

Abstract
Background
Patients with familial hypercholesterolemia who harbored both low‐density lipoprotein receptor (LDLR) and PCSK9 (proprotein convertase subtilisin/kexin type 9) gene variants exhibit severe phenotype associated with substantially high levels of low‐density lipoprotein cholesterol. In this study, we investigated the cardiovascular outcomes in patients with both LDLR and PCSK9 gene variants.
Methods and Results
A total of 232 unrelated patients with LDLR and/or PCSK9 gene variants were stratified as follows: patients with LDLR and PCSK9 (LDLR/PCSK9) gene variants, patients with LDLR gene variant, and patients with PCSK9 gene variant. Clinical demographics and the occurrence of primary outcome (nonfatal myocardial infarction) were compared. The observation period of primary outcome started at the time of birth and ended at the time of the first cardiac event or the last visit. Patients with LDLR/PCSK9 gene variants were identified in 6% of study patients. They had higher levels of low‐density lipoprotein cholesterol (P=0.04) than those with LDLR gene variants. On multivariate Cox regression model, they experienced a higher incidence of nonfatal myocardial infarction (hazard ratio, 4.62; 95% CI, 1.66–11.0; P=0.003 versus patients with LDLR gene variant). Of note, risk for nonfatal myocardial infarction was greatest in male patients with LDLR/PCSK9 gene variants compared with those with LDLR gene variant (86% versus 24%; P<0.001).
Conclusions
Patients with LDLR/PCSK9 gene variants were high‐risk genotype associated with atherogenic lipid profiles and worse cardiovascular outcomes. These findings underscore the importance of genetic testing to identify patients with LDLR/PCSK9 gene variants, who require more stringent antiatherosclerotic management.
Keywords: cardiovascular outcome, familial hypercholesterolemia, LDLR gene, PCSK9 gene
Subject Categories: Lipids and Cholesterol, Genetics, Cardiovascular Disease
Nonstandard Abbreviations and Acronyms
- FH
familial hypercholesterolemia
- HeFH
heterozygous familial hypercholesterolemia
- LDLR
low‐density lipoprotein receptor
- PCSK9
proprotein convertase subtilisin/kexin type 9
- TAUSSIG
Trial Assessing Long Term Use of PCSK9 Inhibition in Subjects With Genetic LDL Disorders
Clinical Perspective
What Is New?
Patients with low‐density lipoprotein receptor (LDLR) and PCSK9 (proprotein convertase subtilisin/kexin type 9) gene variants were familial hypercholesterolemia genotype, which causes an elevated risk of nonfatal myocardial infarction.
What Are the Clinical Implications?
Risk stratification according to sex and genotype may be a potential risk stratification tool to identify high‐risk patients with familial hypercholesterolemia who need more intense antiatherosclerotic therapies.
Familial hypercholesterolemia (FH) is a genetic disorder caused by variants in low‐density lipoprotein receptor (LDLR) gene, PCSK9 (proprotein convertase subtilisin/kexin type 9) gene, and apolipoprotein B (APOB) gene. These genetic variants can cause elevation of low‐density lipoprotein cholesterol (LDL‐C) levels and tendon or skin xanthomas, 1 , 2 , 3 which lead to the higher risk of atherosclerotic cardiovascular disease (ASCVD) in FH. 4 These observations emphasize the clinical importance of the genotype that causes FH because of its association with metabolic and cardiovascular risks.
Recent genetic analyses of FH have identified patients with variants in 2 different causative genes. In recent published analyses from Japan, 5 , 6 patients with both LDLR and PCSK9 gene variants (LDLR/PCSK9 gene variants) were detected in 1.2% to 4.0% of Japanese patients with FH. Although such patients represent a small fraction of patients with FH overall, the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel has proposed that this genotype represents a severe form of FH. 3 The presence of both causative gene variants has the potential to substantially elevate circulating LDL‐C level. Given that LDL‐C is a major driver of atherosclerosis in FH, this feature may be an important atherogenic substrate responsible for ASCVD in patients with LDLR/PCSK9 gene variants. 6 , 7 However, the clinical demographics and outcomes in patients with LDLR/PCSK9 gene variants remain to be fully elucidated. In this study, we investigated the prevalence, clinical characteristics, and cardiovascular outcomes of patients with LDLR and PCSK9 gene variants.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Study Population
This study retrospectively analyzed 377 Japanese unrelated patients with clinically diagnosed heterozygous FH (HeFH) who underwent genetic testing to identify variants in LDLR gene and/or PCSK9 gene at the National Cerebral and Cardiovascular Center, Osaka, Japan, between 2005 and 2016. HeFH was diagnosed on the basis of Japanese Atherosclerosis Society Criteria 2017: having ≥2 of the following factors: LDL‐C ≥180 mg/dL, tendon/skin xanthoma, and a family history of FH or premature coronary artery disease within second‐degree relatives. 8 We excluded 124 patients without LDLR or PCSK9 gene variants. Furthermore, in 253 patients who had gene variant in LDLR gene and/or PCSK9 gene, the following patients were excluded: those with 2 gene variants in LDLR gene (n=1) and PCSK9 gene (n=1), those with both LDLR gene variants and PCSK9 gene loss‐of‐function variants (n=5), and those who were aged <20 years (n=14). The remaining 232 unrelated patients with FH were included into the current analysis (Figure 1). The protocol of this study was approved by the institutional review board of the National Cerebral and Cardiovascular Center (approval No. M17‐56). Each patient gave written informed consent to participate in the study. All clinical investigations were conducted in accordance with the principles of the Declaration of Helsinki.
Figure 1. Flowchart of the study patients.
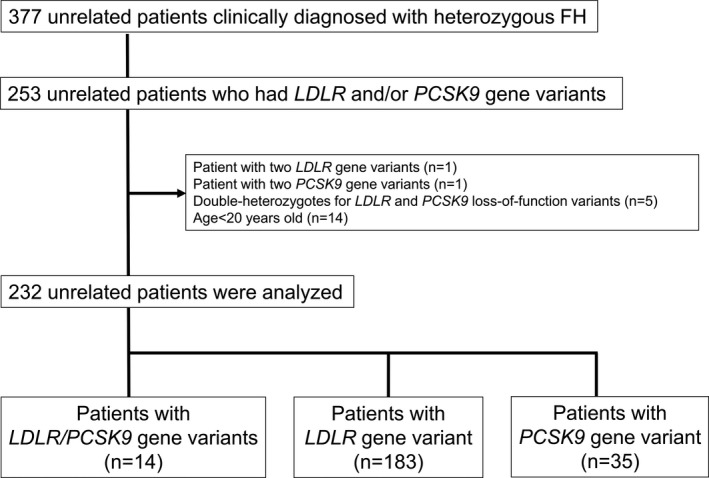
FH indicates familial hypercholesterolemia; LDLR, low‐density lipoprotein receptor; and PCSK9, proprotein convertase subtilisin/kexin type 9.
DNA Analysis
DNA analyses were conducted on genomic DNA extracted from patients’ whole blood using an automated DNA extraction machine (QIAsymphony; QIAGEN, Valencia, CA). All coding regions and the exon‐intron boundary sequence of the LDLR and PCSK9 genes were examined by direct sequencing, as described previously. 6 Samples without variants in both the LDLR gene and the PCSK9 genes were analyzed for large deletions or insertions in the LDLR gene by the multiplex ligation‐dependent probe amplification method using the P062B LDLR multiplex ligation‐dependent probe amplification kit (MRC Holland, Amsterdam, the Netherlands). The pathogenicity of LDLR and PCSK9 gene variants was assessed according to the American College of Medical Genetics criteria. 9 With regard to PCSK9 gene variant, the current analysis included 3 variants (p.Val4Ile, p.Glu32Lys, and p.Arg496Trp) 6 , 10 , 11 that are rare (allele frequency <1%) among East Asian population. 12 PCSK9 p.Val4Ile gene variant is rated as benign using American College of Medical Genetics criteria, 13 whereas its potential association for serum LDL‐C level has been shown by our published study. 6 Therefore, in the current study, we included (1) all of these 3 PCSK9 gene variants and (2) those except PCSK9 p.Val4Ile gene variant into the analysis. All variants were denoted using known and accepted nomenclature based on the full lengths of the splice variants with 860 and 692 amino acids (National Center for Biotechnology Information reference sequence NM_000527.4 for LDLR gene and NM_174936.3 for PCSK9 gene). 14 , 15
Cardiovascular Outcomes
The primary outcome was defined as the occurrence of nonfatal myocardial infarction (MI). The secondary outcome included the occurrence of nonfatal MI and coronary revascularization. Because any cardiac cause of death was not observed in the current study subjects, we did not include this event in either primary or secondary outcomes. MI was defined as the presence of cardiac ischemic symptoms with biomarker evidence of myocardial injury and electrocardiographic changes suggestive of new ischemia (new ST‐T changes or new left bundle‐branch block) or the development of pathological Q waves on electrocardiography. 16 Coronary revascularization was defined as percutaneous coronary intervention or coronary artery bypass grafting for any reason. These outcomes were determined through medical record review and, when necessary, through a mailed questionnaire or telephone follow‐up.
Measurement of Lipid Parameters
LDL‐C levels at baseline were calculated by the Friedewald formula, except for triglyceride levels >400 mg/dL. 17 In patients who had already received lipid‐lowering agents at their first visit, we estimated baseline LDL‐C levels according to the type and dose of their lipid‐lowering medication, applying a correcting factor for LDL‐C based on the reported efficacy of each drug, as performed previously in similar analyses. 18 , 19 , 20 , 21 Fasting serum levels of total cholesterol, triglycerides, high‐density lipoprotein cholesterol, and lipoprotein (a) were measured by enzymatic methods (Sekisui Medical, Tokyo, Japan) using an automated analyzer (Hitachi Labospect 008; Hitachi‐Hitec, Tokyo, Japan). Apolipoproteins were measured by turbidimetric immunoassay (Nittobo Medical, Tokyo, Japan) using an automated analyzer (JCA‐BM8060; JEOL, Tokyo, Japan) by LSI Medience Corporation (Tokyo, Japan).
Statistical Analysis
Two‐group comparison was conducted using the Fisher's exact test for the categorical variables. Normally distributed continuous variables were expressed as mean±SD, and nonnormally distributed continuous data were summarized as the median (interquartile range). Both continuous variables were compared using permutation test with the perm package of R. The Kaplan‐Meier method was used to estimate survival curves for primary and secondary outcomes, and the exact log‐rank test was used to assess differences between patients with LDLR gene variant and those with PCSK9 gene variant and between patients with LDLR gene variant and those with LDLR and PCSK9 gene variants. 22 The current study collected lifetime cardiac events after the birth, but not those after the first visit to our clinic. This is because 82% of primary outcome in study subjects occurred before their first visit (Table S1). To evaluate true cardiac event risks in the current study population, the observation period started after the time of birth, and it ended at the time of the first cardiac event or the last visit. Patients who were free from primary outcome at the last visit were considered as censored observation. Cox proportional hazards model was used to identify high‐risk cause of primary outcome using following covariates determined before the analysis (model 1: sex and genotype; and model 2: sex, history of hypertension, diabetes mellitus, smoking, and genotype). In addition, this analysis further adjusted the following 2 variables: duration until achieving LDL‐C goal (defined as >50% decrease of LDL‐C from baseline or on‐treatment LDL‐C <100 mg/dL) from the time of birth and that during optimal LDL‐C control under the therapy. 8 , 23 Because history of hypertension, diabetes mellitus, smoking, duration until achieving LDL‐C goal from the time of birth, and that during optimal LDL‐C control under the therapy were time varying, we analyzed these covariates as time‐varying covariates. 24 P<0.05 was considered statistically significant. All analyses were performed with SPSS (SPSS Japan, Tokyo, Japan), STATA 13 (StataCorp, College Station, TX), and R 3.6.3 (R Core Team, R Foundation for Statistical Computing, Vienna, Austria).
Results
Genetic Features in Patients With FH
In this study, the prevalence rates of patients with LDLR/PCSK9 gene variants, those with LDLR gene variants, and those with PCSK9 gene variants were 6%, 80%, and 14%, respectively. In patients with LDLR/PCSK9 gene variants, 13 LDLR gene variants and 2 PCSK9 gene variants were identified (Table S2). All variants identified in this cohort were listed in Tables S3 and S4. 13
Clinical Demographics in Patients With Different Gene Variants
We summarize baseline clinical characteristics of the patients with LDLR and/or PCSK9 gene variants (Table 1). There were no significant differences in the percentages of men and the prevalence of coronary risk factors in patients with LDLR gene variant compared with those with PCSK9 gene variant and in patients with LDLR gene variant compared with those with LDLR/PCSK9 gene variants (Table 1). For lipid profiles at baseline, LDL‐C level was highest in patients with LDLR/PCSK9 gene variants (316±75 mg/dL) compared with those with LDLR gene variant (273±72 mg/dL; P=0.04) and those with PCSK9 gene variant (219±58 mg/dL) (Table 2). Baseline triglycerides, apolipoprotein A‐I, apolipoprotein A‐II, and apolipoprotein C‐II were higher in patients with PCSK9 gene variant than patients with LDLR gene variants (P<0.001, P=0.001, P<0.001, and P<0.001, respectively), whereas the levels of lipoprotein (a), apolipoprotein C‐III, and apolipoprotein E were similar.
Table 1.
Clinical Demographics
| Demographics | Patients With LDLR Gene Variant (n=183) | Patients With PCSK9 Gene Variant (n=35) | P Value* | Patients With LDLR/PCSK9 Gene Variants (n=14) | P Value † |
|---|---|---|---|---|---|
| Age, y | 43±16 | 53±13 | <0.001 | 53±13 | 0.66 |
| Male sex, n (%) | 92 (51) | 17 (49) | 0.85 | 7 (50) | 0.98 |
| BMI ≥30 kg/m2, n (%) | 0 (0) | 1 (3) | 0.05 | 1 (7) | 0.02 |
| Hypertension, n (%) | 42 (23) | 7 (20) | 0.68 | 5 (36) | 0.34 |
| Diabetes mellitus, n (%) | 4 (2) | 1 (3) | 0.82 | 0 (0) | 0.43 |
| Smoking history, n (%) | 75 (41) | 15 (43) | 0.36 | 8 (57) | 0.29 |
| Family history of coronary artery disease, n (%) | 53 (29) | 2 (6) | 0.16 | 4 (29) | 0.92 |
| Any xanthomas, n (%) | 138 (76) | 18 (51) | 0.04 | 12 (86) | 0.61 |
| Corneal arcus, n (%) | 68 (37) | 8 (23) | 0.33 | 5 (36) | 0.92 |
Categorical variables (sex, BMI ≥30 kg/m2, history of hypertension, diabetes mellitus, and smoking, family history of coronary artery disease, and presence of any xanthomas and corneal arcus) were expressed as number (percentage) and compared using the Fisher's exact test. Normally distributed continuous variables (age) were expressed as mean±SD and compared using permutation test. BMI indicates body mass index; LDLR, low‐density lipoprotein receptor; and PCSK9, proprotein convertase subtilisin/kexin type 9.
Patients with LDLR gene variant vs those with PCSK9 gene variant.
Patients with LDLR gene variant vs those with LDLR/PCSK9 gene variants.
Table 2.
Baseline Lipid Profiles
| Variable | Patients With LDLR Gene Variant (n=183) | Patients With PCSK9 Gene Variant (n=35) | P Value* | Patients With LDLR/PCSK9 Gene Variants (n=14) | P Value† |
|---|---|---|---|---|---|
| LDL‐C, mg/dL | 273±72 | 219±58 | <0.001 | 316±75 | 0.04 |
| HDL‐C, mg/dL | 52±16 | 58±13 | 0.03 | 50±21 | 0.64 |
| Triglycerides, mg/dL | 91 (70–138) | 139 (88–195) | <0.001 | 133 (68–167) | 0.32 |
| Lp(a), mg/dL | 29±26 | 28±22 | 0.85 | 27±17 | 0.82 |
| Apolipoprotein A‐I, mg/dL | 127±33 | 147±25 | 0.001 | 117±38 | 0.31 |
| Apolipoprotein A‐II, mg/dL | 27±6 | 33±6 | <0.001 | 27±6 | 0.91 |
| Apolipoprotein B, mg/dL | 127±39 | 108±37 | 0.01 | 135±29 | 0.43 |
| Apolipoprotein C‐II, mg/dL | 3.8±1.7 | 5.8±1.9 | <0.001 | 4.1±1.5 | 0.61 |
| Apolipoprotein C‐III, mg/dL | 9.5±8.6 | 11.0±3.2 | 0.25 | 9.0±2.4 | 0.75 |
| Apolipoprotein E, mg/dL | 4.3±1.3 | 4.2±0.8 | 0.68 | 4.2±1.0 | 0.76 |
Normally distributed continuous variables (LDL‐C, HDL‐C, Lp[a], apolipoprotein A‐I, apolipoprotein A‐II, apolipoprotein B, apolipoprotein C‐II, apolipoprotein C‐III, and apolipoprotein E) were expressed as mean±SD, and nonnormally distributed continuous variables (triglycerides) were expressed as median (interquartile range). Both continuous variables were compared using permutation test. HDL‐C indicates high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; LDLR, low‐density lipoprotein receptor; Lp(a), lipoprotein (a); and PCSK9, proprotein convertase subtilisin/kexin type 9.
Patients with LDLR gene variant vs those with PCSK9 gene variant.
Patients with LDLR gene variant vs those with LDLR/PCSK9 gene variants.
Use of Lipid‐Lowering Therapy and On‐Treatment Lipid Parameters
Table 3 shows a comparison of lipid‐lowering therapy and on‐treatment lipid profiles. Over 85% of study patients received statin. Of note, high‐intensity statin and PCSK9 inhibitor, including evolocumab and alirocumab, were more frequently used in patients with LDLR/PCSK9 gene variants (high‐intensity statin: P=0.03; PCSK9 inhibitor: P<0.001; evolocumab: P=0.002; and alirocumab: P=0.05 versus patients with LDLR gene variant). As a consequence, these therapeutic differences were associated with a greater absolute (P<0.001 versus patients with LDLR gene variant) and percentage reduction of LDL‐C (P=0.008 versus patients with LDLR gene variant) in patients with LDLR/PCSK9 gene variants. Furthermore, a higher proportion of these patients achieved LDL‐C <100 mg/dL (P=0.002) and percentage reduction of LDL‐C <50% (P=0.04) in patients with LDLR/PCSK9 gene variants compared with those with LDLR gene variant (Table 4).
Table 3.
On‐Treatment Lipid‐Lowering Therapies
| Variable | Patients With LDLR Gene Variant (n=183) | Patients With PCSK9 Gene Variant (n=35) | P Value* | Patients With LDLR/PCSK9 Gene Variants (n=14) | P Value† |
|---|---|---|---|---|---|
| Statin, n (%) | 160 (88) | 32 (91) | 0.93 | 12 (86) | 0.54 |
| High‐intensity statin, n (%) | 6 (16) | 2 (6) | 0.07 | 6 (43) | 0.03 |
| Ezetimibe, n (%) | 105 (58) | 11 (31) | 0.002 | 6 (43) | 0.21 |
| Cholestyramine, n (%) | 32 (18) | 0 (0) | <0.001 | 2 (14) | 0.71 |
| PCSK9 inhibitor, n (%) | 10 (5) | 0 (0) | 0.06 | 6 (43) | <0.001 |
| Evolocumab, n (%) | 6 (3) | 0 (0) | 0.14 | 4 (29) | 0.002 |
| Alirocumab, n (%) | 4 (2) | 0 (0) | 0.23 | 2 (14) | 0.05 |
| Lipoprotein apheresis, n (%) | 1 (0) | 0 (0) | 0.26 | 0 (0) | 0.70 |
Categorical variables (use of statin, high‐intensity statin, ezetimibe, cholestyramine, PCSK9 inhibitor, evolocumab, alirocumab, and lipoprotein apheresis; patients with LDL‐C <100 mg/dL; and patients with percentage reduction of LDL‐C <50%) were expressed as number (percentage) and compared using the Fisher's exact test. LDLR indicates low‐density lipoprotein receptor; and PCSK9, proprotein convertase subtilisin/kexin type 9.
Patients with LDLR gene variant vs those with PCSK9 gene variant.
Patients with LDLR gene variant vs those with LDLR/PCSK9 gene variants.
Table 4.
On‐Treatment Lipid Levels
| Variable | Patients With LDLR Gene Variant (n=183) | Patients With PCSK9 Gene Variant (n=35) | P Value* | Patients With LDLR/PCSK9 Gene Variants (n=14) | P Value† |
|---|---|---|---|---|---|
| On‐treatment LDL‐C, mg/dL | 136±49 | 116±46 | 0.03 | 110±52 | 0.05 |
| Absolute reduction of LDL‐C, mg/dL | 121±71 | 96±54 | 0.01 | 206±114 | 0.008 |
| Percentage reduction of LDL‐C, % | 44±21 | 45±25 | 0.66 | 61±26 | 0.03 |
| Patients with LDL‐C <100 mg/dL, n (%) | 33 (18) | 13 (37) | 0.01 | 8 (57) | 0.002 |
| Patients with percentage reduction of LDL‐C <50%, n (%) | 87 (48) | 14 (40) | 0.78 | 10 (71) | 0.04 |
Categorical variables (patients with LDL‐C <100 mg/dL and patients with percentage reduction of LDL‐C <50%) were expressed as number (percentage) and compared using the Fisher's exact test. Normally distributed continuous variables (on‐treatment LDL‐C, absolute reduction of LDL‐C, and percentage reduction of LDL‐C) were expressed as mean±SD and compared using permutation test. LDL‐C indicates low‐density lipoprotein cholesterol; LDLR, low‐density lipoprotein receptor; and PCSK9, proprotein convertase subtilisin/kexin type 9.
Patients with LDLR gene variant vs those with PCSK9 gene variant.
Patients with LDLR gene variant vs those with LDLR/PCSK9 gene variants.
Occurrence of Primary and Secondary Outcomes in Patients With LDLR/PCSK9 Gene Variants
The frequencies of primary and secondary outcomes are summarized in Table 5, Table S1, and Table S5. During the observational period (mean, 53±17 years), 39 patients experienced primary outcome (mean time to event occurrence, 50±14 years). Moreover, higher occurrences of primary (nonfatal MI: exact log‐rank: P=0.02) outcomes were observed in patients with LDLR/PCSK9 gene variants compared with those with LDLR gene variant (Figure 2A). Univariate Cox proportional hazards model identified significantly higher incidence of nonfatal MI in male patients (P<0.001), patients with smoking history (P=0.002), and patients with LDLR/PCSK9 gene variants (P=0.03 versus patients with LDLR gene variants). On multivariate Cox proportional hazards model analysis, which included sex and genotype as covariates (model 1), patients with LDLR/PCSK9 gene variants had significantly higher likelihood of experiencing nonfatal MI than those with LDLR gene variant (hazard ratio [HR], 3.21; 95% CI, 1.20–7.20; P=0.02). We further adjusted the history of hypertension, diabetes mellitus, and smoking as covariates. Patients with LDLR and PCSK9 gene variants still had a significantly higher incidence of primary outcome compared with those with LDLR gene variant (HR, 4.26; 95% CI, 1.66–11.0; P=0.003) (model 2) (Table 6). Furthermore, additional analysis adjusted (1) the duration from birth to achieving LDL‐C goal and (2) the duration during optimal LDL‐C control under the therapy as well. The mean values of these variables were 52.0 and 1.8 years, respectively. Even after adjusting these variables, patients with LDLR and PCSK9 gene variants still had higher incidence of nonfatal MI in subjects with HeFH (HR, 6.08; 95% CI, 2.29–16.1; P<0.001 versus patients with LDLR gene variant).
Table 5.
Summary of Primary and Secondary Outcomes
| Variable | All Patients (n=232) | Patients With LDLR/PCSK9 Gene Variants (n=14) | Patients With LDLR Gene Variant (n=183) | Patients With PCSK9 Gene Variant (n=35) |
|---|---|---|---|---|
| Primary outcome: nonfatal MI, n (%) | 39 (17) | 6 (43) | 30 (16) | 3 (9) |
| Secondary outcome: a composite of nonfatal MI and coronary revascularization, n (%) | 69 (30) | 7 (50) | 55 (30) | 7 (20) |
Categorical variables were expressed as number (percentage). LDLR indicates low‐density lipoprotein receptor; MI, myocardial infarction; and PCSK9, proprotein convertase subtilisin/kexin type 9.
Figure 2. Comparison of prognostic influence of genotype.
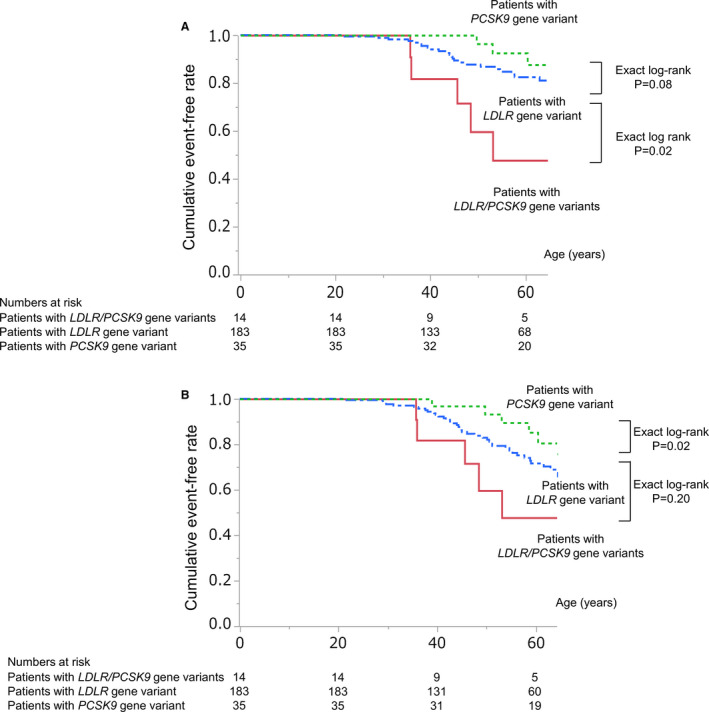
Prognostic influence of genotype in patients with familial hypercholesterolemia on primary outcome (nonfatal myocardial infarction [MI]) (A) and secondary outcome (nonfatal MI and coronary revascularization) (B). Solid red, blue dash‐dotted, and green dotted lines indicate event‐free survival curves for patients with low‐density lipoprotein receptor (LDLR)/PCSK9 (proprotein convertase subtilisin/kexin type 9) gene variants, patients with LDLR gene variant, and patients with PCSK9 gene variant, respectively.
Table 6.
Cox Proportional Hazards Model for Primary Outcome (Nonfatal MI)
| Variable | Unadjusted | Adjusted | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Model 1 | Model 2 | ||||||||
| HR | 95% CI | P Value | HR | 95% CI | P Value | HR | 95% CI | P Value | |
| Men | 4.30 | (2.07–10.1) | <0.001 | 4.73 | (2.28–11.1) | <0.001 | 32.3 | (0.22–4700) | 0.17 |
| Hypertension | 0.92 | (0.46–1.84) | 0.81 | … | … | … | 0.33 | (0.01–7.57) | 0.49 |
| Diabetes mellitus | 1.93 | (0.46–8.04) | 0.37 | … | … | … | 0.12 | (0.0001–120) | 0.55 |
| Smoking history | 3.42 | (1.55–7.54) | 0.002 | … | … | … | 6.74 | (0.09–510) | 0.68 |
| LDLR gene variants | Reference | … | … | Reference | … | … | Reference | ||
| vs PCSK9 gene variant | 0.41 | (0.10–1.15) | 0.10 | 0.39 | (0.09–1.10) | 0.09 | 0.45 | (0.13–1.53) | 0.21 |
| vs LDLR/PCSK9 gene variants | 2.97 | (1.11–6.68) | 0.03 | 3.21 | (1.20–7.20) | 0.02 | 4.26 | (1.66–11.0) | 0.003 |
Unadjusted HRs for nonfatal MI were calculated by a univariate Cox proportional hazards model. Adjusted HRs were calculated by a multivariate Cox proportional hazards model using (model 1: sex and genotype; and model 2: sex, history of hypertension, diabetes mellitus, and smoking, and genotype) as covariates listed before analysis. HR indicates hazard ratio; LDLR, low‐density lipoprotein receptor; MI, myocardial infarction; and PCSK9, proprotein convertase subtilisin/kexin type 9.
We evaluated the study population after excluding the patients with PCSK9 p.Val4Ile gene variant. Similar to the aforementioned results, patients with LDLR/PCSK9 gene variants had higher LDL‐C level (Figure S1) and worse cardiovascular outcomes than those with LDLR or PCSK9 gene variants (Figure S2A and S2B).
Risk Stratification of Primary Outcome, According to Sex and Genotype
The incidence of primary outcome was further investigated in subgroups stratified according to sex and genotype (Figure 3). Men and patients with LDLR/PCSK9 gene variants were associated with more frequent occurrence of primary outcome ( male patients versus female patients, P=0.006; patients with LDLR/PCSK9 gene variants versus patients with LDLR gene variant, P=0.03). Of note, in men, patients with LDLR/PCSK9 gene variants had a higher risk of primary outcome than patients with LDLR gene variant (P<0.001), whereas the incidence of primary outcome was not statistically significant in female subjects (Figure 3).
Figure 3. Risk for primary outcome (nonfatal myocardial infarction [MI]), stratified according to sex and genotype.
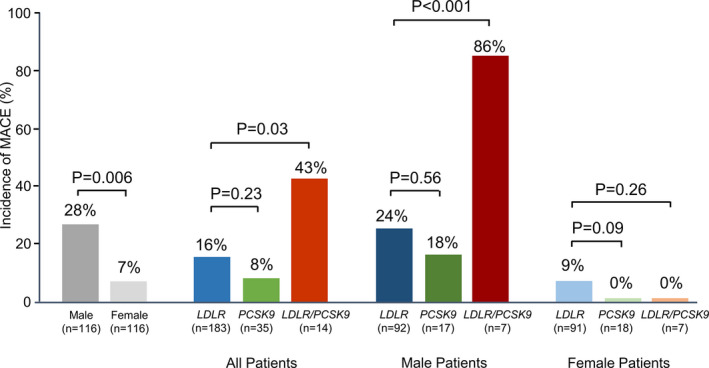
Genotype alone or in combination with sex difference and rate of nonfatal MI is shown. Low‐density lipoprotein receptor (LDLR) indicates patients with LDLR gene variant; LDLR/PCSK9 (proprotein convertase subtilisin/kexin type 9), patients with LDLR and PCSK9 gene variants; and PCSK9, patients with PCSK9 gene variant; MACE indicates major adverse cardiovascular event.
Discussion
Our analyses provide clinical evidence of cardiovascular risk in patients with LDLR/PCSK9 gene variants. Although the prevalence of this genotype was 6% in our Japanese cohort, the lipid profiles and cardiovascular outcomes of the patients were distinct, characterized by higher levels of LDL‐C as well as more frequent occurrence of nonfatal MI. The current findings support a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel that proposed patients with 2 different causative gene variants as a severe genotype of FH. 3
The poor cardiovascular outcomes in patients with LDLR/PCSK9 gene variants could be explained by their more atherogenic lipid profiles based on genetic characteristics. LDLR is a major contributor to low‐density lipoprotein (LDL) metabolism. LDLR gene variant elevates LDL particles via the diminished quality and/or quantity of LDLR 25 and promotes production of apolipoprotein B‐100 from hepatocytes. 26 PCSK9 itself induces an elevation of LDL particles in circulation because of the degradation of LDLR. 27 In addition to LDLR gene variant, the concomitance of PCSK9 gain‐of‐function gene variant could further promote atherogenicity. These basic mechanisms suggest the concomitance of both LDLR and PCSK9 gene variants as a considerably atherogenic genetic phenotype that exhibits substantially high LDL‐C level and a greater frequency of ASCVD.
Differences in baseline triglycerides and apolipoprotein C‐II across the groups are another interesting observation in the current analysis. We observed higher triglyceride levels in patients with PCSK9 gene variant alone and those with LDLR/PCSK9 gene variants. Patients with PCSK9 gain‐of‐function gene variant are characterized as having degradation of LDLR because of higher PCSK9 affinity for LDLR. Given that PCSK9 has been shown to degrade LDLR as well as LDL receptor‐related protein‐1 (LRP1) and very‐LDLR, 28 this property of PCSK9 may result in an elevated level of triglyceride‐rich lipoprotein. In response to this PCSK9‐mediated elevation of triglyceride‐rich lipoprotein, which apolipoprotein C‐II is a component of (eg, very‐LDL), apolipoprotein C‐II might be elevated.
The clinical significance of PCSK9 gene variants, especially PCSK9 p.Val4Ile gene variant, has not been fully annotated. Although this variant has been reported to elevate LDL‐C level in patients with LDLR gene variant, 6 the American College of Medical Genetics guidelines classified it as benign one. In Korean subjects, those with PCSK9 p.Val4Ile gene variant did not necessarily exhibit an elevated LDL‐C level, 29 suggesting that the pathogenicity of PCSK9 p.Val4Ile gene variant is still inconsistent. However, in our analysis, a substantially heightened cardiac risk of LDLR/PCSK9 gene variants existed regardless of including or excluding PCSK9 p.Val4Ile gene variant. This finding underscores the concomitance of LDLR and PCSK9 gene variants to considerably modify atherogenic properties of HeFH.
The current observation highlights the importance of sex difference in cardiac outcomes of HeFH. Similar to our findings, previous reports showed a higher cardiovascular risk in male subjects with HeFH than female subjects with HeFH. 30 , 31 One of possible mechanisms behind these observations could be atheroprotective properties of sex hormones, such as estrogen. Because estrogen has been shown to modulate inflammation and oxidative stress, these estrogen‐mediated effects may account for different cardiovascular outcomes between male and female patients with HeFH. Our findings as well as data from previous reports indicate sex as an important clinical characteristic to stratify future cardiovascular risks in subjects with HeFH.
The current observation provides additional evidence that supports genetic testing to refine risk stratification of cardiovascular events in subjects with HeFH. Accumulating findings through numerous genetic studies have shown that pathogenic variants and their severity, causing HeFH, were associated with the degree of hypercholesterolemia and the risk for the development of coronary artery diseases. However, possibly because of its costing issue as well as ethical reasons, genetic testing is underused in the clinical settings. 32 Our analysis demonstrated patients with LDLR/PCSK9 gene variants as another prognostic genotype exhibiting high LDL‐C and worse cardiovascular risk as well. In particular, cardiac event rate markedly increased in “male” with HeFH with this genetic variant. This approach could help to identify patients with high‐risk HeFH who require intensified lipid‐lowering therapies.
The high cardiovascular risk in patients with LDLR/PCSK9 gene variants emphasizes the need to adopt more stringent lipid management in these patients. In the TAUSSIG (Trial Assessing Long Term Use of PCSK9 Inhibition in Subjects With Genetic LDL Disorders), patients with LDLR/PCSK9 gene variants responded well to PCSK9 inhibitor, evolocumab. 33 This observation indicates the importance of commencing PCSK9 inhibitors earlier to prevent future ASCVD in patients with LDLR/PCSK9 gene variants.
Lower apolipoprotein A‐I level in patients with LDLR/PCSK9 gene variants indicates this apolipoprotein as a potential therapeutic target to mitigate their cardiovascular risks. Apolipoprotein A‐I harbors a variety of atheroprotective properties, including cholesterol efflux capacity. 34 Because of these attractive antiatherosclerotic effects, a variety of apolipoprotein A‐I mimetic peptides have been developed, and their clinical benefit has been investigated. However, recent clinical trials did not find any favorable benefit of these agents to halt coronary atherosclerosis in patients with ASCVD. 35 In addition, Ditiatkovski et al reported that in vivo functional properties of apolipoprotein A‐I mimetic peptide were not necessarily the same as their in vitro ones. 36 These findings suggest the complexity of apolipoprotein A‐I functionality, which requires a better approach to evaluate its in vivo efficacy on atherosclerotic plaques. Difficulties still exist to translate antiatherosclerotic effects of apolipoprotein A‐I into the clinical settings.
Several caveats should be noted. First, this was an observational study conducted in a single center, and the numbers of unrelated patients with LDLR/PCSK9 gene variants and those who experienced cardiovascular events were relatively small. In addition, the use and selection of lipid‐lowering therapy were conducted according to individual physicians’ discretion. The current study analyzed lifetime cardiac events after the birth, but not those after the first visit to our clinic. This is because 82% of primary outcomes occurred before their first visit. These analyses may induce immortal time bias. The CIs of HRs in some variables are wide. This is possibly because of small numbers of study subjects in patients with PCSK9 gene variants alone (n=35) and with LDLR/PCSK9 gene variants (n=14). We stratified subjects according to types of gene variants but not their putative functions. The patients with LDLR/PCSK9 gene variants and those with LDLR gene variant may include a wide range of biologic properties, which may also cause their wide CI (model 1: 95% CI, 1.20–7.20; model 2: 95% CI, 1.66–11.0). Because APOB gene variant has not been identified in the Japanese population, 5 clinical demographics and outcomes in patients with LDLR and APOB or APOB and PCSK9 gene variants remain unknown.
In conclusion, patients with LDLR/PCSK9 gene variants were associated with more atherogenic lipid profiles and a greater likelihood of experiencing ASCVD. Our findings suggest that patients with LDLR/PCSK9 gene variants are a high‐risk FH category who warrant intensive and personalized management.
Sources of Funding
This work was partly supported by grants from the Ministry of Health, Labour and Welfare of Japan for Clinical Research on Intractable Diseases (H26‐nanji‐ippan‐056, H30‐nanji‐ippan‐003), by grant‐in‐aid for scientific research (KAKENHI) from the Japan Society for the Promotion of Science (JP17K08681), and by grants from the Japan Agency for Medical Research and Development (16ek0210075h0001), from the Intramural Research Fund for Cardiovascular Diseases of the National Cerebral and Cardiovascular Center (28‐2‐2; 29‐6‐11), from a Japan Heart Foundation and Astellas Grant for Research on Atherosclerosis Update, from the Takeda Science Foundation, and from the Japanese Circulation Society.
Disclosures
Dr Harada‐Shiba is an outside director of Liid Pharmaceuticals; has received grants from Aegerion, Astellas Pharma, Kaneka Medics, MSD, and Takeda; and has received lecture honoraria for her lectures from Astellas Pharma, Astellas Amgen, Kowa, MSD, and Sanofi. Dr Kataoka has received grants from Cerenis Therapeutics; and has received lecture honoraria for his lectures from Astellas Amgen, Kowa, MSD, and Sanofi. Dr Ogura has received lecture honoraria for his lectures from Astellas, Astellas Amgen, and Sanofi. Dr Son has received personal fees from MSD, Takeda, Mitsubishi Tanabe Pharma, Ono, and Taisho Toyama; and has received grants from MSD, Sanofi, Eli Lilly, Novartis, Takeda, and Mitsubishi Tanabe. Dr Shimokawa has received grants from Bayer; has received scholarship fund from Astellas Pharma, Daiichi‐Sankyo, Mitsubishi Tanabe Pharma, Nippon Shinyaku, Shionogi, Sumitomo Dainippon Pharma, and Teijin Pharma; has an affiliation with endowed department with Astellas Pharma, AstraZeneka, Boehringer Ingelheim, Kowa, Mochida, Nippon Shinyaku, Ono, Otsuka, and Takeda; and has received lecture honoraria for his lectures from Bayer, Boehringer Ingelheim, and Daiichi‐Sankyo. Dr Yasuda has received grants from Takeda; and has received lecture honoraria for his lectures from Bristol Meyers and Daiichi‐Sankyo.
Supporting information
Tables S1–S5
Figures S1–S2
Acknowledgments
We thank Naotaka Ohta, Hiroaki Masuda, Rieko Isoda, and Suguru Yamamoto for DNA analysis.
(J Am Heart Assoc. 2021;10:e018263. DOI: 10.1161/JAHA.120.018263.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.018263
For Sources of Funding and Disclosures, see page 10.
References
- 1. Watts GF, Gidding S, Wierzbicki AS, Toth PP, Alonso R, Brown WV, Bruckert E, Defesche J, Lin KK, Livingston M, et al. Integrated guidance on the care of familial hypercholesterolemia from the International FH Foundation. J Clin Lipidol. 2014;8:148–172. DOI: 10.1016/j.jacl.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 2. Gidding SS, Ann Champagne M, de Ferranti SD, Defesche J, Ito MK, Knowles JW, McCrindle B, Raal F, Rader D, Santos RD, et al. The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association. Circulation. 2015;132:2167–2192. DOI: 10.1161/CIR.0000000000000297. [DOI] [PubMed] [Google Scholar]
- 3. Santos RD, Gidding SS, Hegele RA, Cuchel MA, Barter PJ, Watts GF, Baum SJ, Catapano AL, Chapman MJ, Defesche JC, et al. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016;4:850–861. DOI: 10.1016/S2213-8587(16)30041-9. [DOI] [PubMed] [Google Scholar]
- 4. Khera AV, Won H‐H, Peloso GM, Lawson KS, Bartz TM, Deng X, van Leeuwen EM, Natarajan P, Emdin CA, Bick AG, et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–2589. DOI: 10.1016/j.jacc.2016.03.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mabuchi H, Nohara A, Noguchi T, Kobayashi J, Kawashiri M‐A, Inoue T, Mori M, Tada H, Nakanishi C, Yagi K, et al. Genotypic and phenotypic features in homozygous familial hypercholesterolemia caused by proprotein convertase subtilisin/kexin type 9 (PCSK9) gain‐of‐function mutation. Atherosclerosis. 2014;236:54–61. DOI: 10.1016/j.atherosclerosis.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 6. Ohta N, Hori M, Takahashi A, Ogura M, Makino H, Tamanaha T, Fujiyama H, Miyamoto Y, Harada‐Shiba M. Proprotein convertase subtilisin/kexin 9 V4I variant with LDLR mutations modifies the phenotype of familial hypercholesterolemia. J Clin Lipidol. 2016;10:547–555. DOI: 10.1016/j.jacl.2015.12.024. [DOI] [PubMed] [Google Scholar]
- 7. Sjouke B, Defesche JC, Hartgers ML, Wiegman A, Roeters van Lennep JE, Kastelein JJ, Hovingh GK. Double‐heterozygous autosomal dominant hypercholesterolemia: clinical characterization of an underreported disease. J Clin Lipidol. 2016;10:1462–1469. DOI: 10.1016/j.jacl.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 8. Harada‐Shiba M, Arai H, Ishigaki Y, Ishibashi S, Okamura T, Ogura M, Dobashi K, Nohara A, Bujo H, Miyauchi K, et al. Guidelines for diagnosis and treatment of familial hypercholesterolemia 2017. J Atheroscler Thromb. 2018;25:751–770. DOI: 10.5551/jat.CR003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier‐Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. DOI: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fasano T, Sun XM, Patel DD, Soutar AK. Degradation of LDLR protein mediated by “gain of function” PCSK9 mutants in normal and ARH cells. Atherosclerosis. 2009;203:166–171. DOI: 10.1016/j.atherosclerosis.2008.10.027. [DOI] [PubMed] [Google Scholar]
- 11. Noguchi T, Katsuda S, Kawashiri MA, Tada H, Nohara A, Inazu A, Yamagishi M, Kobayashi J, Mabuchi H. The E32K variant of PCSK9 exacerbates the phenotype of familial hypercholesterolaemia by increasing PCSK9 function and concentration in the circulation. Atherosclerosis. 2010;210:166–172. DOI: 10.1016/j.atherosclerosis.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 12. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell‐Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. DOI: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hori M, Ohta N, Takahashi A, Masuda H, Isoda R, Yamamoto S, Son C, Ogura M, Hosoda K, Miyamoto Y, et al. Impact of LDLR and PCSK9 pathogenic variants in Japanese heterozygous familial hypercholesterolemia patients. Atherosclerosis. 2019;289:101–108. DOI: 10.1016/j.atherosclerosis.2019.08.004. [DOI] [PubMed] [Google Scholar]
- 14. Sudhof TC, Goldstein JL, Brown MS, Russell DW. The LDL receptor gene: a mosaic of exons shared with different proteins. Science. 1985;228:815–822. DOI: 10.1126/science.2988123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Du F, Hui Y, Zhang M, Linton MF, Fazio S, Fan D. Novel domain interaction regulates secretion of proprotein convertase subtilisin/kexin type 9 (PCSK9) protein. J Biol Chem. 2011;286:43054–43061. DOI: 10.1074/jbc.M111.273474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD; the Writing Group on behalf of the Joint ESC/ACCF/AHA/WHF Task Force for the Universal Definition of Myocardial Infarction . Third universal definition of myocardial infarction. Circulation. 2012;126:2020–2035. DOI: 10.1161/CIR.0b013e31826e1058. [DOI] [PubMed] [Google Scholar]
- 17. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low‐density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499–502. DOI: 10.1093/clinchem/18.6.499. [DOI] [PubMed] [Google Scholar]
- 18. Besseling J, Kindt I, Hof M, Kastelein JJ, Hutten BA, Hovingh GK. Severe heterozygous familial hypercholesterolemia and risk for cardiovascular disease: a study of a cohort of 14,000 mutation carriers. Atherosclerosis. 2014;233:219–223. DOI: 10.1016/j.atherosclerosis.2013.12.020. [DOI] [PubMed] [Google Scholar]
- 19. Law MR, Wald NJ, Rudnicka AR. Quantifying effect of statins on low density lipoprotein cholesterol, ischaemic heart disease, and stroke: systematic review and meta‐analysis. BMJ. 2003;326:1423. DOI: 10.1136/bmj.326.7404.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Masana L. Pitavastatin ‐ from clinical trials to clinical practice. Atheroscler Suppl. 2010;11:15–22. DOI: 10.1016/S1567-5688(10)71065-5. [DOI] [PubMed] [Google Scholar]
- 21. Nanchen D, Gencer B, Muller O, Auer R, Aghlmandi S, Heg D, Klingenberg R, Räber L, Carballo D, Carballo S, et al. Prognosis of patients with familial hypercholesterolemia after acute coronary syndromes. Circulation. 2016;134:698–709. DOI: 10.1161/CIRCULATIONAHA.116.023007. [DOI] [PubMed] [Google Scholar]
- 22. Vandin F, Papoutsaki A, Raphael BJ, Upfal E. Accurate computation of survival statistics in genome‐wide studies. PLoS Comput Biol. 2015;11:e1004071. DOI: 10.1371/journal.pcbi.1004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella‐Tommasino J, Forman DE, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139:e1046–e1081. [DOI] [PubMed] [Google Scholar]
- 24. Fisher LD, Lin DY. Time‐dependent covariates in the Cox proportional‐hazards regression model. Annu Rev Public Health. 1999;20:145–157. DOI: 10.1146/annurev.publhealth.20.1.145. [DOI] [PubMed] [Google Scholar]
- 25. Jeon H, Blacklow SC. Structure and physiologic function of the low‐density lipoprotein receptor. Annu Rev Biochem. 2005;74:535–562. DOI: 10.1146/annurev.biochem.74.082803.133354. [DOI] [PubMed] [Google Scholar]
- 26. Twisk J, Gillian‐Daniel DL, Tebon A, Wang L, Barrett PH, Attie AD. The role of the LDL receptor in apolipoprotein B secretion. J Clin Invest. 2000;105:521–532. DOI: 10.1172/JCI8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32:71–77. DOI: 10.1016/j.tibs.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Banerjee Y, Santos RD, Al‐Rasadi K, Rizzo M. Targeting PCSK9 for therapeutic gains: have we addressed all the concerns? Atherosclerosis. 2016;248:62–75. DOI: 10.1016/j.atherosclerosis.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 29. Lee CJ, Lee Y, Park S, Kang SM, Jang Y, Lee JH, Lee SH. Rare and common variants of APOB and PCSK9 in Korean patients with extremely low low‐density lipoprotein‐cholesterol levels. PLoS One. 2017;12:e0186446. DOI: 10.1371/journal.pone.0186446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ahmad Z, Li X, Wosik J, Mani P, Petr J, McLeod G, Murad S, Song L, Adams‐Huet B, Garg A. Premature coronary heart disease and autosomal dominant hypercholesterolemia: increased risk in women with LDLR mutations. J Clin Lipidol. 2016;10:101–108.e3. DOI: 10.1016/j.jacl.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Alonso R, Andres E, Mata N, Fuentes‐Jimenez F, Badimon L, Lopez‐Miranda J, Padro T, Muniz O, Diaz‐Diaz JL, Mauri M, et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 2014;63:1982–1989. [DOI] [PubMed] [Google Scholar]
- 32. Sturm AC, Knowles JW, Gidding SS, Ahmad ZS, Ahmed CD, Ballantyne CM, Baum SJ, Bourbon M, Carrie A, Cuchel M, et al. Clinical genetic testing for familial hypercholesterolemia: JACC scientific expert panel. J Am Coll Cardiol. 2018;72:662–680. [DOI] [PubMed] [Google Scholar]
- 33. Raal FJ, Hovingh GK, Blom D, Santos RD, Harada‐Shiba M, Bruckert E, Couture P, Soran H, Watts GF, Kurtz C, et al. Long‐term treatment with evolocumab added to conventional drug therapy, with or without apheresis, in patients with homozygous familial hypercholesterolaemia: an interim subset analysis of the open‐label TAUSSIG study. Lancet Diabetes Endocrinol. 2017;5:280–290. DOI: 10.1016/S2213-8587(17)30044-X. [DOI] [PubMed] [Google Scholar]
- 34. Ogura M, Hori M, Harada‐Shiba M. Association between cholesterol efflux capacity and atherosclerotic cardiovascular disease in patients with familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2016;36:181–188. DOI: 10.1161/ATVBAHA.115.306665. [DOI] [PubMed] [Google Scholar]
- 35. Nicholls SJ, Puri R, Ballantyne CM, Jukema JW, Kastelein JJP, Koenig W, Wright RS, Kallend D, Wijngaard P, Borgman M, et al. Effect of infusion of high‐density lipoprotein mimetic containing recombinant apolipoprotein A‐I Milano on coronary disease in patients with an acute coronary syndrome in the MILANO‐PILOT Trial: a randomized clinical trial. JAMA Cardiol. 2018;3:806–814. DOI: 10.1001/jamacardio.2018.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ditiatkovski M, Palsson J, Chin‐Dusting J, Remaley AT, Sviridov D. Apolipoprotein A‐I mimetic peptides: discordance between in vitro and in vivo properties‐brief report. Arterioscler Thromb Vasc Biol. 2017;37:1301–1306. DOI: 10.1161/ATVBAHA.117.309523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S5
Figures S1–S2