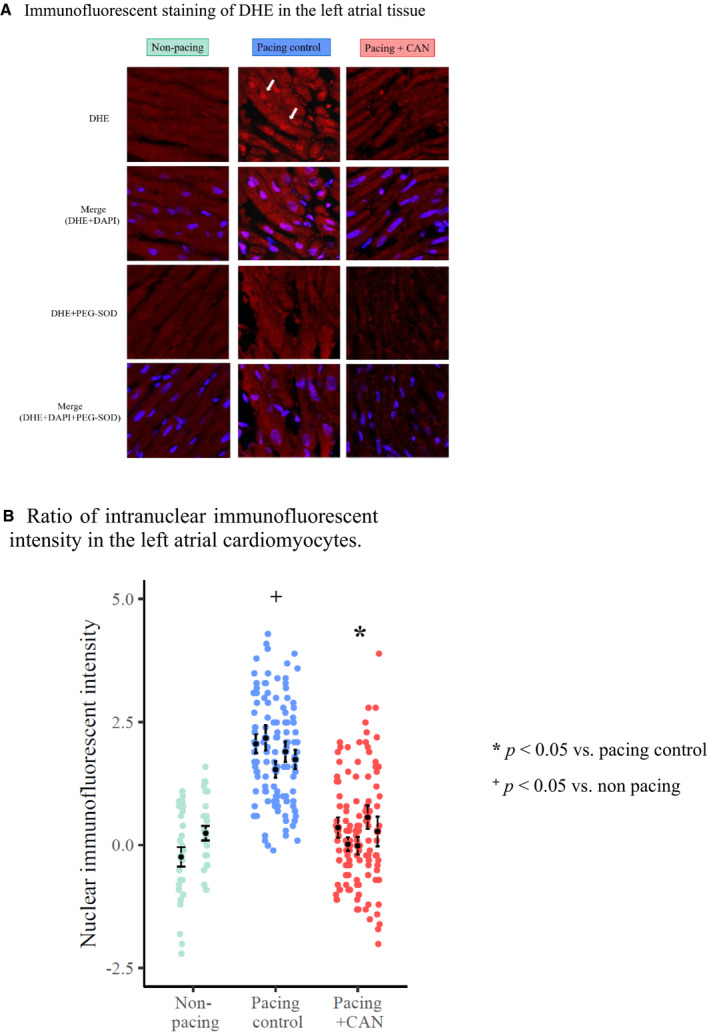
DHE is clearly observable in the myocardial nucleus (indicated by solid arrows) in the pacing control group when compared with the non‐pacing group and could be localized by a merged image of DHE and DAPI staining. As this expression is negated by PEG‐SOD, it is considered to reflect enhanced ROS expression. In contrast, this expression of DHE was suppressed in the pacing+CAN group relative to in the pacing control group (A). The NII (an indicator of ROS expression) was higher in the pacing control group than in the nonpacing group (P<0.05) and lower in the pacing+CAN group than in the pacing control group (P<0.05) (B). The ΔNII was calculated by subtracting the mean NII of the non‐pacing group. In hierarchical statistical analysis, the response variable, ΔNII, was modeled with Gaussian distribution and an identity link function. The fixed effect and the random effect were the experimental conditions and subject ID, respectively. CAN indicates canagliflozin; DAPI, 4,6‐diamidino‐2‐phenylindole; DHE, dihydroethydium; NII, nuclear immunofluorescent intensity; PEG‐SOD, polyethylene glycol–superoxide dismutase; and ROS, reactive oxygen species.
Discussion
Main Findings
The present study has demonstrated several important findings. Three‐week rapid atrial pacing caused atrial electrical remodeling changes, such as AERP shortening, CV decrease, and AF inducibility increase. Furthermore, atrial structural remodeling changes, such as interstitial fibrosis and enhanced oxidative stress, were confirmed. The administration of CAN suppressed the electrophysiological changes as well as the degree of interstitial fibrosis and the extent of oxidative stress.
Role of a Hyperoxidative State in the Atrial Remodeling Process of AF
Previous reports have suggested the possible roles of oxidative stress and inflammation in the mechanisms of the promotion of electrical and structural substrates for AF. 3 , 15 In these reports, the direct generation of ROS in the myocardium was speculated through the actions of NOXs (NADPH [nicotinamide adenine dinucleotide phosphate] oxidases), mitochondria, xanthine oxidase, and “uncoupled” nitrous oxide. These hyper‐ROS were considered to play crucial roles in the development of an AF substrate. 1 , 3 , 16 Atrial tachycardia–induced calcium accumulation causes an increase in such oxidative stress and the resulting changes in the cellular redox state facilitate the genesis and perpetuation of atrial arrhythmias. 1 , 17 Because increased oxidative stress can promote tissue damage and the proliferation of interstitial fibrosis as structural atrial remodeling, such can cause a decrease in CV, possibly resulting in an increase in AF inducibility. 18
Mitochondria have been recognized as a major source of ROS owing to electron leakage from the respiratory chain to oxygen, resulting in the formation of superoxide. Lin et al investigated the oxidative damage of mitochondrial DNA in the right atrial appendages of patients with chronic AF undergoing cardiac surgery. 15 They found that the degree of mitochondrial DNA damage in patients with AF was higher than that in control patients who presented in sinus rhythm. 15 It appears that initial Ca2+ overloading in the mitochondrial matrix eventually alters the mitochondrial membrane potential to reduce adenosine triphosphate synthesis and produce excess ROS. 15 These findings suggest that oxidative injury and the deletion of mitochondrial DNA in cardiac muscles are increased in patients with AF, which might contribute to the impairment of mitochondrial bioenergetic functioning and the induction of an oxidative vicious cycle involved in the pathogenesis of atrial myopathy in AF. 15
Interestingly, one recent report suggested that atrial sources of ROS vary with the duration and substrate of AF, 19 whereas mitochondria have been proposed as the major ROS source for long‐term AF. Xie et al suggested that the atrial intracellular Ca2+ release channel/ryanodine receptor is a specific molecular target of oxidative stress that is fundamental in the development of AF. 11 , 20 These authors demonstrated the functional importance of Ca2+ release channel/ryanodine receptor oxidation in AF pathophysiology, showing that mitochondria‐derived ROS oxidize Ca2+ release channel/ryanodine receptor in atrial myocytes, which leads to an increase in intracellular Ca2+ leakage. Importantly, reducing mitochondrial ROS production attenuates atrial diastolic sarcoplasmic reticulum Ca2+ leakage and prevents AF. 19 These findings suggest a strong correlation between the production of mitochondrial ROS and the development of an AF substrate. In the present study, we also found an increase in oxidative stress in atrial tissue, suggesting the role of oxidative stress in the promotion of atrial remodeling as an AF substrate in our canine model.
Mechanism of the Suppressive Effects of CAN in Our Canine AF Model
In the present study, CAN suppressed the expression of a hyperoxidative state in the atrial tissue and suppressed an increase in AF inducibility in our canine AF model. In a previous canine atrial rapid pacing model, the direct myocardial generation of ROS associated with NOXs, leakage in the mitochondrial electron transport chain, and “uncoupled” nitrous oxide appeared to play a crucial role in the development of AF substrate. 16 Additionally, we documented that DHE staining exhibited ROS overexpression in the atrial myocardium and this expression was almost completely negated by CAN administration. Furthermore, increased levels of ketone bodies, that is, β‐HA and acetoacetic acid, were observed in the vascular tissues in our animal model. Ferrannini et al and Mudaliar et al 21 , 22 hypothesized that the removal of large amounts of glucose from the body and the subsequent reduction of the insulin/glucagon ratio by SGLT2 inhibitor treatment might boost lipid mobilization and oxidation in the liver, stimulating ketogenesis. The resulting metabolic condition, characterized by a mildly hyperketonemic state in prolonged fasting, increased the myocardial uptake of β‐HA, which competes with fatty acid oxidation. This substrate shift is considered cardioprotective because of the high metabolic efficiency of ketone body oxidation.
Finally, β‐HA might be linked to in vitro antioxidative 23 and antiarrhythmic properties. 24 A metabolic switch favoring cardiac ketone body oxidation was proposed as a major driver of such properties via the suppression of mitochondrial ROS production. Li et al reported that NOX subunit NOX2/4, which mediates mitochondrial and cardiac dysfunctions, 25 is a major source of ROS in the failing heart. On the other hand, Kimura et al demonstrated that canagliflozin significantly reduced messenger RNA content and levels of NOX2 and NOX4 in myocardial infarction rats with type 2 diabetes mellitus. 26 They concluded that the elevation of blood β‐HA levels by treatment with canagliflozin was related to the suppressed expression of NOXs. 26 Therefore, we thought that transcriptional remodeling of ROS sources by treatment with canagliflozin might be associated with the suppression of cardiomyocyte ROS. Although no data directly revealed about the transcriptional remodeling of cardiomyocyte ROS sources in this study, our experimental data still indicate the therapeutic potential of SGLT2 inhibitors for treating a myocardial oxidative state.
Limitations
The present study has several limitations that should be noted. First, the influence of tachycardia–induced heart failure cannot be ruled out completely. We selected our model setting, that is, no AV block, which mimicked clinical AF, to evaluate the effects of the drug in a real AF situation and we believe that the abovementioned influence was absent or small as there was no difference in hemodynamic parameters between the groups with and without CAN, respectively. Second, CV may be influenced by changes in the conduction pathway. Because there is no difference in LA diameter between pre and post pacing in the 2 groups, we evaluated the CVs by calculating as the reciprocal of this conduction time as previously reported. However, we think this to be a major limitation. Third, myocardial ketone use was not assessed in detail. Some reports have shown that cardiac ketone use increased and caused myocardial ketone oxidation among failing hearts in vivo and in vitro. 27 , 28 Therefore, the antioxidative mechanism of CAN‐induced hyperketonemia should be understood as a possible speculation. Fourth, as a single dose of CAN was used in this study, the dose‐dependency could not be determined. Finally, as the expressions of ionic channels, connexin expression, and/or phosphorylation were not examined, the effect of CAN on cellular electrical remodeling could not be adequately discerned. Furthermore, another report found that the administration of dapagliflozin imparted an antiarrhythmogenic effect by increasing the expression of gap junction protein p‐Cx43 S368, which could be mainly responsible for reducing the arrhythmia vulnerability in rats with cardiac ischemia–reperfusion injury. 29 Similar clinical effects might be expected through the same pharmacological class effect. This study describes the protective effects of CAN but does not provide information about the molecular mechanisms underlying the reduced electrical and structural remodeling. These mechanisms should be studied in future research with different designs.
Conclusions
We found that CAN suppressed AF inducibility, AERP shortening, and CV decrease in our canine AF model, representing effects that were associated with the suppression of tissue fibrosis and oxidative stress. CAN and possibly other SGLT2 inhibitors might be useful for preventing AF and suppressing the promotion of atrial remodeling as an AF substrate.
Sources of Funding
None.
Disclosures
The author has no financial conflicts of interest to disclosure to report.
(J Am Heart Assoc.2021;10:e017483. DOI: 10.1161/JAHA.119.017483.)
For Sources of Funding and Disclosures, see page 11.
References
- 1. Mihm MJ, Yu F, Carnes CA, Reiser PJ, McCarthy PM, Van Wagoner DR, Bauer JA. Impaired myofibrillar energetics and oxidative injury during human atrial fibrillation. Circulation. 2001;104:174–180. 10.1161/01.CIR.104.2.174 [DOI] [PubMed] [Google Scholar]
- 2. Korantzopoulos P, Kolettis TM, Dimitrios G, Goudevenos JA. The role of oxidative stress in the pathogenesis and perpetuation of atrial fibrillation. Int J Cardiol. 2007;115:135–143. 10.1016/j.ijcard.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 3. Pinho‐Gomes AC, Reilly S, Brandes RP, Casadei B. Targeting inflammation and oxidative stress in atrial fibrillation. Role of 3‐hydroxy‐3‐methylglutaryl‐coenzyme a reductase inhibition with statins. Antioxid Redox Signal. 2013;20:1268–1285. 10.1089/ars.2013.5542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kishihara J, Niwano S, Niwano H, Aoyama Y, Satoh A, Oikawa J, Kiryu M, Fukaya H, Masaki Y, Tamaki H, et al. Effect of carvedilol on atrial remodeling in canine model of atrial fibrillation. Cardiovasc Diagn Ther. 2014;4:28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kiryu M, Niwano S, Niwano H, Kishihara J, Aoyama Y, Fukaya H, Masaki Y, Izumi T. Angiotensin II‐mediated up‐regulation of connective tissue growth factor promotes atrial tissue fibrosis in the canine atrial fibrillation model. Europace. 2012;14:1206–1214. 10.1093/europace/eus052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Igarashi T, Niwano S, Niwano H, Yoshizawa T, Nakamura H, Fukaya H, Fujiishi T, Ishizue N, Satoh A, Kishihara J, et al. Linagliptin prevents atrial electrical and structural remodeling in a canine model of atrial fibrillation. Heart Vessels. 2018;33:1258–1265. 10.1007/s00380-018-1170-0 [DOI] [PubMed] [Google Scholar]
- 7. Neal B, Perkovic V, Mahaffey KW, De Zeeuw D, Fulcher G, Erondu N, Shaw W, Law G, Desai M, Matthews DR. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644–657. 10.1056/NEJMoa1611925 [DOI] [PubMed] [Google Scholar]
- 8. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, Silverman MG, Zelniker TA, Kuder JF, Murphy SA, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347–357. 10.1056/NEJMoa1812389 [DOI] [PubMed] [Google Scholar]
- 9. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117–2128. 10.1056/NEJMoa1504720 [DOI] [PubMed] [Google Scholar]
- 10. McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, Ponikowski P, Sabatine MS, Anand IS, Bělohlávek J, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. 2019;381:1995–2008. 10.1056/NEJMoa1911303 [DOI] [PubMed] [Google Scholar]
- 11. Satoh A, Niwano S, Niwano H, Kishihara J, Aoyama Y, Oikawa J, Fukaya H, Tamaki H, Ako J. Aliskiren suppresses atrial electrical and structural remodeling in a canine model of atrial fibrillation. Heart Vessels. 2017;32:90–100. 10.1007/s00380-016-0874-2 [DOI] [PubMed] [Google Scholar]
- 12. Fukaya H, Niwano S, Satoh D, Masaki Y, Niwano H, Kojima J, Moriguchi M, Izumi T. Inhomogenic effect of bepridil on atrial electrical remodeling in a canine rapid atrial. Circ J. 2008;72:318–326. [DOI] [PubMed] [Google Scholar]
- 13. Sikkel MB, Francis DP, Howard J, Gordon F, Rowlands C, Peters NS, Lyon AR, Harding SE, MacLeod KT. Hierarchical statistical techniques are necessary to draw reliable conclusions from analysis of isolated cardiomyocyte studies. Cardiovasc Res. 2017;113:1743–1752. 10.1093/cvr/cvx151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuznetsova A, Brockhoff PB, Christensen RHB. lmerTest Package: tests in linear mixed effects models. J Stat Softw. 2017;82:1548–7660. [Google Scholar]
- 15. Lin PH, LeebSH WYH. Oxidative damage to mitochondrial DNA in atrial muscle of patients with atrial fibrillation. Free Radic Biol Med. 2003;35:1310–1318. [DOI] [PubMed] [Google Scholar]
- 16. Cangemi R, Celestini A, Calvieri C, Carnevale R, Pastori D, Nocella C, Vicario T, Pignatelli P, Violi F. Different behaviour of NOX2 activation in patients with paroxysmal/persistent or permanent atrial fibrillation. Heart. 2012;98:1063–1066. 10.1136/heartjnl-2012-301952 [DOI] [PubMed] [Google Scholar]
- 17. Van Wagoner DR. Redox modulation of cardiac electrical activity. J Cardiovasc Electrophysiol. 2001;12:183–184. 10.1046/j.1540-8167.2001.00183.x [DOI] [PubMed] [Google Scholar]
- 18. Wijffels MC, Kirchhof CJ, Dorland R, Allessie MA. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation. 1995;92:1954–1968. 10.1161/01.CIR.92.7.1954 [DOI] [PubMed] [Google Scholar]
- 19. Reilly SN, Jayaram R, Nahar K, Antoniades C, Verheule S, Channon KM, Alp NJ, Schotten U, Casadei B. Atrial sources of reactive oxygen species vary with the duration and substrate of atrial fibrillation: implications for the antiarrhythmic effect of statins. Circulation. 2011;124:1107–1117. 10.1161/CIRCULATIONAHA.111.029223 [DOI] [PubMed] [Google Scholar]
- 20. Xie W, Santulli G, Reiken SR, Yuan Q, Osborne BW, Chen BX, Marks AR. Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep. 2015;5:1–11. 10.1038/srep11427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA‐REG OUTCOME trial: a thrifty substrate hypothesis. Diabetes Care. 2016;39:1108–1114. 10.2337/dc16-0330 [DOI] [PubMed] [Google Scholar]
- 22. Mudaliar S, Alloju S, Henry RR. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA‐REG OUTCOME study? A unifying hypothesis. Diabetes Care. 2016;39:1115–1122. 10.2337/dc16-0542 [DOI] [PubMed] [Google Scholar]
- 23. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Moan NL, Grueter CA, Lim H, Saunders LR, Stevens RD, et al. Supression of oxidative stress and β‐OHB as endogenous histone deactetylase. Science. 2013;339:211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cotter DG, Schugar RC, Crawford PA. Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol. 2013;304:H1060–H1076. 10.1152/ajpheart.00646.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li B, Tian J, Sun Y, Xu TR, Chi RF, Zhang XL, Hu XL, Zhang YA, Qin FZ, Zhang WF. Activation of NADPH oxidase mediates increased endoplasmic reticulum stress and left ventricular remodeling after myocardial infarction in rabbits. Biochem Biophys Acta. 2015;1852:805–815. 10.1016/j.bbadis.2015.01.010 [DOI] [PubMed] [Google Scholar]
- 26. Kimura Y, Kuno A, Tanno M, Sato T, Ohno K, Shibata S, Nakata K, Sugawara H, Abe K, Igaki Y, et al. Canagliflozin, a sodium–glucose cotransporter 2 inhibitor, normalizes renal susceptibility to type 1 cardiorenal syndrome through reduction of renal oxidative stress in diabetic rats. J Diabetes Investig. 2019;10:933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. 2016;133:706–716. 10.1161/CIRCULATIONAHA.115.017545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Krüger M, Hoppel CL, et al. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133:698–705. 10.1161/CIRCULATIONAHA.115.017355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tanajak P, Sa‐Nguanmoo P, Sivasinprasasn S, Thummasorn S, Siri‐Angkul N, Chattipakorn SC, Chattipakorn N. Cardioprotection of dapagliflozin and vildagliptin in rats with cardiac ischemia reperfusion injury. J Endocrinol. 2018;236:69–84. 10.1530/JOE-17-0457 [DOI] [PubMed] [Google Scholar]