

Abstract
While calcium imaging has become a mainstay of modern neuroscience, the spectral properties of current fluorescent calcium indicators limit deep tissue imaging as well as simultaneous use with other probes. Using two monomeric near-infrared fluorescent proteins, we engineered a near-infrared FRET-based genetically encoded calcium indicator (iGECI). iGECI exhibits high brightness, high photostability, and up to 600% increase in fluorescence response to calcium. In dissociated neurons, iGECI detects spontaneous neuronal activity, and electrically and optogenetically induced firing. We validated iGECI performance up to a depth of almost 400 μm in acute brain slices using one-photon light-sheet imaging. Applying hybrid photoacoustic and fluorescence microscopy, we simultaneously monitored neuronal and hemodynamic activities in the mouse brain through an intact skull, with ~3 μm lateral and ~25–50 μm axial resolution. Using two-photon imaging, we detected evoked and spontaneous neuronal activity in the mouse visual cortex, with fluorescence changes of up to 25%. iGECI allows biosensors and optogenetic actuators to be multiplexed without spectral crosstalk.
One of the primary goals of neuroscience is to link complex neural phenomena to the structure and function of neural circuits. An essential step toward this goal is simultaneous recording of large numbers of neurons within defined populations, without disrupting their connectivity. Traditional electrophysiological approaches provide excellent sensitivity and temporal resolution but are limited by the number of cells that can be recorded simultaneously1. Moreover, assigning functional activity to specific cells is quite difficult, limiting the ability to resolve circuit maps. Genetically encoded biosensors for neuronal activity2 based on fluorescent proteins (FPs), including genetically encoded calcium indicators (GECIs), combined with modern in vivo imaging techniques3–5 overcome these limitations and allow high-resolution functional imaging in vivo.
Two major scaffolds of GECIs have been developed. Both have a calcium-binding module, mainly consisting of a calmodulin-M13 pair or a troponin C domain. The first scaffold, for intensiometric GECIs6, 7, carries a single FP whose fluorescence is modulated upon calcium binding. The second scaffold, for ratiometric GECIs8–10, consists of two FPs, where calcium binding causes distance- and orientation-dependent changes in Förster resonance energy transfer (FRET) between them. Currently available red-shifted GECIs, including RCaMPs6, 11 and RGECOs11, 12, suffer from high scattering and absorbance of light and autofluorescence in the visible spectral range, making them suboptimal for deep-brain imaging with one-photon excitation. Although this limitation can be circumvented by two-photon excitation, it requires more expensive imaging systems and typically limits the 3D field of view. Shifting GECIs further into the near-infrared (NIR) spectral range would solve this problem and also allow the combination of GECIs with optogenetic actuators using one-photon light sources. This could enable the study of complex processes in large numbers of neurons, correlating activities across neuronal populations, and neural circuit mapping.
GECIs with both excitation and emission in an NIR tissue transparency window are preferable for functional imaging in vivo. Several bacterial phytochromes have been engineered into monomeric NIR FPs13–15. Bacterial phytochromes use biliverdin (BV), an abundant product of heme catabolism in mammalian cells, as a chromophore linear tetrapyrrole. The spectral properties of NIR FPs make them attractive building blocks to engineer fully NIR GECIs. However, the only reported intensiometric indicator NIR-GECO1 based on a mIFP NIR FP14 is dim, has low efficiency of BV chromophore binding and, consequently, shows limited contrast for in vivo imaging16.
Here, we developed a fully NIR FRET-based calcium indicator on the basis of a cameleon-like GECI scaffold and the recently described bright monomeric NIR FPs, miRFP67013, 15 and miRFP72017. We validated this NIR indicator using single objective-based scanned oblique plane illumination (SOPi) one-photon microscopy, benchmarking the new indicator against GCaMP6s. With this indicator we detected neural activity in response to electrical or optogenetic stimulation from large regions of the motor cortex using a low power one-photon excitation laser. Next, we built a hybrid photoacoustic and fluorescence microscope and demonstrated the feasibility of the developed NIR indicator to simultaneously monitor neuronal and hemodynamic activities in the mouse brain through an intact skull. Lastly, we applied this indicator for detection of visually evoked and spontaneous neuronal activity in the mouse brain at depth using two-photon microscopy.
Results
Engineering of near-infrared fluorescent iGECI.
We started engineering NIR GECI variants using standard design of ratiometric calcium indicators, where the FRET donor and acceptor FPs are located around a Ca2+-sensing module (Supplementary Fig. 1). We tested relative positions of the miRFP670 donor and miRFP720 acceptor and several sensing modules, including troponin C from the Twitch indicators, a pair of mutated calmodulin (CaM) and M13 peptide from the D3cpV indicator, and a CaM-M13 pair from the YC3.6 indicator. We found that the original CaM-M13 sensing module from the YC3.6 fused N-terminally with the miRFP670 and C-terminally with the miRFP720 weakly responded to Ca2+ ions in mammalian cell lysates. To further optimize the response, we next systematically truncated the N-terminus of CaM and the C-terminus of M13, as well as the C-terminus of miRFP670 and the N-terminus miRFP720. As a result, we found a NIR GECI variant with the miRFP670 truncated by one amino acid residue from the C-terminus, the miRFP720 truncated by 17 amino acid residues from the N-terminus, and flexible 2 amino acid residue linkers between the miRFP670 and CaM (L1 linker), and the M13 and miRFP720 (L2 linker). This variant showed ~40% decrease of miRFP670 fluorescence in response to Ca2+ in HeLa cell lysates and was further used as a template to optimize L1 and L2 linkers.
Bacteria were transformed with a library of random mutants of the linkers and sorted using a fluorescence activated cell sorter (FACS) to select clones that fluoresce in both miRFP670 and miRFP720 channels. The FACS-collected bacterial clones were then grown on Petri dishes and transferred to nitrocellulose membrane for screening of permeabilized colonies. Next, the NIR GECI variants with stronger Ca2+ responses were grown in multi-well plates, and bacterial lysate was screened again. The best performing NIR GECI clones were recloned to a mammalian expression vector and tested in HeLa cell lysates. A mixture of several advanced clones provided templates for a new library used in the next round of screening.
After several rounds of molecular evolution, a NIR GECI variant containing -YT- linker L1 and -VV- linker L2 and performing best in HeLa cells was selected. To achieve larger responses in dissociated neurons we further tuned Ca2+ affinity of the selected NIR GECI variant. For this we varied the linker L3 between the CaM and M13 parts, which affected Ca2+ affinity in YC-Nano indicators18, using either the original YC3.6 CaM with 3 Ca2+-binding sites or the wild-type CaM with all 4 Ca2+ binding sites. We found that CaM with 4 Ca2+ sites and Gly7Ser linker L3 resulted in the highest changes in miRFP670 fluorescence in both HeLa cells and neurons. This NIR GECI variant was termed iGECI and chosen for further validation (Fig. 1a and Supplementary Fig. 2).
Figure 1. Characterization of iGECI in vitro and in HeLa cells.
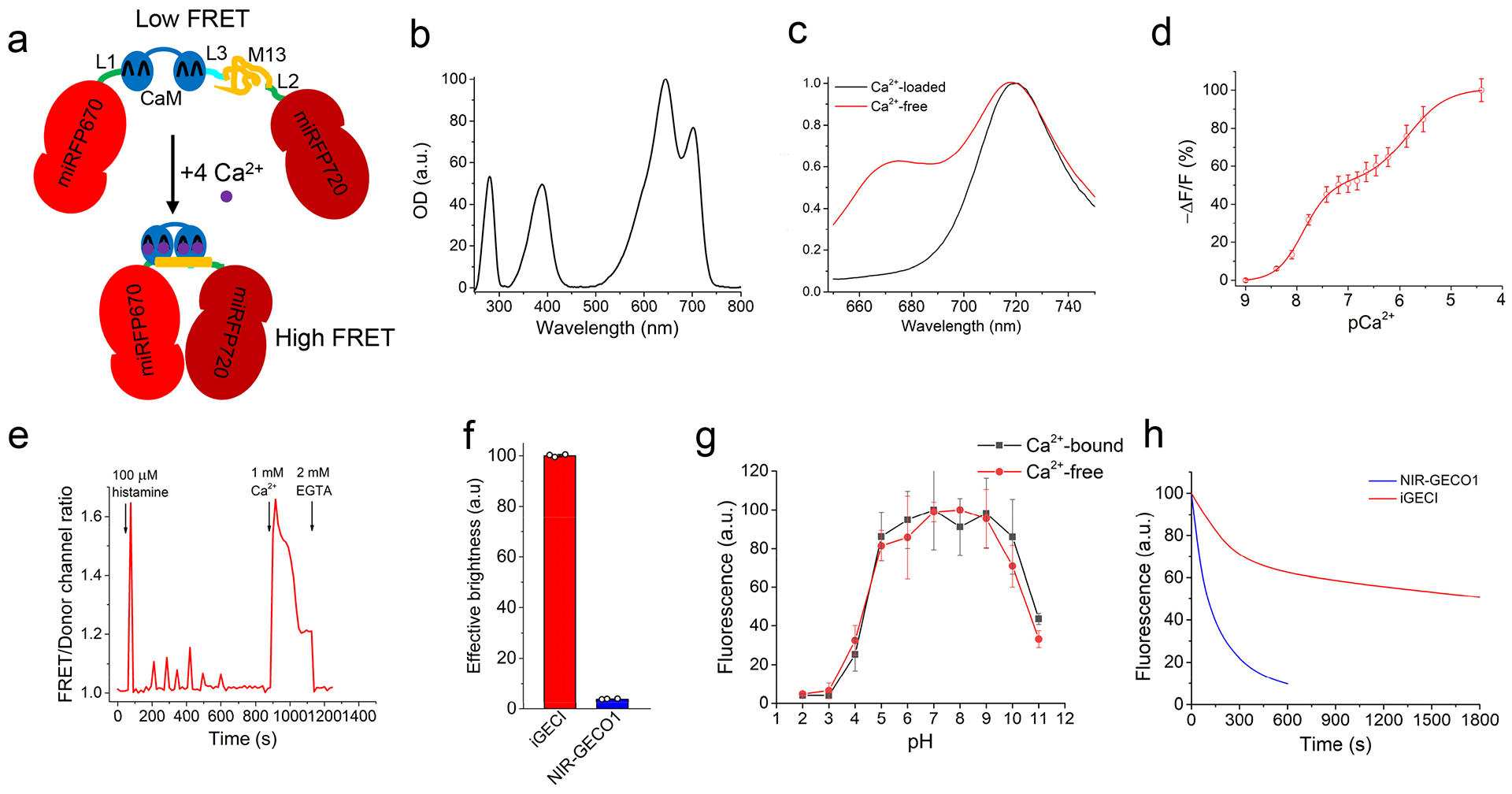
(a) Schematic representation of iGECI and its mechanism of Ca2+ response. miRFP670, the FRET donor, is colored red; miRFP720, the FRET acceptor, is colored dark-red. Ca2+-sensing module is represented by calmodulin (CaM, blue) and the M13 peptide (yellow). Linkers L1 and L2 between the donor and sensing module, and the sensing module and the acceptor, respectively, are colored green. Linker L3 between CaM and M13 is colored cyan. (b) Absorbance spectrum of purified iGECI. (c) Fluorescence spectra of iGECI expressed in HeLa cells in the absence (red line) and presence (black line) of 1 mM Ca2+ measured in cell lysate. Cells were treated with 25 μM of BV chromophore for 24 h before lysis. Spectra were normalized against miRFP720 acceptor emission peak. (d) Response of purified iGECI measured at 670 nm as a function of Ca2+ concentration. (e) Typical Ca2+ transients reported by iGECI in live HeLa cells. Ratio changes of FRET (ex. 605 nm, em. 725/40 nm) to the donor (ex. 605 nm, em. 680/20 nm) fluorescence intensities upon treatment with 100 μM of histamine, in the absence of extracellular Ca2+, followed by changing the media to one containing 1 mM Ca2+ and then 2 mM EGTA. (f) Comparison of iGECI and NIR-GECO1 brightness in live HeLa cells measured using flow cytometry. The 640 nm laser was used for excitation, and 647 nm longpass edge filter to detect fluorescence. Fluorescence intensities were normalized to absorption efficiencies of indicators at 640 nm. (g) Dependence of purified iGECI fluorescence on pH value in the presence of 2 mM EGTA (red), or 1 mM Ca2+ (black). (h) Photobleaching curves of iGECI and NIR-GECO1 in live HeLa cells excited using a 605/30 nm bandpass and imaged using a 647 nm longpass filter. Photobleaching data were normalized to absorption efficiencies of indicators at 605 nm. No exogenous BV was supplied. n=10 cells. In (d, f, g) error bars represent SEM, n=3 experiments.
Characterization of iGECI in vitro and in non-neuronal mammalian cells.
iGECI had a minor absorption peak at 390 nm, which corresponds to the overlaid Soret bands of the miRFP670 donor and the miRFP720 acceptor, and two major absorption maxima at 640 nm and 700 nm, which correspond to the absorption of miRFP670 and miRFP720 (Fig. 1b). iGECI fluorescence peaked at 670 nm and 720 nm, respectively (Fig. 1c). iGECI exhibited 600% change of the FRET/donor fluorescence ratio between the Ca2+-free and Ca2+-saturated states, mainly attributed to the decrease in miRFP670 donor fluorescence (ΔR/R≈-ΔF/F). Linear unmixing of the iGECI spectra revealed that the ~38% increase of miRFP720 emission was largely compensated by the decrease of miRFP670 cross-bleed emission in the FRET channel, resulting in only ~8% apparent FRET-channel changes (Supplementary Fig. 3). This feature simplified iGECI imaging setup to a single miRFP670 donor emission channel. As a result of four Ca2+-binding sites, iGECI showed two affinity constants, Kd1=15 nM and Kd2=890 nM (Fig. 1d). These constants were close to those of the YC-Nano15 indicator (23 nM and 930 nM, respectively)18. iGECI enabled reliable recording of histamine-evoked Ca2+ oscillations in live mammalian cells (Fig. 1e).
We next compared iGECI with NIR-GECO1 expressed in mammalian cells under the CMV promoter. In transiently transfected HeLa cells NIR-GECO1 was 19.2-fold dimmer than iGECI (Fig. 1f and Supplementary Fig. 4). Incubation of NIR-GECO1-expressing cells with the saturating 25 μM concentration of exogenous BV for 24 h resulted in 8.1-fold increase in brightness (Supplementary Fig. 5). This indicated that only 12% of NIR-GECO1 molecules had bound endogenous BV chromophore and fluoresced, whereas the remaining 88% of NIR-GECO1 remained available for undesirable Ca2+ buffering. Although exogenous BV dramatically increased the brightness of the NIR-GECO1 expressing cells, they remained 5-fold dimmer than the iGECI-expressing cells similarly treated with BV (Supplementary Fig. 4).
iGECI showed the same pH stability in both Ca2+-free and Ca2+-loaded states, with pKa=4.5 and pKa2=10.5 (Fig. 1g). The pH stability of iGECI was similar to that observed for NIR-GECO1 in the Ca2+-loaded state, but substantially broader than for NIR-GECO1 in the Ca2+-free state (Supplementary Table 1). Moreover, iGECI was 18-fold more photostable (t0.5 =1795 s) than NIR-GECO1 (t0.5 =100 s) (Fig. 1h). iGECI photobleaching kinetics was biphasic, likely reflecting the difference in photostability of the Ca2+-free (lower, because of lower FRET from the miRFP670 donor) and the Ca2+-bound (higher, because of higher FRET) molecules. Overall, we concluded that iGECI possesses favorable characteristics for imaging neuronal activity.
Characterization of iGECI in cultured neurons.
For further characterization of iGECI, we transduced dissociated mouse hippocampal neurons with AAV2/9-CaMKII-iGECI and stimulated neuronal activity with an electrical field. We delivered 1–160 electrical pulses to stimulate the cells while imaging fluorescence of cell bodies (Fig. 2). iGECI response amplitudes (Fig. 2a) measured as -ΔF/F of miRFP670 donor increased with the number of stimuli, from 5.7% for a single pulse to 22.6% for train of 160 pulses. Incubation with 25 μM BV for 3 h increased the amplitudes by 1.4–2.2-fold (12.9% for 1 pulse, 30.4% for 160 pulses). Consequently, the signal-to-noise ratio increased with the number of stimuli and with the addition of BV (Fig. 2b). Rise time, measured as the period from the offset of the stimulus to the peak of fluorescence change (as previously described7), decreased with the number of stimuli: from 0.7 s for single pulse to 0.2 s for train of 160 pulses (Fig. 2c). Half-time of the signal decay after the peak was 14.4 s for single pulses, increasing to 30.7 s for 160 pulses (Fig. 2d). Exogenous BV did not change rise time or decay rate.
Figure 2. Characterization of iGECI in dissociated mouse neurons.
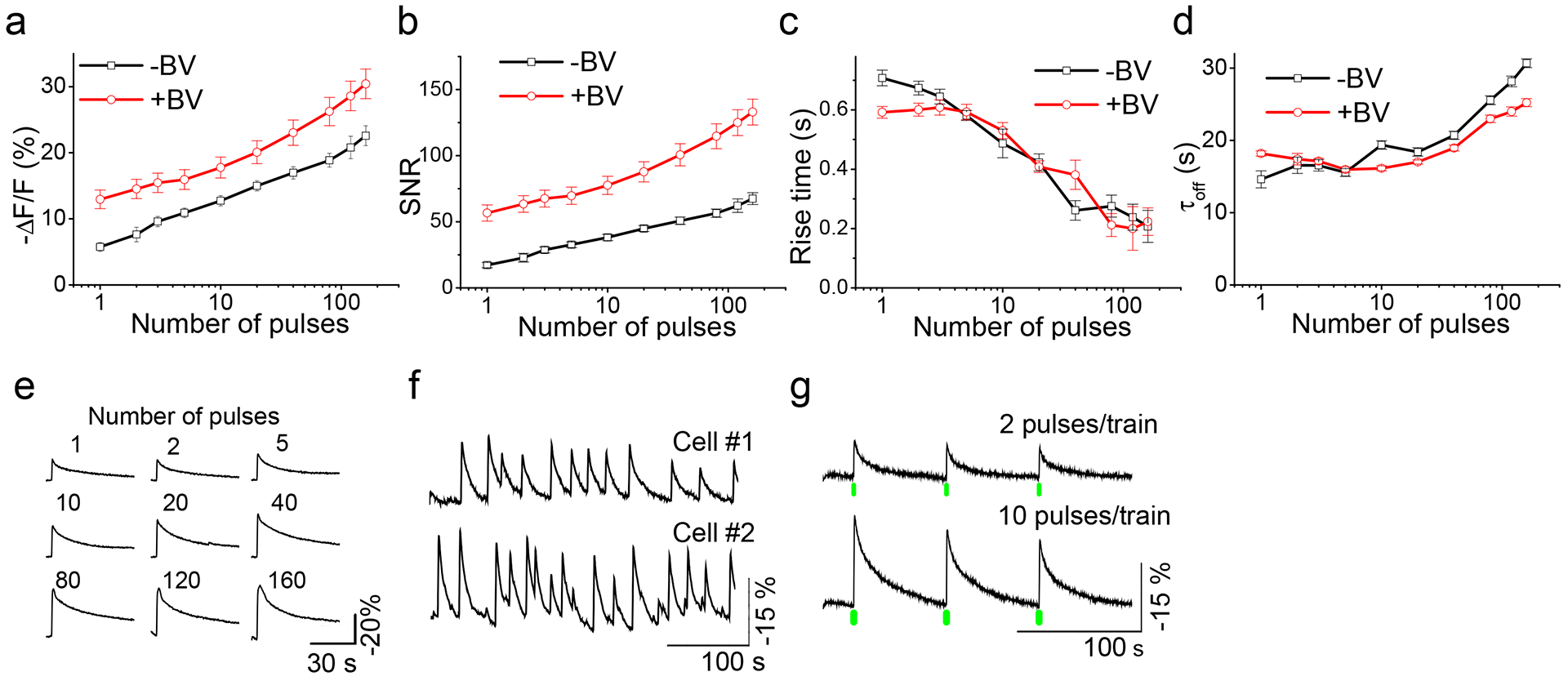
(a) Response amplitudes of iGECI as a function of the number of field stimulation pulses. Spontaneous activity was suppressed by synaptic inhibitors. (b) Signal-to-noise ratios. The noise was defined as the standard deviation of optical signal in the 5 s preceding stimulation. (c) Rise time (time to peak, i.e. the minimum of fluorescence value). Rise times were calculated starting from stimulus offset. (d) Half-time of signal decay. In (a-d) black lines represent data obtained without BV, and red lines after 3 h long pre-incubation with saturated 25 μM concentration of the BV chromophore. In (a-d), error bars represent SEM, n=20 cells. (e) Representative traces of single trial iGECI fluorescence responses to field stimulation in a cultured mouse hippocampal neuron. (f) Spontaneous calcium activity of cultured neurons monitored by iGECI fluorescence. Examples cells were from different cultures. (g) iGECI responses to the activation of CheRiff channelrhodopsin with either 2 or 10 short (4 ms) pulses of 505 nm light. Green bars indicate trains of 505 nm pulses. In (e, f, g) normalization and exponential subtraction were applied to the traces and the y-scale was inverted.
To compare iGECI with NIR-GECO1 in neurons, we made an AAV2/9-CaMKII-NIR-GECO1. iGECI exhibited higher signal-to-noise ratio, shorter rise time, similar response amplitude for small number of pulses, and slower off kinetics, as compared to NIR-GECO1 (Supplementary Fig. 6 and Supplementary Table 1). In addition to stimulated activity, (Fig. 2e) iGECI also detected spontaneous activity in neuronal cultures (Fig. 2f).
Photostability of iGECI in neurons (t0.5 =1735 s) was similar to that in HeLa cells (Fig. 1h). In contrast, NIR-GECO1 exhibited notably higher photostability in neurons than in non-neuronal cells (t0.5 =134 s) (Supplementary Fig. 7 and Supplementary Table 1), likely reflecting the difference in the basal levels of Ca2+ in HeLa cells (14 nM)19 and neurons (50–100 nM)20. In HeLa cells the vast majority of NIR-GECO1 molecules (Kd=215 nM) were in the Ca2+-free state, while in neurons more NIR-GECO1 molecules were Ca2+-bound. Because extinction coefficient of Ca2+-bound NIR-GECO1 is 3-fold lower than in the Ca2+-free state16, this consequently decreased light absorption efficiency and increased the total photostability of the NIR-GECO1 molecules.
To test the suitability of iGECI for spectral multiplexing with blue-green light-activatable actuators, we transduced iGECI-expressing neurons with AAV2/9-hSyn1-CheRiff channelrhodopsin. Activation of CheRiff with 505 nm light caused calcium transients, which were detected by iGECI with high fidelity (Fig. 2g). Increasing the number of 505 nm light pulses led to larger changes in iGECI fluorescence. Green light did not elicit calcium signals in neurons expressing iGECI alone, while the same neurons reliably responded to electrical stimuli (Supplementary Fig. 8), indicating that iGECI can be easily combined with actuators of neuronal activity for multiplexed measurement and control of neural firing.
Functional imaging of iGECI in acute mouse brain slices.
To evaluate the performance of iGECI in acute brain slices, we injected AAV2/9-CaMKII-iGECI unilaterally into the motor cortex of C57BL/6 neonatal mice (age P2–4). Expression was validated in fixed tissue (Fig. 3a). A modified setup of the SOPi microscope21 was used for direct, rapid imaging of optically sectioned oblique planes in the samples (Fig. 3b and Online Methods). A 632.8 nm laser beam was scanned with one galvanometer-based planar scan mirror to create the light-sheet22. Another galvanometer mirror, conjugated to the back focal plane of the microscope objective, was used for fine adjustment of the oblique image plane23. Oblique plane illumination at 45° angle was created in the sample volume for optically sectioned imaging across depth. Imaging of the illuminated sample plane was achieved by a sCMOS camera coupled with a 676/37 emission filter.
Figure 3. Oblique light-sheet functional imaging of iGECI in acute brain slices.
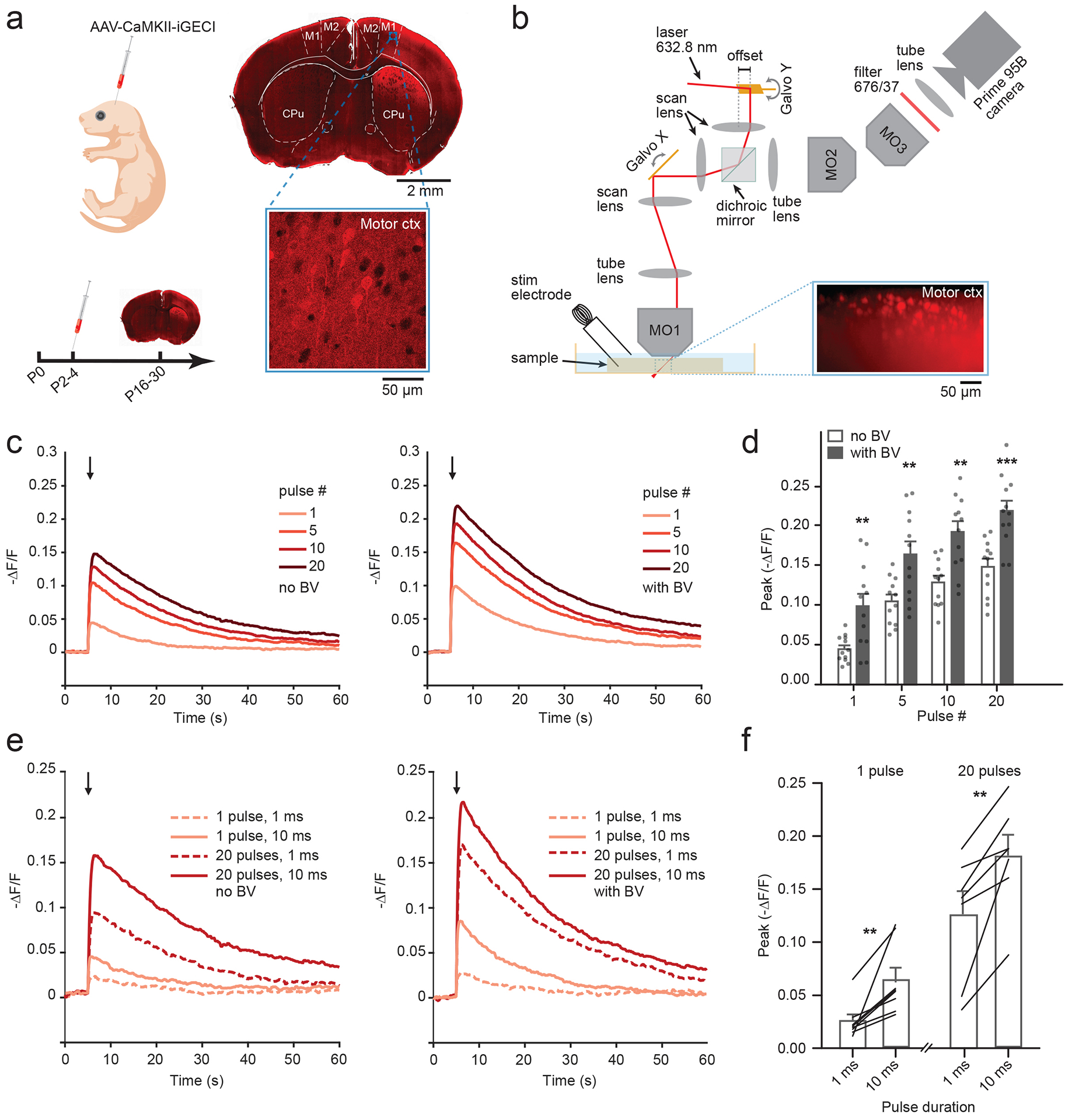
(a) Left, neonatal viral transduction schematic, with experimental timeline. Right, an epifluorescence image of iGECI expression 4 weeks after a unilateral injection. Close up, confocal image of viral expression in the motor cortex (10 images, 3 mice). (b) Schematic illustrating SOPi-based light-sheet imaging of iGECI in acute mouse brain sections in response to electrical stimulation, using 632.8 nm laser, with detection centered at 676 nm. Inset shows an example of oblique light-sheet image of iGECI expression. (c) Fluorescence traces (-ΔF/F) following electrical stimulation in acute coronal brain slices of the motor cortex. Left, average traces across experiments (n=12 experiments/condition, single full field ROI/experiment) for different pulse numbers (1 mA, 10 ms pulse width, 20 Hz; imaging rate, 20 frames per second, fps). Arrow marks onset of stimulation. Right, same as left, following 2 h pre-incubation with saturated 25 μM concentration of the BV chromophore. (d) Summary data for response amplitude to varying stimulation pulse number presented in (c). Each column represents the mean from multiple experiments, where each experiment contains a single full field ROI. ± BV data are unpaired and derived from separate experiments. Two-way repeated measures ANOVA: BV p=0.0008; pulse number p<0.0001, interaction p=0.2745, error bars represent SEM, n=12 for each column. Asterisks represent with Bonferroni post hoc tests, **p<0.001, *** p<0.0001. (e) Left, average traces (-ΔF/F), comparing responses to two stimulation pulse durations for single pulses and trains of 20 stimuli (20 Hz). Right, same as left, following 2 h pre-incubation with saturated 25 μM concentration of the BV chromophore. (f) Summary data for response amplitude to varying pulse length presented in (e), from experiments ± BV incubation. Each line reflects a paired comparison from one experiment varying pulse length. Bars represent mean with SEM. Paired t-test: 1 pulse, n=8, p=0.0077; 20 pulses n=7 p=0.0065. **p<0.001, ***p<0.0001.
Acute brain slices were prepared 2–8 weeks post injection, and monopolar electrical stimulation was used to induce neuronal activity. First, iGECI response amplitudes were compared to those after 2 h incubation of acutely prepared brain slices in BV (Fig. 3c,d). A 10 ms pulse duration was chosen because it consistently evoked the iGECI responses to a single stimulus. The frequency of stimulation (20 Hz) was selected to match the typical firing rate of cortical pyramidal neurons24. Without exogenous BV, -ΔF/F of miRFP670 donor ranged from 5% after a single pulse to 15% after a train of 20 pulses. Exogenous BV enhanced the iGECI functional response 1.5–2 fold, depending on the pulse number (two-way repeated measures ANOVA: p=0.0008; pulse number, p<0.0001; interaction, p=0.2745). Then, we compared the 1 ms pulse width, as was done for cell culture experiments, with 10 ms one for single pulses and trains of 20 stimuli (Fig. 3e,f). iGECI reliably detected both narrow and wide pulses, and as expected, longer stimulation led to larger fluorescence changes (Paired t-test: 1 pulse, p=0.0077; 20 pulses p=0.0065).
Because iGECI excitation and emission wavelengths are red-shifted, compared to the widely used GCaMP6 family of indicators, we expected less optical scattering of signal from deeper regions of the tissue for one-photon imaging. To directly compare depth penetration of iGECI and GCaMP6s, we co-expressed the two AAVs under the control of the CaMKII promoter into the motor cortex. Functional changes in response to electrical stimulation were observed for up to ~400 μm in depth for both sensors (Fig. 4a–c). We compared the relative changes in absolute fluorescence signal with depth and the -ΔF/F signal between iGECI and GCaMP6s (Fig. 4b,c). All values were normalized to the signal from an ROI at 50 μm depth. Relative baseline intensity of iGECI decreased less with depth than for GCaMP6s (Fig. 4c, left) (two-way repeated measures ANOVA: depth F14,197=30.91 ***p<0.0001; GCaMP6s vs. iGECI F1,197=60.07 ***p<0.0001; interaction F14,197=5.74 ***p<0.0001). The initial increase in intensity and responses with depth is likely due to the cellular damage that occurs in the superficial layer of acute slice preparations. While for iGECI the ΔF/F peak was smaller than that of GCaMP6s, the iGECI functional signals did not demonstrate the rapid depth-dependent decline observed for GCaMP6s (Fig. 4c, right) (two-way repeated measures ANOVA: Depth F14,197=3.65 ***p<0.0001; GCaMP6s vs. iGECI F1,197=58.84 ***p<0.0001; interaction F14,197=4.40 ***p<0.0001). Therefore, the NIR wavelength shift of iGECI translates into depth stability, which is useful for in vivo applications.
Figure 4. Spectral multiplexing of iGECI with GCaMP6s or ChR2 optogenetic actuator in acute brain slices.
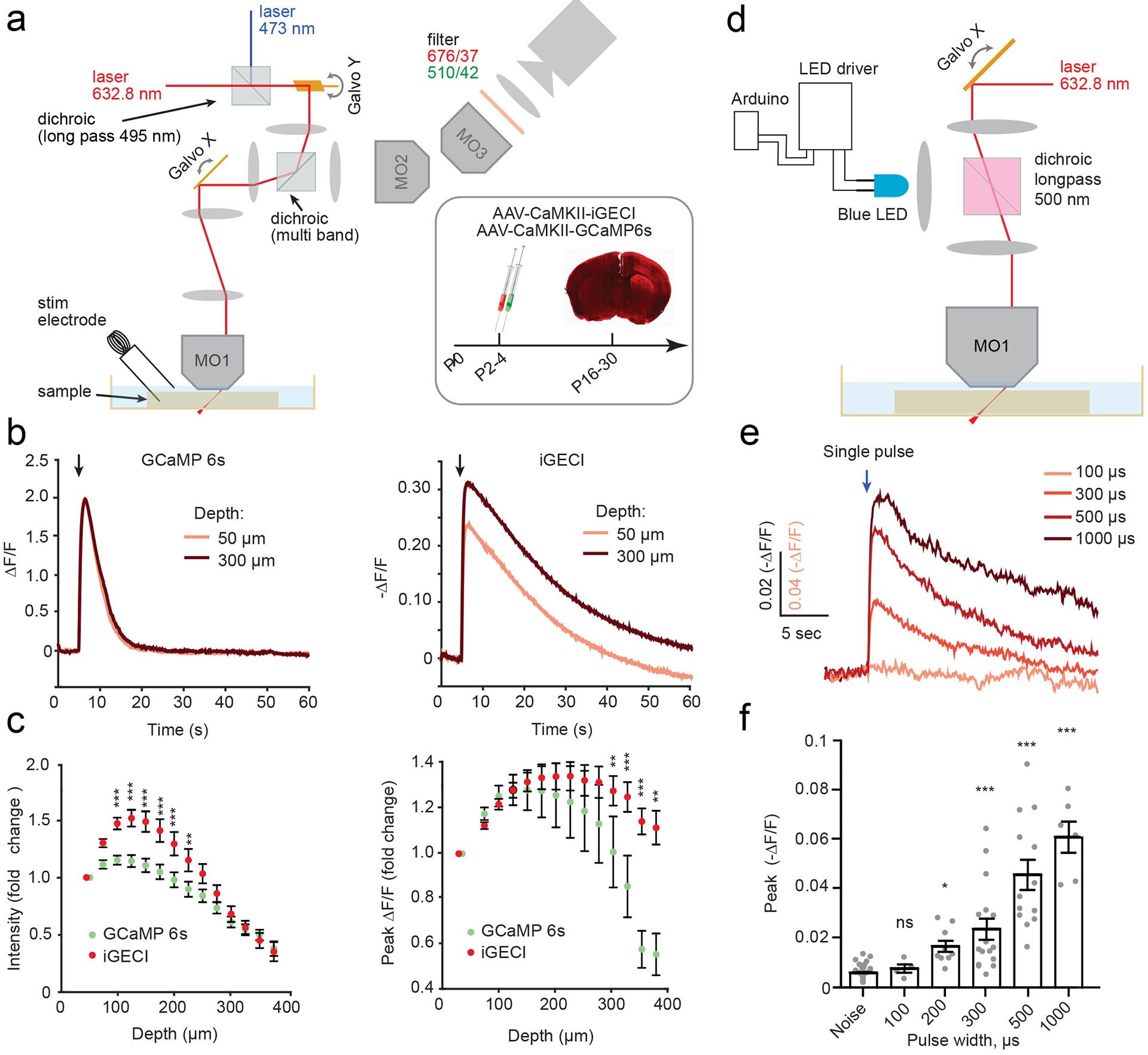
(a) SOPi imaging system showing modifications for dual sensor imaging. Inset: schematics of AAV injections and experimental timeline. (b) Example data from a single experiment using electrical stimulation, analyzed at two depths, as noted. Left, GCaMP6s signal (ΔF/F) in response to electrical stimulation. Right, same but for iGECI. Stimulation protocols: 10 pulses, 1 mA, 10 ms pulse width, 20 Hz; imaging rate, 20 fps. (c) Quantification of fluorescence changes for iGECI and GCaMP6s with increasing depth across experiments (n=16, separate acute slices). All values were normalized to fluorescence intensity at 50 μm depth. Left, raw intensity values. Right, peak ΔF/F in response to electrical stimulation. Error bars reflect SEM. Two-way ANOVA, fluorophore p<0.0001, depth p<0.0001, interaction, p<0.0001. Bonferroni post hoc tests, **p<0.001, *** p<0.0001. (d) SOPi imaging system showing modifications for optogenetic stimulation using a 470 nm LED. Animals were injected with AAVs driving ChR2 and iGECI expression under CaMKII promoter. (e) Average traces for iGECI motor cortex responses to single optogenetic stimulation pulses widths (0.1–1 ms) using 470 nm light (n=15). Arrow indicates the time of stimulation. (f) Quantification of peak fluorescence changes. (Error bars reflect SEM. One-Way ANOVA. Kruskal-Wallis test, p<0.0001. Asterisks denote Dunn’s multiple comparisons tests to baseline noise fluctuations, * p<0.005, **p<0.001, *** p<0.0001. Noise n=35, 100 μs n=4, 200 μs n=9, 500 μs n=13, 1000 μs n=6).
To evaluate the compatibility of iGECI for spectral multiplexing in brain slices, we co-expressed iGECI with the ChR2 channelrhodopsin actuator, encoded by an AAV under CaMKII promoter, in the motor cortex of neonatal mouse pups (Fig. 4d–f). One strength of optical stimulation is the ability to finely control power, enabling the titration of the smallest, including potentially subthreshold iGECI responses, in the presence of blockers for fast neurotransmitters to disrupt circuit level propagation of activity. ChR2 evokes single spikes in different genetically targeted neuronal classes using stimuli in the range of 0.1–1 ms25. We applied single pulses of 470 nm light ranging from 100 μs to 1 ms in duration, detecting responses with sub-millisecond stimuli which increased in amplitude with pulse duration (Fig. 4e,f). Stimuli of ≥200 μs showed a statistically significant response of iGECI, compared to baseline fluctuations. Therefore, iGECI can likely detect single spike responses and could in principle be suitable for studying subthreshold and subcellular Ca2+ dynamics with appropriate AAV targeting.
In vivo iGECI imaging by hybrid photoacoustic and fluorescence microscopy.
To study the performance of iGECI in vivo, we have developed a hybrid photoacoustic and fluorescence microscopy system capable of imaging brain oxygenation and neural activity simultaneously (Fig. 5a and Online Methods). Photoacoustic microscopy has been proven effective for quantifying blood oxygenation with high sensitivity, using only hemoglobin in the red blood cells as the endogenous probe26–28. Photoacoustic and fluorescence detection are functionally complementary, and they can be seamlessly integrated without signal interference. This hybrid photoacoustic and fluorescence system was able to achieve a lateral resolution of ~3 μm, and an axial resolution of 25 μm for photoacoustic imaging and 50 μm for fluorescence signals.
Figure 5. In vivo imaging of iGECI using hybrid photoacoustic and fluorescence microscopy.
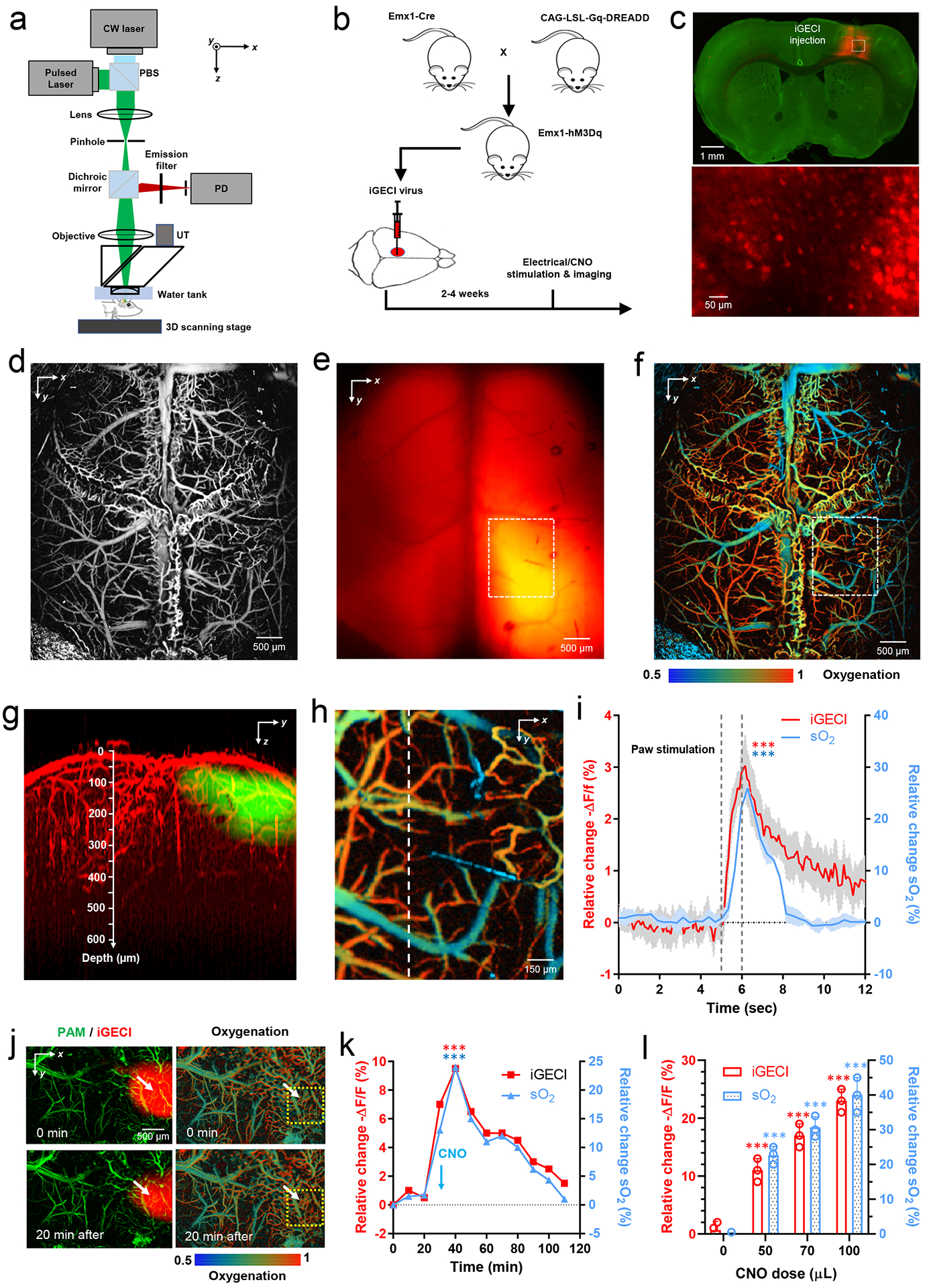
(a) Hybrid photoacoustic and fluorescence microscopy. PBS, polarizing beam splitter; PD, photodiode; UT, ultrasonic transducer. (b) Transgenic mouse cross that expresses an excitatory hM3Dq DREADD in the forebrain pyramidal neurons. Clozapine-N-oxide (CNO) can be administered to enhance neuronal firing. (c) Fluorescence images of the mouse brain slice, showing the full-view iGECI expression (top, red) and iGECI-expressing neurons (bottom). (d-e) In vivo hybrid photoacoustic image of the brain vasculature (d) and fluorescence image of the iGECI expression (e) through the intact skull. (f) In vivo photoacoustic imaging of the blood oxygenation. (g) Depth projection of overlaid PAM and fluorescence images, showing a ~600 μm penetration depth. (h) A close-up image of the dashed region in (f), showing the oxygenation of the cortical hindlimb region. Data shown in (c-h) are representative of three independent experiments with similar results. (i) Oxygenation and calcium responses to electrical stimulations of the mouse left hindlimb. Time traces of the averaged iGECI signals (-ΔF/F) and blood oxygenation (sO2) along the dashed line, across the region shown in (h). The stimulation lasted for 1 s. n=4 biologically independent mice. Shaded area: ±SEM. *** p value <0.001; paired two-sided student’s t-test; Exact p values: iGECI, 0.000323; sO2, 0.000275. (j) Simultaneous imaging of the iGECI (left, red) and blood oxygenation (right) before and 20 min after i.p. injection of indicated volume of CNO (1 μg/ml). (k) Time traces of the averaged iGECI signals (-ΔF/F) and sO2 within the yellow dashed circle in (j), following i.p. injection of CNO. Data in (j) and (k) are representative of three independent experiments with similar results. *** p value <0.001; paired two-sided student’s t-test. Exact p values: iGECI, 0.000148; sO2, 0.000836. (l) The peak changes in iGECI signals (-ΔF/F) and sO2 with different CNO doses. n=3 biologically independent mice at each dose. *** p value <0.001; paired two-sided student’s t-test. Exact p values: iGECI for dose 50, 70, 100 μL, 0.000317, 0.000538, 0.000791, respectively; sO2 for dose 50, 70, 100 μL, 0.000874, 0.000683, 0.000592, respectively. In all experiments, no exogenous BV was supplied.
For imaging brain oxygenation and neuronal activity, we used a transgenic Emx1-hM3Dq mouse model, which expresses the excitatory chemogenetic actuator hM3Dq predominantly in forebrain excitatory neurons (Fig. 5b)29. Pyramidal neurons in the Emx1-hM3Dq mice were infected with an AAV encoding iGECI (Fig. 5c and Supplementary Fig. 9). hM3Dq is a modified form of the human M3 muscarinic (hM3) receptor and can be activated by the inert clozapine metabolite clozapine-N-oxide (CNO), engaging the Gq signaling pathway30 and enhancing neuronal excitability31.
First, we imaged the whole mouse cortex in vivo using the hybrid system, with the skull intact. The photoacoustic and fluorescence images were automatically co-registered. Cortex vasculature and blood oxygenation were resolved by photoacoustic microscopy at a single-vessel level (Fig. 5d,f). Fluorescence images reflected the strong expression of iGECI in the hindlimb sensory region (Fig. 5e). Using photoacoustic microscopy, penetration depth of ~600 μm into the cortex was achieved through the intact skull (Fig. 5g).
Using the hybrid imaging system, we then performed electrical stimulation on the left hindlimb of Emx1-hM3Dq mice (Fig. 5h). We observed a 3% of -ΔF/F of the miRFP670 donor channel in iGECI, accompanied by a 25% increase in the blood oxygenation (Fig. 5i). The latter readout was consistent with previous results for hindlimb stimulation28. Taking advantage of the high-resolution vessel image from the photoacoustic imaging, we analyzed the fluorescence signals only from regions without blood vessels, which largely mitigated the potential impact of hemodynamics on the fluorescence measurement32. We observed that the blood oxygenation changes peaked ~200 ms after the calcium signal changes, indicating the delay of hemodynamics in neurovascular coupling. We also observed that the blood oxygenation changes decayed faster than the calcium signals, which may be partially due to the relatively slow decay kinetics of iGECI. These results demonstrated the feasibility of using the hybrid imaging system with iGECI to simultaneously monitor neuronal and hemodynamic activities in the brain.
We next performed chemical stimulations in the Emx1-hM3Dq mice by i.p. injection of CNO. Similar to the electrical stimulations, after the CNO injection we observed substantial increase in the cortical oxygenation (~25%) and decrease in the iGECI signals (~10% of miRFP670 -ΔF/F) (Fig. 5j). Both the oxygenation and iGECI changes peaked at 20 min after the CNO injection, and slowly reduced to the baseline at 90 min after the CNO injection (Fig. 5k). The dynamics of the iGECI fluorescence were consistent with oxygenation when neuronal activity was chemogenetically elevated. The overall blood flow in the cortex also increased (Supplementary Fig. 10), confirming neurovascular coupling. Since CNO-induced neural activity is dose-dependent, we observed iGECI signal change up to 23% (-ΔF/F) with the higher CNO dose (Fig. 5l). We also observed a similar dose dependence of the blood oxygenation changes (Fig. 5l). No exogenous BV was supplemented in these experiments.
In vivo iGECI two-photon imaging in mouse visual cortex.
We next imaged iGECI in the mouse primary visual cortex transduced with AAV2/9-CaMKII-iGECI at subcellular resolution. In a subset of mice, we also co-expressed GCaMP6s by co-injecting AAV2/1-hSyn-GCaMP6s. After recovery from virus injection and cranial window implantation, we imaged their two-photon excited fluorescence using 900 nm excitation light on a custom-built two-photon microscope (Supplementary Fig. 11). It has been shown that the NIR FPs engineered from bacterial phytochromes exhibit optimal two-photon excitation in 800–900 nm range33. In the mouse brains expressing iGECI alone or co-expressed with GCaMP6s, we observed a large population of neurons with iGECI throughout the cortical volume to a depth of 600 μm (Supplementary Fig. 12). iGECI was mainly excluded from the nucleus, which is expected for the 86 kDa iGECI construct that exceeds the maximal size of ~55 kDa for proteins freely diffusing through the nuclear pore. iGECI positive neurons were observed even after >3 months of incubation, indicating that iGECI can be stably expressed over months.
Next, to test the functionality of iGECI in detecting visually-evoked neuronal activity, we recorded iGECI signals in layer 2/3 neurons of the primary visual cortex, in response to drifting grating visual stimuli (Fig. 6) and compared them with those of the co-expressed GCaMP6s (Fig. 6a). In cells co-expressing iGECI and GCaMP6s (Fig. 6b, c, cells i-iii), we observed calcium transients in both iGECI and GCaMP6s signals (Fig. 6d, left panel), with the elevated calcium level increasing GCaMP6s fluorescence while decreasing iGECI’s miRFP670 donor fluorescence. iGECI signal change reached 5% of miRFP670 -ΔF/F, which was 3–6-fold smaller than ΔF/F observed with GCaMP6s. We also observed visually evoked calcium transients (Fig. 6d, right panel) in cells expressing iGECI alone (Fig. 6b, cells 1–6). Because of the lower signal-to-noise ratio and short recording periods, in vivo measurements did not permit reliable estimation of iGECI kinetics parameters.
Figure 6. iGECI reports visually evoked and spontaneous neuronal activity in vivo.
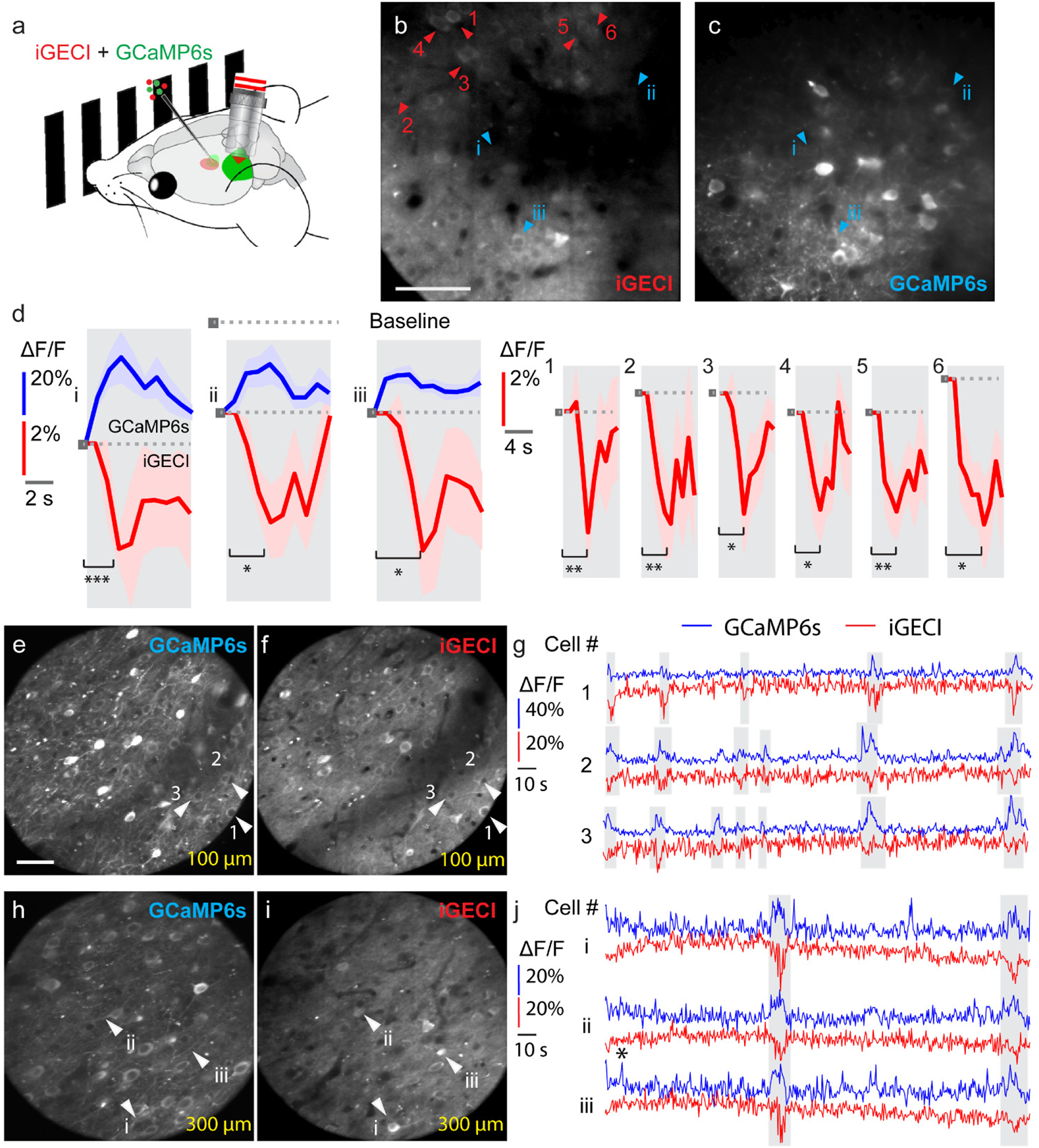
(a) Schematics of the in vivo calcium imaging experiments with visual stimulation on head-fixed awake mice. (b, c) Two-photon fluorescence images (256 × 256 pixels at 1 μm pixel size) of the same neurons in the mouse primary visual cortex transfected with iGECI and GCaMP6s. Images were acquired simultaneously at 200 μm below dura. Scale bar, 50 μm. (d) Example ΔF/F calcium transients (10-trial average) for neurons co-expressing GCaMP6s and iGECI (left panel, i-iii) and neurons only expressing iGECI (right panel, 1–6). Shaded area: ±SEM. * p value<0.05; ** p value<0.01; *** p value<0.001; paired two-sided student’s t-test. Exact p values (cells i-iii): 0.000451, 0.0135, and 0.0146; (cells 1–6): 0.00280, 0.00368, 0.0133, 0.0188, 0.00721, and 0.0108. Data are representative of five independent experiments with similar results. (e-f, h-i) Two-photon fluorescence images (225 × 225 pixels at 1.5 μm pixel size) of neurons in the mouse primary visual cortex at (e, f) 100 μm and (h, i) 300 μm below dura. Scale bar: 50 μm. (g, j) Example ΔF/F calcium transients for neurons co-expressing GCaMP6s and iGECI at 100 μm (1–3 in (e, f), white arrowheads) and 300 μm (i-iii in h, i, white arrowheads). Data are from one experiment. In all experiments, no exogenous BV was supplied.
We next applied iGECI to probe spontaneous activity of neurons located 100 μm and 300 μm below the dura in the primary visual cortex. In neurons co-expressing GCaMP6s and iGECI, we observed highly correlated calcium transients from the two indicators (shaded area, Fig. 6g, j). The averages of the iGECI signal peaks of all recorded calcium events indicated their similar amplitudes (11.0% ±3.3% at 100 μm depth and 12.7% ±5.7% at 300 μm depth) (Supplementary Fig. 13). Compared to visually evoked activity measured by iGECI, here the iGECI signal had substantially larger peak responses of up to 25% of -ΔF/F, suggesting that these spontaneous events correspond to burst firing of neurons.
To further quantitatively compare iGECI and GCaMP6s, we calculated the relative response rate of iGECI for 60 neurons (Fig. 6e–i) using the GCaMP6s-reported events as a reference. The distribution of the relative response rate (Supplementary Fig. 14) showed that in ~60% of the neurons iGECI detected at least 20% of GCaMP6s-reported events, and in ~30% of the neurons iGECI detected at least 40% of GCaMP6s-reported events. Although iGECI had a lower detection sensitivity than GCaMP6s, the most sensitive and robust calcium indicator to date, it performed as a capable reporter of neuronal activity in vivo. No BV was introduced in any in vivo experiments.
Discussion
Using rational design followed by random mutagenesis and screening in both bacteria and mammalian cells, we have developed the first fully NIR FRET-based indicator for calcium ions, iGECI (Supplementary Fig. 1). The iGECI uses monomeric NIR FPs engineered from bacterial phytochromes, miRFP670 and miRFP720, characterized by improved cellular brightness (Fig. 1a and Supplementary Fig. 2). Unlike conventional fluorescent biosensors, NIR indicators allow for deeper penetration in tissue, making them emerging powerful tools for functional imaging.
When compared to NIR-GECO1, the only other NIR GECI consisting of a single NIR FP and operating in intensiometric mode, iGECI is substantially brighter in cells, exhibits broader pH stability in the Ca2+-free state, better photostability in non-neuronal cells and neurons, and higher signal-to-noise ratio (Fig. 1, 2 and Supplementary Table 1). Importantly, iGECI is substantially more efficient in incorporating endogenous BV chromophore.
While GECI possesses two emission bands at 670 nm and 720 nm (Fig. 1c), the latter channel responds to calcium changes weakly (Supplementary Fig. 3). We anticipate that various applications could benefit from simultaneous “dynamic” (670 nm channel) and “static” (720 nm channel) signals measured from the same iGECI probe excited with a single source wavelength. For example, imaging and fiber photometry applications could utilize the longer wavelength emission at 720 nm as a built-in bright control to correct for motion-related intensity changes or laser power fluctuations.
We extensively characterized iGECI and demonstrated its efficient performance in non-neuronal mammalian cells (Fig. 1), in dissociated cultured neurons (Fig. 2), in acute mouse brain slices (Fig. 3, 4), in the mouse brain at a mesoscale level through an intact skull (Fig. 5), and in the visual cortex through a cranial window with subcellular resolution (Fig. 6). In vivo the iGECI-transduced neurons maintained normal morphology and high level of expression over months after AAV injection. Furthermore, for iGECI validation we applied a wide range of imaging techniques, from conventional epifluorescence microscopy (Fig. 2) and two-photon in vivo microscopy (Fig. 6) to emerging imaging approaches, such as an one-photon SOPi microscopy (Fig. 3, 4) and hybrid photoacoustic-fluorescence microscopy (Fig. 5). Notably, combination of one-photon SOPi technique with iGECI demonstrated its ability to image deeper in optically scattering tissue due to a wavelength-dependent decrease in optical scattering (Fig. 4c). Moreover, the NIR wavelength shift enabled crosstalk-free spectral multiplexing of iGECI with GFP-like biosensors (Fig. 5) and with channelrhodopsin optogenetic actuators (Fig. 2g, 4e,f).
Importantly, the advantages of iGECI enabled efficient detection of calcium transients deep in acute brain slices and in the mouse brain in vivo without supplying exogenous BV (Fig. 5 and 6), which was not possible for NIR-GECO1. Moreover, in one-photon and two-photon imaging systems that provide different spatial resolution, mesoscale (Fig. 5) and subcellular (Fig. 6), iGECI exhibited functional response of 23–25% in the mouse brain under chemogenetically induced (Fig. 5) and spontaneous (Fig. 6) brain activity states.
We performed multiparameter functional imaging using the hybrid photoacoustic-fluorescence microscopy (Fig. 5). The activity of neurons is closely associated with local hemodynamics via neurovascular coupling, where elevated neuronal firing leads to a corresponding increase in blood perfusion and oxygenation34. In particular, blood oxygenation reflects the enhanced oxygen consumption in the activated brain regions, which is the foundation of functional magnetic resonance imaging35. To study neural activity and blood oxygenation simultaneously, traditional optical imaging approaches have to rely on intrinsic optical signals or on oxygen-sensitive fluorescent dyes36, which suffer from low sensitivity or spectral overlap with visible-range GECIs. iGECI enabled us to detect neuronal and hemodynamic activities simultaneously (Fig. 5). We anticipate that with enhanced sensitivity, deep-penetrating photoacoustic imaging may directly detect iGECI in the photoacoustic mode, which should further increase iGECI imaging depth37, 38.
Using two-photon microscopy to image iGECI we efficiently detected changes in visually-evoked neuronal activity in the primary visual cortex of head-fixed awake mice in response to drifting grating visual stimuli, indicating that iGECI reliably reported calcium transients in vivo (Fig. 6).
In conclusion, across all biological samples and imaging techniques we tested, iGECI exhibited sufficient brightness and good functional performance. These results suggest that iGECI should be an extremely useful, easily and broadly applicable tool for the broad life science community.
Online Methods
Design of bacterial and mammalian plasmids.
Truncated version of miRFP670 (deletion of 13 N-terminal amino acids, miRFP670Δ13N) and miRFP720 (deletion of 17 N-terminal amino acids, miRFP720Δ17N) were PCR amplified from pmiRFP670-C113 and pmiRFP720-C117 plasmids. A calmodulin-M13 (CaM-M13) sensing module was PCR amplified from pYC3.6-C1 plasmid (Addgene #67899). A NIR-GECO1 gene was PCR amplified from pDuEx2-NIR-GECO1 (Addgene #113680)16. A plasmid pUCmini-iCAP-PHP.eB encoding modified AAV2/9 capsid was kindly provided by V. Gradinaru (California Institute of Technology, Addgene #103005). A pHelper plasmid was from AAV-Helper Free System kit (Agilent #240071).
For bacterial expression of GECI variants, pBAD/HisD vector (Life Technologies/Invitrogen) was used. Mammalian expression plasmids were based on a pEGFP-N1 vector (Clontech) with a standard CMV promoter. For expression in dissociated neurons and live mice iGECIs or NIR-GECO1 genes were cloned into pAAV-CW3SL-EGFP (Addgene #61463) instead of EGFP.
Molecular evolution of iGECI.
DNA fragments encoding miRFP670Δ13N, CaM-M13 and miRFP720Δ17N were PCR amplified and ligated into pEGFP-N1 vector. After rational designing of L1 and L2 linkers, the promising variant was cloned into pBAD/His-D vector and subjected to random mutagenesis and screening in E.coli. BL21 AI host (ThermoFisher Scientific) containing a pWA23h plasmid encoding heme oxygenase (HO) for BV synthesis in E.coli39, was electroporated with library DNA, grown overnight in LB medium containing 0.02% rhamnose and 0.05% arabinose for induction of HO and iGECI synthesis, respectively. Library of clones was sorted with FACS, using double positive gating for eliminating non-fluorescent clones resulting from stop-codons and frame shifts. Pre-sorted library was plated on Petri dishes containing 0.02% rhamnose and 0.05% arabinose. Dishes were incubated overnight at 37°C, then for 12 h at 30°C, and for 24 h at 18°C. Colonies were transferred to nitrocellulose membranes and permeabilized by spraying with Ca2+-free solution (30 mM MOPS, pH 7.5, 100 mM KCl, 50 μg/ml poly-L-lysine, 50 μg/ml ionomycin). Membranes were incubated for 5 min, and basal fluorescence in the donor (ex. 605 nm, em. 680 nm) and FRET (ex. 605 nm, em. 720 nm) channels was acquired using an IVIS instrument (Perkin Elmer/Caliper Life Sciences). Then membranes were treated by spraying with high Ca2+ solution (30 mM MOPS, pH 7.5, 100 mM KCl, 50 μg/ml poly-L-lysine, 50 μg/ml ionomycin, 100 mM CaCl2), incubated for 5 min, and Ca2+-loaded state of fluorescence was recorded using the same filter sets. Data were analyzed using Living Image v.3.0 software (Perkin Elmer/Caliper Life Sciences).
Clones with the best Ca2+-loaded/basal fluorescence ratio were subjected to the next step of screening. They were transferred to 5 ml LB liquid culture in 24 deep-well plates containing 0.02% rhamnose and incubated for 8 h at 37°C on a rotating shaker. Then arabinose was added to 0.05%, temperature decreased to 30°C, and the cultures were incubated overnight. Next morning the temperature was changed to 18°C and the cultures were incubated for 24 h. Then the bacterial cultures were lysed with B-PER (ThermoFisher Scientific) and pelleted. Supernatants were transferred to 96-well plates and divided, one part was loaded with 1 mM CaCl2, another one with 2 mM EGTA. Fluorescence in the donor (ex. 605 nm, em. 680 nm) and FRET (ex. 605 nm, em. 720 nm) channels was acquired using the IVIS instrument.
The best performing clones were re-cloned into a mammalian expression vector and evaluated in HeLa cell lysates. HeLa cells were transiently transfected using Effectene (Qiagen), 48 h after transfection cells were harvested and lysed with M-PER (ThermoFisher Scientific). Lysates were clarified by spinning and divided in two samples, one part was loaded with 1 mM CaCl2, another one with 2 mM EGTA. Fluorescence spectra were recorded with the FluoroMax-3 spectrofluorometer. The best performing clones were subjected to a new round of L1 and L2 evolution in E.coli and HeLa cells. About 4000 clones were analyzed in each round of screening.
Lastly, we performed mutagenesis of a L3 linker between CaM and M13 peptide by introducing a modification similar to YC-Nano14018. Then we introduced the mutation in CaM sequence, reverting glutamic acid to glutamine and making all four Ca2+-binding EF-hand active, similar to YC2.68. Then, we added two glycine residues and a serine, similar to YC-Nano15, which showed the highest ΔF/F in HeLa cells.
Protein purification and in vitro characterization.
iGECIs with polyhistidine tags on the N-terminus were expressed in BL21 AI host (Life Technologies/Invitrogen) containing a pWA23h plasmid. Bacteria were grown in LB medium supplemented with ampicillin, kanamycin, and 0.02% rhamnose for 6–8 h, followed by an induction of the protein expression by adding 0.05% arabinose. The proteins were purified using a Ni-NTA agarose (Qiagen).
For absorbance measurements, a Hitachi U-2000 spectrophotometer was used. Fluorescence spectra in the range of 660–780 nm were recorded with the FluoroMax-3 spectrofluorometer. Ca2+ titrations were carried out using EGTA-buffered Ca2+ solutions (Calcium Calibration Buffer Kit, Life Technologies). We prepared buffers by mixing a Ca-EGTA buffer and an EGTA buffer to give free Ca2+ concentrations ranging from zero to 39 μM at 25°C. Fluorescence intensities were plotted against Ca2+ concentrations and fitted by a double sigmoidal binding function to determine Kd.
To measure Ca2+ transitions evoked by histamine, HeLa cells were transiently transfected with iGECI1.0 using Effectene (Qiagen) and cultured 48 h. Then the medium was changed to Live cell imaging solution (Life Technologies/Invitrogen), supplemented with 1 mM CaCl2, 100 mM KCl and 1 mM D-glucose, and basal fluorescence in the donor (ex. 605 nm, em. 680/20 nm) and FRET channels (ex. 605 nm, em. 725/40 nm) was recorded. Time-lapse imaging was performed using an Olympus IX81 inverted epifluorescence microscope, equipped with a 200 W Xenon lamp (Sutter Instruments) and a 60× 1.35 NA oil immersion objective lens (UPlanSApo, Olympus). The microscope was operated with a SlideBook v.6.0.8 software (Intelligent Imaging Innovations). Histamine solution was added to the cells to a final concentration of 100 μM and fluorescence was recorded for 10 min. After that, the imaging solution with calcium and histamine was replaced by a Ca2+-free imaging solution, and cells were incubated for 5 min equilibrate calcium. Then, an imaging solution containing 2 mM EDTA was added to the cells and fluorescence was recorded for another 5 min. Solution changes were done using MPII peristaltic pump perfusion system (Warner Instruments).
Photobleaching measurements of iGECI and NIR-GECO1 in live HeLa cells and in dissociated mouse neurons were performed with the 100× 1.4 NA oil immersion objective lens (UPlanSApo, Olympus), 605/30 nm excitation, and 647 nm longpass emission filters at 14 mW/cm2 light power density measured at the back aperture of the objective lens (~8.3 W/cm2 at the specimen plane) and normalized to the efficiency of absorption at 605 nm by each indicator.
Brightness comparison of iGECI and NIR-GECO1 was performed in HeLa cells transiently co-transfected with the corresponding calcium indicator and EGFP at 10:1 plasmid ratio. 48 h after transfection, cells were analyzed with BD LSRII flow cytometer using 488 nm and 640 nm excitation lasers and 520/40 nm emission filter for EGFP, and 647 nm longpass edge emission filter. The cells were first gated using the EGFP signal and then NIR fluorescence intensity of the calcium indicators was quantified. The NIR fluorescence intensity was normalized to the efficiency of absorption at 640 nm for each indicator. To study the dependence of brightness on the BV chromophore, saturated 25 μM concentration of exogenous BV was added to cells for 24 h prior to flow cytometry. Flow cytometry gating was performed using intact cells, single cells and live cells. The live cells were further gated in the NIR channel (Supplementary Fig. 15).
pH stability was studied using a series of Hydrion buffers (Micro Essential Laboratory) in the presence of either 2 mM EGTA or 1 mM of Ca2+. Fluorescence was excited at 620 nm, and emission was recorded at 640–760 nm. The area under the spectra at different pH values was quantified.
Preparation of high-titer AAVs.
AAV particles were obtained as described40. Briefly, plasmid DNA for AAV production was purified with NucleoBond Xtra Maxi EF kit (Macherey-Nagel) and AAV-293T cells (Agilent) were co-transfected with AAV2/9 genome plasmid, pAAV2-CaMKII-iGECI, pAAV2-CaMKII-NIRGECO1 or pAAV2-hSyn1-CheRiff, AVV capsid plasmid pUCmini-iCAP-PHP.eB and pHelper using polyethyleneimine (PEI, Santa Cruz). Cell media was collected 72 h after transfection. 120 h after transfection cells and media were collected and combined with the media collected at 72 h. Cells were harvested by centrifugation and then lysed with a salt-active nuclease (HL-SAN, Arcticzyme). 8% PEG was added to the media, incubated for 2 h on ice and then pelleted. PEG pellet was treated with SAN and combined with lysed cells. Cell suspension was clarified by centrifugation. Supernatant was applied on iodixanol gradient and subjected to ultracentrifugation 2 h 25 min at 350,000 g. Virus fraction was collected, washed and enriched on Amicon 15 100,000 MWCO centrifuge device. Purified virus stored at 4°C. Virus titer was defined by qPCR. Aliquot of virus was consequently treated with DNAse I and proteinase K and then used as a template for qPCR. A NheI digested pAAV2-CaMKII-iGECI plasmid with known concentration was used as a reference. AAV9-CaMKIIa-hChR2(H134R)-EYFP and AAV9-CaMKII-GCaMP6s.WPRE.SV40 were obtained from Addgene.
Imaging in dissociated neuron cultures.
Neurons were isolated from the hippocampi of postnatal (P0-P1) Swiss-Webster mice using a published protocol41 and cultured in Neurobasal Plus Medium with B-27 Plus Supplement (Gibco), additional 1 mM GlutaMAX (Gibco), 100 U/ml penicillin and 100 μg/ml streptomycin, on poly-D-lysine (EMD Millipore) coated glass coverslips (thickness 0.13–0.17 mm, diameter 12 mm, ThermoFisher Scientific) at the density of ~70,000 cells per coverslip. Half of the medium was exchanged twice a week. Transfection was performed on DIV10 using Calcium Phosphate Transfection Kit (Invitrogen) and a previously published protocol42. For experiments with AAVs, neurons were transduced on DIV7 with 109 viral genomes per well (in 24-well plate) and recorded on DIV16–18 at 37°C. Grass S48 stimulator (Grass Instruments) and custom platinum electrodes (0.5 mm diameter) were used for field stimulation (1–160 square pulses per stimulus, 1 ms pulse width, 85 Hz, 50 V). Synaptic transmission inhibitors were applied43: 10 μM CNQX (R&D Systems), 10 μM gabazine (SantaCruz Biotechnology), 10 μM R-CPP (Enzo Life Sciences), and 1 μM S-MCPG (Cayman Chemicals). The 617 nm LED (Mightex Systems) was used for fluorescence excitation. The excitation filter was 620/15 nm, with a 640LP dichroic mirror, and the emission filter was 667/30 nm for iGECI and 720/40 nm for NIR-GECO1. The frame rate was 10 Hz for NIR-GECO1 and 5 Hz for iGECI. Fluorescence was recorded using an Orca Flash 4.0LT camera (Hamamatsu), Olympus IX81 microscope, and LUCPlanFLN 20× 0.45 NA air objective lens (Olympus). Light power density at the specimen plane was 1.4 W/cm2 (6-fold lower than in the photobleaching experiments), and total duration of imaging was less than 0.5 h. Bath solution contained (in mM): 125 NaCl, 2.5 KCl, 1 MgCl2, 10 HEPES, 3 CaCl2, 30 glucose, pH 7.3, 305–307 mOsm44.
For experiment with CheRiff, neurons were co-transduced with iGECI AAV and CheRiff AAV and incubated with 25 μM BV and 2 μM all-trans retinal for 3 h before recording. Synaptic activity was blocked by inhibitors, as described before43: 10 μM CNQX (R&D Systems), 10 μM gabazine (Santa Cruz Biotechnology), 10 μM R-CPP (Enzo Life Sciences), and 1 μM S-MCPG (Cayman Chemicals). Trains of 2 or 10 pulses of green light (505 nm Mightex Systems LED, 510/20 nm optical filter, 4 ms pulse width, 50 ms interval between pulses, using 3 mW/cm2 measured at the back aperture of a 40× 0.75 NA UPLFLN dry objective lens) were used for CheRiff activation. The stimulator, camera and LEDs were controlled by Master-8 (AMPI) and MatLab R2018b (MathWorks).
Neonatal injections.
P2–5 neonates were cryo-anesthetized, mounted on a stereotaxic frame (David Kopf Instruments), and maintained under anesthesia for the duration of the procedure, as described previously45. AAV2/9-CaMKII-iGECI (2.5×1012 vg/ml) was injected into the motor cortex through a pulled glass pipette (100 nl/min, 500 nl) with an UltraMicroPump controller (World Precision Instruments). For experiments using optogenetic stimulation and dual sensor comparison, AAV9-CaMKIIa-hChR2(H134R)-EYFP.WPRE.hGH (3.9×1012 vg/ml) or AAV9-CaMKII-GCaMP6s.WPRE.SV40 (1×1012 vg/ml), respectively, were co-injected along with AAV2-CaMKII-iGECI. The pipette was held in place for 5 min after injection ended. Experiments were carried out 2–8 weeks after the injection.
Acute brain slice preparation.
P16-P55 mice were deeply anesthetized with isoflurane, followed by a transcardial perfusion using ice cold ACSF (artificial cerebrospinal fluid) containing (mM) 127 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 20 glucose, 2 CaCl2, and 1 MgCl2. Brain was removed, blocked, mounted, and placed into a chamber containing 34°C ACSF oxygenated with 95% O2, 5% CO2. Coronal 250 μm cortical brain slices were made using a Leica VT1200s vibratome, as previously described46, 47. Slices were transferred to a holding chamber containing 34°C ACSF and recovered for 30 min before being cooled to room temperature (22–24°C). For a subset of experiments, slices were incubated in 25 μM BV (0.1% DMSO, Sigma-Aldrich) for 1–2 h at room temperature. Spectral multiplexing experiments with ChR2 or GCaMP6s were carried out in the presence of 10 μM Gabazine, 10 μM NBQX, 10 μM CPP (Tocris) and 25 μM BV.
Scanned oblique plane illumination (SOPi) microscopy.
A single objective based light-sheet microscope was modified for iGECI imaging. In the illumination path, a HeNe laser (632.8 nm, HNL100L, Thorlabs) was scanned rapidly with a galvanometer based planar scan mirror (GVSM001, Thorlabs) and an achromatic doublet lens as scan lens (AC254–100A-ML, Thorlabs) to create the light-sheet. A dichroic beam-splitter (Di03-R405/488/532/635-t1, Semrock) and another achromatic doublet lens (AC254–100A-ML, Thorlabs) were used to conjugate the plane containing first galvanometer’s rotation axis onto second galvanometer (QS-12, Nutfield Technology). A telescope formed by two achromatic doublet lenses (AC508–100A-ML, AC508–200A-ML, Thorlabs) conjugated the galvanometer rotation axes to the back focal plane of the main microscope objective (20x, 1 W, XLUMPLFLN20XW, Olympus). A lateral offset in the incident laser beam introduced the desired 45° tilt in the illumination light-sheet in the sample volume. In the detection path, the fluorescence signal reverse traced the same path as the illumination beam until the dichroic beam-splitter followed by a tube lens (AC254–150A-ML) and a microscope objective (20x, 0.75 NA, UPLSAPO20X, Olympus), to form an intermediate image of the illuminated plane. This intermediate image was then magnified and imaged on a scientific CMOS camera (Prime 95B, Photometrics) with the help of a microscope objective (20× 0.45 NA, LUCPLFLN20X, Olympus), a filter (FF01 676/37–25, Semrock) and a tube lens (AC254–150A-ML). Functionally, the first galvanometer scanner provided rapid scanning for light-sheet creation, and the second galvanometer scanner enabled tilt-invariant lateral scan of the oblique light-sheet in the sample volume. For dual biosensor imaging, a dichroic beam splitter (FF495-Di03, Semrock) was used to integrate a blue laser (473 nm, Dragonlasers) in the illumination path for GCaMP6s excitation. For optogenetic stimulation, a blue LED (CREE, 470 nm) was used with a collimating lens and a 500 nm long pass dichroic beam splitter (#69–899, Edmund Optics). The beam splitter was placed between the tube lens and the scan lens of the main microscope objective.
Functional light-sheet imaging.
Acute brain slices were placed in a chamber containing room temperature ACSF, recirculated at a rate of 1–2 ml/min. A monopolar glass pipette electrode was positioned using a Siskiyou manual manipulator. The electrode was placed within the motor cortex, 200–400 μm away from the imaging region. Electrical stimulation was done with a DS3 isolated current stimulator (Digitimer). Stimulation parameters: 1 mA, 20 Hz, 1 or 10 ms pulse width. Two digital output pins on an Arduino board (UNO Rev3, Arduino) were used to produce TTL compatible trigger signals for electrical stimulation and image acquisition through the camera. Total number of camera trigger signals, start and end of electrical stimulation trigger signal, and the frequency of both trigger signals were controlled through custom C++ code. μManager was used for image acquisition on the camera48. For experiments involving optogenetic stimulation, a blue LED (470 nm, CREE) with a current driver circuit was used. This LED was controlled through the same Arduino board, where its pulse width was varied from 100 μs to1 ms. Power was measured at 3.75 mW/cm2 and maintained constant for all experiments.
A custom Matlab GUI was used to control both galvanometer scanners. The sample holder was mounted on a XYZ translation stage (PT3, Thorlabs). A manual manipulator was attached to the main objective arm of the SOPi system. A flip mirror was placed behind the first tube lens of the SOPi setup to visualize electrode placement in bright-field mode prior to functional imaging at 20 fps. Image intensity analysis was carried out using Fiji ROI Manager4. Data were analyzed using a custom Matlab script. Traces were converted into -ΔF/F; a 1 s long period immediately prior to electrical or optogenetic stimulation was used to calculate baseline fluorescence.
Histology.
Mice were deeply anesthetized with isoflurane, followed by a transcardial perfusion using PBS containing 2% paraformaldehyde and 2% glutaraldehyde. Brains were extracted, postfixed for 24 h at 4°C, and sectioned at a thickness of 60 μm on a vibratome (Leica VT1200s). Slices were mounted, dried and coverslipped in glycerol:TBS with Hoechst. Large scale images were acquired on an epifluorescence motorized-stage microscope (Olympus VS120). Confocal images were acquired with a Leica SP5 confocal microscope (Leica Microsystems).
Statistical analyses for in vivo experiments.
Group statistical analyses were done using GraphPad Prism (GraphPad). For group sizes, both the number of experiments and the number of animals is provided. All data are expressed as mean SEM or individual plots. For two-group comparisons, statistical significance was determined by two-tailed Student’s t-tests. For multiple group comparisons, two-way analysis of variance (ANOVA) tests were used, followed by Bonferroni post hoc comparisons. Pearson regression was used for correlation analyses. p<0.05 was considered statistically significant (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001).
Hybrid photoacoustic and fluorescence microscopy.
In photoacoustic microscopy, as photons travel in tissue, some are absorbed by biomolecules, and their energy is partially or completely converted into heat. The heat-induced pressure wave propagates in tissue, and is detected outside the tissue by an ultrasonic transducer or transducer array to form an image that maps the original optical energy deposition in the tissue37. To induce photoacoustic signals, a dye laser at 610 nm (Credo, Spectral Physics) pumped by a 3 ns pulsed laser beam at 532 nm (INNOSLAB, Edgewave; pulse repetition rate: up to 30 kHz) is combined with another 3 ns pulsed laser beam at 532 nm (VGEN-G, Spectral Physics; pulse repetition rate: up to 700 kHz) via a dichroic mirror. Laser energy fluctuations are monitored by a fast photodiode that samples a small portion of the laser beams. A continuous-wave laser at 635 nm (Civil Laser) is combined with the pulsed laser beams via a polarizing beam combiner. The two pulsed laser beams are used for photoacoustic microscopy, and the continuous laser beam is used for fluorescence microscopy. The three laser beams are focused by a plano-convex lens (LA1131, Thorlabs), then spatially filtered by a 50-μm-diameter pinhole (P50C, Thorlabs). The filtered laser beams are focused to ~3 μm spots by an objective lens (AC127–050-A, Thorlabs; NA: 0.1 in air). An optical-acoustic beam combiner, composed of a right-angled prism (32332, Edmund) and a rhomboid prism (49419, Edmund), provides optical-acoustic coaxial alignment. Here, a thin layer of silicone oil sandwiched between the two prisms reflects ultrasound but transmits light. An optical correction lens attached to the top surface of the combiner corrects the optical aberration due to the prism. The photoacoustic waves are focused by an acoustic lens and detected by an ultrasonic transducer (V214-BB-RM, Olympus-NDT; central frequency: 50 MHz; one-way −6 dB bandwidth: 100%). The fluorescent signals (>670 nm) are reflected by a dichroic mirror (DMSP650, Thorlabs), filtered by an emission filter (DMLP650, Thorlabs), and then detected by a Si-amplified photodiode (PDA100A2, Thorlabs). De-ionized water in a water tank is required to provide acoustic coupling from the sample surface to the acoustic lens. The bottom of the water tank is sealed with a piece of membrane that is both optically and acoustically transparent. Single-depth imaging is provided by 2D linear motor-stage scanning of the sample along the x- and y-axis at a speed of up to 20 mm/s. When necessary, additional depth-scanning of the optical focal zone along the z-axis extends the focal range of the photoacoustic/fluorescence system at the expense of imaging speed, and can provide 3D images of the mouse brain. The two pulsed lasers at 532 nm and 610 nm are triggered with a time interval of 500 ns. The 500 ns delay allows the first photoacoustic signal excited by the 532 nm pulse to travel 0.75 mm, which is approximately the maximum penetration depth of photoacoustic microscopy in the brain. An FPGA-based Labview program synchronizes the laser firing, motor scanning, and data acquisition.
Because blood has relatively weak but still non-negligible absorption at 670 nm, the fluorescence signals underneath the major blood vessels might be underestimated. Therefore, to analyze the fluorescence signals, we only chose the regions (pixels) outside the major blood vessels, based on the automatically co-registered photoacoustic images of the blood vessels (e.g., Fig. 5g and 5j). The high spatial resolution of the system allows us to identify each individual vessel to avoid signal confounding, mitigating the potential impact of the blood oxygenation change on the fluorescence measurement.
Electrical paw stimulation.
Electrical stimulations were introduced by one pair of needle electrodes inserted under the skin of the left hindlimb. The electrodes were connected to an isolated pulse stimulator (A-M 2100) synchronized with the integrated photoacoustic/fluorescence system. The procedure consisted of ten trials and lasted for 5 min. Each trial lasted for 10 s. The first 5 s were resting state, while the 6th s was the left hindlimb stimulation, followed by 4 s of recovery. Each stimulation period consisted of a train of electrical pulses with an amplitude of 2 mA, a pulse width of 0.25 ms and a repetition rate of 50 Hz. The stimulation period and intensity were controlled to avoid inducing any paw motions. At least ten trials were performed on each mouse (n = 4).
Emx1-hM3Dq mouse model and the injection of iGECI-expressing AAV. Mouse lines CAG-LSL-Gq-DREADD (#026220) and Emx1Cre/Cre (#005628) were purchased from the Jackson Laboratory. We then generated CAG-LSL-Gq-DREADD:Emx1-Cre (Emx1-hM3Dq) mice to restrict excitatory hM3Dq DREADD expression predominantly to forebrain excitatory neurons. Genotyping was performed by PCR analysis using mouse-tail genomic DNA samples.
Emx1-hM3Dq mice were anesthetized with an intraperitoneal injection of a ketamine-xylazine solution, and then placed on a stereotaxic frame. A skin incision was cut on the middle line and one burr hole (~1 mm in diameter) made on the skull. A pulled glass pipette containing iGECI-expressing AAV was inserted into the targeted brain region (AP = 0.0 mm, ML= −2.0 mm, DV = 2.0 mm). Virus (1 μl) was slowly delivered into the target site using an UltraMicroPump3 with SMARTouch Controller (World Precision Instruments). After completion of the injection, the glass pipette was left in place for 5 min before it was slowly withdrawn. The burr hole was sealed with bone wax, and the incision closed with suture. After 3 weeks post viral injection, mice were used for imaging experiments.
For monitoring the fast dynamic response to the paw stimulation (Fig. 5i), we did not perform depth scanning (i.e., z-scanning), instead we focused only at the depth of ~150 μm beneath the cortical surface where the fluorescence signal was the strongest. Then, we were able to monitor one line of the cortex with a scanning rate of 10 Hz over a 1 mm scanning range. Similarly, for monitoring the relatively slow dynamic response to CNO challenge, we monitored a 1×1 mm region of the cortex with an imaging time of 5 min per frame.
Virus injection and cranial window implantation.
For in vivo preparations, C57BL/6J mice were at least 9 week old at the time of stereotactic virus injection and cranial window implantation. Virus injection pipettes were made by pulling (Dual-stage Glass Micropipette Puller, Narishige, PC-10) regular glass pipettes (Wiretrol II) followed by beveling (Micro grinder, Narishige, EG-401) at ~35° with a 17–23 μm opening. Injection pipettes were back-filled with mineral oil (Fisher Scientific). A hydraulic manipulator (Narishige, MO-10) together with a fitted plunger were used for virus loading and injection. Mice were given the analgesic buprenorphine (subcutaneous, 0.3 mg/kg) and were deeply anesthetized under isoflurane (2.0% by volume in O2) during the process of virus injection. Following a 3.5 mm diameter craniotomy over the left cortex and keeping the brain wet with sterile phosphate buffered saline (PBS, Invitrogen), either iGECI (AAV2/2-CaMKII-iGECI, 3.35×1013 vg/mL) alone or a mixture of iGECI (AAV2/2-CaMKII-iGECI, 3.35 × 1013 vg/mL) and GCaMP6s (AAV2/1-hSyn1-GCaMP6s, 6.64×1013 vg/mL) at a 10:1 (v/v) ratio was slowly (25 nL/min) injected at 350 μm and 550 μm below the dura in 3–5 sites within the mouse primary visual cortex. A total of 200–400 nL was injected at each depth of each site. A cranial window made by gluing (Norland 68 Optical Adhesive) together a glass ring (inner diameter: 3 mm; outer diameter: 4.5 mm) and a glass disk (diameter: 3.5 mm), both laser cut from standard microscope cover glass (Fisherbrand, No. 1.5, 0.16–0.19 mm thick) was embedded into the craniotomy and bonded with the skull by Vetbond (3M). As a final step, a titanium head-bar was fixed on the skull with Vetbond and fast curing orthodontic acrylic resin (Lang Dental).
Visual stimulation for the mouse.
A custom-modified DLP projector and a Teflon-made film screen were used to present visual stimuli. The screen was oriented at ~ 50° to the long axis of the mouse and was placed 14 cm from the mouse’s right eye, covering ~70° × 70° of its visual space. An LED light (450–495 nm, SugarCUBE) was used as the light source of the projector. Oriented gratings had 100% contrast, a spatial frequency of 0.15 cycle per degree, and drifted at 2 cycles/s. Four oriented gratings at 10 repetitions each were presented in a pseudo-random sequence. In each trial, grey screen was first shown to head-fixed awake mice for 6 s for baseline fluorescence measurement. In this period, the brain was imaged in the first 3 s, followed by another 3 s without exposure to excitation light. Drifting gratings of four motion directions were presented in a pseudo-random sequence for 8 s, accompanied by fluorescence imaging. After that, we waited another 17 s before starting the next round of imaging. Visual stimuli and image acquisition were synchronized using custom written code.
Two-photon imaging in vivo.
In vivo structural and functional imaging was performed with a custom two-photon fluorescence microscope (Supplementary Figure 10). Mice were habituated for experimental handling for at least one week before imaging. Mice were head-fixed and anesthetized (by 1% isoflurane in O2) during structural and spontaneous activity imaging, and they were awake during visually-evoked activity imaging. 900 nm excitation wavelength was used for both iGECI and GCaMP6s excitation. Spontaneous activity was recorded continuously for 500 frames at 2.2 frames per second. Visually-evoked activity was imaged at ~200 μm below dura at 1.3 frames per second. Power under objective used for in vivo imaging ranged 35–170 mW, with higher power for deeper imaging.
Animal use.
Animals were handled according to the protocols approved by the Northwestern University, Duke University, and University of California at Berkeley Animal Care and Use Committees. Approximately equal numbers of males and females were used for every experiment. Mice were group-housed, with standard feeding, light-dark cycle, and enrichment procedures; littermates were randomly assigned to conditions. Wild-type C57BL/6J mice (stock 000664) were ordered from Jackson Laboratory, and Swiss Webster mice were acquired from Charles River.
Data availability.
The main data supporting the findings of this study are available within the article and its supplemental information. The additional data are available from the corresponding author on reasonable request. The GenBank accession numbers are MT997078 and MT997079 for iGECI and iGECI-NES (nuclear exclusion sequence) constructs, respectively. Plasmids encoding these constructs will be available on Addgene. Acquisition and analysis code will be available on GitHub or on reasonable request.
Supplementary Material
Acknowledgments
We thank Olena Oliinyk (University of Helsinki, Finland) and Andrii Kaberniuk (Albert Einstein College of Medicine) for the useful suggestions, George Robertson (Keyence Corporation of America) for the technical support, and Biological Imaging Facility of Northwestern University for the access to confocal microscope. This work was supported by the grants GM122567, NS103573, NS115581 (all to V.V.V.), EY030705 (to D.M.S.), EB028143, NS111039, EB027304, CA243822 (all to J.Y.) and MH117111, NS107539 (both to Y.K.) from the National Institutes of Health; 18CSA34080277 from American Heart Association (to J.Y.); Beckman Young Investigator Award, Searle Scholar Award, and Rita Allen Foundation Award (all to Y.K). J. C-J. is a T32 NS041234 fellow.
Footnotes
Online Methods
Methods and any associated references are available in the online version of the paper.
Competing interests
The authors declare no competing interests.
References
- 1.Scanziani M & Hausser M Electrophysiology in the age of light. Nature 461, 930–939 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Leopold AV, Shcherbakova DM & Verkhusha VV Fluorescent Biosensors for Neurotransmission and Neuromodulation: Engineering and Applications. Front Cell Neurosci 13, 474 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ji N, Milkie DE & Betzig E Adaptive optics via pupil segmentation for high-resolution imaging in biological tissues. Nat Methods 7, 141–147 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Debarre D et al. Image-based adaptive optics for two-photon microscopy. Opt Lett 34, 2495–2497 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rueckel M, Mack-Bucher JA & Denk W Adaptive wavefront correction in two-photon microscopy using coherence-gated wavefront sensing. Proc Natl Acad Sci U S A 103, 17137–17142 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akerboom J et al. Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogenetics. Front Mol Neurosci 6, 2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen TW et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499, 295–300 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagai T, Yamada S, Tominaga T, Ichikawa M & Miyawaki A Expanded dynamic range of fluorescent indicators for Ca(2+) by circularly permuted yellow fluorescent proteins. Proc Natl Acad Sci U S A 101, 10554–10559 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palmer AE et al. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol 13, 521–530 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Thestrup T et al. Optimized ratiometric calcium sensors for functional in vivo imaging of neurons and T lymphocytes. Nat Methods 11, 175–182 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Dana H et al. Sensitive red protein calcium indicators for imaging neural activity. Elife 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao Y et al. An expanded palette of genetically encoded Ca(2)(+) indicators. Science 333, 1888–1891 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shcherbakova DM et al. Bright monomeric near-infrared fluorescent proteins as tags and biosensors for multiscale imaging. Nat Commun 7, 12405 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu D et al. A naturally monomeric infrared fluorescent protein for protein labeling in vivo. Nat Methods 12, 763–765 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matlashov ME et al. A set of monomeric near-infrared fluorescent proteins for multicolor imaging across scales. Nat Commun 11, 239 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qian Y et al. A genetically encoded near-infrared fluorescent calcium ion indicator. Nat Methods 16, 171–174 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shcherbakova DM, Cox Cammer N, Huisman TM, Verkhusha VV & Hodgson L Direct multiplex imaging and optogenetics of Rho GTPases enabled by near-infrared FRET. Nat Chem Biol 14, 591–600 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Horikawa K et al. Spontaneous network activity visualized by ultrasensitive Ca(2+) indicators, yellow Cameleon-Nano. Nat Methods 7, 729–732 (2010). [DOI] [PubMed] [Google Scholar]
- 19.Bootman MD & Berridge MJ Subcellular Ca2+ signals underlying waves and graded responses in HeLa cells. Current Biology 6, 855–865 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Grienberger C & Konnerth A Imaging Calcium in Neurons. Neuron 73, 862–885 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Kumar M, Kishore S, Nasenbeny J, McLean DL & Kozorovitskiy Y Integrated one- and two-photon scanned oblique plane illumination (SOPi) microscopy for rapid volumetric imaging. Opt Express 26, 13027–13041 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar M & Kozorovitskiy Y Tilt-invariant scanned oblique plane illumination microscopy for large-scale volumetric imaging. Opt Lett 44, 1706–1709 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar M & Kozorovitskiy Y Tilt (in)variant lateral scan in oblique plane microscopy: a geometrical optics approach. Biomed Opt Express 11, 3346–3359 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herman AM, Huang L, Murphey DK, Garcia I & Arenkiel BR Cell type-specific and time-dependent light exposure contribute to silencing in neurons expressing Channelrhodopsin-2. Elife 3, e01481 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brunker J, Yao J, Laufer J & Bohndiek SE Photoacoustic imaging using genetically encoded reporters: a review. J Biomed Opt 22 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Yao J et al. Multiscale photoacoustic tomography using reversibly switchable bacterial phytochrome as a near-infrared photochromic probe. Nat Methods 13, 67–73 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yao J et al. High-speed label-free functional photoacoustic microscopy of mouse brain in action. Nat Methods 12, 407–410 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu H et al. Cre-dependent DREADD (Designer Receptors Exclusively Activated by Designer Drugs) mice. Genesis 54, 439–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manvich DF et al. The DREADD agonist clozapine N-oxide (CNO) is reverse-metabolized to clozapine and produces clozapine-like interoceptive stimulus effects in rats and mice. Sci Rep 8, 3840 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Armbruster BN, Li X, Pausch MH, Herlitze S & Roth BL Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci U S A 104, 5163–5168 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bouchard MB, Chen BR, Burgess SA & Hillman EM Ultra-fast multispectral optical imaging of cortical oxygenation, blood flow, and intracellular calcium dynamics. Opt Express 17, 15670–15678 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Piatkevich KD et al. Near-Infrared Fluorescent Proteins Engineered from Bacterial Phytochromes in Neuroimaging. Biophys J 113, 2299–2309 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Girouard H & Iadecola C Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol (1985) 100, 328–335 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Fabiani M et al. Neurovascular coupling in normal aging: a combined optical, ERP and fMRI study. Neuroimage 85 Pt 1, 592–607 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liao LD et al. Neurovascular coupling: in vivo optical techniques for functional brain imaging. Biomed Eng Online 12, 38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang LV & Yao J A practical guide to photoacoustic tomography in the life sciences. Nat Methods 13, 627–638 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gottschalk S et al. Rapid volumetric optoacoustic imaging of neural dynamics across the mouse brain. Nat Biomed Eng 3, 392–401 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piatkevich KD, Subach FV & Verkhusha VV Far-red light photoactivatable near-infrared fluorescent proteins engineered from a bacterial phytochrome. Nat Commun 4, 2153 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Challis RC et al. Systemic AAV vectors for widespread and targeted gene delivery in rodents. Nat Protoc 14, 379–414 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Beaudoin GM 3rd et al. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc 7, 1741–1754 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Jiang M & Chen G High Ca2+-phosphate transfection efficiency in low-density neuronal cultures. Nat Protoc 1, 695–700 (2006). [DOI] [PubMed] [Google Scholar]
- 43.Wardill TJ et al. A neuron-based screening platform for optimizing genetically-encoded calcium indicators. PLoS One 8, e77728 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hochbaum DR et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat Meth 11, 825–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kozorovitskiy Y, Peixoto R, Wang W, Saunders A & Sabatini BL Neuromodulation of excitatory synaptogenesis in striatal development. Elife 4, e10111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiao L, Priest MF, Nasenbeny J, Lu T & Kozorovitskiy Y Biased Oxytocinergic Modulation of Midbrain Dopamine Systems. Neuron 95, 368–384 e365 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiao L, Priest MF & Kozorovitskiy Y Oxytocin functions as a spatiotemporal filter for excitatory synaptic inputs to VTA dopamine neurons. Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Edelstein AD et al. Advanced methods of microscope control using muManager software. J Biol Methods 1 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The main data supporting the findings of this study are available within the article and its supplemental information. The additional data are available from the corresponding author on reasonable request. The GenBank accession numbers are MT997078 and MT997079 for iGECI and iGECI-NES (nuclear exclusion sequence) constructs, respectively. Plasmids encoding these constructs will be available on Addgene. Acquisition and analysis code will be available on GitHub or on reasonable request.