

Abstract
Purpose:
Diffuse intrinsic pontine glioma (DIPG) is among the deadliest of pediatric brain tumors. Radiation therapy is the standard of care treatment for DIPG, but offers only transient relief of symptoms for DIPG patients without providing significant survival benefit. Oncolytic virotherapy (OV) is an anti-cancer treatment that has been investigated for treating various types of brain tumors.
Experimental Design:
Here, we have explored the use of mesenchymal stem cells (MSC) for OV delivery and evaluated treatment efficacy using preclinical models of DIPG. The survivin promoter drives the conditional replication of OV used in our studies. The efficiency of OV entry into the cells is mediated by fiber modification with seven lysine residues (CRAd.S.pK7). Patients’ samples and cell lines were analyzed for the expression of viral entry proteins and survivin. The ability of MSCs to deliver OV to DIPG was studied in the context of a low dose of irradiation.
Results:
Our results show that DIPG cells and tumors exhibit robust expression of cell surface proteins and survivin that enable efficient OV entry and replication in DIPG cells. MSCs loaded with OV disseminate within a tumor and release OV throughout the DIPG brainstem xenografts in mice. Administration of OV-loaded MSCs with radiotherapy to mice bearing brainstem DIPG xenografts results in more prolonged survival relative to that conferred by either therapy alone (p<0.01).
Conclusions:
Our study supports oncolytic virus CRAd.S.pK7 encapsulated within MSCs as a therapeutic strategy that merits further investigation and potential translation for DIPG treatment.
Introduction
Diffuse intrinsic pontine glioma (DIPG) is an incurable pediatric brain tumor with a peak incidence between 6 and 9 years of age (1). The current standard of care is irradiation, which temporarily relieves symptoms but has no significant effect on overall survival (OS), which is less than 18 months. Five-year survival for DIPG patients is less than 1% (2–5). 80% of DIPG have a mutation of genes encoding histone 3 (H3.1/3.3- K27M). Amplification of platelet-derived growth factor receptor alpha (PDGFRA), and mutations affecting activin A receptor type I (ACVR1) and tumor protein p53 (TP53), are also common among DIPG (6–9). Due to their diffuse nature and location within the brainstem, DIPG are not surgically resectable (5). As is the case for most brain tumors, systemically administered chemotherapeutics have limited access to DIPG due to the blood-brain barrier (BBB) (10–12).
The homing capacity of mesenchymal stem cells (MSCs) to tumors makes them excellent carriers of anticancer therapeutics. MSCs have been used as delivery vehicles for anticancer agents such as proteins, suicide gene/enzyme prodrugs, or oncolytic vectors and are known for having tumor tropic properties (13–17). MSCs can cross the BBB and reach brain tumor tissue following systemic administration. Intranasal delivery (IND) is an alternative administration route of therapy-carrying stem cells that has shown effectiveness in preclinical studies for treating neurological disorders including multiple sclerosis, viral brain infections, Parkinson’s disease, ischemic brain injury, and glioblastoma (18–22).
We previously showed that the encapsulation of oncolytic virus (OV) CRAd.S.pK7 by MSCs has the potential to deliver a high therapeutic load to maximize tumor coverage with simultaneous protection from the fast clearance of the OV by the immune system (14). Modification of the viral fiber in CRAd.S.pK7 with a heparin-binding domain, poly-L-lysine (pK7), increases OV entry’s efficiency into tumor cells (23). Once in tumor cells, an expression of viral proteins is driven by a survivin promoter (24), active in numerous cancers, including GBM, but not in normal cells (25,26). Stem cell-mediated delivery of CRAd.S.pK7 ensures virus dissemination throughout the tumor. The stem-cell-based delivery approach also delays the elimination virus by the host immune system (14,27) and decreases neuroinflammatory response, thereby providing neuroprotection to the normal brain (28). The results from our preclinical studies in GBM models led to a phase I clinical trial investigating the safety of stem cell carrying OV for treating GBM patients (NCT03072134). As administered locally to the tumor site or by IND using stem cell carriers, we previously showed that oncolytic virotherapy is an effective treatment when tested against models of glioblastoma. Stem cells are capable of delivering therapeutic cargos to distant parts of the brain (14,22–24,28–32).
This study evaluated CRAd.S.pK7 OV as a potential therapeutic for treating children with DIPG. CRAd.S.pK7 replication was examined in three patient-derived DIPG lines, and MSC migration and delivery of CRAd.S.pK7 were analyzed in vivo using DIPG brainstem xenograft models. Lastly, the efficacy of local and intranasal administration of MSCs carrying OV was determined through survival analysis of mice with brainstem xenografts, both in the presence and absence of radiation.
Materials and Methods
Patients specimens
Patients specimens were collected at postmortem per Children’s National Medical Center with IRB approval (IRB #Pro00001339) as described previously (5), and the U.S. Common Rule. The written consents were obtained from parents (for children under 13), assent from child and consent from a parent if 13–17 y.o., and consent from a patient if 18 y.o. or over.
Cell lines
Primary SF8628, SF7761, and DIPG007 were used in the study and tested for mouse pathogens, including mycoplasma (using the PCR method) by Charles River on June 12, 2019. H3.3K27M DIPG cell lines were generously provided by Dr. Rintaro Hashizume (Northwestern University). Using the Powerplex16HS System (Promega DC2101), DNA fingerprints were obtained to confirm the cell lines’ identity. The establishment of SF8628 from a surgical specimen, and tumor cell modification for expression of firefly luciferase, for in vivo bioluminescence imaging has been described by Hashizume et al.[34]. DIPG007 (H3.3K27M DIPG) cell line (RRID: CVCL_VU70) was kindly provided by Dr. Angel Montero Carcaboso, Hospital Sant Joan de Déu, Barcelona, Spain). This cell line was last tested for mycoplasma using the PCR method on May 15, 2020. After defrosting, cell lines were used up to 3 passages. Human bone marrow-derived MSCs were purchased from the Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine and were grown in MEMalpha supplemented with 10% FBS.
Bioluminescent imaging and MRI
Tumor visualization using MRI was done according to a previously described protocol (22). Detailed information on BLI can be found in supplemental materials..
Radiation Treatment
Experimental mice were exposed to 0.5 Gy radiation 3 times a week for 2 weeks to reach a total dose of 3 Gy. Detailed procedures are described in supplemental materials.
MSC labeling, Histology, Quantitative analysis of MSCs migration in vivo, Immunocytochemistry, Western Blot, Quantitative PCR, Invasion Assay, Cytokine Array, siRNA silencing.
Detailed procedures can be found in supplemental materials.
Flow cytometry
Flow cytometry was performed on the BD Fortessa flow cytometer (Becton Dickinson, Franklin Lakes, NJ) at the Robert H. Lurie Comprehensive Cancer Center Flow Cytometry Core Facility. Detailed procedures can be found in supplemental materials.
Quantitative analysis of the binding CRAd.S.pK7 to the cell surface.
Details are described in supplemental materials.
Evaluation of CRAd.S.pK7 replication and toxicity
To assess the release of viral progeny, DIPG cells expressing firefly luciferase were infected with CRAd.S.pK7 at MOI 10 v.p./cell in DMEM media supplemented with 1%FBS. Detailed procedures for assessment can be found in supplemental materials.
Analysis of Survivin Promoter Activity in DIPG Cells
To assess the survivin promoter activity, we employed a replication-deficient adenoviral vector expressing firefly luciferase under the survivin promoter’s control as previously described (33). Detailed procedures for assessment can be found in supplemental materials.
Animals and Surgical Procedures
All animal procedures were approved by the Northwestern University Institutional Animal Care and Use Committee. Six-week-old female athymic mice (BALB/c background) were purchased from Charles River and housed under aseptic conditions. Pontine injection of tumor cells was performed as described previously (34). Detailed surgical procedures are described in supplemental materials.
Intranasal Delivery
Mice were treated with methimazole (50 mg/kg body weight) 2 days before IN-MSC delivery as previously described (22). Detailed information can be found in supplemental materials
Survival Analysis
Mice bearing intracranial DIPG tumors were randomized for subsequent treatment with saline, control MSCs, or therapeutic MSCs loaded with OV. MSCs bearing OV were delivered using either through IN route or intracranial injection into the brain at a location proximal to the DIPG tumor at a depth of 2.5 mm.
Statistics and data analysis
The collected data analyses were performed using GraphPad Prism 8 Software (RRID: SCR_002798) and SAS 9.4 (Statistical Analysis System, RRID: SCR_008567). RNAseq was analyzed as previously published (35,36). Analysis of RNAseq from The Children’s Brain Tumor Tissue Consortium (CBTTC) and Pediatric Brain Tumor Atlas (PNOC003) obtained from PedcBioPortal (https://pedcbioportal.kidsfirstdrc.org/) as previously described (37,38). Details on statistics and data analysis can be found in supplemental materials.
Results
In vitro and in vivo DIPG models
In order to investigate the potential of OV-carrying MSCs for DIPG treatment, we utilized a patient-derived xenograft (PDX) model of DIPG. Human DIPG cells were directly injected into the brainstem of mice (Fig. 1A) and formed tumors, as confirmed by three-dimensional bioluminescent imaging (BLI) and magnetic resonance imaging (MRI). Histologic analysis of resected mouse brains revealed an infiltrative growth pattern in the brainstem, consistent with that observed in patients (Fig. 1A).
Figure 1. Radiation improves MSC migration toward DIPG tumors.
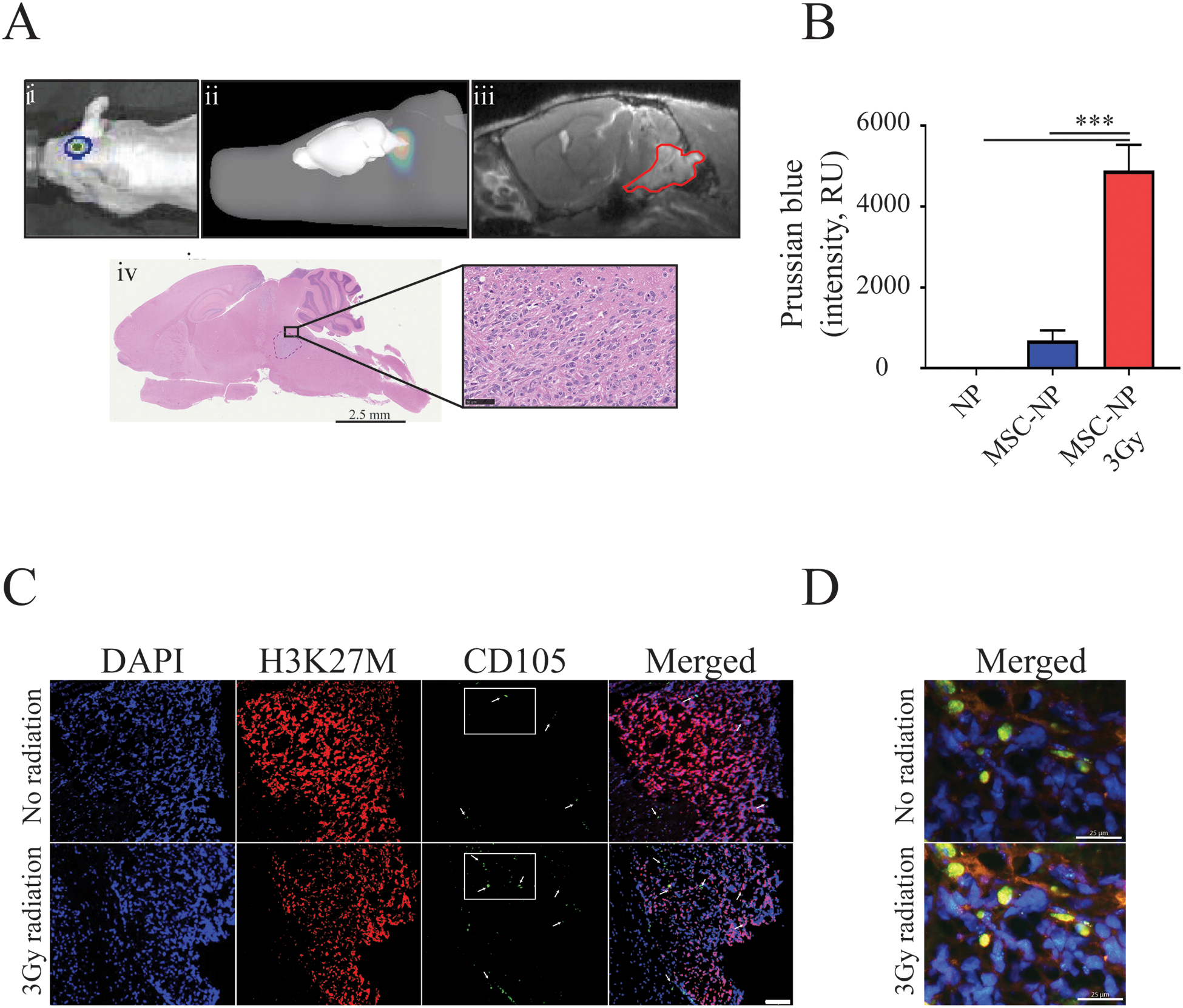
A) Intracranial implantation of SF8628 DIPG cells results in pontine tumor formation in mice. Representative images show i) 2D-bioluminescent (BLI) image of the tumor, ii) 3D-rendered BLI, iii) MR image of the tumor (outlined in red) depicting engraftment of the pontine tumor, and iv) histological visualization of the tumor by H&E stain, shown at low (scale=2.5 mm) and high-power magnification (scale=50μm). B) Irradiation significantly increased the trafficking of iron NP-labeled MSCs to the tumor site (One-way ANOVA with Tukey’s post hoc test, n≤3. Level of significance: *** p <0.001). C) MSC migration to DIPG tumors was further validated using IHC (nuclear staining DAPI (blue), histone H3K27M (red) and MSCs-CD105 (green, indicated by white arrows). Scale= 100 μm. D) High power magnification of regions from C) indicated with rectangles that show colocalization of CD105 expressing MSCs with DIPG cells. Scale=25 μm.
In vitro and in vivo effects of radiation on MSC tropism for DIPG
Initially, we investigated the effects of radiation on DIPG cells in culture. Human SF8628 DIPG cells were treated with fractionated irradiation for three days, resulting in the cumulative dose administration of 3, 6, and 12 Gy (SFig. 1A). Treatment of cells with 6 and 12 Gy irradiation revealed substantial reductions in their viability on days 5 (at 6Gy: 90.2±5.6%; at 12Gy:73.1±0.54%) and 6 (at 6Gy:19.26±6.7%; at 12Gy:29.38±6.76%, SFig. 1A).
DIPG cells were treated with a sublethal dose of 3 Gy to examine the effect of radiation on MSC tropism to DIPG in vitro and in vivo. MSCs labeled with iron nanoparticles (SPIO) were administered by IND to mice with brainstem DIPG xenografts to evaluate radiation-associated effects on MSC migration to the tumor in vivo. Prussian Blue staining revealed a significant increase in iron-labeled MSCs within and around brainstem tumor in irradiated animals, as compared to non-irradiated mice treated with SPIO’s alone (Fig. 1B, One-way ANOVA, p<0.0001). The tumor location of MSCs was confirmed by staining of tissue with anti-CD105 antibody (Fig. 1C–D). Collectively, these data demonstrate that fractionated low-dose radiation increases the migration of MSCs toward DIPG tumors.
Irradiation increases the expression of chemoattractant cytokines in DIPG tissue
Tumor tissue was dissected from irradiated and non-irradiated mice to investigate potential mediators of MSC tropism for DIPG (SFig. 1B). Results from array analysis revealed multiple chemokines/cytokines as being upregulated in DIPG tumors in response to radiation treatment (Fig. 2A). Bone morphogenetic protein (BMPR) 2, and epidermal growth factor (EGFR) receptors, binding partners for two of upregulated proteins, (BMP-4) and betacellulin (BTC), respectively, were expressed by MSCs (Fig. 2B–C). The siRNA suppression of BMP-4 or BTC alone in SF8628 cells did not affect MSC migration toward conditioned media of irradiated DIPG cells (SFig. 1C). In contrast, both chemokines’ concomitant silencing significantly reduced MSC tropism for radiation-treated SF8628 and DIPG007 cells (Fig. 2E–F). These data indicate that combined BMP4 and BTC expression is important for MSC tropism to irradiated DIPG tumors.
Figure 2. Radiation-induced upregulation of betacellulin (BTC) and bone morphogenic protein 4 (BMP-4) contributes to MSC migration to DIPG tumors.
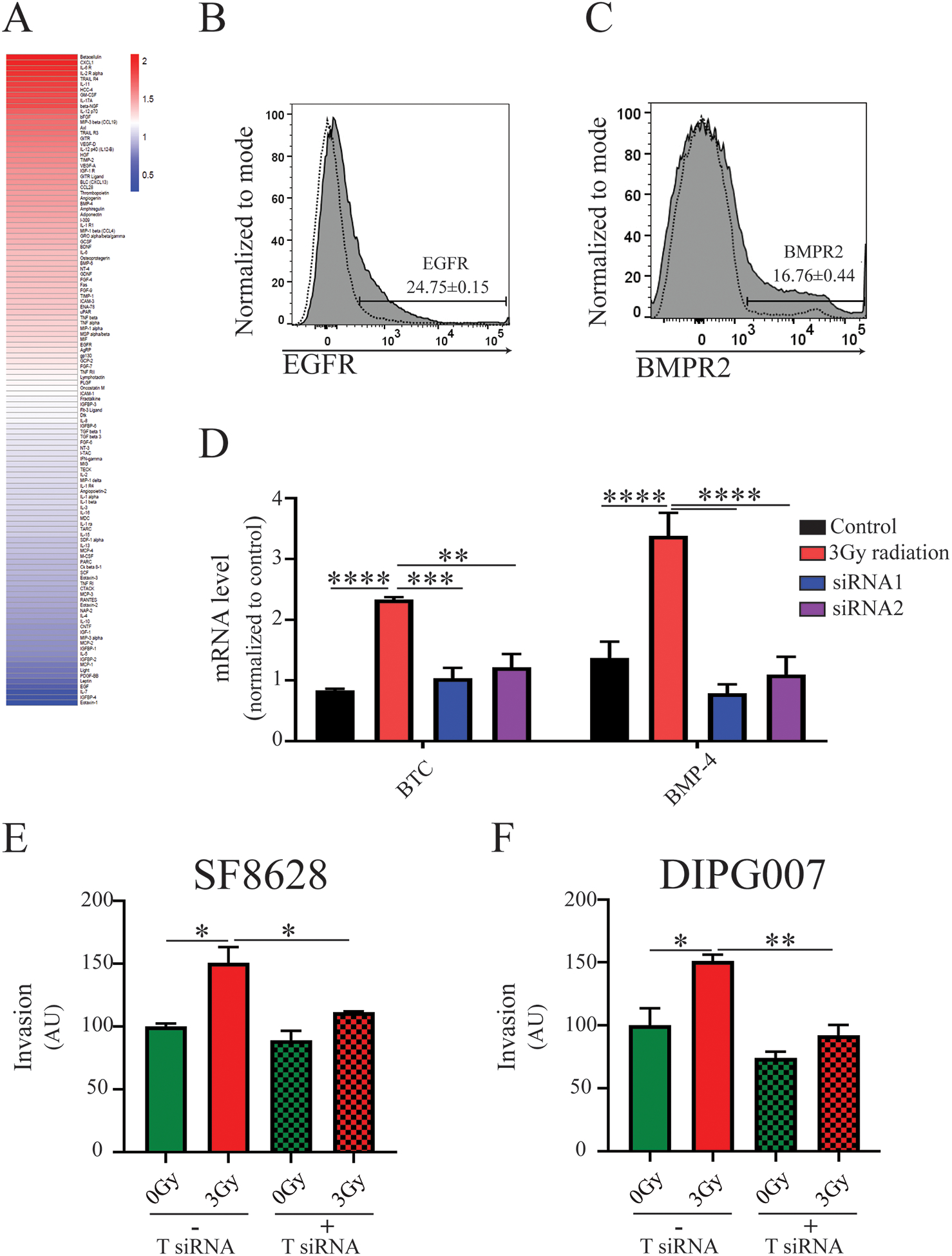
A) Protein array of SF8628 DIPG cells isolated from the brainstem of control and irradiated mice (n=3 per control and irradiated groups). Heat map values for each protein represent an average of two independent reads of the array. Representative flow cytometry histogram of MSCs for expression of B) EGFR-epidermal growth factor (n=3) and C) BMPR2-bone morphogenic protein receptor 2 (n=3), receptors for BTC and BMP-4 ligands, respectively. D) Quantitative PCR assessment of the mRNA levels of expression of BTC and BMP-4 after siRNA-induced gene silencing (n=3, One-way ANOVA with Tukey’s post hoc test. Levels of significance are: ** p <0.01, *** p <0.001 and **** p<0.0001). E) and F) Migration of MSCs toward irradiated SF8628 and DIPG007 cells, respectively, after silencing of both BTC and BMP-4 siRNA in vitro. (One-way ANOVA with Tukey’s post hoc test, n≤4. Level of significance: * p <0.05, ** p <0.01).
OV infectivity, replication in, and cell killing of DIPG in vitro
We compared the efficacy of CRAd.S.pK7 and Ad5delta24RGD that have distinct modifications of their fibers necessary for cellular entry of OV. These viruses also differ in promoters driving viral gene expression: survivin for CRAd.S.pK7, and CMV for Ad5delta24RGD. Both viruses successfully killed DIPG cells in vitro (Fig. 3 A–C), though CRAd.S.pK7 was more effective at a lower multiplicity of infection (MOI) in SF8628 and SF7761 cells (Fig. 3A, B). These findings are consistent with results showing superior binding of the CRAd.S.pK7 OV to the surface of the SF8628 over the SF7761 and DIPG007 cells (Fig. 3D).
Figure 3. Oncolytic activity of CRAd.S.pK7 in patient-derived DIPG cells.
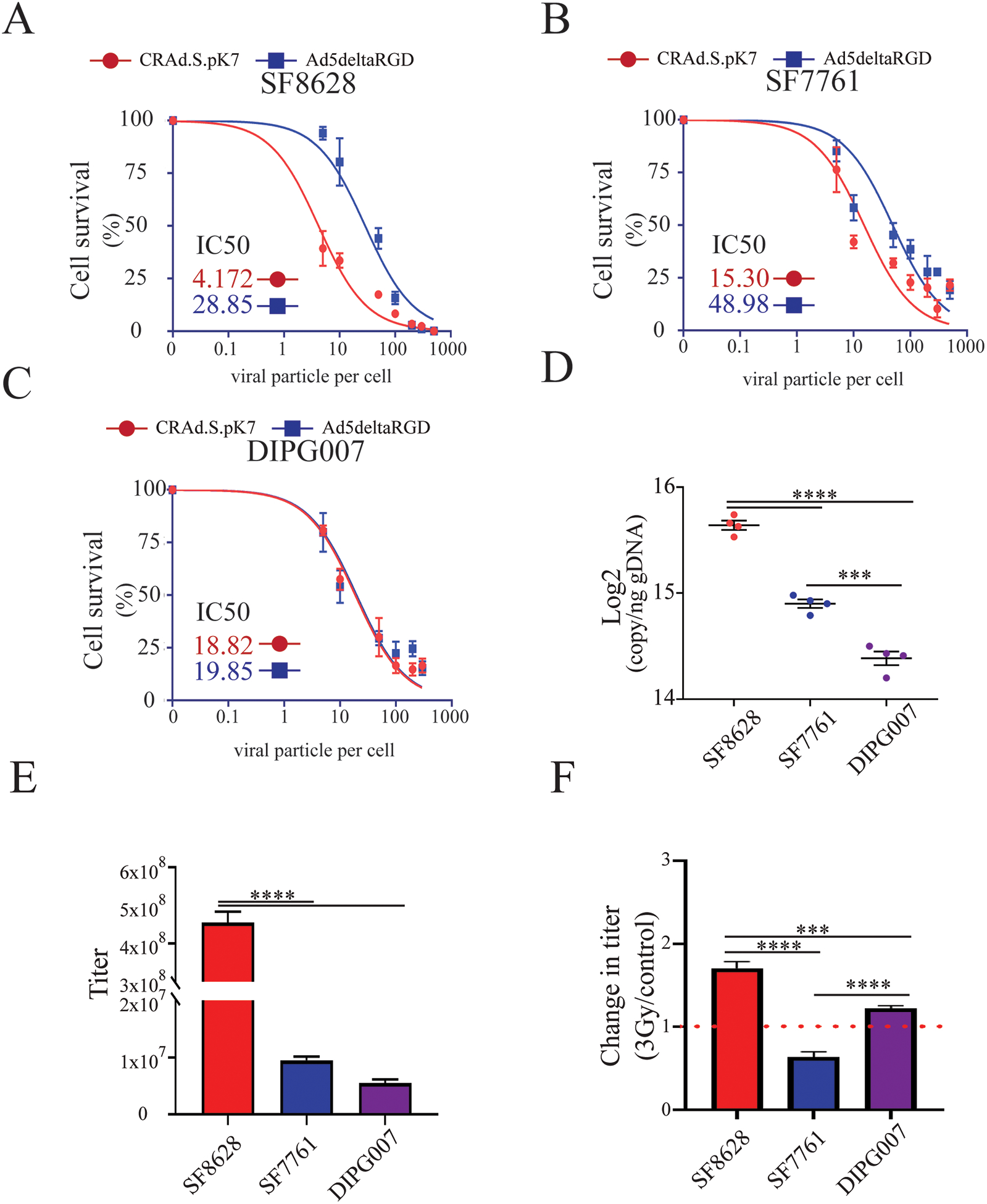
A) Kinetics of killing by CRAd.S.pK7 and Ad5deltaRGD of SF8628 DIPG cells expressing firefly luciferase were measured by the loss of luciferase activity 3 days after treatment of cells with OV (n=4). B) Kinetics of killing by CRAd.S.pK7 and Ad5deltaRGD of SF7761 DIPG cells (n=4) and C) DIPG007 DIPG cells (n=4) were assessed as described in A. D) Binding assay of CRAd.S.pK7 with the surface of DIPG cell lines SF8628, SF7761, DIPG007 was performed to assess the difference in the binding of viral particles to the cell surface using PCR-based quantification of viral DNA (One-way ANOVA with Tukey’s post hoc test, n=4. Levels of significance are: *** p <0.001 and **** p<0.0001). E) The release of CRAd.S.pK7 progeny from SF8628, SF7761, and DIPG007 DIPG cell lines was assessed by determining viral titer using staining for hexon viral protein in 293T cells (Negative binomial model was fitted and followed by Comparisons of Group Least Squares Means with Tukey-Kramer adjustment, n=4). F) Change in the progeny release of CRAd.S.pK7 from in DIPG cells after 3Gy irradiation treatment (One-way ANOVA with Tukey’s post hoc test, n=4. Levels of significance are: * p <0.05, ** p <0.01, *** p <0.001 and **** p<0.0001).
Viral replication in SF8628 cells was 47.94±5.23 times higher than in SF7761 and 82.82±3.03 times higher than in DIPG007 cells (Fig. 3E); though all tested cells robustly generated and released viral progeny. In the context of irradiation, SF8628 and DIPG007 cells exposed to 3Gy irradiation produced 70.12±7.92% and 22.3±3.28%, respectively, more OV than non-irradiated control cells (p<0.0001), whereas a reduction by 44% in was observed in irradiated SF7761 (Fig. 3F).
To identify individual DIPG molecular characteristics that could help predict individual tumor responsiveness to CRAd.S.pK7 therapy, both in the presence and absence of radiation, we used multiple approaches. The analysis of RNAseq data from DIPG tumor tissue (5) revealed robust expression of mRNAs encoding integrin (SFig. 2A–C) and CAR (SFig. 2D), attachment proteins for the virus. Analysis of patients’ DIPG tissue (Fig. 4A, B) and DIPG cell lines (Fig. 4C, D) showed a robust expression of syndecan 1 and perlecan mRNAs, with syndecan 1 expressed at a higher level in SF8628 cells, among all DIPG cell lines (Fig. 4E(i)). Irradiation of SF8628 cells slightly reduced the presence of the syndecan 1 at the cell surface of treated cells by 6.67±0.02% (Fig. 4E(ii)).
Figure 4. Expression of surface heparan sulfate proteoglycans in human DIPG tumor tissue, patient-derived cell lines, and mouse xenograft tumors.
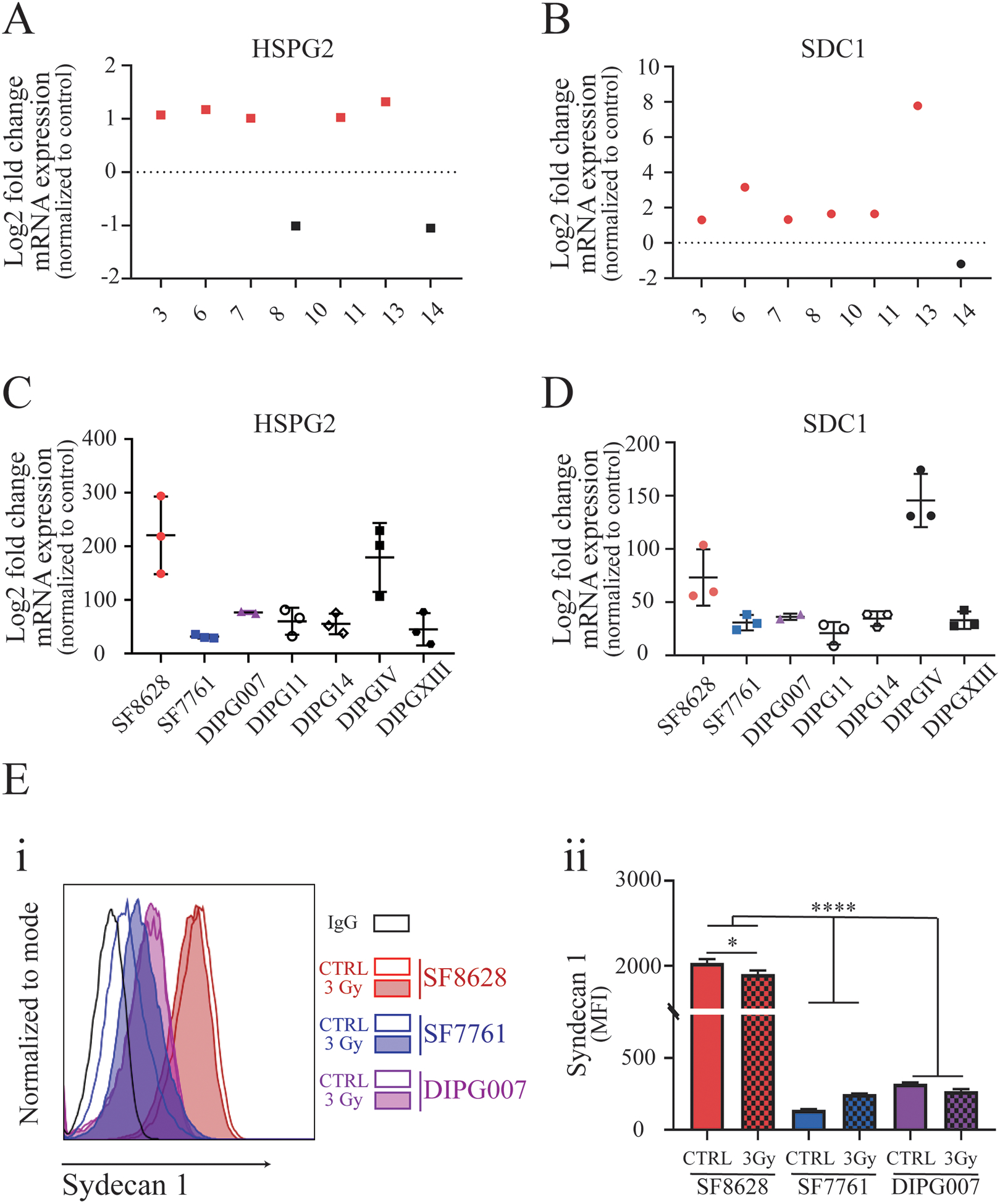
RNAseq analysis for the expression of A) heparan sulfate proteoglycan 2 (HSPG2) and B) syndecan 1(SDC1) in DIPG patient samples, with tumor values indicated in relation to corresponding normal brain tissue expression in the same patient. RNAseq analysis of the patient-derived primary cell lines SF8628, SF7761, DIPG007, DIPG11, DIPG14, DIPGIV, and DIPGXIII for expression of C) HSPG2 and D) SDC1. Data are indicated in relation to HSPG2 and SDC1 RNA expression in normal astrocytes. E) (i) Representative flow cytometry histograms show the surface expression of syndecan 1 in DIPG cell lines SF8628, SF7761, and DIPG007 (n≥3) before and after fractionated irradiation treatment with 3Gy total dose; (ii) Analysis of syndecan 1 levels expressed on the cell surface in control and irradiation-treated DIPG cells detected by flow cytometry (Welch’s ANOVA test with Dunnett’s post hoc test, n=4–7. Levels of significance are: * p <0.05 and **** p<0.0001.
As the survivin promoter drives the replication of CRAd.S.pK7 in cancer cells, we examined survivin expression in silico using RNA microarray data sets available at the GlioVis database (39). This analysis showed that survivin, encoded by the BIRC5 gene, is significantly overexpressed in a range of pediatric brain tumors compared to non-tumor tissues (SFig. 3A, B). Analysis of RNAseq data set from patient tissues (Children’s Brain Tumor Tissue Consortium, CBTTC, https://cbttc.org/) revealed a significantly greater expression of survivin (<0.0001) in high-grade pediatric glioma compared to low-grade glioma (SFig. 3C) (37,38). Analysis of RNAseq data set (5) showed a significant increase of survivin in the majority of human DIPG samples when compared to normal brain tissue controls (Fig. 5A: p-value < 0.05). Analysis of RNAseq data obtained from the CBTTC (n=14) and the Pediatric Brain Tumor Atlas (PNOC003, n=31) also showed robust expression of BIRC5 RNA in DIPG tissue (Fig. 5B, SFig. 4 A–B)(37,38). Immunostaining of DIPG tissues showed a varying degree of survivin expression among DIPG patients, in agreement with variability seen at the mRNA level (Fig. 5C). RNAseq and quantitative PCR analysis revealed robust BIRC5 mRNA expression in all DIPG cell lines (Fig. 5D–E), and Western blot results showed high levels of survivin protein in SF8628 and DIPG007 cells (Fig. 5 F (i and ii)). We observed reduced survivin promoter activity, mRNA expression, and reduced protein levels in DIPG007 and SF7761 cells, but not in SF8628 irradiated cells (Fig. 6A–C). Consistent with the radiation-induced increase in survivin expression observed in SF8628 cells, combinatorial treatment with CRAd.S.pK7 and radiation increased the number of dying cells by an additional 24.78±2.65 % in comparison to control, after 5 days in culture (SFig. 5). These data indicate that CRAd.S.pK7 possesses potent lytic activity in DIPG cells, with its replication being dependent on the expression of survivin and proteins mediating viral entry into DIPG cells.
Figure 5. Survivin expression in human DIPG tumor tissue, patient-derived cell lines, and mouse xenograft tumors.
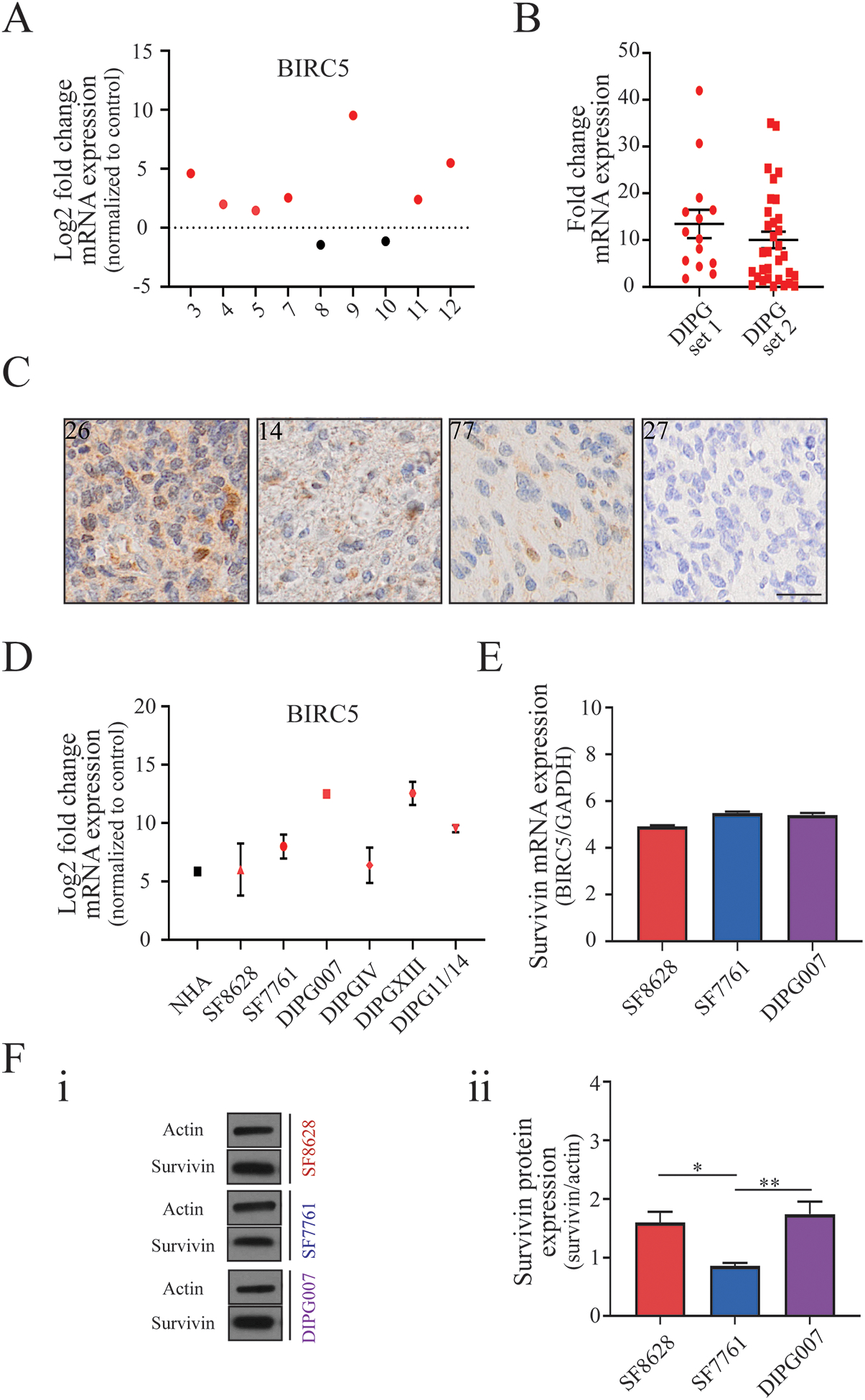
A) Fold change in mRNA expression of the BIRC5 gene encoding the survivin protein in human DIPG tumor tissue samples, normalized to average expression across normal brain tissue from the same patients. B) Fold change in BIRC5 expression in human DIPG tumor tissue. ‘DIPG set 1’ data was acquired from the Children’s Brain Tumor Tissue Consortium (CBTTC) (n=14), and ‘DIPG set 2’ data was acquired from the Pediatric Brain Tumor Atlas (PNOC003) (n=31) dataset (PedcBioPortal). C) Immunohistochemistry of DIPG patient samples stained for survivin representing strong (score=III, patient 26), medium-strong (score=II, patient 14), weak (score=I, patient 77) and absent (score=0, patient number 27) expression (scale: 50μm). D) RNA expression of BIRC5 gene in normal human astrocytes (NHA) and patient-derived DIPG cell lines. E) qPCR analysis of basal BIRC5 mRNA expression in DIPG cell lines SF8628, SF7761, DIPG007. F) (i) Representative Western blot images and (ii) quantification of survivin protein expression normalized to actin expression in DIPG cell lines (One-way ANOVA with Tukey’s post hoc test, n=3. Levels of significance are: * p <0.05 and ** p <0.01).
Figure 6. The expression of survivin is affected by irradiation in patient-derived cell lines.
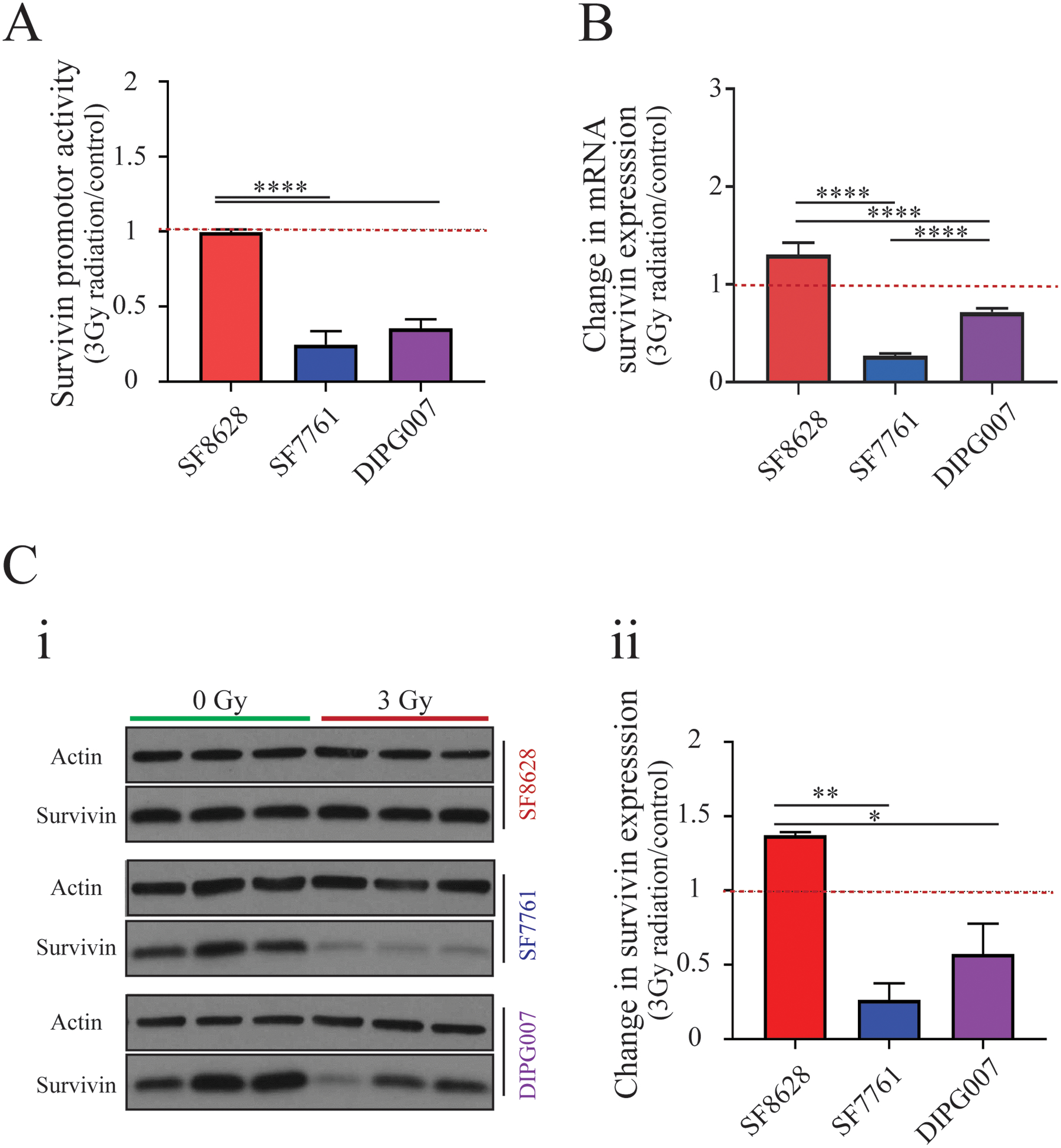
A) Survivin promoter activity in irradiated SF8628, SF7761, and DIPG007 cell lines normalized to untreated controls (One-way ANOVA with Tukey’s post hoc test, n=6. Levels of significance: **** p<0.0001). B) qPCR analysis of survivin mRNA in response to irradiation (One-way ANOVA with Tukey’s post hoc test, n=4. Levels of significance: **** p<0.0001). C) (i) Western blot showing survivin expression in SF8628, SF7761, and DIPG007 DIPG cell lines before (0Gy) and after (3Gy) irradiation; (ii) change in survivin protein expression in response to irradiation treatment (One-way ANOVA with Tukey’s post hoc test, n=3. Levels of significance are: * p <0.05 and ** p <0.01).
CRAd.S.pK7 treatment mediated by MSC delivery improves the survival of mice-bearing DIPG tumors, and survival benefit is enhanced when combined with fractionated irradiation
We performed a longitudinal analysis to establish the effect of irradiation on tumor growth in vivo. Using BLI, we found that 3Gy radiation did not significantly delay tumor growth of SF8628 DIPG PDX (Fig. 7A). Next, SF8628 mice bearing SF8628 brainstem xenografts were treated with CRAd.S.pK7-loaded MSCs following irradiation. There was no survival benefit observed in association with IND administration of cell-based OV (SFig. 6A), even though the expression of hexon, a viral capsid protein, was evident in the DIPG tissue day 5 after treatment (SFig. 6B). Local delivery of the MSCs-CRAd.S.pK7 to SF8628 DIPG xenograft tumors 2.5 mm superficial to the tumor implantation site (SFig. 6C) was not beneficial monotherapy (Fig. 7B). The irradiation of animals improved mice’s survival in saline and control MSCs groups by 3.5–5 days (p<0.001). The animals’ survival was further (p<0.001) improved with MSCs-CRAd.S.pK7 treatment after tumor irradiation (Fig. 7B). Histopathological evaluation of brain tissues revealed the virus’s presence within the tumor for up to days 30–45 post-injection of mice with CRAd.S.pK7-loaded MSCs, as judged by the expression of hexon and E1A viral proteins (Fig. 7C). Consistent with the survival results, viral replication was observed in just one of four brains from mice treated with MSCs-CRAd.S.pK7 in the non-irradiated group. In contrast, all mice in the irradiated group showed viral replication within the intracranial tumor. The virus’s distribution within the tumor was variable among treated animal subjects, with estimated tumor bed coverage ranging from 3.5% to 35% (SFig. 6D). Finally, the analysis of brains from mice with DIPG007 tumors receiving MSCs-CRAd.S.pK7 treatment revealed viral replication and spread in association with radiation, as evidenced by the expression of hexon and E1A viral proteins (Fig. 7D). Collectively, these data confirm the ability of MSCs to successfully deliver CRAd.S.pK7 to DIPG in the context of low dose fractionated irradiation, resulting in a robust therapeutic effect in mice bearing DIPG tumors.
Figure 7. Survival analysis of mice treated with MSC-CRAd.S.pK7 via local versus intranasal delivery following radiation.

A) Dynamics of growth of control (n=7) and irradiated (n=7) (total dose 3Gy) SF8628 DIPG xenograft tumors were analyzed by serial BLI imaging. B) Survival of control, non-irradiated, and irradiated (3Gy fractionated) SF8628 DIPG- bearing mice treated with MSC/CRAd.S.pK7. Injections of saline or MSCs without a virus served as controls. Kaplan-Meier survival curves were compared using log-rank tests with p-values adjusted with Bonferroni correction, n=5–6. Levels of significance * p <0.05, ** p <0.01. The experiment evaluating the effect of MSC/CRAd.S.pK7 on the survival of mice bearing SF8628 DIPG xenograft tumors has been repeated twice. C) Images of H&E-stained brain tissue and corresponding sections stained for viral proteins, hexon, and E1A. D) The expression of hexon and E1A in the brain of irradiated mice bearing DIPG007 tumors. Scale bar=100μm.
Discussion
Despite over 40 years of preclinical evaluation and clinical trials, DIPG persists as an incurable pediatric brain tumor and the number one cause of cancer-related death in children. CRAd.S.pK7 is a potent oncolytic agent presently being evaluated for safety in adult GBM patients (NCT03072134). In this study, we used patient-derived DIPG cell lines and mouse xenograft models (19,34,40) to evaluate MSCs-loaded with the CRAd.S.pK7 for potential translation to the treatment of DIPG patients. Our analyses of patient tumor samples and patient-derived DIPG lines revealed the presence of cellular mediators necessary for the entry and replication of CRAd.S.pK7 in DIPG cells. Further, we have shown successful CRAd.S.pK7 replication in DIPG cells in vitro and showed that low dose fractionated irradiation significantly improves animal survival when treated with MSCs carrying OV.
Delivery systems and platforms that overcome anatomical impediments posed by deep-seated brain tumors, such as DIPG, are in high demand. Stem cells, including MSCs, have low immunogenicity, can circumvent certain anatomic restrictions, such as the BBB, and navigate within the tight structure of the brainstem to home to the tumor site (41–44). Neural and mesenchymal stem cells are well established for safe clinical use in treating the central nervous system’s neurodegenerative and malignant diseases. They can be used as carriers of therapeutic agents, including OVs (13,14,23,30,44,45). In the results presented here, we show that MSCs can deliver a therapeutic payload to brainstem xenografts in the context of fractionated irradiation (46,47). Our results indicate that DIPG cells’ or DIPG-bearing mouse brain irradiation promotes MSC migration toward DIPG in vitro (SFig 1A) and in vivo (Fig. 1C, D). Analysis of cytokine and chemokine expression in untreated and irradiated DIPG tumors revealed many upregulated factors capable of inducing cellular survival and stem cell homing to the tumor (Fig. 2). Our results also show that the survivin promoter’s use to drive OV gene expression is appropriate in that the endogenous survivin gene, BIRC5, is highly expressed in most DIPG (Fig. 5).
OV-mediated tumor killing is also influenced by the virus’s ability to infect tumor cells, and our analysis of patient-derived tumor tissue samples and cell lines showed that all DIPG express mRNA for viral attachment proteins syndecan 1 and perlecan, though at variable levels. High syndecan 1 and perlecan expression in the SF8628 DIPG cells were associated with higher viral progeny, relative to SF7761 and DIPG007 cells expressing lower levels of syndecan 1 and perlecan. Cumulatively, our results suggest that several tumor-specific molecular characteristics influence cell-based OV treatment efficacy.
Importantly, because radiation is the standard treatment for DIPG, we explored its effects in the context of IND and local delivery to the brain, superficial to tumor location. The local, but not IND, delivery of MSC-OV resulted in significantly improved animal survival. This finding suggests a sufficient quantity of oncolytic adenovirus must reach the tumor. Recent studies in preclinical models of DIPG and pediatric glioblastoma (GBM) demonstrated an encouraging efficacy of locally delivered OVs (48). The upregulation of cytokine and chemokine expression in irradiated tumors suggests that modification of MSCs with corresponding receptors may improve their migratory capacity and effective delivery of therapeutic load directly to the tumor site (29).
CRAd.S.pK7 does not replicate in murine glioma cells, precluding its evaluation in an immunocompetent model. Martinez-Velez et al. recently showed that the efficacy of OV treatment in preclinical models of pediatric glioma depends on triggering the adaptive immune response (48,49). The immune system’s contribution to the therapeutic actions of adenovirus encoding for thymidine kinase and fms-like tyrosine kinase 3 has been demonstrated in a murine model of brainstem glioma harboring G328V mutation in the ACVR1 gene (50). These findings are in line with prior studies of multiple tumors types treated with OV that demonstrate a modulated immune response to OV administration that contributes to control of tumor growth (51–54). However, because this immune response is also responsible for the rapid clearance of viral particles, a delay in OV elimination could improve this anticancer treatment strategy (55–59). In that respect, OV delivery using MSCs has been shown to protect OV from rapid viral clearance by the immune system, and consequently to extend the window of oncolytic activity within the tumor, increasing therapeutic efficacy (14). The systemic delivery of autologous MSCs loaded with oncolytic virus Icovir-5 has been recently tested and demonstrated safety in small cohorts of pediatric and adult patients with relapsed/refractory solid tumors (60). Allogeneic bone marrow-derived human MSCs loaded with the oncolytic adenovirus DNX-2401 will be tested in another clinical trial in patients with recurrent glioblastoma using intra-arterial administration (ClinicalTrials.gov Identifier: NCT03896568). These clinical studies indicate substantial interest in the continued development and application of MSC carriers of the oncolytic virus for treating non-CNS and CNS solid tumors.
In conclusion, our study supports oncolytic virus CRAd.S.pK7 encapsulated within MSCs when used in combination with low dose radiation as a therapeutic strategy that merits further investigation and potential translation for DIPG treatment.
Supplementary Material
Statement of Translational Relevance.
The treatment of diffuse pontine intrinsic glioma (DIPG) is particularly challenging due to its infiltration of life-supporting structures in the brainstem, prohibiting tumor resection. Whereas radiation is commonly used, it does not provide significant survival benefits. Thus, this disease that is most prevalent in children is universally lethal. Delivery of drugs and biologicals to these tumors is a major therapeutic challenge. Here we demonstrate the feasibility of using mesenchymal stem cells to deliver oncolytic virus for DIPG. This therapy takes advantage of the virus-induced oncolysis while employs stem cells as carriers to enhance the penetration of these particles into the tumors. Our studies support cellular carrier-mediated delivery of oncolytic viruses as a treatment of DIPG, a strategy that can be translated into a clinical trial for this challenging condition.
Acknowledgments:
This work was supported by NIH R01NS087990, R01NS106379, R33 NS101150 and P50CA221747 SPORE for Translational Approaches to Brain Cancer. We are also grateful to the Nora Redman Endowment Fund of the Community Foundation of Louisville, the IDP Foundation, Inc. and to the Isabella Kerr Molina Foundation for generous gifts to support the study. Some of the materials employed in this work were provided by the Texas A&M Health Science Center College of Medicine Institute for Regenerative Medicine at Scott & White through a grant from ORIP of the NIH, Grant # P40OD011050. This work was also supported by the Northwestern University - Flow Cytometry Core Facility, the Center for Advanced Microscopy, and the Mouse Histology and Phenotyping Laboratory, all supported by NCI P30-CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center. The authors are also grateful to the Pathology Core Facility of the Robert H Lurie Comprehensive Cancer Center for its excellent service.
Footnotes
Conflict of interest statement
M.S.L. and D.T. C. have a patent about the cell-based virotherapy for the treatment of cancer. Other authors declare no conflict of interest.
References
- 1.Gwak HS, Park HJ. Developing chemotherapy for diffuse pontine intrinsic gliomas (DIPG). Crit Rev Oncol Hematol 2017;120:111–9. [DOI] [PubMed] [Google Scholar]
- 2.Berger MS, Edwards MS, LaMasters D, Davis RL, Wilson CB. Pediatric brain stem tumors: radiographic, pathological, and clinical correlations. Neurosurgery 1983;12:298–302. [DOI] [PubMed] [Google Scholar]
- 3.Jansen MH, van Vuurden DG, Vandertop WP, Kaspers GJ. Diffuse intrinsic pontine gliomas: a systematic update on clinical trials and biology. Cancer Treat Rev 2012;38:27–35. [DOI] [PubMed] [Google Scholar]
- 4.Jones C, Karajannis MA, Jones DTW, Kieran MW, Monje M, Baker SJ, et al. Pediatric high-grade glioma: biologically and clinically in need of new thinking. Neuro Oncol 2017;19:153–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saratsis AM, Kambhampati M, Snyder K, Yadavilli S, Devaney JM, Harmon B, et al. Comparative multidimensional molecular analyses of pediatric diffuse intrinsic pontine glioma reveals distinct molecular subtypes. Acta Neuropathol 2014;127:881–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012;44:251–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paugh BS, Qu C, Jones C, Liu Z, Adamowicz-Brice M, Zhang J, et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J Clin Oncol 2010;28:3061–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takami H, Yoshida A, Fukushima S, Arita H, Matsushita Y, Nakamura T, et al. Revisiting TP53 Mutations and Immunohistochemistry--A Comparative Study in 157 Diffuse Gliomas. Brain Pathol 2015;25:256–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paugh BS, Zhu X, Qu C, Endersby R, Diaz AK, Zhang J, et al. Novel oncogenic PDGFRA mutations in pediatric high-grade gliomas. Cancer Res 2013;73:6219–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hendricks BK, Cohen-Gadol AA, Miller JC. Novel delivery methods bypassing the blood-brain and blood-tumor barriers. Neurosurg Focus 2015;38:E10. [DOI] [PubMed] [Google Scholar]
- 11.Meel MH, Kaspers GJL, Hulleman E. Preclinical therapeutic targets in diffuse midline glioma. Drug Resist Updat 2019;44:15–25. [DOI] [PubMed] [Google Scholar]
- 12.Parker Kerrigan BC, Hossain A, Yamashita S, Lang FF. Stem Cell Therapy of Gliomas. Prog Neurol Surg 2018;32:124–51. [DOI] [PubMed] [Google Scholar]
- 13.Hammer K, Kazcorowski A, Liu L, Behr M, Schemmer P, Herr I, et al. Engineered adenoviruses combine enhanced oncolysis with improved virus production by mesenchymal stromal carrier cells. Int J Cancer 2015;137:978–90. [DOI] [PubMed] [Google Scholar]
- 14.Ahmed AU, Rolle CE, Tyler MA, Han Y, Sengupta S, Wainwright DA, et al. Bone marrow mesenchymal stem cells loaded with an oncolytic adenovirus suppress the anti-adenoviral immune response in the cotton rat model. Mol Ther 2010;18:1846–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao D, Najbauer J, Garcia E, Metz MZ, Gutova M, Glackin CA, et al. Neural stem cell tropism to glioma: critical role of tumor hypoxia. Mol Cancer Res 2008;6:1819–29. [DOI] [PubMed] [Google Scholar]
- 16.Choi SA, Lee JY, Wang KC, Phi JH, Song SH, Song J, et al. Human adipose tissue-derived mesenchymal stem cells: characteristics and therapeutic potential as cellular vehicles for prodrug gene therapy against brainstem gliomas. Eur J Cancer 2012;48:129–37. [DOI] [PubMed] [Google Scholar]
- 17.Young JS, Morshed RA, Kim JW, Balyasnikova IV, Ahmed AU, Lesniak MS. Advances in stem cells, induced pluripotent stem cells, and engineered cells: delivery vehicles for anti-glioma therapy. Expert Opin Drug Deliv 2014;11:1733–46. [DOI] [PubMed] [Google Scholar]
- 18.Danielyan L, Schafer R, von Ameln-Mayerhofer A, Buadze M, Geisler J, Klopfer T, et al. Intranasal delivery of cells to the brain. Eur J Cell Biol 2009;88:315–24. [DOI] [PubMed] [Google Scholar]
- 19.Hashizume R, Ozawa T, Gryaznov SM, Bollen AW, Lamborn KR, Frey WH 2nd, et al. New therapeutic approach for brain tumors: Intranasal delivery of telomerase inhibitor GRN163. Neuro Oncol 2008;10:112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danielyan L, Schafer R, von Ameln-Mayerhofer A, Bernhard F, Verleysdonk S, Buadze M, et al. Therapeutic efficacy of intranasally delivered mesenchymal stem cells in a rat model of Parkinson disease. Rejuvenation Res 2011;14:3–16. [DOI] [PubMed] [Google Scholar]
- 21.Moyano AL, Steplowski J, Wang H, Son KN, Rapolti DI, Marshall J, et al. microRNA-219 Reduces Viral Load and Pathologic Changes in Theiler’s Virus-Induced Demyelinating Disease. Mol Ther 2018;26:730–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spencer D, Yu D, Morshed RA, Li G, Pituch KC, Gao DX, et al. Pharmacologic modulation of nasal epithelium augments neural stem cell targeting of glioblastoma. Theranostics 2019;9:2071–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahmed AU, Thaci B, Tobias AL, Auffinger B, Zhang L, Cheng Y, et al. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J Natl Cancer Inst 2013;105:968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ulasov IV, Zhu ZB, Tyler MA, Han Y, Rivera AA, Khramtsov A, et al. Survivin-driven and fiber-modified oncolytic adenovirus exhibits potent antitumor activity in established intracranial glioma. Hum Gene Ther 2007;18:589–602. [DOI] [PubMed] [Google Scholar]
- 25.Altieri DC, Marchisio PC. Survivin apoptosis: an interloper between cell death and cell proliferation in cancer. Lab Invest 1999;79:1327–33. [PubMed] [Google Scholar]
- 26.Takai N, Miyazaki T, Nishida M, Nasu K, Miyakawa I. Expression of survivin is associated with malignant potential in epithelial ovarian carcinoma. Int J Mol Med 2002;10:211–6. [PubMed] [Google Scholar]
- 27.Morshed RA, Gutova M, Juliano J, Barish ME, Hawkins-Daarud A, Oganesyan D, et al. Analysis of glioblastoma tumor coverage by oncolytic virus-loaded neural stem cells using MRI-based tracking and histological reconstruction. Cancer Gene Ther 2015;22:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmed AU, Tyler MA, Thaci B, Alexiades NG, Han Y, Ulasov IV, et al. A comparative study of neural and mesenchymal stem cell-based carriers for oncolytic adenovirus in a model of malignant glioma. Mol Pharm 2011;8:1559–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dey M, Yu D, Kanojia D, Li G, Sukhanova M, Spencer DA, et al. Intranasal Oncolytic Virotherapy with CXCR4-Enhanced Stem Cells Extends Survival in Mouse Model of Glioma. Stem Cell Reports 2016;7:471–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Balyasnikova IV, Prasol MS, Ferguson SD, Han Y, Ahmed AU, Gutova M, et al. Intranasal delivery of mesenchymal stem cells significantly extends survival of irradiated mice with experimental brain tumors. Mol Ther 2014;22:140–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sonabend AM, Ulasov IV, Han Y, Rolle CE, Nandi S, Cao D, et al. Biodistribution of an oncolytic adenovirus after intracranial injection in permissive animals: a comparative study of Syrian hamsters and cotton rats. Cancer Gene Ther 2009;16:362–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonabend AM, Ulasov IV, Tyler MA, Rivera AA, Mathis JM, Lesniak MS. Mesenchymal stem cells effectively deliver an oncolytic adenovirus to intracranial glioma. Stem Cells 2008;26:831–41. [DOI] [PubMed] [Google Scholar]
- 33.Van Houdt WJ, Haviv YS, Lu B, Wang M, Rivera AA, Ulasov IV, et al. The human survivin promoter: a novel transcriptional targeting strategy for treatment of glioma. J Neurosurg 2006;104:583–92. [DOI] [PubMed] [Google Scholar]
- 34.Hashizume R, Ozawa T, Dinca EB, Banerjee A, Prados MD, James CD, et al. A human brainstem glioma xenograft model enabled for bioluminescence imaging. J Neurooncol 2010;96:151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang TY, Piunti A, Lulla RR, Qi J, Horbinski CM, Tomita T, et al. Detection of Histone H3 mutations in cerebrospinal fluid-derived tumor DNA from children with diffuse midline glioma. Acta Neuropathol Commun 2017;5:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qi J, Esfahani DR, Huang T, Ozark P, Bartom E, Hashizume R, et al. Tenascin-C expression contributes to pediatric brainstem glioma tumor phenotype and represents a novel biomarker of disease. Acta Neuropathol Commun 2019;7:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol 2017;19:139–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashizume R, Smirnov I, Liu S, Phillips JJ, Hyer J, McKnight TR, et al. Characterization of a diffuse intrinsic pontine glioma cell line: implications for future investigations and treatment. J Neurooncol 2012;110:305–13. [DOI] [PubMed] [Google Scholar]
- 41.Wang X, Gao J, Ouyang X, Wang J, Sun X, Lv Y. Mesenchymal stem cells loaded with paclitaxel-poly(lactic-co-glycolic acid) nanoparticles for glioma-targeting therapy. Int J Nanomedicine 2018;13:5231–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Timin AS, Peltek OO, Zyuzin MV, Muslimov AR, Karpov TE, Epifanovskaya OS, et al. Safe and Effective Delivery of Antitumor Drug Using Mesenchymal Stem Cells Impregnated with Submicron Carriers. ACS Appl Mater Interfaces 2019;11:13091–104. [DOI] [PubMed] [Google Scholar]
- 43.Layek B, Sadhukha T, Panyam J, Prabha S. Nano-Engineered Mesenchymal Stem Cells Increase Therapeutic Efficacy of Anticancer Drug Through True Active Tumor Targeting. Mol Cancer Ther 2018;17:1196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mendes Filho D, Ribeiro PDC, Oliveira LF, de Paula DRM, Capuano V, de Assuncao TSF, et al. Therapy With Mesenchymal Stem Cells in Parkinson Disease: History and Perspectives. Neurologist 2018;23:141–7. [DOI] [PubMed] [Google Scholar]
- 45.Yong RL, Shinojima N, Fueyo J, Gumin J, Vecil GG, Marini FC, et al. Human bone marrow-derived mesenchymal stem cells for intravascular delivery of oncolytic adenovirus Delta24-RGD to human gliomas. Cancer Res 2009;69:8932–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freese C, Takiar V, Fouladi M, DeWire M, Breneman J, Pater L. Radiation and subsequent reirradiation outcomes in the treatment of diffuse intrinsic pontine glioma and a systematic review of the reirradiation literature. Pract Radiat Oncol 2017;7:86–92. [DOI] [PubMed] [Google Scholar]
- 47.Fontanilla HP, Pinnix CC, Ketonen LM, Woo SY, Vats TS, Rytting ME, et al. Palliative reirradiation for progressive diffuse intrinsic pontine glioma. Am J Clin Oncol 2012;35:51–7. [DOI] [PubMed] [Google Scholar]
- 48.Martinez-Velez N, Garcia-Moure M, Marigil M, Gonzalez-Huarriz M, Puigdelloses M, Gallego Perez-Larraya J, et al. The oncolytic virus Delta-24-RGD elicits an antitumor effect in pediatric glioma and DIPG mouse models. Nat Commun 2019;10:2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martinez-Velez N, Marigil M, Garcia-Moure M, Gonzalez-Huarriz M, Aristu JJ, Ramos-Garcia LI, et al. Delta-24-RGD combined with radiotherapy exerts a potent antitumor effect in diffuse intrinsic pontine glioma and pediatric high grade glioma models. Acta Neuropathol Commun 2019;7:64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mendez F, Kadiyala P, Nunez FJ, Carney S, Nunez FM, Gauss JC, et al. Therapeutic Efficacy of Immune Stimulatory Thymidine Kinase and fms-like Tyrosine Kinase 3 Ligand (TK/Flt3L) Gene Therapy in a Mouse Model of High-Grade Brainstem Glioma. Clin Cancer Res 2020;26:4080–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoo JY, Swanner J, Otani Y, Nair M, Park F, Banasavadi-Siddegowda Y, et al. oHSV therapy increases trametinib access to brain tumors and sensitizes them in vivo. Neuro Oncol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bonora M, Wieckowsk MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L, et al. Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 2015;34:1608. [DOI] [PubMed] [Google Scholar]
- 53.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017;170:1109–19 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jung BK, Oh E, Hong J, Lee Y, Park KD, Yun CO. A hydrogel matrix prolongs persistence and promotes specific localization of an oncolytic adenovirus in a tumor by restricting nonspecific shedding and an antiviral immune response. Biomaterials 2017;147:26–38. [DOI] [PubMed] [Google Scholar]
- 55.Evgin L, Ilkow CS, Bourgeois-Daigneault MC, de Souza CT, Stubbert L, Huh MS, et al. Complement inhibition enables tumor delivery of LCMV glycoprotein pseudotyped viruses in the presence of antiviral antibodies. Mol Ther Oncolytics 2016;3:16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Breitbach CJ, Paterson JM, Lemay CG, Falls TJ, McGuire A, Parato KA, et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol Ther 2007;15:1686–93. [DOI] [PubMed] [Google Scholar]
- 57.Kurozumi K, Hardcastle J, Thakur R, Yang M, Christoforidis G, Fulci G, et al. Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. J Natl Cancer Inst 2007;99:1768–81. [DOI] [PubMed] [Google Scholar]
- 58.Han J, Chen X, Chu J, Xu B, Meisen WH, Chen L, et al. TGFbeta Treatment Enhances Glioblastoma Virotherapy by Inhibiting the Innate Immune Response. Cancer Res 2015;75:5273–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, Yu J, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A 2006;103:12873–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruano D, Lopez-Martin JA, Moreno L, Lassaletta A, Bautista F, Andion M, et al. First-in-Human, First-in-Child Trial of Autologous MSCs Carrying the Oncolytic Virus Icovir-5 in Patients with Advanced Tumors. Mol Ther 2020;28:1033–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.