

Abstract
Background
CDNF (cerebral dopamine neurotrophic factor) belongs to a new family of neurotrophic factors that exert systemic beneficial effects beyond the brain. Little is known about the role of CDNF in the cardiac context. Herein we investigated the effects of CDNF under endoplasmic reticulum‐stress conditions using cardiomyocytes (humans and mice) and isolated rat hearts, as well as in rats subjected to ischemia/reperfusion (I/R).
Methods and Results
We showed that CDNF is secreted by cardiomyocytes stressed by thapsigargin and by isolated hearts subjected to I/R. Recombinant CDNF (exoCDNF) protected human and mouse cardiomyocytes against endoplasmic reticulum stress and restored the calcium transient. In isolated hearts subjected to I/R, exoCDNF avoided mitochondrial impairment and reduced the infarct area to 19% when administered before ischemia and to 25% when administered at the beginning of reperfusion, compared with an infarct area of 42% in the untreated I/R group. This protection was completely abrogated by AKT (protein kinase B) inhibitor. Heptapeptides containing the KDEL sequence, which binds to the KDEL‐R (KDEL receptor), abolished exoCDNF beneficial effects, suggesting the participation of KDEL‐R in this cardioprotection. CDNF administered intraperitoneally to rats decreased the infarct area in an in vivo model of I/R (from an infarct area of ≈44% in the I/R group to an infarct area of ≈27%). Moreover, a shorter version of CDNF, which lacks the last 4 residues (CDNF‐ΔKTEL) and thus allows CDNF binding to KDEL‐R, presented no cardioprotective activity in isolated hearts.
Conclusions
This is the first study to propose CDNF as a new cardiomyokine that induces cardioprotection via KDEL receptor binding and PI3K/AKT activation.
Keywords: cardioprotection, cerebral dopamine neurotrophic factor, KDEL‐receptor, PI3K‐AKT pathway
Subject Categories: Basic Science Research, Myocardial Infarction
Nonstandard Abbreviations and Acronyms
- AKT
protein kinase B
- CDNF
cerebral dopamine neurotrophic factor
- CHOP
CCAAT‐enhancer‐binding protein homologous protein
- ER
endoplasmic reticulum
- GRP78
78 kDa glucose‐regulated protein
- hiPSC‐dCM
human induced pluripotent stem cells differentiated into cardiomyocytes
- I/R
ischemia/reperfusion
- KDEL‐R
KDEL‐receptor
- KHB
Krebs‐Henseleit buffer
- LVDP
left ventricular developed pressure
- LVEDP
left ventricular end‐diastolic pressure
- MANF
mesencephalic astrocyte‐derived neurotrophic factor
- PI3K
phosphoinositide 3‐kinase
- PKC
protein kinase C
- postCDNF
CDNF postconditioning
- preCDNF
CDNF preconditioning
- ROS
reactive oxygen species
- STAT3
signal transducers and activators of transcription 3
- TG
thapsigargin
Clinical Perspective
What Is New?
CDNF (cerebral dopamine neurotrophic factor) operates as a cardiomyokine reducing injuries caused by ischemia/reperfusion and thapsigargin‐induced endoplasmic/sarcoplasmic reticulum stress in cellular and animal models.
Importantly, this effect is seen whether hearts are exposed to CDNF pre or post ischemia/reperfusion; the effect elicited by CDNF is dependent on PI3K‐AKT (protein kinase B) activation, which converges to final effectors of cardioprotection (mitochondria and endoplasmic reticulum).
CDNF induces the cardioprotective effect by activation of the KDEL receptor at the cell membrane of cardiomyocytes.
What Are the Clinical Implications?
CDNF may represent a new therapeutic agent in the treatment of ischemic injuries.
The cardioprotective features of CDNF in the postconditioning model may elect this neurotrophic factor as a potential therapeutic agent in the treatment of revascularization lesions, reducing reperfusion arrhythmias and improving postischemic cardiac hemodynamics.
Furthermore, selective activation of the KDEL receptor may represent a new target in the treatment of ischemic diseases.
Acute myocardial infarction is predominant among health complications in the world, being the main cause of morbidity and mortality. 1 Myocardial ischemia/reperfusion (I/R) injury triggers several adverse intracellular effects including mitochondrial damage and endoplasmic reticulum (ER) stress, 1 , 2 , 3 leading to cell death, tissue impairment, cardiac dysfunction, coronary artery spasm, and cardiac arrest. 1
In the cardiomyocytes context, I/R induces ER stress by impairing protein folding in the ER, and it is likely that ischemia also affects the secretion of several proteins. 2 However, secretion of one group of proteins called cardiomyokines may actually be preserved or even enhanced by virtue of their induction during the ER stress response, protecting cardiac myocytes against I/R damage. 2
CDNF (cerebral dopamine neurotrophic factor) together with MANF (mesencephalic astrocyte‐derived neurotrophic factor) form a new family of neurotrophic factors that are structurally unrelated to the other 3 neurotrophic factor families. 4 Human CDNF shares 59% amino‐acid identity with human MANF and both have signal peptides that address them to the ER/sarcoplasmic reticulum (SR). 4 , 5
Although the precise mechanism(s) of action of MANF and CDNF is(are) not completely understood, ER/SR stress relief seems to be one of their main activities. Several studies have shown that MANF and CDNF are induced by ER/SR stress. 5 , 6 , 7 , 8 , 9 ER/SR stress has been implicated in a series of insults, including reduction in ER/SR calcium stores, oxidative stress, and an altered protein glycosylation pattern, among others. All of these cause the accumulation of misfolded proteins in the ER/SR, triggering a series of cellular events, collectively called the unfolding protein response. This response activates the translation of a set of very specific proteins, which relieve the stress and, if necessary, induce cell apoptosis. 10 , 11
Different from other proteins involved in ER/SR stress, MANF and CDNF are secreted to the extracellular milieu, where they exert autocrine/paracrine/endocrine effects. 5 , 7 , 12 , 13 Interestingly, MANF and CDNF have in their C‐terminal end the sequences RTDL or KTEL, respectively. These sequences closely resemble the classical ER/SR retention signal, KDEL (Lys‐Asp‐Glu‐Leu), which has a high affinity for KDEL‐receptors (KDEL‐R). This sequence is present in several ER/SR‐resident proteins such as GRP78 (glucose‐regulated protein 78), calreticulin, and PDI (protein disulfide isomerase), 14 , 15 , 16 for instance. The KDEL‐R is involved in the retrieval of proteins from Golgi back to ER/SR, avoiding their secretion. The presence of degenerated KDEL sequences in MANF and CDNF suggests that their affinity for the KDEL‐R is diminished, contributing to their increased secretion. 7 , 14 Indeed, deletion of the RTDL sequence from MANF enhances its secretion in mouse mesencephalic astrocytes. 12
The cardioprotective activity of MANF has been investigated in detail, 2 , 7 , 9 but even though the heart and skeletal muscle express relatively high amounts of CDNF, 17 there is only 1 report in the literature showing increased levels of CDNF expression in tunicamycin‐stressed H9C2 cells. 18 Furthermore, no previous study has tested whether the CDNF provides effective cardioprotection in an entire heart.
The main goal of the present study was to investigate CDNF effects in isolated cardiomyocytes, whole hearts, and an in‐vivo animal model of myocardial infarction.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. The authors declare that Western blot supporting data are available within the online supplementary files.
Materials
All chemicals (analytical grade) were obtained from Sigma‐Aldrich (St. Louis, MO, USA) if not otherwise specified. All solutions were freshly prepared and filtered (1.2 μm, Millipore).
Animals
Adult male Wistar rats (300–400 g) and neonatal CD‐1 mice (1–3 days old) were used, following the Guide for the Care and Use of Laboratory Animals, 8th edition (US National Institutes of Health, 2011) and the local Institutional Animal Care and Use Committee (100/16 and 015/17).
CDNF and CDNF‐ΔKTEL Production and Purification
CDNF and CDNF‐ΔKTEL were synthesized and purified according to Latge et al. 19 The plasmid pET‐25b(+) containing a synthetic CDNF or the CDNF‐ΔKTEL cDNA was transformed in Escherichia coli strain Rosetta Gami B (DE3) (Stratagene, San Diego, CA, USA). Cell cultures were grown at 37C and induced with 1 mmol/L isopropyl β‐d‐1‐thiogalactopyranoside for 2 hours. The cells were harvested by centrifugation, resuspended in extraction buffer (20 mmol/L MES at pH 6.0, Complete, EDTA‐free Tabs; Roche, Basel, Switzerland) and lysed by sonication at 4C. After centrifugation, the fraction containing the soluble proteins was loaded onto a 5 mL Hitrap SP XL column (GE Healthcare, Chicago, IL, USA) equilibrated with 20 mmol/L MES at pH 6.0. The bound material was eluted with a linear gradient of NaCl (0–1 mmol/L) at 67 mmol/L per minute. Fractions containing the protein as checked by monitoring absorption at 280 nm and denaturing polyacrylamide gel electrophoresis were loaded onto a Superdex 75 column (169 100 mm; GE Healthcare) equilibrated with 20 mmol/L MES at pH 6.0 and 150 mmol/L NaCl. By mass spectrometry the molecular mass of CDNF‐ΔKTEL was 17 984 (not shown), the exact value expected for CNDF with the last 4 residues deleted. The secondary structure of both proteins was evaluated by circular dichroism, showing identical results (Figure S1).
Differentiation of Human‐Induced Pluripotent Stem Cells Into Cardiomyocytes
Human‐induced pluripotent stem cells were differentiated into cardiomyocytes (hiPSC‐dCM) as previously described. 20 Three healthy volunteers were invited to donate 5 mL of peripheral blood after providing written informed consent. This study was approved by the National Institute of Cardiology's ethics review board under number 27044614.3.0000.5272. All experiments were performed in accordance with relevant guidelines and regulations. Briefly, mononuclear cells were cultivated in enrichment medium for erythroblasts, and, after 12 days, cells were infected with CytoTune‐iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher Scientific, Waltham, MA, USA). iPSCs were maintained in DMEM/F12, GlutaMAX supplement, with 20% KnockOut Serum Replacement, 1% penicillin‐streptomycin (P/S), 100 µmol/L NEEA, 0.1 mmol/L β‐mercaptoethanol, 10 ng/mL of β‐fibroblast growth factor. Briefly, 3×105 hiPSCs were plated on a 48‐plate dish treated with 1% Matrigel® hESC‐Qualified Matrix (Corning, New York, USA) and cultivated on mTeSR1 (STEMCELL Technologies, Vancouver, Canada) for 3 days. On day 0, the cells were cultivated in RPMI 1640 (Gibco, Thermo Fisher Scientific) supplemented with B‐27 (1640 [Gibco] Supplement, without insulin [Gibco] [RB‐] and 9 μmol/L) of CHIR 99021 (R&D Systems, Minneapolis, MN, USA). After 24 hours, the cells were cultivated with RB‐ for the next 2 days and on days 3 and 4, the WNT signaling was inhibited with 10 and 5 μmol/L XAV939 (R&D Systems), respectively. On day 7, we observed the first beating areas and the cells were then cultivated in RPMI 1640 (Gibco) supplemented with B‐27 Supplement (Gibco) (RB+) until day 30. These cells were called hiPSC‐dCM.
For efficient evaluation of cardiomyocyte differentiation, after day 30, hiPSC‐dCM were dissociated using Tryple 1x (Gibco) until the cells detached from the culture plate. Then the cells were centrifuged at 300g/5 min. The cells were fixed with formaldehyde 4% for 20 minutes at room temperature, permeabilized with PBS Triton 0.3% for 30 minutes and stained with troponin T (1:200) (Thermo Fisher Scientific) for 30 minutes at 4ºC. Afterward, the cells were stained with Alexa Fluor 647 goat anti‐mouse IgG (1:1000) secondary antibody (Thermo Fisher Scientific) for 30 minutes at 4ºC. Cells stained only with secondary antibody were used as fluorescence negative control. The data were acquired in a BD Accuri C6 (BD Biosciences, San Jose, CA, USA) and analyzed with the FlowJo v10.1 software (FlowJo, Ashland, OR, USA).
Isolation and Culture of Mouse Cardiomyocytes
Cardiomyocytes were isolated from the hearts of 125 neonatal mice (CD1‐mice) 1 to 3 days after birth, as previously described. 21 For each cell culture, 4 to 6 neonatal mice hearts were used. Briefly, the hearts were dissected out, placed in PBS, and washed. The heart tissue was minced and digested in a dissociation solution (in mmol/L: NaCl, 136.7; KCl, 2.68; Na2HPO4, 0.352; NaHCO3, 11.9; dextrose, 11) containing pancreatin (1.25 mg/mL) and BSA (3 mg/mL) for 5 minutes at 37C with gentle stirring. The supernatant fraction containing cells from each digestion was collected in a conical tube, suspended in growth medium containing 15% fetal bovine serum for inhibition of proteolytic enzymes and spun at 300 g/5 min. The elimination of nonmuscle cells was achieved by preplating for 1 hour. After that, the cells were counted and seeded in 6‐well plates at a density of 105 cells/well and cultured in DMEM‐high glucose containing 10% fetal bovine serum at 37C and 5% CO2. Experimental in‐vitro conditions were established 3 days after plating.
Immunocytochemistry Imaging and Quantification of Fluorescence Intensity
Cells were fixed with 4% paraformaldehyde for 10 minutes at room temperature. Then, cells were permeabilized in 0.3% Triton X‐100 in PBS and incubated with a blocking solution (5% BSA, in PBS pH 7.4) for 1 hour. After incubation with appropriate primary (1:500) and secondary antibodies (1:500) and Hoechst (1:5000), coverslips were mounted with prolong. Images were acquired with an Olympus DS‐Fi2 confocal microscope equipped with ×63 oil immersion objective and with a Nikon DS‐fi2 camera operated with the standard QC capture software (Leica). Quantification was performed with ImageJ software (National Institutes of Health, Baltimore, MD) using the corrected total cell fluorescence method.
[Ca2+] Measurements
Human iPSC‐dCM were plated on glass coverslips for 5 days and loaded with 5 μmol/L Fura‐2AM at 37C in complete culture medium containing 2.5 mmol/L probenecid for 40 to 60 minutes before the intracellular calcium measurements. The cells were then accommodated in a chamber of ≈500 μL whose base was formed by the coverslip supporting the cells that were maintained at 37C in the complete medium. Cytoplasmic calcium concentrations of groups of hiPSC‐dCM (20–30 cells) were measured on a fluorescence imaging spectrofluorimeter (Easy Ratio Pro equipped with a DeltaRAMX Illuminator, an Olympus IX71 microscope, a QuantEM 5125C camera and the Image‐Pro Plus v. 6.3 software; PTI Photon Technology International, Princeton, NJ, USA). Fura‐2 was excited alternately at 340 nm and 380 nm, and the emission was collected at 510 nm. The ratio measurement, which is proportional to the cytoplasmic calcium concentration, was determined every 100 ms for at least 5 minutes. Free intracellular Ca2+ concentration [Ca2+]i was monitored in arbitrary units as the F340/380 nm ratio. The amplitude of variations in [Ca2+]i (∆ F340/F380) for cells without treatment and with treatment was obtained for 5 different intervals of 5 to 10 seconds from ratio measurements.
Treatments Employed for hiPSC‐dCM and Mouse Cardiomyocytes Before Western Blot and Calcium Measurements
Conditions/Groups
Control: mouse cardiomyocytes or hiPSC‐dCM cultures received no treatment before calcium measurement or Western blot analysis; Thapsigargin (TG): mouse cardiomyocytes or hiPSC‐dCM cultures were incubated with TG (1 µmol/L) during 20 hours before calcium measurements or Western blot analyses; CDNF: mouse cardiomyocytes or hiPSC‐dCM cultures were incubated with CDNF (1 µmol/L) during 1 hour before calcium measurements or Western blot analyses. TG+CDNF: hiPSC‐dCM cultures were incubated with CDNF (1 µmol/L) 1 hour before TG (1 µmol/L). The calcium measurements or Western blot analyses were performed after 20 hours of incubation with TG. TG+CDNF+Wortmannin: hiPSC‐dCM cultures were incubated first with wortmannin (0.3 µmol/L) for 15 minutes, then with CDNF (1 µmol/L for 1 hour) and finally with TG (1 µmol/L). The calcium measurements were performed after 20 hours of incubation with TG. CDNF+Wortmannin: mouse cardiomyocytes or hiPSC‐dCM cultures were incubated first with wortmannin (0.3 µmol/L) for 15 minutes, then with CDNF (1 µmol/L) for 1 hour. The Western blot analyses were performed after 20 hours of incubation. Wortmannin: hiPSC‐dCM cultures were incubated with wortmannin (0.3 µmol/L) for 20 hours before the calcium measurements.
The medium was not changed after each substance addition in each group. The calcium measurements and/or Western blot analyses were performed after 20 hours of incubation in all groups.
In Vivo Myocardial I/R
Myocardial infarction experiments were performed in male Wistar rats (9–12 weeks) weighing 300 to 400 g as previously described. 21 , 22 In these experiments, ischemia was induced for 90 minutes by left coronary artery ligation followed by 180 minutes of reperfusion by left coronary artery ligation release. The rats were anesthetized with xylazine 20 mg/kg and ketamine 80 mg/kg, and a 2‐cm incision was made on the left side of the thorax, parallel to the sternum. The fifth and sixth ribs were separated, exposing the heart, to access the left coronary artery. During myocardial infarction induction or before the end of 180 minutes of reperfusion, 12.5% of the animals (4 out of 32) died. The animals were randomly divided into 4 groups before the procedures, all of them with 7 animals each: SHAM, I/R, postCDNF, and postCDNF‐ΔKTEL. Sham‐operated animals underwent the same procedure, but the coronary ligature was left untied. In the groups CDNF‐postconditioning (postCDNF) and CDNF‐ΔKTEL‐postconditioning (postCDNF‐ΔKTEL) the rats received an intraperitoneal injection of recombinant CDNF or CDNF‐ΔKTEL (3.3 µg/g BW) at 80 minutes of ischemia, and then after the full 90 minutes of ischemia the reperfusion was performed for 180 minutes. After myocardial infarction, the animals were euthanized by cervical dislocation under anesthesia. The hearts were excised and cannulated by aorta and washed with Krebs‐Henseleit buffer solution (KHB, described later). The myocardial infarct area was evaluated using Evans Blue triphenyltetrazolium chloride double staining. Following the washing, 2 mL of a 1% solution of Evans Blue dye were retrogradely perfused into the aorta. After perfusion, the hearts were washed with KHB solution and frozen at −20C for at least 30 minutes. The hearts were sliced in 5 cross‐sectional slices and the sections were incubated in 1% triphenyltetrazolium chloride at 37C for 5 minutes. The sections were then fixed in 10% formalin solution for 24 hours. The heart sections were analyzed using Image J software and areas were summed and calculations of White Area (infarct area)/total red+White Area (Risk area) for each heart were determined. The blue area (Evans Blue‐staining) determined the nonischemic region. This gives the myocardial infarction size as a percentage of the area at risk.
Langendorff Experimental Protocols and I/R
The I/R experiments were performed on isolated rat hearts as described previously. 22 , 23 The hearts were rapidly removed and cannulated through the aorta in a modified Langendorff apparatus and perfused at the constant flow of 10 mL/min with KHB solution: in mmol/L: NaCl 118; NaHCO3 25; KCl 4.7; KH2PO4 1.2; MgSO4 1.2; CaCl2 1.25, and glucose 11, at 37C and equilibrated with a gas mixture of 95% O2 and 5% CO2 (pH 7.4). The perfusion temperature was held constant by a heat exchanger located next to the aortic cannula. A fluid‐filled latex balloon was inserted through the left atrium into the left ventricle and connected to a pressure transducer and the PowerLab System (AD Instruments, Australia) for continuous left ventricular pressure recording. The left ventricular end‐diastolic pressure (LVEDP) was set to 10 mm Hg by balloon inflation; during the experiment, the hearts were continuously immersed in 37C warm buffer to avoid hypothermia. Hearts were allowed to stabilize for 20 minutes before a protocol was started. All hearts were subjected to 30 minutes of basal recordings. The ischemia protocol was induced by a full stop of retrograde perfusion for 30 minutes. The reperfusion was performed by full reestablishment of retrograde perfusion during 10 minutes for mitochondria analysis or 60 minutes for infarct size measurements.
Langendorff Experimental Protocols
Conditions/Groups
No I/R: Hearts were perfused with KHB for 70 minutes (only for mitochondria isolation); I/R: Global ischemia was induced for 30 minutes by complete perfusion arrest followed by 10 minutes (for mitochondrial function assessment) or 60 minutes (for infarct size measurements) of reperfusion with KHB solution; CDNF preconditioning (preCDNF): The hearts were perfused with the KHB solution containing CDNF (1 µmol/L) for 5 minutes before I/R protocol; CDNF postconditioning (postCDNF): After 30 minutes of global ischemia, CDNF (1 µmol/L) was perfused during the first 5 minutes of reperfusion, followed by 5 or 55 minutes of reperfusion with KHB solution without CDNF. In these 2 groups the antagonists to previously reported cardioprotective pathways were used and the substances were perfused for 5 minutes before CDNF and along with CDNF (1 µmol/L). The antagonists used were wortmannin 0.3 µmol/L (PI3K‐AKT inhibitor), chelerythrine 10 µmol/L and rottlerin 10 µmol/L (PKC inhibitors), and AG490 10 µmol/L (JAK‐STAT3 inhibitor).
Left Ventricular Developed Pressure, LVEDP, and Infarcted Area Measurements
Left ventricular developed pressure (LVDP) and LVEDP were analyzed at different time points during the experiments, namely, at baseline, every 10 minutes during ischemia, and every 10 minutes during reperfusion until 60 minutes of reperfusion. For this, the mean value of a 30 seconds recording at the respective time point was taken. After 60 minutes reperfusion, the hearts were sliced into 1.5 mm cross‐sections from apex to base and incubated in 1% triphenyltetrazolium chloride for 4 minutes at 37C, followed by incubation in a 10% (v/v) formaldehyde solution for 24 hours to improve the contrast between the stained (viable) and unstained (necrotic) tissues. The infarct area was determined by planimetry using ImageJ software (version 1.22, National Institutes of Health). Infarct size was expressed as a percentage of the area at risk (total).
ECG Recordings in Isolated Hearts
Three silver‐chloride electrodes were used to obtain electrocardiographic recordings. Two electrodes were connected to the differential input of a high‐gain amplifier (BioAmp; AD Instruments) positioned close to the left ventricle and right atrium. The third electrode was grounded. The electrocardiograms were recorded with PowerLab/400 and Chart 4.0 software (AD Instruments). The analysis of ventricular arrhythmias was based on the Lambeth convention. 24
Peptides Used in I/R Experiments
The peptide sequences corresponding to the last 7 amino‐acid residues of the C‐terminal region from human (THPKTEL) and rat (TRPQTEL) CDNF or a scrambled peptide DRATSAL 25 were synthesized by GenOne Biotechnologies with 98% purity and tested separately. Each peptide was perfused at 2 µmol/L in combination with CDNF (1 µmol/L) for 5 minutes before I/R.
Antibody Used in I/R Experiments
To reinforce the evidence that the KDEL receptor is involved in the protection induced by CDNF, 5 µg/mL of the antibody (Sigma‐Aldrich) against peptides from the C‐terminal domain of human CDNF were incubated with CDNF 1 µmol/L for 1 hour before the experiment. After 1 hour of incubation, CDNF (1 µmol/L) plus the antibody (5 µg/mL) were perfused for 5 minutes before the ischemia and reperfusion protocol.
Western Blots
Polyacrylamide gel electrophoresis (PAGE) with sodium dodecyl sulfate (SDS) were loaded with 150 µg of total protein from cell media or 50 µg of total protein from cell or tissue extracts. The proteins were separated by electrophoresis and transferred to polyvinylidene fluoride membranes (Millipore). Membranes were stained with Ponceau S solution (Sigma‐Aldrich). The membranes were incubated with appropriate antibodies directed against CDNF (Sigm‐ Aldrich PRS4343; polyclonal, 1:1000), GAPDH (Invitrogen #MAS15736; monoclonal, 1:10 000), CHOP (CCAAT‐enhancer‐binding protein homologous protein; Santa Cruz Biotechnology Sc‐166682; monoclonal, 1:500), p‐AKT1/2/3ser473 (phosphoprotein kinase B, Cell Signalling #4058;monoclonal, 1:750), total AKT1/2/3 (t‐AKT1/2/3 Cell Signalling #4691; monoclonal, 1:750), GRP 78 (78 kDa glucose‐regulated protein; Santa Cruz Biotecnology SC33575; polyclonal 1:1000), p‐PKCƐser729 (phosphoprotein kinase C epsilon; Abcam ab63387; polyclonal 1:750), t‐PKCƐ (total protein kinase C epsilon; Abcam ab233292; polyclonal; 1:750), Phospho Signal Transducer and Activator of Transcription 3 (p‐STAT3tyr705, Cell Signalling #9145; monoclonal 1:1000), and total‐STAT3 (t‐STAT3, Cell Signalling #12640; monoclonal 1:1000). After incubation with the respective secondary antibodies, immunoreactive signals were detected by chemiluminescence (Pierce Biotechnology, Waltham, MA, USA). The full Western blots photos are presented in the online supplementary files.
Mitochondria Isolation and Measurements of Mitochondrial Function
Mitochondria isolation was performed by differential centrifugation according to. 26 , 27 After ischemia (30 minutes) and reperfusion (10 minutes), the isolated hearts were placed in ice‐cold mitochondria‐isolation buffer containing, in mmol/L: Sucrose, 250; HEPES, 10 and ethylene glycol tetraacetic acid (EGTA), 1; pH 7.4 with 0.5% w/v BSA. The tissue was minced carefully using scissors. Next, the minced tissue was homogenized with a tissue homogenizer (Ultra‐Turrax) using 2 10‐second treatments at a shaft rotation rate of 6500 rpm to release subsarcolemmal mitochondria. This homogenate was further homogenized with proteinase type XXIV (8 IU/mg tissue weight) using a Teflon pestle for the release of the interfibrillar mitochondria from the tissue. The tissue homogenate was centrifuged at 700g for 10 minutes at 4C. The supernatant was collected and centrifuged at 12 300g for 10 minutes. The resulting pellet was resuspended in mitochondria‐isolation buffer without BSA and centrifuged at 10 000g for 10 minutes at 4C. This process was repeated, and the pellet was resuspended in mitochondria‐isolation buffer. The protein concentration of the isolated pellet was verified using a protein assay (Lowry method, Biorad, Hercules, CA, USA) by comparison to a BSA standard (Thermo Fisher Scientific).
Mitochondrial Oxygen Consumption Measurements
Mitochondrial oxygen consumption was evaluated with a Clark‐type electrode (Strathkelvin, Glasgow, UK) at 37C during magnetic stirring in respiration, in mmol/L: 125 KCl, 10 MOPS, 2 MgCl2, 5 KH2PO4, 0.2 EGTA with pyruvate and malate (5 mmol/L/5 mmol/L, respectively) as substrates for complex I. The oxygen electrode was calibrated applying a solubility coefficient of 217 nmol O2/mL at 37C. For the measurement of complex I oxygen consumption, mitochondria (protein amount of 100 µg) were added to 1 mL respiration buffer. After 2 minutes of incubation, 1 mmol/L ADP was added and ADP‐stimulated respiration measured for 2 minutes. Next, mitochondria were used to measure complex IV respiration and maximal uncoupled oxygen uptake or the respiration buffer containing mitochondria was transferred from the respiration chamber to quantify ATP production or extramitochondrial reactive oxygen species (ROS) concentration, respectively. Complex IV respiration was stimulated by adding N,N,N,N`‐tetramethyl‐p‐phenylenediamine (TMPD, 300 μmol/L) combined with ascorbate (3 μmol/L). Maximal uncoupled oxygen uptake was evaluated with 30 nmol/L carbonyl cyanide‐p‐trifluoromethoxyphenyl‐hydrazone. 26 , 27
Mitochondrial Oxygen Consumption Under Simulated Hypoxia/Reoxygenation
The respiration buffer (without pyruvate and malate) was made hypoxic by the insertion of purified nitrogen until the oxygen concentration was <15 nmol O2 mL−1. Mitochondria (200 μg) were added to 0.5 mL hypoxic buffer. After 8 minutes of hypoxia, the O2‐saturated respiration buffer (0.5 mL) was added to achieve reoxygenation for 2 minutes. After simulated hypoxia/reoxygenation or the respective normoxic time control (10 minutes), pyruvate and malate (5 mmol/L/5 mmol/L, respectively) were added as substrates for complex I. Mitochondria were stimulated with ADP and the oxygen consumption was evaluated for 2 minutes Finally, mitochondria were used to evaluate complex IV respiration and maximal uncoupled oxygen uptake, or extramitochondrial ROS concentration, or ATP production. 27 , 28 Time control (n=4): Mitochondria were added to 0.5 mL oxygenated buffer. After 8 minutes, O2‐saturated respiration buffer (0.5 mL) was added for 2 minutes (time control 10 minutes) and substrates for complex I were provided. After all, mitochondria were used to assess complex IV respiration and maximal uncoupled oxygen uptake. CDNF (n=4): Mitochondrial proteins were added to 0.5 mL hypoxic buffer supplemented with 1 μmol/L CDNF. After 8 minutes of hypoxia, O2‐saturated incubation buffer (0.5 mL), again supplemented with 1 μmol/L CDNF, was added to achieve reoxygenation for 2 minutes. After simulated hypoxia/reoxygenation substrates for complex I. Finally, mitochondria were used to either measure complex IV respiration and maximal uncoupled oxygen uptake.
Mitochondrial ATP Production
After the measurement of ADP‐stimulated respiration, the respiration buffer with mitochondria was removed from the respiration chamber and instantly supplemented with the ATP assay mix (diluted 1:5). Mitochondrial ATP production after each respiration measurement was verified immediately and compared with ATP standards using a 96‐well white plate and a spectrofluorometer (SpectraMax® M3; Molecular Devices, EUA) at 560 nm emission wavelength. 27 , 29
Mitochondrial Swelling and Transmembrane Potential
The mitochondrial swelling and transmembrane potential were assessed using a spectrofluorometer (SpectraMax® M3; Molecular Devices, EUA). The mitochondrial swelling was assessed using a spectrophotometric at 540 nm of absorption. A mitochondrial suspension (100 μg/mL) was added to the respiration buffer in the absence of respiratory substrates, at 37C and under continuous stirring. The mitochondrial swelling was induced with 100 nmol/L calcium. The swelling was expressed as a percentage of the absorption of the solution with mitochondria in the presence of cyclosporin A (0% of mitochondrial swelling), in relation to that absorbed after the supplement with carbonyl cyanide‐p‐trifluoromethoxyphenyl‐hydrazone (100% of mitochondrial swelling). For mitochondrial transmembrane potential evaluation the probe tetramethylrhodamine methyl ester (400 nmol/L) was added to the respiration buffer and stimulated at 580 nm. The mitochondrial transmembrane potential was expressed as the percentage of fluorescence produced by tetramethylrhodamine methyl ester ‐labeled mitochondria with cyclosporin A (0% of mitochondrial despolarization), relative to that emitted after the addition of carbonyl cyanide‐p‐trifluoromethoxyphenyl‐hydrazone (100% of mitochondrial despolarization). 27
Extramitochondrial ROS Concentration
The Amplex Red Hydrogen Peroxide Assay Kit (Life Technologies, Carlsbad, CA, USA) was applied to assess the extramitochondrial ROS concentration. The respiration buffer with mitochondria was removed from the respiration chamber and supplemented with 50 µmol/L Amplex UltraRed and 2 U/mL of horseradish peroxidase. The supernatant was collected after 120 minutes incubation in the dark room. Extramitochondrial ROS concentration was verified and compared with H2O2 standards using a 96‐well black plate and a spectrofluorometer (SpectraMax® M3; Molecular Devices, EUA) at 540 nm emission and 580 nm extinction wavelengths. 27
Statistical Analysis
Data are presented as the mean±standard error of the mean (SEM). Data were analyzed by Prism 8.0 software (GraphPad, San Diego, CA, USA) using Student t test (when appropriate) or using 2‐way ANOVA for repeated measures (LVEDP or LVDP) and 1‐way ANOVA (infarct size, Western blot, mitochondrial functions). When a significant difference was detected, 2‐way or 1‐way ANOVA was followed by Bonferroni post hoc tests. P<0.05 was considered statistically significant.
Results
TG Induces ER Stress and Increases CDNF Secretion in Mouse Neonatal Ventricular Cardiomyocytes and in hiPSC‐dCM
Human iPSC‐dCM cells were produced as described previously 20 and their differentiation was confirmed by expression of troponin T. Approximately 61% of troponin T‐positive cells were observed in our differentiation protocol. As seen in Figure 1, in the absence of TG, CDNF was found mainly inside the cells and very little of this neurotrophic factor appeared in the cell media either in mouse cardiomyocytes (controls in Figure 1A) or in hiPSC‐dCM (controls in Figure 1B). However, after 20 hours in the presence of TG, considerable amounts were detected in the cell media whereas the amount of CDNF detected in the cell lysates not changed (Figure 1A and 1B, TG). As seen in Figure 1C, the treatment of hiPSC‐dCM cells with TG significantly increased the expression of GRP78, a hallmark of ER‐stress, to 1.7‐fold when compared with controls, confirming that these cells were under ER/SR stress condition. Interestingly, when the cells were pretreated with exogenous CDNF (exoCDNF) for 15 minutes before TG addition, there was no increase in GRP78 (Figure 1C), suggesting that exoCDNF is preventing TG‐induced ER/SR stress. Figure 1E shows that isolated hearts subjected to I/R had their levels of CDNF increased to 27‐fold the control value, and this was accompanied by an ≈1.7‐fold increase in CHOP, another ER/SR stress marker, suggesting that these hearts were under ER/SR stress. ExoCDNF added before I/R led to a significant decrease in CHOP levels to ≈50% (Figure 1D). Confocal images of mouse cardiomyocytes treated with TG (Figure 1F) showed an increase in CDNF (quantified in f1) and GRP78 (quantified in f2) confirming that these cells were under ER/SR stress. It has to be mentioned that the addition of TG at this concentration causes death in fewer than 15% of mouse cardiomyocytes (data not shown).
Figure 1. Thapsigargin (TG) treatment increases the levels of CDNF in human and mouse cardiomyocytes as well as its secretion to the extracellular media.
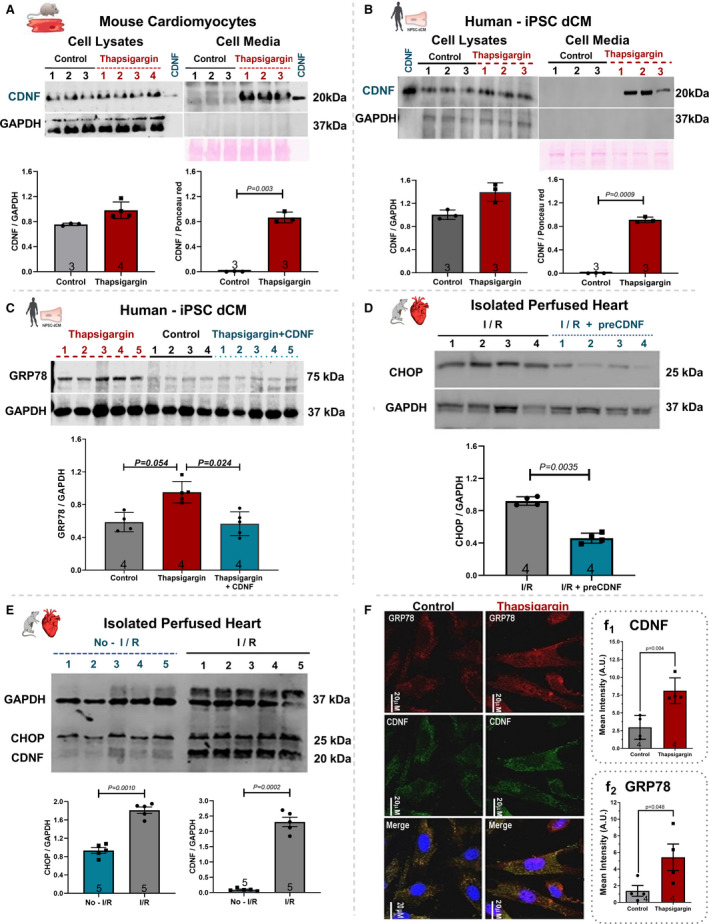
A, Mouse cardiomyocyte or (B) Human iPSC‐dCM were treated for 20 hours with TG (1 µmol/L) and the CDNF contents in cell lysates and cell media were evaluated by Western blot analyses. C, Levels of GRP78 in cell extracts of hiPSC‐dCM cultures treated with TG (1 µmol/L) or with CDNF (1 µmol/L) before TG. D, Levels of CHOP from hearts subjected to I/R or I/R after CDNF treatment (1 µmol/L/5 minutes). E, Levels of CHOP and CDNF from rat hearts subjected to I/R protocol in relation to no‐I/R condition. F, Levels of CDNF and GRP78 in mouse cardiomyocytes before and after TG treatment, measured by confocal imaging. Scale bar represents 20 µm (f1 and f2) immunocytochemistry quantification of CDNF and GRP78, respectively. The levels of CDNF, GRP78, and CHOP were normalized to GAPDH levels in cell lysate and to Ponceau red in extracellular media. Recombinant CDNF was used as control. Each lane represents a different cell culture. The protein expression values are expressed as mean±SEM with 1‐way ANOVA followed by Bonferroni post hoc test. CDNF indicates cerebral dopamine neurotrophic factor; CHOP, CCAAT/‐enhancer‐binding protein homologous protein; iPSC‐dCM, iPSC‐dCM, induced pluripotent stem cells differentiated into cardiomyocytes; GRP78, 78 kDa glucose‐regulated protein; and I/R, ischemia/reperfusion.
Taken together these data suggest that TG and I/R, 2 ER/SR stressors, increase the concentration of CDNF, GRP78, and CHOP in hiPSC‐dCM and neonatal mouse cardiomyocytes as well as in the isolated intact heart. Pretreatment of the cells or the hearts with exoCDNF blocked the increase in the 2 markers of unfolding protein response, GRP78 and CHOP, suggesting that CDNF is an anti‐ER/SR stressor in the cardiac context. Interestingly, TG treatment, in addition to increasing the cellular levels of CDNF in mouse or human cardiomyocytes, also increases its secretion to the extracellular milieu.
ExoCDNF Prevents the Ca2+‐ Transient Depletion Induced by TG
As seen in Figure 2, TG treatment reduced calcium transients of hiPSC‐dCM (reduced spike amplitudes in Figure 2A, quantified in Figure 2B). However, preincubation of these cells with CDNF upon TG addition potentiated the calcium transients to values even greater than the control, suggesting that exoCDNF exerts a direct effect on cardiomyocyte calcium homeostasis under stress conditions (Figure 2). Pretreatment with wortmannin abrogates the effects of CDNF on calcium transients after TG addition (to be described later).
Figure 2. ExoCDNF restores the calcium transient in hiPSC‐dCM perturbed by TG treatment, and wortmannin abrogates this beneficial effect of CDNF.
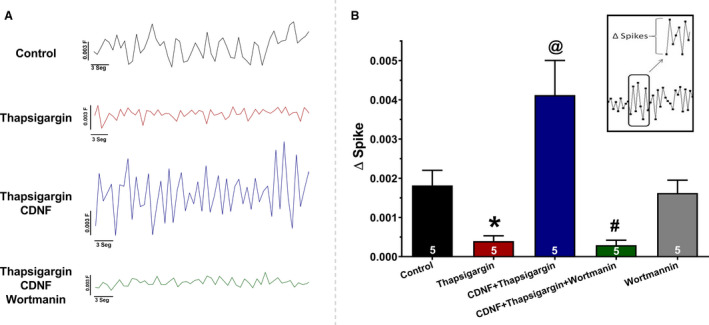
Groups: Control, TG, TG+CDNF, and TG+CDNF+wortmannin (representative traces in A). B, ΔSpike (average of 20 peaks for each experiment) represents the difference between the calcium level in a specific top peak in relation to the previous lower peak (see the inset). The values are expressed as means±SEM with 1‐way ANOVA followed by Bonferroni post hoc test. Each experiment represents the number of hiPSC‐dCM cultures tested. *P<0.01 vs control; @ P<0.01 vs TG and # P<0.01 vs CDNF+TG. CDNF indicates cerebral dopamine neurotrophic factor; hiPSC‐dCM, iPSC‐dCM, human induced pluripotent stem cells differentiated into cardiomyocytes; and TG, thapsigargin.
Cardioprotective Effects of exoCDNF on Isolated Hearts
Initially, we evaluated whether rat heart chambers express endogenous CDNF in significant amounts. As shown in Figure S2 this does occur, with the ventricles expressing greater amounts of CDNF per unit of total protein than the atria.
Next, the cardioprotective activity of CDNF was evaluated by perfusion with exoCDNF as a preconditioning treatment (Figure 3A and 3B, preCDNF), and as a postconditioning treatment (Figure 3C and 3D, postCDNF). Because there was no significant difference in the baseline of the LVDP among the groups (Table S1), all LVDP changes are expressed as a percentage of the baseline values, which were taken as 100%. Under ischemic condition all control hearts exhibited a rapid reduction of LVDP down to zero, recovering poorly (to ≈20%) during reperfusion (Figure 3A and 3C, circles). Interestingly, preCDNF and postCDNF treatments enhanced these recoveries considerably, up to 65% and 53%, respectively (Figure 3A and 3C, squares). Regarding LVEDP (Figure 3B and 3D), preCDNF kept the pressure low during reperfusion (40 mm Hg, squares) in relation to the control, which rose to ≈70 mm Hg at the end of the reperfusion process (circles). However, postCDNF treatment induced no significant alteration in LVEDP compared with controls (Figure 3D).
Figure 3. ExoCDNF protects isolated hearts from injuries provoked by I/R and the protection is prevented by wortmannin, a PI3K/AKT antagonist.
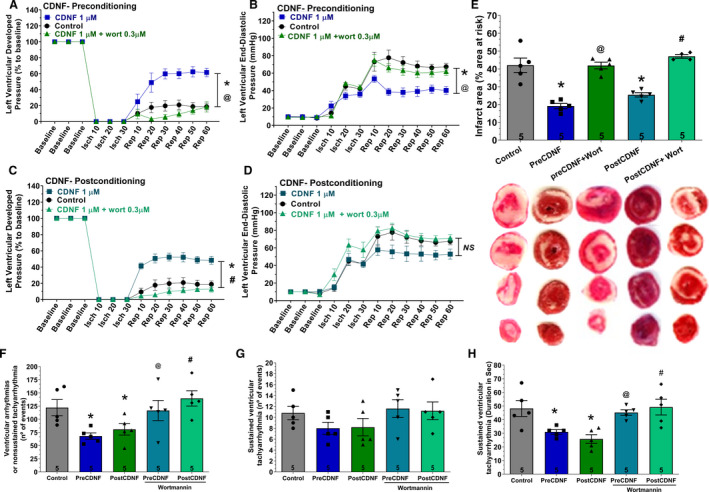
Time courses of left ventricular developed pressure (LVDP) (A and C) and left ventricular end‐diastolic pressure (LVEDP) (B and D) during I/R protocol. In A and B, CDNF (1 µmol/L) was perfused 5 minutes before ischemia (preconditioning) and in C and D, during the first 5 minutes of reperfusion (postconditioning). Wortmannin (0.3 µmol/L) was perfused 5 minutes before CDNF. E, Infarct area of hearts. Representative cross‐section images of TCC‐stained ventricle hearts subjected to I/R. F, The number of ventricular arrhythmias or nonsustained tachyarrhythmia events. G, The number of events and (H) the duration of sustained ventricular tachyarrhythmia produced during reperfusion. Values are expressed as means±SEM. Number in each column is n of hearts. NS, not significant; *P<0.01 vs control; @ P<0.01 vs preCDNF and # P<0.01 vs postCDNF with 1‐way ANOVA followed by Bonferroni post hoc tests for infarct area and arrhythmias analysis and 2‐way ANOVA followed by Bonferroni post hoc tests for LVDP and LVEDP analysis. AKT indicates protein kinase B; CDNF, cerebral dopamine neurotrophic factor; I/R, ischemia/reperfusion; and TCC, triphenyltetrazolium chloride.
Myocardial infarct areas evaluated after 60 minutes of reperfusion were measured in control, preCDNF, and postCDNF hearts (Figure 3E). As seen, in the control hearts the mean infarct area reached ≈42%, whereas the hearts subjected to preCDNF or postCDNF treatments showed a substantial reduction in the infarct area to ≈19% and ≈25%, respectively.
As shown in Figure 3F through 3H), there was a significant reduction in the mean number of ventricular arrhythmias or nonsustained tachyarrhythmia events (Figure 3F) produced during the 60 minutes of reperfusion in preCDNF and postCDNF protocols. The number of sustained ventricular tachyarrhythmias was not significantly reduced by preCDNF or postCDNF treatments (Figure 3G), although the duration of these tachyarrhythmias was reduced from ≈48 seconds to 33 seconds and 25 seconds in preCDNF and postCDNF, respectively (Figure 3H). The data with wortmannin are presented next.
PI3K/AKT Pathway is Mediating the Cardioprotective Effects of exoCDNF
Next, we examined the signaling pathway involved in the cardioprotective effect conferred by exoCDNF. Surprisingly, only wortmannin was able to abrogate the cardioprotective effect of exoCDNF treatment (Figure 3, triangles) whereas the other inhibitors, AG490 (JAK‐STAT3 inhibitor), rottlerin, and chelerythrine (PKC inhibitors) failed, as displayed in Figure S3. As seen in Figure 3, the massive recovery in LVDP (Figure 3A) or the maintenance of the low values of LEVDP (Figure 3B) observed with preCDNF treatment before I/R injury (squares) were completely abolished in the presence of wortmannin (triangles) and values were similar to those of the control groups (circles). Because only wortmannin abrogated CDNF‐induced cardioprotection in preCDNF condition, only this inhibitor was evaluated in postCDNF treatment, and again wortmannin abrogated LVDP recovery (Figure 3B). The pretreatment with wortmannin led to an increase of infarct area in preCDNF and postCDNF to ≈40% and ≈45%, respectively, similar to the control (Figure 3E). It has to be mentioned that wortmannin alone does not exert any effect on infarct area as previously documented. 30 The lack of effect of the other inhibitors on the infarct area is depicted in Figure S3. In addition, treatment with wortmannin in preCDNF and postCDNF protocols abolished the reduction of ventricular arrhythmias in hearts subjected to I/R (Figure 3F and 3H). Besides, as shown in Figure 2, the addition of wortmannin also abrogates the protective effects of CDNF on calcium transients after TG addition.
The levels of p‐AKT were evaluated in hiPSC‐dCM, mouse cardiomyocytes, and isolated hearts before and after the addition of exoCDNF (Figure 4A through 4C, respectively). In all 3 models, the levels of p‐AKT more than doubled after CDNF addition, except when CDNF was added after incubation with wortmannin. Furthermore, CDNF did not increase the phosphorylated STAT3 or phosphorylated PKCƐ, as shown in Figure 4F. Taken together these data strongly suggest that the PI3K/AKT pathway is involved in the beneficial effects conferred by CDNF to cardiomyocytes and heart.
Figure 4. ExoCDNF treatment increases the level of phosphorylated AKT (p‐AKT) in rat isolated hearts and in mouse and human cardiomyocytes.
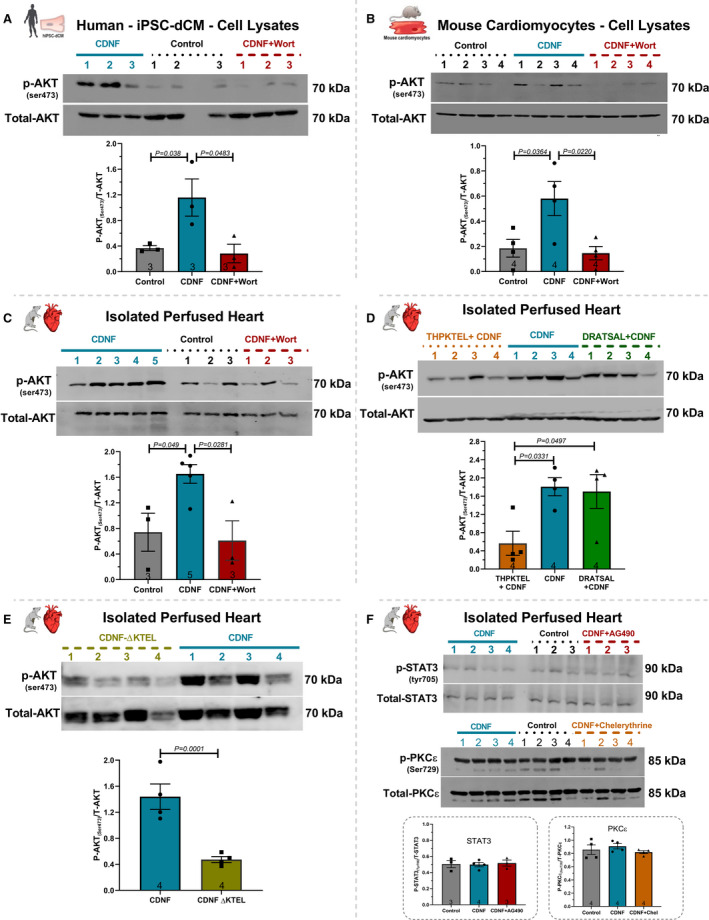
Cultures of (A) hiPSC‐dCM or (B) mouse cardiac myocytes were treated with CDNF (1 µmol/L/20 hours) or with wortmannin (0.3 µmol/L/15 minutes) before CDNF addition. In (C), rat isolated hearts were perfused with CDNF (1 µmol/L/5 minutes, preconditioning) before I/R either alone or in combination with wortmannin (0.3 µmol/L/5 minutes) before CDNF treatment. D, The peptide THPKTEL that binds to the KDEL‐R blocks CDNF‐induced PI3K/AKT activation. Isolated hearts were perfused with CDNF (1 µmol/L/5 minutes, preconditioning) before I/R either alone or in combination with THPKTEL or with the scrambled peptide DRATSAL (peptides added 5 minutes before CDNF treatment and during the 5 minutes CDNF treatment). E, CDNF‐ΔKTEL was unable to activate p‐AKT in isolated perfused hearts. Isolated hearts were perfused with CDNF‐ΔKTEL (1 µmol/L/5 minutes) before I/R. The intensities of the p‐AKT bands were normalized to total‐AKT levels. F, The STAT3 inhibitor (AG490, 10 µmol/L/5 minutes) and the PKC inhibitor (chelerythrine, 10 µmol/L/5 minutes) not abolished the protection generated by CDNF. Isolated hearts were perfused with CDNF (1 µmol/L/5 minutes, preconditioning) before I/R either alone or in combination with AG490, 10 µmol/L/5 minutes or chelerythrine, 10 µmol/L/5 minutes before CDNF treatment. The number of experiments is equal to the number of lanes. The data were expressed as means±SEM, with 1‐way ANOVA followed by Bonferroni post hoc tests. AKT indicates protein kinase B; CDNF, cerebral dopamine neurotrophic factor; and iPSC‐dCM, iPSC‐dCM, induced pluripotent stem cells differentiated into cardiomyocytes.
exoCDNF Exerts a Protective Effect on Mitochondrial Function After Heart I/R
The protection of mitochondrial function during I/R has been reported to be beneficial to cardiomyocytes. 3 In order to investigate whether CDNF could exert any effect on mitochondrial function and whether wortmannin could counteract these effects, mitochondria were isolated from hearts subjected to I/R in the Langendorff model with or without perfusion of CDNF (preCDNF or postCDNF) and CDNF+Wortmannin (Figure 5).
Figure 5. ExoCDNF reduces cardiac mitochondrial impairment induced by I/R and wortmannin abrogates CDNF beneficial effects.
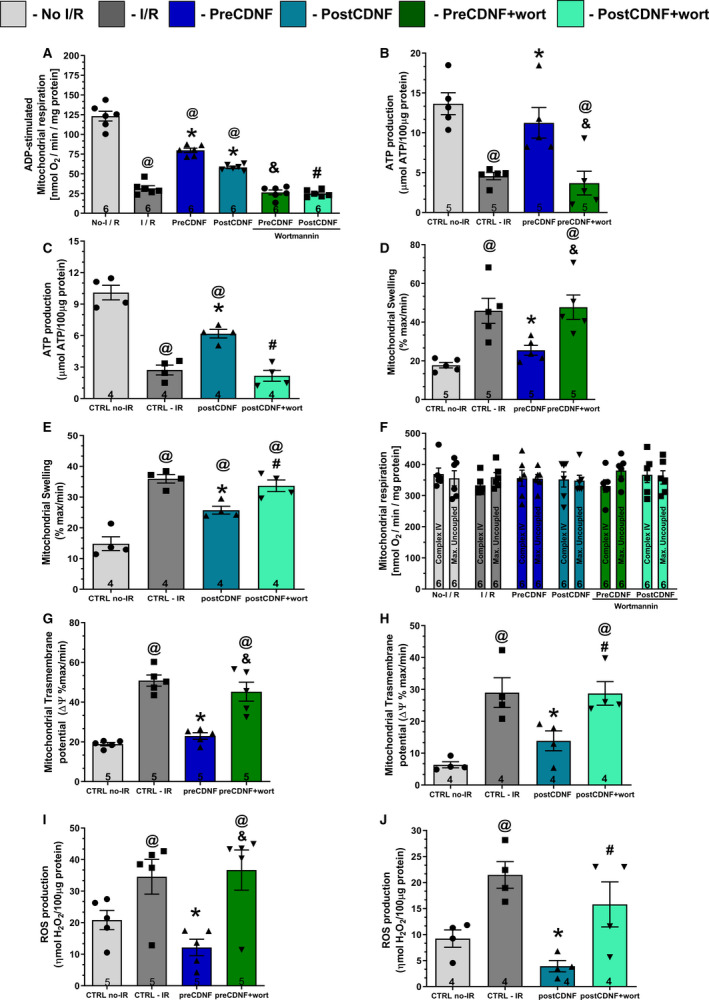
A, ADP‐stimulated complex I respiration; (B and C) ATP production; (D and E) Mitochondrial swelling; (F) Complex IV and maximal uncoupled oxygen respiration; (G and H) Mitochondrial transmembrane potential (ΔΨ); and (I and J) Reactive oxygen species (ROS) production. Groups: No‐I/R, I/R, preCDNF, postCDNF and preCDNF+Wortmannin, and postCDNF+Wortmannin. (Wortmannin 0.3 µmol/L). * P<0.05 vs I/R; @ P<0.05 vs No‐I/R; & P<0.05 vs preCDNF and # P<0.05 vs postCDNF. Number in each column is n of hearts. The data were expressed as means±SEM, with 1‐way ANOVA followed by Bonferroni post hoc tests. CDNF indicates cerebral dopamine neurotrophic factor; CTRL, control; and I/R, ischemia/reperfusion.
As expected, ADP‐stimulated complex I respiration was reduced in the I/R group and this decrease was restored partially, but significantly, by pre‐ or postCDNF treatments; but the pretreatment was more effective (Figure 5A). Wortmannin abrogated the improvement conferred by preCDNF and postCDNF. The same scenario was found for ATP production (Figure 5B and 5C) and mitochondrial swelling (Figure 5D and 5E), indicating that the I/R‐induced oxidative phosphorylation impairment and the increased mitochondrial volume were partially reversed by pre‐ or postCDNF treatments, acting through the PI3K/AKT pathway. Mitochondrial complex IV respiration and maximal oxygen uptake of uncoupled mitochondria were not different between groups, reflecting an equal loading of viable mitochondria (Figure 5F). Mitochondrial transmembrane potential showed an increase due to I/R, reflecting the inner membrane depolarization. PreCDNF and postCDNF treatments ameliorated this noxious mitochondrial transmembrane potential increase and wortmannin abolished the beneficial effects of CDNF (Figure 5G and 5H, respectively). Next, mitochondrial ROS production was measured and, as expected, I/R led to an enhancement in ROS levels, but pre‐ or postCDNF treatments reduced ROS only when wortmannin was absent (Figure 5I and 5J, respectively).
To test whether exoCDNF could exert direct mitochondrial protection independent of PI3K/AKT activation, mitochondria from nonischemic hearts were isolated and subjected to hypoxia/reoxygenation with or without previous incubation with CDNF. As shown in Figure S4A, the ADP‐stimulated complex I respiration was reduced in the hypoxia/reoxygenation group compared with the control group, but in this case CDNF was ineffective in blocking or reversing this effect, confirming that intact cells are necessary for the beneficial effects of CDNF. Mitochondrial complex IV respiration and maximal oxygen uptake of uncoupled mitochondria were not different between groups, reflecting an equal loading of viable mitochondria (Figure S4B).
KDEL‐R in the Membrane Binds CDNF and Mediates Its Cardioprotective Activity
We postulated that the KDEL‐R, which has been found in the cell membrane 25 , 31 , 32 and has on its operator sequence an ER stress‐responsive element, 33 , 34 might function as a CDNF receptor. To investigate this, we perfused hearts before CDNF treatment with 2 heptapeptides, which incorporate the last 7 amino‐acid residues of human (THPKTEL) and rat (TRPQTEL) CDNF. The rationale was to block CDNF's putative binding site on the KDEL‐R, thus impairing the CDNF function. As seen in Figure 6, (TRPQTEL, Figure 6A, 6B and 6E; TKPKTEL, Figure 6C, 6D and 6F), both peptides were able to prevent the cardioprotection induced by preCDNF treatment, abolishing the beneficial recovery of LVDP (Figure 6A and 6C, triangles), the beneficial reduction of LVEDP (Figure 6B and 6D, triangles), and the decrease in infarct areas of the hearts (Figure 6E and 6F) induced by CDNF. As seen in these panels, perfusion with the peptides alone was not able to confer cardioprotection (diamonds in all panels), suggesting that the occupancy of the binding site on the KDEL‐R by these short ligands is not enough for full activation of the downstream pathway and cardioprotection.
Figure 6. The cardioprotective effect of exoCDNF is blocked by heptapeptides that bind to KDEL‐R.
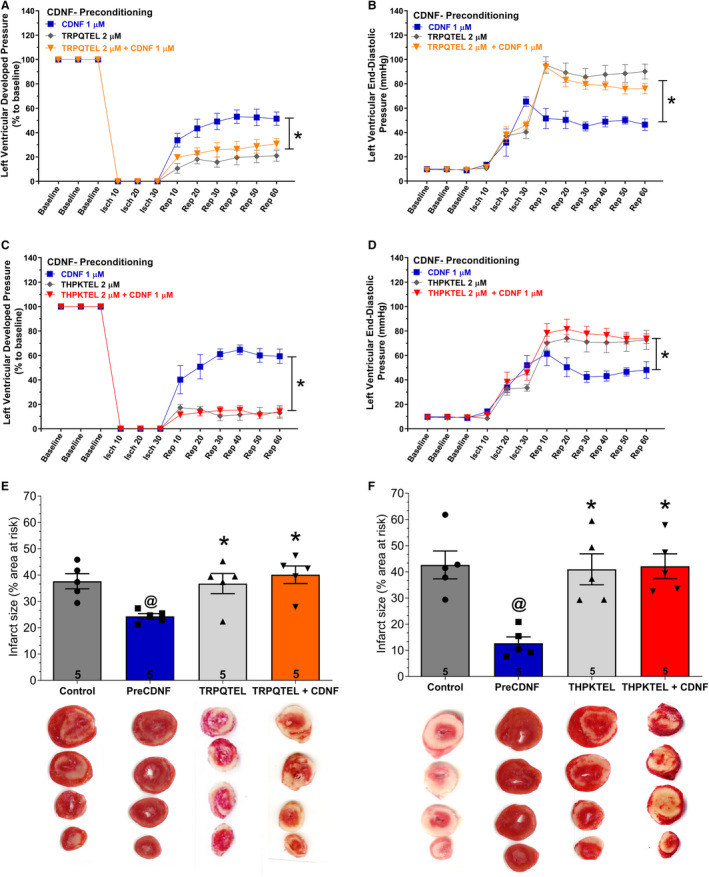
Time courses of left ventricular developed pressure (LVDP) (A and C) and left ventricular end‐diastolic pressure (LVEDP) (B and D) during I/R protocol. CDNF (1 µmol/L), peptide (in [A, B, and E] TRPQTEL and in [C, D and F], THPKTEL L; 2 µmol/L of each peptide) or CDNF (1 µmol/L)+peptide (2 µmol/L) were perfused before ischemia (5 minutes). E and F, Infarct area of hearts. Representative cross‐section images of TCC‐stained ventricle hearts subjected to I/R in the absence or in the presence of CDNF and peptides. Number in each column is n of hearts. The data were expressed as means±SEM. @ P<0.01 vs control; *P<0.01 vs preCDNF. With 1‐way ANOVA followed by Bonferroni post hoc tests for infarct area analysis and 2‐way ANOVA followed by Bonferroni post hoc tests for LVDP and LVEDP analysis. CDNF indicates cerebral dopamine neurotrophic factor; isch, ischemia; rep, reperfusion; and TCC, triphenyltetrazolium chloride.
Figure S5 shows that the peptide DRATSAL, used as a scrambled peptide by Henderson and collaborators, 25 was not able to abolish the protective effect of CDNF in I/R experiments. Interestingly, THPKTEL was able to abolish CDNF‐induced p‐AKT enhancement, an effect that was not observed with the scrambled peptide (Figure 4D). This result reinforces the link between CDNF binding to the KDEL‐R and the activation of the PI3K/AKT signaling pathway.
Figure 7 shows that the preincubation of CDNF with the antibody anti‐CDNF completely abrogates the cardioprotective role of CDNF (triangles in Figure 7A, LVDP; Figure 7B, LVEDP, and Figure 7E, infarct area).
Figure 7. The cardioprotective effect of preconditioning with exoCDNF is blocked by an anti‐CDNF antibody or by CDNF‐ΔKTEL.
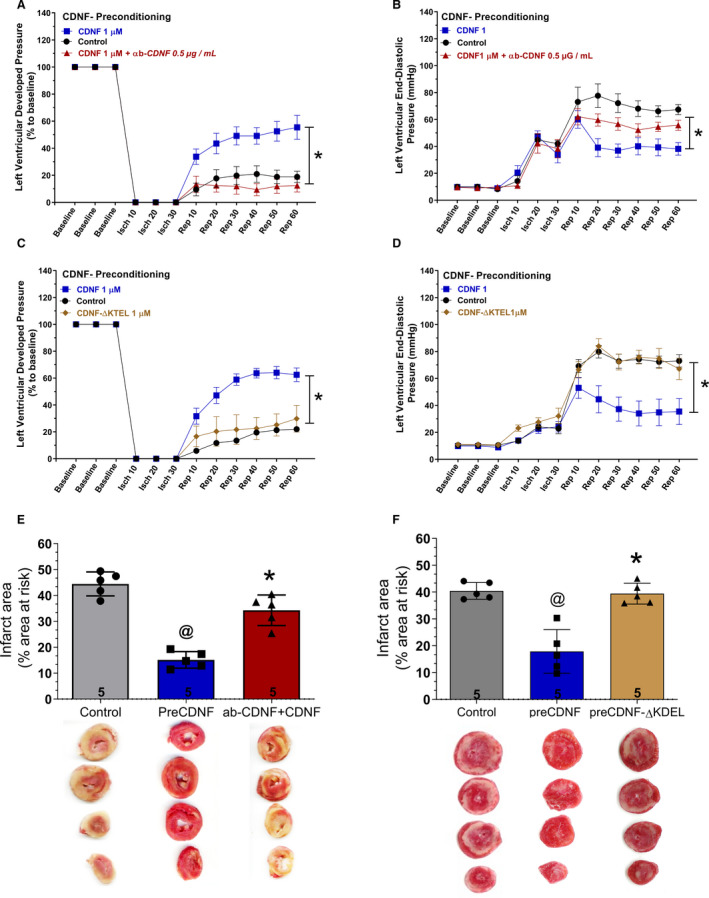
CDNF (1 µmol/L) or CDNF+antibody were perfused before ischemia (5 minutes) (A, B, and E). CDNF‐ΔKTEL was unable to confer cardioprotection to isolated perfused hearts subjected to I/R. Isolated perfused hearts were perfused with CDNF‐ΔKTEL (1 µmol/L/5 minutes) before I/R (C, D, and F). Time courses of (A and C) left ventricular developed pressure (LVDP) and (B and D) left ventricular end‐diastolic pressure (LVEDP) during I/R protocol. Representative cross‐section images of TCC‐stained ventricles hearts subjected to I/R in the absence or in the presence of CDNF, CDNF+anti‐CDNF antibody (E), or CDNF‐ΔKTEL (F). Number in each column is n of hearts. The data were expressed as means±SEM. @ P<0.001 vs control; *P<0.001 vs preCDNF. With 1‐way ANOVA followed by Bonferroni post hoc tests for infarct area analysis and 2‐way ANOVA followed by Bonferroni post hoc tests for LVDP and LVEDP analysis. CDNF indicates cerebral dopamine neurotrophic factor; isch, ischemia; rep, reperfusion; and TCC, triphenyltetrazolium chloride.
Finally, we constructed a CDNF‐ΔKTEL version of the protein in which the last 4 residues were deleted. This construct presented the expected molecular mass and secondary structure of the intact CDNF as evidenced by mass spectrometry (see Methods) and circular dichroism (Figure S1). As seen in Figure 7C, 7D, and 7F, CDNF‐ΔKTEL was ineffective in conferring cardioprotection under preconditioning either the LVDP (Figure 7C, diamonds), LVEDP (Figure 7D, diamonds), or infarct area (Figure 7F). This construct was also unable to activate p‐AKT in isolated perfused hearts (Figure 4E) reinforcing the importance of the KTEL sequence for exogenous CDNF binding to its receptor, probably the KDEL‐R that translocated to the plasma membrane under stress condition. The full Western blots photos are presented in Figure S6.
CDNF, But Not CDNF‐ΔKTEL, is Cardioprotective in Rats Subjected Myocardial Infarction
Because we have strong evidence of the protective action of CDNF in cardiomyocytes in culture or in intact isolated rat hearts subjected to ER/SR stress conditions (TG or I/R), CDNF or CDNF‐ΔKTEL were administered intraperitoneally to rats after ischemia and before reperfusion and the infarct area was evaluated (Figure 8). As seen, whereas the animals subjected to I/R had ≈40% of the heart area infarcted, CDNF reduced the damage significantly to 25%, and the CDNF‐ΔKTEL exerted no effect.
Figure 8. CDNF, but not CDNF‐ΔKTEL, protect in vivo the hearts of rats subjected to I/R.
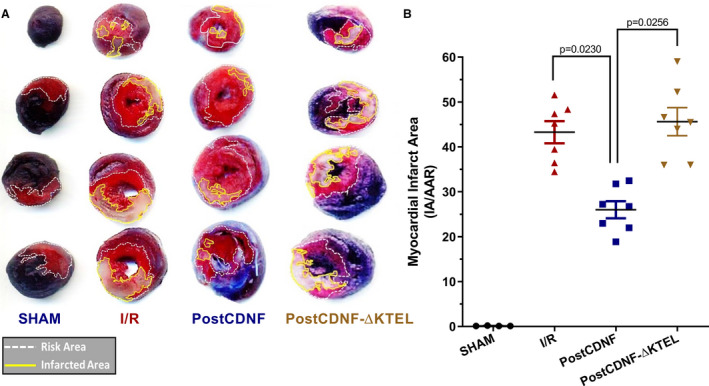
CDNF or CDNF‐ΔKTEL were administered intraperitoneally (IP) after ischemia and before reperfusion and the infarct areas were evaluated. A, Representative images of the heart sections where the red regions represent the area at‐risk areas (TTC stained), the white regions represent the infarct areas, and the blue, nonaffected regions (Evans‐blue). B, Quantitative analysis of hearts showed a significant reduction (P<0.01, vs I/R) in the percentage of white/white+red areas of the hearts. The data were expressed as means±SEM of 7 hearts. With 1‐way ANOVA followed by Bonferroni post hoc tests. CDNF indicates cerebral dopamine neurotrophic factor; IA/AAR, infarct area/area at risk; I/R, ischemia/reperfusion; and TTC, triphenyltetrazolium chloride.
Discussion
Cardiomyokines are proteins secreted by a healthy or a diseased heart and they display a beneficial autocrine/paracrine function. 2 , 23 , 35 Here we present evidence for CDNF as a new cardiomyokine with possible use in the clinics. Our data showed that heart hemodynamic function was significantly preserved upon CDNF treatment under pre‐ or postconditioning regimens after I/R. We also showed that ventricular arrhythmias were significantly reduced by CDNF, as was the infarct area (Figure 3). Interestingly, all these CDNF cardioprotective effects were abolished when its interaction with the KDEL‐R was blocked, either with competing for KTEL containing heptapeptides, with anti‐CDNF antibody or with a CDNF construct in which the last 4 amino acids were deleted (Figures 6 and 7). We thus propose the KDEL‐R as a putative receptor for this neuro/cardiokine protein, and PI3K/AKT as the signaling pathway (Figures 2, 3, 4) that is activated upon receptor binding in the heart.
The KDEL receptor is a 7‐transmembrane‐domain protein whose function is to sort and retrieve proteins bearing a KDEL sequence (calnexin, GRP78, PDI, for instance) and possibly proteins with KDEL‐like sequences (Erp72, MANF, and CDNF) from the Golgi complex to the ER/SR. 31 , 36 , 37 The importance of KDEL‐R to cardiomyocyte homeostasis was demonstrated by studying transgenic mice that express a mutant KDEL‐R in which reverse transport from Golgi to ER/SR was compromised. These animals died sporadically after attaining the age of more than 14 weeks with cardiac hypertrophy. 38 Recent studies, however, have suggested that the KDEL‐R has additional functions and new cellular localizations, including the plasma membrane. In this regard, there is mounting evidence for the presence of KDEL‐R at the surface of mammalian cells, where the receptor binds cargo proteins such as A/B microbial toxins K28, MANF, and other proteins in which a KDEL‐like sequence is present at the C‐terminus. 25 , 31 , 39 , 40 Trychta and collaborators 40 have coined the term “ER exodosis” in an elegant study where they showed a massive departure of proteins containing KDEL‐related sequences upon calcium‐induced ER‐stress in several mammalian cell lines. Even beneficial chaperones were among the proteins that were released.
The importance of the KDEL‐R for MANF trafficking has been investigated in SH‐SY5Y cells by using cells expressing different MANF constructs (with or without the KDEL‐like RTDL sequence) in combination with different isoforms of the KDEL‐R (KDEL‐R1, 2 and 3). 25 Interestingly, that study showed consistently that removal of the C‐terminal RTDL sequence of MANF increased its secretion, reinforcing the idea that the KDEL‐R is an important partner of MANF in ER. In addition, the study conducted by Henderson and collaborators 25 detected the presence of FLAG‐tagged KDEL‐R at the cell surface under ER‐stress, and MANF binding to the cell surface required the RTDL sequence. This study unequivocally showed that KDEL‐R located at the cell membrane modulates the binding of extracellular MANF, leading to the conclusion, as posed here, that the KDEL‐R is the putative receptor for this family of neutrophil factors. However, that study did not relate MANF‐induced cardioprotection with its binding to cell membrane KDEL‐R. Here we have shown that CDNF cardioprotection depends on the KTEL sequence and its ability to bind the KDEL‐R.
Our data show that CDNF binding to KDEL‐R activates PI3K/AKT signaling. We do not know what is(are) the connector(s) between CDNF/KDEL‐R and PI3K/AKT signaling, but we do know that the increase in p‐AKT that takes place in hearts upon CDNF treatment is abrogated by THPKTEL, a peptide that binds to the KDEL‐R at the cell membrane, but not by DRATSAL, a scrambled, ineffective peptide, as well as CDNF construct in which the last 4 amino acids were deleted (CDNF‐ΔKTEL) not induced an increase in p‐AKT (Figure 4 and Figure S5). The connection between the KDEL receptor and AKT phosphorylation can be suggested by previous work, in which KDEL receptor would function as a G protein‐coupled receptor related to Ras/Raf signaling, 15 because Ras has been shown to bind and activate the p110a catalytic subunit of PI3K 41 , 42 , 43 , 44 with consequent AKT activation. Moreover, there are several reports in the literature showing signaling pathways controlled by KDEL‐R, mainly in cancer or neuronal cells. Interestingly, GRP78, which is present at the cell membrane in several cancer cells, colocalizes with both subunits of PI3K, namely p85 and p110, as well as with phosphatidylinositol (3,4,5)‐trisphosphate controlling PI3K/AKT pathway either directly or indirectly. 45
Cardiac ischemia induces calcium transient depletion resulting in dysfunction of the contractile machinery and cell injury. 3 Here, we have shown that CDNF prevents ER/SR stress, by restoring calcium transient in cardiomyocytes (Figure 2). This effect would reduce calcium overload and prevent mitochondrial lesions and cell death. 3 , 29 Although immediate restoration of blood flow (reperfusion) is essential for myocardial survival, paradoxically, reperfusion itself can induce injury mediated by ER/SR stress, cytosolic calcium overload, and mitochondrial impairment. 3 Thus, we showed the postCDNF perfusion reducing the reperfusion lesions, by mitochondrial maintenance and reduction of the infarct area. Moreover, CDNF was able to prevent myocardial injury in the postischemic treatment in vivo model (Figure 8). As reported here, the inhibitor of the PI3K/AKT pathway abolished the protection conferred by pre‐ and postCDNF leading to an increase in the infarct area. The AKT, as part of the RISK (Reperfusion Injury Salvage Kinase) pathway, is an excellent target for preventing lethal reperfusion injury. There is a consensus that the RISK pathway mediates the programmed cell survival, as well as extensive preclinical evidence that activation of this pathway by pharmacological agents reduces the size of myocardial infarction by up to 50%. 46 , 47 Cardioprotection by the RISK pathway has been attributed to the activation of PI3K/AKT signaling pathway inducing inhibition of mitochondrial permeability transition pore opening, SR Ca2+ uptake improvement, and antiapoptotic pathway activation. 3 , 47 These kinases are activated during myocardial reperfusion and confer cardioprotection, avoiding lethal reperfusion injury. 46 AKT inhibitor (wortmannin) was the only tested inhibitor that was able to abolish all the beneficial effects of CDNF, suggesting that STAT3 and PKC do not take place in the observed cardioprotection. Other cardiomyokines can also induce the activation of PI3K/AKT signaling, and now CDNF can be included in this list. 48 , 49 , 50 We thus propose that exogenous CDNF has the potential to prevent revascularization lesions and should be further tested as a potential enhancer of the RISK pathway. It is reasonable to assume that the endogenous, low concentration of CDNF (pico‐nano molar range) is not sufficient to exert cardioprotection under stress conditions.
In conclusion, the present study characterizes CDNF as new cardiomyokine acting through its putative receptor KDEL and activating PI3K/AKT to exert its cardioprotective activity. All these findings are summarized as a model in Figure 9.
Figure 9. Schematic representation of the main findings of the present study.
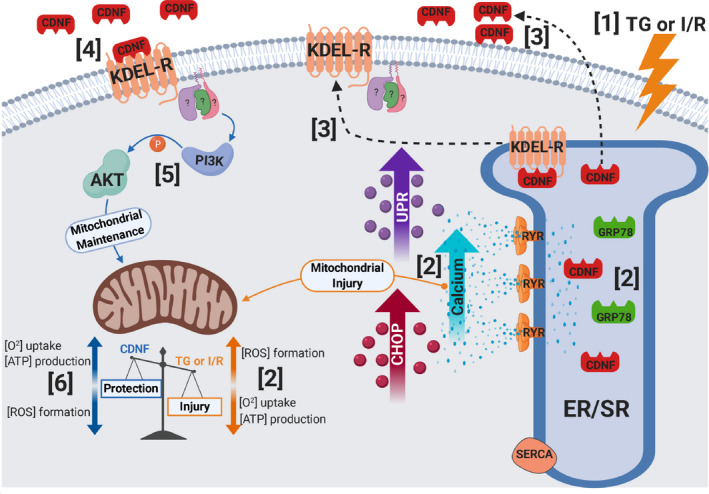
(1) TG or I/R induce cellular injuries causing (2) an increase in cytosolic calcium and UPR activation, upregulation of GRP78, CDNF, and CHOP, as well as mitochondria impairment (decrease in [O2] and [ATP] and an increase in [ROS]). (3) CDNF is an ER/SR‐resident protein remaining bound to KDEL‐R through its degenerated KTEL sequence located at the C‐terminal end. Under stress, KDEL‐R and CDNF dissociate and migrate from the ER/SR to the cell membrane and extracellular milieu, respectively, (4) allowing the binding of CDNF to the KDEL‐R at the cell surface with the subsequent (5) activation of PI3K/AKT signaling pathway either directly or indirectly conferring (6) mitochondrial maintenance and cell survival (cardioprotection). CDNF indicates cerebral dopamine neurotrophic factor; CHOP, CCAAT/‐enhancer‐binding protein homologous protein; ER/SR, endoplasmic reticulum/sarcoplasmic reticulum; I/R, ischemia/reperfusion; ROS, reactive oxygen species; RYR, ryanodine receptor; SERCA, sarco/endoplasmic reticulum Ca2+‐ATPase; TG, thapsigargin; and UPR, unfolding protein response.
Sources of Funding
This work was supported by Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ) (E‐26/010.003018/2014 and E‐26/202.947/2017); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) (fellowships to the students); and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) (402967/2016‐0).
Disclosures
None.
Supporting information
Table S1
Figures S1–S6
Acknowledgments
We are grateful to Martha M. Sorenson for her suggestions and careful reading of the manuscript and Dr Diana Pellizari, Santiago Alonso, and Elisabeth M. Duarte for laboratory technical assistance.
(J Am Heart Assoc. 2021;10:e019685. DOI: 10.1161/JAHA.120.019685.)
Preprint posted on BioRxiv June 27, 2019. doi: https://doi.org/10.1101/683110.
For Sources of Funding and Disclosures, see page 20.
References
- 1. Heusch G. Myocardial ischemia: lack of coronary blood flow, myocardial oxygen supply‐demand imbalance, or what? Am J Physiol Heart Circ Physiol. 2019;316:H1439–H1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Glembotski CC. Functions for the cardiomyokine, MANF, in cardioprotection, hypertrophy and heart failure. J Mol Cell Cardiol. 2011;51:512–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Heusch G. Molecular basis of cardioprotection: signal transduction in ischemic pre‐, post, and remote conditioning. Circ Res. 2015;116:674–699. [DOI] [PubMed] [Google Scholar]
- 4. Lindahl M, Saarma M, Lindholm P. Unconventional neurotrophic factors CDNF and MANF: structure, physiological functions and therapeutic potential. Neurobiol Dis. 2017;97:90–102. [DOI] [PubMed] [Google Scholar]
- 5. Liu H, Tang X, Gong L. Mesencephalic astrocyte‐derived neurotrophic factor and cerebral dopamine neurotrophic factor: new endoplasmic reticulum stress response proteins. Eur J Pharmacol. 2015;750:118–122. [DOI] [PubMed] [Google Scholar]
- 6. Apostolou A, Shen Y, Liang Y, Luo J, Fang S. Armet, a UPR‐upregulated protein, inhibits cell proliferation and ER stress‐induced cell death. Exp Cell Res. 2008;314:2454–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Glembotski CC, Thuerauf DJ, Huang C, Vekich JA, Gottlieb RA, Doroudgar S. Mesencephalic astrocyte‐derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion. J Biol Chem. 2012;287:25893–25904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hartley CL, Edwards S, Mullan L, Bell PA, Fresquet M, Boot‐Handford RP, Briggs MD. Armet/Manf and Creld2 are components of a specialized ER stress response provoked by inappropriate formation of disulphide bonds: implications for genetic skeletal diseases. Hum Mol Genet. 2013;22:5262–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tadimalla A, Belmont PJ, Thuerauf DJ, Glassy MS, Martindale JJ, Gude N, Sussman MA, Glembotski CC. Mesencephalic astrocyte‐derived neurotrophic factor is an ischemia‐inducible secreted endoplasmic reticulum stress response protein in the heart. Circ Res. 2008;103:1249–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol. 2012;13:89–102. [DOI] [PubMed] [Google Scholar]
- 11. Lee AH, Iwakoshi NN, Glimcher LH. XBP‐1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oh‐Hashi K, Tanaka K, Koga H, Hirata Y, Kiuchi K. Intracellular trafficking and secretion of mouse mesencephalic astrocyte‐derived neurotrophic factor. Mol Cell Biochem. 2012;363:35–41. [DOI] [PubMed] [Google Scholar]
- 13. Sun ZP, Gong L, Huang SH, Geng Z, Cheng L, Chen ZY. Intracellular trafficking and secretion of cerebral dopamine neurotrophic factor in neurosecretory cells. J Neurochem. 2011;117:121–132. [DOI] [PubMed] [Google Scholar]
- 14. Capitani M, Sallese M. The KDEL receptor: new functions for. an old protein. FEBS Lett. 2009;583:3863–3871. [DOI] [PubMed] [Google Scholar]
- 15. Giannotta M, Fragassi G, Tamburro A, Vanessa C, Luini A, Sallese M. Prohibitin: a novel molecular player in KDEL receptor signalling. Biomed Res Int. 2015;2015:319454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raykhel I, Alanen H, Salo K, Jurvansuu J, Nguyen VD, Latva‐Ranta M, Ruddock L. A molecular specificity code for the three mammalian KDEL receptors. J Cell Biol. 2007;179:1193–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lindholm P, Voutilainen MH, Laurén J, Peränen J, Leppänen VM, Andressoo JO, Lindahl M, Janhunen S, Kalkkinen N, Timmusk T, et al. Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature. 2007;448:73–77. [DOI] [PubMed] [Google Scholar]
- 18. Liu H, Yu C, Yu H, Zhong L, Wang Y, Liu J, Zhang S, Sun J, Duan L, Gong L, et al. Cerebral dopamine neurotrophic factor protects H9c2 cardiomyocytes from apoptosis. Herz. 2018;43:346–351. [DOI] [PubMed] [Google Scholar]
- 19. Latge C, Cabral KM, de Oliveira GA, Raymundo DP, Freitas JA, Johanson L, Romão LF, Palhano FL, Herrmann T, Almeida MS, et al. The solution structure and dynamics of full‐length human cerebral dopamine neurotrophic factor and its neuroprotective role against α‐synuclein oligomers. J Biol Chem. 2015;290:20527–20540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta‐catenin signaling under fully defined conditions. Nat Protoc. 2013;8:162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bagno LL, Carvalho D, Mesquita F, Louzada RA, Andrade B, Kasai‐Brunswick TH, Lago VM, Suhet G, Cipitelli D, Werneck‐de‐Castro JP, et al. Sustained IGF‐1 secretion by adipose‐derived stem cells improves infarcted heart function. Cell Transplant. 2016;25:1609–1622. [DOI] [PubMed] [Google Scholar]
- 22. Lindsey ML, Bolli R, Canty JM Jr, Du XJ, Frangogiannis NG, Frantz S, Gourdie RG, Holmes JW, Jones SP, Kloner RA, et al. Guidelines for experimental models of myocardial ischemia and infarction. Am J Physiol Heart Circ Physiol. 2018;314:H812–H838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maciel L, de Oliveira DF, Verissimo da Costa GC, Bisch PM, Nascimento JHM. Cardioprotection by the transfer of coronary effluent from ischaemic preconditioned rat hearts: identification of cardioprotective humoral factors. Basic Res Cardiol. 2017;112:52. [DOI] [PubMed] [Google Scholar]
- 24. Walker MJA, Curtis MJ, Hearse DJ, Campbell RWF, Janse MJ, Yellon DM, Cobbe SM, Coker SJ, Harness JB, Harron DWG, et al. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia, infarction, and reperfusion. Cardiovasc Res. 1988;22:447–455. [DOI] [PubMed] [Google Scholar]
- 25. Henderson MJ, Richie CT, Airavaara M, Wang Y, Harvey BK. Mesencephalic astrocyte‐derived neurotrophic factor (MANF) secretion and cell surface binding are modulated by KDEL receptors. J Biol Chem. 2013;288:4209–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bøtker HE, Hausenloy D, Andreadou I, Antonucci S, Boengler K, Davidson SM, Deshwal S, Devaux Y, Di Lisa F, Di Sante M, et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol. 2018;113:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maciel L, de Oliveira DF, Monnerat G, Campos de Carvalho AC, Nascimento JHM. Exogenous 10 kDa‐heat shock protein preserves mitochondrial function after hypoxia/reoxygenation. Front Pharmacol. 2020;11:545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kleinbongard P, Gedik N, Witting P, Freedman B, Klöcker N, Heusch G. Pleiotropic, heart rate‐independent cardioprotection by ivabradine. Br J Pharmacol. 2015;172:4380–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gedik N, Maciel L, Schulte C, Skyschally A, Heusch G, Kleinbongard P. Cardiomyocyte mitochondria as targets of humoral factors released by remote ischemic preconditioning. Arch Med Sci. 2017;13:448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ha T, Hu Y, Liu L, Lu C, McMullen JR, Kelley J, Kao RL, Williams DL, Gao X, Li C. TLR2 ligands induce cardioprotection against ischaemia/reperfusion injury through a PI3K/Akt‐dependent mechanism. Cardiovasc Res. 2010;87:694–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Becker B, Shaebani MR, Rammo D, Bubel T, Santen L, Schmitt MJ. Cargo binding promotes KDEL receptor clustering at the mammalian cell surface. Sci Rep. 2016;6:28940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruggiero C, Grossi M, Fragassi G, Di Campli A, Di Ilio C, Luini A, Sallese M. The KDEL receptor signalling cascade targets focal adhesion kinase on focal adhesions and invadopodia. Oncotarget. 2017;9:10228–10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mizobuchi N, Hoseki J, Kubota H, Toyokuni S, Nozaki J, Naitoh M, Koizumi A, Nagata K. ARMET is a soluble ER protein induced by the unfolded protein response via ERSE‐II element. Cell Struct Funct. 2007;32:41–50. [DOI] [PubMed] [Google Scholar]
- 34. Oh‐Hashi K, Hirata Y, Kiuchi K. Transcriptional regulation of mouse mesencephalic astrocyte‐derived neurotrophic factor in Neuro2a cells. Cell Mol Biol Lett. 2013;18:398–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Doroudgar S, Glembotski CC. The cardiokine story unfolds: ischemic stress‐induced protein secretion in the heart. Trends Mol Med. 2011;17:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dorner AJ, Wasley LC, Raney P, Haugejorden S, Green M, Kaufman RJ. The stress response in Chinese hamster ovary cells. Regulation of ERp72 and protein disulfide isomerase expression and secretion. J Biol Chem. 1990;265:22029–22034. [PubMed] [Google Scholar]
- 37. Voutilainen MH, Arumäe U, Airavaara M, Saarma M. Therapeutic potential of the endoplasmic reticulum located and secreted CDNF/MANF family of neurotrophic factors in Parkinson's disease. FEBS Lett. 2015;589:3739–3748. [DOI] [PubMed] [Google Scholar]
- 38. Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y, Suzuki M, Nishino T, Nakaya H, Koseki H. Aoe T Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol. 2004;24:8007–8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Riffer F, Eisfeld K, Breinig F, Schmitt MJ. Mutational analysis of K28 preprotoxin processing in the yeast Saccharomyces cerevisiae . Microbiology. 2002;148:1317–1328. [DOI] [PubMed] [Google Scholar]
- 40. Trychta KA, Bäck S, Henderson MJ, Harvey BK. KDEL receptors are differentially regulated to maintain the ER proteome under calcium deficiency. Cell Rep. 2018;25:1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314–341. [DOI] [PubMed] [Google Scholar]
- 42. Cao Z, Liao Q, Su M, Huang K, Jin J, Cao D. AKT and ERK dual inhibitors: the way forward? Cancer Lett. 2019;459:30–40. [DOI] [PubMed] [Google Scholar]
- 43. Rodriguez‐Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, Fry MJ, Waterfield MD, Downward J. Phosphatidylinositol‐3‐OH kinase as a direct target of Ras. Nature. 1994;370:527–532. [DOI] [PubMed] [Google Scholar]
- 44. Rodriguez‐Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3‐kinase by interaction with Ras and by point mutation. EMBO J. 1996;15:2442–2451. [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Y, Tseng CC, Tsai YL, Fu X, Schiff R, Lee AS. Cancer cells resistant to therapy promote cell surface relocalization of GRP78 which complexes with PI3K and enhances PI(3,4,5)P3 production. PLoS One. 2013;8:e80071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hausenloy DJ, Yellon DM. New directions for protecting the heart against ischaemia‐reperfusion injury: targeting the Reperfusion Injury Salvage Kinase (RISK)‐pathway. Cardiovasc Res. 2004;61:448–460. [DOI] [PubMed] [Google Scholar]
- 47. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121–1135. [DOI] [PubMed] [Google Scholar]
- 48. Blackwood EA, Thuerauf DJ, Stastna M, Stephens H, Sand Z, Pentoney A, Azizi K, Jakobi T, Van Eyk JE, Katus HA, et al. Proteomic analysis of the cardiac myocyte secretome reveals extracellular protective functions for the ER stress response. J Mol Cell Cardiol. 2020;143:132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li T, Xu W, Gao L, Guan G, Zhang Z, He P, Xu H, Fan L, Yan F, Chen G. Mesencephalic astrocyte‐derived neurotrophic factor affords neuroprotection to early brain injury induced by subarachnoid hemorrhage via activating Akt‐dependent prosurvival pathway and defending blood‐brain barrier integrity. FASEB J. 2019;33:1727–1741. [DOI] [PubMed] [Google Scholar]
- 50. Yuan Y, Lau WB, Su H, Sun Y, Yi W, Du Y, Christopher T, Lopez B, Wang Y, Ma XL. C1q‐TNF‐related protein‐9, a novel cardioprotetcive cardiokine, requires proteolytic cleavage to generate a biologically active globular domain isoform. Am J Physiol Endocrinol Metab. 2015;308:E891–E898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Figures S1–S6