

SUMMARY
All immune systems use precise target recognition to interrogate foreign invaders. During CRISPR-Cas immunity, prokaryotes capture short spacer sequences from infecting viruses and insert them into the CRISPR array. Transcription and processing of the CRISPR locus generates small RNAs containing the spacer and repeat sequences, which guide Cas nucleases to cleave a complementary protospacer in the invading nucleic acids. In most CRISPR systems, sequences flanking the protospacer drastically affect cleavage. Here we investigated the target requirements of the recently discovered RNA-targeting type VI-A CRISPR-Cas system in its natural host, Listeria seeligeri. We discovered that target RNAs with extended complementarity between the protospacer flanking sequence and the repeat sequence of the guide RNA are not cleaved by the type VI-A nuclease Cas13, neither in vivo nor in vitro. These findings establish fundamental rules for the design of Cas13-based technologies and provide a mechanism for preventing self-targeting in type VI-A systems.
Keywords: CRISPR, Cas13, immunity, RNase
Graphical Abstract
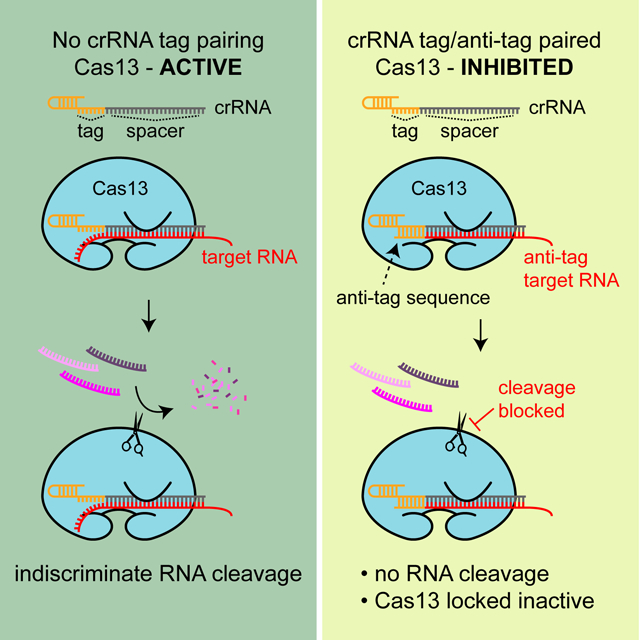
eTOC Blurb:
Type VI CRISPR systems employ the RNA-guided Cas13 nuclease to sense and cleave infecting RNA targets. Meeske et al. show that prevention of autoimmunity is mediated by sensing of extended complementarity between the Cas13 RNA guide and target RNAs that may originate from the CRISPR locus.
INTRODUCTION
Immune systems protect their host by specifically detecting and eliminating foreign agents. To achieve this, an efficient immune response must make use of precise target recognition mechanisms. Clustered, regularly interspaced, short palindromic repeats (CRISPR) systems and their associated (Cas) proteins defend their prokaryotic hosts from invasion by bacteriophage (Barrangou et al., 2007) and plasmids (Marraffini and Sontheimer, 2008). Immunological memories of infection by these genetic elements are captured and preserved by CRISPR systems in an array of short (~30 nucleotide) segments of foreign DNA called spacers, which are flanked by repeat sequences on either side (Barrangou et al., 2007). The array is transcribed as a long precursor CRISPR RNA (pre-crRNA) and then cleaved at each repeat sequence to liberate individual CRISPR RNAs (crRNAs) (Brouns et al., 2008; Deltcheva et al., 2011; Hale et al., 2008). These are small RNAs that contain the spacer sequence and a short region of the repeat, known as the crRNA tag. After processing, crRNAs form a ribonucleoprotein complex with Cas effector nucleases and guide them to destroy foreign nucleic acid targets (called protospacers) that are complementary to the spacer region.
A wide variety of CRISPR-Cas systems have been discovered and categorized into six types on the basis of their cas gene content and mechanism of target interference (Makarova et al., 2015; Shmakov et al., 2017). In all CRISPR types studied to date, the sequences that flank the protospacer are fundamental for target recognition and for the prevention of an immune reaction against the spacer sequences in the CRISPR array itself. In the DNA-targeting Type I, II, and V CRISPR-Cas systems the presence of a short protospacer adjacent motif (PAM) is strictly required for target recognition and cleavage (Deveau et al., 2008; Gasiunas et al., 2012; Jinek et al., 2012; Semenova et al., 2011; Zetsche et al., 2015). Importantly, PAMs are absent from the repeats that flank the spacer in the CRISPR array of these systems, preventing autoimmune cleavage of these targets that are complementary to the crRNA guide and reside within the host genome. In contrast, Type III CRISPR systems are much more flexible in their protospacer-flanking sequence requirements for cleavage: most flanking sequences license targeting except those having high levels of homology to the CRISPR repeat (Elmore et al., 2016; Marraffini and Sontheimer, 2010; Pyenson et al., 2017). Type III CRISPR-Cas systems are unique in that they use the crRNA guide to sense target transcripts and cleave both DNA and RNA (Elmore et al., 2016; Estrella et al., 2016; Kazlauskiene et al., 2016; Samai et al., 2015). Targets with complementarity extending beyond the spacer-protospacer RNA duplex into the crRNA tag region, fail to activate the DNase activity of Cas10 within the type III effector ribonucleoprotein complex (Elmore et al., 2016; Estrella et al., 2016; Kazlauskiene et al., 2016; Samai et al., 2015). It is believed that this targeting rule protects the CRISPR locus from self-cleavage upon anti-sense transcription of the array (Kazlauskiene et al., 2016; Lillestøl et al., 2009).
Type VI-A CRISPR systems encode a large single effector protein called Cas13, which binds to RNA targets in a crRNA-guided manner (Abudayyeh et al., 2016; East-Seletsky et al., 2016; Liu et al., 2017a). Once target-bound, Cas13 undergoes a conformational change, unleashing a nonspecific trans-RNA cleavage activity catalyzed by its dual higher eukaryotes and prokaryotes nucleotide-binding (HEPN) domains. Here, we investigated the targeting rules used by Cas13 in Listeria seeligeri, a natural host of the Type VI-A CRISPR-Cas system. A previous report that investigated the function of the Leptotrichia shahii Type VI-A system via over-expression of this locus in an Escherichia coli heterologous host indicated that Cas13 recognizes a single nucleotide motif adjacent to the target called the protospacer flanking site (PFS) (Abudayyeh et al., 2016), yet another study reported no PFS rules for Cas13 targeting in vitro (East-Seletsky et al., 2016). Our in vivo and in vitro data indicate that, similar to the mechanism employed by type III CRISPR systems, extended complementarity between the crRNA tag and the target flanking sequence (the “anti-tag”) blocks the activation of Cas13, preventing RNA cleavage. We determined that at least seven nucleotides of the anti-tag contribute to target discrimination, and that complementarity at these sites is necessary and sufficient for target protection. Our results show that, similar to Type III systems, Type VI-A systems can use tag:anti-tag RNA pairing to protect the host from toxic Cas13 misactivation by antisense transcription of the CRISPR locus. Furthermore, we found that anti-tag RNAs act as potent inhibitors of Cas13 activity, suggesting a potential regulatory mechanism for this system. Our findings also establish important targeting rules for Cas13-based diagnostics and RNA knock-down technologies (Abudayyeh et al., 2017; East-Seletsky et al., 2016; Gootenberg et al., 2018; Gootenberg et al., 2017; Myhrvold et al., 2018).
RESULTS
The Listeria seeligeri type VI-A CRISPR system restricts conjugative plasmid transfer.
To study type VI-A CRISPR function under conditions that most accurately approximate those in nature, we performed experiments in Listeria seeligeri. Its 2.8 Mb genome contains a type VI-A CRISPR system with five endogenous spacers alongside a cas13 homolog (Fig. 1A). The CRISPR array begins in an unusual orientation, with a degenerate repeat at the 5’ end preceding the spacers. Degenerate repeats are commonly observed at one end of a CRISPR array and flank the oldest, often nonfunctional spacers; we therefore hypothesized that the newest spacers are located at the 3’ end and began our spacer nomenclature at this end of the array (Makarova et al., 2015). We developed a “loop-in, loop-out” allelic replacement method for L. seeligeri and constructed a deletion of the CRISPR locus (see Methods and Fig. S1A), including cas13 and two neighboring ORFs predicted to be in the same operon. Using the E. coli-Listeria shuttle vector pAM8, we engineered pTarget conjugative plasmids each containing a protospacer complementary to one of the endogenous L. seeligeri spacers inserted into the 3’ untranslated region of the chloramphenicol acetyltransferase (cat) gene (Fig. 1B). Each pTarget plasmid was transferred into wild-type and ΔCRISPR recipient strains via conjugative mating with an E. coli donor, and transconjugants were isolated using a combination of nalidixic acid to kill donors and chloramphenicol to select for pTarget (Fig. 1C). All target plasmids gave rise to L. seeligeri transconjugants with the ΔCRISPR recipient, as did a control plasmid lacking a target sequence when mated with a wild-type recipient. Conjugation of plasmids harboring targets matching spc1, spc2, and spc4 was restricted in wild-type, but not ΔCRISPR cells, likely due to cleavage of the cat transcript. Re-incorporation of the CRISPR-Cas locus (including the CRISPR array and cas13 but not orf2 or orf3) into the PSA phage integration site of ΔCRISPR bacteria using the pPL2e vector (Lauer et al., 2002) restored CRISPR targeting of the conjugative plasmid carrying the spc2 protospacer, confirming CRISPR-dependent, anti-conjugation immunity (Fig. 1D). To investigate the lack of restriction of plasmids containing targets matching spc3 or spc5, we looked for the presence of mature, matching crRNAs by Northern blot (Fig. 1E). For all the functional spacers (spc1, spc2, and spc4), crRNAs of the expected length (55 nucleotides) were detected in wild-type, but not in ΔCRISPR hosts. CrRNAs matching spc3 were undetectable, suggesting that this spacer is not functional due to lack of processing or instability of its crRNA. In contrast, spc5 crRNAs were expressed at levels similar to the other, functional crRNAs and therefore we speculate that the resulting crRNA may not be competent for interference due to the presence of the degenerate repeat.
Figure 1: The Listeria seeligeri type VI-A CRISPR system restricts conjugative plasmid transfer.
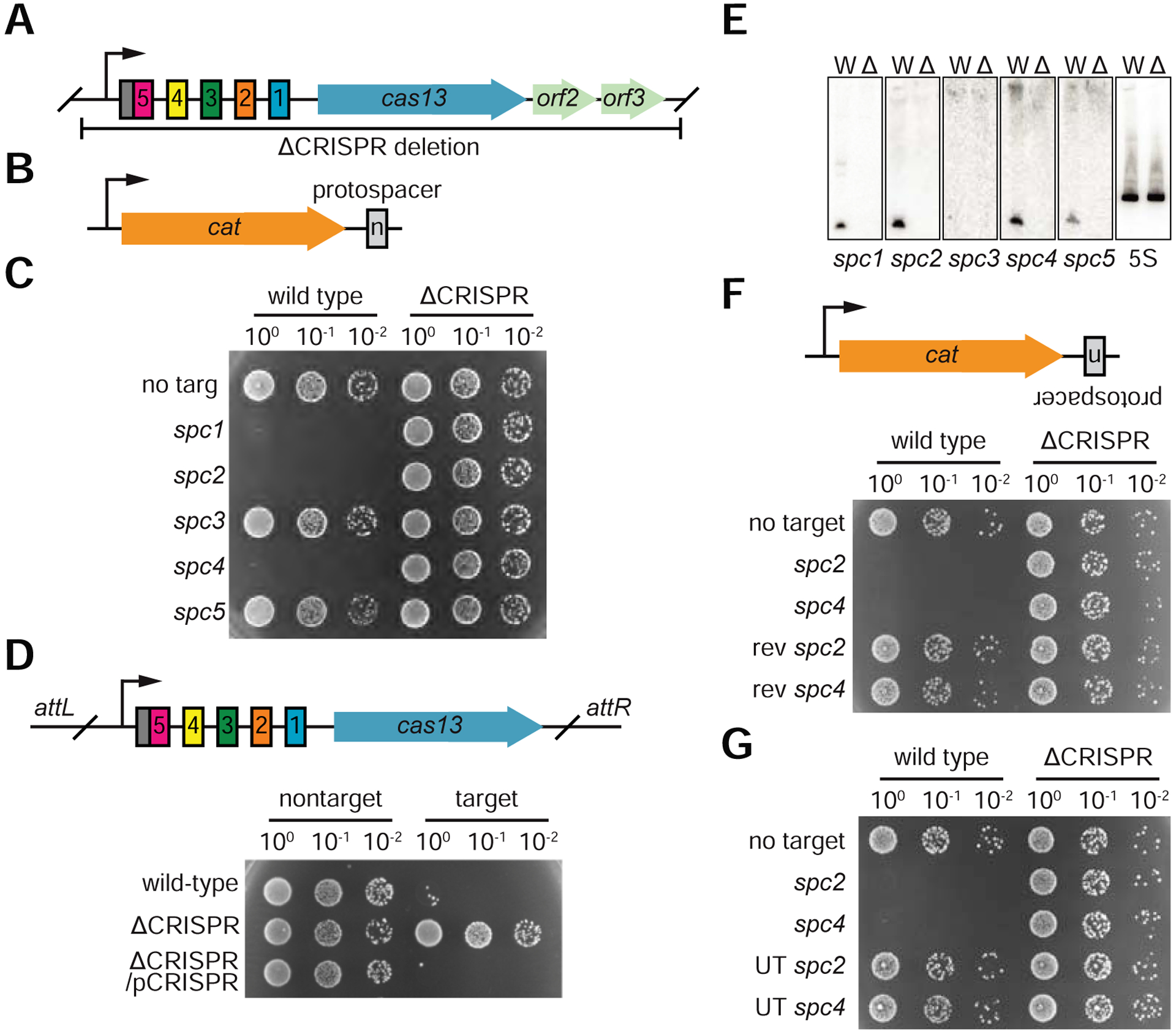
A. Schematic of L. seeligeri type VI CRISPR locus and region deleted for this study, with spacers shown in colors and repeats shown in white. Degenerate repeat is indicated in gray. B. Schematic of pTarget plasmid constructs with a protospacer inserted into the coding strand of the cat gene 3’ UTR. C. Conjugation assay for CRISPR interference. Wild type or ΔCRISPR L. seeligeri recipients were co-cultured with E. coli donor containing a CRISPR target matching the indicated spacer. Transconjugants were selected from diluted cultures on media containing nalidixic acid and chloramphenicol. D. Complementation of CRISPR locus deletion by ectopically integrated pCRISPR construct. E. Northern blots with probes against each spacer in the CRISPR array and 5S rRNA loading control. F. Lack of targeting against protospacers in reverse orientation or (G) placed in untranscribed (UT) regions. See also Figure S1.
Consistent with the reported RNA-targeting mechanism of other type VI CRISPR systems (Abudayyeh et al., 2016), the transfer of plasmids with protospacers inserted in the non-transcribed strand of the cat gene (Fig. 1F and S1B), as well as the conjugation of plasmids with targets inserted into a weakly transcribed region of pTarget (Fig. 1G and S1B), was not restricted. In vitro experiments with purified Cas13 have demonstrated that it catalyzes nonspecific “collateral” RNase activity upon target binding (Abudayyeh et al., 2016; East-Seletsky et al., 2016; Liu et al., 2017b). Furthermore, it was reported that Leptotrichia shahii Cas13 activity elicited a growth defect when expressed in E. coli along with a target RNA (Abudayyeh et al., 2016). However, Cas13 achieves specific RNA knockdown in mammalian cells without apparent collateral activity (Abudayyeh et al., 2017). We noticed the formation of very small transconjugant colonies several days after of conjugation of the spc4 pTarget plasmid into wild-type recipients. We confirmed this severe growth defect by measuring the growth of wild-type and ΔCRISPR transconjugants in liquid cultures in the presence of chloramphenicol (Fig. 2A). To investigate the contribution of collateral RNA damage to this growth defect, we repeated the experiment in the absence of chloramphenicol, where the knock-down of the cat transcript target should not have an effect on cell growth. Again, we observed a marked growth defect of wild-type transconjugants (Fig. 2B), consistent with the observed nonspecific RNA cleavage activity in other type VI CRISPR systems and demonstrating that collateral RNase activity also occurs in natural hosts. Finally, we performed Northern blots to detect the cat target transcript and a control 5S rRNA in the slow-growing, wild-type transconjugants (Fig. 2C). Consistent with the presence of both specific and non-specific Cas13-mediated RNA degradation, we observed cleavage products for both transcripts, which were absent from wild-type cells carrying a targetless plasmid and from ΔCRISPR transconjugant samples. Altogether, these data show that in L. seeligeri, a native host, type VI-A CRISPR-Cas systems provide immunity against plasmid conjugation through the degradation of both spacer-specific and non-related transcripts, the latter resulting in a severe growth defect of the host.
Figure 2: The type VI CRISPR system elicits collateral RNA cleavage in the native host.
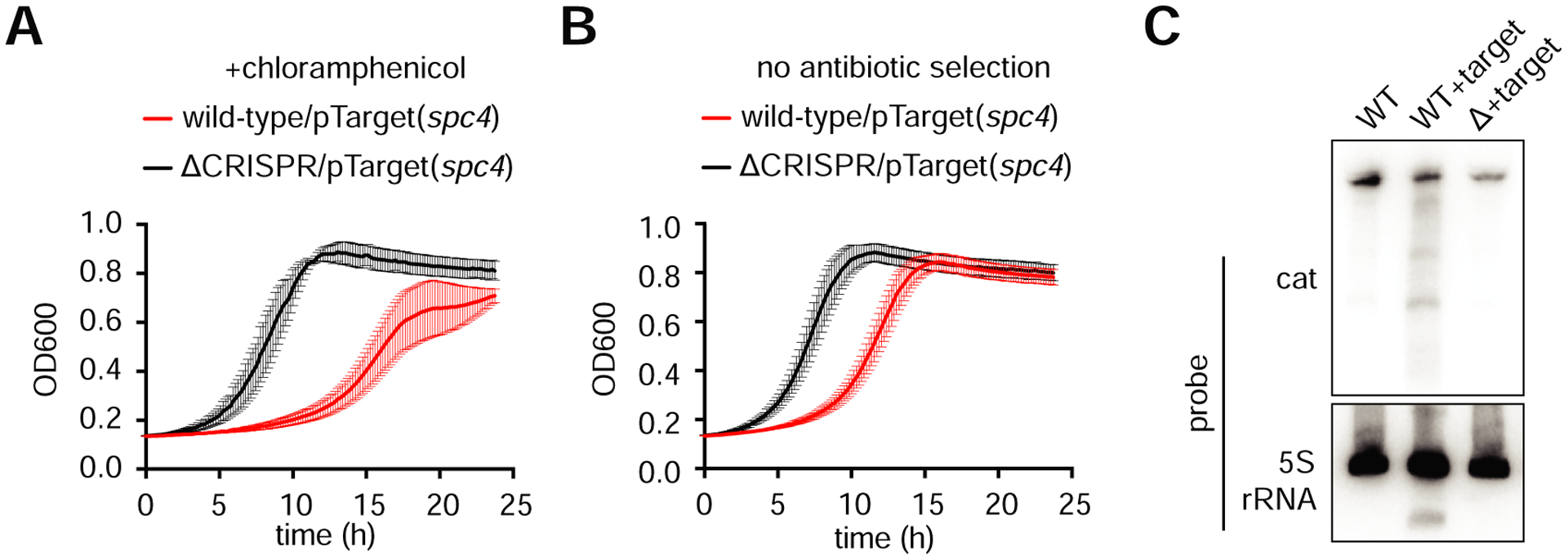
A. Liquid growth assay of the indicated strains illustrating CRISPR-dependent growth defects in the presence of target with or without target selection. Error bars represent standard error of the mean from four biological replicates. B. Northern blot analysis of target (cat) and nontarget (5S rRNA) transcripts in the indicated strains.
A GTT sequence immediately downstream of the target protospacer enables escape from type VI-A CRISPR targeting.
Previous studies in which type VI-A CRISPR systems were overexpressed in a heterologous host have uncovered sequence motifs adjacent to the target RNA protospacer that are required for the activation of Cas13 interference activity in vivo (Abudayyeh et al., 2017; Abudayyeh et al., 2016; Cox et al., 2017; East-Seletsky et al., 2016; Liu et al., 2017b). This motif (referred to as the protospacer flanking site, PFS) is typically found at one end of the RNA protospacer and in the case of type VI-A systems consists of a preference for non-G at the +1 position following the 3’ end of the target. In contrast, two other reports of Cas13a RNA cleavage activity in vitro did not detect any sequence preference at the PFS position (East-Seletsky et al., 2016). Similarly flexible target flanking sequence requirements were reported for other variants of Cas13 (Abudayyeh et al., 2016; Cox et al., 2017; Konermann et al., 2018; Liu et al., 2017b; Yan et al., 2018). Therefore, we wondered whether the PFS or other target sequence preferences of Cas13 in its native host might differ from those observed in transplanted systems.
To probe PFS requirements, we built target (spc2 and spc4) plasmid libraries containing five randomized nucleotides (1024 different sequences) on either the 5’ or 3’ side of the protospacer sequence following the cat gene, and transformed the libraries by conjugation separately into wild-type and ΔCRISPR L. seeligeri, selecting for target expression (Fig. S2A). For each combination of library and recipient strain, 20,000 transformants were pooled, target plasmids were isolated, the target site amplified by PCR and the products subjected to next-generation sequencing. In this experiment, the abundance of flanking sequences that escape targeting would be enriched in the wild-type strain but unaffected in the ΔCRISPR strain, and thus we calculated the enrichment ratio for each PFS as wild-type/ΔCRISPR reads. Upon conjugation into the ΔCRISPR strain both libraries yielded mostly large colonies. In contrast, transconjugants of the wild type strain were heterogeneous with both small and large colonies. When we tabulated PFS representation in the ΔCRISPR strain, each of the 1,024 possible 3’ and 5’ PFS sequences was detected for both spacers. No particular sequences were specifically depleted in the wild-type strain at either side of spc2 (Fig. S2B) or spc4 (Fig. S2C) targets, suggesting that most sequences are efficiently targeted by the type VI-A CRISPR system and that a PAM is not required for targeting as in type I, II, and V systems. We then looked at sequences that were enriched in the wild-type strain. No consistently enriched sequences were detected in the 5’ end of the spacer libraries for either target (Fig. S2D). In contrast, we detected a group of related sequences on the 3’ side of each protospacer that was enriched in the wild-type strain, suggesting that these targets escaped cleavage by Cas13. For the spc2 target the sequences began with GTTT (Fig. 3A), a motif detected when all sequences that were over five-fold enriched in wild type were weighted for representation in the spc2 library (Fig. 3B). A similar sequence (GTT) was also enriched in the spc4 library (Fig. 3C–D). Interestingly, re-analysis of PFS data obtained via expressing Leptotrichia shahii Cas13 in E. coli (Abudayyeh et al., 2016) also showed the presence of a GTT motif (Fig. S2E). In conclusion, our studies corroborate earlier findings indicating the absence of a PAM requirement for Cas13 targeting, but also uncovered a previously unrecognized motif, GTT or GTTT that, when present immediately downstream of the protospacer, prevents targeting.
Figure 3: A GTT sequence immediately downstream of the target protospacer enables escape from type VI-A CRISPR targeting.
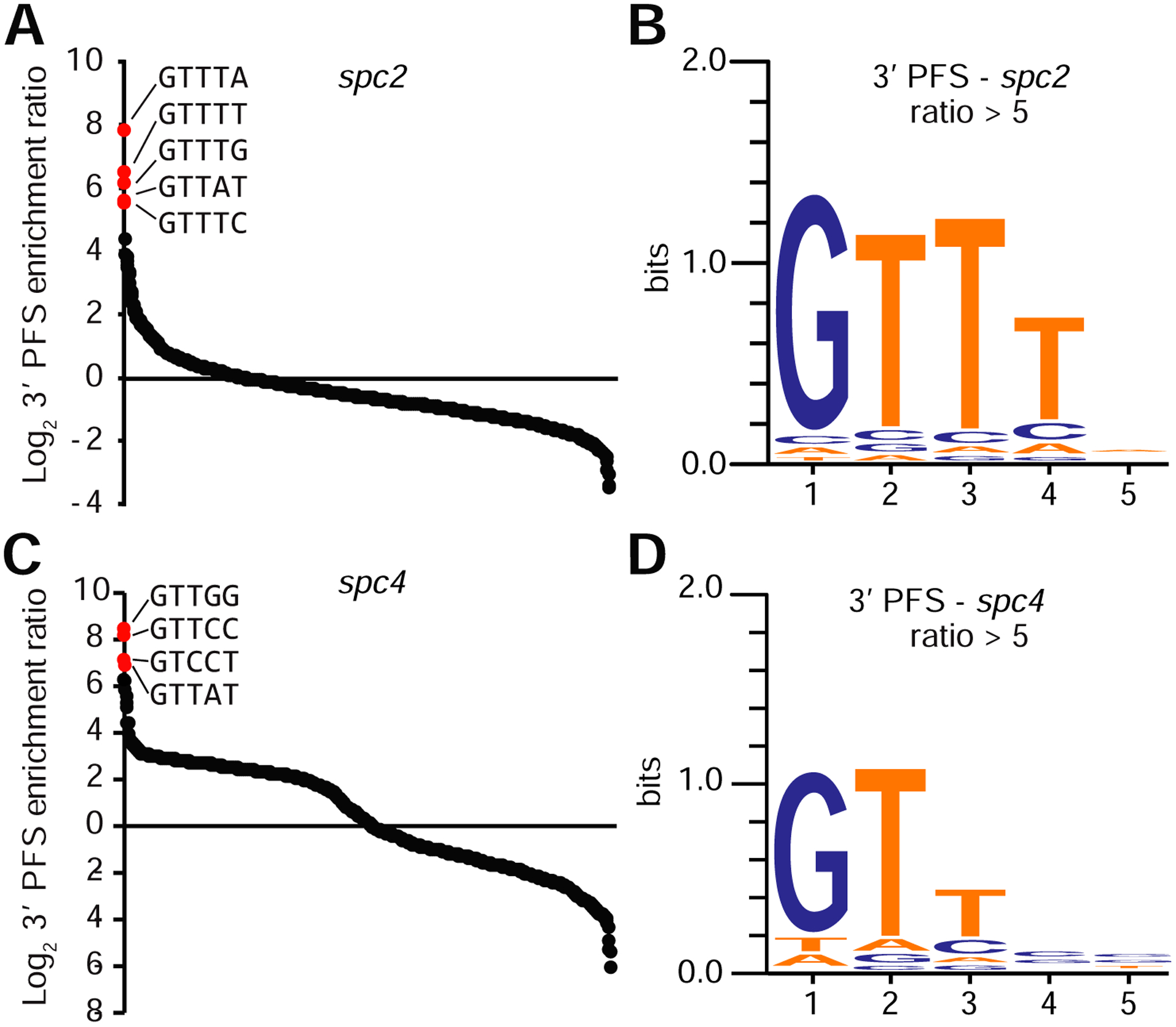
A. Distribution of log2 enrichment ratios (WT/DCRISPR) for all 1024 3’ flanking sequences for the spc2 library. Top enriched sequences are indicated in red. B. Weighted sequence logos for 3’ target flanking sequences enriched more than five fold in the CRISPR+ strain for spc2 library. C. Enrichment ratios and (D) sequence logo for the spc4 library. See also Figure S2.
Repeat-like sequences immediately downstream of the protospacer enable target protection.
Since the repeats of the L. seeligeri type VI-A CRISPR locus begin with the sequence 5’-GTTT-3’, we investigated the effect of the addition of repeat-like sequences downstream of the protospacer on immunity (Fig. 4A). To do this, we used our conjugation assay to test immunity against the transfer of plasmids harboring 0 to 8 nucleotides of repeat sequence downstream of the spc2 protospacer (Fig. 4B). All plasmids were transferred efficiently in the ΔCRISPR strain, and transformation of a target without homology to the CRISPR repeat in the 3’ PFS was restricted and led to the formation of only a few very small transconjugant colonies. Addition of up to three nucleotides of the repeat at the 3’ end of the protospacer resulted in similar restriction. However, a 3’ PFS with four repeat nucleotides conferred partial protection to the target plasmid, giving rise to more and intermediate-sized colonies. This result validated our previous target library findings, confirming that the enrichment for the 5’-GTTT-3’ was a consequence of protection from type VI-A immunity. Addition of five repeat nucleotides improved protection, and six or more made the target plasmid completely resistant to type VI-A targeting, yielding many transconjugant colonies of wild-type size (Fig. 4B). A similar protection was conferred by an 8-nt repeat downstream of the protospacer in pTarget(spc4), and as expected from the result of the library experiment, replacement of the 5’ PFS with 8 nucleotides of repeat sequence did not affect type VI-A immunity against neither spc2 nor spc4 targets (Fig. S3A). As a more sensitive assessment of growth rate of the different transconjugants, we examined their growth in liquid culture in the presence of target selection. As expected from the equivalent size of the ΔCRISPR transconjugant colonies on plates, their growth in liquid media was the same irrespective of the pTarget plasmid present (Fig. 4C). Wild-type transconjugants, on the other hand, displayed different degrees of growth delays (Fig. 4D) that were inversely correlated with the number of repeat-derived nucleotides at the 3’ end of the protospacer in pTarget (except those having 7 and 8 repeat nucleotides, which grew indistinguishably). We also tested an analogous series of 3’ PFS for the spc4 target (Fig. S3B). At least 6 nucleotides were required for full protection from CRISPR-mediated restriction, although even 1 nucleotide of repeat homology conferred some protective effect (Fig S3C–D).
Figure 4: Repeat-like sequences immediately downstream of the protospacer enable target protection.
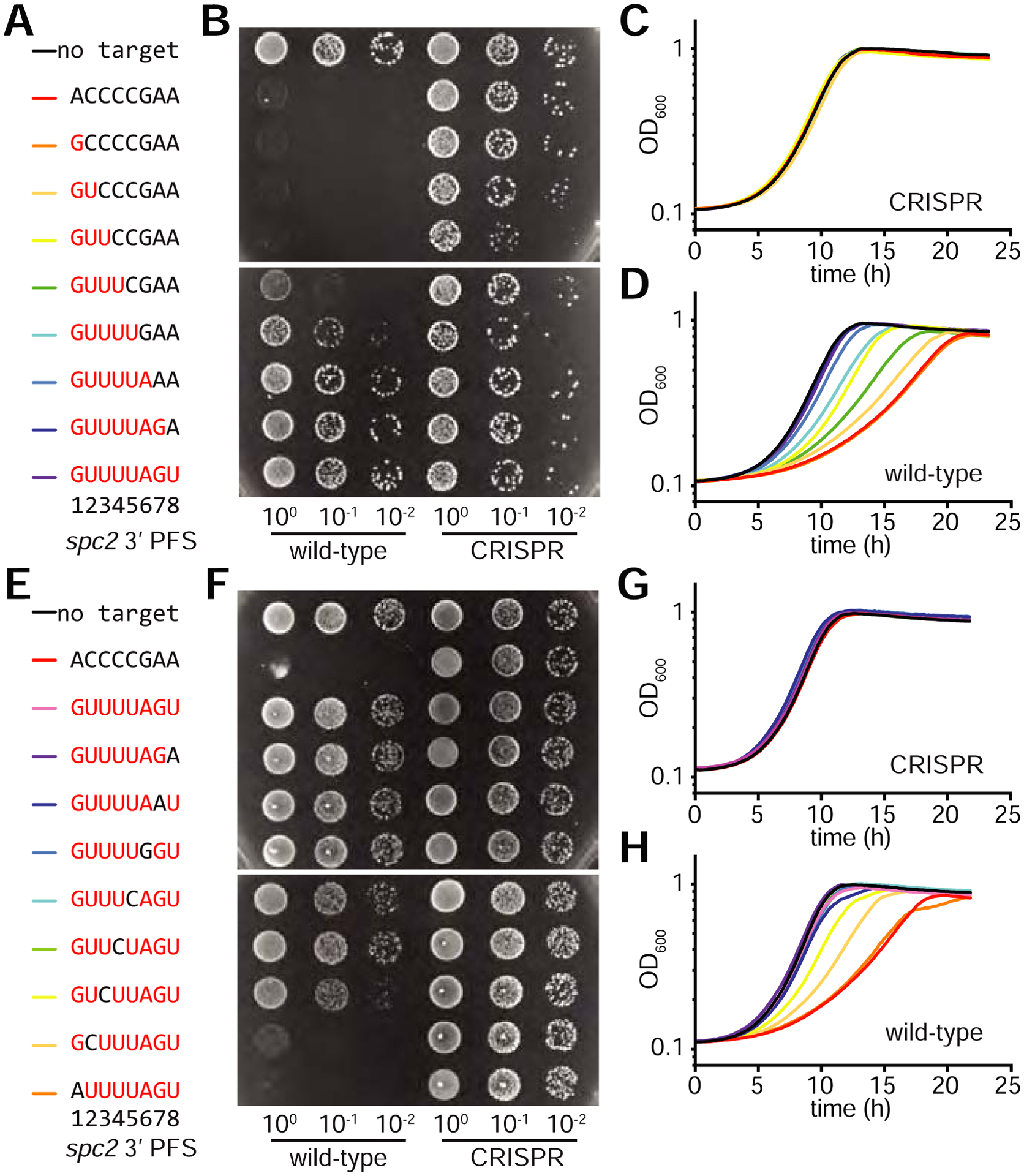
A. Series of spc2 pTarget plasmids containing repeat-derived sequences (shown in red) were tested for type VI CRISPR interference using (B) conjugation assay and (C-D) liquid growth assay. E. Series of spc2 pTarget plasmids containing 8nt repeat sequence with a 1nt mismatch at each position tested via (F) conjugation assay and (G-H) liquid growth assay. Traces represent the mean from four biological replicates. See also Figure S3.
To determine if any individual nucleotide position is critical for protection, we mutated each of the 8 repeat nucleotides flanking spc2 back to that present in the original 3’ PFS (Fig. 4E). First we looked at pTarget conjugation (Fig. 4F) and found that restoration of the original nucleotide at positions 8 to 4 (from the 3’ end of the protospacer) was as protective as the full, 8-nt repeat sequence. However, restoration at position 3 and 2 resulted in partial targeting (stronger in the presence of the position 2 mutation). Finally, restoration at position 1 resulted in wild-type levels of targeting. These results were corroborated after measuring the growth rates of the transconjugants. We found no differences in growth of the control ΔCRISPR transconjugants (Fig. 4F), but for the wild-type transconjugants (Fig. 4G) we observed normal growth for nucleotide restoration at positions 8 to 4, intermediate proliferation for positions 3 and 2 (worse growth for position 2), and the lowest growth rate for restoration of the first nucleotide (same growth as the full original sequence). These results showed that repeat nucleotides at positions 1, 2 and 3 are the most important for the protective effects of a repeat sequence placed downstream of the protospacer.
Extended crRNA-target complementarity prevents type VI-A CRISPR targeting.
The protection afforded by the presence of a repeat sequence downstream of the protospacer could in principle be the result of (i) an extension of the complementarity between the crRNA and the target (5’-GUUUUAGU-3’ in the target RNA, Fig. 5A), or (ii) a specific recognition of repeat-derived bases by the Cas13 nuclease that leads to an inhibition of target cleavage. To distinguish between these two possibilities, we tested whether mutations in the crRNA that disrupted complementarity could be rescued by compensatory mutations in the target 3’ PFS. If specific bases of the 5’-GUUUUAGU-3’ sequence were sensed by Cas13, the mutant targets would be protected from cleavage in spite of the restoration of complementarity by the compensatory mutation.
Figure 5: Extended crRNA-target complementarity prevents type VI-A CRISPR targeting.
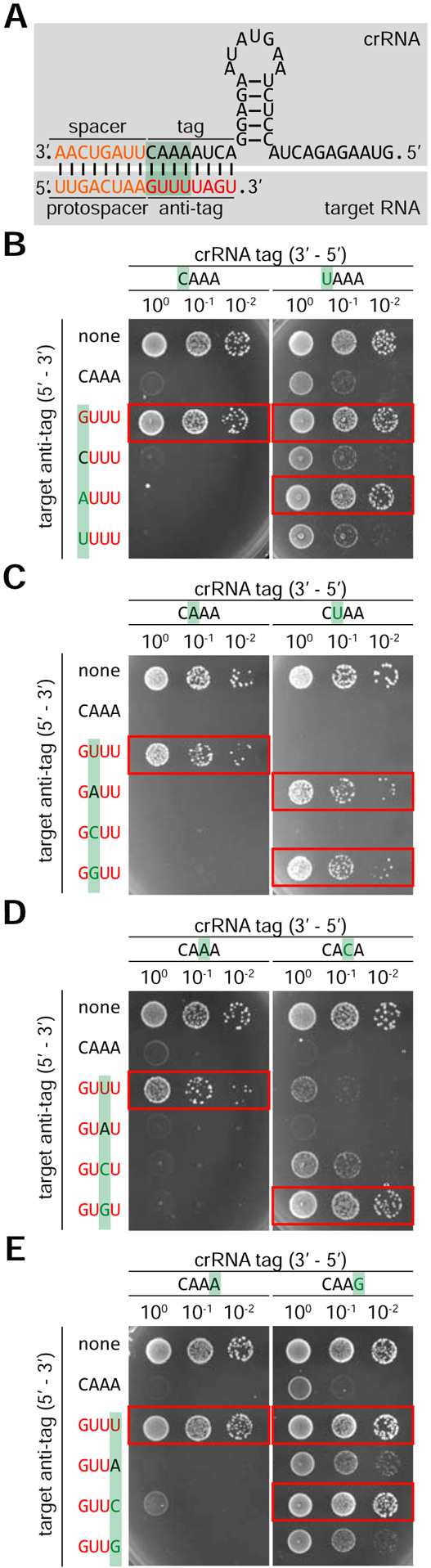
A. Schematic of type VI crRNA binding to RNA targets with crRNA tag complementarity. The spacer and protospacer are shown in orange, crRNA tag in black, and complementary anti-tag in red. Nucleotides in the green boxed region were tested for participation in sensing tag/anti-tag complementarity. B-E. Conjugation assays were carried out with either wild type L. seeligeri recipients or ones carrying mutations in the spc2 crRNA tag at positions 1 (B), 2 (C), 3 (D), or 4 (E), with the mutated position highlighted in green. pTarget plasmids carried spc2-matching targets with an anti-tag sequence (shown in red), except for mutations in the highlighted position. Combinations resulting in full tag/anti-tag complementarity (allowing G-U RNA wobble base pairing) are boxed in red. See also Figure S4.
We constructed a set of target plasmids containing a six-nucleotide repeat sequence downstream of the protospacer in pTarget(spc2), with all possible bases at positions 1 to 4, where single nucleotide deviations from the repeat sequence lead to restoration of targeting in our previous experiment (Fig. 4F). None of these flanking sequences affected plasmid conjugation into ΔCRISPR Listeria cells (Fig. S4). In Listeria recipients expressing the wild-type crRNA, except for the target with a six-nucleotide, repeat-derived 3’ PFS (5’-GUUUUA-3’), the transfer of all target plasmids harboring single-nucleotide mutations within this region was efficiently restricted (Fig. 5B–E). This result corroborates the data obtained for single nucleotide mutations in Figure 4F in the context of a six-nucleotide repeat sequence downstream of the protospacer (different than the eight-nucleotide repeat construct in our previous experiment) and expands the observation to all possible base changes in positions 1, 2, 3 and 4 of the repeat-derived 3’ PFS. We then tested the transfer of each of these target plasmids into Listeria recipients expressing crRNAs with mutations in the corresponding positions of the tag region. When compared to the wild-type crRNA, the mutation C>U in position 1 (Fig. 5B) provided incomplete immunity against the transfer of wild-type pTarget(spc2) (with the non-repeat 3’PFS 5’-CAAA-3’), suggesting that the mutation partially affects crRNA processing and/or RNA cleavage by Cas13. The presence of a C or U in position 1 of the 3’ PFS resulted in similarly incomplete targeting and therefore disrupted the protection conferred by the 6-nt repeat 3’ PFS. However, the presence of a G or A in this position restored complete protection. These results are consistent with a role for complementarity-mediated protection, since the pairing U:G (a wobble base pair present in dsRNA (Varani and McClain, 2000)) or U:A, but not the non-complementary U:C nor U:U, prevented type VI-A immunity. The mutation A>U in position 2 of the crRNA tag (Fig. 5C) did not affect Cas13 activity, since it provided full immunity against the transfer of wild-type pTarget(spc2). Similar targeting was observed with targets with mismatches (U:T and U:C) in this position, but protection was restored when base pairing was allowed (U:A and U:G). In the case of the A>C substitution in position 3 of the crRNA tag (Fig. 5D), the mutation completely restricted the transfer of wild-type pTarget(spc2), but failed in the presence of the C:G compensatory mutation; the non-complementary targets at this position were either fully (C:A) or partially targeted (C:U and C:C). Finally, the A>G mutation in position 4 (Fig. 5E) provided incomplete immunity against the transfer of wild-type pTarget(spc2) when compared to the transfer of a target-less plasmid, suggesting a partial deficiency in Cas13 crRNA processing and/or targeting. Non-complementary targets in this position (G:A or G:G) were equally targeted, but the restoration of complementarity (G:U and G:C) completely disrupted type VI-A immunity against conjugation. Taken together, these results indicate that extended complementarity between the crRNA tag and the 3’ PFS (hereafter referred as the “anti-tag”) abrogates type VI-A CRISPR-Cas immunity in L. seeligeri.
Extended complementarity inhibits Cas13 cis- and trans-cleavage in vitro.
To investigate the molecular details of the inhibitory effects of tag/anti-tag complementarity on Cas13 activity we decided to test our findings using biochemical assays. Because we were unable to purify L. seeligeri Cas13, we used Leptotrichia buccalis Cas13 (LbuCas13). We expressed and purified the nuclease from E. coli and incubated it with a synthetic crRNA (harboring 21 nucleotides of spacer sequence) and radiolabeled RNA targets as previously described (East-Seletsky et al., 2016; Liu et al., 2017a). We tested three different target sequences: a non-specific target, one containing a protospacer and another in which the protospacer was followed by an 8-nt anti-tag (Fig. 6A). We observed cleavage of the protospacer target but not of the non-specific or anti-tag targets (Fig. 6B and Fig. S5A). We then tested the previously reported trans-RNA cleavage activity of LbuCas13 (East-Seletsky et al., 2016; Liu et al., 2017a) by radiolabeling the non-specific RNA and adding unlabeled target or anti-target activating RNAs. We detected trans-RNA degradation only in the absence of the anti-tag sequence (Fig. 6C). Altogether these results demonstrate that the lack of type VI-A CRISPR-Cas immunity in vivo is due to the inactivation of Cas13 cis- and trans-RNase activity by the anti-tag target.
Figure 6: Extended complementarity inhibits Cas13 cis- and trans-cleavage in vitro.
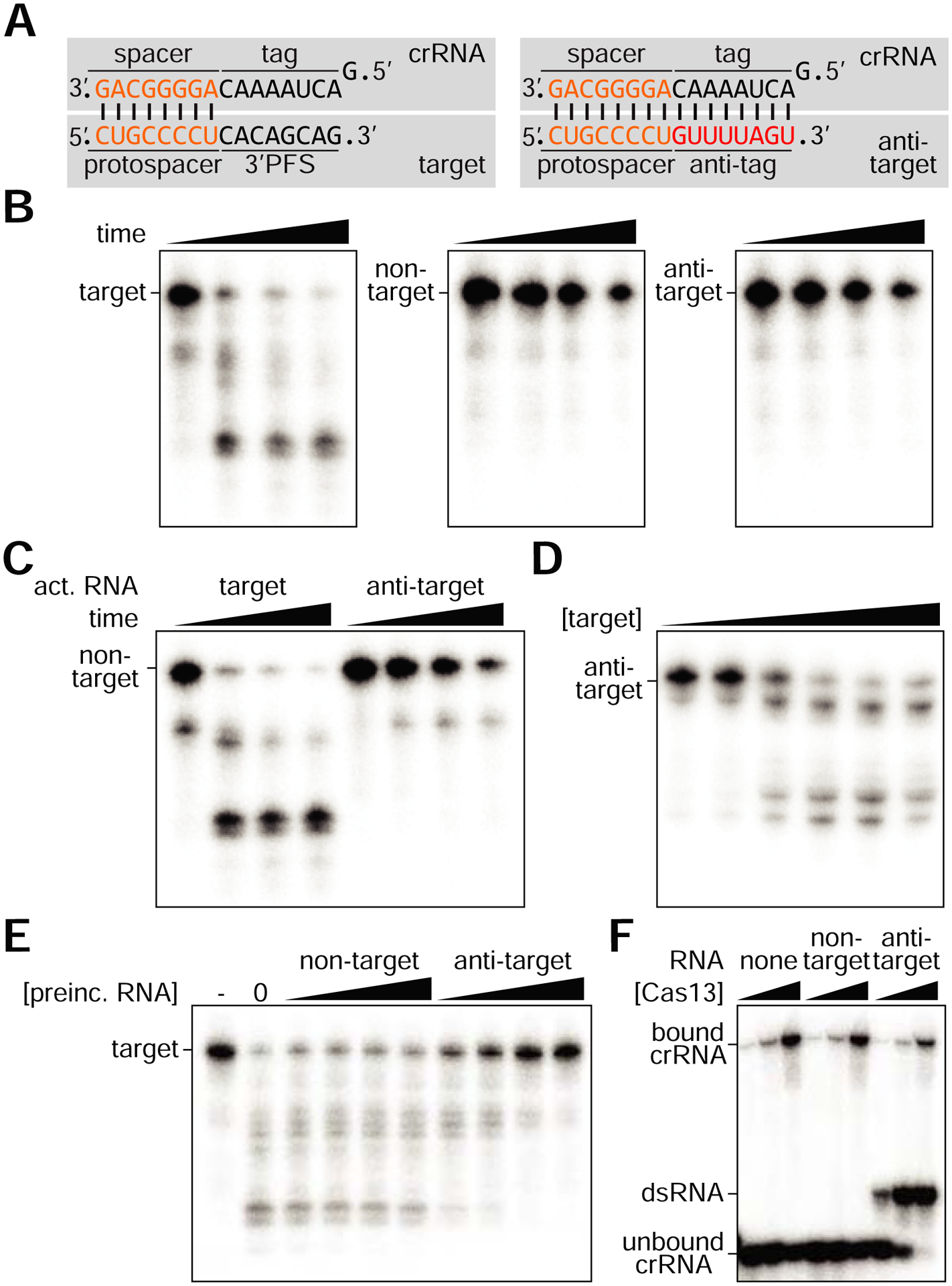
A. A schematic showing protospacer-containing target RNA (left) with noncomplementary 3’ extension, and anti-target RNAs bearing 8nt anti-tag that extends the crRNA:target duplex. B. In vitro cis-RNA cleavage time course with 10 nM purified LbuCas13 and 10 nM labeled target, non-target or anti-target RNA substrates. Reaction products were analyzed at 0, 5, 20, and 60 minutes incubation. C. Cas13 trans-RNA cleavage assay. Labeled nontarget RNA substrates (10 nM) were incubated with 10 nM Cas13 RNP and the indicated unlabeled activating RNA target (10 nM). D. Trans-cleavage of 10 nM labeled anti-target by preincubation of 10 nM Cas13 with increasing amounts (0.01, 0.1, 1, 10, 100, 1000 nM) of activating target RNA. E. Inhibition of target cleavage by addition of competing anti-target RNA. 10 nM target RNA was cleaved by Cas13 RNP in reactions pre-incubated with 10, 25, 50, or 100 nM nontarget or anti-target RNA. F. EMSA testing interaction between 1 nM crRNA and 0, 10, 100 nM Cas13 in the presence of 100 nM competitor RNA. See also Figure S5.
Next, we explored the mechanism behind the inhibition of Cas13 by the anti-tag target. Since in vitro assays only contain Cas13 and target and guide RNAs, the results above demonstrate that additional host factors are not required for Cas13 inhibition in the presence of the anti-tag. First we tested whether, as our in vivo data suggests, Cas13 inhibition requires the formation of an extended target RNA:crRNA duplex in the presence of the anti-tag. To address this, we tested whether anti-tag targets without access to the crRNA were substrates for activated Cas13 by initiating targeting reactions with LbuCas13 and an unlabeled target RNA and then adding a radiolabeled anti-tag target (Fig. 6D). We observed cleavage of the anti-tag target under these conditions, indicating that inhibition of targeting mediated by the anti-tag only occurs when this sequence is paired with the crRNA tag. Next, we determined whether the tag/anti-tag interaction locks Cas13 in an inactive form incapable of cleaving a protospacer-only target. We pre-incubated the Cas13/crRNA complex with an unlabeled anti-tag protospacer or with unlabeled non-target RNA to control for the amount of total RNA in each reaction. Then, we added a radiolabeled target RNA to the reaction and looked for cleavage products. While preincubation with a non-specific RNA could not prevent the cleavage of the target RNA (the cleavage products were indistinguishable from the control without unlabeled RNA), preincubation with the anti-tag RNA inhibited target cleavage in a concentration-dependent manner (Fig. 6E). To determine if this inhibition, which was strongest at the higher concentrations of anti-tag RNA, was also possible at excess concentrations of the target RNA, we repeated the experiment with a constant concentration of non-specific or anti-tag RNA (100 nM) and tested the inhibition of Cas13a trans-RNase activity when activated using different concentrations of target RNA (up to 1000 nM). We observed Cas13a inhibition even at the highest target RNA concentration (Fig. S5B), a result that suggests that the anti-tag inhibitor locks Cas13a in an inactive form. Finally, to rule out the possibility that anti-tag inhibition is achieved through the sequestration of the crRNA guide away from Cas13a; i.e. during pre-incubation the anti-tag RNA forms a dsRNA with the crRNA outside of the nuclease, we performed an electrophoretic mobility shift assay to assess whether the crRNA interaction with Cas13 was preserved after the addition of a large excess of anti-tag target RNA (anti-tag RNA:crRNA ratio of 100:1). Although we observed the formation of an anti-tag RNA:crRNA duplex, this was at the expense of the free crRNA (Fig. 6F). Compared to the addition of a non-target RNA control, most of the Cas13a-bound crRNA remained bound in the presence of the anti-tag target, demonstrating that anti-tag inhibition does not work through the separation of the nuclease from its guide. Collectively, our in vivo and in vitro experiments demonstrate that RNA targets harboring an anti-tag sequence immediately downstream of the protospacer form a ribonucleoprotein complex that contains an extended dsRNA duplex with the tag sequence of the crRNA. This additional RNA complementarity locks the Cas13a complex into an inactive conformation unable to perform both cis and trans RNA degradation.
Anti-tag inactivation of Cas13 prevents self-targeting of antisense CRISPR transcripts.
Our findings indicate that RNAs originating from antisense transcription of the CRISPR array will have a perfect anti-tag and should be spared from Cas13 cleavage. This would be similar to the mechanism employed by type III systems to avoid self-targeting (Elmore et al., 2016; Kazlauskiene et al., 2016; Marraffini and Sontheimer, 2010). We checked for the presence of antisense CRISPR transcription in L. seeligeri by both Northern blot and β-galactosidase transcriptional fusion assays (Fig. S6). We did not detect antisense transcripts in either of these experiments, neither in the presence nor absence of a spc4 target. Therefore, to test if tag/anti-tag pairing could prevent self-targeting of the CRISPR array, we engineered plasmids with the CRISPR locus in the sense and anti-sense orientations with respect to the constitutive ldh promoter and followed by reverse-facing terminators to isolate the CRISPR sequences from transcription from other promoters (Fig. 7A). As expected from our findings, the presence of an anti-tag sequence prevented the targeting of the perfectly matching protospacers within the anti-sense CRISPR transcript, which we confirmed was expressed (Fig. 7B and S6B). In contrast, the introduction of a G>C mutation in position 1 of the anti-tag, which generates a mismatch with the crRNA tag, resulted in a strong type VI-A CRISPR-Cas response against the anti-sense CRISPR plasmid (Fig. 7AB). These results exemplify the importance of the tag/anti-tag inhibition of Cas13 to prevent the otherwise potentially toxic bidirectional transcription of the CRISPR array.
Figure 7: Anti-tag inactivation of Cas13 prevents self-targeting of antisense CRISPR transcripts.
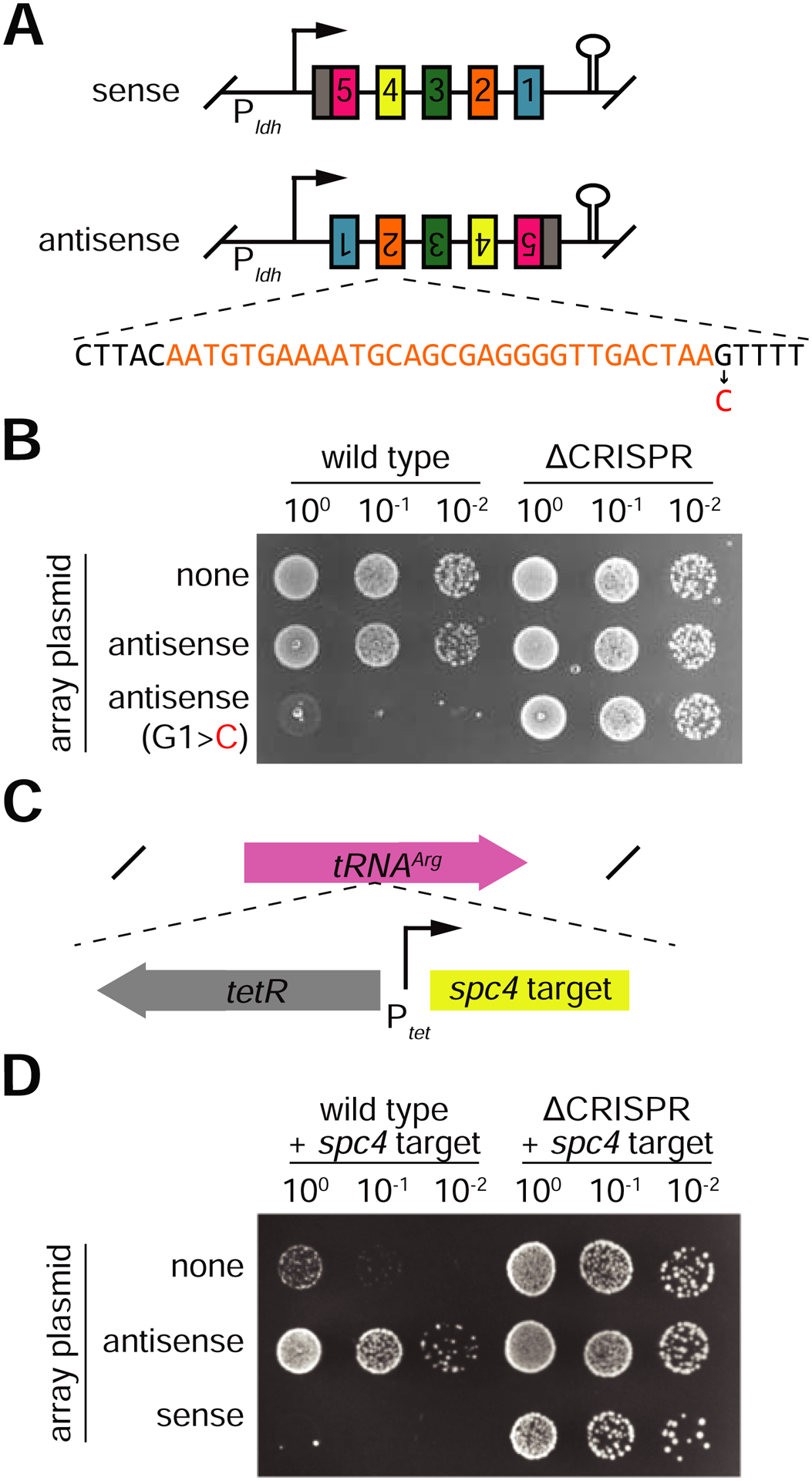
A. Plasmids engineered to constitutively express the CRISPR array in sense or anti-sense orientations. A variant of the antisense plasmid was constructed with a single mutation in the PFS flanking spc2 (spc sequence in orange, repeat sequences in black, mismatch highlighted in red). B. Conjugation assay showing CRISPR-dependent toxicity of a single mismatch in antisense CRISPR array transcript C. An aTc-inducible target construct matching spc4 was integrated into the tRNAArg locus of the L. seeligeri genome. D. Conjugation assay demonstrating inhibition of CRISPR interference by expression of antisense CRISPR array. Plasmids bearing sense or antisense array were conjugated into WT or ΔCRISPR recipients carrying the inducible target, and plated in the presence of 100 ng/mL aTc.
Given that (i) our in vitro findings showed that anti-tag targets prevent both cis and trans Cas13a RNase activities (Fig. 6), and (ii) anti-sense CRISPR transcripts contain anti-tags and are tolerated in vivo, we wondered whether anti-sense transcription of the CRISPR locus could inhibit the trans RNA degradation by type VI-A systems. To test this we integrated an anhydrotetracycline (aTc)-inducible, spc4-matching target into the tRNAArg gene of both wild-type and ΔCRISPR L. seeligeri strains (Fig. 7C). Addition of aTc elicited a strong growth defect of wild-type, but not ΔCRISPR cells (Fig. 7D). Therefore we used this assay to determine if anti-sense CRISPR transcripts could interfere with type VI-A immunity against the chromosomal target. We introduced the plasmids expressing the CRISPR locus in the sense and anti-sense orientation (Fig. 7A) and added aTc. We found no immunity against the chromosomal target in the presence of the anti-sense CRISPR transcript (Fig. 7D). These results show that anti-sense transcription of the CRISPR locus can inhibit Cas13a activity in vivo and therefore it could be used for the regulation of the type VI-A CRISPR-Cas immune response.
DISCUSSION
Here we have shown that type VI-A CRISPR-Cas immunity is inhibited by extended complementarity between the crRNA tag and the 3’ protospacer flanking sequence (here named the “anti-tag”, Fig. 5A). Our data indicate that this complementarity prevents targeting by L. seeligeri Cas13 in vivo, as well as by L. buccalis Cas13 in vitro. Biochemical assays indicate that anti-tag targets interfere with the activation of the Cas13:crRNA ribonucleoprotein complex through the direct annealing to the crRNA guide within the nuclease, preventing both cis- and trans-cleavage of RNA molecules. The presence of the same inhibitory mechanism in both LseCas13 and LbuCas13, which are only 20.8% identical, suggests it is a widespread feature of type VI-A CRISPR-Cas systems. Given the recent repurposing of Cas13a as a tool for nucleic acid detection and RNA knockdown in eukaryotic cells (Abudayyeh et al., 2017; East-Seletsky et al., 2016; Gootenberg et al., 2018; Gootenberg et al., 2017), our findings highlight important targeting rules for the design of effective crRNA guides in these technologies.
A previous screen reported that a single G in position 1 of the 3’ PFS is sufficient to protect targets from cleavage by LshCas13a in an E. coli heterologous host (Abudayyeh et al., 2016). Our re-analysis of the LshCas13a 3’ PFS screen data in E. coli indicated that highly protected targets contain extended repeat sequences in the 3’ PFS, beyond the first G (Fig S2E). Therefore we believe that both studies produced consistent screen results. However, during the validation of the screen data with individual targets we found that 5–7 complementary nucleotides are required for full protection in the L. seeligeri natural host (Fig. 6A–D), whereas a single complementary G nucleotide is required in E. coli (Abudayyeh et al., 2016). We believe that this discrepancy could be due to (i) improper folding or activity of Cas13a in the heterologous host, resulting in the production of a weaker nuclease for which less tag/anti-tag pairing has a greater inhibitory effect, and/or (ii) the use of different target sequences, whose nucleotide composition could influence the effect of the 3’PFS on cleavage. Indeed, we found some spacer-specific differences when we compared the degrees of tag/anti-tag complementarity required to eliminate the immunity against spc2 and spc4 targets (Figs. 4 and S3). These discrepancies highlight the importance of studying the biology of type VI systems in their natural hosts.
Whereas Cas13a cleaves upstream of the stem-loop structure formed by repeat sequences in the pre-crRNA of type VI-A systems, Cas13b cleaves downstream of this structure in type VI-B systems (Smargon et al., 2017). As a consequence of these cleavage differences, the crRNA tag sequence is located in the 5’ end of type VI-A crRNAs and in the 3’ of type VI-B crRNAs. The PFS requirements reported for type VI-B systems are also switched in orientation, and the presence of C on the 5’ side of the protospacer prevents efficient targeting in heterologous hosts (Cox et al., 2017; Smargon et al., 2017). We speculate that, since type VI-B repeats described so far begin with G, this nucleotide will be part of the crRNA tag and therefore the presence of a 3’ end anti-tag in type VI-B targets may inhibit Cas13b nuclease activity in the same way that a 5’ end anti-tag inhibits Cas13a. Although for some Cas13 homologs no particular PFS requirements have been identified (Abudayyeh et al., 2017; Konermann et al., 2018; Yan et al., 2018), it remains to be determined if the presence of an anti-tag flanking the protospacer prevents target cleavage. We suspect that the inhibition of Cas13a generated by the extension of complementarity between the crRNA and the target RNA could be a general feature of all type VI CRISPR-Cas systems. Understanding the effect of anti-tag sequences in other Cas13 homologs will be important for the investigation of the function of type VI systems both in their native bacteria and heterologous eukaryotic hosts.
The tag/anti-tag inhibition of Cas13a targeting has parallels in type III CRISPR-Cas systems. In these systems recognition of a complementary transcript by the crRNA guide within the Cas10-Csm (type III-A) or Cas10-Cmr (III-B) nuclease complex results in the specific cleavage of the target RNA and non-specific degradation of the transcribed DNA (Elmore et al., 2016; Estrella et al., 2016; Kazlauskiene et al., 2016; Samai et al., 2015). In contrast with the effect of the anti-tag in type VI-A CRISPR-Cas immunity, which prevents both specific and non-specific cleavage of RNA, tag/anti-tag pairing in type III systems does not affect cleavage of the complementary RNA by the Csm3/Cmr4 subunit, but completely abolishes non-specific DNA degradation by the Cas10 nuclease (Kazlauskiene et al., 2016; Samai et al., 2015). Type III systems also express an RNase, Csm6 (III-A) or Csx1 (III-B), which, similarly to Cas13, contains a HEPN RNA degradation domain that hydrolyzes non-specific RNAs after recognition of an RNA target by the crRNA guide (Jiang et al., 2016; Niewoehner and Jinek, 2016). Csm6/Csx1, however, are not part of the type III effector complexes and, as opposed to Cas13, they do not carry a guide RNA themselves. Instead, the non-specific RNase activity is activated indirectly through the generation of a second messenger: upon crRNA recognition of the target RNA, the PALM domain of Cas10 converts ATP into a cyclic oligoadenylate product which allosterically activates Csm6 (Kazlauskiene et al., 2017; Niewoehner et al., 2017). It remains to be determined if the tag/anti-tag interaction in type III systems abolishes this activation.
What role might crRNA tag/anti-tag inhibition play during type VI-A CRISPR-Cas immunity? We can speculate about two possible scenarios where this inhibition will be beneficial to the host. First, similar to type III systems (Kazlauskiene et al., 2016; Marraffini and Sontheimer, 2010), we showed that type VI-A systems can use tag/anti-tag inhibition to prevent the triggering of Cas13a activity by antisense transcription of the CRISPR array (Fig. 7A–B). Although we did not detect antisense CRISPR transcripts in L. seeligeri, such transcription could arise after the horizontal gene transfer of CRISPR systems (Godde and Bickerton, 2006). The random integration of a laterally transferred type VI-A CRISPR locus into the genome of the new host could result in toxic head-on transcription across the CRISPR array. If such scenarios were common, then type VI-A systems may benefit from a mechanism to avoid this form of auto-immunity. A second use of the tag/anti-tag inhibition could be for the regulation of type VI-A immunity. Under circumstances where Cas13 activity is undesirable, controlled antisense CRISPR transcription could rapidly abrogate Cas13a activity, as we showed using an inducible promoter upstream of an inverted CRISPR array (Fig. 7C–D). This mode of inactivation would be faster than the direct turn off of crRNA or cas13a gene expression, since in this case the elimination of CRISPR activity will not be immediate but will depend on the turnover rate of the type VI-A ribonuclease complex. While it remains to be determined if this type of regulation naturally occurs in microorganisms, tag/anti-tag inhibition of Cas13a activity could certainly be used to quickly turn off Cas13a-mediated knock-down of transcripts in eukaryotic cells.
STAR METHODS
Contact for Reagents and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Luciano Marraffini (marraffini@rockefeller.edu).
Experimental Model and Subject Details
Bacterial strains and growth conditions:
All L. seeligeri strains were derived from ATCC strain 35967 (Rocourt and Grimont), and were propagated in Brain Heart Infusion (BHI) broth or agar at 30°C. Constructs were cloned in E. coli DH5α, miniprepped, and transformed into the conjugative donor strain E. coli SM10 for conjugative mating. All E. coli strains were cultured in Lysogeny Broth (LB) at 37°C.
Method Details
Plasmid construction
The broad host range conjugative vector pAM401-oriT (Sauer et al., 2010) was used to construct pAM8 by replacement of its Gram-negative chloramphenicol resistance cassette by the ampicillin resistant cassette of pUC19. pTarget plasmids were constructed by the addition of a protospacer downstream of the Gram-positive chloramphenicol resistance gene of pAM8. To construct pCRISPR plasmids, the L. seeligeri CRISPR locus was inserted into the PSA site-specific integrating vector pPL2e (Lauer et al., 2002). Lists of strains, plasmids, and oligonucleotide primers, along with details of plasmid construction can be found in Supplementary Data 1.
E. coli – L. seeligeri conjugation:
Donor E. coli SM10 strains carrying E. coli-Listeria shuttle vectors were cultured in LB containing 100 μg/mL ampicillin (for pAM8-derived vectors) or 25 μg/mL chloramphenicol (for pPL2e-derived vectors). Recipient L. seeligeri strains were cultured in BHI. 100 μL each of donor and recipient culture were combined in 10 mL BHI, and concentrated onto a 0.45 μm membrane filter, which was then overlaid onto a BHI agar plate containing 8 μg/ml oxacillin, which weakens the cell wall, enhancing conjugation. Mating plates were incubated for 4 hr at 37°C, then cells were resuspended in 2 mL BHI, diluted as indicated, and plated at 30°C on selective media containing 50 μg/mL nalidixic acid (selects against donor). Selection for plasmid recipients was done with either 15 μg/mL chloramphenicol (for pAM8-derived vectors) or 1 μg/mL erythromycin (for pPL2e-derived vectors). Plates were incubated for 72 hr before imaging.
L. seeligeri strain construction:
Allelic replacement in L. seeligeri was conducted using a “loop in, loop out” strategy, illustrated in Figure S1. 1 kb homologous sequences flanking each side of the region to be replaced were cloned into a suicide plasmid unable to replicate in L. seeligeri. The plasmid carries a gram negative origin of replication, an oriT sequence for conjugative transfer, a lacZ gene (G. stearothermophilus bgaB), and a gram positive chloramphenicol resistance marker. This plasmid was transferred to L. seeligeri via conjugation as described above, and 100 μL of resuspended cells was plated for selection on 50 μg/mL nalidixic acid and 15 μg/mL chloramphenicol. Isolated transconjugants (integrants) were streak-purified and confirmed lacZ+ by patching on BHI plates containing 100 μg/mL 5-Bromo-4-Chloro-3-Indolyl β-D-Galactopyranoside (X-gal). Integrants were passaged (grown to saturation, then diluted 1000 fold) four times in BHI without selection. Passaged cultures were diluted and plated on BHI X-gal, and incubated 2–3 days at 37°C. White ( lacZ-) colonies (excisants) were isolated, cultured in BHI, and chromosomal DNA was prepared by lysozyme treatment followed by phenol-chloroform extraction. Each excisant was checked for the deletion by PCR using primers flanking the desired deletion, and Sanger-sequenced.
Northern blot analysis:
RNA was isolated from 1 mL of L. seeligeri culture at OD600 0.5. Cell pellets were resuspended in 80 μL RNase-free PBS, and lysed by a 5 min treatment with lysozyme at 20 μg/mL, followed by the addition of 1% sarkosyl. RNA was purified from these lysates using the Zymo Research Direct-zol RNA MiniPrep Plus kit according to the manufacturer’s instructions. For Northern blot analysis, 20 μg RNA per sample was diluted in RNA loading dye and separated by gel electrophoresis on a 6% denaturing TBE-urea polyacrylamide gel. RNA was transferred to an Invitrogen BrightStar Plus nylon membrane using a semi-dry transfer apparatus. RNA was fixed to membranes using a Stratalinker UV crosslinker (Optimal Crosslinking setting). Membranes were pre-hybridized 30 min at 42°C in 2X SSC 1% SDS + 0.1 mg /mL denatured salmon sperm DNA. 250 pmol ssDNA probe was labeled with 20 μCi γ−32P-ATP, purified using an Illustra MicroSpin G-50 column, and hybridized to the membrane overnight at 42°C. Membranes were washed twice with 2X SSC 0.1% SDS, once with 1X SSC 0.1% SDS, then exposed to a phosphoscreen for 24 hr, and imaged using a Beckman Coulter FLA7000IP Typhoon storage phosphorimager.
RNA sequencing:
L. seeligeri RNA samples isolated as described above were prepared for RNA sequencing by depletion of ribosomal RNA using the Ribo-Zero rRNA removal (Gram Positive Bacteria) kit (Illumina) according to the manufacturer’s instructions. The remaining RNA was prepared for deep sequencing with the TruSeq Stranded mRNA Library Prep kit (Illumina), and sequenced on the MiSeq platform using a 150 cycle cartridge (v3 reagents) on a single-end run. Reads were quality-trimmed and mapped to the pAM8 plasmid sequence using STAR (Dobin et al., 2013), and coverage was visualized with Artemis (Carver et al., 2012). Raw sequencing reads have been deposited to the Sequence Read Archive under accession SRP142551.
PFS library construction and deep sequencing:
Templates for library construction were amplified from pAM8 using oAM288+oAM185 (first fragment) and oAM288+oAM183 (second fragment). A second round of amplification introduced the protospacer and five adjacent randomized nucleotides. PFS libraries were constructed by amplification of the first round product with oAM290+oAM185 (spc2, 3’ PFS), oAM291+oAM183 (spc2, 5’ PFS), oAM292+oAM185 (spc4, 3’ PFS), or oAM293+oAM183 (spc4, 5’ PFS). These products were ligated to the appropriate acceptor (amplified with oAM246+oAM183 (spc2, 3’ PFS), oAM245+oAM185 (spc2, 5’ PFS), oAM250+oAM183 (spc4, 3’ PFS), or oAM249+oAM185 (spc4, 5’ PFS) via Gibson assembly to circularize the plasmid. Gibson products were transformed into NEB5α competent cells selecting on ampicillin and 20,000 transformants were isolated for each library. Transformants were pooled, plasmids were isolated and transformed into E. coli SM10 (selecting on ampicillin) for conjugation. Transformants were pooled and inoculated into LB amp for mating. 100 μL donor and recipient cultures were mated as described above, and transconjugants were selected on BHI agar containing nalidixic acid and chloramphenicol at 30°C. 20,000 transconjugants were selected and pooled. Target plasmids were isolated by resuspending pooled cell pellets in 250 μL Qiagen Miniprep buffer P1, adding 1mg lysozyme, incubating 37°C 10 min and proceeding with the miniprep protocol according to the manufacturer’s instructions. Target amplicon was amplified with oAM334 and oAM336 using 10 ng plasmid as template. PCR products were prepared for sequencing using the TruSeq Nano DNA LT kit and sequencing was carried out with an Illumina MiSeq system using a 150 cycle cartridge (v3 reagents) on a single-end run. Target sequences were extracted and PFS sequences tabulated and sorted using standard UNIX commands. Raw sequence reads have been uploaded to SRA under accession SRP142551. Normalized PFS counts for each target and strain are available in Supplementary Data 1.
Protein purification:
His6-MBP-LbuCas13 was purified essentially as previously described. Briefly, the E. coli BL21 (DE3) Rosetta2 strain were transformed with the LbuCas13 expression plasmid (Addgene), and fresh transformants were grown to OD600=0.4 in LB + ampicillin, then diluted in 1L 2×YT broth + ampicillin and grown at 37°C to OD600=0.6. Then, IPTG was added to 1mM and expression was carried out overnight at 16°C. Cells were pelleted and lysed by lysozyme treatment and sonication in lysis buffer (50mM Tris pH 7.0, 0.5M NaCl, 5% glycerol, 1mM DTT). Clarified lysates were applied to pre-equilibrated amylose resin (NEB), washed with 25 column volumes of lysis buffer, and eluted with the same buffer containing 10mM maltose. Eluate was cleaved with TEV protease at a 100:1 molar ratio overnight at 4°C, and untagged Lb uCas13 was isolated from cleaved tag by gel filtration chromatography (isocratic elution). A single disperse peak contained purified untagged LbuCas13. Fractions were concentrated to 1 μM and flash-frozen.
In vitro RNA cleavage assays:
crRNA and ssRNA targets were commercially synthesized (IDT, sequences available in Supplementary Data 1), labeled at 1 μM with T4 polynucleotide kinase and 10 μCi γ−32P-ATP, and purified using Illustra MicroSpin G-50 columns. Cleavage reactions were carried out in 10 mM HEPES pH 7.0, 50 mM KCl, 5 mM MgCl2, and 5% glycerol. LbuCas13, crRNA and labeled targets were combined, each at 10 nM, and reactions were performed at room temperature for 20–60 min (as indicated). An equal volume of 2X formamide RNA sample buffer was added to each reaction, samples were boiled 5 min, cooled on ice 2 min, and cleavage products were resolved on 15% TBE-urea denaturing PAGE gels. Gels were exposed to a phosphoscreen for 1 hr and imaged using a Beckman Coulter FLA7000IP Typhoon storage phosphorimager.
Electrophoretic mobility shift assay:
Commercially synthesized crRNA was 5’ end-labeled as described above. 1 nM labeled crRNA was combined with 0, 10, or 100 nM LbuCas13 and when indicated, 100 nM unlabeled competitor RNA. Binding reactions were performed for 90 min at room temperature in 20 mM HEPES pH 7.0, 50 mM KCl, 10 μg/mL bovine serum albumin, 100 μg/mL salmon sperm DNA, 0.01% Triton-X-100, and 5% glycerol. Samples were resuspended in native PAGE sample buffer, and resolved by 8% nondenaturing TBE PAGE at 4°C. Gels were exposed to a phosphoscreen f or 1 hr and imaged using a Beckman Coulter FLA7000IP Typhoon storage phosphorimager.
β-galactosidase assay:
Gene expression experiments were performed using 1 mL samples of exponentially growing cultures resuspended in 1 mL Z buffer (100 mM phosphate buffer pH 7.4, 10 mM KCl, 1 mM MgSO4, 50 mM β-mercaptoethanol, 1 mg/mL lysozyme). Samples were incubated at 37°C 5 min before adding 200 μL 4 mg/m L o-nitrophenyl-β-D-galactopyranoside (ONPG) and then transferred to 55°C for measurements of heat-stable β-galactosidase activity. Reactions were quenched with 500 μL 1M Na2CO3, insoluble debris pelleted, and absorbance measurements were taken at 420 nm and 550 nm.
Quantification and Statistical Analysis
For quantification of deep sequencing reads in the L. seeligeri PFS screen, flanking sequences were counted in the wild type and ΔCRISPR libraries, and normalized to the total number of reads for each library. For each of the 1024 flanking sequences, the WT:ΔCRISPR normalized abundance ratio was calculated and plotted. For growth curves in Fig. 2, error bars represent standard error of the mean from four biological replicates. For β-galactosidase assays in Fig S6, error bars represent standard error of the mean from two biological replicates.
Data and Software Availability
The raw PFS screen data related to Figure 3 and raw RNA-seq reads related to Figure S1 have been deposited in the Sequence Read Archive under ID code SRP142551.
Supplementary Material
Data S1: Lists of normalized PFS abundances, plasmids, and oligonucleotides used in the study. Related to Figure 3 and STAR Methods.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Listeria seeligeri Rocourt and Grimont | ATCC | 35967 |
| Listeria seeligeri ΔCRISPR | This paper | N/A |
| Escherichia coli SM10 | Darren Higgins, Harvard Medical School | N/A |
| Deposited Data | ||
| PFS screen and RNA sequencing of L. seeligeri samples | This paper | Sequence Read Archive SRP142551 |
| PFS screen from Leptotrichia shahii type VI locus in E. coli | Abudayyeh et al., 2016 | Sequence Read Archive SRX1820082, SRX1820086 |
| Oligonucleotides | ||
| See Data S1 | This paper | N/A |
| Recombinant DNA | ||
| See Data S1 | This paper | N/A |
Highlights:
Target-activated Cas13 cleaves RNA indiscriminately in its native bacterial host
Extended complementarity between guide and target RNA blocks cleavage
Targets with extended guide pairing lock Cas13 into an inhibited state
Inhibition by extended complementarity prevents self-targeting
ACKNOWLEDGEMENTS:
We thank all members of the Marraffini Lab for helpful discussion and encouragement, Daniel Portnoy for sharing E. coli-Listeria shuttle vectors pAM401-oriT and pPL2e, Darren Higgins and Daniel Grubaugh for Listeria advice and sharing allelic exchange and conjugation protocols. AJM was supported by a Merck fellowship and a Helen Hay Whitney postdoctoral fellowship. Support for this work comes from the National Institute of Health Director’s Pioneer Award 1DP1GM128184-01 (LAM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS: The authors declare no competing interests.
REFERENCES
- Abudayyeh OO, Gootenberg JS, Essletzbichler P, Han S, Joung J, Belanto JJ, Verdine V, Cox DBT, Kellner MJ, Regev A, et al. (2017). RNA targeting with CRISPR-Cas13. Nature 550, 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abudayyeh OO, Gootenberg JS, Konermann S, Joung J, Slaymaker IM, Cox DB, Shmakov S, Makarova KS, Semenova E, Minakhin L, et al. (2016). C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Brouns SJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJ, Snijders AP, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T, Harris SR, Berriman M, Parkhill J, and McQuillan JA (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, and Zhang F (2017). RNA editing with CRISPR-Cas13. Science 358, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, and Charpentier E (2011). CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471, 602–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, and Moineau S (2008). Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol 190, 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- East-Seletsky A, O’Connell MR, Knight SC, Burstein D, Cate JH, Tjian R, and Doudna JA (2016). Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore JR, Sheppard NF, Ramia N, Deighan T, Li H, Terns RM, and Terns MP (2016). Bipartite recognition of target RNAs activates DNA cleavage by the Type III-B CRISPR-Cas system. Genes Dev. 30, 447–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrella MA, Kuo FT, and Bailey S (2016). RNA-activated DNA cleavage by the Type III-B CRISPR-Cas effector complex. Genes Dev. 30, 460–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, and Siksnys V (2012). Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. U.S.A 109, E2579–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godde JS, and Bickerton A (2006). The repetitive DNA elements called CRISPRs and their associated genes: evidence of horizontal transfer among prokaryotes. J. Mol. Evol 62, 718–729. [DOI] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung J, Collins JJ, and Zhang F (2018). Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 360, 439–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, et al. (2017). Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 356, 438–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale C, Kleppe K, Terns RM, and Terns MP (2008). Prokaryotic silencing (psi)RNAs in Pyrococcus furiosus. RNA 14, 2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Samai P, and Marraffini LA (2016). Degradation of phage transcripts by CRISPR-associated RNases enables type III CRISPR-Cas immunity. Cell 164, 710–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskiene M, Kostiuk G, Venclovas C, Tamulaitis G, and Siksnys V (2017). A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science 357, 605–609. [DOI] [PubMed] [Google Scholar]
- Kazlauskiene M, Tamulaitis G, Kostiuk G, Venclovas C, and Siksnys V (2016). Spatiotemporal Control of Type III-A CRISPR-Cas Immunity: Coupling DNA Degradation with the Target RNA Recognition. Mol. Cell 62, 295–306. [DOI] [PubMed] [Google Scholar]
- Konermann S, Lotfy P, Brideau NJ, Oki J, Shokhirev MN, and Hsu PD (2018). Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676 e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer P, Chow MY, Loessner MJ, Portnoy DA, and Calendar R (2002). Construction, characterization, and use of two Listeria monocytogenes site-specific phage integration vectors. J. Bacteriol 184, 4177–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillestøl RK, Shah SA, Brugger K, Redder P, Phan H, Christiansen J, and Garrett RA (2009). CRISPR families of the crenarchaeal genus Sulfolobus: bidirectional transcription and dynamic properties. Mol. Microbiol 72, 259–272. [DOI] [PubMed] [Google Scholar]
- Liu L, Li X, Ma J, Li Z, You L, Wang J, Wang M, Zhang X, and Wang Y (2017a). The Molecular Architecture for RNA-Guided RNA Cleavage by Cas13a. Cell 170, 714–726 e710. [DOI] [PubMed] [Google Scholar]
- Liu L, Li X, Wang J, Wang M, Chen P, Yin M, Li J, Sheng G, and Wang Y (2017b). Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities. Cell 168, 121–134 e112. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, Barrangou R, Brouns SJ, Charpentier E, Haft DH, et al. (2015). An updated evolutionary classification of CRISPR-Cas systems. Nat. Rev. Microbiol 13, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, and Sontheimer EJ (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, and Sontheimer EJ (2010). Self versus non-self discrimination during CRISPR RNA-directed immunity. Nature 463, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myhrvold C, Freije CA, Gootenberg JS, Abudayyeh OO, Metsky HC, Durbin AF, Kellner MJ, Tan AL, Paul LM, Parham LA, et al. (2018). Field-deployable viral diagnostics using CRISPR-Cas13. Science 360, 444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewoehner O, Garcia-Doval C, Rostol JT, Berk C, Schwede F, Bigler L, Hall J, Marraffini LA, and Jinek M (2017). Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature 548, 543–548. [DOI] [PubMed] [Google Scholar]
- Niewoehner O, and Jinek M (2016). Structural basis for the endoribonuclease activity of the type III-A CRISPR-associated protein Csm6. RNA 22, 318–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyenson NC, Gayvert K, Varble A, Elemento O, and Marraffini LA (2017). Broad Targeting Specificity during Bacterial Type III CRISPR-Cas Immunity Constrains Viral Escape. Cell Host Microbe 22, 343–353 e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samai P, Pyenson N, Jiang W, Goldberg GW, Hatoum-Aslan A, and Marraffini LA (2015). Co-transcriptional DNA and RNA Cleavage during Type III CRISPR-Cas Immunity. Cell 161, 1164–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, and Portnoy DA (2010). Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJ, and Severinov K (2011). Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc. Natl. Acad. Sci. U.S.A 108, 10098–10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, Abudayyeh OO, Gootenberg JS, Makarova KS, Wolf YI, et al. (2017). Diversity and evolution of class 2 CRISPR-Cas systems. Nat. Rev. Microbiol 15, 169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smargon AA, Cox DB, Pyzocha NK, Zheng K, Slaymaker IM, Gootenberg JS, Abudayyeh OA, Essletzbichler P, Shmakov S, Makarova KS, et al. (2017). Cas13b Is a Type VI-B CRISPR-Associated RNA-Guided RNase Differentially Regulated by Accessory Proteins Csx27 and Csx28. Mol. Cell 65, 618–630 e617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varani G, and McClain WH (2000). The G × U wobble base pair. A fundamental building block of RNA structure crucial to RNA function in diverse biological systems. EMBO Rep 1, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan WX, Chong S, Zhang H, Makarova KS, Koonin EV, Cheng DR, and Scott DA (2018). Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Mol. Cell 70, 327–339 e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, et al. (2015). Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163, 759–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Lists of normalized PFS abundances, plasmids, and oligonucleotides used in the study. Related to Figure 3 and STAR Methods.
Data Availability Statement
The raw PFS screen data related to Figure 3 and raw RNA-seq reads related to Figure S1 have been deposited in the Sequence Read Archive under ID code SRP142551.