

Abstract
Background
Abnormal endothelial function in the lungs is implicated in the development of pulmonary hypertension; however, there is little information about the difference of endothelial function between small distal pulmonary artery (PA) and large proximal PA and their contribution to the development of pulmonary hypertension. Herein, we investigate endothelium‐dependent relaxation in different orders of PAs and examine the molecular mechanisms by which chronic hypoxia attenuates endothelium‐dependent pulmonary vasodilation, leading to pulmonary hypertension.
Methods and Results
Endothelium‐dependent relaxation in large proximal PAs (second order) was primarily caused by releasing NO from the endothelium, whereas endothelium‐dependent hyperpolarization (EDH)–mediated vasodilation was prominent in small distal PAs (fourth–fifth order). Chronic hypoxia abolished EDH‐mediated relaxation in small distal PAs without affecting smooth muscle–dependent relaxation. RNA‐sequencing data revealed that, among genes related to EDH, the levels of Cx37, Cx40, Cx43, and IK were altered in mouse pulmonary endothelial cells isolated from chronically hypoxic mice in comparison to mouse pulmonary endothelial cells from normoxic control mice. The protein levels were significantly lower for connexin 40 (Cx40) and higher for connexin 37 in mouse pulmonary endothelial cells from hypoxic mice than normoxic mice. Cx40 knockout mice exhibited significant attenuation of EDH‐mediated relaxation and marked increase in right ventricular systolic pressure. Interestingly, chronic hypoxia led to a further increase in right ventricular systolic pressure in Cx40 knockout mice without altering EDH‐mediated relaxation. Furthermore, overexpression of Cx40 significantly decreased right ventricular systolic pressure in chronically hypoxic mice.
Conclusions
These data suggest that chronic hypoxia‐induced downregulation of endothelial Cx40 results in impaired EDH‐mediated relaxation in small distal PAs and contributes to the development of pulmonary hypertension.
Keywords: cardiovascular disease, connexin, endothelial cell, gap junction, hypoxia‐induced pulmonary hypertension
Subject Categories: Pulmonary Hypertension
Nonstandard Abbreviations and Acronyms
- Cx37
connexin 37
- Cx40
connexin 40
- Cx43
connexin 43
- Cx40KO
connexin 40 knockout
- EC
endothelial cell
- EDH
endothelium‐dependent hyperpolarization
- EDR
endothelium‐dependent relaxation
- GJ
gap junction
- IK
intermediate‐conductance Ca2+‐activated potassium
- MPEC
mouse pulmonary endothelial cell
- PGI2
prostaglandin I2
- PH
pulmonary hypertension
- RVSP
right ventricular systolic pressure
- SMC
smooth muscle cell
- Wt
wild type
Clinical Perspective
What Is New?
Endothelium‐dependent relaxation in small distal pulmonary arteries (fourth‐fifth order) is primarily caused by endothelium‐dependent hyperpolarization–mediated vasodilation instead of releasing NO; chronic hypoxia abolishes endothelium‐dependent hyperpolarization–mediated relaxation in small distal pulmonary arteries, but not in large proximal pulmonary arteries.
The protein levels are significantly lower for connexin 40 (Cx40) and higher for connexin 37 in mouse pulmonary endothelial cells from hypoxic mice than in normoxic mice.
Cx40 knockout mice exhibited significant attenuation of endothelium‐dependent hyperpolarization–mediated relaxation and marked increase in right ventricular systolic pressure, a surrogate measure of pulmonary arterial systolic pressure in mice; overexpression of Cx40 significantly decreased right ventricular systolic pressure in chronically hypoxic mice.
What Are the Clinical Implications?
Cx40 plays a crucial role in the development of hypoxia‐induced pulmonary hypertension by regulating endothelium‐dependent hyperpolarization–mediated relaxation in small distal pulmonary arteries, and that overexpression of Cx40 ameliorates hypoxia‐induced pulmonary hypertension.
Overexpression or stimulation of Cx40 is potentially a novel therapeutic strategy for patients with pulmonary hypertension who do not respond to conventional treatment.
Hypoxia‐induced pulmonary hypertension (PH) is a risk factor for right‐sided heart failure and death in patients with hypoxic cardiopulmonary diseases (eg, chronic obstructive pulmonary disease, obstructive sleep apnea, and interstitial lung disease) and inhabitants living in high altitude. 1 , 2 , 3 Despite the progress in developing medications for PH, the morbidity and mortality of PH associated with hypoxia and cardiopulmonary diseases still remain high.
PH is characterized by increased pulmonary vascular resistance, and the primary cause of increased pulmonary vascular resistance is a decrease in intraluminal diameter caused by sustained pulmonary vasoconstriction, concentric pulmonary arterial wall thickening, and obliterative lesions in small pulmonary arteries (PAs). Alveolar hypoxia causes pulmonary vasoconstriction by inducing smooth muscle contraction 4 , 5 and attenuating endothelium‐dependent relaxation in PAs. 6 , 7 Lung vascular endothelial dysfunction is implicated in the development and progression of hypoxia‐induced PH. 7 , 8 , 9
In small distal or resistance arteries, the endothelium contributes to vasodilation mainly by inducing hyperpolarization of smooth muscle cells (SMCs), referred to as endothelium‐dependent hyperpolarization (EDH)–mediated vascular relaxation, in addition to releasing NO and prostaglandin I2 (PGI2). However, in large proximal or conduit arteries, the endothelium contributes to relaxing vessels primarily by releasing NO. 10 , 11 Common strategies to enhance pulmonary vasodilation in patients with PH are focused on the NO and PGI2 signaling cascades, and therefore, may not be sufficient to relax small distal PAs. EDH is initiated by activation of small‐conductance Ca2+‐activated K+ channels in endothelial cells (ECs); the hyperpolarizing electrical signal in ECs is then propagated to SMCs through the gap junctions (GJs), a tunnel between ECs and between EC and SMC. 12 , 13 Of note, there are more myoendothelial GJs in small vessels than in large vessels 14 , 15 , 16 ; thus, small distal vessels can be rapidly and efficiently relaxed through EDH.
GJs are cell membrane channels made of connexin proteins, with >20 connexin genes identified in the human genome. 17 So far, connexin 37 (Cx37), connexin 40 (Cx40), and connexin 43 (Cx43) (connexin 26, connexin 32, and connexin 45 in some organs) are found in ECs. Cx40 is predominantly expressed in vascular ECs, 12 , 13 whereas Cx37 and Cx43 are expressed in ECs and SMCs. GJs are the pathways for the intercellular electrical propagation and transportation of small molecules <1 kDa (eg, ATP). 18 Disturbing GJ activity by knockdown or inhibition of Cx40 attenuates vascular relaxation 19 and inhibits EC proliferation and migration. 20 , 21 Systemic Cx40 knockout (Cx40KO) mice exhibit a significant increase in blood pressure, 22 , 23 whereas Cx37 knockout mice show normal blood pressure. 22 Systemic Cx43 knockout mouse is embryonic lethal because of the developmental failure of the heart, 24 whereas endothelial‐specific Cx43 knockout results in hypotension. 25 Cx40 expression is decreased in the lungs 26 , 27 and PAs 28 in animals of experimental PH, and pulmonary ECs from patients with PH. 29 However, the pathophysiological role of Cx40 in PH is still unrevealed. In this study, we investigated EDH‐mediated relaxation in small distal PAs from a hypoxia‐induced PH mouse model and examined the pathogenic role of Cx40 in the development of hypoxia‐induced PH.
METHODS
The detailed Methods section is included in Data S1. All supporting data are available within the article and its Data Supplement. The information of materials can also be found in Table S1.
Animals
All experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee at The University of Arizona and the University of California, San Diego, and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health. C57BL/6J male mice were purchased from Jackson Laboratory (Bar Harbor, ME). Systemic Cx40KO mice were kindly provided by Dr Janis Burt from The University of Arizona, 30 and C57BL/6J mice were used as a wild‐type (Wt) control. Tie2‐driven Cx40 Wt overexpressing mice and Tie2‐driven Cx40 negative mutant overexpressing mice were provided by Dr Anthony Ashton from the University of Sydney. 31 Mice without Tie2‐driven Cx40 Wt gene or Tie2‐driven Cx40 negative mutant gene were used as Wt control. These mice were bred in the animal facility of The University of Arizona and University of California, San Diego. Male mice were used for experiments in this study. The primer sequence information for genotyping and copy number assessment is listed in Table S2. Mice were randomly allocated to experimental groups, and hypoxia‐induced PH was generated by placing mice (8 weeks old) in the normobaric hypoxic chamber (10% O2) for 4 weeks. Sugen‐hypoxia PH mouse model was generated by injections of Sugen 5416 (20 mg/kg, once a week) during the 4 weeks of hypoxic exposure. Cx40 overexpression was achieved by a single intravenous injection of cytomegalovirus (CMV)‐Cx40 adenovirus 32 or control adenovirus (adenovirus vector without gene insertion) at 3×109 plaque‐forming unit/mouse. Three weeks after hypoxic exposure, mice that received either Cx40 adenovirus or control adenovirus were kept in the hypoxic chamber for an additional 1 week before experimentation. Lung dissection was performed under anesthesia with sodium pentobarbital (130 mg/kg, intraperitoneally), and all efforts were made to minimize pain.
Statistical Analysis
We conducted data analysis in a blinded manner wherever possible and set proper controls for every experimental plan. The mouse number and independent experiment number are described in the figure legends. Statistical analysis was performed using GraphPad Prism 7.04. After the data passed a normality test, the 2‐tailed Student t‐test was used for comparisons of 2 groups, and 1‐way ANOVA was used for multiple comparisons. If the data did not pass the normality test, a nonparametric test (Mann‐Whitney for 2 groups, Kruskal‐Wallis for multiple comparisons) was used. Statistical comparison between dose‐response curves was made by 2‐way ANOVA with Bonferroni post hoc test. Differences were considered to be statistically significant when P<0.05. Data are presented as mean±SEM. In RNA‐sequencing data, q values are obtained from adjusted P values using the Benjamini‐Hochberg false discovery rate approach to correct for multiple testing. The fold changes with q values <0.05 are considered significant.
RESULTS
Endothelium‐Dependent Relaxation in Small Distal PAs Is Primarily Induced by EDH‐Mediated Relaxation
Large proximal PA (second order) and small distal PA (fourth‐fifth order) were dissected from the same lobe of the lung, and endothelium‐dependent relaxation (EDR) was assessed by administrating acetylcholine in a dose‐dependent manner. The diameters of large proximal PAs and small distal PAs were 137.1±17.9 µm and 113.2±6.1 µm, respectively (nmice=6 per group; P=0.03). Small distal PAs exhibited significantly greater EDR than large proximal PAs (Figure 1A). EDR is composed of NO‐, PGI2‐, and EDH‐mediated relaxations 32 , 33 ; therefore, acetylcholine‐induced relaxation in the presence of N‐nitro‐L‐arginine methyl ester (an endothelial NO synthase inhibitor; 100 μmol/L) and indomethacin (a cyclooxygenase inhibitor; 10 μmol/L) represents EDH‐mediated relaxation. Small distal PAs showed strong EDH‐mediated relaxation (>50%), whereas EDH‐mediated relaxation was hardly seen in large proximal PAs (≤20%) (Figure 1B). There was little difference in smooth muscle–dependent relaxation, assessed by sodium nitroprusside (an NO donor)–induced vasodilation, between small distal PAs and large proximal PAs (Figure 1C). Those data suggest that large proximal PAs relax mainly via NO‐ and PGI2‐dependent relaxation, whereas small distal PAs relax largely through EDH‐mediated relaxation.
Figure 1. Vascular relaxation in different orders of pulmonary arteries (PAs).
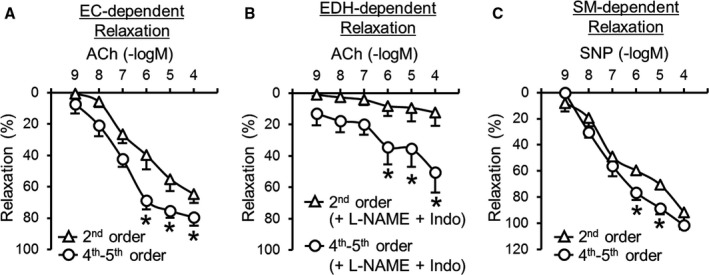
A, Endothelial cell (EC)–dependent relaxation evaluated by acetylcholine (ACh) administration in PAs. Large proximal PAs (second order), nmice=6; small distal PAs (fourth‐fifth order), nmice=6. B, Endothelium‐dependent hyperpolarization (EDH)–mediated relaxation in PAs determined by ACh administration in the presence of N‐nitro‐L‐arginine methyl ester (L‐NAME) (an endothelial NO synthase inhibitor; 100 µmol/L) and indomethacin (Indo) (a cyclooxygenase inhibitor; 10 µmol/L). Nmice=6 per group. C, Smooth muscle (SM)–dependent relaxation evaluated by sodium nitroprusside (SNP) administration in PAs. Nmice=6 per group. Data are mean±SEM. *P<0.05 vs second order. Statistical comparison between dose‐response curves was made by 2‐way ANOVA with Bonferroni post hoc test.
Chronic Hypoxia Attenuates EDR and EDH‐Mediated Relaxation in Small Distal PAs, Causing PH
Small distal PAs isolated from chronically hypoxic mice displayed attenuated EDR and EDH‐mediated relaxation without any change in smooth muscle–dependent relaxation (Figure 2A). Mice exposed to hypoxia (10% O2) for 4 weeks developed PH, evidenced by a significant increase in right ventricular systolic pressure (RVSP), a surrogate measure of pulmonary arterial systolic pressure (Figure 2B), 34 and Fulton Index, a measurement of right ventricular hypertrophy attributable to increased afterload (Figure 2C).
Figure 2. Effect of hypoxic exposure on endothelial function, right ventricular systolic pressure (RVSP), and connexin (Cx) expression in pulmonary endothelial cells.
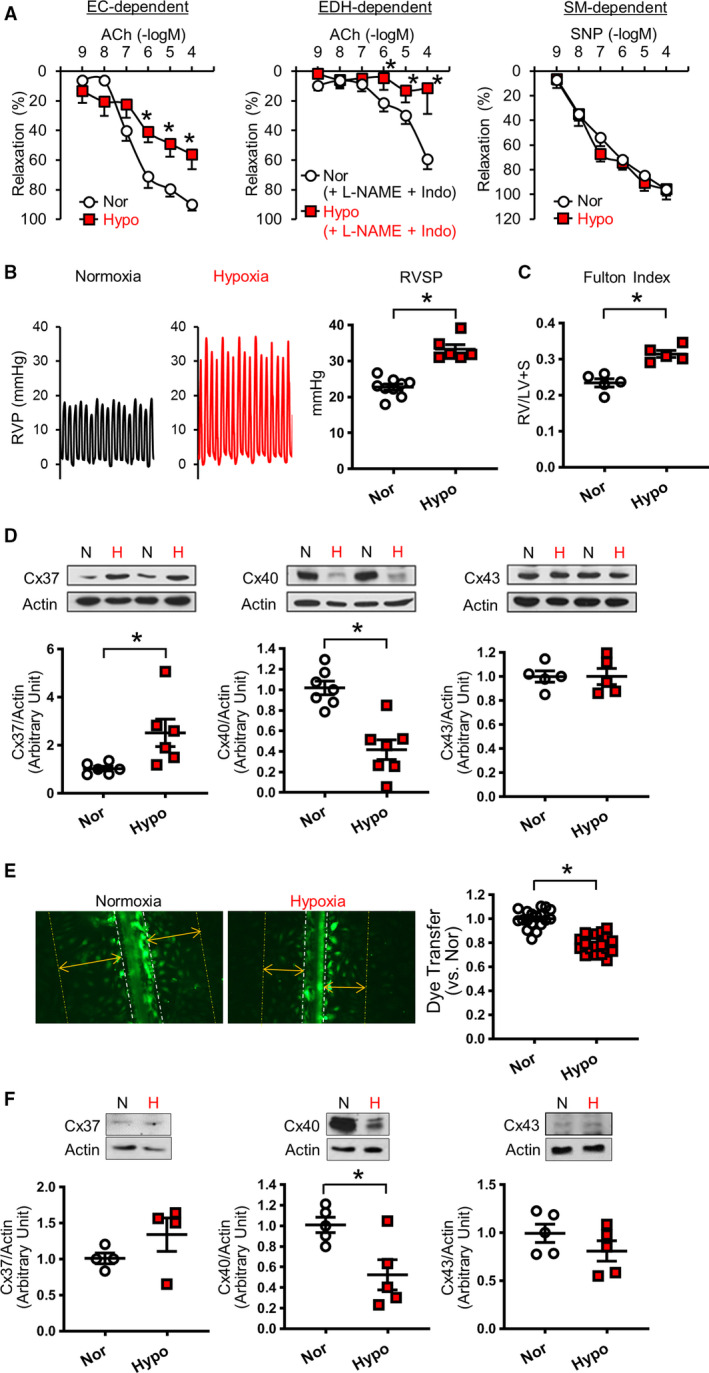
A, Vascular response in small distal pulmonary arteries (fourth‐fifth order). Left: Endothelial cell (EC)–dependent relaxation. Normoxia (Nor), nmice=8; Hypoxia (Hypo), nmice=7. Middle: Endothelium‐dependent hyperpolarization (EDH)–mediated relaxation. Nor, nmice=8; Hypo, nmice=5. Right: Smooth muscle (SM)–dependent relaxation. Nor, nmice=8; Hypo, nmice=7. B, Left shows typical records of right ventricular pressure (RVP). Dot plot shows RVSP in mice exposed to 4 weeks of hypoxia (Hypo; nmice=6) or normoxia (Nor; nmice=9). C, Fulton Index (right ventricle [RV]/(left ventricle [LV]+septum [S])). nmice=5 per group. D, Protein levels of Cxs in mouse pulmonary endothelial cells (ECs) isolated from normoxia‐ or hypoxia‐exposed mice. Cx37, nmice=6 per group; Cx40, nmice=7 per group; Cx43, nmice=5 per group. E, Gap junction activity in human pulmonary ECs. Representative photographs showing the images of Lucifer yellow dye transfer in ECs. The dot plot shows summarized data of dye transfer (the distance compared with control) in normoxia‐exposed ECs (Nor; nexperiments=18) and hypoxia‐exposed ECs (Hypo; nexperiments=18). F, Protein levels of Cxs in human pulmonary ECs exposed to normoxia or hypoxia for 48 hours. Cx37, nexperiments=4 per group; Cx40, nexperiments=5 per group; Cx43, nexperiments=5 per group. Data are mean±SEM. *P<0.05 vs Nor. Unpaired Student t‐test (2 tailed) was used for comparisons of 2 experimental groups. Statistical comparison between dose‐response curves was made by 2‐way ANOVA with Bonferroni post hoc test. In the data of Cx37 in Figure 2F, Mann‐Whitney was used for the statistical comparison. ACh indicates acetylcholine; H, hypoxia; Indo, indomethacin; L‐NAME, N‐nitro‐L‐arginine methyl ester; N, normoxia; and SNP, sodium nitroprusside.
Cx40 Expression Is Decreased, and Cx37 Expression Is Increased, in Mouse Pulmonary ECs Isolated From Hypoxia‐Induced PH Mice
To investigate the target genes affected by chronic hypoxia, we conducted RNA sequencing using mouse pulmonary ECs (MPECs) isolated from normoxic and hypoxic mice. Because chronic hypoxia significantly decreased EDH‐mediated relaxation (Figure 2A), we focused on genes related to EDH, such as small‐conductance Ca2+‐activated K+ channels and GJ proteins, 33 and the result was shown in the Table. RNA‐sequencing data revealed that the expression levels of intermediate‐conductance Ca2+‐activated potassium (IK) channel (KCNN4) and Cx40 (Gja5) were significantly decreased, whereas Cx37 (Gja4) and Cx43 (Gja1) were significantly increased in MPECs from chronically hypoxic mice compared with normoxic control mice. Furthermore, we found that Cx40 protein level was decreased, and Cx37 protein level was increased, in MPECs in hypoxia‐exposed mice; there was no difference in protein levels of Cx43 (Figure 2D) and IK channels (Figure S1) between the groups. We also used a severe PH mouse model, mice with Sugen/hypoxia–induced PH, to compare the expression level of Cx40; we found that MPECs from Sugen/hypoxia–induced PH mice exhibited a significant decrease in Cx40 protein expression compared with the control (Figure S2).
Table 1.
Comparison of EDH‐Related Gene Levels in MPECs Between Normoxia Control Mice and Chronically Hypoxic Mice
| Gene | Status | Log2 (Fold Change) | P Value | q Value | Significant |
|---|---|---|---|---|---|
| SK Channels and IK Channel | |||||
| SK1 (Kcnn1) | NOTEST | ||||
| SK2 (Kcnn2) | NOTEST | ||||
| SK3 (Kcnn3) | OK | 0.337 | 0.013 | 0.088 | No |
| SK4, IK (Kcnn4) | OK | −0.581 | 0.001 | 0.009 | Yes |
| GJ/Connexin Proteins | |||||
| Gja1 (Cx43) | OK | 0.599 | <0.001 | 0.001 | Yes |
| Gja3 (Cx46) | NOTEST | ||||
| Gja4 (Cx37) | OK | 0.569 | <0.001 | 0.001 | Yes |
| Gja5 (Cx40) | OK | −0.613 | <0.001 | 0.005 | Yes |
| Gja6 (Cx33) | NOTEST | ||||
| Gja8 (Cx50) | NOTEST | ||||
| Gja10 (Cx59, Cx62) | NOTEST | ||||
| Gjb1 (Cx32) | NOTEST | ||||
| Gjb2 (Cx26) | NOTEST | ||||
| Gjb3 (Cx31) | NOTEST | ||||
| Gjb4 (Cx30.3) | NOTEST | ||||
| Gjb5 (Cx30.1) | NOTEST | ||||
| Gjb6 (Cx30) | NOTEST | ||||
| Gjc1 (Cx45) | OK | 0.261 | 0.047 | 0.210 | No |
| Gjc2 (Cx47) | OK | −0.151 | 0.502 | 0.817 | No |
| Gjc3 (Cx31.3) | NOTEST | ||||
| Gjd2 (Cx26) | NOTEST | ||||
| Gjd3 (Cx31.9) | NOTEST | ||||
| Gjd4 (Cx40.1) | OK | 0.065 | 0.491 | 0.810 | No |
| Gje1 (Cx23) | NOTEST | ||||
| Muscarinic acetylcholine receptors (Chrm) | |||||
| Chrm1 | OK | 0.080 | 0.847 | 0.968 | No |
| Chrm1 | NOTEST | ||||
| Chrm1 | NOTEST | ||||
| Chrm2 | OK | −0.244 | 0.097 | 0.333 | No |
| Chrm3 | OK | −0.706 | 0.029 | 0.153 | No |
| Chrm4 | OK | 0.324 | 0.257 | 0.586 | No |
| Chrm5 | NOTEST | ||||
The data were obtained by RNA sequencing by QIAGEN. If RNA expression level was too low, the status indicates “NOTEST.” Nmice=6 per group. Chrm indicates cholinergic receptor muscarinic; Cx23, connexin 23; Cx26, connexin 26; Cx30, connexin 30; Cx30.1, connexin 30.1; Cx30.3, connexin 30.3; Cx31, connexin 31; Cx31.3, connexin 31.3; Cx31.9, connexin 31.9; Cx32, connexin 32; Cx33, connexin 33; Cx37, connexin 37; Cx40, connexin 40; Cx40.1, connexin 40.1; Cx43, connexin 43; Cx45, connexin 45; Cx46, connexin 46; Cx50, connexin 50; Cx59, connexin 59; Cx62, connexin 62; EDH, endothelium‐dependent hyperpolarization; GJ, gap junction; IK, intermediate‐conductance Ca2+‐activated potassium; MPEC, mouse pulmonary endothelial cell; and SK, small‐conductance Ca2+‐activated K+.
Acute Hypoxic Exposure Decreases GJ Activity in Pulmonary ECs
Connexins are the component of GJ that propagates the electrical signals between cells. Because we observed the change of connexin expression levels, we examined whether hypoxia regulated the activity of GJ in vitro. Human pulmonary ECs were first exposed to hypoxia (3% O2) for 48 hours, and GJ activity was then assessed by dye transfer experiment. 35 As shown in Figure 2E, GJ activity was significantly attenuated by hypoxia in ECs. Short‐term exposure of ECs to hypoxia also decreased Cx40 protein expression, but did not change Cx37 and Cx43 levels (Figure 2F), suggesting that Cx40 is more susceptive or sensitive to hypoxia than Cx37 and Cx43. To examine whether the inhibition of NO and prostacyclin would affect GJ activity, we tested the effect of N‐nitro‐L‐arginine methyl ester and indomethacin on GJ activity with or without exposure of ECs to hypoxia. As shown in Figure S3, NO/PGI2 inhibition did not affect GJ activity regardless of hypoxic exposure. These data suggest that hypoxia is sufficient to inhibit GJ activity in ECs through the downregulation of Cx40 and attenuate EDH‐mediated relaxation.
Cx40 Deletion Leads to Attenuated EDH‐Mediated Relaxation in Small Distal PAs and Increases RVSP
To investigate the specific role of Cx40 in the development of PH, we obtained Cx40KO mice 30 and compared vascular response and RVSP in Wt and Cx40KO mice. MPECs from Cx40KO mice exhibited significant decreases in Cx40 and Cx37 protein levels, but showed no difference in Cx43 level compared with MPECs from Wt mice (Figure 3A). Deletion of Cx40 significantly attenuated EDR and EDH‐mediated relaxation in small distal PAs (Figure 3C); however, it did not affect vasodilation in large proximal PAs (Figure 3B). Furthermore, Cx40 deletion slightly, but with statistical significance, increased RVSP (Figure 3D), and it decreased Fulton Index (Figure 3E). In line with the result from Cx40KO mice, Tie2‐driven Cx40 negative mutant overexpressing mice 31 exhibited a significant increase in RVSP without changing Fulton Index (Figure S4).
Figure 3. Characterization of connexin 40 knockout (Cx40KO) mice.
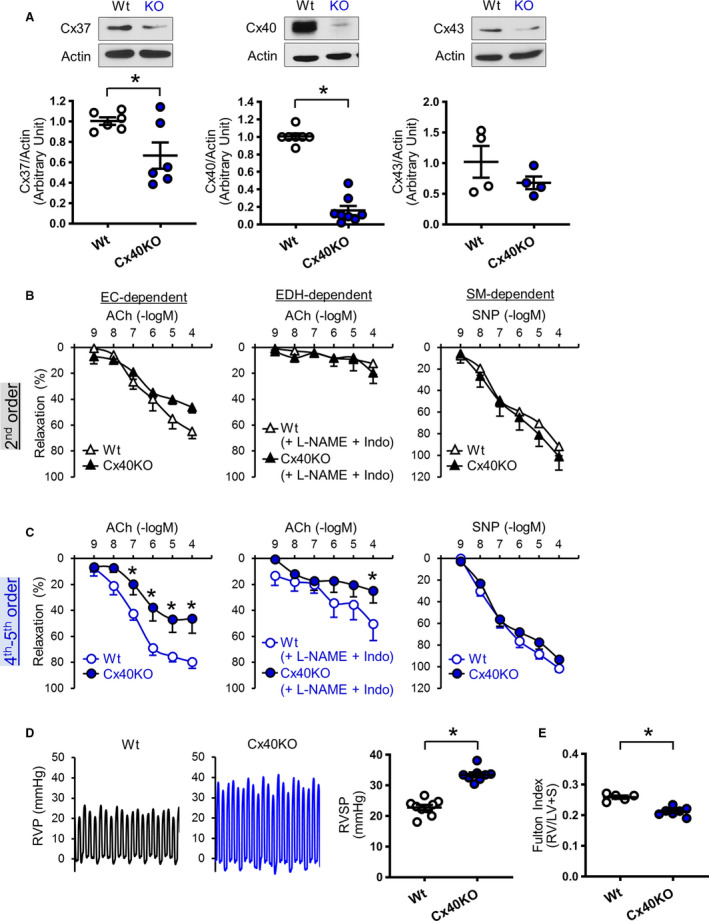
A, Cx protein levels determined by Western blot. Left: Cx37. Nmice=6 per group. Middle: Cx40. Wild type (Wt), nmice=7; Cx40KO mice, nmice=8. Right: Cx43. Nmice=4 per group. B, Vascular relaxation in large proximal pulmonary arteries (PAs) (second order). C, Vascular relaxation in small distal PAs (fourth‐fifth order). Left: Endothelial cell (EC)–dependent relaxation. Nmice=6 per group. Middle: Endothelium‐dependent hyperpolarization (EDH)–mediated relaxation. Nmice=6 per group. Right: Smooth muscle (SM)–dependent relaxation. Nmice=6 per group. D, Typical record of right ventricular pressure (RVP) and summarized data of right ventricular systolic pressure (RVSP). Wt, nmice=9; Cx40KO, nmice=8. E, Fulton Index (right ventricle [RV]/(left ventricle [LV]+septum [S])). Wt, nmice=5; Cx40KO, nmice=7. Data are mean±SEM. *P<0.05 vs Wt. Unpaired Student t‐test (2 tailed) was used for comparisons of 2 experimental groups. Statistical comparison between dose‐response curves was made by 2‐way ANOVA with Bonferroni post hoc test. In the data of Cx40 and Cx43 in Figure 3A, Mann‐Whitney was used for the statistical comparison. ACh indicates acetylcholine; Indo, indomethacin; L‐NAME, N‐nitro‐L‐arginine methyl ester; and SNP, sodium nitroprusside.
In the Absence of Cx40, Chronic Hypoxia Does Not Affect EDH‐Mediated Relaxation, but Still Increases RVSP
We found that chronic hypoxia decreased Cx40 expression in MPECs, and Cx40 deletion attenuated EDH‐mediated relaxation and increased RVSP. Therefore, we examined the effect of chronic hypoxia on vasodilation and RVSP in Cx40KO mice. Figure 4A demonstrates that chronic hypoxia did not alter EDR, EDH‐mediated relaxation, or smooth muscle–dependent relaxation in small distal PAs in Cx40KO mice. However, chronic hypoxia was still able to increase RVSP in Cx40KO mice (Figure 4B), accompanied with increased Fulton Index (Figure 4C). Neither hypoxia nor Cx40KO affected body weight and heart rate; however, Cx40KO mice exhibited higher mean arterial pressure than Wt mice, whereas chronic hypoxia slightly decreased mean arterial pressure in Cx40KO mice without significant difference (Figure 5A through 5C).
Figure 4. Exposure to chronic hypoxia in connexin 40 knockout (Cx40KO) mice does not alter vascular response; however, it further increases right ventricular systolic pressure (RVSP).
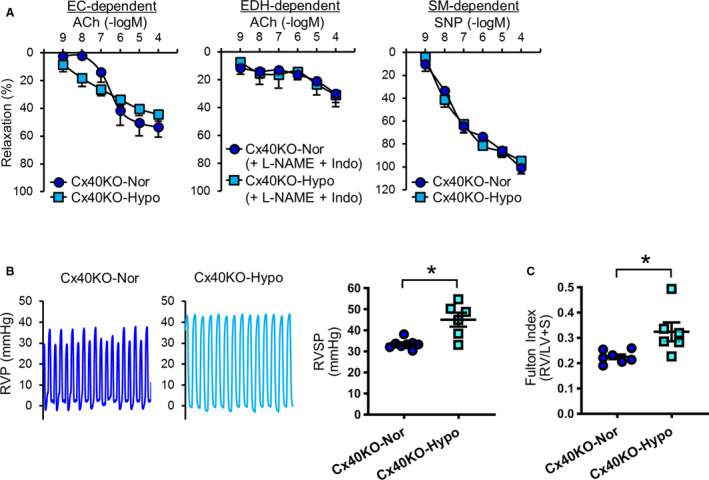
A, Vascular response in small distal (fourth‐fifth order) pulmonary arteries. Left: Endothelial cell (EC)–dependent relaxation. Normoxic Cx40KO mice (Cx40‐Nor), nmice=7; Cx40KO mice exposed to hypoxia (Cx40KO‐Hypo), nmice=8. Middle: Endothelium‐dependent hyperpolarization (EDH)–mediated relaxation. Cx40‐Nor, nmice=7; Cx40KO‐Hypo, nmice=5. Right: Smooth muscle (SM)–dependent relaxation. Cx40‐Nor, nmice=7; Cx40KO‐Hypo, nmice=8. B, Typical record of right ventricular pressure (RVP) and summarized data of right ventricular systolic pressure (RVSP). Cx40‐Nor, nmice=8; Cx40KO‐Hypo, nmice=6. C, Fulton Index (right ventricle [RV]/(left ventricle [LV]+septum [S])). Cx40‐Nor, nmice=7; Cx40KO‐Hypo, nmice=6. Data are mean±SEM. *P<0.05 vs Cx40‐Nor. Unpaired Student t‐test (2 tailed) was used for comparisons of 2 experimental groups. Statistical comparison between dose‐response curves was made by 2‐way ANOVA with Bonferroni post hoc test. ACh indicates acetylcholine; Indo, indomethacin; L‐NAME, N‐nitro‐L‐arginine methyl ester; and SNP, sodium nitroprusside.
Figure 5. Characterization of wild‐type (Wt) and connexin 40 knockout (Cx40KO) mice with or without 4 weeks of hypoxic exposure.
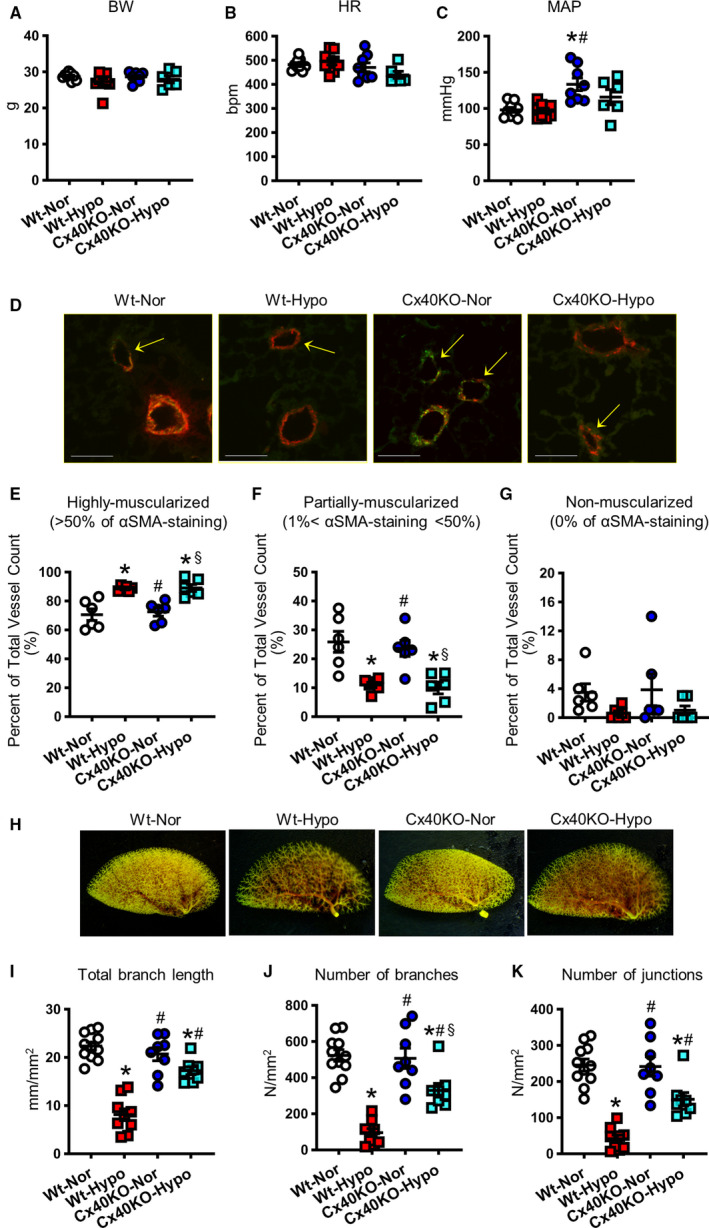
A–C, Hemodynamic parameters. A, Body weight (BW). B, Heart rate (HR). C, Mean arterial pressure (MAP). Wt mice exposed to normoxia (Wt‐Nor), nmice=9; Wt mice exposed to hypoxia (Wt‐Hypo), nmice=9; Cx40KO mice exposed to normoxia (Cx40KO‐Nor), nmice=8; Cx40KO mice exposed to hypoxia (Cx40KO‐Hypo), nmice=6. D‐G, Muscularization of the pulmonary arterioles. D, Representative images. Pulmonary arteries were stained with von Willebrand factor AF488 (an endothelial cell marker) and anti–α‐smooth muscle actin–Cy3 (αSMA; a smooth muscle cell marker). Bar=70 µm. Peripheral pulmonary arteries (20 µm<diameter<70 µm) were classified into highly muscularized (>50% of αSMA staining; E), partially muscularized (1%<αSMA staining<50%; F), and nonmuscularized (0% of αSMA staining; G). Nmice=6 per group. H‐K, Lung angiograph. H, Representative images. I, Total branch length. J, Number of branches. K, Number of junctions. Wt‐Nor, nmice=11; Wt‐Hypo, nmice=9; Cx40KO‐Nor, nmice=8; Cx40KO‐Hypo, nmice=8. Data are mean±SEM. *P<0.05 vs Wt‐Nor. # P<0.05 vs Wt‐Hypo. § P<0.05 vs Cx40KO‐Nor. Statistical comparison between 4 groups was made by 1‐way ANOVA with Bonferroni post hoc test. In the data of Figure 5A, 5G, and 5K, Kruskal‐Wallis test was used for the statistical comparison.
We found that chronic hypoxia led to pulmonary arteriole muscularization, as shown by a significant increase in the number of highly muscularized PAs and a decrease in the number of partially muscularized PAs. However, the deletion of Cx40 had a negligible effect on pulmonary arteriole muscularity regardless of hypoxic exposure (Figure 5D through 5G).
To further examine the effect of Cx40 on hypoxia‐induced structural changes of the pulmonary vasculature, we conducted angiography experiments and compared the total branch length and the number of branches and junctions between normoxic and hypoxic Wt and Cx40KO mice (Figure 5H through 5K). Chronic hypoxia significantly decreased the total branch length and the number of branches and junction in Wt mice and Cx40KO mice; however, the hypoxia‐induced loss of branches was significantly attenuated in Cx40KO mice in comparison to Wt mice. These data suggest that further increase in RVSP, seen in chronically hypoxic Cx40KO mice, would be caused primarily by increased PA muscularization.
Cx40 Overexpression Increases EDH‐Mediated Relaxation and Ameliorates Hypoxia‐Induced PH
Next, we examined whether overexpression of Cx40 was able to restore hypoxia‐mediated inhibition of EDR and EDH‐mediated relaxation in small distal PAs and to reverse hypoxia‐induced PH. Cx40 overexpression in MPECs by adenoviral induction of Cx40 gene was confirmed using Western blot analysis (Figure 6A). Cx40 overexpression in hypoxia‐exposed mice significantly augmented EDR and EDH‐mediated relaxation in small distal PAs (Figure 6B). In addition, Cx40 overexpression ameliorated hypoxia‐induced PH (Figure 6C) without changing Fulton Index (Figure 6D). To further confirm the protective effect of Cx40 overexpression on hypoxia‐induced PH, we obtained mice in which Cx40 was genetically overexpressed in ECs (Tie2‐driven Cx40 Wt overexpressing mice). 31 As shown in Figure S5, endothelial‐specific overexpression of Cx40 (Tie2‐driven Cx40 Wt overexpressing mice) prevented the development of hypoxia‐induced PH. Our data strongly suggest that Cx40 overexpression could be a novel therapeutic strategy for PH by increasing EDH‐mediated relaxation in small distal PAs, which has never been focused as a target for PH treatment.
Figure 6. Connexin 40 (Cx40) overexpression increases vascular relaxation and decreases right ventricular systolic pressure (RVSP) in hypoxia‐exposed mice.
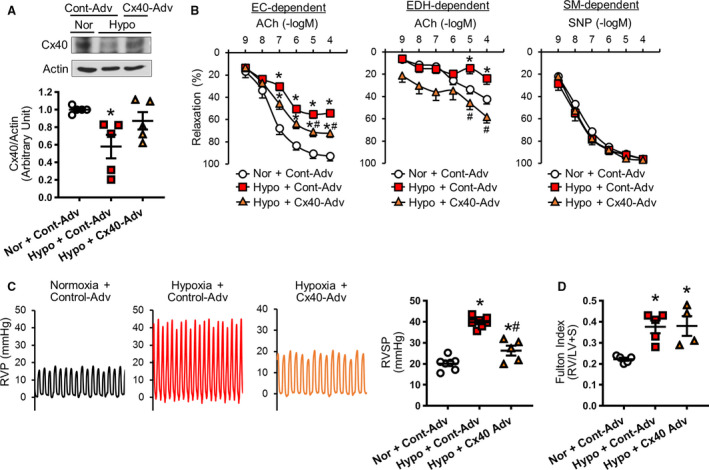
A, Cx40 protein level in mouse pulmonary endothelial cells (ECs) from normoxia‐exposed control adenovirus (Cont‐Adv)–injected mouse (Nor+Cont‐Adv), hypoxia‐exposed Cont‐Adv–injected mouse (Hypo+Cont‐Adv), and hypoxia‐exposed Cx40‐Adv–injected mouse (Hypo+Cx40‐Adv). Dot plot shows summarized data. Nmice=5 per group. B, Vascular response in small distal (fourth‐fifth order) pulmonary arteries. Nor+Cont‐Adv, nmice=10; Hypo+Cont‐Adv, nmice=11; Hypo+Cx40‐Adv, nmice=10. Left: EC‐dependent relaxation. Middle: Endothelium‐dependent hyperpolarization (EDH)–mediated relaxation. Right: Smooth muscle (SM)–dependent relaxation. C, Typical record of right ventricular pressure (RVP) and summarized data of RVSP. Nor+Cont‐Adv, nmice=7; Hypo+Cont‐Adv, nmice=7; Hypo+Cx40‐Adv, nmice=5. D, Fulton Index (right ventricle [RV]/(left ventricle [LV]+septum [S])). Nor+Cont‐Adv, nmice=5; Hypo+Cont‐Adv, nmice=5; Hypo+Cx40‐Adv, nmice=4. Data are mean±SEM. *P<0.05 vs Nor+Cont‐Adv. # P<0.05 vs Hypo+Cont‐Adv. Statistical comparison of dose‐response curves among 3 groups was made by 2‐way ANOVA with Bonferroni post hoc test. Statistical comparison of RVSP among 3 groups was made by 1‐way ANOVA with Bonferroni post hoc test. ACh indicates acetylcholine; and SNP, sodium nitroprusside.
DISCUSSION
In this study, we focused on endothelial function in small distal PAs and investigated whether restoration of endothelial function in small distal PAs, evaluated by EDH‐mediated relaxation, had an inhibitory effect on the development and progression of hypoxia‐induced PH. The contractile effect of hypoxia on pulmonary SMCs has been studied for decades; however, there is little information on the effect of hypoxia on EDR, especially EDH‐mediated relaxation in small distal PAs. PAs isolated from patients with chronic obstructive pulmonary disease show decreased EDR attributable to impaired NO production. 36 Chronic hypoxia attenuates EDR by reducing NO production or bioavailability in rat 37 , 38 and mouse 8 , 39 PAs. In those studies, the authors may have used large PAs (main or first‐second order of conduit PA) because the blockade of NO abolishes EDR. As shown in our study (Figure 1), acetylcholine‐induced vasodilation in large proximal PAs is predominantly dependent on NO and/or PGI2, whereas EDH‐mediated relaxation largely contributed to EDR in small distal PAs, evidenced by strong acetylcholine‐induced relaxation in the presence of NO and PGI2 inhibitors. Furthermore, we demonstrated that chronic hypoxia significantly attenuated EDH‐mediated relaxation in small distal PAs accompanied by increased RVSP and Fulton Index (Figure 2A through 2C). Our RNA‐sequencing data revealed that mRNA levels of IK channel and Cx40 were significantly decreased, whereas Cx37 and Cx43 were increased in MPECs isolated from hypoxia‐induced PH mice (Table). Contrary to our expectations, the protein level of IK channel showed no significant difference between normoxic control mice and hypoxic mice (Figure S1). Wandall‐Frostholm et al also reported that 4 weeks of hypoxia exposure did not alter the level of IK channel in mice, and suppression of IK and small‐conductance Ca2+‐activated K+ channels did not change RVSP. 40 However, Kroigaard et al demonstrated that chronic hypoxia attenuated EDH‐mediated relaxation in rat PAs (third‐fourth order) because of downregulation of IK channel. 41 Further study is required to identify the role of IK channel in the development of PH.
Several groups showed the decrease of Cx40 levels in the lungs of patients with PH and animals with experimental PH, 26 , 27 , 28 , 29 as did we in MPECs isolated from mice with hypoxia‐induced PH (Figure 2B). However, the functional role of Cx40 in the development of PH was not clear until now. In this study, we found that the deletion of Cx40 in mice significantly inhibited EDH‐mediated relaxation in small distal PAs, but not in large PAs (Figure 3B and 3C). Although Cx40 deletion attenuated endothelial function only in small distal PAs, Cx40KO mice exhibited a significant increase in RVSP (Figure 3D). Endothelium‐specific Tie2‐driven Cx40 negative mutant overexpressing mice also showed an increase in RVSP (Figure S4), which was in agreement with the result from Cx40KO mice. Without Cx40, hypoxia was no longer able to inhibit EDH‐mediated relaxation (Figure 4A). However, chronic exposure of Cx40KO mice to hypoxia further enhanced PH (Figure 4B), accompanied with increased arteriole muscularization (Figure 5D through 5G). These data suggest that hypoxia can lead to PH by, at least, 2 independent mechanisms: (1) vascular remodeling and (2) attenuated EDH‐dependent relaxation in small distal PAs through Cx40 downregulation in pulmonary ECs. In other words, Cx40 downregulation is one of the causes of hypoxia‐induced PH, but not an absolute cause. We initially thought that the change in protein levels of other connexins in MPECs or hearts might be involved in the further increase of RVSP in hypoxia‐exposed Cx40KO mice. Figure S6 showed that the expression levels of those proteins were not altered in hypoxic‐exposed Cx40KO mice compared with normoxic Cx40KO mice. These data imply that chronic hypoxia augmented RVSP in Cx40KO mice primarily through increased arteriole muscularization without obvious contribution of changes in other connexins.
Overexpression of Cx40 by adenoviral induction of Cx40 gene restored EDR and EDH‐mediated relaxation in small distal PAs and ameliorated hypoxia‐induced PH (Figure 6). In line with this result, constitutive Cx40 overexpression in ECs prevented the development of hypoxia‐induced PH (Figure S5). Those data suggest that Cx40 overexpression is useful for both restoration and prevention of hypoxia‐induced PH. We believe Cx40 is a novel alternative target for the drug discovery in hypoxia‐induced PH because of its selective effect on endothelial function in small distal PAs.
Recent studies have also highlighted the change of Cx43 levels in PH. Bullaud et al demonstrated the increase in Cx43 level in pulmonary SMCs of hypoxia‐induced PH rats, but not in monocrotaline‐induced PH rats. 42 Bouvard et al also showed the increase of Cx43 expression in PAs of patients with PH attributable to lung diseases and hypoxia (but not in patients with idiopathic PAH) and mice with hypoxia‐induced PH. 43 They, however, found that Cx43 inhibition did not decrease RVSP in hypoxia‐induced PH mice, although PA muscularization was reduced. On the other hand, Htet et al showed that hypoxic exposure to mice for 2 weeks decreased Cx43 levels in PAs. 28 The opposite results of changes in Cx43 level shown in this study would be likely caused by short‐time exposure of hypoxia in mice. They also showed that Cx43 knockout mice (heterozygous) exhibited a further decrease in Cx43 after hypoxic exposure. 28 McNair et al demonstrated that acute hypoxic exposure increased Cx43 expression in pulmonary fibroblast, but not in pulmonary SMCs, and led to fibroblast proliferation ex vivo. 44 On the basis of the reports mentioned above, hypoxia‐mediated Cx43 overexpression in pulmonary SMCs or fibroblasts might have a maladaptive effect on pulmonary arterial remodeling, which potentially results in PH. The protein level of endothelial Cx43 was not tested in those studies, and we found that the protein level of Cx43 in MPECs was not altered in hypoxia‐induced PH mice (Figure 2C). Cx43 is expressed ubiquitously in different tissues and cells; therefore, it is important to study cell‐specific expression pattern of Cx43 and its potential role in pulmonary vascular disease.
It had been reported that increased Cx37 level leads to cell apoptosis 45 and cell cycle arrest. 46 , 47 We found that hypoxia reduced GJ activity, whereas Cx40 was significantly decreased, and Cx37 was increased, in hypoxia‐induced PH mice (Figure 2C and 2D). These data suggest that increased Cx37 is probably not the cause for hypoxia‐mediated reduction of GJ activity; what is the role of increased Cx37 in PH then? The key data from this study are the basal RVSP in Cx40KO mice was higher than in Wt mice, although Cx37 protein level in MPECs was slightly decreased in Cx40KO mice in comparison to MPECs from Wt mice (Figure 3). This means that only Cx40 downregulation is sufficient to increase RVSP shown in hypoxia‐induced PH mice. We will conduct further experiments to investigate the role of Cx37 in the development of PH in the future.
Inhaled NO and intravenous infusion of PGI2, along with medicines for increasing NO bioavailability and PGI2 analogs, are major drugs used for the treatment of patients with PH; however, the efficacy of these medicines can vary among patients with PH, and some patients do not even respond to these medications. Herein, we demonstrate that restoration of EDH‐mediated relaxation in small distal artery by increasing Cx40 expression or function is an efficient stratagem to ameliorate PH. Drugs that specifically open Cx40 or upregulate Cx40 in pulmonary ECs would provide a new type of treatment for pulmonary arterial hypertension and PH associated with lung disease and hypoxia.
Sources of Funding
This work was supported by grants from the National Heart, Lung, and Blood Institute of the National Institutes of Health (HL142214 and HL146764 to Dr Makino).
Disclosures
None.
Supporting information
Data S1
Tables S1–S2
Figures S1–S6
Acknowledgments
We wish to thank Dr Janis Burt at The University of Arizona for providing the mice lacking Cx40 gene for this study.
(J Am Heart Assoc. 2020;9:e018327. DOI: 10.1161/JAHA.120.018327.)
Supplementary Material for this article is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.018327
For Sources of Funding and Disclosures, see page 13.
References
- 1. Zangiabadi A, De Pasquale CG, Sajkov D. Pulmonary hypertension and right heart dysfunction in chronic lung disease. Biomed Res Int. 2014;2014:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guvenc TS, Huseyinoglu N, Ozben S, Kul S, Cetin R, Ozen K, Dogan C, Balci B. Right ventricular geometry and mechanics in patients with obstructive sleep apnea living at high altitude. Sleep Breath. 2016;20:5–13. [DOI] [PubMed] [Google Scholar]
- 3. Naeije R, Dedobbeleer C. Pulmonary hypertension and the right ventricle in hypoxia. Exp Physiol. 2013;98:1247–1256. [DOI] [PubMed] [Google Scholar]
- 4. Wanstall JC, Hughes IE, O'Donnell SR. Reduced relaxant potency of nitroprusside on pulmonary artery preparations taken from rats during the development of hypoxic pulmonary hypertension. Br J Pharmacol. 1992;107:407–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wan J, Yamamura A, Zimnicka AM, Voiriot G, Smith KA, Tang H, Ayon RJ, Choudhury MS, Ko EA, Wang J, et al. Chronic hypoxia selectively enhances L‐ and T‐type voltage‐dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav3.2. Am J Physiol Lung Cell Mol Physiol. 2013;305:L154–L164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aaronson PI, Robertson TP, Ward JP. Endothelium‐derived mediators and hypoxic pulmonary vasoconstriction. Respir Physiol Neurobiol. 2002;132:107–120. [DOI] [PubMed] [Google Scholar]
- 7. Tanaka S, Shiroto T, Godo S, Saito H, Ikumi Y, Ito A, Kajitani S, Sato S, Shimokawa H. Important role of endothelium‐dependent hyperpolarization in the pulmonary microcirculation in male mice: implications for hypoxia‐induced pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2018;314:H940–H953. [DOI] [PubMed] [Google Scholar]
- 8. Jaitovich A, Jourd'heuil D. A brief overview of nitric oxide and reactive oxygen species signaling in hypoxia‐induced pulmonary hypertension. Adv Exp Med Biol. 2017;967:71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wolin MS, Gupte SA, Mingone CJ, Neo BH, Gao Q, Ahmad M. Redox regulation of responses to hypoxia and NO‐cGMP signaling in pulmonary vascular pathophysiology. Ann N Y Acad Sci. 2010;1203:126–132. [DOI] [PubMed] [Google Scholar]
- 10. Garland CJ, Dora KA. EDH: endothelium‐dependent hyperpolarization and microvascular signalling. Acta Physiol (Oxf). 2017;219:152–161. [DOI] [PubMed] [Google Scholar]
- 11. Ellinsworth DC, Sandow SL, Shukla N, Liu Y, Jeremy JY, Gutterman DD. Endothelium‐derived hyperpolarization and coronary vasodilation: diverse and integrated roles of epoxyeicosatrienoic acids, hydrogen peroxide, and gap junctions. Microcirculation. 2016;23:15–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Figueroa XF, Duling BR. Gap junctions in the control of vascular function. Antioxid Redox Signal. 2009;11:251–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sohl G, Willecke K. Gap junctions and the connexin protein family. Cardiovasc Res. 2004;62:228–232. [DOI] [PubMed] [Google Scholar]
- 14. Hill CE, Rummery N, Hickey H, Sandow SL. Heterogeneity in the distribution of vascular gap junctions and connexins: implications for function. Clin Exp Pharmacol Physiol. 2002;29:620–625. [DOI] [PubMed] [Google Scholar]
- 15. Sandow SL, Hill CE. Incidence of myoendothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium‐derived hyperpolarizing factor‐mediated responses. Circ Res. 2000;86:341–346. [DOI] [PubMed] [Google Scholar]
- 16. Straub AC, Zeigler AC, Isakson BE. The myoendothelial junction: connections that deliver the message. Physiology. 2014;29:242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oyamada M, Oyamada Y, Takamatsu T. Regulation of connexin expression. Biochim Biophys Acta. 2005;1719:6–23. [DOI] [PubMed] [Google Scholar]
- 18. Saez JC, Berthoud VM, Branes MC, Martinez AD, Beyer EC. Plasma membrane channels formed by connexins: their regulation and functions. Physiol Rev. 2003;83:1359–1400. [DOI] [PubMed] [Google Scholar]
- 19. de Wit C, Roos F, Bolz SS, Kirchhoff S, Kruger O, Willecke K, Pohl U. Impaired conduction of vasodilation along arterioles in connexin40‐deficient mice. Circ Res. 2000;86:649–655. [DOI] [PubMed] [Google Scholar]
- 20. Gartner C, Ziegelhoffer B, Kostelka M, Stepan H, Mohr FW, Dhein S. Knock‐down of endothelial connexins impairs angiogenesis. Pharmacol Res. 2012;65:347–357. [DOI] [PubMed] [Google Scholar]
- 21. Alonso F, Domingos‐Pereira S, Le Gal L, Derre L, Meda P, Jichlinski P, Nardelli‐Haefliger D, Haefliger JA. Targeting endothelial connexin40 inhibits tumor growth by reducing angiogenesis and improving vessel perfusion. Oncotarget. 2016;7:14015–14028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krattinger N, Capponi A, Mazzolai L, Aubert JF, Caille D, Nicod P, Waeber G, Meda P, Haefliger JA. Connexin40 regulates renin production and blood pressure. Kidney Int. 2007;72:814–822. [DOI] [PubMed] [Google Scholar]
- 23. de Wit C, Roos F, Bolz SS, Pohl U. Lack of vascular connexin 40 is associated with hypertension and irregular arteriolar vasomotion. Physiol Genomics. 2003;13:169–177. [DOI] [PubMed] [Google Scholar]
- 24. Reaume AG, de Sousa PA, Kulkarni S, Langille BL, Zhu D, Davies TC, Juneja SC, Kidder GM, Rossant J. Cardiac malformation in neonatal mice lacking connexin43. Science. 1995;267:1831–1834. [DOI] [PubMed] [Google Scholar]
- 25. Liao Y, Day KH, Damon DN, Duling BR. Endothelial cell‐specific knockout of connexin 43 causes hypotension and bradycardia in mice. Proc Natl Acad Sci USA. 2001;98:9989–9994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li N, Dai DZ, Dai Y. CPU86017 and its isomers improve hypoxic pulmonary hypertension by attenuating increased ETA receptor expression and extracellular matrix accumulation. Naunyn Schmiedebergs Arch Pharmacol. 2008;378:541–552. [DOI] [PubMed] [Google Scholar]
- 27. Yang L, Yin N, Hu L, Fan H, Yu D, Zhang W, Wang S, Feng Y, Fan C, Cao F, et al. Sildenefil increases connexin 40 in smooth muscle cells through activation of BMP pathways in pulmonary arterial hypertension. Int J Clin Exp Pathol. 2014;7:4674–4684. [PMC free article] [PubMed] [Google Scholar]
- 28. Htet M, Nally JE, Shaw A, Foote BE, Martin PE, Dempsie Y. Connexin 43 plays a role in pulmonary vascular reactivity in mice. Int J Mol Sci. 2018;19:1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim J, Hwangbo C, Hu X, Kang Y, Papangeli I, Mehrotra D, Park H, Ju H, McLean DL, Comhair SA, et al. Restoration of impaired endothelial myocyte enhancer factor 2 function rescues pulmonary arterial hypertension. Circulation. 2015;131:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fang JS, Angelov SN, Simon AM, Burt JM. Compromised regulation of tissue perfusion and arteriogenesis limit, in an AT1R‐independent fashion, recovery of ischemic tissue in Cx40‐/‐ mice. Am J Physiol Heart Circ Physiol. 2013;304:H816–H827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Morton SK, Chaston DJ, Howitt L, Heisler J, Nicholson BJ, Fairweather S, Broer S, Ashton AW, Matthaei KI, Hill CE. Loss of functional endothelial connexin40 results in exercise‐induced hypertension in mice. Hypertension. 2015;65:662–669. [DOI] [PubMed] [Google Scholar]
- 32. Makino A, Platoshyn O, Suarez J, Yuan JX, Dillmann WH. Downregulation of connexin40 is associated with coronary endothelial cell dysfunction in streptozotocin‐induced diabetic mice. Am J Physiol Cell Physiol. 2008;295:C221–C230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vanhoutte PM, Shimokawa H, Feletou M, Tang EH. Endothelial dysfunction and vascular disease: a 30th anniversary update. Acta Physiol (Oxf). 2017;219:22–96. [DOI] [PubMed] [Google Scholar]
- 34. Pan M, Han Y, Si R, Guo R, Desai A, Makino A. Hypoxia‐induced pulmonary hypertension in type 2 diabetic mice. Pulm Circ. 2017;7:175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang Q, Tsuji‐Hosokawa A, Willson C, Watanabe M, Si R, Lai N, Wang Z, Yuan JX, Wang J, Makino A. Chloroquine differentially modulates coronary vasodilation in control and diabetic mice. Br J Pharmacol. 2020;177:314–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dinh‐Xuan AT, Pepke‐Zaba J, Butt AY, Cremona G, Higenbottam TW. Impairment of pulmonary‐artery endothelium‐dependent relaxation in chronic obstructive lung disease is not due to dysfunction of endothelial cell membrane receptors nor to L‐arginine deficiency. Br J Pharmacol. 1993;109:587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Christou H, Hudalla H, Michael Z, Filatava EJ, Li J, Zhu M, Possomato‐Vieira JS, Dias‐Junior C, Kourembanas S, Khalil RA. Impaired pulmonary arterial vasoconstriction and nitric oxide‐mediated relaxation underlie severe pulmonary hypertension in the sugen‐hypoxia rat model. J Pharmacol Exp Ther. 2018;364:258–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murata T, Sato K, Hori M, Ozaki H, Karaki H. Decreased endothelial nitric‐oxide synthase (eNOS) activity resulting from abnormal interaction between eNOS and its regulatory proteins in hypoxia‐induced pulmonary hypertension. J Biol Chem. 2002;277:44085–44092. [DOI] [PubMed] [Google Scholar]
- 39. Dubois M, Delannoy E, Duluc L, Closs E, Li H, Toussaint C, Gadeau AP, Godecke A, Freund‐Michel V, Courtois A, et al. Biopterin metabolism and eNOS expression during hypoxic pulmonary hypertension in mice. PLoS One. 2013;8:e82594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wandall‐Frostholm C, Skaarup LM, Sadda V, Nielsen G, Hedegaard ER, Mogensen S, Kohler R, Simonsen U. Pulmonary hypertension in wild type mice and animals with genetic deficit in KCa2.3 and KCa3.1 channels. PLoS One. 2014;9:e97687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kroigaard C, Kudryavtseva O, Dalsgaard T, Wandall‐Frostholm C, Olesen SP, Simonsen U. KCa3.1 channel downregulation and impaired endothelium‐derived hyperpolarization‐type relaxation in pulmonary arteries from chronically hypoxic rats. Exp Physiol. 2013;98:957–969. [DOI] [PubMed] [Google Scholar]
- 42. Billaud M, Dahan D, Marthan R, Savineau JP, Guibert C. Role of the gap junctions in the contractile response to agonists in pulmonary artery from two rat models of pulmonary hypertension. Respir Res. 2011;12:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bouvard C, Genet N, Phan C, Rode B, Thuillet R, Tu L, Robillard P, Campagnac M, Soleti R, et al. Connexin‐43 is a promising target for pulmonary hypertension due to hypoxaemic lung disease. Eur Respir J. 2020;55:1900169. [DOI] [PubMed] [Google Scholar]
- 44. McNair AJ, Wilson KS, Martin PE, Welsh DJ, Dempsie Y. Connexin 43 plays a role in proliferation and migration of pulmonary arterial fibroblasts in response to hypoxia. Pulm Circ. 2020;10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seul KH, Kang KY, Lee KS, Kim SH, Beyer EC. Adenoviral delivery of human connexin37 induces endothelial cell death through apoptosis. Biochem Biophys Res Commun. 2004;319:1144–1151. [DOI] [PubMed] [Google Scholar]
- 46. Jacobsen NL, Pontifex TK, Li H, Solan JL, Lampe PD, Sorgen PL, Burt JM. Regulation of Cx37 channel and growth‐suppressive properties by phosphorylation. J Cell Sci. 2017;130:3308–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fang JS, Coon BG, Gillis N, Chen Z, Qiu J, Chittenden TW, Burt JM, Schwartz MA, Hirschi KK. Shear‐induced Notch‐Cx37‐p27 axis arrests endothelial cell cycle to enable arterial specification. Nat Commun. 2017;8:2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Tables S1–S2
Figures S1–S6