

Abstract
Background
Anacetrapib is the only cholesteryl ester transfer protein inhibitor proven to reduce coronary heart disease (CHD). However, its effects on reverse cholesterol transport have not been fully elucidated. Macrophage cholesterol efflux (CEC), the initial step of reverse cholesterol transport, is inversely associated with CHD and may be affected by sex as well as haptoglobin copy number variants among patients with diabetes mellitus. We investigated the effect of anacetrapib on CEC and whether this effect is modified by sex, diabetes mellitus, and haptoglobin polymorphism.
Methods and Results
A total of 574 participants with CHD were included from the DEFINE (Determining the Efficacy and Tolerability of CETP Inhibition With Anacetrapib) trial. CEC was measured at baseline and 24‐week follow‐up using J774 macrophages, boron dipyrromethene difluoride–labeled cholesterol, and apolipoprotein B–depleted plasma. Haptoglobin copy number variant was determined using an ELISA assay. Anacetrapib increased CEC, adjusted for baseline CEC, risk factors, and changes in lipids/apolipoproteins (standard β, 0.23; 95% CI, 0.05–0.41). This CEC‐raising effect was seen only in men (P interaction=0.002); no effect modification was seen by diabetes mellitus status. Among patients with diabetes mellitus, anacetrapib increased CEC in those with the normal 1‐1 haptoglobin genotype (standard β, 0.42; 95% CI, 0.16–0.69) but not the dysfunctional 2‐1/2‐2 genotypes (P interaction=0.02).
Conclusions
Among patients with CHD, anacetrapib at a dose linked to improved CHD outcomes significantly increased CEC independent of changes in high‐density lipoprotein cholesterol or other lipids, with effect modification by sex and a novel pharmacogenomic interaction by haptoglobin genotype, suggesting a putative mechanism for reduced risk requiring validation.
Keywords: cholesteryl ester transfer protein, coronary heart disease, diabetes mellitus, haptoglobin genotype, sex
Subject Categories: Lipids and Cholesterol
Nonstandard Abbreviations and Acronyms
- ACCORD
Action to Control Cardiovascular Risk in Diabetes
- BODIPY
boron dipyrromethene difluoride
- CEC
cholesterol efflux capacity
- CETP
cholesteryl ester transfer protein
- CODAM
Cohort on Diabetes and Atherosclerosis Maastrich
- DEFINE
Determining the Efficacy and Tolerability of CETP Inhibition With Anacetrapib
- EPIC
European Prospective Investigation into Cancer
- JUPITER
Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin
- Lp(a)
lipoprotein (a)
- MESA
Multi‐Ethnic Study of Atherosclerosis
- PREDIMED
Prevention With Mediterranean Diet
- PREVEND
Prevention of Renal and Vascular End‐Stage Disease
- RCT
reverse cholesterol transport
- REVEAL
Randomized Evaluation of the Effects of Anacetrapib Through Lipid‐Modification
Clinical Perspective
What Is New?
We demonstrate that among patients with coronary heart disease, anacetrapib increases cholesterol efflux capacity independent of changes in high‐density lipoprotein cholesterol or other lipids.
This increase in cholesterol efflux capacity is modified by sex along with a pharmacogenomic interaction by haptoglobin genotype in patients with diabetes mellitus.
What Are the Clinical Implications?
Determining a patient's haptoglobin genotype may be a strategy to identify patients most likely to benefit from interventions targeting reverse cholesterol transport in those with diabetes mellitus.
In addition, interventions targeting high‐density lipoprotein function may have differing effects based on demographics including sex.
Low high‐density lipoprotein (HDL) cholesterol (HDL‐C) is a significant risk factor for atherosclerotic cardiovascular disease (ASCVD). 1 , 2 However, HDL has many important functions that are not reflected by the cholesterol content and that may serve as potential therapeutic targets. 3 The main antiatherosclerotic function ascribed to HDL is reverse cholesterol transport (RCT), the process by which cholesterol moves from the periphery to the liver and out of the body. 4 , 5 , 6 Impaired RCT is inversely associated with atherosclerosis in both human and animal studies. 7 Macrophage‐specific cholesterol efflux capacity (CEC) is the key first step of RCT. CEC is inversely associated with both prevalent and incident ASCVD independent of HDL‐C level.9, 10, 11, 12
However, studies assessing the effects of interventions on CEC have been mixed, involved heterogeneous populations, and often included medications not yet proven to improve ASCVD risk. 13 , 14 , 15 Anacetrapib is the only CETP (cholesteryl ester transfer protein) inhibitor that has been shown to reduce ASCVD. 16 , 17 Its effects on lipids, not unlike other CETP inhibitors, are diverse, including marked increases in HDL‐C and apolipoprotein A‐I (ApoA‐I) as well as decreases in apolipoprotein B (ApoB) and lipoprotein (a) (Lp[a]). In a small study, anacetrapib 300 mg was shown to improve CEC 18 ; however, its effect on CEC at the dose proven to reduce ASCVD (100 mg) among patients with coronary heart disease (CHD) is not well described.
Intriguingly, recent observational studies have suggested that CEC levels may not only differ by sex but that sex may modify the association between CEC and ASCVD. 19 Whether this is relevant to anacetrapib's effects on CEC is unknown. Beyond sex, haptoglobin is emerging as an HDL‐associated protein that may impact CEC. 20 Normally, haptoglobin binds free and glycosylated hemoglobin, preventing oxidative damage. However, in the context of hyperglycemia, a common structural variant (“2” allele) leads to increased haptoglobin‐hemoglobin complexes with impaired clearance. These complexes bind to ApoA‐I and other HDL‐associated enzymes, enhancing their oxidation and reducing CEC and overall RCT. 21 In addition, among those with diabetes mellitus (DM), the “2” allele has been linked to impaired coronary endothelial function 22 , 23 and modifies the efficacy of intensive glucose control in reducing the risk for CHD. 24 It remains unknown whether such pharmacogenomic interaction is relevant to a CETP inhibitor such as anacetrapib.
Thus, we sought to test the effect of anacetrapib at a dose proven to reduce ASCVD outcomes on CEC within the context of a randomized controlled trial (DEFINE [Determining the Efficacy and Tolerability of CETP Inhibition With Anacetrapib]) and whether this effect is modified by sex, DM, and haptoglobin polymorphism status.
Methods
This study was conducted via an agreement with Merck through an investigator‐initiated proposal. The data will be made available to any investigator after request and approval by both the senior author and Merck subject to the terms of the agreement. As previously reported in the DEFINE trial, the institutional review boards at each participating center approved the trial protocol. The UT Southwestern Medical Center institutional review board determined that this substudy was exempt.
Study Population
The DEFINE trial was a double‐blind, randomized, placebo‐controlled trial of anacetrapib 100 mg in participants at high risk on statin therapy. It enrolled 1623 participants with CHD or CHD risk equivalents from 153 centers in 20 countries from 2008 to 2010. Participants were treated with either anacetrapib 100 mg or placebo for 18 months. The details about the trial design along with inclusion/exclusion criteria have been previously reported. 25 For this current study, the main inclusion criteria were history of CHD, clinical risk factors, lipid measurements, and available plasma at baseline and at 24 weeks. Exclusions included anyone without data at both time points and anyone developing an incident cardiovascular event before the follow‐up visit. The inclusion of only participants with CHD restricted the number of participants to 888. Given that the DEFINE trial enrolled mostly White men with a small number of women and non‐White participants, we selected a random sampling of white men from each treatment group matched for age, sex, DM status, and type of statin based on prior work regarding the variability of the assay. All women and all non‐White participants were included. This selection process ensured adequate information in important subgroups. A total of 600 participants were selected a priori. Of these, 574 participants had complete data and were assessed at baseline and 24‐week follow‐up (Figure S1).
Clinical and Anthropometric Measurements
Demographic characteristics, presence of DM and CHD, and statin use were obtained at enrollment during the trial. Fasting lipid and lipoprotein measurements (HDL‐C, low‐density lipoprotein cholesterol [LDL‐C], ApoA‐I, ApoB, Lp(a), and triglycerides) were collected at baseline and 24‐week follow‐up and performed by the study core laboratory. 25 LDL‐C was calculated using the Friedewald equation; if triglycerides were >400 mg/dL, LDL‐C was directly measured. The remainder of lipid measurement methods can be found as previously reported in the DEFINE trial.
Measurement of CEC
CEC was assessed from fasting EDTA plasma samples obtained at baseline and at 24‐week follow‐up stored at −80°C. CEC measured the efflux of boron dipyrromethene difluoride (BODIPY) fluorescently labeled cholesterol from J774 macrophages to ApoB‐depleted plasma from study participants using a previously described method 26 (Table S1). Baseline and follow‐up specimens were measured on the same plate. CEC was normalized to efflux elicited by pooled human plasma run on every plate. Study personnel were blinded to treatment group and demographic and clinical information.
Measurement of Haptoglobin
Haptoglobin copy number variant status was ascertained in all participants using a commercially available ELISA assay (Savyon Diagnostics, Ltd.) from the same baseline frozen EDTA plasma used for CEC. This copy number variant (termed allele 2) results in polymerization of haptoglobin and a structurally abnormal protein, which can be reliably detected by ELISA. Using the assay, each participant was identified as having the 1‐1 (normal), 2‐1 (heterozygous for abnormal “2” allele), or 2‐2 (homozygous for abnormal “2” allele) copy number variant genotypes. 27 Prior studies have established that allele 2 confers impaired HDL function only among patients with DM and not among those without DM. Thus, the focus of this analysis in our study was on participants with DM. Study personnel were blinded to treatment group, CEC measurements, demographics, and clinical information.
Statistical Analysis
Study participants were stratified by treatment status (placebo versus anacetrapib). Clinical and demographic characteristics along with CEC were compared among groups using Wilcoxon rank sum test for unpaired variables and Wilcoxon matched‐pairs signed rank test for paired variables comparing patient data between time points. Unadjusted changes in CEC were reported as percent change in each treatment group. In multivariate models, the main outcome was follow‐up CEC adjusted for baseline CEC. Multivariable adjusted linear regression analyses were performed to evaluate the independent associations between lipoproteins and follow‐up CEC in the placebo and anacetrapib groups separately, adjusted for baseline CEC, covariates of sex, ethnicity, DM status, and type of statin, along with baseline and change in HDL‐C, ApoA‐I, Apo B, and triglycerides. Each lipoprotein, for example HDL‐C (baseline and change) was tested in separate models and then all lipoproteins were combined into one model. The association between anacetrapib 100 mg versus placebo and follow‐up CEC was tested in linear regression models adjusted for baseline CEC, sex, ethnicity, DM, and type of statin, along with HDL‐C, ApoB, ApoA‐I, and triglycerides (baseline and change). Effect modification of the effect of anacetrapib on CEC was tested for sex and DM with subsequent stratified multivariate models to report the effect sizes in each subgroup. Among patients with DM, the interaction between anacetrapib and haptoglobin copy number variant status was tested. The haptoglobin 2‐1/2‐2 groups were combined given smaller numbers in each respective group while allowing for the comparison between the “1” allele in the haptoglobin 1‐1 homozygotes versus the “2” allele in the combined haptoglobin 2‐1 heterozygotes and haptoglobin 2‐2 homozygotes. All statistical analyses were performed in SAS version 9.4 (SAS Institute Inc.).
Results
Baseline Characteristics
Baseline characteristics of the study participants in the placebo and anacetrapib groups are shown in Table 1. There were no significant differences at baseline between the placebo and anacetrapib groups with regards to age, race, BMI, or presence of DM. However, there was a greater percentage of women in the placebo group compared with the anacetrapib group (46% versus 35%, respectively; P=0.008). The type of statin significantly differed between groups such that in the placebo group more participants took simvastatin while fewer participants took atorvastatin, rosuvastatin, pravastatin, or lovastatin (overall P=0.007). Due to the selection criteria for this substudy resulting in inclusion of all women and non‐White participants with CHD, baseline characteristics differ from the overall DEFINE trial participants (Table S2). The percentage of women in this substudy is greater than in the overall DEFINE trial (35%–46% versus 22%–24%, respectively). The percentage of White participants is lower than in the overall DEFINE trial (77%–79% versus 82%–85%, respectively). The percentage of participants with DM was greater than in the overall DEFINE trial (60%–65% versus 53%, respectively). Finally, the percentage of those on each statin differed from the overall DEFINE trial (higher percentage of rosuvastatin and lower percentage of simvastatin in the anacetrapib group, respectively).
Table 1.
Baseline Characteristics Comparing the Placebo Group Versus the Anacetrapib Group
| Placebo (n=289) | Anacetrapib (n=285) | P Value* | |
|---|---|---|---|
| Age, median (IQR), y | 63.1 (56.9–69.6) | 62.1 (55.2–68.1) | 0.24 |
| Women | 132 (46) | 99 (35) | 0.008 |
| White race | 223 (77) | 226 (79) | 0.55 |
| BMI, median (IQR), kg/m2 | 29.6 (27.0–33.4) | 29.4 (26.3–33.2) | 0.56 |
| DM | 188 (65) | 171 (60) | 0.23 |
| Atorvastatin | 97 (33.6) | 101 (35.4) | 0.007† |
| Simvastatin | 132 (45.7) | 95 (33.3) | |
| Rosuvastatin | 49 (17.0) | 62 (21.8) | |
| Pravastatin | 4 (1.4) | 13 (4.6) | |
| Lovastatin | 6 (2.1) | 13 (4.6) |
Values are presented as number (percentage) unless otherwise indicated. BMI indicates body mass index; DM, diabetes mellitus; and IQR, interquartile range.
P values are obtained using Wilcoxon rank sum test assuming nonparametric distributions for continuous variables and chi‐square test for categorical variables.
The P value for statins is shown as an overall P value.
Effect of Anacetrapib on Lipids and Apolipoproteins
Baseline and follow‐up lipids, apolipoprotein levels, and CEC are shown in Table 2. At baseline, there was a trend toward higher levels of triglycerides in the placebo group compared with the anacetrapib group (131 mg/dL versus 125 mg/dL, respectively; P value 0.06). There was no significant difference between the remainder of lipoproteins/apolipoproteins at baseline. After 24 weeks, in participants treated with anacetrapib, HDL‐C increased by 60 mg/dL (145%) and ApoA‐I increased by 65 mg/dL (45%); LDL‐C decreased by 40 mg/dL (49%) and ApoB decreased by 17 mg/dL (21%); triglycerides decreased by 11 mg/dL (9%); and Lp(a) decreased by 11 nmol/L (15%). In the placebo group, HDL‐C increased by 5 mg/dL (12%), ApoA‐I and ApoB remained unchanged; LDL‐C decreased by 6 mg/dL (7%); triglycerides decreased by 1 mg/dL (3%); and Lp(a) increased by 4 nmol/L (7%).
Table 2.
Baseline, Follow‐Up, and Percent Change in Lipids, Apolipoproteins, and CEC
| Placebo (n=289) | Anacetrapib (n=285) | P Value* | |||||
|---|---|---|---|---|---|---|---|
| Week 0 | Week 24 | Median Percent Change | Week 0 | Week 24 | Median Percent Change | ||
| LDL‐C, mg/dL | 81 (68–94) | 75 (65–87) | −7.0 (−18.8 to 5.0) | 82 (69–94) | 42 (32–54) | −48.9 (−59.0 to −33.2) | <0.0001 |
| HDL‐C, mg/dL | 40 (34–47) | 45 (38–51) | 11.5 (2.1 to 26.3) | 40 (34–47) | 100 (82–117) | 145.2 (118.8 to 188.1) | <0.0001 |
| Triglycerides, mg/dL | 131 (101–194) | 130 (94–177) | −3.3 (−21.2 to 16.4) | 125 (93–172) | 114 (90–146) | −8.7 (−25.1 to 13.7) | 0.09 |
| ApoA‐I, mg/dL | 145 (130–159) | 145 (132–159) | 0 (−7.8 to 9.2) | 142 (127–157) | 207 (183–233) | 45.3 (31.6 to 59.6) | <0.0001 |
| ApoB, mg/dL | 88 (77–101) | 89 (77–101) | 0 (−8.1 to 11.7) | 86 (76–98) | 69 (60–78) | −20.9 (−30.7 to −8.7) | <0.0001 |
| Lipoprotein (a), nmol/L | 27 (12–66) | 31 (13–98) | 6.7 (−3.4 to 42.5) | 27 (10–59) | 16 (5–55) | −15.1 (−49.7 to 0) | <0.0001 |
| CEC normalized | 0.87 (0.77–1.01) | 0.84 (0.72–0.95) | −3.9 (−20.4 to 12.8) | 0.85 (0.76–0.97) | 0.89 (0.79–1.01) | 4.6 (−13.6 to 28.4) | 0.0001 |
Values are presented as median (interquartile range). ApoA‐I indicates apolipoprotein A‐I; ApoB, apolipoprotein B; CEC, cholesterol efflux capacity; HDL‐C, high‐density lipoprotein cholesterol; and LDL‐C, low‐density lipoprotein cholesterol.
P values for median percent change are obtained using Wilcoxon rank sum test assuming nonparametric distributions.
Effect of Anacetrapib on CEC
Compared with placebo, anacetrapib was associated with an 8.6% median increase in unadjusted CEC (P=0.0001). When stratified based on sex and DM status in univariate analysis, a 12.5% and 15.5% median increase was seen in men and those without DM, respectively (P<0.0001 for both men versus women and DM versus no DM) (Figure 1). In multivariable models, anacetrapib was associated with increased CEC (standard β, 0.23; 95% CI, 0.05–0.41) (Figure 2). When stratified based on sex, anacetrapib was associated with increased CEC in men (standard β, 0.36; 95% CI, 0.13–0.58) but not in women (standard β, −0.04; 95% CI, −0.35 to 0.26) (P for interaction=0.002) (Figure 2). However, when stratified based on the presence of DM in multivariable models, there was no significant interaction between anacetrapib and DM status (P for interaction=0.23) with similar standard β coefficients (Figure 2).
Figure 1. Median percent change in cholesterol efflux capacity (CEC) in anacetrapib vs placebo.
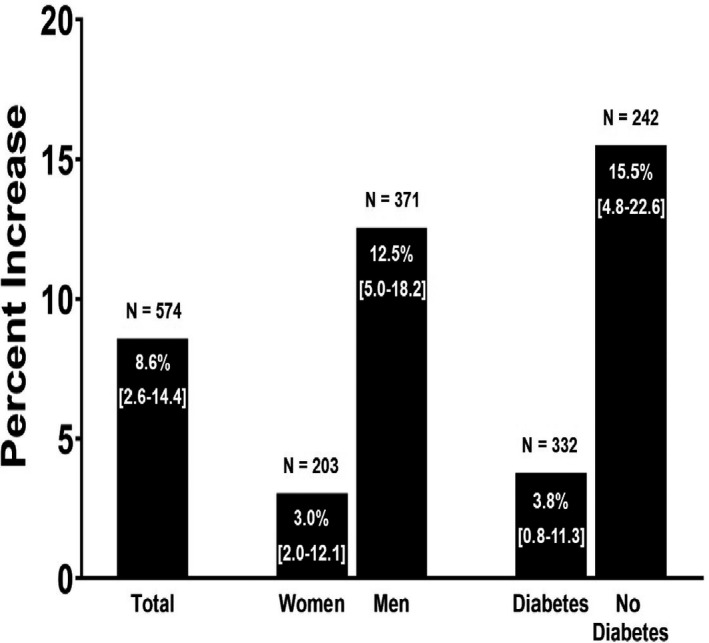
The percent change was calculated separately in the anacetrapib and placebo groups by taking the difference between the week 24 and week 0 unadjusted values for CEC and lipids and dividing by week 0. The median percent increase was the median difference between the anacetrapib group and the placebo group. 95% CIs for median percent change are shown on each bar in brackets. P<0.0001 for women vs men and diabetes mellitus (DM) vs no DM.
Figure 2. Association between anacetrapib and change in cholesterol efflux capacity (CEC).
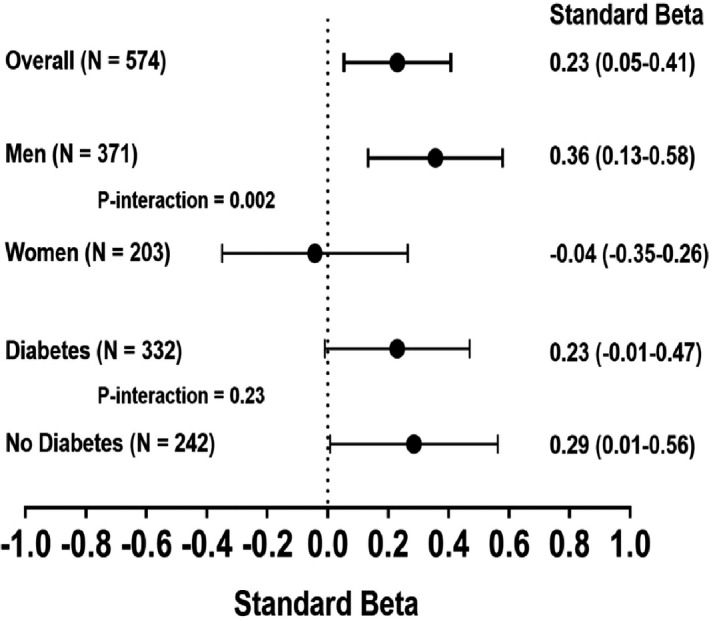
The standardized β estimate represents the SD change in efflux in the anacetrapib group vs placebo presented as standard β (95% CI). Estimates derived from multivariable linear regression models including baseline CEC, covariates of sex, ethnicity, diabetes mellitus (DM) status, and type of statin, along with baseline and change in high‐density lipoprotein cholesterol, apolipoprotein A‐I, apolipoprotein B, and triglycerides. A separate model was constructed for each group (ie, overall, men). P interaction terms were calculated to determine the presence of effect modification by sex and DM status (P interaction for the product of sex×anacetrapib [0.002], presence of DM×anacetrapib [0.23]).
Associations Between Changes in Apolipoproteins and Lipids With Change in CEC
In the placebo group, change in CEC was positively associated with changes in ApoA‐I (standard β, 0.21; P=0.003), ApoB (standard β, 0.26; P=0.0002), and triglycerides (standard β, 0.24; P=0.0006) (Table 3). In the fully adjusted model including all lipids/lipoproteins (HDL‐C, ApoA‐I, ApoB, and triglycerides), change in CEC remained positively associated with ApoA‐I (standard β, 0.21, P=0.02) and ApoB (standard β, 0.16; P=0.04). Of note, in the placebo group there was no significant association between change in HDL‐C and change in CEC in either the separate (standard β, 0.05; P=0.39) or fully adjusted (standard β, −0.01; P=0.89) models.
Table 3.
Association Between Change in Lipids and Apolipoproteins and Change in CEC
| Placebo | Anacetrapib | |||
|---|---|---|---|---|
| Individual Lipoprotein | All Lipoproteins | Individual Lipoproteins | All Lipoproteins | |
| Standard β (P Value)* | Standard β (P Value)† | Standard β (P Value) | Standard β (P Value) | |
| ΔHDL‡ | 0.05 (0.39) | −0.01 (0.89) | 0.03 (0.64) | 0.08 (0.37) |
| ΔApoA‐I | 0.21 (0.003) | 0.21 (0.02) | −0.04 (0.48) | −0.09 (0.29) |
| ΔApoB | 0.26 (0.0002) | 0.16 (0.04) | 0.12 (0.09) | 0.17 (0.02) |
| ΔTriglycerides | 0.24 (0.0006) | 0.15 (0.053) | −0.05 (0.48) | −0.08 (0.27) |
ApoA‐I indicates apolipoprotein A‐I; and ApoB, apolipoprotein B.
A separate linear regression model was used for each set of change in lipoproteins adjusted for baseline cholesterol efflux capacity (CEC), sex, race, presence of diabetes mellitus, and type of statin.
For the combined model, all lipoproteins were included together in separate models.
ΔHigh‐density lipoprotein (HDL) is defined as 24‐week HDL adjusted for baseline values.
In the anacetrapib group, there remained a positive association between change in ApoB and change in CEC (standard β, 0.17; P=0.02) in the fully adjusted model (Table 3). There were no significant associations between change in HDL‐C, ApoA‐I, or triglycerides and change in CEC.
Association Between Haptoglobin Copy Number and CEC
In 332 participants with DM, 51 (15%) had the 1‐1 genotype, 162 (49%) had the 2‐1 genotype, and 119 (36%) had the 2‐2 haptoglobin genotype. Anacetrapib was positively associated with a significant increase in CEC in those with the 1‐1 genotype (standard β, 0.42; P=0.003) but not in those with either 2‐1, 2‐2, or the combined 2‐1/2‐2 genotypes (P for interaction=0.02) after adjustment for baseline CEC (Figure 3). After adjusting for baseline risk factors and change in lipoproteins, the positive association between anacetrapib and CEC in those with the “1” allele remained borderline significant (standard β, 0.55; P=0.05) (P for interaction=0.08). In contrast, among those without DM, there was no significant interaction between anacetrapib and haptoglobin genotype for CEC (P for interaction=0.36) (data not shown).
Figure 3. Association between anacetrapib and cholesterol efflux capacity (CEC) based on haptoglobin genotype.
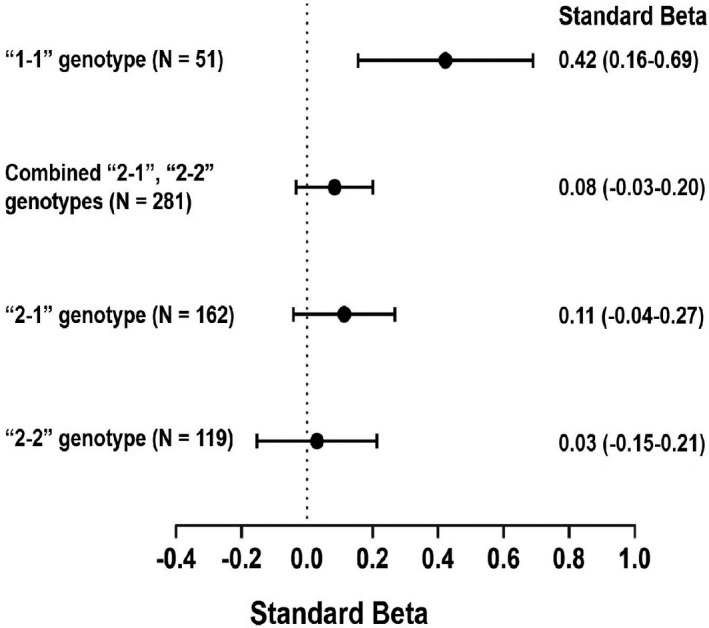
The standardized β estimate represents the SD change in efflux in the anacetrapib group vs placebo presented as standard β (95% CI). Estimates derived from multivariable linear regression models including baseline CEC. “1‐1” indicates haptoglobin “1‐1” genotype; “2‐1”: haptoglobin “2‐1” genotype; and “2‐2”: haptoglobin “2‐2” genotype.
Discussion
In this post hoc substudy of patients with CHD enrolled in the DEFINE trial, anacetrapib 100 mg was associated with increased CEC independent of changes in lipids and apolipoproteins. The effect of anacetrapib was blunted in women and those with DM. Increases in CEC were linked more to changes in apolipoprotein levels than cholesterol levels. Among patients with DM, the effect of anacetrapib on CEC was blunted in those with the haptoglobin “2” allele. These findings suggest that anacetrapib improves a key HDL function, CEC, with heterogeneous effects by sex, DM status, and haptoglobin polymorphism.
CETP inhibitors cause marked increases in HDL‐C levels and have also been shown to increase CEC, with varying effects by transporter‐specific efflux. Transporter‐specific CEC has typically been described for ABCA1‐, ABCG1‐, and SR‐B1–mediated CEC; however, the consistency and accuracy of assay methodology has varied among studies. 28 In patients with dyslipidemia, evacetrapib, when combined with statin therapy, increased both ABCA1‐specific (27%) and non‐ABCA1–specific CEC (15%) compared with statin monotherapy. 29 When compared against placebo, evacetrapib demonstrated a larger increase in both ABCA1‐specific (26%) and non‐ABCA1–specific CEC (47%). In small studies involving patients with dyslipidemia, torcetrapib increased non‐ABCA1–specific CEC. 30 , 31 , 32 In a small number of patients with low to average HDL‐C levels on pravastatin treated for 12 weeks, dalcetrapib 600 mg and 900 mg significantly increased SRB1 efflux 19% and 21%, respectively, with no effect on ABCA1‐mediated CEC. 33 A Study of Dalcetrapib in Patients Hospitalized for an Acute Coronary Syndrome (Dal‐ACUTE), a multicentered trial of patients post–acute coronary syndrome, total and non‐ABCA1–specific CEC was increased with 600 mg of dalcetrapib at 4, 12, and 20 weeks; however, ABCA1‐specific CEC was not significantly increased. 34 In 187 patients with dyslipidemia, TA‐8995 10 mg plus rosuvastatin 10 mg increased non‐ABCA1–specific (67%) and total CEC (31%) compared with placebo; ABCA1‐specific CEC increased by 16%, although nonsignificantly. 35 These studies varied in type of CETP inhibitor, intensity of CETP inhibition, baseline risk and demographics of study populations, and duration of treatments, making comparisons of results related to CEC challenging.
In a small sample of 20 patients, treatment with anacetrapib 300 mg increased radiolabeled CEC by 2.4‐fold. 18 In our substudy involving 574 participants with CHD enrolled within a larger randomized placebo‐controlled trial, anacetrapib 100 mg, a dose shown to improve cardiovascular outcomes, increased ApoA‐I levels 45% while decreasing LDL‐C levels 42%, ApoB levels 21%, Lp(a) levels 22%, and triglyceride levels 6%, results similar to the complete DEFINE trial. 16 Since CETP inhibitors have varied effects on lipids including HDL‐C, ApoA‐I, and ApoB, it is not surprising that there may be varied effects on HDL function. We showed that anacetrapib at a dose proven to improve ASCVD outcomes (100 mg) increased CEC 8.6% compared with placebo on a background of statin therapy. We used fluorescently labeled cholesterol (BODIPY) in our assay system, which has been shown by others to be mostly driven by lipid‐poor particles via ABCA1. 26 However, defining ABCA1‐specific CEC remains challenging and varies by assay methodology; thus, direct comparisons with prior studies may be limited.
Overall, changes in CEC were associated with changes in apolipoprotein levels but not in HDL‐C. Prior studies have reported variations in these associations, likely related to differences in study populations and assay methodology. 28 Studies using radiolabeled cholesterol to assess CEC typically report modest correlations with HDL‐C (baseline and change), reflecting movement of cholesterol via ABCG1 and SR‐BI transporters to larger, mature HDL particles. In the assay system used in this study, BODIPY cholesterol was used, showing primarily transport via ABCA1 to lipid‐poor acceptor particles, hence the poor or negligent correlation with HDL‐C. 26 With respect to apolipoproteins, increased CEC in our study among patients with CHD was associated with increased ApoA‐I and ApoB in the placebo group and only an increase in ApoB in the anacetrapib group. These findings are somewhat consistent with prior studies, most of which used radiolabeled cholesterol. In a case‐control substudy of the JUPITER (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) trial assessing the efficacy of rosuvastatin in patients free of CHD, baseline CEC was positively associated with ApoA‐I and ApoB in addition to HDL‐C and LDL‐C. 36 After 12 months, positive associations were seen between change in CEC and change in HDL‐C and ApoA‐I. In a nested case‐control substudy from the EPIC‐Norfolk study (patients free of CHD), CEC was positively associated with baseline HDL‐C and ApoA‐I but not ApoB. 10 In the dal‐ACUTE study in patients post–acute coronary syndrome, baseline ApoA‐I but not HDL‐C was positively associated with ABCA1‐specific CEC, similar to our results. 34 After 4 weeks, there was no association between ApoA‐I or HDL‐C levels and ABCA1‐specific CEC in the placebo group. However, in the dalcetrapib group, there was a positive association between week 4 ApoA‐I levels in addition to HDL‐C levels and ABCA1‐specific CEC. Associations between CEC and ApoB levels were not tested. The dal‐ACUTE study population differed from our study in that they were within 1‐week post–acute coronary syndrome. In addition, they found that dalcetrapib increased CEC primarily through the non‐ABCA1–mediated pathway. Thus, apolipoproteins are clearly linked to change in CEC and either directly mediate CEC or reflect a lipid‐protein milieu associated with CEC functionality. Comparison of the effects of CETP inhibitors on CEC is likely to reflect differences in effects on apolipoprotein levels, study population, and background statin therapy.
Intriguingly, we found that anacetrapib increased CEC in men but not in women, reflecting a statistically significant interaction by sex. To our knowledge, the impact of sex on CETP inhibition and CEC has not been thoroughly studied thus far. Baseline associations between sex and CEC have not been consistent among studies. Large studies including the JUPITER trial and EPIC (European Prospective Investigation into Cancer) Norfolk population‐based cohort studies have shown that women have higher CEC than men using radiolabeled cholesterol. 10 , 36 On the other hand, in ≈3000 participants from the Dallas Heart Study, there was no association between sex and CEC using BODIPY cholesterol assay. 9 Similar null associations were seen in controls from the PREVEND (Prevention of Renal and Vascular End‐Stage Disease) and MESA (Multi‐Ethnic Study of Atherosclerosis) trials using differentiated THP human macrophages. 19 , 37 However, in patients who developed incident ASCVD in the PREVEND cohort, CEC was significantly decreased in men compared with women. 19 There was a significant interaction with sex such that CEC was inversely associated with ASCVD in men while it was directly associated in women. In a prospective case‐control study from EPIC‐Norfolk, an interaction between CEC and sex was seen with regard to incident cardiovascular disease such that there was an inverse association with men, while no association was seen with women. 10 Addressing transporter‐specific CEC, one study of healthy volunteers found that women had greater CEC via the SR‐BI pathway but lower CEC compared with men via the ABCA1 pathway; there was no significant difference with regards to the ABCG1 pathway. 38 The differences seen between CEC and sex in the literature may be attributable to the study population including the presence or absence of CHD in addition to the method used to obtain CEC. Our study examined the difference in anacetrapib‐mediated CEC by sex among patients with CHD, adding to the current literature on sex and CEC. In our study involving patients with CHD, BODIPY cholesterol was employed, primarily reflecting the ABCA1 pathway and resulting in increased CEC with anacetrapib only among men, consistent with the transporter‐specific investigation of baseline CEC. Thus, future investigation of therapies affecting CEC may need to account for possible sex‐based effect modifications.
In addition to sex, we also explored the impact of DM on anacetrapib‐mediated changes in CEC. Several prior studies except for one have shown little to no correlation between DM status and CEC. 10 In almost 300 participants from the PREDIMED (Prevention With Mediterranean Diet) trial, the presence of DM at baseline visit was not associated with CEC adjusted for HDL‐C. 39 In the MESA cohort, Shea et al 37 also found no significant correlation between DM and CEC. Similar null associations were seen in the CODAM (Cohort on Diabetes and Atherosclerosis Maastrich) study. 40 In addition, in vitro and mouse studies have shown that nonenzymatic glycation of HDL does not diminish CEC. 41 , 42 Similar to sex, differences seen between CEC and DM in the literature may be attributable to the study population including the presence or absence of CHD in addition to the method used to obtain CEC. This relationship may also be influenced by the duration of DM, something not uniformly captured in studies. 43 Our study examined the difference in anacetrapib‐mediated CEC by DM status adding information concerning the relationship between DM and CEC. In our study, although there appeared to be a blunted effect of anacetrapib on CEC among patients with DM, in adjusted analyses there was no significant interaction, consistent with prior reports. Although this lack of effect modification by DM status is consistent with prior reports, it remains possible that the study was underpowered to detect an interaction in this analysis. Thus, DM by itself may not signify differential effects on CEC.
Despite this lack of effect modification by DM status, we sought to explore the potential for a pharmacogenomic interaction among patients with DM. Haptoglobin is an HDL‐associated protein and binds to HDL and hemoglobin in a complex, primarily scavenging free hemoglobin to protect against oxidative damage. In the human genome, haptoglobin genes have been shown to exhibit copy number variance, with duplication events of exons 3 and 4 of the haptoglobin 1 allele, leading to the formation of 2 major common classes of alleles of haptoglobin (referred to as Hp1 and Hp2). The Hp2 category is further split into Hp 2‐1 and Hp 2‐2, depending on the number of Hp1 or Hp2 alleles that are expressed. 44 Of importance to our study, the Hp2 allele (both 2‐1 and 2‐2 variants) can lead to altered HDL protein structure and function, specifically impaired antioxidative function and impaired cholesterol efflux. 21 , 23 The abnormal “2” allele has been linked to impaired coronary endothelial function and increased cardiovascular risk in patients with DM. 22 , 44 , 45 , 46 This allele has also been shown to result in decreased CEC in both mice and humans with DM. 47 It has been hypothesized that the complex between glycosylated haptoglobin 2‐2 protein and hemoglobin binds and oxidizes HDL along with the related lecithin‐cholesterol acyltransferase, decreasing the formation of mature HDL in the process of RCT. 43 It is plausible that the decreased formation of mature HDL from lipid‐poor HDL via the ABCA1 pathway may result in decreased CEC. In non‐Hispanic Whites from the ACCORD (Action to Control Cardiovascular Risk in Diabetes) trial, it was shown that haptoglobin genotype could be helpful in guiding glycemic targets in DM to lower CHD risk. In this analysis, intensive glycemic control (glycated hemoglobin target <6.0%) decreased risk of CHD in the haptoglobin 2‐2 genotype but not in those with the other genotypes. 24 Some investigations have suggested that this haptoglobin‐dependent HDL dysfunction can be improved with vitamin E supplementation, 44 suggesting that haptoglobin genotype may signal potential pharmacogenomic interactions, but only in patients with DM.
Thus, we sought to investigate whether such an interaction exists for anacetrapib. Indeed, we found that the effect of anacetrapib on CEC was significantly blunted in individuals with DM and the haptoglobin “2” allele, whereas it was preserved among patients homozygous for the normal “1” allele. No such effect modification was noted in patients without DM. These findings support the notion that in the milieu of genetically impaired HDL structure/function, the beneficial effects of anacetrapib on CEC are mitigated, which may lead to differential protection against cardiovascular events. Pharmacogenomic interactions have been identified for another CETP inhibitor, dalcetrapib, albeit a different genetic polymorphism. In the dal‐OUTCOMES and dal‐PLAQUE‐2 trials, polymorphisms in the ADCY9 gene, an isoform of adenylate cyclase, were associated with impaired CEC and cardiovascular events. 48 , 49 Different genotypes of the ADCY9 gene were associated with varying outcomes with dalcetrapib treatment showing either increased or decreased cardiovascular events. This same polymorphism did not translate into a pharmacogenomic interaction with evacetrapib or anacetrapib on lipid levels or outcomes. 50 , 51 Thus, just as CETP inhibitors vary in their effects on lipids, apolipoproteins, and CEC, they may also vary with respect to pharmacogenomic interactions. Whether such an interaction between anacetrapib and haptoglobin “2” allele is linked to differential cardiovascular outcomes remains unknown, as does whether this pharmacogenomic interaction exists for other CETP inhibitors or alternative lipid‐modifying drugs. Identifying a patient's haptoglobin genotype suggests a strategy to identify patients most likely to benefit from interventions targeting RCT, specifically in patients with DM.
Several limitations in our study merit remark. First, this study was a substudy of a randomized trial and may not be generalizable to other populations. Second, whether anacetrapib‐mediated increase in CEC is linked to the cardiovascular reductions seen in the REVEAL (Randomized Evaluation of the Effects of Anacetrapib Through Lipid Modification) trial is unknown. Third, this subset of patients was predominantly White, limiting generalizability. Fourth, we used an assay system that highlights the role of lipid‐poor cholesterol acceptors primarily via ABCA1, thus direct comparisons with studies employing other assay systems may be limited. However, ABCA1‐mediated CEC is thought to be the predominant pathway linked to CHD risk. Fifth, given the limited size of this study population, we are unable to draw firm conclusions on the interaction between DM status and haptoglobin genotype on CEC. Sixth, the restriction on the sample size to include only patients with complete data introduces the possibility of selection bias. In addition, excluding patients with incident cardiovascular events restricts the population to those with stable CHD, limiting generalizability.
Conclusions
Among patients with CHD, anacetrapib at a dose linked to improved CHD outcomes increased CEC. The effect of anacetrapib on CEC appears to be blunted in women. This CEC‐raising effect was also blunted among patients with DM and concurrent polymorphism in the haptoglobin gene linked to dysfunctional HDL and increased cardiovascular risk.
Sources of Funding
This study was supported in part by a research grant from Investigator‐Initiated Studies Program of Merck Sharp & Dohme Corp. The opinions expressed in this article are those of the authors and do not necessarily represent those of Merck Sharp & Dohme Corp. Dr Rohatgi is supported by the National Institutes of Health/National Heart, Lung, and Blood Institute (R01HL136724 and K24HL146838).
Disclosures
Rohatgi reports consulting for HDL Diagnostics and CSL Limited, and research support from Merck. The remaining authors have no disclosures to report.
Supporting information
Tables S1–S2
Figure S1
(J Am Heart Assoc 2020;9:e018136. DOI: 10.1161/JAHA.120.018136.)
Supplementary Material for this article are available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.018136
For Sources of Funding and Disclosures, see page 9.
References
- 1. Barter P, Gotto AM, LaRosa JC, Maroni J, Szarek M, Grundy SM, Kastelein JJ, Bittner V, Fruchart JC; Treating to New Targets I . HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med. 2007;1301–1310. [DOI] [PubMed] [Google Scholar]
- 2. Gordon DJ, Knoke J, Probstfield JL, Superko R, Tyroler HA. High‐density lipoprotein cholesterol and coronary heart disease in hypercholesterolemic men: the Lipid Research Clinics Coronary Primary Prevention Trial. Circulation. 1986;1217–1225. [DOI] [PubMed] [Google Scholar]
- 3. Rader DJ, Hovingh GK. HDL and cardiovascular disease. Lancet. 2014;618–625. [DOI] [PubMed] [Google Scholar]
- 4. Kosmas CE, Martinez I, Sourlas A, Bouza KV, Campos FN, Torres V, Montan PD, Guzman E. High‐density lipoprotein (HDL) functionality and its relevance to atherosclerotic cardiovascular disease. Drugs Context. 2018;212525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ono K. Current concept of reverse cholesterol transport and novel strategy for atheroprotection. J Cardiol. 2012;339–343. [DOI] [PubMed] [Google Scholar]
- 6. deGoma EM, deGoma RL, Rader DJ. Beyond high‐density lipoprotein cholesterol levels evaluating high‐density lipoprotein function as influenced by novel therapeutic approaches. J Am Coll Cardiol. 2008;2199–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rader DJ, Alexander ET, Weibel GL, Billheimer J, Rothblat GH. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J Lipid Res. 2009;127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khera AV, Cuchel M, de la Llera‐Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. Cholesterol efflux capacity, high‐density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;2383–2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saleheen D, Scott R, Javad S, Zhao W, Rodrigues A, Picataggi A, Lukmanova D, Mucksavage ML, Luben R, Billheimer J, et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: a prospective case‐control study. Lancet Diabetes Endocrinol. 2015;507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu C, Zhang Y, Ding D, Li X, Yang Y, Li Q, Zheng Y, Wang D, Ling W. Cholesterol efflux capacity is an independent predictor of all‐cause and cardiovascular mortality in patients with coronary artery disease: a prospective cohort study. Atherosclerosis. 2016;116–124. [DOI] [PubMed] [Google Scholar]
- 12. Ritsch A, Scharnagl H, Marz W. HDL cholesterol efflux capacity and cardiovascular events. N Engl J Med. 2015;1870–1871. [DOI] [PubMed] [Google Scholar]
- 13. Barter PJ, Caulfield M, Eriksson M, Grundy SM, Kastelein JJ, Komajda M, Lopez‐Sendon J, Mosca L, Tardif JC, Waters DD, et al. Effects of torcetrapib in patients at high risk for coronary events. N Engl J Med. 2007;2109–2122. [DOI] [PubMed] [Google Scholar]
- 14. Schwartz GG, Olsson AG, Abt M, Ballantyne CM, Barter PJ, Brumm J, Chaitman BR, Holme IM, Kallend D, Leiter LA, et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N Engl J Med. 2012;2089–2099. [DOI] [PubMed] [Google Scholar]
- 15. Lincoff AM, Nicholls SJ, Riesmeyer JS, Barter PJ, Brewer HB, Fox KAA, Gibson CM, Granger C, Menon V, Montalescot G, et al. Evacetrapib and cardiovascular outcomes in high‐risk vascular disease. N Engl J Med. 2017;1933–1942. [DOI] [PubMed] [Google Scholar]
- 16. Cannon CP, Shah S, Dansky HM, Davidson M, Brinton EA, Gotto AM, Stepanavage M, Liu SX, Gibbons P, Ashraf TB, et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N Engl J Med. 2010;2406–2415. [DOI] [PubMed] [Google Scholar]
- 17. Group HTRC , Bowman L, Hopewell JC, Chen F, Wallendszus K, Stevens W, Collins R, Wiviott SD, Cannon CP, Braunwald E, Sammons E, et al. Effects of anacetrapib in patients with atherosclerotic vascular disease. N Engl J Med. 2017;1217–1227. [DOI] [PubMed] [Google Scholar]
- 18. Yvan‐Charvet L, Kling J, Pagler T, Li H, Hubbard B, Fisher T, Sparrow CP, Taggart AK, Tall AR. Cholesterol efflux potential and antiinflammatory properties of high‐density lipoprotein after treatment with niacin or anacetrapib. Arterioscler Thromb Vasc Biol. 2010;1430–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ebtehaj S, Gruppen EG, Bakker SJ, Dullaart RP, Tietge UJ. HDL (high‐density lipoprotein) cholesterol efflux capacity is associated with incident cardiovascular disease in the general population. Arterioscler Thromb Vasc Biol. 2019;1874–1883. [DOI] [PubMed] [Google Scholar]
- 20. Cahill LE, Levy AP, Chiuve SE, Jensen MK, Wang H, Shara NM, Blum S, Howard BV, Pai JK, Mukamal KJ, et al. Haptoglobin genotype is a consistent marker of coronary heart disease risk among individuals with elevated glycosylated hemoglobin. J Am Coll Cardiol. 2013;728–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Asleh R, Miller‐Lotan R, Aviram M, Hayek T, Yulish M, Levy JE, Miller B, Blum S, Milman U, Shapira C, et al. Haptoglobin genotype is a regulator of reverse cholesterol transport in diabetes in vitro and in vivo. Circ Res. 2006;1419–1425. [DOI] [PubMed] [Google Scholar]
- 22. Asleh R, Levy AP, Levy NS, Asleh A, Goldenstein H, Segol I, Gulati R, Lerman LO, Lerman A. Haptoglobin phenotype is associated with high‐density lipoprotein‐bound hemoglobin content and coronary endothelial dysfunction in patients with mild nonobstructive coronary artery disease. Arterioscler Thromb Vasc Biol. 2019;774–786. [DOI] [PubMed] [Google Scholar]
- 23. Melamed‐Frank M, Lache O, Enav BI, Szafranek T, Levy NS, Ricklis RM, Levy AP. Structure‐function analysis of the antioxidant properties of haptoglobin. Blood. 2001;3693–3698. [DOI] [PubMed] [Google Scholar]
- 24. Carew AS, Levy AP, Ginsberg HN, Coca S, Lache O, Ransom T, Byington R, Rimm EB, Sapp J, Gardner M, et al. Haptoglobin phenotype modifies the influence of intensive glycemic control on cardiovascular outcomes. J Am Coll Cardiol. 2020;512–521. [DOI] [PubMed] [Google Scholar]
- 25. Cannon CP, Dansky HM, Davidson M, Gotto AM Jr, Brinton EA, Gould AL, Stepanavage M, Liu SX, Shah S, Rubino J, et al. Design of the DEFINE trial: determining the EFficacy and tolerability of CETP INhibition with AnacEtrapib. Am Heart J. 2009;513–519.e3. [DOI] [PubMed] [Google Scholar]
- 26. Sankaranarayanan S, Kellner‐Weibel G, de la Llera‐Moya M, Phillips MC, Asztalos BF, Bittman R, Rothblat GH. A sensitive assay for ABCA1‐mediated cholesterol efflux using BODIPY‐cholesterol. J Lipid Res. 2011;2332–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Levy NS, Vardi M, Blum S, Miller‐Lotan R, Afinbinder Y, Cleary PA, Paterson AD, Bharaj B, Snell‐Bergeon JK, Rewers MJ, et al. An enzyme linked immunosorbent assay (ELISA) for the determination of the human haptoglobin phenotype. Clin Chem Lab Med. 2013;1615–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brownell N, Rohatgi A. Modulating cholesterol efflux capacity to improve cardiovascular disease. Curr Opin Lipidol. 2016;398–407. [DOI] [PubMed] [Google Scholar]
- 29. Nicholls SJ, Ruotolo G, Brewer HB, Kane JP, Wang MD, Krueger KA, Adelman SJ, Nissen SE, Rader DJ. Cholesterol efflux capacity and pre‐beta-1 HDL concentrations are increased in dyslipidemic patients treated with evacetrapib. J Am Coll Cardiol. 2015;2201–2210. [DOI] [PubMed] [Google Scholar]
- 30. Yvan‐Charvet L, Matsuura F, Wang N, Bamberger MJ, Nguyen T, Rinninger F, Jiang XC, Shear CL, Tall AR. Inhibition of cholesteryl ester transfer protein by torcetrapib modestly increases macrophage cholesterol efflux to HDL. Arterioscler Thromb Vasc Biol. 2007;1132–1138. [DOI] [PubMed] [Google Scholar]
- 31. Catalano G, Julia Z, Frisdal E, Vedie B, Fournier N, Le Goff W, Chapman MJ, Guerin M. Torcetrapib differentially modulates the biological activities of HDL2 and HDL3 particles in the reverse cholesterol transport pathway. Arterioscler Thromb Vasc Biol. 2009;268–275. [DOI] [PubMed] [Google Scholar]
- 32. Bellanger N, Julia Z, Villard EF, El Khoury P, Duchene E, Chapman MJ, Fournier N, Le Goff W, Guerin M. Functionality of postprandial larger HDL2 particles is enhanced following CETP inhibition therapy. Atherosclerosis. 2012;160–168. [DOI] [PubMed] [Google Scholar]
- 33. Ballantyne CM, Miller M, Niesor EJ, Burgess T, Kallend D, Stein EA. Effect of dalcetrapib plus pravastatin on lipoprotein metabolism and high‐density lipoprotein composition and function in dyslipidemic patients: results of a phase IIb dose‐ranging study. Am Heart J. 2012;515–521, 521.e511‐513. [DOI] [PubMed] [Google Scholar]
- 34. Ray KK, Ditmarsch M, Kallend D, Niesor EJ, Suchankova G, Upmanyu R, Anzures‐Cabrera J, Lehnert V, Pauly‐Evers M, Holme I, et al. The effect of cholesteryl ester transfer protein inhibition on lipids, lipoproteins, and markers of HDL function after an acute coronary syndrome: the dal‐ACUTE randomized trial. Eur Heart J. 2014;1792–1800. [DOI] [PubMed] [Google Scholar]
- 35. van Capelleveen JC, Kastelein JJ, Zwinderman AH, van Deventer SJ, Collins HL, Adelman SJ, Round P, Ford J, Rader DJ, Hovingh GK. Effects of the cholesteryl ester transfer protein inhibitor, TA‐8995, on cholesterol efflux capacity and high‐density lipoprotein particle subclasses. J Clin Lipidol. 2016;1137–1144.e3. [DOI] [PubMed] [Google Scholar]
- 36. Khera AV, Demler OV, Adelman SJ, Collins HL, Glynn RJ, Ridker PM, Rader DJ, Mora S. Cholesterol efflux capacity, high‐density lipoprotein particle number, and incident cardiovascular events: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation. 2017;2494–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shea S, Stein JH, Jorgensen NW, McClelland RL, Tascau L, Shrager S, Heinecke JW, Yvan‐Charvet L, Tall AR. Cholesterol mass efflux capacity, incident cardiovascular disease, and progression of carotid plaque. Arterioscler Thromb Vasc Biol. 2019;89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Catalano G, Duchene E, Julia Z, Le Goff W, Bruckert E, Chapman MJ, Guerin M. Cellular SR‐BI and ABCA1‐mediated cholesterol efflux are gender‐specific in healthy subjects. J Lipid Res. 2008;635–643. [DOI] [PubMed] [Google Scholar]
- 39. Hernaez A, Soria‐Florido MT, Schroder H, Ros E, Pinto X, Estruch R, Salas‐Salvado J, Corella D, Aros F, Serra‐Majem L, et al. Role of HDL function and LDL atherogenicity on cardiovascular risk: a comprehensive examination. PLoS One. 2019;9:e0218533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Annema W, Dikkers A, de Boer JF, van Greevenbroek MM, van der Kallen CJ, Schalkwijk CG, Stehouwer CD, Dullaart RP, Tietge UJ. Impaired HDL cholesterol efflux in metabolic syndrome is unrelated to glucose tolerance status: the CODAM study. Sci Rep. 2016;27367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rashduni DL, Rifici VA, Schneider SH, Khachadurian AK. Glycation of high‐density lipoprotein does not increase its susceptibility to oxidation or diminish its cholesterol efflux capacity. Metabolism. 1999;139–143. [DOI] [PubMed] [Google Scholar]
- 42. Passarelli M, Shimabukuro AF, Catanozi S, Nakandakare ER, Rocha JC, Carrilho AJ, Quintao EC. Diminished rate of mouse peritoneal macrophage cholesterol efflux is not related to the degree of HDL glycation in diabetes mellitus. Clin Chim Acta. 2000;119–134. [DOI] [PubMed] [Google Scholar]
- 43. Cahill LE, Jensen MK, Chiuve SE, Shalom H, Pai JK, Flint AJ, Mukamal KJ, Rexrode KM, Levy AP, Rimm EB. The risk of coronary heart disease associated with glycosylated hemoglobin of 6.5% or greater is pronounced in the haptoglobin 2–2 genotype. J Am Coll Cardiol. 2015;1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Asleh R, Briasoulis A, Berinstein EM, Wiener JB, Palla M, Kushwaha SS, Levy AP. Meta‐analysis of the association of the haptoglobin genotype with cardiovascular outcomes and the pharmacogenomic interactions with vitamin E supplementation. Pharmgenomics Pers Med. 2018;71–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Levy AP, Hochberg I, Jablonski K, Resnick HE, Lee ET, Best L, Howard BV, Strong HS. Haptoglobin phenotype is an independent risk factor for cardiovascular disease in individuals with diabetes: the Strong Heart Study. J Am Coll Cardiol. 2002;1984–1990. [DOI] [PubMed] [Google Scholar]
- 46. Suleiman M, Aronson D, Asleh R, Kapeliovich MR, Roguin A, Meisel SR, Shochat M, Sulieman A, Reisner SA, Markiewicz W, et al. Haptoglobin polymorphism predicts 30‐day mortality and heart failure in patients with diabetes and acute myocardial infarction. Diabetes. 2005;2802–2806. [DOI] [PubMed] [Google Scholar]
- 47. Schwartz A, Blum S, Asleh R, Pollak M, Kalet‐Litman S, Levy AP. Pharmacogenomic application of the haptoglobin genotype in the treatment of HDL dysfunction. Pharmgenomics Pers Med. 2009;1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tardif JC, Rheaume E, Lemieux Perreault LP, Gregoire JC, Feroz Zada Y, Asselin G, Provost S, Barhdadi A, Rhainds D, L'Allier PL, et al. Pharmacogenomic determinants of the cardiovascular effects of dalcetrapib. Circ Cardiovasc Genet. 2015;372–382. [DOI] [PubMed] [Google Scholar]
- 49. Tardif JC, Rhainds D, Brodeur M, Feroz Zada Y, Fouodjio R, Provost S, Boule M, Alem S, Gregoire JC, L'Allier PL, et al. Genotype‐dependent effects of dalcetrapib on cholesterol efflux and inflammation: concordance with clinical outcomes. Circ Cardiovasc Genet. 2016;340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nissen SE, Pillai SG, Nicholls SJ, Wolski K, Riesmeyer JS, Weerakkody GJ, Foster WM, McErlean E, Li L, Bhatnagar P, et al. ADCY9 genetic variants and cardiovascular outcomes with evacetrapib in patients with high‐risk vascular disease: a nested case‐control study. JAMA Cardiol. 2018;401–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hopewell JC, Ibrahim M, Hill M, Shaw PM, Braunwald E, Blaustein RO, Bowman L, Landray MJ, Sabatine MS, Collins R, et al. Impact of ADCY9 genotype on response to anacetrapib. Circulation. 2019;891–898. DOI: 10.1161/CIRCULATIONAHA.119.041546 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S2
Figure S1