

Abstract
Background
Observational studies have indicated that depression is associated with coronary artery disease (CAD) and myocardial infarction. Nevertheless, causal associations between depression and cardiovascular diseases remain controversial. Hence, we conducted a Mendelian randomization and mediation analysis to evaluate the associations of depression‐related genetic variants with CAD and myocardial infarction.
Methods and Results
Summary statistics from genome‐wide association studies of depression (807 553 individuals), and CAD (60 801 cases, including 43 676 with myocardial infarction, and 123 504 controls) were used. We pooled Mendelian randomization estimates using a fixed‐effects inverse‐variance weighted meta‐analysis and multivariable Mendelian randomization. The mediation effects of potential cardiovascular risk factors on depression‐CAD and myocardial infarction risk were investigated by using mediation analysis. We also explored the relationship of genetic liability to depression with heart failure, atrial fibrillation, and ischemic stroke. Genetic liability to depression was associated with higher CAD (odds ratio [OR], 1.14; 95% CI, 1.06–1.24; P=1.0×10−3) and myocardial infarction (OR, 1.21; 95% CI, 1.11–1.33; P=4.8×10−5) risks. Results were consistent in all sensitivity analyses. Type 2 diabetes mellitus and smoking demonstrated significant mediation effects. Furthermore, our Mendelian randomization analyses revealed that the genetic liability to depression was associated with higher risks of heart failure and small vessel stroke.
Conclusions
Genetic liability to depression is associated with higher CAD and myocardial infarction risks, partly mediated by type 2 diabetes mellitus and smoking. The potential preventive value of depression treatment on cardiovascular diseases should be investigated in the future.
Keywords: cardiovascular disease, coronary artery disease, depression, Mendelian randomization, myocardial infarction
Subject Categories: Cardiovascular Disease, Epidemiology, Risk Factors
Nonstandard Abbreviations and Acronyms
- IV
instrumental variable
- IVW
inverse‐variance weighted
- MR
Mendelian randomization
Clinical Perspective
What Is New?
-
・
Genetically instrumented depression was associated with higher risk of coronary artery disease, myocardial infarction, heart failure, and small vessel stroke.
-
・
Type 2 diabetes mellitus and smoking demonstrated significant mediation effects in the association of depression with coronary artery disease and myocardial infarction.
What Are the Clinical Implications?
-
・
The potential preventive value of depression treatment for reducing the risk of cardiovascular diseases requires further exploration.
Depression is a common mental illness worldwide, affecting >264 million people in 2017. 1 As the leading cause of disability, it places an immense burden on public health systems worldwide. 2 Observational studies have suggested that depression is associated with cardiovascular disease risk, including myocardial infarction. 3 , 4
However, observational studies are not appropriate for causal inferences, because they are susceptible to confounding and reverse causality bias. Furthermore, the typically short duration of follow‐up in observational studies may not accurately reveal the association of long‐term exposure to depression with coronary artery disease (CAD) and myocardial infarction risk. Evidence from a meta‐analysis of 4 prospective cohort studies has indicated that there was no relationship between depression and CAD when only individuals with >15 years follow‐up were included in the analyses. 5
Mendelian randomization (MR) is a method used to infer causality using genetic variants associated with exposures of interest. 6 Since germline genetic variants are randomly assigned to the offspring and remain constant after conception, they are not affected by environmental factors and, therefore, diminish interference of potential confounding factors and reverse causality between exposures and disease outcomes. 7 , 8 In addition, the data of MR can be extracted from 2 independent data sets, known as 2‐sample MR, which improves the availability and efficacy of the study. 7
Hence, to examine the potential causal relationship of genetic liability to depression with the risk of CAD and myocardial infarction, we carried out a 2‐sample MR analysis, using single nucleotide polymorphisms (SNPs) associated with genetic liability to depression as instrumental variables (IVs). Mediation analysis was conducted to investigate whether the effect of depression on CAD and myocardial infarction was potentially mediated.
Methods
Study Design
The diagram of this MR analysis is displayed in Figure 1. In brief, genetic variants were used as IVs to explore the association between depression on CAD and myocardial infarction based on 3 assumptions. First, the genetic variants should directly affect risk of depression (genetic IVs for the depression were selected at a genome‐wide significance level [P<5×10−8]). Second, the genetic variants should not be associated with any possible known confounders. Third, the genetic variants should affect the outcome only through the exposure. Depression‐associated IVs were searched in the GWAS (genome‐wide association study) of the outcome by querying the matched SNPs. Where SNPs were not available in the outcome GWAS, proxies were found via a search through the European population genotype data, originating from Phase 3 (Version 5) of the 1000 Genomes Project (linkage disequilibrium r 2> 0.8; identified using online tool SNiPa, available at: http://snipa.helmholtz‐muenchen.de/snipa3/). MR analyses were conducted using publicly available data (Table S1). All original studies included have obtained ethical review approval and informed consent from the participants. The data and statistical coding that support the findings of this study are available from the corresponding author upon reasonable request.
Figure 1. Diagram of the Mendelian randomization assumptions of the association between depression and cardiovascular diseases.
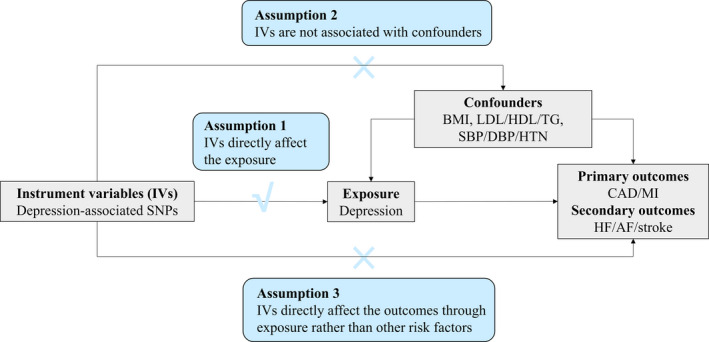
AF indicates atrial fibrillation; BMI, body mass index; CAD, coronary artery disease; DBP, diastolic blood pressure; HDL, high‐density lipoprotein; HF, heart failure; HTN, hypertension; IVs, instrument variables; LDL, low‐density lipoprotein; MI, myocardial infarction; SBP, systolic blood pressure; SNPs, single nucleotide polymorphisms; and TG, triglyceride.
Data Sources
Genetic IVs for depression were obtained from the largest published GWAS meta‐analysis to date, 9 which included the UK Biobank (127 552 cases, 233 763 controls), 10 23andMe_307k (75 607 cases, 231 747 controls), 11 and PGC_139k (43 204 cases, 95 680 controls). 12 Depression was defined based on responses to web‐based surveys, structured diagnostic interviews, or electronic medical records, with individuals who self‐reported as having received a clinical diagnosis of or treatment for depression (see Data S1 for more detail). The GWAS on depression identified 102 independent‐lead SNPs located at 101 loci, identified by linkage disequilibrium r 2<0.1 across a 3 Mb window. The estimate of instrumental variables explained 8.9% of the heritability of depression in up to 807 553 individuals (Table S2).
The primary outcomes were CAD and myocardial infarction. Summary statistics for the association of depression‐related SNPs with CAD and myocardial infarction were obtained from the Coronary Artery Disease Genome‐Wide Replication and Meta‐analysis plus the Coronary Artery Disease Genetics (CardiogramplusC4D) consortium, 13 including 60 801 patients with CAD (among whom were 43 676 myocardial infarction cases), and 123 504 controls. We further explored the associations between depression and other vascular outcomes, including heart failure, atrial fibrillation, and ischemic stroke and its subtypes, as secondary outcomes. Summary‐level data were extracted from the UK Biobank Heart Failure GWAS for heart failure (6504 cases; 387 652 controls), 14 the Atrial Fibrillation Haplotype Reference Consortium for atrial fibrillation (65 446 cases; 522 744 controls), 15 and the MEGASTROKE consortium for ischemic stroke and stroke subtypes (34 217 cases and 404 630 controls). 16 Based on the Trial of Org 10172 in Acute Stroke Treatment criteria, 17 stroke subtypes were categorized as large‐artery stroke (n=4373), small‐vessel stroke cases (n=5386), and cardioembolic‐stroke cases (n=7193) (Table S1).
Statistical Analysis
After extracting the data and harmonizing the direction of estimates via the effect alleles of IVs on depression and the outcomes, we generated effect estimates using the Wald estimator and standard errors with the Delta method. 6 A fixed‐effects inverse‐variance weighted (IVW) meta‐analysis was used to combine the MR estimates as standard analysis. Sensitivity analyses using the simple median, weighted median, MR‐robust adjusted profile score, 18 and MR‐pleiotropy residual sum and outlier 19 were also adopted. The MR‐robust adjusted profile score corrected for horizontal pleiotropy in the IVW analysis using robust adjusted profile scores. 18 The MR‐pleiotropy residual sum and outlier test was used to detect and correct for horizontal pleiotropic outliers in the IVW method, and to explore significant differences in the causal estimates before and after correction for outliers. 19 Heterogeneity statistics were calculated by means of IVW methods, I 2>25% or Cochran Q‐derived P<0.05 was considered as horizontal pleiotropy. 20 The result estimate of the IVW method was considered as the most credible if there was no pleiotropy. 21 , 22 Scatter plots depicting the associations of genetically determined depression with CAD and myocardial infarction were also provided. We performed power calculations to evaluate the minimum effect of >80% power as the significance according to the sample size of each outcome. 23
In addition, we conducted multivariable MR, performing multivariable weighted linear regression with the intercept term set to 0, 24 to intervene on influence of the potential cardiovascular risk factors on causal estimates. We used publicly available summarized data for genetic association of instruments with smoking and alcohol use from the GWAS & Sequencing Consortium of Alcohol and Nicotine use (n=1 232 091 individuals), 25 type 2 diabetes mellitus from the Diabetes Genetics Replication and Meta‐analysis (n=149 821 individuals), 26 body mass index from the Genetic Investigation of Anthropometric Traits (n=322 154 individuals), 27 circulating lipid levels (low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, and triglycerides) from the Global Lipids Genetics Consortium (n=188 578 individuals), 28 and blood pressure measurements (systolic and diastolic blood pressure, and hypertension) from the UK Biobank (n=317 754 individuals), published by the Neale laboratory, 29 respectively.
Cardiovascular risk factors that significantly weakened the association of depression with CAD and myocardial infarction were subsequently explored via mediation analysis, to investigate the mediation effects on the causal pathway from depression to CAD and myocardial infarction (Figure S1). 30
A 2‐sided P value<0.05 was considered statistically significant. For the primary analyses (association of depression with CAD and myocardial infarction), we adjusted the thresholds by Bonferroni correction for number of outcomes. Therefore, we set 2‐sided P values of <0.025 (=0.05/2 outcomes) as the thresholds for significance. For secondary outcomes, the statistical significance thresholds were set at P<0.05/6=0.0083 for the 6 cardiovascular outcomes. MR analyses were conducted using the TwoSampleMR, MendelianRandomization and MR‐pleiotropy residual sum, and outliers R packages. All data analyses were conducted with R version 3.6.1.
Results
Genetically Determined Depression With CAD and Myocardial Infarction
For the primary outcomes, there was >80% power to detect significant differences at an odds ratio (OR) of 1.10 or higher for CAD and myocardial infarction. In the standard IVW analyses, genetically instrumented depression was associated with a higher risk of both CAD (OR, 1.14; 95% CI, 1.06–1.24; P=1.0×10−3) and myocardial infarction (OR, 1.21; 95% CI, 1.11–1.33; P=4.8×10−5) (Figure 2 and Figure S2). There was no indication of heterogeneity in the IVW analyses as measured by I 2 and Cochran Q (I 2=8% for CAD, and I 2=9% for myocardial infarction, respectively). The MR estimates of depression on CAD and myocardial infarction were robust and consistent in all sensitivity analyses (Figure 2). For CAD, the ORs ranged from 1.15 to 1.25, and for myocardial infarction, the ORs ranged from 1.23 to 1.24, (all P<0.01) in the sensitivity analyses. No outlier SNPs were detected with the MR‐pleiotropy residual sum and outlier test. There was no causality between genetically instrumented CAD or myocardial infarction and depression (Table S3).
Figure 2. Mendelian randomization association of genetically predicted depression with coronary artery disease and myocardial infarction.
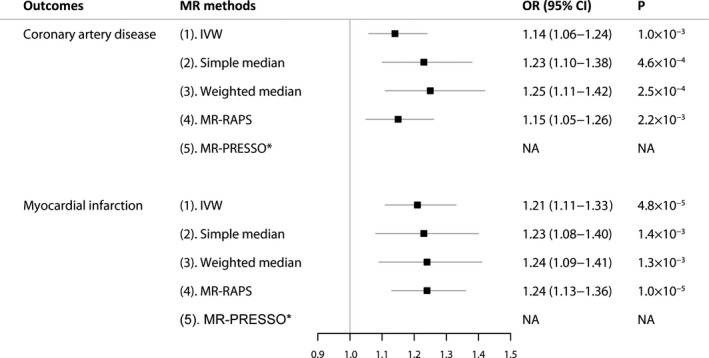
Odds ratios are scaled per genetically predicted 2.72‐fold (1 log‐odds unit) increase in the liability to depression. *No outlier detected. IVW indicates the inverse‐variance weighted method; MR, Mendelian randomization; MR‐PRESSO, MR‐pleiotropy residual sum and outlier; MR‐RAPS, MR‐robust adjusted profile scores; NA, not applicable; and OR, odds ratio.
Multivariable MR and Meditation Analysis of the Depression‐CAD and Myocardial Infarction Risk
The association of genetically predicted depression with CAD and myocardial infarction was robust in the multivariable MR analyses adjusted for genetically determined alcohol use, body mass index, circulating lipid levels, or blood pressure separately. However, after adjusting for smoking or type 2 diabetes mellitus, our MR results demonstrated that there was no causal effect of depression on CAD or myocardial infarction (Table 1).
Table 1.
Multivariable Mendelian Randomization Associations of Depression With Coronary Artery Disease and Myocardial Infarction Risk Adjusting for Cardiovascular Risk Factors
| Model | Coronary Artery Disease (n=60 801) | Myocardial Infarction (n=43 676) | ||
|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | |
| Unadjusted model | 1.14 (1.06–1.24) | 1.0×10−3 | 1.21 (1.11–1.33) | 4.8×10−5 |
| Adjusted for alcohol | 1.15 (1.06–1.26) | 1.5×10−5 | 1.22 (1.10–1.34) | 7.9×10−5 |
| Adjusted for smoking | 1.03 (0.93–1.14) | 0.57 | 1.06 (0.95–1.19) | 0.31 |
| Adjusted for T2D | 1.09 (0.96–1.24) | 0.20 | 1.14 (0.98–1.32) | 0.08 |
| Adjusted for BMI | 1.15 (1.01–1.31) | 0.03 | 1.23 (1.06–1.42) | 5.9×10−3 |
| Adjusted for LDL‐C | 1.17 (1.03–1.33) | 0.02 | 1.25 (1.08–1.45) | 3.6×10−3 |
| Adjusted for HDL‐C | 1.15 (1.01–1.29) | 0.03 | 1.22 (1.06–1.40) | 5.3×10−3 |
| Adjusted for TG | 1.15 (1.01–1.32) | 0.04 | 1.21 (1.04–1.42) | 0.02 |
| Adjusted for SBP | 1.15 (1.06–1.25) | 1.1×10−3 | 1.22 (1.11–1.34) | 2.0×10−5 |
| Adjusted for DBP | 1.15 (1.06–1.25) | 1.2×10−3 | 1.22 (1.11–1.33) | 2.4×10−5 |
| Adjusted for hypertension | 1.15 (1.05–1.25) | 1.7×10−3 | 1.22 (1.11–1.34) | 3.6×10−5 |
Results are scaled per genetically predicted standard deviation increase of liability to depression. BMI indicates body mass index; DBP, diastolic blood pressure; HDL‐C, high‐density lipoprotein cholesterol; LDL‐C, low‐density lipoprotein cholesterol; OR, odds ratio; SBP, systolic blood pressure; T2D, type 2 diabetes mellitus; and TG, triglyceride.
We conducted a mediation analysis to investigate whether the effects of depression on CAD and myocardial infarction were mediated by type 2 diabetes mellitus and smoking (Table 2). The mediation effect of type 2 diabetes mellitus was 0.056 (95% CI, 0.024–0.087; P=5.4×10−4) with a mediated proportion of 41.2% (95% CI, 17.9%–64.4%) on CAD, and 0.041 (95% CI, 0.017–0.065; P=9.1×10−4) with a mediated proportion of 24.1% (95% CI, 9.4%– 38.8%) on myocardial infarction, respectively. Likewise, the mediation effect of smoking was 0.047 (95% CI, 0.018–0.075; P=1.3×10−3) with a mediated proportion of 30.5% (95% CI, 12.5%– 48.5%) on CAD, and 0.048 (95% CI, 0.022–0.074; P=2.7×10−4) with a mediated proportion of 24.9% (95% CI, 11.5%–38.3%) on myocardial infarction, respectively.
Table 2.
Mediation Analysis of the Mediation Effect of Depression on Coronary Artery Disease and Myocardial Infarction via Type 2 Diabetes Mellitus, Smoking, or Alcohol Use
| Outcome | Mediator | Total Effect* | Direct Effect A † | Direct Effect B ‡ | Mediation Effect § | Mediated Proportion | |
|---|---|---|---|---|---|---|---|
| Effect Size (95% CI) | Effect Size (95% CI) | Effect Size (95% CI) | Effect Size (95% CI) | P | (%) (95% CI) | ||
| Coronary artery disease | Type 2 diabetes mellitus | 0.135 (0.054–0.215) | 0.500 (0.272–0.729) | 0.111 (0.074–0.148) | 0.056 (0.024–0.087) | 5.4×10−4 | 41.2 (17.9, 64.4) |
| Smoking | 0.135 (0.054 –0.215) | 0.235 (0.203–0.266) | 0.175 (0.074–0.276) | 0.041 (0.017–0.065) | 9.1×10−4 | 30.5 (12.5, 48.5) | |
| Myocardial infarction | Type 2 diabetes mellitus | 0.135 (0.054 –0.215) | 0.500 (0.272–0.729) | 0.093 (0.056–0.131) | 0.047 (0.018–0.075) | 1.3×10−3 | 24.1 (9.4, 38.8) |
| Smoking | 0.135 (0.054 –0.215) | 0.235 (0.203–0.266) | 0.206 (0.099–0.313) | 0.048 (0.022–0.074) | 2.7×10−4 | 24.9 (11.5, 38.3) | |
Total effect: the effect of depression on the outcome.
Direct effect A: the effect of the depression on the mediator.
Direct effect B: the effect of the mediator on the outcome after adjusting for depression.
Mediation effect: the effect of depression on the outcome acting through the mediator.
Genetically Determined Depression and Other Cardiovascular Diseases
We further examined the effects of depression on other cardiovascular diseases (Figure 3). For the secondary outcomes, power calculations indicated that depression instruments provided adequate statistical power (>80%) to detect significant differences at an OR of 1.10 or higher for heart failure, atrial fibrillation, and any ischemic stroke, and 1.20 or higher for ischemic stroke subtypes, respectively. Genetic liability to depression was associated with a higher risk of small‐vessel stroke, with an OR of 1.36 (95% CI, 1.11–1.65; P=2.4×10−3). A suggestive association (P=0.05) was found between depression and heart failure (OR, 1.20; 95% CI, 1.03–1.39; P=0.02). No indication of heterogeneity was found in associations of small vessel stroke or heart failure with depression as measured by I 2 and Cochran Q. The IVW estimate showed there was no association of the genetically determined depression with atrial fibrillation (OR, 1.00; 95% CI, 0.94–1.06; P=0.95), any ischemic stroke (OR, 1.06; 95% CI, 0.98–1.16; P=0.17), large‐artery stroke (OR, 1.12; 95% CI, 0.91–1.39; P=0.29), or cardioembolic stroke (OR, 0.94; 95% CI, 0.80–1.11; P=0.49). No causal effect of cardiovascular diseases on depression was detected (Table S3).
Figure 3. Mendelian randomization association between genetically predicted depression and other cardiovascular diseases.
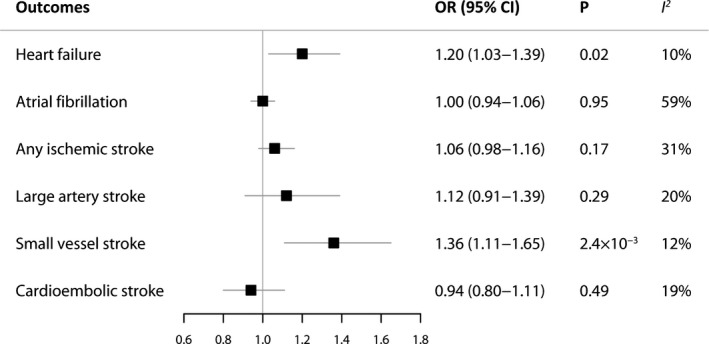
ORs are scaled to per genetically predicted 2.72‐fold (1 log‐odds unit) increase in the liability to depression. Estimates were obtained using the inverse variance‐weighted method under a fixed‐effects model. OR indicates odds ratio.
Discussion
This MR study demonstrated significant associations of genetic liability to depression with CAD and myocardial infarction risk. Genetically instrumented depression was also associated with higher risks of heart failure and small‐vessel stroke.
Our findings were in line with a meta‐analysis of 30 prospective studies demonstrating that depression was associated with both CAD (relative risk, 1.30; 95% CI, 1.22–1.40) and myocardial infarction (relative risk, 1.30; 95% CI, 1.18–1.44) risk. 5 However, the relationship between depression and CAD in the meta‐analysis disappeared after >15 years of follow‐up. Our MR study revealed long‐term and stable effects of depression on this risk. In addition, previous research showed that there was no genetic causal effect of CAD on depression (OR, 1.01; 95% CI, 1.00–1.03; P=0.11), 31 which was consistent with our results.
The causality from depression to CAD and myocardial infarction has a biological basis. Previous studies have shown that genetic liability to depression was associated with higher body mass index and obesity, 32 , 33 hypertension, 34 increased sympathetic excitability, 35 and endothelial dysfunction. 36 A recent MR study reported that depression and CAD shared common risk factors such as interleukin‐6, C‐reactive protein, and triglycerides, 31 which was in line with clinical findings. 37
Previous observational studies and MR analyses yielded inconsistent results on causal relationship from depression to an increased risk of stroke. Wium‐Andersen et al 38 performed a large prospective cohort study including 93 076 participants. After a follow‐up period of 20.6 years, 11 787 stroke and 2276 depression cases were identified. They found that depression was associated with a higher risk of stroke (hazard ratio, 1.94; 95% CI, 1.63–2.30). In contrast, Gill et al 39 showed that there was no genetic causal relationship from depression (56 SNPs explaining 1.2% of the variance) to ischemic stroke or poor functional outcome 90 days after ischemic stroke (60 341 cases and 454 450 controls; the analysis of functional outcome 3 months after ischemic stroke, based on analysis of 6021 patients). A more recent MR study conducted by Cai et al, 40 using 72 SNPs of P<1×10−6 identified in a study of 135 458 depression cases and 344 901 controls, reported that depression was associated with a 33% increased risk of small‐vessel stroke (95% CI, 1.08–1.65), but not with other stroke types, which was in line with our findings. The discrepancy might be the result of selection bias, because the use of antidepressants also increases the risk of stroke. 41 Our MR estimates were less affected by bias for causal inference between depression and stroke risk with depression‐associated SNPs. Moreover, the 102 SNPs used as IVs in our study, explaining up to 8.9% of the variance, yielded more reliable results than the above 2 MR studies.
Daskalopoulou et al 42 performed a retrospective cohort study of 1 937 360 participants who were free from cardiovascular disease at baseline, using UK electronic health records. After a median follow‐up period of 6.9 years, 367 117 (19.0%) patients with a history of depression, and 14 359 heart failure events were identified in the study. Compared with the controls, patients diagnosed with depression had a higher risk of heart failure (OR, 1.18; 95% CI, 1.13–1.24) in a fully adjusted Cox regression model. Another study, including 3 500 570 patients admitted with heart failure (9.7% with depression), reported that the presence of depression was associated with a higher risk of 30‐day readmission rate (19.7% versus 18.5%; P<0.001). 43
Our study is the first to use MR analyses to explore the relationship of depression with CAD and myocardial infarction, with sensitivity analyses and intervening on potential cardiovascular risk factor for robustness evaluation. The study included several strengths. First, a total of 102 SNPs were used as IVs, which explained 8.9% of the variance, in an analysis of 807 553 individuals. Second, the study used a large sample size, with up to 60 801 CAD cases and 123 504 controls by using the 2‐sample MR analysis. Third, we included comprehensive cardiovascular diseases as outcomes. Fourth, the influence of potential confounders and bias in observational studies was minimized by using MR analyses, particularly after correcting for pleiotropy. However, our MR analyses were subject to some limitations. First, the definitions of depression were different in the 3 included studies of GWAS, which weakened the causal effect of genetic instruments to some extent. However, there were strong genetic correlations (>0.85) between them, and the large sample size ensured the directionality agreement. Second, patient‐level data, such as sex, were lacking; therefore, we could not assess the association between depression and cardiovascular diseases in different sexes, considering there have been studies reporting increased risk of myocardial infarction in women, but not in men. 5 Third, the majority of the participants in this study were of European descent, which might limit the scope of our findings. Fourth, there was a sample overlap (UK Biobank mainly) in the GWAS of depression and secondary outcomes of heart failure and atrial fibrillation, which might lead to some model overfitting. However, the analysis of depression‐related SNPs (P<1×10−6) from only the 23andMe_307K data set yielded similar results (Figure S3). Fifth, the potential pleiotropy mediated through unknown causal pathways in the association between depression and cardiovascular diseases may affect the results, despite the lack of evidence for pleiotropy in the analyses, or the consistent results in the multivariable MR analyses.
In conclusion, our MR study supports the causal associations of genetic liability to depression with risks of CAD, myocardial infarction, heart failure, and small‐vessel stroke. Future studies are warranted to elucidate the potential preventive value of depression treatment on cardiovascular outcomes.
Sources of Funding
This work was supported by the National Key Research and Development Program of China (Grant no. 2016YFC1301003); the National Science Foundation of China (Grant nos. 30900612, 81800231, and 81873484); the Natural Science Foundation of Zhejiang Province, Zhejiang, China (Grant no. LZ16H020001); the grant of Medical Science Research Foundation of Zhejiang Province (Grant no. 2018ZD017), and the grants from the Zhejiang Provincial Natural Science Foundation (Grant nos. LY15H020002 and Y17H020020). Marios Georgakis has received funding from the Onassis Foundation and the German Academic Exchange Service (DAAD).
Disclosures
None.
Supporting information
Data S1
Tables S1–S3
Figure S1–S3
Acknowledgments
The authors thank the CARDIoGRAMplusC4D Consortium, UK Biobank Heart Failure GWAS Consortium, AF HRC Consortium, MEGASTROKE Consortium, GIANT Consortium, DIAGRAM Consortium, and GLGC Consortium for making the GWAS data publicly available.
Author contributions: Conceptualization: YL and ZW. Methodology: YL, ZW, and MKG. Resources and data curation: YL, and HL. Writing – Original Draft: YL. Writing ‐ Review and Editing: YL, MKG, and LZ.
(J Am Heart Assoc. 2021;10:e017986. DOI: 10.1161/JAHA.120.017986.)
For Sources of Funding and Disclosures, see page 7.
See Editorial by de Geus
References
- 1. GBD 2017 Disease and Injury Incidence and Prevalence Collaborators . Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392:1789–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization . Depression. 2020. Available at: https://www.who.int/news‐room/fact‐sheets/detail/depression. Accessed April 5, 2020.
- 3. Whang W, Kubzansky LD, Kawachi I, Rexrode KM, Kroenke CH, Glynn RJ, Garan H, Albert CM. Depression and risk of sudden cardiac death and coronary heart disease in women: results from the Nurses' Health Study. J Am Coll Cardiol. 2009;53:950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scherrer JF, Garfield LD, Chrusciel T, Hauptman PJ, Carney RM, Freedland KE, Owen R, True WR, Lustman PJ. Increased risk of myocardial infarction in depressed patients with type 2 diabetes. Diabetes Care. 2011;34:1729–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gan Y, Gong Y, Tong X, Sun H, Cong Y, Dong X, Wang Y, Xu X, Yin X, Deng J, et al. Depression and the risk of coronary heart disease: a meta‐analysis of prospective cohort studies. BMC Psychiatry. 2014;14:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey SG. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27:1133–1163. [DOI] [PubMed] [Google Scholar]
- 7. Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89–R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holmes MV, Ala‐Korpela M, Smith GD. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol. 2017;14:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Howard DM, Adams MJ. Genome‐wide meta‐analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Howard DM, Adams MJ. Genome‐wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat Commun. 2018;9:1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet. 2016;48:1031–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, Adams MJ, Agerbo E, Air TM, Andlauer TMF, et al. Genome‐wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, et al. A comprehensive 1,000 Genomes‐based genome‐wide association meta‐analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aragam KG, Chaffin M, Levinson RT, McDermott G, Choi SH, Shoemaker MB, Haas ME, Weng LC, Lindsay ME, Smith JG, et al. Phenotypic refinement of heart failure in a National biobank facilitates genetic discovery. Circulation. 2019;139:489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roselli C, Chaffin MD. Multi‐ethnic genome‐wide association study for atrial fibrillation. Nat Genet. 2018;50:1225–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Malik R, Chauhan G, Traylor M. Multiancestry genome‐wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50:524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Adams HP Jr, Bendixen BH, Kappelle LJ, Biller J, Love BB, Gordon DL, Marsh EE III. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of Org 10172 in ac ute stroke treatment. Stroke. 1993;24:35–41. [DOI] [PubMed] [Google Scholar]
- 18. Zhao Q, Wang J, Hemani G, Bowden J, Small DS. Statistical inference in two‐sample summary‐data Mendelian randomization using robust adjusted profile score. arXiv preprint arXiv:1801.09652. 2018.
- 19. Verbanck M, Chen CY. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greco MF, Minelli C, Sheehan NA, Thompson JR. Detecting pleiotropy in Mendelian randomisation studies with summary data and a continuous outcome. Stat Med. 2015;34:2926–2940. [DOI] [PubMed] [Google Scholar]
- 21. White J, Swerdlow DI, Preiss D, Fairhurst‐Hunter Z, Keating BJ, Asselbergs FW, Sattar N, Humphries SE, Hingorani AD, Holmes MV. Association of lipid fractions with risks for coronary artery disease and diabetes. JAMA Cardiol. 2016;1:692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun D, Zhou T, Heianza Y, Li X, Fan M, Fonseca VA, Qi L. Type 2 diabetes and hypertension. Circ Res. 2019;124:930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brion MJ, Shakhbazov K, Visscher PM. Calculating statistical power in Mendelian randomization studies. Int J Epidemiol. 2013;42:1497–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Burgess S, Thompson SG. Multivariable Mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am J Epidemiol. 2015;181:251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu M, Jiang Y, Wedow R. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, et al. Large‐scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013;45:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Neale lab . UK Biobank GWAS results. 2018. Available at: http://www.nealelab.is/uk‐biobank/. Accessed March 28, 2018.
- 30. Burgess S, Daniel RM, Butterworth AS, Thompson SG. Network Mendelian randomization: using genetic variants as instrumental variables to investigate mediation in causal pathways. Int J Epidemiol. 2015;44:484–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Khandaker GM, Zuber V, Rees JMB, Carvalho L. Shared mechanisms between coronary heart disease and depression: findings from a large UK general population‐based cohort. Mol Psychiatry. 2020;25:1477–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mulugeta A, Zhou A, Vimaleswaran KS, Dickson C, Hypponen E. Depression increases the genetic susceptibility to high body mass index: evidence from UK Biobank. Depress Anxiety. 2019;36:1154–1162. [DOI] [PubMed] [Google Scholar]
- 33. Speed MS, Jefsen OH. Investigating the association between body fat and depression via Mendelian randomization. Transl Psychiatry. 2019;9:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meng L, Chen D, Yang Y, Zheng Y, Hui R. Depression increases the risk of hypertension incidence: a meta‐analysis of prospective cohort studies. J Hypertens. 2012;30:842–851. [DOI] [PubMed] [Google Scholar]
- 35. Veith RC, Lewis N, Linares OA, Barnes RF, Raskind MA, Villacres EC, Murburg MM, Ashleigh EA, Castillo S, Peskind ER, et al. Sympathetic nervous system activity in major depression. Basal and desipramine‐induced alterations in plasma norepinephrine kinetics. Arch Gen Psychiatry. 1994;51:411–422. [DOI] [PubMed] [Google Scholar]
- 36. Sherwood A, Hinderliter AL, Watkins LL, Waugh RA, Blumenthal JA. Impaired endothelial function in coronary heart disease patients with depressive symptomatology. J Am Coll Cardiol. 2005;46:656–659. [DOI] [PubMed] [Google Scholar]
- 37. Osimo EF, Pillinger T, Rodriguez IM, Khandaker GM, Pariante CM, Howes OD. Inflammatory markers in depression: a meta‐analysis of mean differences and variability in 5,166 patients and 5,083 controls. Brain Behav Immun. 2020;87:901–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wium‐Andersen MK, Wium‐Andersen IK, Prescott EIB, Overvad K, Jorgensen MB, Osler M. An attempt to explain the bidirectional association between ischaemic heart disease, stroke and depression: a cohort and meta‐analytic approach. Br J Psychiatry. 2020;217:434–441. [DOI] [PubMed] [Google Scholar]
- 39. Gill D, James NE, Monori G, Lorentzen E, Fernandez‐Cadenas I, Lemmens R, Thijs V, Rost NS, Scott R, Hankey GJ, et al. Genetically determined risk of depression and functional outcome after ischemic stroke. Stroke. 2019;50:2219–2222. [DOI] [PubMed] [Google Scholar]
- 40. Cai H, Cai B, Zhang H, Sun W, Wang Y, Zhou S, Ye Z, Zhang Z, Liang J. Major depression and small vessel stroke: a Mendelian randomization analysis. J Neurol. 2019;266:2859–2866. [DOI] [PubMed] [Google Scholar]
- 41. Trajkova S, d'Errico A, Soffietti R, Sacerdote C, Ricceri F. Use of antidepressants and risk of incident stroke: a systematic review and meta‐analysis. Neuroepidemiology. 2019;53:142–151. [DOI] [PubMed] [Google Scholar]
- 42. Daskalopoulou M, George J, Walters K, Osborn DP, Batty GD, Stogiannis D, Rapsomaniki E, Pujades‐Rodriguez M, Denaxas S, Udumyan R, et al. Depression as a risk factor for the initial presentation of twelve cardiac, cerebrovascular, and peripheral arterial diseases: data linkage study of 1.9 million women and men. PLoS One. 2016;11:e0153838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Patel N, Chakraborty S, Bandyopadhyay D, Amgai B, Hajra A, Atti V, Das A, Ghosh RK, Deedwania PC, Aronow WS, et al. Association between depression and readmission of heart failure: a national representative database study. Prog Cardiovasc Dis. 2020. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Tables S1–S3
Figure S1–S3