

Abstract
Background
Heart pathological hypertrophy has been recognized as a predisposing risk factor for heart failure and arrhythmia. DUSP (dual‐specificity phosphatase) 26 is a member of the DUSP family of proteins, which has a significant effect on nonalcoholic fatty liver disease, neuroblastoma, glioma, and so on. However, the involvement of DUSP26 in cardiac hypertrophy remains unclear.
Methods and Results
Our study showed that DUSP26 expression was significantly increased in mouse hearts in response to pressure overload as well as in angiotensin II–treated cardiomyocytes. Cardiac‐specific overexpression of DUSP26 mice showed attenuated cardiac hypertrophy and fibrosis, while deficiency of DUSP26 in mouse hearts resulted in increased cardiac hypertrophy and deteriorated cardiac function. Similar effects were also observed in cellular hypertrophy induced by angiotensin II. Importantly, we showed that DUSP26 bound to transforming growth factor‐β activated kinase 1 and inhibited transforming growth factor‐β activated kinase 1 phosphorylation, which led to suppression of the mitogen‐activated protein kinase signaling pathway. In addition, transforming growth factor‐β activated kinase 1–specific inhibitor inhibited cardiomyocyte hypertrophy induced by angiotensin II and attenuated the exaggerated hypertrophic response in DUSP26 conditional knockout mice.
Conclusions
Taken together, DUSP26 was induced in cardiac hypertrophy and protected against pressure overload induced cardiac hypertrophy by modulating transforming growth factor‐β activated kinase 1–p38/ c‐Jun N‐terminal kinase–signaling axis. Therefore, DUSP26 may provide a therapeutic target for treatment of cardiac hypertrophy and heart failure.
Keywords: cardiac hypertrophy, DUSP26, heart failure, TAK1
Subject Categories: Hypertrophy, Heart Failure
Nonstandard Abbreviations and Acronyms
- BW
body weight
- CKO
conditional knockout
- DMSO
dimethyl sulfoxide
- DUSP
dual‐specificity phosphatase
- ERK
extracellular signal‐regulated kinase
- HW
heart weight
- iTAK1
transforming growth factor‐β activated kinase 1–specific inhibitor
- JNK
c‐Jun N‐terminal kinase
- LV
left ventricular
- LVEDD
left ventricular end‐diastolic diameter
- MAPK
mitogen‐activated protein kinase
- p38
p38 kinase
- TAC
transverse aortic constriction
- TAK1
transforming growth factor‐β activated kinase 1
Clinical Implications
What Is New?
Dual‐specificity phosphatase 26 expression was increased in hypertrophic hearts and cardiomyocytes.
Dual‐specificity phosphatase 26 bound to transforming growth factor‐β activated kinase 1 and inhibited transforming growth factor‐β activated kinase 1 phosphorylation, which led to suppression of mitogen‐activated protein kinase signaling pathway.
Transforming growth factor‐β activated kinase 1–specific inhibitor inhibited cardiomyocyte hypertrophy and attenuated the exaggerated hypertrophic response in dual‐specificity phosphatase 26 conditional knockout mice.
What Are the Clinical Implications?
Dual‐specificity phosphatase 26 exerted a protective effect against pressure overload–induced cardiac hypertrophy and may provide a therapeutic target for treatment of cardiac hypertrophy and heart failure.
Enlargement of the heart in response to abnormal stresses, such as hypertension, myocardial injury, or excessive neurohumoral activation is described as “pathological hypertrophy.” 1 Cardiac function is reserved in the early phase of cardiac hypertrophy, which is described as the compensatory phase. However, sustained stress stimulation promotes the transition from the compensatory to the decompensated hypertrophy phase, which is characterized by dilated left ventricular (LV) and reduced contractile dysfunction, generally predisposing to the development of heart failure and arrhythmia. 2 , 3 To date, both clinical and basic studies showed that molecular therapy targeting suppression of LV hypertrophy at the compensatory phase could maintain the cardiac function, resulting in declined mortality. 4 , 5 , 6 As is well known, on hypertrophic stresses, signaling pathways are pivotal regulators of cardiac hypertrophy, which initiate various pathophysiological processes and LV dilation. Therefore, molecules that can modulate the activity of specific signaling pathways could be the promising therapeutic targets for preventing cardiac hypertrophy.
The greater MAPK (mitogen‐activated protein kinase) signaling is the most conversant signaling pathway, which is activated and participates in hypertrophy, remodeling, contractility, and heart failure on abnormal stimulation. The MAPK consists of 3 main branches, which are p38 kinase (p38)/MAPK, ERK (extracellular signal‐regulated kinase) 1/2 and JNKs (c‐Jun N‐terminal kinases). Nearly all of the MAPK signaling components are activated in hypertrophic heart failure of humans and animal models. 7 , 8 , 9 Studies showed that p38 appeared to have both pathological and compensatory functions in the heart. Because overexpression of activated mitogen‐activated protein kinase kinase 3/p38 or mitogen‐activated protein kinase kinase 6/p38 in hearts promote cardiomyopathy with hypertrophy and early lethality in transgenic mice. 10 However, specifically, deletion of the primary p38 gene in the heart also predisposed to heart failure because mice showed a worse phenotype after transverse aortic constriction. 11 The ERK1/2 signaling pathway is generally considered prohypertrophic. MKK1 transgenic mice with activation of ERK1/2 showed concentric growth (wall thickening) of the heart. 12 JNK mainly has an antihypertrophic effect and is reported to repress cardiac hypertrophy through inhibition of calcineurin/nuclear factor of activated T cells signaling. 13 Therefore, the regulation of p38, JNK, and ERK1/2 phosphorylation is critical in determining the response to any given stress stimulation in hearts.
DUSPs (dual‐specificity phosphatases) are a kind of family of proteins, which dephosphorylate the threonine/serine and tyrosine residues of their substrates. Some of them with a regulatory region at N‐terminus are responsible for the dephosphorylation of MAPKs and have been referred to as MAPK phosphatases. Studies showed that overexpression of DUSP1 in the heart alleviated the induction of hypertrophy by aortic banding or catecholamine injection through down‐regulation of p38, MAPK, JNK1/2, and ERK1/2 in mice. 14 DUSP6 knockout mice had a larger heart size because of much greater myocyte proliferation and activation of the ERK1/2 signaling pathway at the molecular level. 15 A recent study showed that DUSP1 and DUSP4 double‐null mice had a lower survival rate associated with more serious cardiac dysfunction and LV dilatation by elevation of the phosphorylation of p38/MAPK. 16 DUSP26 was reported to control the activity of ERK, leading to phosphorylation/dephosphorylation of heat shock transcription factor‐4b and altering its ability to bind DNA. 17 DUSP26 has been proved to regulate cell differentiation, proliferation, and apoptosis through the MAPK pathway. Our resent study demonstrated that DUSP26 expressed in hepatocytes protected against nonalcoholic fatty liver disease by regulation of the TAK1–p38/JNK signaling axis. 18 However, the function and the mechanism of DUSP26 in heart hypertrophy are unclear. In this article, we study in detail the regulation of DUSP26 expression and the effect of DUSP26 on pathological hypertrophy in vitro and in vivo.
Methods
The data, analytic methods, and study materials will not be made available to other researchers for purposes of reproducing the results or replicating the procedure. Other researchers may contact the corresponding author regarding the data and methodological questions.
Mice
All mice used in this study were maintained and bred in the Division of Laboratory Animal Resources at the Tongji Medical College. All animal protocols conformed to the Guidelines for the Care and Use of Laboratory Animals and were approved by the Animal Care and Use Committees of the Central Hospital of Wuhan, Tongji Medical College, Huazhong University of Science and Technology.
Mouse models were divided into 12 groups: DUSP26–conditional knockout (CKO) sham (n=10), DUSP26‐CKO transverse aortic constriction (TAC) (n=10), DUSP26‐Flox sham (n=10), DUSP26‐Flox TAC (n=10), DUSP26‐transgenic sham (n=10), DUSP26‐transgenic TAC (n=10), DUSP26‐nontransgenic sham (n=10), DUSP26‐nontransgenic TAC (n=10), DUSP26‐CKO TAC dimethyl sulfoxide (DMSO) (n=10), DUSP26‐Flox TAC DMSO (n=10), DUSP26‐CKO TAC iTAK1 (transforming growth factor‐β activated kinase 1–specific inhibitor) (n=10), DUSP26‐Flox TAC iTAK1 (n=10).
DUSP26‐floxed mice were obtained as previously reported. 18 Then, the DUSP26‐floxed mice were mated with the tamoxifen‐inducible transgenic mice ([Myh6‐cre/Esr1*]1Jmk/J) that express MerCreMer driven by the cardiomyocyte‐specific α‐myosin heavy chain promoter (α‐myosin heavy chain–MerCreMer, The Jackson Laboratory, stock No. 005650) to obtain Dusp26Flox/Flox‐α‐myosin heavy chain‐MerCreMer mice. Cre‐mediated recombination of floxed alleles was induced in 6‐week‐old mice through an intraperitoneal injection of tamoxifen (25 mg/kg per day, T‐5648; Sigma‐Aldrich, St. Louis, MO) for 5 consecutive days, leading to the generation of cardiac‐specific DUSP26 CKO mice. DUSP26‐floxed mice were also treated with equal doses of tamoxifen injection as the controls.
To produce cardiac‐specific DUSP26 transgenic mice, the linearized α‐myosin heavy chain–DUSP26 plasmid was constructed and then microinjected into fertilized mice embryos to produce cardiac‐specific DUSP26‐transgenic mice identified by polymerase chain reaction (PCR) analysis of tail genomic DNA. The PCR primers were as follows: forward, 5′‐ATCTCCCCCATAAGAGTTTGAG‐3′; and reverse, 5′‐TGAAAGTGGATGCTCATATC‐3′. DUSP26 expression was evaluated by western blotting. Male mice and their wild‐type littermates (nontransgenic) aged 8 to 10 weeks (24–27 g) were used in all subsequent experiments.
iTAK1 5Z‐7‐Oxozeaenol (5Z‐7‐ox; O9890–1 MG; Sigma‐Aldrich) was administered intraperitoneally to mice 5 mg/kg every 3 days to inhibit TAK1 activation.
TAC Surgery
Cardiac hypertrophy was induced in mice through partial TAC of the aortic arch, as previously described with some adaptations. 19 Briefly, 8‐ to 10‐week‐old male mice were fixed in a supine position after being anesthetized with sodium pentobarbital via an intraperitoneal injection, and the skin in the middle of the chest was opened to expose the aortic arch through the right side of clavicle after the toe pinch reflex disappeared. Body temperature was maintained as close as possible to 37.0°C throughout the experiment using a self‐regulating heating pad. Subsequently, a specific needle (26‐G for body weights [BWs] of 25–27 g) was placed on the aortic arch and ligating with 7‐0 silk suture; then, the needle was removed rapidly before the closure of the skin. Mice were observed until recovery in a 37.0°C heated cage. The sham group underwent a similar procedure just without constriction of the aortic arch.
Echocardiographic Analysis of Cardiac Function
Echocardiography was performed to evaluate cardiac function at 4 weeks following sham and TAC using a MyLabGamma ultrasound system (Esaote, Genoa, Italy) with an 18‐MHz transducer. Mice were anesthetized with isoflurane (1.5%–2%) and fixed on the plate. The end‐systole and end‐diastole were defined as the phases in which the smallest or largest LV area was obtained, respectively. The LV end‐diastolic diameter (LVEDD), LV end‐systolic diameter, LV fractional shortening, and LV ejection fraction were measured from M‐mode records at the time of the end systole and end diastole, respectively, with a sweep of 50 mm/s at the midpapillary muscle level. LV fractional shortening was calculated as follow: LV fractional shortening % = ([LVEDD−LV end‐systolic diameter]/LVEDD×100%). All measurements were averaged over 3 consecutive cardiac cycles. Heart rates of mice were monitored continuously, and the echocardiographic examination would be conducted when the heart rates reached 400 bmp (Figure S1).
Cardiac Histology
Hearts were obtained from experimental animals 4 weeks after TAC surgery that had been perfused with a 10% potassium chloride solution to induce cardiac arrest at the end of diastole and then harvested and fixed with a 10% formalin solution. After being embedded in paraffin, the hearts were cut into 5‐μm transverse sections. Sections were stained with hematoxylin and eosin to measure the myocyte cross‐sectional area, and the abundance of collagen was assessed after Picrosirius red staining. Fibrosis was expressed as a percentage of the average positively stained area relative to the total area. More than 40 fields per group were examined. A quantitative digital image analysis system (Image‐Pro Plus 6.0; Media Cybernetics, Inc., Rockville, MD) was used for the image measurements.
Cardiomyocyte Culture and Infection With Recombinant Lentiviral Vectors
The DUSP26 gene was amplified by nested PCR and then cloned into the pHAGE‐3×flag vector. shRNAs targeting DUSP26 were cloned to pLKO.1 vector to knockdown DUSP26 gene. For transfection and lentiviral infection, recombinant constructs were cotransfected into HEK‐293T cells with lentiviral helper plasmids (pMD2.G and psPAX2) and polyethylenimine transfection reagent. After 6 to 16 hours, fresh media were replaced and lentiviral particles were harvested 48 hours later and then used for target cell infection. Briefly, viruses containing DUSP26 and control particles were added to H9C2 cells in the presence of polybrene (8 μg/mL). Infected cells were selected by puromycin (2 μg/mL) for 48 hours and then used for indicated assays.
Immunofluorescence
Immunofluorescence staining was performed to determine the surface area of the cell. H9C2 cells were infected with the indicated lentivirus for 24 hours, stimulated with phosphate‐buffered saline (PBS), angiotensin II (1 μmol/L) or iTAK1 NG25 (2.5 μmol/L, HY‐15434, MCE) for 48 hours under a condition of 37.0°C, 5% CO2 and fixed with 4% formaldehyde. After permeabilization with 0.1% Triton X‐100 in PBS and blocking with a 10% bovine serum albumin solution at room temperature, cells were immunostained with an α‐actinin antibody (1:100 dilution, 05‐384; Merck Millipore, Burlington, MA), followed by staining with a fluorescent secondary antibody (donkey anti‐mouse IgG [H+L] secondary antibody, 1:200, A21202; Invitrogen, Carlsbad, CA). Image‐Pro Plus 6.0 software was used to measure the surface area of the cell.
Quantitative Real‐Time PCR and Western Blotting
TRIzol reagent (15596018, Thermo Fisher Scientific, Waltham, MA) was used to extract total RNA from indicated cells or tissues according to the manufacturer's instructions. RNA was reverse transcribed into cDNA using a Transcriptor First Strand cDNA Synthesis Kit (04896866001; Roche, Basel, Switzerland), and then quantitative real‐time PCR amplification was performed using SYBR Green PCR Master Mix (04887352001, Roche) to detect the gene expression. Glyceraldehyde‐3‐phosphate dehydrogenase was used as the reference gene. The primer pairs used in this study are listed in Table S1.
For the western blot analysis, total protein samples were extracted from ventricular tissues or cell samples using RIPA lysis buffer (50 mmol/L Tris‐HCl PH7.4, 150 mmol/L NaCl, 1% Triton X‐100, 1% sodium deoxycholate, 0.1% SDS, 1 mmol/L EDTA), containing protease inhibitor cocktail (4963124001, Roche) and the protein concentration was determined with a BCA Protein Assay Kit (23225, Thermo Fisher Scientific). Protein samples were separated by SDS‐PAGE, then transferred to polyvinylidene fluoride membranes and continuously blocked with 5% nonfat milk at room temperature for 1 hour. After incubation with multiple antibodies overnight at 4°C, the secondary antibodies were added the next day, and bands were visualized using the ChemiDoc XRS+ system (Bio‐Rad Laboratories, Hercules, CA). The levels of specific proteins were normalized to the levels of glyceraldehyde‐3‐phosphate dehydrogenase. The antibodies used in this study are listed in Table S2.
Plasmid Constructs
Coding region sequences of DUSP26 were amplified by PCR and cloned to pHAGE‐Flag lentiviral vector. One independent shRNAs targeting DUSP26 was designed, synthesized, and cloned to pLKO.1‐puro vector. Expression plasmids of DUSP26 and TAK1 were obtained by PCR and then inserted into pcDNA5 vectors with indicated tag. Primers used to generate these constructs are listed in Table S3.
Immunofluorescence Staining in Cells
293T cells with pcDNA5‐Flag‐ DUSP26 /pcDNA5‐HA‐TAK1 plasmids were fixed with 4% paraformaldehyde followed by permeabilization with 0.1% Triton X‐100 and then incubated with indicated primary antibodies. The cells were then incubated with an Alexa Fluor 488–conjugated goat anti‐mouse IgG (A11031, 1:500 dilution, Invitrogen) and Alexa Fluor 568–conjugated goat anti‐rabbit IgG (A11036, 1:500 dilution, Invitrogen) secondary antibody for 1 hour. Images were acquired by confocal microscopy (LCS‐SP8‐STED, Leica Camera, Wetzlar, Germany).
Coimmunoprecipitation Assays
Immunoprecipitation was performed to determine protein–protein interactions. For c‐immunoprecipitation experiments, HEK293T cells were cotransfected with the indicated plasmids. After transfection for 24 hours, cells were washed with cold PBS and resuspended in ice‐cold immunoprecipitation buffer (20 mmol/L Tris‐HCl, pH 7.4, 150 mmol/L NaCl, 1 mmol/L EDTA, and 0.5% NP‐40) containing protease inhibitor cocktail (04693132001, Roche). The obtained cell lysates were incubated with protein A/G‐agarose beads (P4691, Sigma‐Aldrich) and indicated antibodies for 3 hours at 4°C. The beads were washed 5 to 6 times with immunoprecipitation buffer containing 150 or 300 mmol/L NaCl, then the immunoprecipitated proteins boiled with SDS loading buffer and subjected to immunoblotting for analysis. Endogenous immunoprecipitation of DUSP26 and TAK1 in DUSP26 overexpressed cells was performed similarly using indicated antibodies.
Statistical Analysis
Data are presented as means±SD. Comparisons between 2 groups were performed using a 2‐tailed Student t test. Differences among more than 2 groups were assessed using 1‐way ANOVA, followed by the Bonferroni test (equal variances assumed) or Tamhane's T2 (equal variances not assumed) test. Small‐sample‐size groups (n<4) and nonnormally distributed data were analyzed with the nonparametric Kruskal–Wallis test followed by Dunn's test. A value of P<0.05 was considered to indicate a statistically significant difference. All statistical analyses were performed using SPSS software, version 21.0 (IBM, Armonk, NY).
Results
DUSP26 Expression Was Increased in Hypertrophic Hearts and Cardiomyocytes
To explore the potential role of DUSP26 in the development of cardiac hypertrophy and heart failure, we first examined whether the expression levels of DUSP26 were altered in pathological hearts. TAC in the mouse is a commonly used experimental model for pressure overload–induced cardiac hypertrophy and heart failure. 19 At 4 or 8 weeks after C57BL/6 mice underwent TAC surgery, western blotting, and real‐time PCR analysis revealed that the DUSP26 protein expression and messengers RNA (mRNA) levels were significantly increased in TAC hearts, compared with sham group (Figure 1A and 1B). Furthermore, using H9C2 cells treated with angiotensin II (1 μmol/L) for 48 hours in vitro to induce hypertrophy, we found that the DUSP26 protein expression level was also increased in these hypertrophic cardiomyocytes (Figure 1C). Together, DUSP26 was significantly increased in pressure overload–induced hypertrophic hearts in vivo as well as in angiotensin II–treated cardiomyocytes in vitro.
Figure 1. DUSP26 expression was increased in hypertrophic hearts and cardiomyocytes.
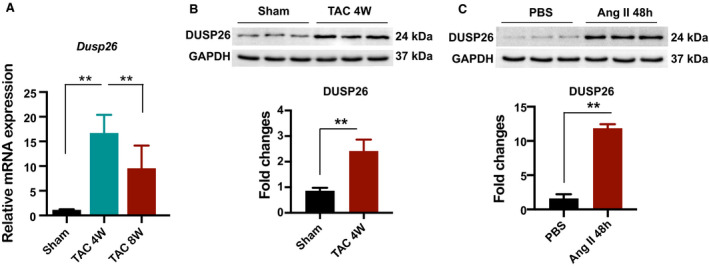
1A, Real‐time PCR analysis of Dusp26 mRNA levels in the hearts of mice subjected to transverse aortic constriction or sham for the indicated time (n=3 per group). B, Western blot analysis of DUSP26 protein levels in the hearts of mice subjected to transverse aortic constriction or sham for the indicated time (n=3 per group). C, Western blot analysis of DUSP26 protein levels in H9C2 cells treated with angiotensin II (1 μmol/L) for 48 hours. Data are presented as the mean±SD. **P<0.01. DUSP26 indicates dual‐specificity phosphatase 26; PBS, phosphate‐buffered saline; PCR, polymerase chain reaction; and TAC, transverse aortic constriction.
DUSP26 Suppressed Angiotensin II–Induced Cardiomyocyte Hypertrophy
To further explore the effects of DUSP26, we performed gain‐ and loss‐of‐function experiments on cardiomyocytes. H9C2 cells were infected with shRNAs cloned to the pLKO.1 vector to knock down DUSP26 or nontargeting pLKO.1 vector as a control (Figure 2A and 2B). These cells were treated with angiotensin II (1 μmol/L) to induce cellular hypertrophy or PBS control for 48 hours and were immunostained with α‐actinin. In the angiotensin II–treated groups, cardiomyocyte hypertrophy was much more serious in DUSP26‐KD H9C2 cells as indicated by obviously increased cell surface area (Figure 2C) and mRNA levels of hypertrophic markers (Anp and Myh7) were also higher compared with H9C2 cells infected with the control pLKO.1 vector (Figure 2D). Conversely, overexpression of DUSP26 with Flag‐DUSP26 protected cardiomyocytes against hypertrophy induced by angiotensin II, which was demonstrated by reduced cellular surface area and mRNA levels of Anp and Myh7 (Figure 2E through 2G). These results indicated that DUSP26 negatively regulated cardiac hypertrophy in vitro.
Figure 2. DUSP26 suppressed angiotensin II–induced cardiomyocyte hypertrophy.
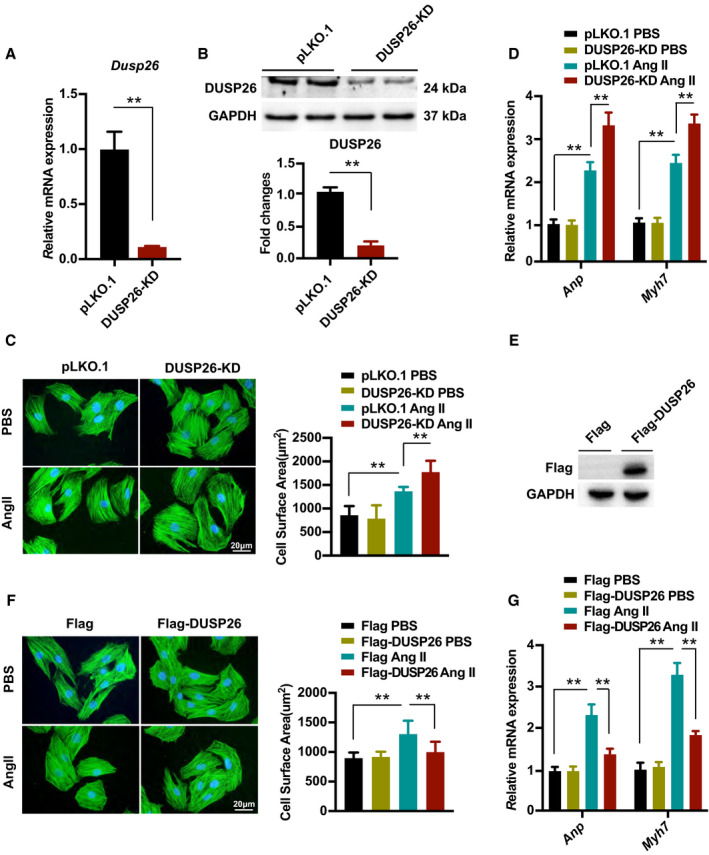
A and B, Real‐time PCR analysis (A) and western blot (B) of DUSP26 levels in H9C2 cells infected with lentiviruses targeting DUSP26 or nontargeting pLKO.1 vector. C, Representative images of H9C2 cells infected with lentiviruses targeting DUSP26 or nontargeting pLKO.1 vector and treated with angiotensin II (1 μmol/L) or PBS for 48 hours. Cardiomyocytes were stained with antibody against α‐actinin. Quantitative data of average myocyte surface area. (blue, nucleus; green, α‐actinin; scale bar, 20 μm). D, Real‐time PCR analysis of hypertrophic markers expression in H9C2 cells infected with lentiviruses targeting DUSP26 or nontargeting pLKO.1 vector followed by PBS or angiotensin II (1 μmol/L) treatment for 48 hours. E, Western blot analysis of DUSP26 levels in H9C2 cells infected with lentiviruses containing Flag‐DUSP26 or Flag‐control. F, Representative images of H9C2 cells infected with indicated lentiviral vectors followed by PBS or angiotensin II (1 μmol/L) treatment for 48 hours and quantitative data of average myocyte surface area. (blue, nucleus; green, α‐actinin; scale bar, 20 μm). G, Quantification results for mRNA levels of the hypertrophic marker genes (Anp, Myh7) in H9C2 cells infected with lentiviruses containing Flag‐DUSP26 or Flag‐control followed by PBS or angiotensin II (1 μmol/L) treatment for 48 hours. Data are presented as the mean±SD. **P<0.01. Anp indicates atrial natriuretic peptide; DUSP26, dual‐specificity phosphatase 26; Myh7, myosin heavy chain 7; PBS, phosphate‐buffered saline; and PCR, polymerase chain reaction.
Cardiac‐Specific DUSP26 Deletion Aggravated Pressure Overload–Induced Hypertrophy In Vivo
To evaluate the function of DUSP26 in pathological hypertrophy in vivo, we used a mouse model with cardiac‐specific DUSP26‐CKO mice. The DUSP26 protein expression in DUSP26‐CKO hearts was confirmed by western blot (Figure 3A). These mice had no pathological alterations in cardiac structure and function at baseline. After 4 weeks of TAC surgery, DUSP26‐CKO mice exhibited a remarkable deterioration in cardiac hypertrophy compared with control littermates (Flox) mice, as evinced by increased ratio of heart weight (HW) to body weight (Figure 3B), ratio of lung weight to BW (Figure 3C), and also HW to tibia length (Figure 3D). Histological examination revealed an increased cross‐sectional area of cardiomyocytes in DUSP26‐CKO mice compared with Flox mice (Figure 3E). Accordingly, the mRNA levels of several hypertrophic markers, including Anp, Bnp and Myh7, were much higher in DUSP26‐CKO mice than in Flox mice (Figure 3K). Additionally, with no significant difference in heart rates (Figure S1A), echocardiographic and hemodynamic measurements demonstrated that DUSP26‐CKO mice showed more serious left ventricular dilation and cardiac dysfunction than Flox mice in response to pressure overload, as indicated by LVEDD, LV end‐systolic dimension, LV fractional shortening, and LV ejection fraction (Figure 3F through 3I). Meanwhile, cardiac DUSP26 deficiency promoted pressure overload–mediated cardiac fibrosis, as evinced by much more fibrosis signal both in the interstitial and perivascular regions (Figure 3J). The mRNA levels of several fibrosis markers, including Collagen Iα, Collagen III and Ctgf, were much higher in DUSP26‐CKO mice than in Flox mice (Figure 3L). Taken together, these results demonstrated that cardiac‐specific absence of DUSP26 in mice aggravates pathological cardiac hypertrophy induced by pressure overload.
Figure 3. Cardiac‐specific DUSP26 deletion aggravated pressure overload–induced hypertrophy in vivo.
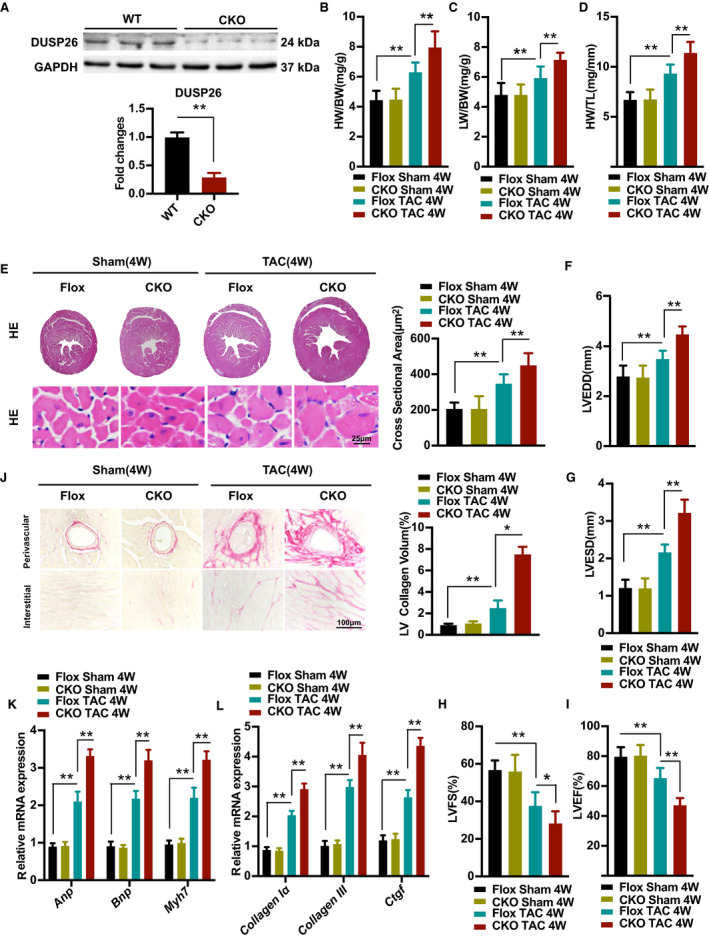
A, Western blot analysis of protein expression of DUSP26 in hearts of 8 week‐old WT or DUSP26 knockout mice (n=3 per group). B through D, The ratios of heart weight to body weight (HW/BW; B), lung weight to body weight (LW/BW; C) and heart weight to tibia length (HW/TL; D) in Flox and DUSP26‐CKO mice after 4 weeks of TAC or sham surgery. (n=10 per group). E, Representative images of heart sections with histological examination and quantification of myocyte cross‐sectional area in Flox and DUSP26‐CKO mice after 4 weeks of TAC or sham surgery as indicated by groups (scale bar, 25 μm; n=6 per group). F through I, Echocardiographic measurements of indicated groups. Average data of left ventricular end‐diastolic diameter (LVEDD; F), left ventricular end‐systolic diameter (LVESD; G), left ventricular fractional shortening (LVFS; H) and left ventricular ejection fraction (LVEF; I) (n=10 per group). J, Representative images of histological analysis of cardiac perivascular and interstitial fibrosis and quantification for the fibrotic area in different genotype groups. (scale bar, 100 μm; n=6 per group). K and L, Real‐time PCR analysis of mRNA levels of multiple hypertrophic marker genes (Anp, Bnp, and Myh7) (K) and fibrotic marker genes (collagen Iα, collagen III, and Ctgf) (L) in indicated groups (n=4 per group). Data are presented as the mean ±SD. *P<0.05, **P<0.01. Anp indicates atrial natriuretic peptide; Bnp, brain natriuretic peptide; BW, body weight; CKO, conditional knockout; Ctgf, connective tissue growth factor; DUSP26, dual‐specificity phosphatase 26; Myh7, myosin heavy chain 7; and TAC, transverse aortic constriction.
Cardiac‐Specific DUSP26 Overexpression Attenuated Pressure Overload–Induced Cardiac Hypertrophy
We next investigated whether elevated DUSP26 levels in the heart would attenuate cardiac hypertrophy. To examine this issue, transgenic mice with cardiac‐specific overexpression of DUSP26 were generated using α‐myosin heavy chain promoter (Figure 4A). DUSP26‐transgenic mice were established and verified by western blotting and real‐time PCR (Figure 4B and 4C). Under basal conditions, the cardiac‐specific DUSP26 transgenic mice were healthy and showed no apparent cardiac morphologic or pathological abnormalities (data not showed). Cardiac‐specific DUSP26‐transgenic mice and their wild‐type littermates (referred to as nontransgenic) were subjected to TAC surgery or sham operation. Concordant with our hypothesis, cardiac‐specific DUSP26 overexpression significantly inhibited cardiac hypertrophy in response to pressure overload, as evinced by lower ratios of HW/BW, lung weight/BW, HW/tibia length in transgenic mice than in nontransgenic mice (Figure 4D through 4F). The cardiac‐specific DUSP26‐transgenic mice also showed a smaller cross‐sectional area of cardiomyocytes (Figure 4G), as well as decreased mRNA levels of hypertrophic markers (Anp, Bnp, and Myh7) following TAC simulation compared with nontransgenic mice (Figure 4M). Meanwhile, with no significant difference in heart rates (Figure S1B), TAC‐triggered LV dilation and dysfunction as determined by echocardiographic and hemodynamic measurements were significantly improved in the cardiac‐specific DUSP26‐transgenic mice compared with the nontransgenics (Figure 4H through 4K).
Figure 4. Cardiac‐specific DUSP26 overexpression attenuated pressure overload–induced cardiac hypertrophy.
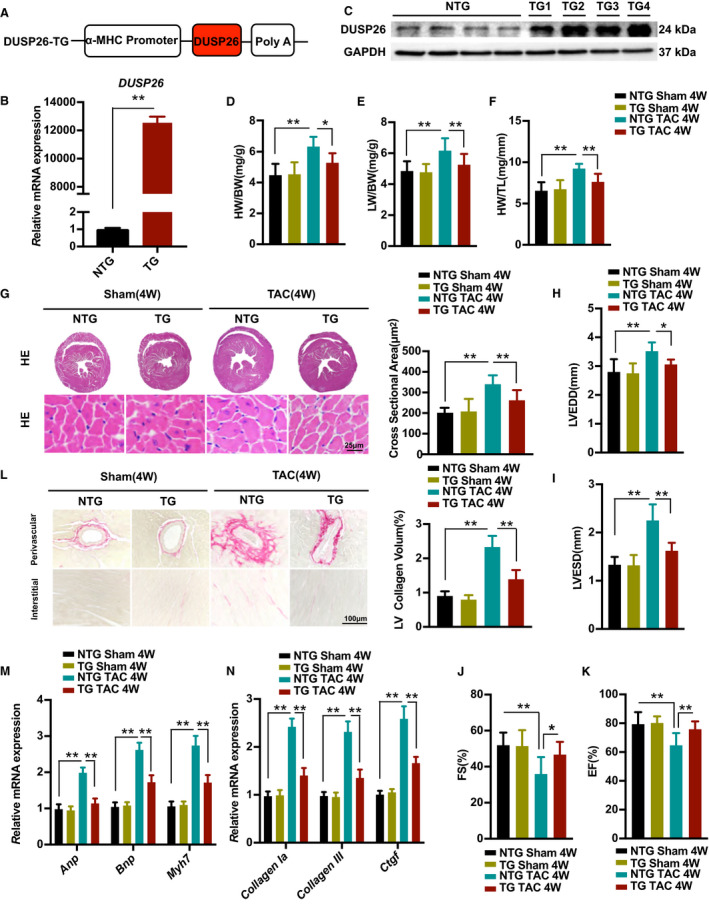
A, Schematic diagram for the construction of the cardiac‐specific expression of the DUSP26‐transgenic mice. B and C, Real‐time PCR analysis (B) and western blot (C) of DUSP26 levels in the DUSP26‐transgenic mice and their nontransgenic littermates (n=4 per group). D through F, The ratios of heart weight to body weight (HW/BW; D), lung weight to body weight (LW/BW; E), heart weight to tibia length (HW/TL; F) in DUSP26‐TG and nontransgenic mice after 4 weeks of TAC or sham surgery. (n=10 per group). G, Representative images of the hematoxylin and eosin staining, quantification of myocyte cross sectional area in indicated groups. (scale bar, 25 μm; n=6 per group). H through K, Echocardiographic measurements in indicated groups. Average data of left ventricular end‐diastolic diameter (LVEDD; H), left ventricular end‐systolic diameter (LVESD; I), left ventricular fractional shortening (LVFS; J) and left ventricular ejection fraction (LVEF; K) (n=10 per group). L, Representative images of histological analysis of cardiac perivascular and interstitial fibrosis and quantification for the fibrotic area in different genotype groups. (scale bar, 100 μm; n=6 per group). M and N, Real‐time PCR analysis of mRNA levels of multiple hypertrophic marker genes (Anp, Bnp, and Myh7) and fibrotic marker genes (collagen Iα, collagen III, and Ctgf) in indicated groups (n=4 per group) Data are presented as the mean±SD. *P<0.05, **P<0.01. Anp indicates atrial natriuretic peptide; Bnp, brain natriuretic peptide; Ctgf, connective tissue growth factor; DUSP26, dual‐specificity phosphatase 26; Myh7, myosin heavy chain 7; and TAC, transverse aortic constriction.
We next assessed the effect of cardiac‐specific DUSP26 overexpression on TAC‐triggered cardiac fibrosis. Cardiac‐specific DUSP26‐TG hearts consistently exhibited decreased fibrosis signals in the interstitial and perivascular spaces, decreased collagen volumes (Figure 4L), as well as decreased mRNA levels of fibrotic markers (collagen Iα, collagen III, and ctgf) after 4 weeks of TAC surgery, compared with nontransgenics (Figure 4N). Collectively, these results indicated that DUSP26 overexpression attenuated cardiac hypertrophy and fibrosis in response to pressure overload.
DUSP26 Inhibited TAK1‐p38/JNK Signaling Pathway In Vivo and In Vitro
To elucidate the molecular mechanisms by which DUSP26 plays a protection role in pathological cardiac hypertrophy, we determined to evaluate the expression of TAK1, ERK, JNK, and p38, which were well‐established regulators of cardiac hypertrophy. 20 , 21 The phosphorylation levels of TAK1, ERK, JNK, and p38 were increased in mice after TAC stimulation, compared with sham controls. However, only the phosphorylation levels of TAK1, JNK, and p38 were affected by DUSP26 in hypertrophic mouse hearts. The pressure overload–induced phosphorylation of TAK1, JNK, and p38 was enhanced in DUSP26‐CKO mice compared with Flox mice but was decreased in DUSP26‐transgenic mice following TAC simulation compared with nontransgenic mice. (Figure 5A and 5B). Consistent with the results in vivo, DUSP26 knockdown resulted in increased phosphorylation levels of TAK1, JNK, and p38 in H9C2 cells after angiotensin II administration, whereas overexpression of DUSP26 markedly inhibited phosphorylation of TAK1, JNK, and p38 (Figure 5C and 5D). These findings suggested that the TAK1‐p38/JNK signaling pathway was regulated by DUSP26 during cardiac hypertrophy in vivo and in vitro.
Figure 5. DUSP26 inhibited TAK1‐p38/JNK signaling pathway in vivo and in vitro.
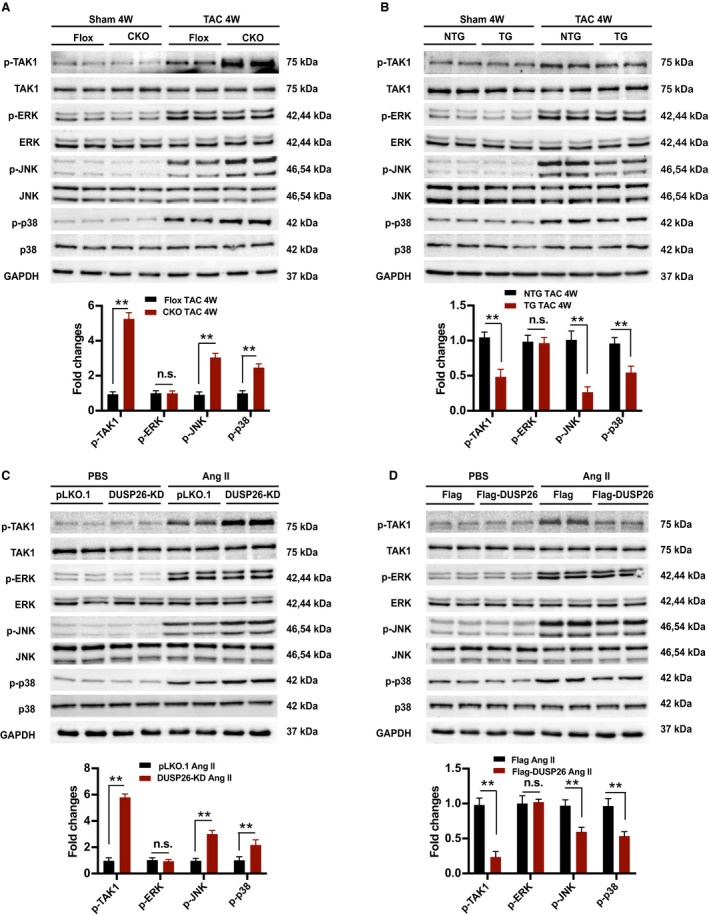
A, Western blot and quantification results of the phosphorylation and total protein levels of TAK1, ERK, JNK, and p38 in the hearts of Flox and DUSP26‐CKO mice 4 weeks after sham or TAC operation (n=4 per group). B, Western blot and quantification results of the phosphorylation and total protein levels of TAK1, ERK, JNK, and p38 in the hearts of DUSP26‐TG and nontransgenic mice 4 weeks after sham or TAC operation (n=4 per group). C, H9C2 cells were infected with lentiviruses targeting DUSP26 or nontargeting pLKO.1 vector and treated with angiotensin II (1 μmol/L) or PBS for 48 hours. D, Western blot measurement and quantification results of phosphorylation and total protein levels of TAK1, ERK, JNK, and p38 in H9C2 cells infected with Flag‐DUSP26 or Flag‐control and treated with angiotensin II (1 μmol/L) or PBS for 48 hours. Data are presented as the mean±SD. **P<0.01. CKO indicates conditional knockout; ERK, extracellular signal‐regulated kinase; JNK, c‐Jun N‐terminal kinase; n.s., no significance between the 2 indicated groups; p38, p38 kinase; TAC, transverse aortic constriction; and TAK1, transforming growth factor‐β activated kinase 1.
DUSP26 Bound to TAK1 and Blocked Its Phosphorylation to Attenuate DUSP26‐Regulated Cardiac Hypertrophy
To further determine the interaction between DUSP26 and TAK1, we transfected HA‐tagged TAK1 and Flag‐tagged DUSP26 into HEK293T cells and then performed immunostaining, immunoprecipitation, and coimmunoprecipitation experiments. Immunostaining showed that DUSP26 and TAK1 colocalized in the cytoplasm (Figure 6A). Immunoprecipitation results demonstrated that DUSP26 interacted with TAK1 and vice versa (Figure 6B). Then, we did coimmunoprecipitation experiments to investigate endogenous protein interactions in H9C2 cells with endogenous TAK1 and infected Flag‐tagged DUSP26, and similar results were observed. (Figure 6C). Furthermore, DUSP26 overexpression resulted in decreased phosphorylation levels of TAK1 at T187, but not at S412 (Figure 6G). These results demonstrated the direct interaction between DUSP26 and TAK1, and TAK1 is dephosphorylated by DUSP26 at T187. As DUSP26 is a MAPK phosphatase, we hypothesized that DUSP26 interacted with TAK1 and then dephosphorylated TAK1 to exert antihypertrophic action.
Figure 6. DUSP26 bound to TAK1 and blocked its phosphorylation to attenuate DUSP26‐regulated cardiac hypertrophy.
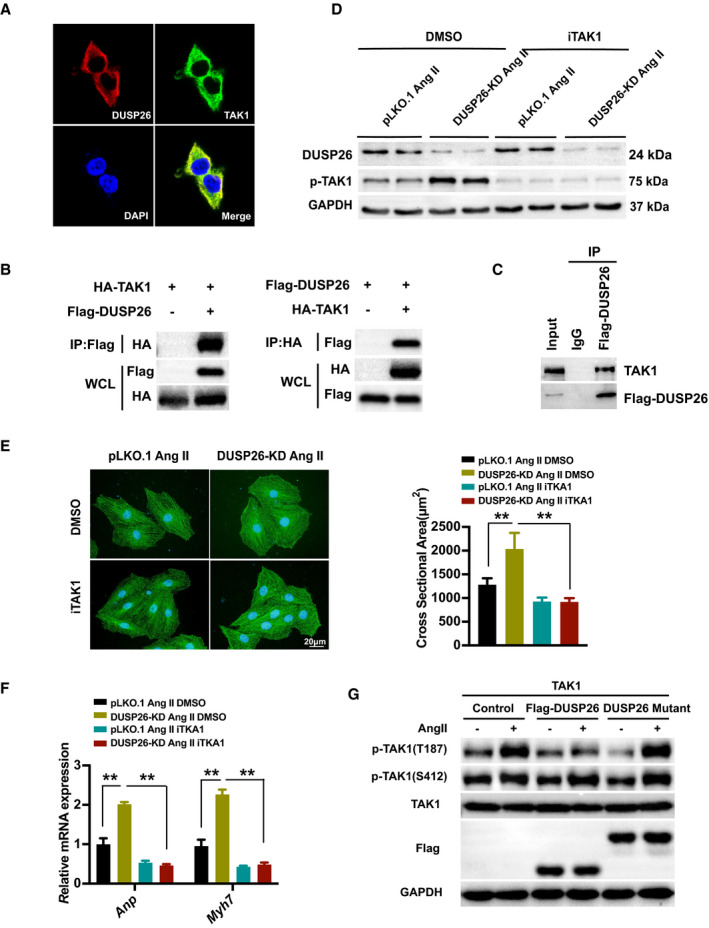
A, Representative images of colocalization of DUSP26 and TAK1 proteins in HEK293T cells with double immunofluorescent staining. (blue, nucleus; green, TAK1; red, DUSP26). B, Total protein extracted from HEK293T cells cotransfected with HA‐TAK1 and Flag‐DUSP26 to perform immunoprecipitation for examining the interaction between TAK1 and DUSP26. C, Total protein extracted from H9C2 cells infected with Flag‐DUSP26 to perform coimmunoprecipitation experiment for examining the interaction between DUSP26 and endogenous TAK1. D, Western blot analysis of protein levels of p‐TAK1 and DUSP26 in H9C2 cells infected with viruses targeting DUSP26 or nontargeting pLKO.1 vector and then treated with iTAK1 (2.5 μmol/L) or DMSO along with angiotensin II (1 μmol/L) stimulation for 48 hours. E, Representative images of H9C2 cells infected with lentiviruses targeting DUSP26 or nontargeting pLKO.1 vector and then treated with iTAK1 (2.5 μmol/L) or DMSO and angiotensin II (1 μmol/L) stimulation for 48 hours. Cardiomyocytes were stained with antibody against α‐actinin. (blue, nucleus; green, α‐actinin; scale bar, 20 μm). F, Quantification results of mRNA levels of the hypertrophic marker genes (Anp, Myh7) in cardiomyocytes infected with viruses targeting DUSP26 or nontargeting pLKO.1 vector and then treated with iTAK1 (2.5 μmol/L) or DMSO and angiotensin II (1 μmol/L) stimulation for 48 hours. G, Western blot results of the phosphorylation protein levels of TAK1 at T187 and S412 and total protein levels of TAK1 in H9C2 cells infected with Flag‐DUSP26 or DUSP26 mutant and treated with angiotensin II (1 μmol/L) or PBS for 30 minutes. **P<0.01. Anp indicates atrial natriuretic peptide; ERK, extracellular signal‐regulated kinase; iTAK1, TAK1‐specific inhibitor; JNK, c‐Jun N‐terminal kinase; Myh7, myosin heavy chain 7; p38, p38 kinase; and TAK1, transforming growth factor‐β activated kinase 1.
The iTAK1 could specifically inhibit the phosphorylation of TAK1. We checked whether the exaggerated hypertrophic response in DUSP26‐KD H9C2 cells could be rescued by blocking TAK1 activity. Compared with DMSO‐treated controls, the phosphorylation of TAK1 induced by angiotensin II in DUSP26‐KD H9C2 cells was largely diminished by treatment of iTAK1 (Figure 6D). As predicted, iTAK1 reversed the worsened hypertrophic response, which were evidenced by inhibition of hypertrophic cardiomyocytes and reduction of the transcript levels of the fetal genes Anp and Myh7 in DUSP26‐KD H9C2 cells treated with angiotensin II and iTAK1 (Figure 6E and 6F). These data further indicated that the DUSP26 played an antihypertrophic role through suppressing the activity of TAK1.
Pharmacological Inhibition of TAK1 Attenuated the Exaggerated Hypertrophic Response in DUSP26‐CKO Mice
We next elucidated whether the exaggerated hypertrophic response could be rescued by blocking TAK1 activity in DUSP26‐CKO mice. Compared with the DMSO control, iTAK1 administration reversed the worsened hypertrophic response, which was evidenced by lower ratios of HW/BW, lung weight/BW, HW/tibia length (Figure 7A through 7C). The mice treated with iTAK1 also exhibited a smaller cross‐sectional area of cardiomyocytes (Figure 7H), as well as decreased mRNA levels of hypertrophic markers (Anp, Bnp, and Myh7) following TAC simulation compared with DMSO control (Figure 7J). With no significant difference in heart rate (Figure S1C), TAC‐triggered LV dilation and dysfunction as determined by echocardiographic and hemodynamic measurements were significantly improved in iTAK1‐treated mice (Figure 7D through 7G).
Figure 7. Pharmacological inhibition of TAK1 attenuated the exaggerated hypertrophic response in DUSP26‐CKO mice.
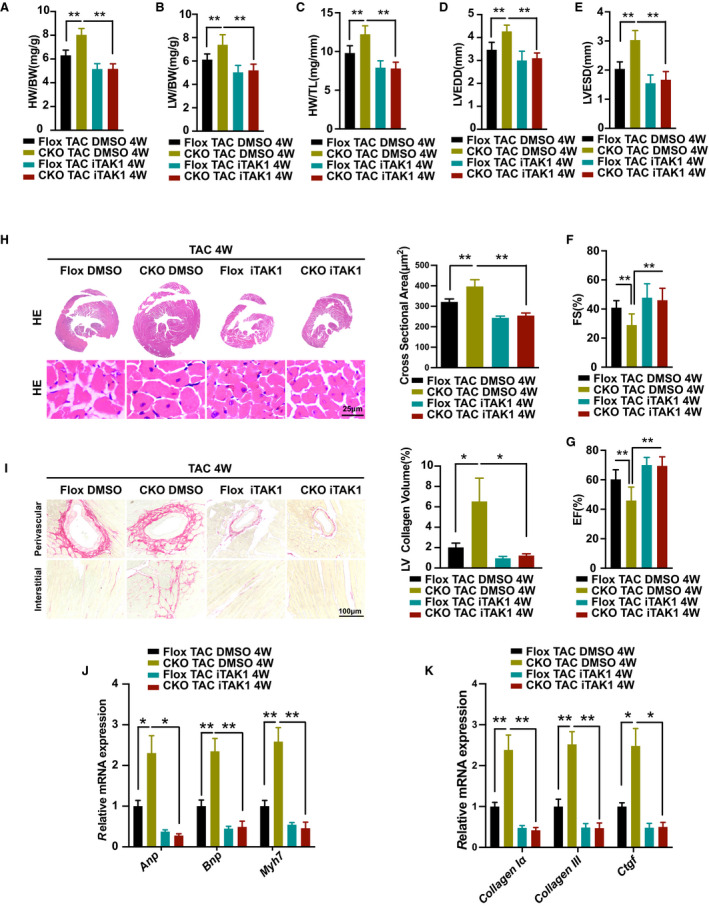
A through C, The ratios of heart weight to body weight (HW/BW; A), lung weight to body weight (LW/BW; B), heart weight to tibia length (HW/TL; C) in DUSP26‐CKO mice treated with TAK1‐specific inhibitor iTAK1 (5 mg/kg) or DMSO by intraperitoneal administration after of TAC surgery. (n=10 per group). D through G, Echocardiographic measurements in indicated groups. Average data of left ventricular end‐diastolic diameter (LVEDD; D), left ventricular end‐systolic diameter (LVESD; E), left ventricular fractional shortening (LVFS; F) and left ventricular ejection fraction (LVEF; G) (n=10 per group). H, Representative images of the hematoxylin and eosin staining, quantification of myocyte cross‐sectional area in indicated groups. (scale bar, 25 μm; n=6 per group). I, Representative images of histological analysis of cardiac perivascular and interstitial fibrosis and quantification for the fibrotic area in different genotype groups (scale bar, 100 μm; n=6 per group). J and K, Real‐time PCR analysis of mRNA levels of multiple hypertrophic marker genes (Anp, Bnp, and Myh7) and fibrotic marker genes (collagen Iα, collagen III, and Ctgf) in indicated groups. (n=4 per group) Data are presented as the mean±SD. *P<0.05, **P<0.01.; Anp indicates atrial natriuretic peptide; Bnp, brain natriuretic peptide; CKO, conditional knockout; Ctgf, connective tissue growth factor; DMSO, dimethyl sulfoxide; DUSP26, dual‐specificity phosphatase 26; Myh7, myosin heavy chain 7; TAC, transverse aortic constriction; and TL, tibia length.
Furthermore, we assessed the effect of pharmacological inhibition of TAK1 on TAC‐triggered cardiac fibrosis in vivo. Compared with DMSO control, hearts of DUSP26‐CKO mice treated with iTAK1 showed decreased fibrosis signal in the interstitial and perivascular spaces, decreased collagen volumes (Figure 7I), as well as decreased mRNA levels of fibrotic markers (collagen Iα, collagen III, and ctgf) after 4 weeks of TAC surgery (Figure 7K).
These findings suggested that pharmacological inhibition of TAK1 attenuated the exaggerated hypertrophic response in DUSP26‐CKO mice.
Discussion
Heart failure is associated with pathological cardiac hypertrophy, which is characterized by increased cardiomyocyte size and cardiac fibrosis, with reduced systolic and diastolic function. Accumulating evidence indicates that the MAPK signaling pathway plays a central role in the development and progression of cardiac hypertrophy. The DUSPs modulate activation of the terminal MAPKs (ERK1/2, JNK, and p38) and hence have been referred to as MAPK phosphatases. 21 , 22 , 23 , 24 The involvement of DUSPs in cardiac hypertrophy remains unclear. In our study, we observed that DUSP26 expression was predominantly increased in mouse hearts in response to pressure overload. Cardiac‐specific overexpression of DUSP26 mice showed attenuated cardiac hypertrophy and fibrosis after 4 weeks of TAC compared with control. Conversely, deficiency of DUSP26 in mouse hearts resulted in increased cardiac hypertrophy and deteriorated cardiac function. These results clearly demonstrate that DUSP26 plays a cardioprotective role in cardiac hypertrophy induced by pressure overload.
DUSP26 is one of a family of dual‐specificity MAPK phosphatases, which modulates the level of phosphorylation of all 3 major MAPK terminal effectors. The activation of ERK was inhibited by DUSP26 in the development of lung cancer. Our data implicated that cardiac‐specific deficiency of DUSP26 up‐regulated the phosphorylation of p38 and JNK in pressure‐overloaded mice, without effect on the phosphorylation of ERK in the heart. And the phosphorylation of p38 and JNK were inhibited in the hearts of cardiac‐specific DUSP26‐TG mice. DUSP26 selectively regulated MAPK substrates of p38/JNK in hypertrophic hearts. This profile is reminiscent of results obtained with other DUSPs gene‐targeted mice. DUSP1/4 double‐null mice inhibited activity of p38/MAPK in the heart, with no alterations in activation of JNK or ERK, which showed cardiomyopathy with aging. 16 The phosphorylation of ERK1/2 was down‐regulated in hearts of cardiac‐specific DUSP6 TG mice, with no effect on p38 or JNK activation at baseline or following stimulation. 25 Constitutive expression of DUSP1 in mouse hearts resulted in inactivation of 3 MAPKs, with attenuated cardiac hypertrophy after aortic banding (AB)‐induced stimulation. 14 Collectively, each member of the DUSP family specialized regulation of the MAPK terminal effector proteins in cardiomyocytes.
In our study, it has been proved that DUSP26 exerted antihypertrophic effects by suppressing activation of MAPK signaling pathway in TAC mice. We conducted further experiments to explore the specific mechanisms between DUSP26 and MAPK signaling pathway in cardiomyocytes. TAK1 is a key upstream molecule of MAPK‐JNK/p38, which plays an indispensable role in the development of cardiac hypertrophy and heart failure. 26 It has been reported that constitutively activated TAK1 induced cardiac hypertrophy, fibrosis, fetal gene expression, and severe myocardial dysfunction response to pressure overload. 27 Our results indicated that phosphorylation of TAK1 was increased in heart of cardiac‐specific DUSP26‐knockout mice. Moreover, we showed that DUSP26 bound to TAK1 to inhibit TAK1 phosphorylation, which led to suppression of the MAPK signaling pathway and attenuated cardiac hypertrophy. Suppression of TAK1 protected against angiotensin II–induced cardiomyocyte hypertrophy. 28 TAK1 inhibition by small pharmacologic molecules has shown promising therapeutic value in inflammatory disorders and cancer. 29 In the present study, the chemical inhibitor of TAK1 inhibited cardiomyocyte hypertrophy induced by angiotensin II and attenuated the exaggerated hypertrophic response in DUSP26‐CKO mice. Our work was the first evidence to identify DUSP26 binding with TAK1 at T187 to inhibit its phosphorylation. Previous work indicated that a TAK1–TAB1 fusion protein is available for enzyme sources and structure‐based drug design. 30 The interaction between DUSP26 and TAK1 may be a new potential target for treatment of heart failure.
Collectively, DUSP26 was induced in cardiac hypertrophy. DUSP26 exerted a protective effect against pressure overload–induced cardiac hypertrophy by modulating TAK1–p38/ JNK signaling axis in vitro and in vivo. These findings strongly suggest that DUSP26 may provide a therapeutic target for treatment of cardiac hypertrophy and heart failure.
Sources of Funding
This work was supported by grants from the National Natural Science Foundation of China (81730015, 81470482, and 81600186).
Disclosures
None.
Supporting information
Tables S1–S3
Figure S1
(J Am Heart Assoc. 2021;10:e014311.. DOI: 10.1161/JAHA.119.014311.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.119.014311
For Sources of Funding and Disclosures, see page 15.
Contributor Information
Manhua Chen, Email: cmh_centre@163.com.
Jiahong Xia, Email: jiahong.xia@hust.edu.cn.
References
- 1. Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–1380. DOI: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]
- 2. Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. DOI: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 3. Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med. 1991;114:345–352. DOI: 10.7326/0003-4819-114-5-345. [DOI] [PubMed] [Google Scholar]
- 4. Zou Y, Hiroi Y, Uozumi H, Takimoto E, Toko H, Zhu W, Kudoh S, Mizukami M, Shimoyama M, Shibasaki F, et al. Calcineurin plays a critical role in the development of pressure overload‐induced cardiac hypertrophy. Circulation. 2001;104:97–101. DOI: 10.1161/01.CIR.104.1.97. [DOI] [PubMed] [Google Scholar]
- 5. Ichihara S, Senbonmatsu T, Price E Jr, Ichiki T, Gaffney FA, Inagami T. Angiotensin II type 2 receptor is essential for left ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II‐induced hypertension. Circulation. 2001;104:346–351. DOI: 10.1161/01.CIR.104.3.346. [DOI] [PubMed] [Google Scholar]
- 6. Senbonmatsu T, Ichihara S, Price E Jr, Gaffney FA, Inagami T. Evidence for angiotensin II type 2 receptor‐mediated cardiac myocyte enlargement during in vivo pressure overload. J Clin Invest. 2000;106:R25–R29. DOI: 10.1172/JCI10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gutkind JS, Offermanns S. A new G(q)‐initiated MAPK signaling pathway in the heart. Dev Cell. 2009;16:163–164. DOI: 10.1016/j.devcel.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 8. Haq S, Choukroun G, Lim H, Tymitz KM, del Monte F, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T, et al. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–677. DOI: 10.1161/01.CIR.103.5.670. [DOI] [PubMed] [Google Scholar]
- 9. Toischer K, Rokita AG, Unsöld B, Zhu W, Kararigas G, Sossalla S, Reuter SP, Becker A, Teucher N, Seidler T, et al. Differential cardiac remodeling in preload versus afterload. Circulation. 2010;122:993–1003. DOI: 10.1161/CIRCULATIONAHA.110.943431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, Saffitz J, Chien K, Xiao R‐P, Kass DA, et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001;98:12283–12288. DOI: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishida K, Yamaguchi O, Hirotani S, Hikoso S, Higuchi Y, Watanabe T, Takeda T, Osuka S, Morita T, Kondoh G, et al. P38alpha mitogen‐activated protein kinase plays a critical role in cardiomyocyte survival but not in cardiac hypertrophic growth in response to pressure overload. Mol Cell Biol. 2004;24:10611–10620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kehat I, Davis J, Tiburcy M, Accornero F, Saba‐El‐Leil MK, Maillet M, York AJ, Lorenz JN, Zimmermann WH, Meloche S, et al. Extracellular signal‐regulated kinases 1 and 2 regulate the balance between eccentric and concentric cardiac growth. Circ Res. 2011;108:176–183. DOI: 10.1161/CIRCRESAHA.110.231514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liang Q, Bueno OF, Wilkins BJ, Kuan CY, Xia Y, Molkentin JD. C‐Jun N‐terminal kinases (JNK) antagonize cardiac growth through cross‐talk with calcineurin‐NFAT signaling. EMBO J. 2003;22:5079–5089. DOI: 10.1093/emboj/cdg474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bueno OF, De Windt LJ, Lim HW, Tymitz KM, Witt SA, Kimball TR, Molkentin JD. The dual‐specificity phosphatase MKP‐1 limits the cardiac hypertrophic response in vitro and in vivo. Circ Res. 2001;88:88–96. DOI: 10.1161/01.RES.88.1.88. [DOI] [PubMed] [Google Scholar]
- 15. Maillet M, Purcell NH, Sargent MA, York AJ, Bueno OF, Molkentin JD. DUSP6 (MKP3) null mice show enhanced ERK1/2 phosphorylation at baseline and increased myocyte proliferation in the heart affecting disease susceptibility. J Biol Chem. 2008;283:31246–31255. DOI: 10.1074/jbc.M806085200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Auger‐Messier M, Accornero F, Goonasekera SA, Bueno OF, Lorenz JN, van Berlo JH, Willette RN, Molkentin JD. Unrestrained p38 MAPK activation in Dusp1/4 double‐null mice induces cardiomyopathy. Circ Res. 2013;112:48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu Y, Mivechi NF. Association and regulation of heat shock transcription factor 4b with both extracellular signal‐regulated kinase mitogen‐activated protein kinase and dual‐specificity tyrosine phosphatase DUSP26. Mol Cell Biol. 2006;26:3282–3294. DOI: 10.1128/MCB.26.8.3282-3294.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ye P, Liu J, Xu W, Liu D, Ding X, Le S, Zhang H, Chen S, Chen M, Xia J. Dual‐specificity phosphatase 26 protects against nonalcoholic fatty liver disease in mice through transforming growth factor beta‐activated kinase 1 suppression. Hepatology. 2019;69:1946–1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11:214–222. DOI: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 20. Javadov S, Jang S, Agostini B. Crosstalk between mitogen‐activated protein kinases and mitochondria in cardiac diseases: therapeutic perspectives. Pharmacol Ther. 2014;144:202–225. DOI: 10.1016/j.pharmthera.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rose BA, Force T, Wang Y. Mitogen‐activated protein kinase signaling in the heart: angels versus demons in a heart‐breaking tale. Physiol Rev. 2010;90:1507–1546. DOI: 10.1152/physrev.00054.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dickinson RJ, Keyse SM. Diverse physiological functions for dual‐specificity MAP kinase phosphatases. J Cell Sci. 2006;119:4607–4615. DOI: 10.1242/jcs.03266. [DOI] [PubMed] [Google Scholar]
- 23. Garrington TP, Johnson GL. Organization and regulation of mitogen‐activated protein kinase signaling pathways. Curr Opin Cell Biol. 1999;11:211–218. DOI: 10.1016/S0955-0674(99)80028-3. [DOI] [PubMed] [Google Scholar]
- 24. Baines CP, Molkentin JD. Stress signaling pathways that modulate cardiac myocyte apoptosis. J Mol Cell Cardiol. 2005;38:47–62. DOI: 10.1016/j.yjmcc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 25. Purcell NH, Wilkins BJ, York A, Saba‐El‐Leil MK, Meloche S, Robbins J, Molkentin JD. Genetic inhibition of cardiac ERK1/2 promotes stress‐induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc Natl Acad Sci U S A. 2007;104:14074–14079. DOI: 10.1073/pnas.0610906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shimizu I, Minamino T. Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol. 2016;97:245–262. DOI: 10.1016/j.yjmcc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 27. Zhang D, Gaussin V, Taffet GE, Belaguli NS, Yamada M, Schwartz RJ, Michael LH, Overbeek PA, Schneider MD. TAK1 is activated in the myocardium after pressure overload and is sufficient to provoke heart failure in transgenic mice. Nat Med. 2000;6:556–563. DOI: 10.1038/75037. [DOI] [PubMed] [Google Scholar]
- 28. Watkins SJ, Borthwick GM, Oakenfull R, Robson A, Arthur HM. Angiotensin II‐induced cardiomyocyte hypertrophy in vitro is TAK1‐dependent and Smad2/3‐independent. Hypertens Res. 2012;35:393–398. DOI: 10.1038/hr.2011.196. [DOI] [PubMed] [Google Scholar]
- 29. Sakurai H. Targeting of TAK1 in inflammatory disorders and cancer. Trends Pharmacol Sci. 2012;33:522–530. DOI: 10.1016/j.tips.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 30. Brown K, Vial SC, Dedi N, Long JM, Dunster NJ, Cheetham GM. Structural basis for the interaction of TAK1 kinase with its activating protein TAB1. J Mol Biol. 2005;354:1013–1102. DOI: 10.1016/j.jmb.2005.09.098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Figure S1