

Abstract
Background
Caffeine is the most widely consumed psychostimulant and is associated with lower risk of coronary artery disease (CAD) and type 2 diabetes mellitus (T2DM). However, whether these associations are causal remains unknown. This study aimed to identify genetic variants associated with caffeine intake, and to investigate evidence for causal links with CAD or T2DM. In addition, we aimed to replicate previous observational findings.
Methods and Results
Observational associations were tested within UK Biobank using Cox regression analyses. Moderate observational caffeine intakes from coffee or tea were associated with lower risks of CAD or T2DM, with the lowest risks at intakes of 121 to 180 mg/day from coffee for CAD (hazard ratio [HR], 0.77 [95% CI, 0.73–0.82; P<1×10−16]), and 301 to 360 mg/day for T2DM (HR, 0.76 [95% CI, 0.67–0.86]; P=1.57×10−5). Next, genome‐wide association studies were performed on self‐reported caffeine intake from coffee, tea, or both in 407 072 UK Biobank participants. These analyses identified 51 novel genetic variants associated with caffeine intake at P<1.67×10−8. These loci were enriched for central nervous system genes. However, in contrast to the observational analyses, 2‐sample Mendelian randomization analyses using the identified loci in independent disease‐specific cohorts yielded no evidence for causal links between genetically determined caffeine intake and the development of CAD or T2DM.
Conclusions
Mendelian randomization analyses indicate genetically determined higher caffeine intake might not protect against CAD or T2DM, despite protective associations in observational analyses.
Keywords: caffeine intake, coronary artery disease, genetics, Mendelian randomization, type 2 diabetes mellitus
Subject Categories: Basic Science Research
Nonstandard Abbreviations and Acronyms
- CARDIoGRAMplusC4D
Coronary Artery Disease Genome wide Replication and Meta‐analysis plus The Coronary Artery Disease Genetics
- DIAGRAM
Diabetes Genetics Replication And Meta‐analysis
- eQTL
expression quantitative trait locus
- MR
Mendelian randomization
- T2DM
type 2 diabetes mellitus
Clinical Perspective
What Is New?
Leveraging data from >400 000 individuals, we identified 51 novel genetic loci associated with caffeine intake.
We confirmed phenotypic associations between caffeine intake and the development of coronary artery disease or type 2 diabetes mellitus, but by exploiting instrumental variable analyses we found no evidence for causality of this association.
What Are the Clinical Implications?
Our data do not support recommending caffeine intake to protect against the development of coronary artery disease of type 2 diabetes mellitus.
Caffeine is the most commonly consumed psychostimulant in the world and is readily available in coffee, tea, and other food products. 1 Previous observational studies and meta‐analyses have generally reported beneficial associations between moderate intake of coffee, the main dietary source of caffeine, 1 and risk of cardiovascular disease 2 and type 2 diabetes mellitus (T2DM), 3 as well as cardiovascular and all‐cause mortality. 4 , 5 Contrasting results have been reported as well for cardiovascular disease outcomes, including coronary artery disease (CAD), 2 , 6 , 7 , 8 , 9 and therefore coffee and tea are not generally included in dietary guidelines. 10 Given its widespread consumption, altering caffeine intake might be an interesting way to influence population‐wide risk of developing CAD and T2DM.
Because of the observational design of previous studies, which include many cross‐sectional and case‐control studies, it is difficult to provide insight into causal relationships. Genome‐wide association studies (GWASs) have identified several single‐nucleotide polymorphisms (SNPs) associated with caffeine or coffee intake through genes such as AHR and CYP1A2, which affect the metabolism of caffeine. 11 , 12 , 13 , 14 , 15 , 16 , 17 Unlike traditional observational studies, Mendelian randomization (MR) analyses have the advantageous applicability of uncovering causal links using genetic variants, which are randomly allocated at conception, as instrumental variables for modifiable risk factors to test potential causal links with disease outcomes. So far, MR analyses between genetically determined higher caffeine intake and risk of CAD 7 , 18 or T2DM 19 failed to provide support for a causal link. However, these studies used only few SNPs and investigated coffee as the sole source of caffeine.
Here, we investigated the observational associations between habitual caffeine intake from coffee, tea, or both with new‐onset CAD and T2DM in a large prospective observational cohort. To further our knowledge of the genetic architecture underlying caffeine intake, we carried out GWASs for caffeine intake from coffee, tea, or both in over 400 000 participants from the UK Biobank to identify novel variants for caffeine intake. Using this set of SNPs, we aimed to investigate the causal relationship between caffeine intake with CAD and T2DM in large independent cohorts.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. GWAS summary statistics generated during the present study will be made available in the following repository: https://doi.org/10.17632/d8nwkm7p9p.1.
Study Population
The UK Biobank study is a population‐based prospective cohort whose design and population have been described previously. 20 From 2006 to 2010, >500 000 individuals between the ages of 40 and 69 years were recruited in the United Kingdom. All participants gave informed consent, 21 and the UK Biobank study was approved by the North West Multi‐centre Research Ethics committee. 22 Details regarding the UK Biobank study population are provided in Data S1.
Ascertainment of Coffee and Tea Intake
During the first visit to the assessment center, daily coffee and tea intake were assessed by asking participants, “How many cups of coffee do you drink each day? (Include decaffeinated coffee)” and “How many cups of tea do you drink each day? (Include black and green tea).” In addition, coffee drinkers were asked what type of coffee they usually drink. Caffeine intake was calculated as the number of cups of coffee or tea multiplied by the caffeine content per cup. 23 Combined caffeine intake from both coffee and tea was calculated as the sum of the daily caffeine intake from coffee and tea from individuals who provided data on both. Full details on the ascertainment of coffee, tea, and daily caffeine intake are provided in Data S1.
CAD and T2DM Prevalence and Incidence in the UK Biobank
Prevalence at baseline and incidence of new‐onset CAD and T2DM cases within UK Biobank were, per prior analysis, 24 based on self‐reported data, International Classification of Diseases, Ninth Revision (ICD‐9) and Tenth Revision (ICD‐10) 25 coded primary and secondary diagnoses, operation codes, 26 and death attributable to either condition from inclusion in the UK Biobank until end of follow‐up (March 31, 2017, for participants from England; February 29, 2016, for Wales; and October 31, 2016, for Scotland) as described in Data S1. Incident cases that were based on self‐reported diagnoses during follow‐up visits were included only if there were no events recorded according to the ICD‐9 or ICD‐10 or operation codes data and only if the participant did not report this in the previous visit. If the participant was the same age as the reported age of diagnosis, the median date between the visit and the participant's birthday was taken as date of event. If the age of diagnosis was before the participant's current age, we took the median date of the year of the reported age of diagnosis counted from the participant's birthday. If age of diagnosis was not available, we took the median date between the visit of the first self‐reported diagnosis and the previous visit. Individuals with a history of CAD or T2DM at inclusion were excluded from the respective observational analyses.
Covariates
At the first visit, weight (in kilograms) and height (in centimeters) were measured and used to calculate the body mass index (in kilograms per square meter). Age was calculated as the difference between date of birth and date of inclusion in the UK Biobank. Sex, ethnicity, weekly alcohol intake (UK units) and active smoking at inclusion were self‐reported. Weekly alcohol intake was right‐skewed and therefore log2 transformed for participants who provided this data. For participants without these accurate data on the number of units, we estimated the weekly alcohol intake using a more crude questionnaire of alcohol intake frequency where participants were asked, “About how often do you drink alcohol?” For this, we fitted a linear regression between with the log2‐transformed weekly alcohol intake and alcohol intake frequency in participants with both measures, and predicted weekly alcohol intake on the remaining individuals. The Townsend Deprivation Index, a proxy for socioeconomic status, was provided by the UK Biobank and inverse rank normalized because of a right‐skewed distribution. 24
Genotyping and Imputation in UK Biobank
UK Biobank participants were genotyped using custom Affymetrix Axiom (UK Biobank Lung Exome Variant Evaluation 27 or UK Biobank) arrays. The genotyping methods, arrays, and quality‐control procedures have been described previously in detail 28 , 29 and are briefly described in Data S1.
Statistical Analysis
We performed multivariable Cox regression analyses to test the association of observational caffeine intake per 60 mg caffeine (equivalent to the caffeine content of 1 cup of instant coffee or 2 cups of tea) with new‐onset CAD and T2DM in the UK Biobank. Hazard ratios with 95% CIs were calculated for 1 to 60, 61 to 120, 121 to 180, 181 to 240, 241 to 300, 301 to 360, or >360 mg of caffeine from coffee or combined, compared with individuals who drank 0 mg. Because of the lower caffeine content per cup of tea compared with caffeinated coffee, the hazard ratios and 95% CIs for caffeine from tea were calculated for 1 to 60, 61 to 120, 121 to 180, or >180 mg (equivalent to >6 cups of tea) of caffeine compared with individuals who had 0‐mg intake from tea. The time scale for the Cox regression analyses was from inclusion in the UK Biobank until the outcome of interest, death or end of follow‐up. Cox regression analyses were performed unadjusted and adjusted for age, sex, body mass index, active smoking, Townsend Deprivation Index, and weekly alcohol intake using Stata version 15 (StataCorp, College Station, TX).
All genetic analyses were adjusted for age, sex, genotyping array, and the first 30 genetic principal components to adjust for population stratification. We performed separate GWASs for inverse rank normalized combined caffeine intake, caffeine from coffee, and caffeine from tea in 19 400 838 SNPs using BOLT‐LMM version 2.3.1 software (Broad Institute, Cambridge, MA). 30 A Bonferroni corrected P<1.67×10−8 (traditional GWAS significance threshold of 5×10−8/3) was considered genome‐wide significant. This significance threshold is conservative, considering that our phenotypes are correlated with Spearman's rank correlation coefficients between phenotype pairs ranging from r=−0.33 to 0.71 (Table S1). Details of the GWAS analyses, functional annotation of candidate genes, 31 , 32 , 33 , 34 , 35 and biological pathways are provided in Data S1.
We performed MR analyses using previously published summary statistics from the CARDIoGRAMplusC4D (Coronary Artery Disease Genome wide Replication and Meta‐analysis plus The Coronary Artery Disease Genetics) consortium (123 504 controls and 60 801 [33.0%] cases) 36 and the DIAGRAM Diabetes Genetics Replication And Meta‐analysis)) consortium (132 532 controls and 26 676 [16.8%] cases) 37 to gain insight into potential causal relationships between caffeine intake and CAD or T2DM, respectively. Lead SNPs of each caffeine intake trait that reached P<1.67×10−8 were used to create a weighted genetic risk score and were also used as instrumental variables in the MR. Each genetic risk score was created using an additive model per GWAS, summing the number of effect alleles (0, 1, or 2) per individual after multiplying it with the effect size between the SNP and the GWAS phenotype. Statistical power for the MR with a binary outcome was calculated using an alpha of 0.05 and the explained variance of each genetic risk score, as described previously. 38 For the MR, SNPs that were not available in CARDIoGRAMplusC4D or DIAGRAM were replaced with proxies with R 2>0.8, and were otherwise excluded from the MR analyses if no eligible proxies were available. SNP effects were harmonized across studies using the built‐in feature of the TwoSampleMR package in R (R Foundation for Statistical Computing, Vienna, Austria). The association between genetically determined higher caffeine intake and CAD or T2DM was assessed using fixed‐effects inverse‐variance weighted meta‐analyses. Odds ratios (ORs) with 95% CIs are presented for the MR outcomes. To maximize the likelihood of reporting true findings, α was set at 0.005 instead of 0.05. 39 Associations with P<0.05 were considered suggestively significant. We assessed potential weak instrument bias per SNP using the F‐statistic 40 and I2 GX. 41 We determined the I2 index. 42 Cochran's Q, Rücker's Q′, and Q‐Q′ 43 to test for heterogeneity and thus potential pleiotropy. MR‐Egger, 43 MR Pleiotropy Residual Sum and Outlier 44 and MR inverse‐variance weighted random effects 43 were used as pleiotropy analyses. MR‐Steiger filtering 45 was performed to remove variants more strongly associated with the outcome than the exposure. Weighted median and weighted mode‐based estimator MR analyses 46 were performed as additional sensitivity analyses. Details of the MR analyses are provided in Data S1.
Results
Cohort Characteristics
Of 502 525 UK Biobank individuals, 362 316 were available for the combined caffeine intake analyses, 373 522 for caffeine from coffee, and 395 866 for caffeine from tea (Figure S1). Baseline characteristics are shown in Table, per caffeine intake trait in Table S2, and stratified by caffeine intake in Tables S3 through S5. Median (interquartile range) combined caffeine intake was 205 (120–290) mg/day, from coffee 85 (3–180) mg/day, and from tea 90 (60–150) mg/day.
Table 1.
Baseline Characteristics of All Included 407 072 UK Biobank Participants
| Characteristics | Men | Women |
|---|---|---|
| Total, N | 186 968 | 220 104 |
| Age, y, mean (SD) | 57.16 (8.08) | 56.72 (7.92) |
| Daily caffeine intake, mg/d, median (IQR) | ||
| Combined caffeine | 210 (150–300) | 180 (120–270) |
| Caffeine from coffee | 85 (6–180) | 60 (3–170) |
| Caffeine from tea | 90 (60–150) | 90 (60–150) |
| Blood pressure, mm Hg, mean (SD) | ||
| Systolic | 139.60 (16.15) | 128.74 (17.88) |
| Diastolic | 84.69 (8.22) | 79.94 (8.20) |
| Active smoker, N (%) | ||
| No | 164 791 (88.1) | 200 946 (91.3) |
| Yes | 22 177 (11.9) | 19 158 (8.7) |
| Body mass index , kg/m2, mean (SD) | 27.85 (4.23) | 27.05 (5.13) |
| Weekly alcohol intake, UK units, median (IQR) | 15.40 (5.50, 28.40) | 6.40 (1.60, 13.20) |
| Hypertension, N (%) | ||
| No | 119 965 (64.2) | 160 881 (73.1) |
| Yes | 67 003 (35.8) | 59 223 (26.9) |
| Hyperlipidemia, N (%) | ||
| No | 139 471 (74.6) | 188 444 (85.6) |
| Yes | 47 497 (25.4) | 31 660 (14.4) |
Combined caffeine intake was calculated as the sum of caffeine intake from coffee and tea. Body mass index was calculated as weight in kilograms divided by height in meters squared. Smoking status and weekly alcohol intake were self‐reported at inclusion. IQR indicates interquartile range.
Associations of Observational Caffeine Intake With CAD and T2DM
During nearly 10 years (median, 8.1 years; interquartile range, 7.5–8.6) of follow‐up in 345 809 participants without history of CAD and 347 718 participants without history of T2DM, 14 681 (4.2%) individuals developed CAD, and 6982 (2.0%) developed T2DM in the combined caffeine cohort. Results for unadjusted analyses are presented in Tables S6 and S7. In multivariable adjusted analyses (Tables S8 and S9), combined caffeine intake was very modestly or not associated with CAD or T2DM. However, the individual components, caffeine from coffee or tea, did show associations with lower risks of new‐onset CAD and T2DM (Figure 1A and 1B, respectively). Overall, the associations between caffeine from coffee or tea with CAD and T2DM followed U‐curve–type shapes, with the highest protective effects of caffeine intake from coffee on CAD at moderate intakes (121–180 mg/day), compared with no, lower, or higher intakes. Associations between caffeine from coffee with CAD or T2DM were not appreciably different when additionally adjusted for caffeine from tea, nor were the associations for caffeine from tea when additionally adjusted for caffeine from coffee (Table S10). Overall, caffeine intake from coffee was associated with lower risks of CAD and T2DM compared with caffeine from tea or combined. To determine whether this may be attributable to confounding by other, noncaffeine, substances, we stratified the analyses by cups of decaffeinated or caffeinated coffee and found similar results. Both caffeinated and decaffeinated coffee were associated with lower risk of CAD and T2DM compared with no or high (>6 cups for caffeinated coffee; >3 for decaffeinated coffee) intake (Table S11).
Figure 1. Associations between observational caffeine intake with new‐onset coronary artery disease (A) and type 2 diabetes mellitus (B).
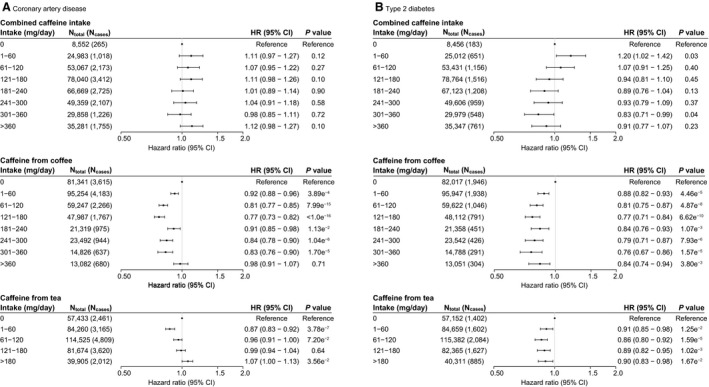
Hazard ratios (HR) with 95% CIs were calculated using Cox regression analyses, adjusted for age, sex, active smoking, body mass index, and log‐transformed weekly alcohol intake. Estimates <1 indicate a beneficial association between caffeine intake and outcome. Sixty milligrams of caffeine is equivalent to 1 cup of instant coffee or 2 cups of tea.
GWAS on Caffeine Intake Traits
We identified 62 SNPs in 37 loci: 32 novel, associated with combined caffeine intake (Figure 2; Table S12); 27 SNPs in 24 loci (20 novel) with caffeine from coffee (Figure S2; Table S13); and 27 SNPs in 24 loci (21 novel) with caffeine from tea (Figure S3; Table S14). When combined on the basis of the lowest P value over all traits, 73 unique SNPs in 5 known and 51 novel loci were associated with ≥1 caffeine trait (Figure S4, Table S15). In total, 15 of 20 previously reported SNPs were replicated within 1 MB of our sentinel SNPs (Table S16). Regional association plots for each independent locus per trait are presented in Figures S5 through S7 and QQ plots in Figures S8 through S10. The sentinel SNPs identified in the combined caffeine, caffeine from coffee, and caffeine from tea GWAS explained 1.32%, 0.59%, and 0.45% of variance in caffeine intake of their respective trait. The heritability rate () for all SNPs in the GWAS was 8.2% for combined caffeine intake, 6.1% for caffeine from coffee, and 7.1% for caffeine from tea.
Figure 2. Manhattan plot for combined caffeine intake.
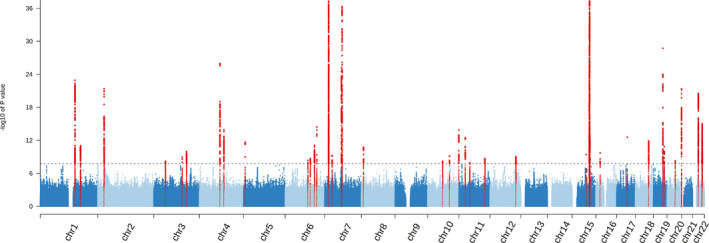
Manhattan plot showing the results for the genome‐wide associations with combined caffeine intake in the UK Biobank with the −log10 P value on the vertical axis. The sentinel single nucleotide polymorphisms that reached genome‐wide significance (P<1.67×10−8) are colored red.
Using the genetic risk score of each GWAS, each unit change in genetically determined caffeine intake was consistent with 131.6 mg combined caffeine intake, 134.5 mg caffeine intake from coffee, and 86.1 mg caffeine intake from tea. In coffee drinkers, depending on the type of coffee usually drunk, each unit related from 1.5 cup of decaffeinated coffee to 2.1 cups of instant coffee (Table S17).
Candidate Genes and Deeper Insights Into Biology
We explored the potential biology of the sentinel SNPs per GWAS by prioritizing potentially causal genes in these loci based on proximity, expression quantitative trait locus (eQTL) analyses, and data‐driven expression‐prioritized integration for complex traits. In total, we identified 48 candidate genes for combined caffeine intake, 27 for caffeine from coffee, and 40 for caffeine from tea (Figure 3). We identified the previously reported AHR, CYP1A1, and POR genes in all 3 GWASs. In addition, 2 novel genes, GOLPH3L and HORMAD1, were associated with all caffeine traits.
Figure 3. Venn diagram of candidate genes associated with caffeine intake.
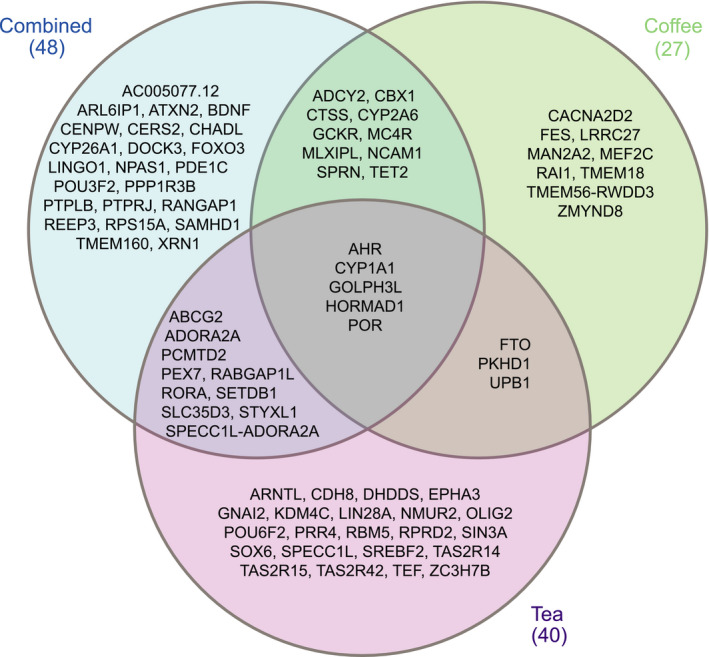
Candidate genes were prioritized based on proximity, data‐driven expression‐prioritized integration for complex traits, and expression quantitative trait locus mapping for combined caffeine intake, caffeine from coffee, and caffeine from tea.
Across 209 tissue and cell types, central nervous system tissues were most enriched for SNPs associated with caffeine from tea and combined, but none with caffeine from coffee (Table S18). Furthermore, 6 combined caffeine intake loci, and 3 loci each of caffeine from coffee or tea, contained variants with eQTLs in at least 1 tissue. The strongest associations were found for rs768283768 near HORMAD1 and GOLPH3L, which tagged multiple tissues (Table S19).
Genetically Determined Caffeine Intake and CAD
The association between genetically determined caffeine intake and CAD was tested in the independent CARDIoGRAMplusC4D cohort (123 504 controls and 60 801 [33.0%] cases). In total, 35 SNPs from caffeine for combined caffeine intake, 22 for caffeine from coffee (rs2298527 excluded based on intermediate allele frequency in CARDIoGRAMplusC4D), and 24 for caffeine from tea (Table S20 through S22). F‐statistics indicated low chances of weak instrument bias (Table S23) and I2 GX indicated low chances of measurement error in MR‐Egger (Table S24). However, I2 and Cochran's Q indicated heterogeneity, and thus potential pleiotropy, for all caffeine traits (Table S24). Using the random effects inverse‐variance weighted method as indicated by the nonsignificant Q‐Q′ and MR‐Egger intercepts, we found that genetically determined caffeine intake from combined or coffee were not associated with CAD (OR, 1.12 [95% CI, 0.80–1.40], P=0.31; OR 1.26 [95% CI, 0.82–1.93], P=0.28, respectively). MR‐Egger was used for caffeine from tea because the Q‐Q′ was significant; however, also for caffeine from tea, no association with CAD was indicated (OR, 1.60 [95% CI, 0.75–3.44], P=0.24). MR Pleiotropy Residual Sum and Outlier analyses corroborated these findings for all traits, with and without trimming outlier SNPs (Table S25). MR‐Steiger filtering also did not attenuate the results for any caffeine trait (Table S26). Finally, weighted median and mode‐based analyses also indicated no association between genetically determined caffeine intake and CAD. Individual SNP effects are shown in Figures S11 through S13 and the MR analyses in Figure 4A.
Figure 4. Mendelian randomization results for genetically determined higher caffeine intake (per SD) on coronary artery disease (A) and type 2 diabetes mellitus (B).
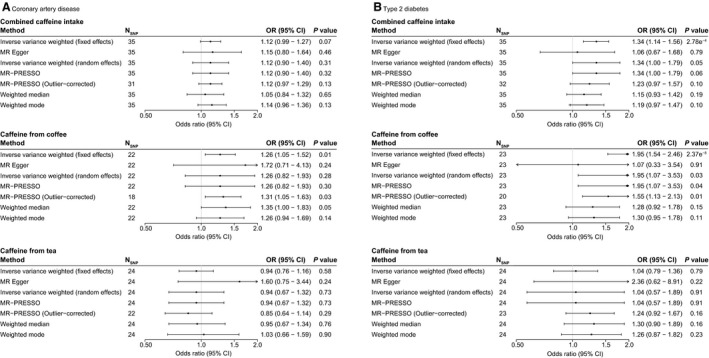
Odds ratios (OR) with 95% CIs are provided per standard deviation increase in genetically determined caffeine intake from combined, coffee, or tea. Number of single‐nucleotide polymorphisms (SNPs) included are shown per method. Estimates <1.0 indicate a beneficial association between genetically determined caffeine intake and outcome. MR‐PRESSO indicates Mendelian Randomization Pleiotropy Residual Sum and Outlier.
Genetically Determined Caffeine Intake and T2DM
The association between genetically determined caffeine intake and T2DM was investigated in the DIAGRAM cohort (132 532 controls and 26 676 [16.8%] cases). In DIAGRAM, 35 SNPs for combined caffeine intake, 23 SNPs for caffeine from coffee, and 24 SNPs for caffeine from tea were used (Tables S27 through S29). Also here, I2 indices and Cochran's Q indicated pleiotropy for all traits, and the MR‐Egger intercept was not significant. However, because the Q‐Q′ was significant for all traits, we focused on the MR‐Egger estimate for the causal effect. The MR‐Egger analyses indicated no association between genetically determined higher caffeine intake from any trait with risk of T2DM (OR, 1.06 [95% CI, 0.67–1.68], P=0.79 for combined caffeine intake; OR, 1.07 [95% CI, 0.33–3.54], P=0.91 for caffeine from coffee; OR, 2.36 [95% CI, 0.62–8.91], P=0.22 for caffeine from tea; Figure 4B; estimates per SNP in Figures S14 through S16). Additional analyses using MR Pleiotropy Residual Sum and Outlier and MR‐Steiger also found no associations between caffeine intake with T2DM after respectively trimming outliers and filtering (Tables S25 and S26). Finally, also weighted and mode‐based estimator MR analyses were in line with these findings and indicated no association with T2DM.
Combined Caffeine Intake–Specific Variants
In total, 18 variants were associated with combined caffeine intake, of which the annotated genes do not overlap with those of caffeine from coffee or caffeine from tea. However, these variants were most strongly associated with combined caffeine intake compared with caffeine from tea or coffee and had concordant betas across all traits (Table S15). This suggests that these variants act on both caffeine from coffee and caffeine from tea. We repeated the MR analyses using these variants or their proxies available in CARDIoGRAMplusC4D and DIAGRAM. Similar to the MR using all combined caffeine intake variants, we found no associations with CAD or T2DM.
Moderate Versus Extreme Caffeine Intakes From Coffee or Tea
Because of the U‐shaped curve observed in the observational analyses between caffeine from coffee and caffeine from tea with CAD or T2DM, we performed exploratory analyses to investigate variants associated with moderate caffeine intake from coffee or tea separately. Extremes of caffeine intake (0 and >360 mg/day for coffee and 0 and >120 mg/day for tea) were taken together and values between the extremes as moderate intake. A total of 373 522 individuals (99 427 [26.6%] with moderate intake) were included in the GWAS for moderate caffeine consumption from coffee, and 395 866 (188 013 [47.8%] with moderate intake) in the GWAS for moderate caffeine consumption from tea. However, GWAS on either phenotype found no variants at P<1.67×10−8 or P<5×10−8.
Discussion
In this large prospective study, we observed U‐type associations between observational caffeine intake with CAD and T2DM, although similar intakes from different sources had dissimilar effect sizes. In addition, we identified 51 novel genetic loci associated with caffeine intake, more than tripling the number of known loci. 11 , 12 , 13 , 14 , 15 , 16 , 17 In contrast to the observational analyses, genetic causal inference analyses indicated that genetically determined caffeine intake was not associated with CAD or T2DM.
Our observational findings are concordant with previous studies showing inverse or U‐type associations between caffeine intake with CAD 2 , 47 and T2DM. 3 , 47 , 48 A meta‐analysis in 1 283 685 individuals (28 347 CAD cases) estimated a relative risk of 0.89 (95% CI, 0.85–0.94) for CAD at 3 to 5 cups of coffee daily and a neutral effect at higher intakes (>360 mg or >6 cups of coffee) compared with no intake. 2 A plausible explanation for the U‐type shape of the association is that coffee is a liquid extract of coffee beans and it contains a complex chemical mixture of biologically active compounds, some with beneficial and others with harmful effects. 49 At moderate intakes, the beneficial effects could outweigh or counteract the harmful effects, whereas at higher intakes the harmful effects may counterbalance this. 2 Our results for T2DM are in line with the most recent meta‐analysis, which reported a relative risk of 0.70 (95% CI, 0.65–0.75) in individuals who consumed 5 cups of coffee per day compared with nondrinkers, although they reported no U‐type associations. 50 The hypothesis that moderate caffeine intake may have beneficial effects compared with extreme intakes is also not supported by our findings for combined caffeine intake. The null findings of the observational analyses for combined caffeine intake indicate that caffeine by itself is unlikely to affect disease risk. The current study used the largest number of caffeine SNPs to date from different dietary sources, which is relevant for this UK population, where tea is the second‐largest source of caffeine 1 and may confound the association. Using these SNPs in robust causal inference analyses, we found no associations between genetically determined higher or lower caffeine intake and CAD or T2DM. These findings are in line with previous MR studies of caffeine intake on CAD and T2DM. 7 , 18 , 19 The null findings of the combined caffeine intake SNPs can be considered a negative control for the observational findings. There is accumulating evidence that previous beneficial associations between caffeine intake with outcomes were attributable to residual confounding, most likely because of other compounds found in coffee 3 , 7 , 18 , 19 or smoking, 51 since no difference in outcomes is reported between decaffeinated and caffeinated coffee for CAD 8 or T2DM. 3 Also, in the current study, we found that observational decaffeinated coffee consumption was associated with similar effect sizes compared with caffeinated coffee. Caffeinated coffee was more robustly associated with outcomes, but this is likely attributable to the larger number of caffeinated coffee drinkers. Furthermore, caffeine from coffee was generally associated with lower estimates compared with caffeine from tea or combined, arguing against an independent effect of caffeine. In addition, both previous and the current MR analyses consistently lack evidence for causality, providing further argument against a protective effect of genetically determined higher caffeine intake.
To our knowledge, this is the largest study to date to investigate the association of both observational and genetically determined caffeine intake from multiple sources with CAD and T2DM. This study also reports the largest number of caffeine intake–associated SNPs, while also replicating previously reported SNPs. These newly identified variants were then used in independent disease‐specific cohorts for both CAD and T2DM in 2‐sample MR analyses. The explained variance of the sentinel SNPs is comparable with previously published GWASs on coffee 7 , 12 or alcohol 52 intake, which range between 0.6% and 1.3%. However, the explained variance was of little influence on the statistical power for the MR.
This study has some limitations. In the current analyses, caffeine intake was calculated on the basis of self‐reported data at a single time point at baseline, which does not take into account possible changes in coffee‐ and tea‐drinking habits. Furthermore, because the caffeine content of coffee may differ depending on the method of preparation, 53 , 54 use of filter, 55 and type of coffee bean, 1 and individuals may drink several types of coffee, the actual caffeine intake per day may differ from our calculation. We did not take into account caffeine intake from other sources such as cola or energy drinks, as this information was not available. In addition, the main MR analyses assume linear associations, whereas the causal associations might be nonlinear, with higher risks at low and high intakes, such as the U‐shaped–curve associations observed in the observational analyses. However, it was not possible to examine nonlinear associations in the MR analyses because these require individual‐level data in the outcome cohorts, which were not available. The MR analyses should therefore be interpreted with caution at the extremes of caffeine intake. It remains unclear which genetic variants are responsible for the specific parts of the potential U‐shaped–curve association, and we cannot exclude the possibility that the variants associated with caffeine intake from coffee or tea could have bidirectional effects on the association. Exploratory analyses to investigate the nonlinear association within the UK Biobank, however, indicate that there may be no genetic variants solely associated with moderate or extreme caffeine intake from coffee or tea.
Also, despite our sensitivity analyses to test for and minimize bias, especially from genetic pleiotropy in which the instrumental variables may act on the outcome through other pathways than caffeine, this cannot be completely excluded. We found evidence for heterogeneity in the MR for CAD and T2DM for all caffeine traits, indicating that pleiotropy cannot be ruled out. We therefore report the correct model per degree of pleiotropy as the main results and performed several other sensitivity analyses to take this into account. Finally, the present analyses were performed in individuals of White British ancestry, which may limit the generalizability of the results to other populations.
In conclusion, this large prospective study showed inverse associations between observational caffeine intake with CAD and T2DM. However, effect sizes were similar between caffeinated and decaffeinated coffee; similar caffeine intakes from tea were associated with fewer inverse effects compared with caffeine from coffee. Furthermore, MR analyses in independent cohorts yielded no evidence for causality between genetically determined caffeine intake with CAD or T2DM. The main MR analysis results suggest that increasing caffeine intake may not be protective against the development of CAD or T2DM. However, these do not take into account the nonlinear association observed within the observational analyses. We therefore encourage reanalysis of the results when more advanced methods to study nonlinear associations within a summary‐based 2‐sample MR setting emerge, without individual‐level exposure data in the outcome cohort.
Sources of Funding
Dr Verweij is supported by a Dutch Research Council (Nederlandse Organisatie voor Wetenschappelijk Onderzoek) VENI grant (016.186.125).
Disclosures
None.
Supporting information
Data S1
Tables S1–S29
Figures S1–S16
Acknowledgments
This research was conducted using the UK Biobank Resource under Application Number 12006 and 15031. We thank the CARDIoGRAMplusC4D and DIAGRAM investigators for making their data publicly available. We thank Ruben N. Eppinga, MD; Tom Hendriks, MD; M. Yldau van der Ende, MD; Hilde E. Groot, MD; Yanick Hagemeijer, MSc; and Jan Walter Benjamins, BEng, University of Groningen, University Medical Center Groningen, Department of Cardiology, for their contributions to the extraction and processing of data in the UK Biobank. None of the mentioned contributors received compensation, except for their employment at the University Medical Center Groningen. We also thank the Center for Information Technology of the University of Groningen for their support and for providing access to the Peregrine high‐performance computing cluster.
(J Am Heart Assoc 2020;9:e016808. DOI: 10.1161/JAHA.120.016808.)
Supplementary Materials for this article are available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.016808
For Sources of Funding and Disclosures, see page 9.
References
- 1. Fitt E, Pell D, Cole D. Assessing caffeine intake in the United Kingdom diet. Food Chem. 2013;140:421–426. [DOI] [PubMed] [Google Scholar]
- 2. Ding M, Bhupathiraju SN, Satija A, van Dam RM, Hu FB. Long‐term coffee consumption and risk of cardiovascular disease: a systematic review and a dose‐response meta‐analysis of prospective cohort studies. Circulation. 2014;129:643–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ding M, Bhupathiraju SN, Chen M, van Dam RM, Hu FB. Caffeinated and decaffeinated coffee consumption and risk of type 2 diabetes: a systematic review and a dose‐response meta‐analysis. Diabetes Care. 2014;37:569–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Freedman ND, Park Y, Abnet CC, Hollenbeck AR, Sinha R. Association of coffee drinking with total and cause‐specific mortality. N Engl J Med. 2012;366:1891–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Loftfield E, Cornelis MC, Caporaso N, Yu K, Sinha R, Freedman N. Association of coffee drinking with mortality by genetic variation in caffeine metabolism: findings from the UK Biobank. JAMA Intern Med. 2018;178:1086–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cornelis MC, El‐Sohemy A, Kabagambe EK, Campos H. Coffee, CYP1A2 genotype, and risk of myocardial infarction. JAMA. 2006;295:1135–1141. [DOI] [PubMed] [Google Scholar]
- 7. Nordestgaard AT, Nordestgaard BG. Coffee intake, cardiovascular disease and all‐cause mortality: observational and Mendelian randomization analyses in 95 000–223 000 individuals. Int J Epidemiol. 2016;45:1938–1952. [DOI] [PubMed] [Google Scholar]
- 8. Lopez‐Garcia E, van Dam RM, Willett WC, Rimm EB, Manson JE, Stampfer MJ, Rexrode KM, Hu FB. Coffee consumption and coronary heart disease in men and women: a prospective cohort study. Circulation. 2006;113:2045–2053. [DOI] [PubMed] [Google Scholar]
- 9. Sofi F, Conti AA, Gori AM, Eliana Luisi ML, Casini A, Abbate R, Gensini GF. Coffee consumption and risk of coronary heart disease: a meta‐analysis. Nutr Metab Cardiovasc Dis. 2007;17:209–223. [DOI] [PubMed] [Google Scholar]
- 10. Mozaffarian D. Dietary and policy priorities for cardiovascular disease, diabetes, and obesity: a comprehensive review. Circulation. 2016;133:187–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pirastu N, Kooyman M, Robino A, van der Spek A, Navarini L, Amin N, Karssen LC, Van Duijn CM, Gasparini P. Non‐additive genome‐wide association scan reveals a new gene associated with habitual coffee consumption. Sci Rep. 2016;6:31590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coffee and Caffeine Genetics Consortium , Cornelis MC, Byrne EM, Esko T, Nalls MA, Ganna A, Paynter N, Monda KL, Amin N, Fischer K, Renstrom F, et al. Genome‐wide meta‐analysis identifies six novel loci associated with habitual coffee consumption. Mol Psychiatry. 2015;20:647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Amin N, Byrne E, Johnson J, Chenevix‐Trench G, Walter S, Nolte IM; kConFab Investigators , Vink JM, Rawal R, Mangino M, Teumer A, et al. Genome‐wide association analysis of coffee drinking suggests association with CYP1A1/CYP1A2 and NRCAM. Mol Psychiatry. 2012;17:1116–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sulem P, Gudbjartsson DF, Geller F, Prokopenko I, Feenstra B, Aben KK, Franke B, den Heijer M, Kovacs P, Stumvoll M, et al. Sequence variants at CYP1A1‐CYP1A2 and AHR associate with coffee consumption. Hum Mol Genet. 2011;20:2071–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nakagawa‐Senda H, Hachiya T, Shimizu A, Hosono S, Oze I, Watanabe M, Matsuo K, Ito H, Hara M, Nishida Y, et al. A genome‐wide association study in the Japanese population identifies the 12q24 locus for habitual coffee consumption: the J‐MICC Study. Sci Rep. 2018;8:1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cornelis MC, Monda KL, Yu K, Paynter N, Azzato EM, Bennett SN, Berndt SI, Boerwinkle E, Chanock S, Chatterjee N, et al. Genome‐wide meta‐analysis identifies regions on 7p21 (AHR) and 15q24 (CYP1A2) as determinants of habitual caffeine consumption. PLoS Genet. 2011;7:e1002033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cornelis MC, Kacprowski T, Menni C, Gustafsson S, Pivin E, Adamski J, Artati A, Eap CB, Ehret G, Friedrich N, et al. Genome‐wide association study of caffeine metabolites provides new insights to caffeine metabolism and dietary caffeine‐consumption behavior. Hum Mol Genet. 2016;25:5472–5482. [DOI] [PubMed] [Google Scholar]
- 18. Kwok MK, Leung GM, Schooling CM. Habitual coffee consumption and risk of type 2 diabetes, ischemic heart disease, depression and Alzheimer’s disease: a Mendelian randomization study. Sci Rep. 2016;6:36500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nordestgaard AT, Thomsen M, Nordestgaard BG. Coffee intake and risk of obesity, metabolic syndrome and type 2 diabetes: a Mendelian randomization study. Int J Epidemiol. 2015;44:551–565. [DOI] [PubMed] [Google Scholar]
- 20. Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. UK Biobank: protocol for a large‐scale prospective epidemiological resource 2007. UK Biobank. Available at: http://www.ukbiobank.ac.uk/wp‐content/uploads/2011/11/UK‐Biobank‐Protocol.pdf. Accessed December 15, 2015. [Google Scholar]
- 22. UK Biobank Ethics and Governance Framework 2007. UK Biobank. Available at: https://www.ukbiobank.ac.uk/wp‐content/uploads/2011/05/EGF20082.pdf. Accessed December 15, 2015. [Google Scholar]
- 23. Cafeine . Netherlands Nutrition Centre. Available at: https://www.voedingscentrum.nl/encyclopedie/cafeine.aspx. Accessed January 29, 2018.
- 24. Said MA, Verweij N, van der Harst P. Associations of combined genetic and lifestyle risks with incident cardiovascular disease and diabetes in the UK Biobank Study. JAMA Cardiol. 2018;3:693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. International Classifcation of Diseases (ICD) . WHO. Available at: http://www.who.int/classifications/icd/en/. Accessed January 10, 2016. [Google Scholar]
- 26. OPCS‐4 Classification 2014. National Health Service. Available at: https://digital.nhs.uk/data‐and‐information/information‐standards/information‐standards‐and‐data‐collections‐including‐extractions/publications‐and‐notifications/standards‐and‐collections/dcb0084‐opcs‐classification‐of‐interventions‐and‐procedures. Accessed January 16, 2016. [Google Scholar]
- 27. Wain LV, Shrine N, Miller S, Jackson VE, Ntalla I, Soler Artigas M, Billington CK, Kheirallah AK, Allen R, Cook JP, et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir Med. 2015;3:769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O'Connell J, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. UK Biobank phasing and imputation documentation. UK Biobank, Marchini J. Updated 2015. Available at: https://biobank.ctsu.ox.ac.uk/crystal/docs/impute_ukb_v1.pdf. Accessed August 18, 2017.
- 30. Loh PR, Tucker G, Bulik‐Sullivan BK, Vilhjalmsson BJ, Finucane HK, Salem RM, Chasman DI, Ridker PM, Neale BM, Berger B, et al. Efficient Bayesian mixed‐model analysis increases association power in large cohorts. Nat Genet. 2015;47:284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pers TH, Karjalainen JM, Chan Y, Westra HJ, Wood AR, Yang J, Lui JC, Vedantam S, Gustafsson S, Esko T, et al. Biological interpretation of genome‐wide association studies using predicted gene functions. Nat Commun. 2015;6:5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. GTEx Consortium, Laboratory, Data Analysis &Coordinating Center (LDACC)‐Analysis Working Group, Statistical Methods groups‐Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH, NIH/NIDA, Biospecimen Collection Source Site‐NDRI , et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Qi T, Wu Y, Zeng J, Zhang F, Xue A, Jiang L, Zhu Z, Kemper K, Yengo L, Zheng Z, et al. Identifying gene targets for brain‐related traits using transcriptomic and methylomic data from blood. Nat Commun. 2018;9:2282, 018‐04558‐1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Westra HJ, Peters MJ, Esko T, Yaghootkar H, Schurmann C, Kettunen J, Christiansen MW, Fairfax BP, Schramm K, Powell JE, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lloyd‐Jones LR, Holloway A, McRae A, Yang J, Small K, Zhao J, Zeng B, Bakshi A, Metspalu A, Dermitzakis M, et al. The genetic architecture of gene expression in peripheral blood. Am J Hum Genet. 2017;100:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, et al. A comprehensive 1,000 genomes‐based genome‐wide association meta‐analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Scott RA, Scott LJ, Magi R, Marullo L, Gaulton KJ, Kaakinen M, Pervjakova N, Pers TH, Johnson AD, Eicher JD, et al. An expanded genome‐wide association study of type 2 diabetes in Europeans. Diabetes. 2017;66:2888–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Brion MJ, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. 2013;42:1497–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Benjamin DJ, Berger JO, Johannesson M, Nosek BA, Wagenmakers E‐J, Berk R, Bollen KA, Brembs B, Brown L, Camerer C, et al. Redefine statistical significance. Nat Hum Behav. 2018;2:6–10. [DOI] [PubMed] [Google Scholar]
- 40. Palmer TM, Lawlor DA, Harbord RM, Sheehan NA, Tobias JH, Timpson NJ, Davey Smith G, Sterne JA. Using multiple genetic variants as instrumental variables for modifiable risk factors. Stat Methods Med Res. 2012;21:223–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two‐sample Mendelian randomization analyses using MR‐Egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45:1961–1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Greco MFD, Minelli C, Sheehan NA, Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34:2926–2940. [DOI] [PubMed] [Google Scholar]
- 43. Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two‐sample summary data Mendelian randomization. Stat Med. 2017;36:1783–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hemani G, Bowden J, Davey SG. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum Mol Genet. 2018;27:R195–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46:1985–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Poole R, Kennedy OJ, Roderick P, Fallowfield JA, Hayes PC, Parkes J. Coffee consumption and health: umbrella review of meta‐analyses of multiple health outcomes. BMJ. 2017;359:j5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. van Dam RM, Hu FB. Coffee consumption and risk of type 2 diabetes: a systematic review. JAMA. 2005;294:97–104. [DOI] [PubMed] [Google Scholar]
- 49. Spiller G. Chapter 6. the chemical components of coffee. In: Caffeine. 1st ed. Boca Raton, CA: CRC Press; 1998:97. [Google Scholar]
- 50. Carlstrom M, Larsson SC. Coffee consumption and reduced risk of developing type 2 diabetes: a systematic review with meta‐analysis. Nutr Rev. 2018;76:395–417. [DOI] [PubMed] [Google Scholar]
- 51. Ding M, Satija A, Bhupathiraju SN, Hu Y, Sun Q, Han J, Lopez‐Garcia E, Willett W, van Dam RM, Hu FB. Association of coffee consumption with total and cause‐specific mortality in 3 large prospective cohorts. Circulation. 2015;132:2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Clarke TK, Adams MJ, Davies G, Howard DM, Hall LS, Padmanabhan S, Murray AD, Smith BH, Campbell A, Hayward C, et al. Genome‐wide association study of alcohol consumption and genetic overlap with other health‐related traits in UK Biobank (N=112 117). Mol Psychiatry. 2017;22:1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ludwig IA, Clifford MN, Lean ME, Ashihara H, Crozier A. Coffee: biochemistry and potential impact on health. Food Funct. 2014;5:1695–1717. [DOI] [PubMed] [Google Scholar]
- 54. Gloess AN, Schönbächler B, Klopprogge B, D`Ambrosio L, Chatelain K, Bongartz A, Strittmatter A, Rast M, Yeretzian C. Comparison of nine common coffee extraction methods: instrumental and sensory analysis. Eur Food Res Technol. 2013;236:607–627. [Google Scholar]
- 55. van Dusseldorp M, Katan MB, van Vliet T, Demacker PN, Stalenhoef AF. Cholesterol‐raising factor from boiled coffee does not pass a paper filter. Arterioscler Thromb. 1991;11:586–593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Tables S1–S29
Figures S1–S16