

Abstract
Background
Release of neutrophil extracellular traps (NETs) after percutaneous coronary intervention (PCI) in acute coronary syndrome (ACS) is associated with periprocedural myocardial infarction, as a result of microvascular obstruction via pro‐inflammatory and prothrombotic pathways. Colchicine is a well‐established anti‐inflammatory agent with growing evidence to support use in patients with coronary disease. However, its effects on post‐PCI NET formation in ACS have not been explored.
Methods and Results
Sixty patients (40 ACS; 20 stable angina pectoris) were prospectively recruited and allocated to colchicine or no treatment. Within 24 hours of treatment, serial coronary sinus blood samples were collected during PCI. Isolated neutrophils from 10 patients with ACS post‐PCI and 4 healthy controls were treated in vitro with colchicine (25 nmol/L) and stimulated with either ionomycin (5 μmol/L) or phorbol 12‐myristate 13‐acetate (50 nmol/L). Extracellular DNA was quantified using Sytox Green and fixed cells were stained with Hoechst 3342 and anti‐alpha tubulin. Baseline characteristics were similar across both treatment and control arms. Patients with ACS had higher NET release versus patients with stable angina pectoris (P<0.001), which was reduced with colchicine treatment (area under the curve: 0.58 versus 4.29; P<0.001). In vitro, colchicine suppressed unstimulated (P<0.001), phorbol 12‐myristate 13‐acetate–induced (P=0.009) and ionomycin‐induced (P=0.002) NET formation in neutrophils isolated from patients with ACS post‐PCI, but not healthy controls. Tubulin organization was impaired in neutrophils from patients with ACS but was restored by colchicine treatment.
Conclusions
Colchicine suppresses NET formation in patients with ACS post‐PCI by restoring cytoskeletal dynamics. These findings warrant further investigation in randomized trials powered for clinical end points.
Registration
URL: https://anzctr.org.au; Unique identifier: ACTRN12619001231134.
Keywords: acute coronary syndrome, inflammation, percutaneous coronary intervention, pharmacology
Subject Categories: Acute Coronary Syndromes, Percutaneous Coronary Intervention, Mechanisms, Inflammation
Nonstandard Abbreviations and Acronyms
- CS
coronary sinus
- MTOC
microtubule organization center
- NE
neutrophil elastase
- NET
neutrophil extracellular trap
- ROS
reactive oxygen species
- SAP
stable angina pectoris
Clinical Perspective
What Is New?
This study demonstrates that colchicine inhibits the formation of neutrophil extracellular traps in patients with acute coronary syndrome after percutaneous coronary intervention by modifying the dynamics of the neutrophil cytoskeleton.
What Are the Clinical Implications?
This study provides further rationale for the beneficial use of colchicine in patients with acute coronary syndrome, particularly in the periprocedural period.
Percutaneous coronary intervention (PCI) in patients with acute coronary syndrome (ACS) is associated with a higher incidence of periprocedural myocardial infarction (MI), because of coronary no‐reflow and microvascular plugging by inflamed plaque fragments. 1 This process is believed to be mediated, at least in part, by an overabundance of activated polymorphonuclear neutrophils in vulnerable coronary plaque that release pro‐thrombotic and pro‐inflammatory products post‐PCI. 2 These products include neutrophil extracellular traps (NETs), a network of extracellular DNA, histones, neutrophil elastase (NE), and myeloperoxidase. 3 , 4 Activated neutrophils secrete NETs via a process known as NETosis, which drives pro‐thrombotic and pro‐inflammatory cascades by promoting neutrophil–platelet aggregate formation and inflammatory cytokine release from activated endothelial cells, macrophages, and T‐lymphocytes. 3 , 5 , 6 , 7 , 8 , 9 , 10 Classically, NETosis is facilitated by the generation of intracellular reactive oxygen species (ROS) that activate protein kinase pathways and transcription factors, ultimately leading to chromatin decondensation and NET formation. 11 Increased NET release after primary PCI has been shown to correlate positively with infarct size and negatively with ST‐segment resolution. 3
Despite the introduction of high‐dose statin pre‐PCI, periprocedural MI is still common and affects 1 in 4 patients undergoing PCI. 12 It is also associated with a significantly increased risk of in‐hospital major adverse cardiac events and death. 13 To date, no study investigating the role of a dedicated anti‐inflammatory agent in this setting has been published. Colchicine, a well‐established anti‐inflammatory drug, has emerged as a potential therapeutic tool in patients with coronary disease. Results of the recent LoDoCo (Low‐Dose Colchicine) 14 and COLCOT (Colchicine Cardiovascular Outcomes Trial) 15 trials have demonstrated that long‐term colchicine treatment significantly reduces the risk of secondary ischemic cardiovascular events. Colchicine treatment has also been shown to promote stabilization of atherosclerotic plaque. 16 Although colchicine inhibits neutrophil chemotaxis, 17 its effect on NETosis, particularly in the post‐PCI setting, remains to be evaluated.
Therefore, in this study, we investigated the effects of short‐term colchicine treatment in patients with ACS and patients with stable angina pectoris (SAP) undergoing PCI. We found that it significantly reduced NET release into the coronary sinus (CS) of patients with ACS, in part via direct cytoskeletal fixation in primed neutrophils, thereby arresting chromatin swelling and NET release.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Patient Population
This study was approved by the Sydney Local Health District research ethics and governance office and all participants provided written informed consent. The project was registered with the Australian New Zealand Clinical Trials Registry (ACTRN12619001231134). The samples used in this study were from 60 participants (40 ACS, 20 SAP 18 ) previously obtained from the Department of Cardiology, Royal Prince Alfred Hospital, Sydney, NSW, Australia (Figure 1A). 19 , 20 Classification of patients with ACS was based on concerning symptoms, ECG findings, and cardiac biomarkers, as per the Fourth Universal Definition. 18 Group sizes were based on our previous findings that colchicine reduced transcoronary IL‐1β and IL‐6 levels in patients with ACS levels by 33% and 80%, respectively. 19 Further, as NET release in the CS after PCI in patients with ACS has not been measured previously, we used the release of neutrophil‐derived microparticles in patients with ACS post‐PCI as a surrogate marker of NET release. 20 As such, based on a conservative reduction of 40% in NETs post‐PCI with an alpha of 0.05, power of 80%, a total of 40 patients (20 in each group) were required. Patients were allocated in a 1:1 ratio upon presentation to either no treatment, or treatment with periprocedural colchicine using a previously described regimen 20 : 1 mg followed by 0.5 mg after 1 hour, 6 to 24 hours before coronary angiography. This nonrandom treatment approach was used to ensure equal group sizes. PCI was performed within 30 minutes of commencing coronary angiography. Patients were recruited consecutively and allocated to treatment or control groups in alternating fashion. For the ex vivo aspect of this study, a further 16 colchicine‐naïve patients with ACS and 7 healthy controls were recruited (Figure 1B). Venous blood samples were collected from these patients with ACS within 24 hours of PCI.
Figure 1. Participant flow.
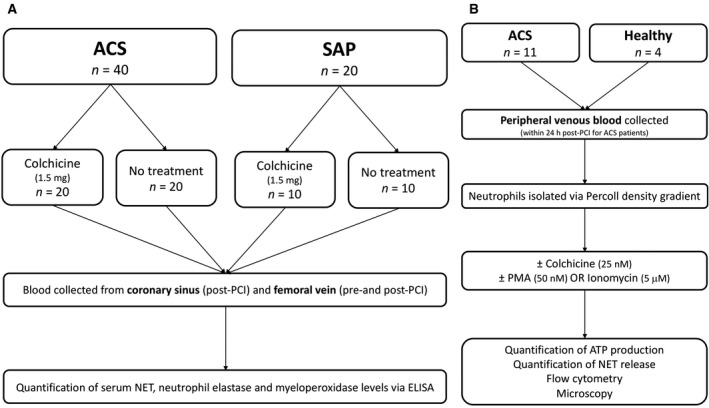
(A) In vivo study: Patients with ACS and SAP were randomized to either colchicine (total dose 1.5 mg) or no treatment. Blood samples were collected from the coronary sinus and femoral vein of patients undergoing PCI for quantification of NET, NE, and MPO levels. (B) Ex vivo study: Peripheral venous blood was collected from healthy volunteers and patients with ACS (within 24 hours post‐PCI). Neutrophils were isolated using a Percoll density gradient and incubated with colchicine (25 nmol/L) or vehicle control, followed by stimulation with PMA (50 nmol/L) or ionomycin (5 μmol/L) and assessment of NET release, cytoskeletal organization, chromatin swelling, ROS production, and cell viability. ACS indicates acute coronary syndrome; MPO, myeloperoxidase; NE, neutrophil elastase; NET, neutrophil extracellular trap; PCI, percutaneous coronary intervention; PMA, phorbol 12‐myristate 13‐acetate; SAP, stable angina pectoris; and ROS, reactive oxygen species.
All included patients were >18 years of age with a clinical indication for a coronary angiogram and PCI. Patients with a >50% stenosis in the left main coronary artery, cardiogenic shock, or hemodynamic instability, pregnant or lactating women, known colchicine hypersensitivity, or patients already taking colchicine, were excluded from this study.
Blood Sampling
During PCI, CS blood samples were collected at 45‐s intervals, as previously described 20 (Table S1). Peripheral venous blood was also collected, pre‐ and postprocedure from the common femoral vein. CS sampling has been shown in multiple prospective studies to offer an enhanced method of assessing the local cardiac milieu and its associated pathophysiology. 21 Our technique to safely cannulate the CS ostium has previously been described. 22 All patients received unfractionated heparin (70 U/kg). As soon as the activated clotting time was confirmed to be >250 s, coronary angioplasty and CS sampling were performed. In all cases, stents were deployed at nominal pressure for 30 s, followed by noncompliant balloon pressure dilatation (size and pressure at operator's discretion).
Blood Processing
Plasma was separated from whole blood by centrifugation at 1500g for 10 minutes and stored at −80°C. Myeloperoxidase (Hycult Biotech) and NE (Abcam) concentrations were qualified by ELISA. Levels of NETs were assessed using a myeloperoxidase‐DNA ELISA as described previously. 23 Procedures were performed and samples collected at variable times of the day based on clinical priority.
Neutrophil Isolation
Peripheral venous blood was collected from patients with ACS up to 24 hours after PCI and healthy volunteers. Blood was collected in EDTA vacutainers and neutrophil isolation commenced within 1 hour of sampling. Neutrophils were isolated using a Percoll‐density gradient, as previously described. 24 This protocol resulted in a granulocyte layer with >95% neutrophils, as confirmed by surface expression of CD66b via flow cytometry and observation using light microscopy and Romanowsky stain. Viability and maturity of isolated neutrophils were determined by staining with Annexin V‐FITC and a PE‐labeled anti‐CXCR4 antibody, respectively.
NETosis Assay
A plate reader–based assay was used to measure NETosis. 11 Isolated neutrophils were resuspended in RPMI medium with 2% (vol/vol) fetal bovine serum at 5×105 cells/mL. Sytox Green was added to the cell suspension (5 µmol/L final concentration) and 200 µL of the resulting solution was seeded in 96‐well black clear‐bottom plates (Corning). Subsequently, colchicine (25 nmol/L final concentration; Sigma‐Aldrich) or vehicle control was added to the wells followed by incubation for 1.5 hour at 37°C. NETosis was then induced with 50 nmol/L phorbol 12‐myristate 13‐acetate (PMA) (Sigma‐Aldrich) or 5 µmol/L ionomycin (Sigma‐Aldrich). Control cells were not stimulated. Fluorescence was measured at 2‐minute intervals for 4 hours using a fluorescence plate reader (504 nm excitation, 523 nm emission; SpectraMax, Molecular Devices). Cells were subsequently lysed using 1% (vol/vol) Triton‐X‐100 and fluorescence was measured again to determine total DNA content. All values were normalized to total DNA at each time point.
Cell Viability Assay
Cell viability was assessed via quantification of ATP levels following stimulation, as previously described. 25 Isolated neutrophils were seeded in white 96‐well plates (Corning) at a density of 2×104 cells/well in RPMI medium with 2% (vol/vol) fetal bovine serum. NETosis was induced with either 50 nmol/L PMA or 5 µmol/L ionomycin for 0, 15, 30, 60, and 120 minutes. CellTiter‐Glo Reagent (Promega) was added in equal volume to the cell culture medium present in each well; the plate was shaken for 2 minutes to induce cell lysis and then incubated at room temperature for 10 minutes to allow stabilization of the luminescent signal. Luminescence was measured (CLARIOstar, BMG Labtech) and values were normalized to unstimulated cells.
Reactive Oxygen Species
Intracellular ROS generation was measured using dihydrorhodamine 123 (Invitrogen). Neutrophils (1×106) were isolated from patients with ACS and incubated at 37°C for 20 minutes with 5 µmol/L dihydrorhodamine 123 in the presence or absence of PMA or ionomycin as previously described. Fluorescence was quantified by flow cytometry (BD LSRFortessa SORP X‐20, Biological Resources Imaging Laboratory, UNSW Sydney).
Microscopy
Neutrophils were isolated from patients with ACS post‐PCI and seeded on Nunc Lab‐Tek II 8‐well chamber slides (ThermoFisher) at a density of 2×104 cells/well. Cells were incubated for 1.5 hours at 37°C with either colchicine (25 nmol/L final concentration) or vehicle control, stimulated with either PMA or ionomycin then fixed with 2% (wt/vol) paraformaldehyde for 10 minutes at room temperature. Cells were permeabilized with PBS containing 1% (wt/vol) BSA and 0.3% (vol/vol) Triton‐X100 at 4°C overnight and then stained with Alexa647 conjugated anti‐α‐tubulin (1:100, Abcam) at 4°C overnight. Chromatin was stained with Hoechst 3342 (1.62 µmol/L final concentration) for 5 minutes at room temperature. Slides were then mounted on coverslips using ProLong Gold (ThermoFisher) and imaged using a 63×/1.4 Plan‐Apochromat lens with 405 nm and 647 nm laser lines on a Zeiss LSM 880 confocal microscope with Airy detector (Biomedical Imaging Facility, UNSW Sydney).
Image Analysis
Images were loaded in custom‐written Matlab (Mathworks) scripts. A semi‐automatic approach was used to identify individual cells and determine the chromatin area per cell. To characterize the tubulin radial intensity profile from the center of microtubule organization center (MTOC), individual cells and the corresponding MTOC were manually selected. For each cell, a circular average of intensity was performed at each radial distance from the center of the defined MTOC. For each radial intensity profile, the maximum intensity at the MTOC was extracted. Multiple fields of view were analyzed for each condition and the average and SEM intensity was calculated at each radius.
Statistical Analysis
Continuous variables were reported as mean±SD and categorical variables as number (percentage). The release of NETs, NE, and myeloperoxidase over 5 sampling time points at 45‐s intervals during PCI was quantified by area under the curve (AUC). Results of the Sytox Green NETosis assay were quantified by plotting fluorescence intensity against time and calculating the AUC for each sample. Normality of distribution was tested using the Shapiro‐Wilk test (P>0.05) as well as skewness and kurtosis. Differences in continuous variables were tested by unpaired or paired Student t test or Mann–Whitney U test, as appropriate. Proportional differences in categorical variables were tested using the χ 2 test or the Fisher exact test for association. A 2‐tailed P value of <0.05 was considered statistically significant. Statistical analysis was performed using SPSS software version 22.0 (IBM).
Results
Baseline Characteristics
From March 2015 to April 2017, 60 patients were recruited into the study, of which 40 presented with an ACS and 20 had SAP. Baseline characteristics were similar between both the treatment group and controls, with no statistically significant difference for any of the recorded demographic, clinical, or angiographic variables (Table) except number of diseased vessels in the patients with ACS (P=0.03). The mean age was 63.3 (±11.9) years and 49 (81.7%) patients were male. Nearly half of the patients were diabetic (45%), and most had hypertension (73.3%), dyslipidemia (81.7%), and were on a statin (80%). There was no difference in peripheral neutrophil count between colchicine‐treated and colchicine‐naïve patients with ACS (P=0.13) or SAP (P=0.50). In the treatment arm, 2 patients underwent vein graft PCI and 4 patients underwent multivessel PCI, compared with no patients in the control arm; however, CS sampling was only performed during the first (culprit lesion) PCI. Mean corrected TIMI frame count (cTFC)—a standardized, quantitative index of assessing coronary flow and a surrogate measure of microcirculatory resistance (higher cTFC associates with greater resistance) 26 —was normal (≤27) for all patients except the untreated ACS cohort (mean cTFC 27.5±4.7). Mean cTFC was higher in patients with ACS versus patients with SAP (27.2±5.2 versus 23.9±1.7), indicating more pronounced microvascular dysfunction. There was no significant difference in mean cTFC between untreated and treated cohorts in either group. Finally, the mean Gensini score 27 , a measure of extent of coronary atherosclerosis on angiography, was slightly higher in patients with ACS (50.7±12.9) versus patients with SAP (47.6±12.7), with no difference between untreated and treated cohorts for either group.
Table 1.
Baseline Demographic and Clinical Characteristics
| Baseline Characteristics | Patients With ACS | Patients With SAP | ||||
|---|---|---|---|---|---|---|
| No Treatment (n=20) | Colchicine (n=20) | P Value | No Treatment (n=10) | Colchicine (n=10) | P Value | |
| Age (y) (n=59) | 66.0 (±13.4) | 61.1 (±13.3) | 0.27 | 62.4 (±8.8) | 63.3 (±8.8) | 0.82 |
| Sex (male) (n=60) | 15 (75%) | 16 (80%) | 1.00* | 8 (80%) | 10 (100%) | 0.47* |
| Time from presentation to PCI (n=60) | 0.87 | N/A | N/A | N/A | ||
| <24 h | 2 (10%) | 3 (15%) | ||||
| 24–48 h | 11 (55%) | 11 (55%) | ||||
| >48 h | 7 (35%) | 6 (30%) | ||||
| Body mass index (kg/m2) (n=57) | 27.7 (±4.1) | 30.1 (±4.6) | 0.11 | 27.8 (±4.9) | 28.9 (±4.7) | 0.60 |
| Blood pressure (mm Hg) (n=57) | ||||||
| Systolic | 131.9 (±16.5) | 126.3 (±41.1) | 0.85 | 153.8 (±37.2) | 146.5(±29.4) | 0.68 |
| Diastolic | 75.7 (±11.2) | 66.0 (±23.4) | 0.17 | 84.2 (±24.6) | 75.3 (±16.1) | 0.53 |
| Diabetes mellitus (n=60) | 8 (40%) | 11 (55%) | 0.34 | 3 (30%) | 5 (50%) | 0.65* |
| Hypertension (n=60) | 15 (75%) | 12 (60%) | 0.31 | 9 (90%) | 8 (80%) | 1.00* |
| Dyslipidemia (n=60) | 15 (75%) | 16 (80%) | 1.00* | 9 (90%) | 9 (90%) | 1.00* |
| Positive family history (n=59) | 6 (30%) | 7 (35%) | 0.74 | 5 (50%) | 3 (33.3%) | 0.65* |
| Smoking history (n=60) | 14 (70%) | 12 (60%) | 0.51 | 8 (80%) | 7 (70%) | 1.00* |
| Current smoker (n=60) | 6 (30%) | 7 (35%) | 0.74 | 2 (20%) | 2 (20%) | 1.00* |
| Previous MI (n=60) | 3 (15%) | 6 (30%) | 0.45* | 3 (30%) | 5 (50%) | 0.65* |
| Previous PCI (n=60) | 4 (20%) | 6 (30%) | 0.47 | 4 (40%) | 4 (40%) | 1.00* |
| Previous CABG (n=60) | 1 (5%) | 2 (10%) | 1.00* | 1 (10%) | 1 (10%) | 1.00* |
| Previous CVA (n=60) | 1 (5%) | 1 (5%) | 1.00* | 0 | 1 (10%) | 1.00* |
| Renal impairment (eGFR<45) (n=56) | 3 (15.8%) | 1 (5.3%) | 0.60* | 0 | 0 | 1.00* |
| Medications (n=56) | ||||||
| Aspirin | 18 (100%) | 20 (100%) | 1.00* | 9 (100%) | 9 (100%) | 1.00* |
| Clopidogrel/prasugrel/ticagrelor | 17 (94.4%) | 19 (95%) | 1.00* | 9 (100%) | 8 (88.9%) | 1.00* |
| Statin | 15 (83.3%) | 17 (85%) | 1.00* | 7 (77.8%) | 9 (100%) | 0.47* |
| Beta‐blocker | 14 (77.8%) | 11 (55%) | 0.14 | 6 (66.7%) | 6 (66.7%) | 1.00* |
| ACEI/ARB | 8 (44.4%) | 12 (60%) | 0.34 | 6 (66.7%) | 7 (77.8%) | 1.00* |
| Biochemistry | ||||||
| Hs‐CRP (mg/L) (n=44) | 21.72 (±29.19) | 15.22 (±24.10) | 0.62 | 3.14 (±1.88) | 1.84 (±1.29) | 0.16 |
| Total cholesterol (mmol/L) (n=47) | 4.53 (±1.83) | 4.82 (±1.43) | 0.38 | 4.03 (±0.70) | 3.96 (±1.23) | 0.71 |
| Absolute neutrophil count (×103/mL) (n=59) | 4.47 (±1.66) | 5.64 (±2.93) | 0.13 | 3.88 (±1.36) | 4.24 (±0.91) | 0.50 |
| Baseline hs‐TnT (ng/L) (n=54) | 554.50 (±1011.75) | 882.0 (±1639.27) | 0.92 | 12.88 (±11.63) | 15.78(±15.10) | 0.42 |
| Angiographic characteristics | ||||||
| Diseased vessels no. (n=57) | 0.03 | 0.97 | ||||
| 1 | 7 (35%) | 5 (25%) | 4 (50%) | 5 (55.6%) | ||
| 2 | 13 (65%) | 9 (45%) | 3 (37.5%) | 3 (33.3%) | ||
| 3 | 0 | 6 (30%) | 1 (12.5%) | 1 (11.1%) | ||
| Coronary artery(s) treated (n=58) | 0.42 | 0.65 | ||||
| LAD | 9 (45%) | 7 (35%) | 5 (55.6%) | 4 (44.4%) | ||
| LCX | 4 (20%) | 4 (20%) | 1 (11.1%) | 2 (22.2%) | ||
| RCA | 7 (35%) | 4 (20%) | 3 (33.3%) | 2 (22.2%) | ||
| SVG | 0 | 1 (5%) | 0 | 1 (11.1%) | ||
| LAD and RCA | 0 | 1 (5%) | 0 | 0 | ||
| LCX and RCA | 0 | 1 (5%) | 0 | 0 | ||
| LAD and LCX | 0 | 2 (10%) | 0 | 0 | ||
| Stent characteristics | ||||||
| Length (mm) (n=45) | 18.1 (±7.4) | 18.1 (±6.8) | 0.83 | 20.1 (±10.1) | 20.1 (±8.2) | 0.90 |
| Size (mm) (n=46) | 2.9 (±0.5) | 2.9 (±0.4) | 0.60 | 2.9 (±0.3) | 3.3 (±0.5) | 0.21 |
| Stent type (n=48) | 0.06 | 0.37 | ||||
| DES | 15 (88.2%) | 12 (66.7%) | 3 (60%) | 7 (87.5%) | ||
| BMS | 2 (11.8%) | 1 (5.6%) | 1 (20%) | 1 (12.5%) | ||
| Bioabsorbable scaffold | 0 | 5 (27.8%) | 1 (20%) | 0 | ||
| Lesion type (n=60) | 0.53 | 0.87 | ||||
| A | 8 (40%) | 10 (50%) | 2 (20%) | 3 (30%) | ||
| B | 12 (60%) | 10 (50%) | 6 (60%) | 5 (50%) | ||
| C | 0 (0%) | 0 (0%) | 2 (20%) | 2 (20%) | ||
| Corrected TFC (n=60) | 27.5 (±4.7) | 26.9 (±5.7) | 0.62 | 24.0 (±1.7) | 23.8 (±1.7) | 0.97 |
ACEI/ARB indicates angiotensin‐converting enzyme inhibitor/angiotensin receptor blocker; ACS, acute coronary syndrome; BMS, bare metal stent; CABG, coronary artery bypass grafting; CVA, cerebrovascular accident; DES, drug‐eluting stent; eGFR, estimated glomerular filtration rate; hs‐CRP, high‐sensitivity C‐reactive protein; hs‐TnT, high‐sensitivity troponin T; LAD, left anterior descending; LCX, left circumflex; MI, myocardial infarction; N/A, not applicable; PCI, percutaneous coronary intervention; RCA, right coronary artery; SAP, stable angina pectoris; SVG, saphenous vein graft; and TFC, TIMI Frame Count.
Fisher exact test used.
For the ex vivo component of this study, a further 16 patients with ACS were recruited from July 2019 to June 2020 (Table S2). This cohort was not subject to oral colchicine treatment before PCI. The mean age of these participants was 68.2 (±14.4) years and 12 (75%) participants were male. The majority of these patients were hypertensive (9; 56.3%), and 10 (62.5%) were dyslipidemic. Only 1 (6.3%) patient had diabetes mellitus.
Events
There were no adverse events related to administration of colchicine, sampling from the CS, or the PCI procedure itself in any of patients in this study. None of the 20 patients with SAP experienced a periprocedural MI. Of the patients with ACS there were 7 (35%) periprocedural MIs in the untreated cohort and 3 (15%) in the treated cohort.
Patients With ACS Have Elevated Markers of Neutrophil Activation in CS Levels Pre‐PCI
Mean baseline (pre‐PCI) levels of NETs (0.205±0.139% versus 0.060±0.047% 23 ; P<0.001; Figure 2A), NE (204.40±54.49 versus 132.70±29.04 ng/mL; P<0.001; Figure 2B), and myeloperoxidase (164.67±37.08 versus 127.25±35.70; P=0.004; Figure 2C) were significantly higher in patients with ACS versus stable controls. There was no significant difference in the baseline values of NETs, NE, or myeloperoxidase between patients who did or did not receive colchicine (Table S3).
Figure 2. Patients with ACS have enhanced neutrophil activity that is acutely suppressed by colchicine.
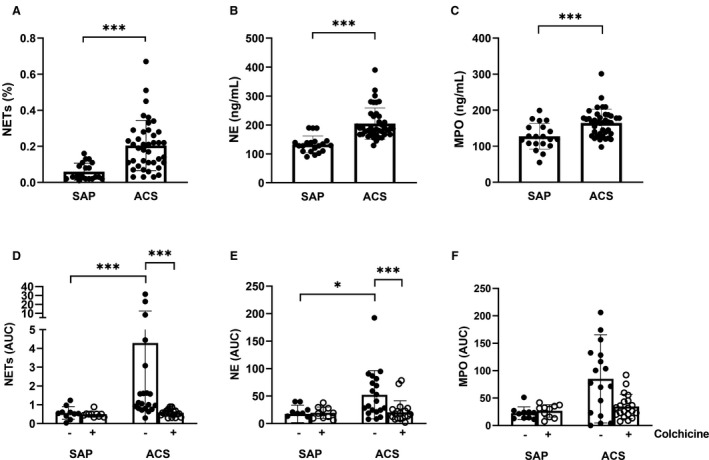
Blood was collected from the coronary sinus before percutaneous coronary intervention and levels of (A) NETs, (B) NE, and (C) MPO quantified. Patients with SAP or ACS were or were not treated with colchicine. Blood was collected from the coronary sinus during the procedure. (D) NETs, (E) NE and (F) MPO levels were measured over time and area under the curve was calculated. Error bars represent SD of the mean. ***P<0.005;*P<0.05. ACS indicates acute coronary syndrome; MPO, myeloperoxidase; NE, neutrophil elastase; NETs, neutrophil extracellular traps; and SAP, stable angina pectoris.
Colchicine Inhibits Neutrophil Activation During PCI in Patients With ACS
In the untreated patients, both NET (AUC, 4.29±8.17 versus 0.57±0.34; P<0.001; Figure 2D) and NE (AUC, 52.41±43.86 versus 17.61±15.91; P=0.01; Figure 2E) levels were significantly elevated in the CS during PCI of culprit lesions in patients with ACS compared with SAP. While myeloperoxidase levels were also increased, this did not reach statistical significance (AUC, 85.06±80.46 versus 22.42±11.68; P=0.10; Figure 2F).
Treatment with colchicine resulted in a significant reduction of both NET and NE release in the CS of patients with ACS. NET release was >7 times lower (AUC, 0.58±0.19 versus 4.29±8.17; P<0.001; Figure 2D) and NE release was >2.5 times lower (AUC, 20.27±21.21 versus 52.41±43.86; P=0.002; Figure 2E) in colchicine‐treated patients with ACS compared with controls during PCI. Myeloperoxidase release was also >2‐fold lower in the treated patients compared with controls (AUC, 34.75±22.13 versus 85.06±80.46; Figure 2F); however, this difference did not reach statistical significance (P=0.09). In contrast, colchicine treatment had no impact on the level of NETs, NE, or myeloperoxidase in patients with SAP. While colchicine reduced pre‐PCI CS IL‐6 levels in patients with ACS as we have described previously, 19 there was no impact on periprocedural IL‐6 levels in either SAP or ACS cohorts (Figure S1). Post‐PCI high‐sensitivity troponin T levels were decreased in colchicine‐treated patients with ACS compared with baseline (−207.65±770.83 ng/L) compared with increased levels in the untreated cohort (164.00±841.01 ng/L; P<0.05), suggestive of decreased periprocedural myocardial injury.
Colchicine Does Not Impact Peripheral Levels of Activated Neutrophil Mediators
To evaluate whether release of neutrophil‐derived products was a systemic process or localized to the coronary circulation (as measured by CS sampling), and whether colchicine suppressed these enzymes and protein networks, we also compared changes in peripheral venous NETs, NE, myeloperoxidase, and IL‐6 levels (post‐PCI minus pre‐PCI). There was no difference in peripheral venous levels of all mediators either pre‐ or post‐PCI in both the colchicine‐treated and nontreated groups. This was true for both patients with SAP and ACS (Figure S2).
ACS Neutrophils Are Primed for NETosis
To further understand the mechanisms of NET production post‐PCI, NET release was measured from neutrophils isolated from both patients with ACS post‐PCI and healthy participants. Neutrophils isolated from patients with ACS were morphometrically similar to those isolated from patients with ACS under unstimulated conditions and PMA treatment for 2 hours induced NETosis in both groups (Figure 3A). To further investigate any differences, time course plate–based NETosis assays were utilized. In the absence of stimulation, neutrophils isolated from patients with ACS displayed increased unstimulated NET formation (AUC, 1.05±0.60 versus 0.23±0.17, respectively; P=0.026; Figure 3B). Similarly, in the presence of PMA, neutrophils isolated from patients with ACS demonstrated augmented NET formation (AUC, 2.05±0.56 versus 0.88±0.50; P=0.006; Figure 3C). A similar trend was observed with ionomycin stimulation, although the difference did not reach statistical significance (AUC, 2.41±1.02 versus 1.58±0.69; P=0.18; Figure 3D). Disease status had no impact on neutrophil viability (Figure S3A) or age as determined by CXCR4 expression (Figure S3B,C).
Figure 3. Colchicine inhibits NETosis in primed neutrophils from patients with ACS independently of ROS generation.
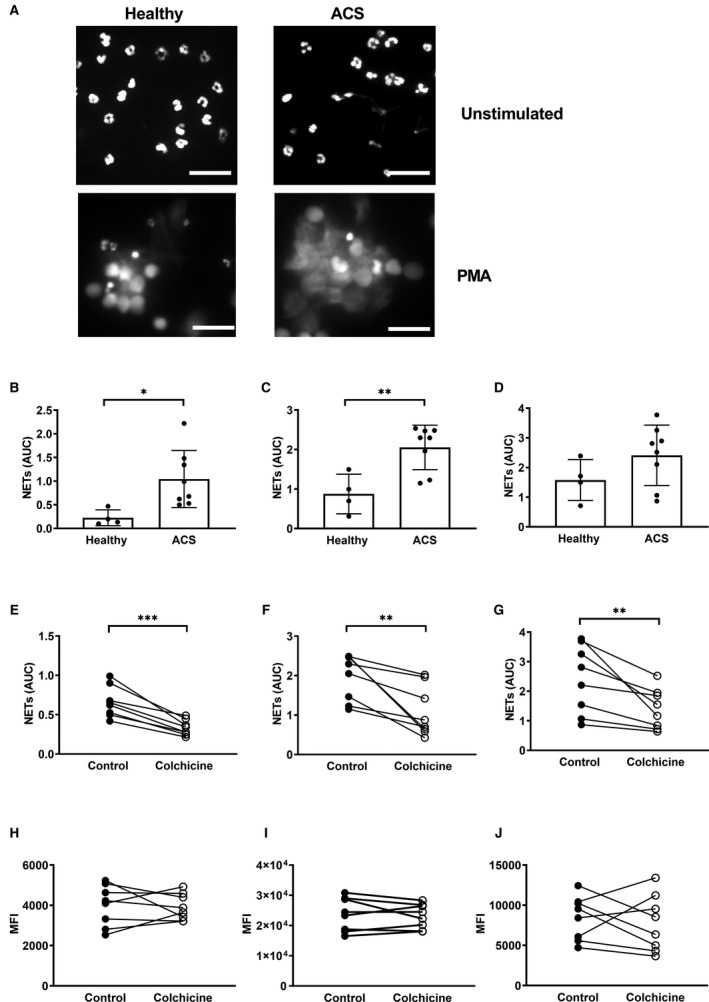
Neutrophils from patients with ACS and healthy participants were isolated and (A) NETosis imaged after 2 hours in unstimulated or PMA‐stimulated cells. NET release was measured using a fluorescent plate–based method in (B) unstimulated cells or in cells stimulated with (C) PMA or (D) ionomycin. Neutrophils from patients with ACS were isolated and treated with 25 nmol/L colchicine for 1.5 hours before measurement of NET release in (E) unstimulated cells or in cells stimulated with (F) PMA or (G) ionomycin. Neutrophils from patients with ACS were isolated and treated with 25 nmol/L colchicine for 1.5 hours before quantification of reactive oxygen species production using dihydrorhodamine 123 in (H) unstimulated, (I) PMA‐, and (J) ionomycin‐stimulated cells. Error bars represent SD from the mean. Scale bar=20 µm. ***P<0.001; **P<0.01; *P<0.05. ACS indicates acute coronary syndrome; AUC, area under the curve; MFI, median fluorescence intensity; PMA, phorbol 12‐myristate 13‐acetate; and NETs, neutrophil extracellular trap.
Colchicine Inhibits NETosis in ACS‐Primed Neutrophils
To examine the direct effect of colchicine on NETosis, neutrophils isolated from patients with ACS post‐PCI were subjected to colchicine treatment in vitro at equivalent physiologic doses. In the absence of stimulation, colchicine markedly reduced unstimulated NETosis (AUC, 0.66±0.20 versus 0.33±0.10; P<0.001; Figure 3E). Similar results were obtained when isolated neutrophils were stimulated with PMA (AUC, 1.95±0.58 versus 1.08±0.64; P=0.009; Figure 3F) or ionomycin (AUC, 2.40±1.16 versus 1.41±0.68; P=0.002; Figure 3G) following colchicine pretreatment. Colchicine had no effect on unstimulated, PMA‐ or ionomycin‐induced NET formation in neutrophils isolated from healthy participants (P=0.16, P=0.28, P=0.64, respectively; Figure S4).
Effect of Colchicine Is Independent of ROS Signaling and ATP Production
As the NET signaling cascade is largely dependent upon ROS generation, we assessed effects of colchicine on ROS in neutrophils derived from patients with ACS. Colchicine had no effect on unstimulated (P=0.81; Figure 3H), PMA‐ (P=0.59; Figure 3I) or ionomycin‐induced (P=0.60; Figure 3J) ROS production, indicating that the effect of colchicine on NET formation was downstream of ROS generation.
To ensure that the effect of colchicine was not a consequence of cellular toxicity, we measured ATP levels in neutrophils following colchicine treatment. Colchicine had no effect on cellular ATP production in either the presence or absence of stimulation, even up to the predefined time point of 2 hours (Figure S5).
Colchicine Inhibits Premature Chromatin Swelling in Neutrophils Isolated From Patients with ACS
Downstream of ROS production, chromatin decondensation and swelling acts as the point of no return for NET formation. As such, we directly visualized the effect of colchicine on this process. In the absence of stimulation, there was no significant difference in chromatin area between heathy, ACS, untreated, or colchicine‐treated neutrophils (Figure 4A and 4B all P>0.6). After 1 hour stimulation with PMA, there were clear differences between the groups (Figure 4C and 4D). Neutrophils isolated from patients with ACS had 100% greater chromatin area compared with those isolated from healthy participants (103.7±34.9 versus 52.1±17.4 μm2; P<0.001). Colchicine pretreatment resulted in a 35% reduction in the chromatin area of neutrophils isolated from patients with ACS (103.7±34.9 versus 66.8±39.0 μm2; P<0.001).
Figure 4. Colchicine inhibits premature chromatin swelling and restores α‐tubulin organization in neutrophils from patients with ACS.
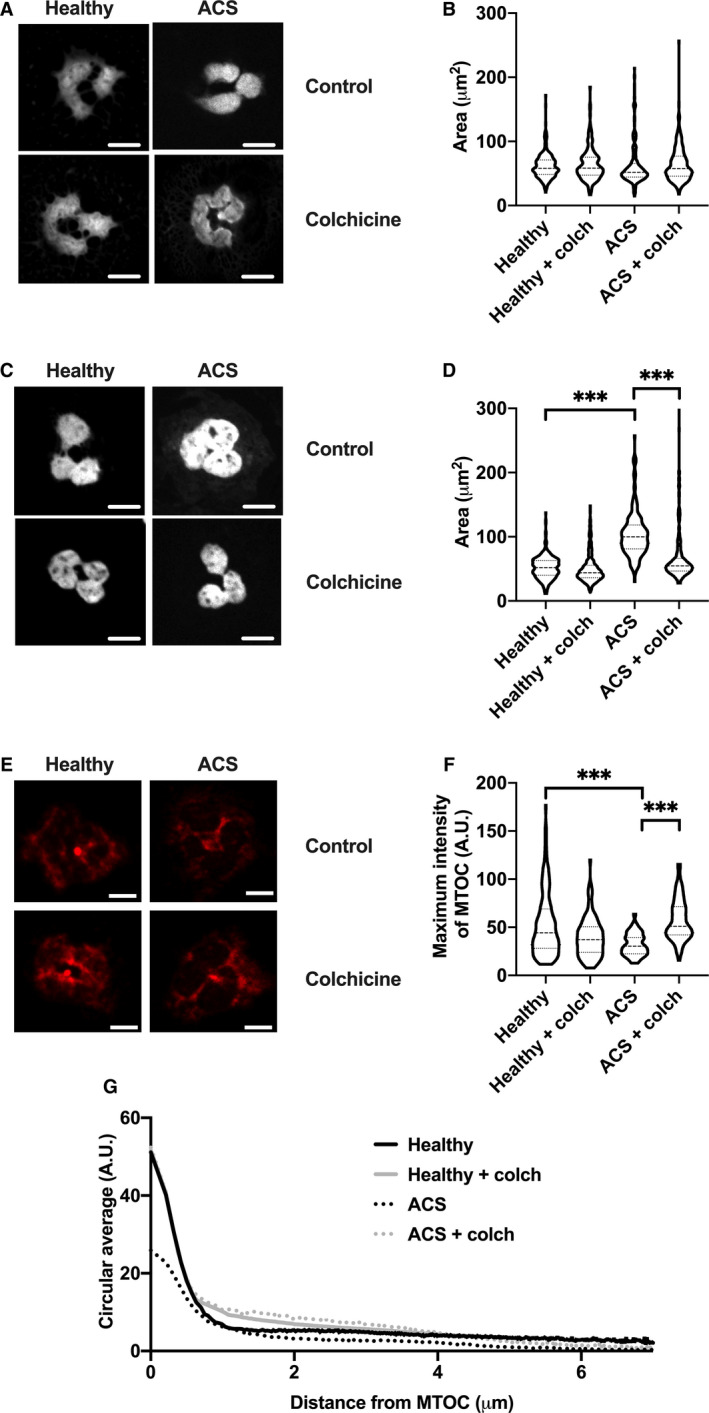
For quantification of chromatin swelling, neutrophils isolated from healthy participants and patients with ACS were treated with colchicine (colch; 25 nmol/L) or vehicle control for 1.5 hours. These cells were then fixed immediately (A, B) or stimulated with PMA (50 nmol/L) for 1 hour then fixed (C, D). All cells were stained with Hoechst 3342 before colchicine treatment and visualized by confocal microscopy. Chromatin area for individual cells was recorded in μm2 and compared between groups. For quantification of MTOC dynamics, neutrophils from healthy participants and patients with ACS were isolated, treated with colchicine (25 nmol/L) or vehicle control for 1.5 hours, then fixed and stained with anti‐α‐tubulin and visualized by confocal microscopy (E). Maximum intensity of the MTOC (F) and intensity as a function of distance from the MTOC (G) were quantified for each condition. Scale bar=5 µm. ***P<0.005. ACS indicates acute coronary syndrome; A.U., arbitrary units; MTOC, microtubule‐organizing center; and PMA, phorbol 12‐myristate 13‐acetate.
Colchicine Restores Disrupted Tubulin Organization in Neutrophils From Patients With ACS
A recent study demonstrated that rearrangement of the neutrophil cytoskeleton occurs during NETosis and may play a role in the early stages of NET release. 24 Under normal conditions, neutrophils have a well‐defined MTOC that, upon activation, rearrange into structures resembling mitotic centrosomes. As colchicine markedly reduced chromatin swelling in neutrophils of patients with ACS, without altering ROS production, we hypothesized that colchicine‐induced changes in cytoskeletal architecture may be responsible for halting NETosis. Neutrophils from healthy participants displayed a clear and well‐defined MTOC (Figure 4E). In contrast, α‐tubulin organization in neutrophils isolated from patients with ACS appeared to be disrupted, with a more diffuse staining pattern that was corrected by colchicine treatment. The MTOC maximal intensity was significantly reduced in neutrophils isolated from patients with ACS (32.16±32.73 A.U.) relative to both cells from healthy subjects (56.1±32.7 A.U.; P<0.005) and colchicine‐treated ACS neutrophils (57.94±21.50 A.U.; P<0.005). Quantification of α‐tubulin staining intensity as a function of distance from the MTOC of each cell demonstrated close similarity between the signatures of healthy and colchicine‐treated ACS neutrophils, particularly within 1 µm of the MTOC (Figure 4G).
Discussion
This study demonstrates, for the first time, that colchicine acutely suppresses post‐PCI local coronary release of key activated neutrophil‐derived mediators in patients with ACS. Furthermore, we demonstrate that neutrophils isolated from patients with ACS post‐PCI are primed to undergo NETosis and that colchicine treatment acts directly on these cells to stabilize the cytoskeleton, thereby inhibiting NET release.
The clinical arm of this study identified several key findings. First, untreated patients with ACS had significantly higher baseline (pre‐PCI) CS levels of all 3 activated neutrophil products (myeloperoxidase, NE, and NETs), and higher local release (post‐PCI) of NETs and NE, compared with patients with SAP. This indicates enhanced neutrophil activation and inflammatory mediator release from unstable plaques versus stable lesions at baseline and after PCI. Second, acute colchicine treatment markedly suppressed CS NETs and NE release post‐PCI in patients with ACS, compared with the untreated cohort. This suggests that colchicine acutely suppresses intraplaque neutrophil activation after angioplasty‐induced plaque disruption.
The ex vivo arm of this study was designed to investigate the direct effect of colchicine on neutrophils isolated from patients with ACS. These experiments yielded several novel findings. Neutrophils of patients with ACS post‐PCI had increased unstimulated and induced NET release, when compared with those of healthy control subjects. Low‐dose colchicine treatment attenuated NET release from neutrophils isolated from patients with ACS but not healthy controls. Quantification of α‐tubulin staining indicated that this “selectivity” of colchicine for ACS neutrophils may be related to microtubule disruption, which was evident in neutrophils isolated from patients with ACS compared with healthy controls.
Neutrophil products are overabundant in vulnerable coronary plaque and play a key role in peri‐PCI MI in patients with ACS. Purported mechanisms include PCI‐induced disruption of the plaque–thrombus interface leading to release of pre‐activated neutrophil mediators in vulnerable plaque and/or PCI‐induced de novo neutrophil activation. 20 , 28 In support of this, we previously reported a substantially higher release of neutrophil‐derived microparticles and myeloperoxidase into the coronary circulation after PCI of unstable lesions (versus stable lesions). 20 This study corroborates these findings; PCI results in a marked release of NETs, and neutrophils from patients with ACS post‐PCI are primed to undergo NETosis. This phenomenon is likely because of a combination of factors precipitated by PCI‐induced plaque disruption including increased pro‐inflammatory cytokine levels, release of cholesterol crystals into the lumen, and mechanical stress. 10 , 29 , 30 Priming of neutrophils increases NETosis, thereby predisposing individuals to a pro‐inflammatory and pro‐thrombotic cascade involving neutrophil‐platelet aggregate formation and complement cascade activation, cytokine release, ROS generation causing oxidative damage, and local tissue degradation. 3 , 5 , 6 , 7 , 8 , 9
Colchicine is an inexpensive and widely available anti‐inflammatory medication with an increasing evidence base demonstrating its efficacy in the secondary prevention of cardiovascular disease. 15 , 16 , 19 , 31 , 32 A small placebo‐controlled randomized trial showed lower cardiac enzyme release and reduced infarct size in colchicine‐treated patients with ST‐segment–elevation myocardial infarction versus controls. 33 Two recent preclinical studies corroborate the inhibitory effect of colchicine on NET formation described in this study. First, in vivo colchicine administration limited cholesterol crystal‐induced NETosis by inhibiting macropinocytosis (a microtubule‐dependent process essential for mediating the pro‐inflammatory effects of cholesterol crystals). 34 Second, ex vivo colchicine treatment of neutrophils isolated from individuals with Beçhet disease was shown to markedly suppress unstimulated NETosis. 35 The present study demonstrates that colchicine inhibits NETosis in patients with ACS by stabilizing the cytoskeleton, thereby attenuating chromatin swelling and subsequent NET release. Chromatin swelling is the major physical force driving NETosis and it has been shown to induce rupture of both the nuclear envelope and plasma membrane, leading to extracellular DNA accumulation. 25 Once initiated, chromatin swelling is irreversible and pharmacological inhibition of NETosis beyond this point is unlikely. 25 By limiting this process, colchicine provides a means of reducing NET‐associated complications in ACS.
This study examines neutrophil activation products in the CS during PCI and circulating neutrophils post‐PCI. It is important to consider that NETs, myeloperoxidase, and NE measured in the CS during PCI were largely a result of intraplaque neutrophil activation, induced by a combination of localized inflammation and direct mechanical stress. 36 Nevertheless, in vitro data from this study further demonstrate systemic neutrophil “priming” post‐PCI. Although these circulating neutrophils are not terminally activated to undergo NETosis, they demonstrate enhanced reactivity to stimuli: a “primed” state. 37 , 38 We show that colchicine not only attenuates the localized release of neutrophil activation products in the coronary circulation but also that colchicine promotes transition of circulating “primed” neutrophils back to a basal state.
In patients with ACS who underwent PCI, colchicine acutely and markedly suppressed local neutrophil activation by directly inhibiting NET release from primed neutrophils. These results provide further evidence of the beneficial role of colchicine in patients with ACS, particularly in the periprocedural period. Larger randomized trials powered for hard clinical outcomes are required to evaluate whether colchicine is effective at reducing periprocedural MI and major adverse cardiac events in patients with ACS.
Sources of Funding
This work was supported by a Ramaciotti Health Investment Grant and a UNSW Sydney School of Medical Sciences Accelerator Grant. SP was supported by a New South Wales Early‐Mid Career Health Fellowship. GM was supported by a research grant from Comisión Nacional de Investigación Científica y Tecnológica Fondo Nacional de Desarrollo Científico y Tecnologico (FONDECYT) Iniciación 11170205.
Disclosures
None.
Supporting information
Tables S1–S3
Figures S1–S5
Acknowledgments
The authors thank the cardiac catheterization laboratory staff at the Royal Prince Alfred Hospital and the Biomedical Imaging Facility staff at UNSW Sydney for their assistance in performing these studies.
(J Am Heart Assoc. 2021;10:e018993. DOI: 10.1161/JAHA.120.018993.)
Preprint posted on MedRxiv April 24, 2020. doi: https://doi.org/10.1101/2020.04.20.20034025.
For Sources of Funding and Disclosures, see page 12.
Contributor Information
Blake J. Cochran, Email: b.cochran@unsw.edu.au.
Sanjay Patel, Email: sanjay.patel@hri.org.au.
References
- 1. Kereiakes DJ. Adjunctive pharmacotherapy before percutaneous coronary intervention in non‐ST‐elevation acute coronary syndromes: the role of modulating inflammation. Circulation. 2003;108:Iii22‐Iii27. DOI: 10.1161/01.CIR.0000086951.09881.51. [DOI] [PubMed] [Google Scholar]
- 2. Bekkers SCAM, Yazdani SK, Virmani R, Waltenberger J. Microvascular obstruction: underlying pathophysiology and clinical diagnosis. J Am Coll Cardiol. 2010;55:1649–1660. [DOI] [PubMed] [Google Scholar]
- 3. Mangold A, Alias S, Scherz T, Hofbauer T, Jakowitsch J, Panzenbock A, Simon D, Laimer D, Bangert C, Kammerlander A, et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST‐elevation acute coronary syndrome are predictors of ST‐segment resolution and infarct size. Circ Res. 2015;116:1182–1192. [DOI] [PubMed] [Google Scholar]
- 4. Papayannopoulos V, Zychlinsky A. Nets: a new strategy for using old weapons. Trends Immunol. 2009;30:513–521. DOI: 10.1016/j.it.2009.07.011. [DOI] [PubMed] [Google Scholar]
- 5. Carbone F, Mach F, Montecucco F. Update on the role of neutrophils in atherosclerotic plaque vulnerability. Curr Drug Targets. 2015;16:321–333. [DOI] [PubMed] [Google Scholar]
- 6. Carbone F, Nencioni A, Mach F, Vuilleumier N, Montecucco F. Pathophysiological role of neutrophils in acute myocardial infarction. Thromb Haemost. 2013;110:501–514. DOI: 10.1160/TH13-03-0211. [DOI] [PubMed] [Google Scholar]
- 7. Döring Y, Drechsler M, Soehnlein O, Weber C. Neutrophils in atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:288–295. [DOI] [PubMed] [Google Scholar]
- 8. Naruko T, Ueda M, Haze K, van der Wal AC, van der Loos CM, Itoh A, Komatsu R, Ikura Y, Ogami M, Shimada Y, et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation. 2002;106:2894–2900. DOI: 10.1161/01.CIR.0000042674.89762.20. [DOI] [PubMed] [Google Scholar]
- 9. Rothmeier AS, Marchese P, Petrich BG, Furlan‐Freguia C, Ginsberg MH, Ruggeri ZM, Ruf W. Caspase‐1‐mediated pathway promotes generation of thromboinflammatory microparticles. J Clin Investig. 2015;125:1471–1484. DOI: 10.1172/JCI79329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science (New York, N.Y.). 2015;349:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Khan MA, Palaniyar N. Transcriptional firing helps to drive netosis. Sci Rep. 2017;7:41749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stone GW, Maehara A, Muller JE, Rizik DG, Shunk KA, Ben‐Yehuda O, Genereux P, Dressler O, Parvataneni R, Madden S, et al. Plaque characterization to inform the prediction and prevention of periprocedural myocardial infarction during percutaneous coronary intervention: the canary trial (coronary assessment by near‐infrared of atherosclerotic rupture‐prone yellow). JACC Cardiovasc Interv. 2015;8:927–936. [DOI] [PubMed] [Google Scholar]
- 13. Testa L, Van Gaal WJ, Biondi Zoccai GG, Agostoni P, Latini RA, Bedogni F, Porto I, Banning AP. Myocardial infarction after percutaneous coronary intervention: a meta‐analysis of troponin elevation applying the new universal definition. QJM. 2009;102:369–378. DOI: 10.1093/qjmed/hcp005. [DOI] [PubMed] [Google Scholar]
- 14. Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low‐dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol. 2013;61:404–410. [DOI] [PubMed] [Google Scholar]
- 15. Tardif J‐C, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, Pinto FJ, Ibrahim R, Gamra H, Kiwan GS, et al. Efficacy and safety of low‐dose colchicine after myocardial infarction. N Engl J Med. 2019;381:2497–2505. [DOI] [PubMed] [Google Scholar]
- 16. Vaidya K, Arnott C, Martinez GJ, Ng B, McCormack S, Sullivan DR, Celermajer DS, Patel S. Colchicine therapy and plaque stabilization in patients with acute coronary syndrome: a CT coronary angiography study. JACC Cardiovasc Imaging. 2018;11:305–316. [DOI] [PubMed] [Google Scholar]
- 17. Leung YY, Yao Hui LL, Kraus VB. Colchicine–update on mechanisms of action and therapeutic uses. Semin Arthritis Rheum. 2015;45:341–350. DOI: 10.1016/j.semarthrit.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, White HD. Fourth universal definition of myocardial infarction (2018). Glob Heart. 2018;13:305–338. DOI: 10.1016/j.gheart.2018.08.004. [DOI] [PubMed] [Google Scholar]
- 19. Martinez GJ, Robertson S, Barraclough J, Xia Q, Mallat Z, Bursill C, Celermajer DS, Patel S. Colchicine acutely suppresses local cardiac production of inflammatory cytokines in patients with an acute coronary syndrome. J Am Heart Assoc. 2015;4:e002128. DOI: 10.1161/JAHA.115.002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Martínez GJ, Barraclough JY, Nakhla S, Kienzle V, Robertson S, Mallat Z, Celermajer DS, Patel S. Neutrophil‐derived microparticles are released into the coronary circulation following percutaneous coronary intervention in acute coronary syndrome patients. Bioscience Reports. 2017;37. DOI: 10.1042/bsr20160430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jaumdally R, Varma C, Macfadyen RJ, Lip GYH. Coronary sinus blood sampling: an insight into local cardiac pathophysiology and treatment? Eur Heart J. 2007;28:929–940. DOI: 10.1093/eurheartj/ehm015. [DOI] [PubMed] [Google Scholar]
- 22. Martínez GJ, Bailey BP, Celermajer DS, Patel S. A safe and easy technique to sample the coronary sinus—facilitating a closer look at cardiac disease. Int J Cardiol. 2014;176:1321–1322. [DOI] [PubMed] [Google Scholar]
- 23. Sil P, Yoo D, Floyd M, Gingerich A, Rada B. High Throughput Measurement of Extracellular DNA Release and Quantitative NET Formation in Human Neutrophils In Vitro . Journal of Visualized Experiments. 2016;112. DOI: 10.3791/52779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dooley DC, Simpson JF, Meryman HT. Isolation of large numbers of fully viable human neutrophils: a preparative technique using percoll density gradient centrifugation. Exp Hematol. 1982;10:591–599. [PubMed] [Google Scholar]
- 25. Neubert E, Meyer D, Rocca F, Gunay G, Kwaczala‐Tessmann A, Grandke J, Senger‐Sander S, Geisler C, Egner A, Schon MP, et al. Chromatin swelling drives neutrophil extracellular trap release. Nat Commun. 2018;9:3767. DOI: 10.1038/s41467-018-06263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gibson CM, Cannon Christopher P, Daley William L, Dodge JT, Alexander B, Marble Susan J, McCabe Carolyn H, Raymond L, Fortin T, Poole WK, et al. Timi frame count. Circulation. 1996;93:879–888. DOI: 10.1161/01.CIR.93.5.879. [DOI] [PubMed] [Google Scholar]
- 27. Gensini GG. A more meaningful scoring system for determining the severity of coronary heart disease. Am J Cardiol. 1983;51:606. DOI: 10.1016/S0002-9149(83)80105-2. [DOI] [PubMed] [Google Scholar]
- 28. Hansson GK, Libby P, Tabas I. Inflammation and plaque vulnerability. J Intern Med. 2015;278:483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meher AK, Spinosa M, Davis JP, Pope N, Laubach VE, Su G, Serbulea V, Leitinger N, Ailawadi G, Upchurch GR Jr. Novel role of IL (interleukin)‐1beta in neutrophil extracellular trap formation and abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2018;38:843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yu X, Tan J, Diamond SL. Hemodynamic force triggers rapid netosis within sterile thrombotic occlusions. J. Thromb. Haemost. 2018;16:316–329. DOI: 10.1111/jth.13907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tucker B, Kurup R, Barraclough J, Henriquez R, Cartland S, Arnott C, Misra A, Martinez G, Kavurma M, Patel S. Colchicine as a novel therapy for suppressing chemokine production in patients with an acute coronary syndrome: a pilot study. Clin Ther. 2019;41:2172–2181. [DOI] [PubMed] [Google Scholar]
- 32. Robertson S, Martinez GJ, Payet CA, Barraclough JY, Celermajer DS, Bursill C, Patel S. Colchicine therapy in acute coronary syndrome patients acts on caspase‐1 to suppress nlrp3 inflammasome monocyte activation. Clin Sci. 2016;130:1237–1246. [DOI] [PubMed] [Google Scholar]
- 33. Deftereos S, Giannopoulos G, Angelidis C, Alexopoulos N, Filippatos G, Papoutsidakis N, Sianos G, Goudevenos J, Alexopoulos D, Pyrgakis V, et al. Anti‐inflammatory treatment with colchicine in acute myocardial infarction. Circulation. 2015;132:1395–1403. DOI: 10.1161/CIRCULATIONAHA.115.017611. [DOI] [PubMed] [Google Scholar]
- 34. Muñoz LE, Boeltz S, Bilyy R, Schauer C, Mahajan A, Widulin N, Grüneboom A, Herrmann I, Boada E, Rauh M, et al. Neutrophil extracellular traps initiate gallstone formation. Immunity. 2019;51:443–450.e4. DOI: 10.1016/j.immuni.2019.07.002. [DOI] [PubMed] [Google Scholar]
- 35. Safi R, Kallas R, Bardawil T, Mehanna CJ, Abbas O, Hamam R, Uthman I, Kibbi AG, Nassar D. Neutrophils contribute to vasculitis by increased release of neutrophil extracellular traps in behcet's disease. J Dermatol Sci. 2018;92:143–150. [DOI] [PubMed] [Google Scholar]
- 36. Rudolph V, Steven D, Gehling UM, Goldmann B, Rudolph TK, Friedrichs K, Meinertz T, Heitzer T, Baldus S. Coronary plaque injury triggers neutrophil activation in patients with coronary artery disease. Free Radic Biol Med. 2007;42:460–465. DOI: 10.1016/j.freeradbiomed.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 37. Geng S, Zhang Y, Lee C, Li L. Novel reprogramming of neutrophils modulates inflammation resolution during atherosclerosis. Sci Adv. 2019;5(2):eaav2309. DOI: 10.1126/sciadv.aav2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vogt KL, Summers C, Chilvers ER, Condliffe AM. Priming and de‐priming of neutrophil responses in vitro and in vivo. Eur J Clin Invest. 2018;48(Suppl 2):e12967. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3
Figures S1–S5