

Brassica napus (B. napus) was originally formed ~7500 years ago by interspecific hybridization between B. rape and B. oleracea (Chalhoub et al., 2014), which supplies approximately 13%–16% of the vegetable oil globally. B. napus serves as an excellent model for polyploid genomics and evolutionary research in plants. Brassica database (BRAD) has long been used for rapeseed genomic research, which provides genome browser and syntenic relationship for multiple Brassicaceae genomes (Wang et al., 2015). In addition, some widely used plant genomic databases such as Genoscope (http://www.genoscope.cns.fr/brassicanapus/) and EnsemblPlants (http://plants.ensembl.org/) also include B. napus genomes. However, these databases are based on the genome of primarily assembled Brassica cultivar Darmor‐bzh, which lack multi‐omics data and rapeseed population information. In recent years, more and more B. napus genomes have been sequenced, and a single reference genome is not sufficient to perform the genetic difference analysis for high‐profile species (Gan et al., 2011); therefore, pan‐genome has been proposed to solve this problem. Pan‐genome is a collection of different individual genomes of a species, which provides a new vision in understanding the genome complexity and a map of the presence/absence variations (PAVs) of genes among these genomes. Recently, eight representative rapeseed cultivars were sequenced by PacBio technology and assembled into pseudo‐chromosomes, which provides new resource for rapeseed genomic research (Song et al., 2020). Based on the above eight B. napus reference genomes, and a collection of 1688 rapeseed re‐sequencing data, we constructed a comprehensive database, B. napus pan‐genome information resource (BnPIR, http://cbi.hzau.edu.cn/bnapus), which is based on gene information module, with Pan‐genome Browser and Gbrowse Synteny as the core, and containing multi‐omics data and common bioinformatics tools.
Similar to the method proposed in rice pan‐genome (Wang et al., 2018a, 2018b), we constructed the pan‐genome of B. napus by ‘PVs + map‐to‐pan’ strategy based on well‐assembled ZS11 reference genome (Figure 1a, Song et al., 2020). Firstly, we collected re‐sequencing data of 1688 rapeseed accessions with an average depth of 8× (Lu et al., 2019; Wang et al., 2018a, 2018b; Wu et al., 2019). Among them, seven representative accessions had deep re‐sequencing (104×–132×) and PacBio sequencing data (Song et al., 2020). The phylogenetic relationship of 1689 accessions including ZS11 is shown in Figure 1b, which was divided into spring‐type oilseed rape (SOR), semi‐winter oilseed rape (SWOR)‐I and SWOR‐II, and winter‐type oilseed rape (WOR) sub‐populations. BnPIR was built on Apache Tomcat HTTP web server (http://tomcat.apache.org/). All the genomic data, collinear data, homologs, gene expression, gene PAVs, metabolic pathways, accession information and related literature were organized and stored in MySQL database (http://www.mysql.com/). Most information can be dynamically accessed through user interactive queries with Highcharts (https://www.highcharts.com/) and Javascripts (https://www.javascript.com/). The web page was constructed and displayed through a popular front‐end component library, Bootstrap (https://getbootstrap.com/). Figure 1c showed the representative resource and tools for constructing BnPIR. The rapeseed pan‐genome was displayed in JBrowse (Buels et al., 2016), providing 1770 tracks for user‐selective display, including genes, transposable elements (TEs), expression profile data, presence frequency and coverage of different accessions (Figure 1d). Considering the assess speed and best performance, we recommend to select less than 30 tracks each time. Furthermore, users can filter the tracks in batches according to the region, country, subgroup, sequencing quality and so on.
Figure 1.
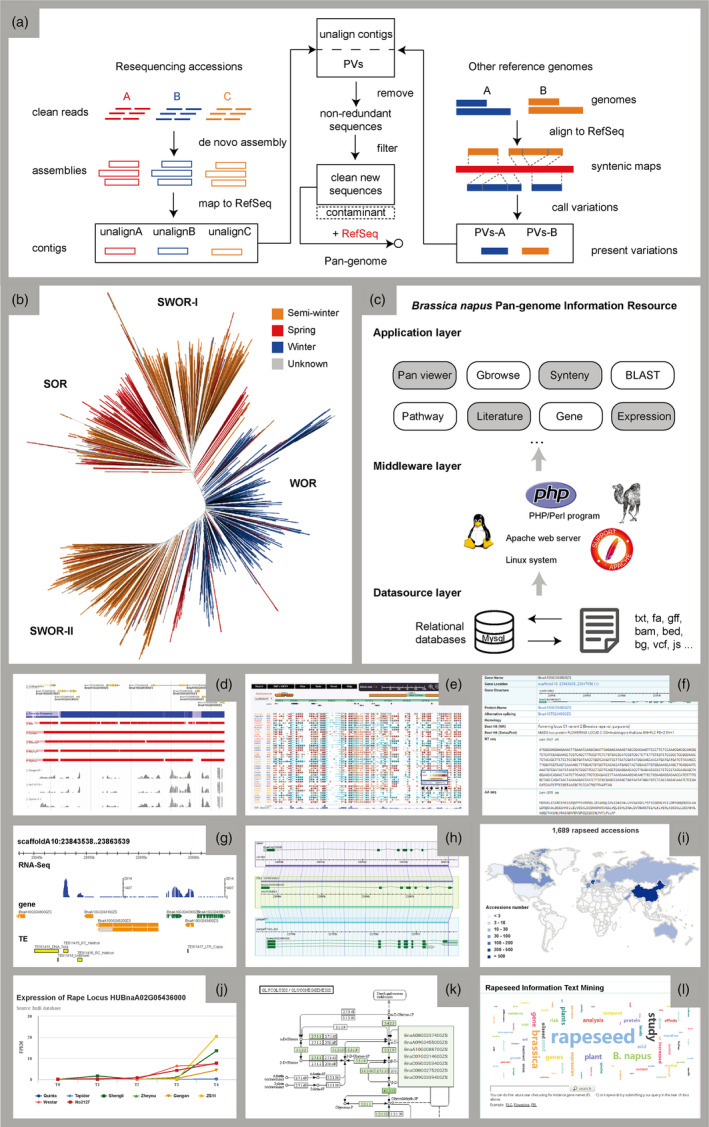
The architecture and representative resources of BnPIR. (a) The pipeline of ‘PVs + map‐to‐pan’ strategy to construct the pan‐genome of B. napus. (b) The phylogenetic tree of 1689 rapeseed accessions. (c) The three‐layer architecture of BnPIR. (d) Pan‐genome browser. (e) Population variations. (f) Gene information page. (g) Gbrowser. (h) Gbrowse synteny of multiple genomes. (i) 1689 rapeseed accessions. (j) Gene expression. T0: 24 days postsowing; T1: 54 days postsowing; T2: 82 days postsowing; T3: 115 days postsowing; T4: 147 days postsowing. (k) KEGG pathway. (l) Literature of rapeseed.
Compared with ZS11 reference genome, the B. napus pan‐genome adds 781.9 Mb sequences and 21 020 protein‐coding genes, which are classified into ‘core genes’ (exist in ≥95% of all rapeseed accessions) and ‘distributed genes’ (exist in <95% of all accessions) by their presence in each variety. Distributed genes are further divided into ‘subspecies imbalance genes’ (frequency in one subspecies is significantly higher than in other subspecies, P value < 0.05), ‘subspecies specific genes’ (>95% in one subspecies) and ‘random genes’ (other distributed genes) according to the frequency of gene existence in different subspecies. Users can quickly query the gene classification and display PAVs in different rapeseed population on phylogenetic tree. Breeders are supposed to focus on the accessions with gene presence in selected donors for their breeding purpose. Except large PAVs in the pan‐genome, we also identified 43 633 669 SNPs and 7 809 506 InDels. In addition, we provided interactive interface to effectively display sequence variation information in 159 high‐coverage accessions with TASUKE (https://tasuke.dna.affrc.go.jp/). The frequency of variations, depth of coverage and annotation of multiples genomes were shown in web‐based genome browser (Figure 1e).
We developed flexible query pages to efficiently retrieve and visualize various types of resources. For example, a keyword‐based search engine by inputting a gene locus (e.g. BnaA10G0244800ZS) or gene name (e.g. FLOWERING LOCUS C) can link to a gene detail information page. In total, 773 065 protein‐coding genes were provided in the gene information page. Basic genetic information includes chromosomal location, coding sequence length, exon number, gene structure, alternative splicing, nucleic acid sequence, the encoded protein sequence, expression data, gene ontology, functional domain, gene classification (core/distributed), frequency in subspecies. (Figure 1f). Moreover, users can access the Gbrowse (https://www.gbrowse.org/) to visualize detailed gene context and upstream/downstream features (Figure 1g). Gbrowse synteny shows collinearity and structure variations comparing to other genomes (Figure 1h). Basic local alignment search tool (BLAST) (Altschul et al., 1990) is provided as a sequence‐based search engine, and homologs can be obtained in multiple B. napus, B. rape and B. oleracea genomes by presenting alignment results in graphical and textual formats. Users can view and download 1689 accessions including subgroup, region and sequencing depth in rapeseed accession table page (Figure 1i). Gene expression module can be used to visualize the gene expression in different accessions throughout flowering period (Figure 1j). The metabolic pathways based on KEGG orthologs (Kanehisa et al., 2009) of the eight rapeseed accessions with reference genomes are provided in BnPIR (Figure 1k). To facilitate gene comparison and retrieval of target genes in different reference genomes, BnPIR provided a unique gene index based on collinear orthologs in nine rapeseed genomes including two SORs (Westar and No2127), four SWORs (ZS11, Gangan, Zheyou7 and Shengli) and three WORs (Darmor‐bzh, Tapidor and Quinta), covering a total of 88 345 protein‐coding genes. Users can compare the gene structural difference in the nine accessions by combining Gbrowse synteny model.
BnPIR also contains practical calculation tools for comparison, evolution and functional analysis of rapeseed and closely related species. OrthoMCL (https://orthomcl.org/) was used to identify homologs in plant genomes, including eight newly sequenced rapeseed accessions (Song et al., 2020), B. napus Darmor‐bzh, Arabidopsis thaliana, B. rape and B. oleracea. We performed OrthoMCL (e‐value: 1e‐5) to identify putative orthologs and paralogs, and closely related gene clusters were obtained in the above species. A total of 109 001 putative homologous groups were identified and stored in BnPIR for query and download. Sub‐genomes A and C inherit the B. rape and B. oleracea genomes, respectively. We use Mummer (http://mummer.sourceforge.net/) to determine the collinear regions between sub‐genomes and visualize the statistical results. A text mining tool is available in BnPIR, which allows to search references by gene names or keywords in 9971 rapeseed‐related articles obtained from PubMed (Figure 1l). For example, The NOD‐like receptor (NLR) gene families play important roles in plant growth and crop breeding. We have comprehensively identified and annotated related genes in different accessions and stored them in BnPIR.
In summary, we have established a comprehensive functional genomic platform, BnPIR, as a new tool for querying and visualizing rapeseed genomes and the pan‐genome based on 1689 accessions. BnPIR contains genomic sequences, gene annotations, phylogenetic relationship, expression data, PAV information, gene classification and common multi‐omics tools for 1689 rapeseed accessions and provides an integration of quick search and visualization. BnPIR will be a rich resource for rapeseed molecular biology and breeding, which will help rapeseed researchers to search and visualize their results in a pan‐genome context, and provide a valuable template for pan‐genome analyses in other species.
Conflict of interest
No conflict of interest declared.
Author contributions
J.‐M. S., Q.‐Y. Y., L. G., K. L. and L.‐L. C. conceived and designed the study. J.‐M. S., D.‐X. L., W.‐Z. X., and Z. Y. performed the analysis and constructed the website. J.‐M. S., D.‐X. L., Q.‐Y. Y. and L.‐L. C. wrote the paper.
Acknowledgements
This project was supported by Hubei Provincial Natural Science Foundation of China (2019CFA014), and Fundamental Research Funds for the Central Universities (2662018PY068).
Song, J.‐M. , Liu, D.‐X. , Xie, W.‐Z. , Yang, Z. , Guo, L. , Liu, K. , Yang, Q.‐Y. and Chen, L.‐L. (2021) BnPIR: Brassica napus pan‐genome information resource for 1689 accessions. Plant Biotechnol. J., 10.1111/pbi.13491
Contributor Information
Qing‐Yong Yang, Email: yqy@mail.hzau.edu.cn.
Ling‐Ling Chen, Email: llchen@mail.hzau.edu.cn.
References
- Altschul, S.F. , Gish, W. , Miller, W. , Myers, E.W. and Lipman, D.J. (1990) Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Buels, R. , Yao, E. , Diesh, C.M. , Hayes, R.D. , Munoz‐Torres, M. , Helt, G. et al. (2016) JBrowse: a dynamic web platform for genome visualization and analysis. Genome Biol. 17, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub, B. , Denoeud, F. , Liu, S. , Parkin, I.A. , Tang, H. , Wang, X. et al. (2014) Early allopolyploid evolution in the post‐Neolithic Brassica napus oilseed genome. Science, 345, 950–953. [DOI] [PubMed] [Google Scholar]
- Gan, X. , Stegle, O. , Behr, J. , Steffen, J.G. , Drewe, P. , Hildebrand, K.L. et al. (2011) Multiple reference genomes and transcriptomes for Arabidopsis thaliana . Nature, 477, 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa, M. , Goto, S. , Furumichi, M. , Tanabe, M. and Hirakawa, M. (2009) KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 38, D355–D360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, K. , Wei, L. , Li, X. , Wang, Y. , Wu, J. , Liu, M. et al. (2019) Whole‐genome resequencing reveals Brassica napus origin and genetic loci involved in its improvement. Nat. Commun. 10, 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J.M. , Guan, Z. , Hu, J. , Guo, C. , Yang, Z. , Wang, S. , et al.(2020) Eight high‐quality genomes reveal pan‐genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 6, 34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, W. , Mauleon, R. , Hu, Z. , Chebotarov, D. , Tai, S. , Wu, Z. et al. (2018a) Genomic variation in 3,010 diverse accessions of Asian cultivated rice. Nature, 557, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, B. , Wu, Z. , Li, Z. , Zhang, Q. , Hu, J. , Xiao, Y. et al. (2018b) Dissection of the genetic architecture of three seed‐quality traits and consequences for breeding in Brassica napus . Plant Biotechnol. J. 16, 1336–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Wu, J. , Liang, J. , Cheng, F. and Wang, X. (2015) Brassica database (BRAD) version 2.0: integrating and mining Brassicaceae species genomic resources. Database, 2015, bav093. [DOI] [PMC free article] [PubMed]
- Wu, D. , Liang, Z. , Yan, T. , Xu, Y. , Xuan, L. , Tang, J. et al. (2019) Whole‐genome resequencing of a worldwide collection of rapeseed accessions reveals the genetic basis of ecotype divergence. Mol. Plant, 12, 30–43. [DOI] [PubMed] [Google Scholar]