

Abstract
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder with multifactorial etiology. The role of microglia in the pathogenesis of AD has been increasingly recognized in recent years; however, the detailed mechanisms shaping microglial phenotypes in AD-relevant pathological settings remain largely unresolved. Myocyte-specific enhancer factor 2C (Mef2C) is a transcription factor with versatile functions. Recent studies have attributed aging-related microglial changes to type I interferon (IFN-I)-associated Mef2C deregulation. In view of the close relationship between brain aging and AD, it is of great interest to determine microglial Mef2C changes in AD-related conditions. In this study, we have found that suppressed Mef2C nuclear translocation was an early and prominent microglial phenotype in a mouse model of brain amyloidosis (5×FAD mice), which exacerbated with age. Echoing the early Mef2C deregulation and its association with microglial activation, transcriptional data showed elicited IFN-I response in microglia from young 5×FAD mice. Amyloid beta 42 (Aβ42) in its oligomeric forms promoted Mef2C deregulation in microglia on acute organotypic brain slices with augmented microglial activation and synapse elimination via microglial phagocytosis. Importantly, these oligomeric Aβ42-mediated microglial changes were substantially attenuated by blocking IFN-I signaling. The simplest interpretation of the results is that Mef2C, concurring with activated IFN-I signaling, constitutes early microglial changes in AD-related conditions. In addition to the potential contribution of Mef2C deregulation to the development of microglial phenotypes in AD, Mef2C suppression in microglia may serve as a potential mechanistic pathway linking brain aging and AD.
Keywords: Type I interferon, Myocyte-specific enhancer factor 2C, Microglia, Amyloid beta, Alzheimer’s disease
1. Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder characterized by insidious onset and progressive cognitive decline primarily attacking the aging population (Querfurth and LaFerla, 2010; Yegambaram et al., 2015). The heterogenous etiology of AD complicates our understanding of the pathogenesis of this neurological disorder and adds a layer of difficulty to early diagnosis and treatment (Querfurth and LaFerla, 2010; Yegambaram et al., 2015). Together with other well-documented AD pathological features, including amyloid beta (Aβ) deposition and Tau hyperphosphorylation, neuroinflammation has been implicated in AD paradigms (Cummings, 2019; Dubois et al., 2010; Querfurth and LaFerla, 2010) and thus emerged as a critical early mediator of neuronal lesions in this neurological disorder (Heneka et al., 2015; Sims et al., 2017; Venegas et al., 2017). Long-regarded as the principal innate immune cells in the brain (Colonna and Butovsky, 2017), microglia play a central role in the initiation and propagation of neuroinflammation, leading to inflammatory neuronal injuries in the AD context (Heneka et al., 2015; Sims et al., 2017; Venegas et al., 2017). In addition to their neuroinflammatory-modulating aspects, activated microglia also exhibit enhanced phagocytosis of synapses, constituting a critical pathway of synapse loss in the early stage of AD (Hong et al., 2016; Rajendran and Paolicelli, 2018). Moreover, microglia are reported to be involved in Aβ plaque buildup and Tauopathies in AD-relative pathological settings (Perea et al., 2018; Venegas et al., 2017). In line with the intertwined relationship between microglia and AD pathology, recent identification of several innate immunity-related genes as genetic risk factors for AD further highlights a microglia-related mechanism of AD etiopathogenesis (Malik et al., 2015). Although the role of microglia as an early and active player in the development of AD has received increasing recognition, the detailed mechanisms that drive microglial changes in AD, especially in its early or prodromal stages remain an unresolved scientific issue.
Advancing age is the strongest risk factor for AD (Swerdlow, 2011). In view of the close relationship between brain aging and AD (Deak et al., 2016; Swerdlow, 2011), pathological changes in the aging brain are informative for our understanding of AD pathogenesis. Brain aging and AD share many aspects of brain pathology, including inflammatory microglial changes (Mosher and Wyss-Coray, 2014). Myocyte-specific enhancer factor 2C (Mef2C) is a transcription factor with versatile downstream signaling (Dong et al., 2017). Recent studies have implicated a decisive role of Mef2C in inflammatory microglial activation in an aging context supported by observations that Mef2C loss-of-function as an aging-effect confers vulnerability to inflammatory mediator-induced microglial activation (Deczkowska et al., 2017). Intriguingly, age-related Mef2C dysfunction is negatively regulated by microglial type I interferon (IFN-I) signaling responses (Deczkowska et al., 2017). Type I interferons are pleiotropic cytokines involved in antiviral immunity and autoimmunity (Pestka et al., 2004; Taniguchi and Takaoka, 2001). Excess IFN-I signaling activation has been linked with a spectrum of diseases known as type I interferonopathies and may cause neurological sequalae (Lee-Kirsch et al., 2015). In recent years, enhanced IFN-I signaling has been accentuated in the development of neuroinflammation and microglial phenotypes in brain aging and AD-related conditions (Deczkowska et al., 2017; Gorle and Vandenbroucke, 2019; Minter et al., 2016; Moore et al., 2020; Roy et al., 2020). In this context, the association of Mef2C with IFN-I signaling may leave open the possibility that Mef2C deregulation is an AD phenotype and contributes to the pathogenesis of this neurological disorder, which is further supported by single-nucleotide polymorphism of Mef2C in sporadic AD (Lambert et al., 2013; Tang et al., 2016). However, this Mef2C-associated mechanism of microglial activation in AD is suffering from lack of evidence. The limited information of the functional status of microglial Mef2C in AD-related conditions thus warrants our current study.
In this study, we found that decreased Mef2C nuclear translocation was an early microglial change in a mouse model that mimics AD brain amyloidopathy with inflammatory changes (5×FAD mice) (Eimer and Vassar, 2013). With the development of microglial activation, reduced microglial Mef2C nuclear translocation was exacerbated in 5×FAD mice. Further unbiased transcriptomics studies confirmed an early microglial IFN-I response in this mouse model. In consistency with the impact of IFN-I signaling on microglial activation, IFNα, an important member of the IFN-I family (Pestka et al., 2004; Taniguchi and Takaoka, 2001), induced Mef2C deregulation in microglia in acute organotypic brain slices. Further in vitro experiments showed that oligomeric Aβ42 suppressed microglial Mef2C nuclear translocation and augmented synapse elimination via microglial phagocytosis in an IFN-I signaling-dependent manner. Although a cause-effect relationship between Mef2C deregulation and microglial phenotypes in AD needs to be established, this proof-of-concept study suggests the incidence of microglial IFN-I response and the associated Mef2C deregulation, which may contribute to microglial activation in AD-related conditions. Our findings further provide us with a novel insight into mechanistical links between brain aging and AD.
2. Methods and materials
2.1. Mice
Animal studies were approved by the University of Kansas Animal Care and Use Committee (IACUC), the University of Texas at Dallas IACUC and were performed in accordance with the National Institutes of Health guidelines for animal care. C57BL/6 and 5×FAD mice [B6SJL-Tg (APP-SwFlLon, PSEN1*M146L*L286 V) 6799Vas/Mmjax] were obtained from Jackson Laboratory. Genotypes of animals were confirmed using PCR and/or amyloid plaque staining. The number of mice used was determined by our previous data and power calculation to ensure that the minimum number of mice required was used in the experiments.
2.2. Immunoblotting
Brain tissues were homogenized in urea buffer containing 50 mM Tris-HCl, pH 6.8, 8 M urea (Fisher Scientific), 2% SDS (Fisher Scientific), 10% glycerol (Fisher Scientific), and protease inhibitors (Millipore). The brain homogenates were separated in 12% Bis-Tris Gels (Thermo Fisher Scientific), then electrotransferred to polyvinylidine fluoride (PVDF) membranes (BioRad). After blocking in 5% non-fat milk (Labscientific Inc) for 1 h at room temperature, the membranes were probed with appropriate primary antibodies overnight at 4 °C followed by incubation with the corresponding secondary antibodies for 1 h at room temperature. The following antibodies were used: rabbit anti-Mef2C (Proteintech, 10,056–1-AP, 1:1000), mouse anti-β-actin (Sigma-Aldrich, #5441, 1:10,000), goat anti-rabbit IgG HRP-conjugated, and goat anti-mouse IgG HRP -conjugated secondary antibodies (Thermo Fisher Scientific, #31460 and #31430). Images were collected on a Bio-Rad Chemidoc Imaging System. Image J software (NIH) was used for analysis.
2.3. Fluorescence-activated cell sorting (FACS) of CD11b+ microglia
FACS purification of CD11b+ microglia were performed according to previously described methods (Srinivasan et al., 2016) with some minor modifications. Briefly, nonTg and 5×FAD mice were perfused with ice-cold PBS under isoflurane anesthetization. Mice cortices and hippocampi were then dissected and incised into ice-cold DPBS (Gibco, 14190144). The brain tissue was dissociated with Accutase (Innovative Cell Technologies, NC9839010) and debris and myelin were removed with Debris Removal Solution (Miltenyi Biotec, 130–109-398). Cells were washed with DPBS followed by centrifuging at 300 ×g at 4 °C for 10 min. The cell pellet was fixed in 50% ice-cold ethanol for 15 min and centrifuged at 500 ×g at 4 °C for 10 min. The pellet was then resuspended in ice-cold DPBS and subjected to immunostaining at 4 °C for 20 min. The following antibodies and reagents were used: Alexa Flour 488-conjugated anti-NeuN (Millipore, MAB377X, 1:1000), PE-conjugated anti-GFAP (BD Biosciences, #561483, 1:50), APC-conjugated anti-CD11b (BD Biosciences, #561690, 1:250), and DAPI (Invitrogen, D21490, 5 ng/ml). The cells were washed twice with DPBS, then resuspended in DPBS. DAPI+ /singlet cells were selected for parent gating to exclude cell debris. CD11b+/NeuN− /GFAP− cell subset was sorted and collected for RNA extraction.
2.4. Quantitative RT-PCR (qRT-PCR)
The FACS-purified microglia were fully lysed in TRizol (Invitrogen). Total RNA was purified using ZYMO Direct-zol RNA Microprep Kits (ZYMO, R2063). The cDNA was synthesized with PrimeScript RT Master Mix (TaKaRa, RR036A) and qPCR reactions were performed on an Applied Biosystems StepOnePlus Real-Time PCR System using PowerUp™ SYBR™ Green Master Mix (Applied Biosystems, A25742) with ROX reference dye (Invitrogen, #12223012). All RNA experiments were performed in an RNase/DNase-free environment to avoid RNA degradation and DNA contamination. The relative quantitation data was acquired with the ΔΔCT method using mouse Gapdh as an internal control. Then the regulation of Mef2C in 5×FAD microglia was determined by calculating fold change with reference to nonTg control samples. The following oligo primers were used: mouse Mef2C (forward primer: ATGCCATCAGTGAATCAAAGGAT, reverse primer: GTGGTACGGTCTCCCAACT); mouse Gapdh (forward primer: GACTTCAACAGCAACTCCCAC, reverse primer: TCCACCACCCTGTTGCTGTA).
2.5. RNAseq library preparation and sequencing
10,000 sorted CD11b+ brain myeloid cells were fully lysed in TRizol (Invitrogen, 15596018) and total RNA was purified using ZYMO Direct-zol RNA Microprep Kits (ZYMO, R2063). Purified total RNA was quantified with a Qubit 3.0 Fluorometer and RNAseq libraries were prepared using SMART-Seq® v4 Ultra® Low Input RNA Kits (TaKaRa, 634889) and Nextera XT DNA Library Preparation Kits (Illumina, FC-131–1024) according to manufacturer’s protocols. The quality of the libraries was analyzed with Fragment Analyzer (Advanced Analytical Technologies, Inc.). The libraries were multiplexed and then sequenced on Illumina Nextseq 500 (Illumina) to generate 33 M of paired end 150 base pair reads per library. The RNAseq raw data was deposited in NCBI-SRA with the accession numbers: PRJNA683892.
2.6. Transcriptomics data analysis
RNAseq reads were aligned to the gene annotation of Mus musculus UCSC mm10 reference genome using STAR aligner Version 2.0.2 (Illumina Basespace). Differential gene expression analysis was performed using the rlog Transformation function from DESeq2 software Version 1.1.0 (Illumina Basespace). Gene regulation significance was assessed with a two-tailed t-Test and the significant regulations (P < 0.05) were selected for bioinformatic analysis. Gene regulation statistics (Valcano and Venn Diagrams) and enriched pathway comparisons were calculated using the FunRich: Functional Enrichment analysis tool (Pathan et al., 2015) and Metascape (http://metascape.org/gp/index.html#/main/step1) (Zhou et al., 2019). Interferon Stimulated Genes (ISGs) were analyzed with Interferome (http://www.interferome.org/interferome/home.jspx) (Rusinova et al., 2013). Gene expression cluster analysis of FPKM (fragments per kilobase of exon model per million reads mapped) of major gene regulations were showed by Morpheus (https://software.broadinstitute.org/morpheus).
2.7. Oligomeric Aβ preparation
Oligomeric Aβ was prepared as previously described (Beck et al., 2016). Aβ42 peptide (GenicBio, A-43-T-1000) was diluted in 1,1,1,3,3,3-hexafluoro-2-propanol (Sigma-Aldrich) to 1 mM. After centrifugation, the supernatant was aliquoted in microcentrifuge tubes and dried overnight in the fume hood to generate a peptide film. The peptide film was then diluted in DMSO to 5 mM and resuspended in cold HAM’S F-12 (Sigma-Aldrich) to 100 μM. The solution was then incubated at 4°C for 24h to prepare oligomeric Aβ42.
2.8. Acute brain slices preparation and treatment
Acute brain slices were prepared following the published protocol with some modifications (Ribeiro et al., 2014). Briefly, 2–3-month-old C57BL/6 mice were anesthetized with isoflurane and decapitated. Brains were quickly extracted and put in ice-cold cutting solution containing the following: 4 mM KCl (Fisher Scientific), 234 mM sucrose (Fisher Scientific), 0.5 mM MgCl2 (Fisher Scientific), 24 mM NaHCO3, 0.7 mM CaCl2 (Sigma-Aldrich), and 10 mM glucose (Fisher Scientific), pH 7.4. Transverse sections (170 μm) were cut on a vibratome (VT1200S, Leica) and carefully transferred to pre-warmed slice culture medium (DMEM (Sigma-Aldrich) supplemented with 20% FBS (Fisher Scientific), 0.5% glucose (Fisher Scientific), 1 mM glutamine (Corning), and 2.5 mM HEPES (Fisher Scientific), pH 7.3) at 37 °C. Brain slices were treated with 1 μM oligomeric Aβ42, and/or anti-IFNAR antibody (MAR1–5A3, BD, 561183) for 2 h. Slices were fixed in 4% paraformaldehyde (PFA, Sigma-Aldrich) for 24 h at 4 °C then subjected to immunostaining.
2.9. Immunofluorescent staining
The frozen tissue sections from mouse brains were prepared as previously described (Tian et al., 2019). For acute organotypic brain slice cultures, samples were fixed overnight in 4% PFA at 4 °C, then subjected to immunostaining. The slices were blocked with blocking buffer (5% donkey serum, 0.3% Triton X-100 in PBS, pH 7.4) for 1 h, then incubated with primary antibodies at room temperature overnight. The following primary antibodies were used: goat anti-Iba1 (Abcam, Ab5076, 1:600), mouse anti-PSD95 (Cell Signaling Technology, #36233, 1:400), and rabbit anti-Mef2C (Proteintech, 10056–1-AP, 1:200). After washing with PBS, the slices were then probed with their appropriate cross-adsorbed secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 594 (Thermo Fisher Scientific, 1:500). DAPI was used for nuclear staining. Images were collected under a Nikon confocal microscope or Leica TCS SPE Laser Scanning Confocal.
2.10. Image analysis
The images were analyzed and reconstructed in three dimensions using Nikon-Elements Advanced Research software. The quantification of synapse engulfment was performed as previously described with some modifications (Andoh et al., 2019). The microglia-engulfed synapses were defined by colocalization of Iba1 and PSD95, which was analyzed by using the “AND” operation in the “binary operation” dialog of NIS elements software to overlap two binary layers. The volume of engulfed synapses and microglia was measured. To determine the synapse engulfment by microglia, the following calculation was used: Volume of internalized synaptic puncta (μm3)/ Volume of microglia (μm3).
To quantify the translocation of Mef2C in microglia, we defined Mef2C in the nucleus by colocalization of DAPI and Mef2C using the “AND” operation in the “binary operation” dialog of NIS elements software to overlap two binary layers. The volume of total Mef2C and nuclear Mef2C was defined by ROIs and measured in each microglia. The volume of cytosolic Mef2C was calculated by subtracting nuclear Mef2C from total Mef2C. The ratio between nuclear Mef2C to cytoplasmic Mef2C was defined as the ratio of nuclear Mef2C volume (μm3) to cytoplasmic Mef2C volume (μm3).
The quantification of convex hull area, number of nodes, and total process length of microglia was performed as previously described (Kongsui et al., 2014; Torres-Platas et al., 2014). The convex hull area and total process length of microglia was analyzed after a maximum intensity projection of z-dimension stacks in the NIS elements software. The number of nodes was counted using these three-dimensional reconstructed images.
2.11. Statistics
Statistical comparisons were performed using GraphPad Prism 8 software. Unpaired two-way Student’s t-test or two-way ANOVA followed by Bonferroni post hoc analysis were applied in data analysis. Numbers of replicates and P values are stated in each figure legend. All data were expressed as the mean ± s.e.m. Significance was concluded when the P value was less than 0.05. Significance was indicated by symbols including * (P < 0.05), ** (P < 0.010), *** (P < 0.001). NS (not significant) denotes P > 0.05.
3. Results
3.1. Decreased microglial Mef2C nuclear translocation in 5×FAD mice
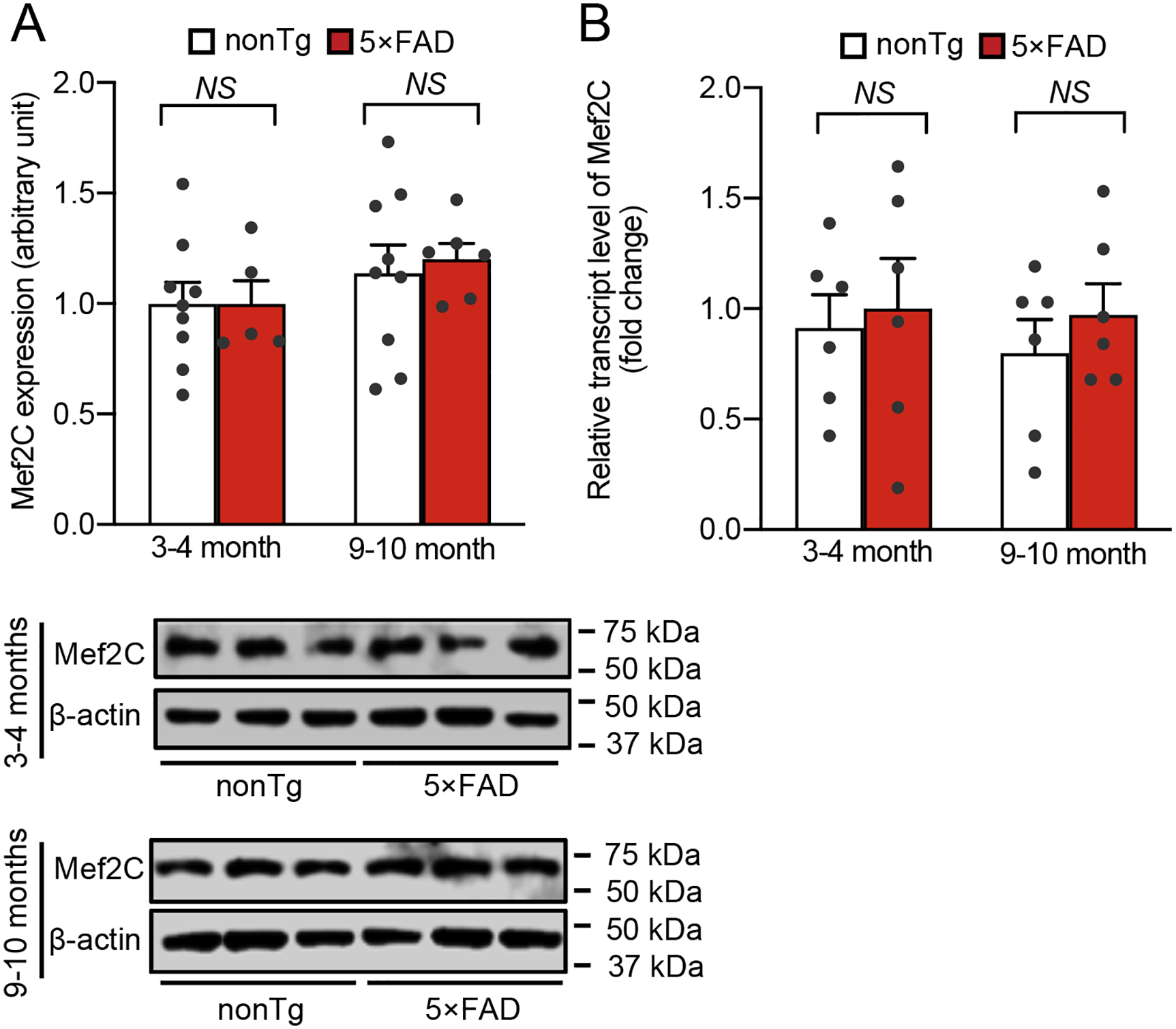
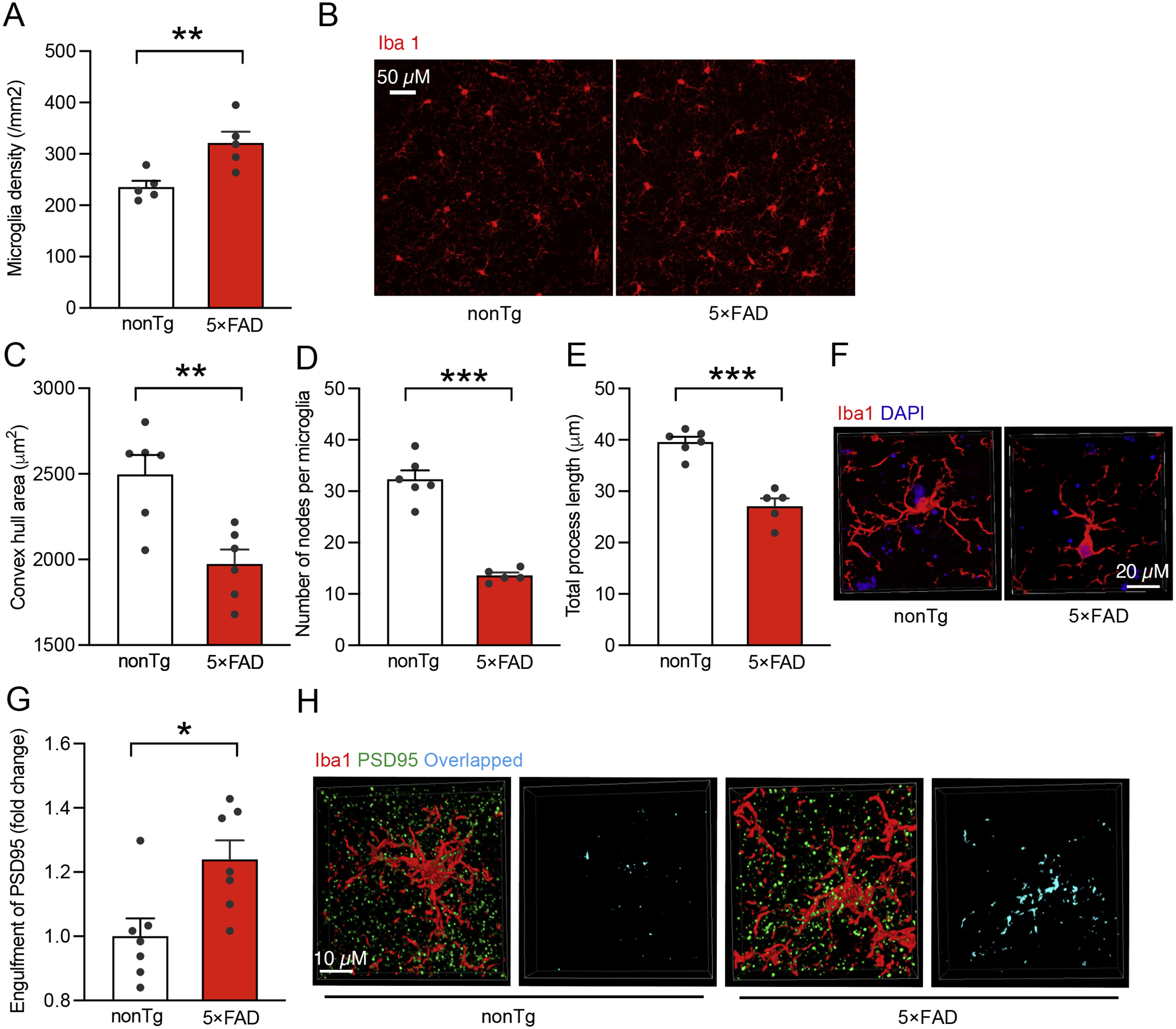
To determine the status of Mef2C in AD-related conditions, we subjected brain extracts from age- and gender-matched nonTg mice and their 5×FAD littermates at 3–4 and 9–10 months old to immunoblotting for Mef2C expression. Selection of the different age groups of mice was based on previous observations of phenotypic changes in 5×FAD mice. 5×FAD mice at 3–4 months old demonstrate mild brain Aβ deposition and neuroinflammation with modest cognitive defects mimicking early-stage AD while 5×FAD mice at the older age have heavy Aβ burden and severely impaired cognition (Beck et al., 2016; Eimer and Vassar, 2013; Tian et al., 2019). Densitometry analysis of the immunoreactive bands showed no significant difference in Mef2C expression between nonTg and 5×FAD mice at the tested ages (Supplementary Fig. 1A). In view of the importance of Mef2C in microglial biology (Deczkowska et al., 2017), we then sought to specifically determine Mef2C levels in microglia. To this end, we isolated microglia from the neocortex of nonTg and 5×FAD mice at 3–4 and 9–10 months old (Bordt et al., 2020) and first examined Mef2C transcript levels with a reverse transcription polymerase chain reaction (RT-PCR) assay. Microglia from mice of both genotypes exhibited comparable levels of Mef2C mRNA (Supplementary Fig. 1B). Because mRNA copies may not reflect the protein levels, we then examined Mef2C expression in microglia by performing immunofluorescent staining for Mef2C on brain slices from mice of both genotypes. This was done to avoid the technical challenge of collecting a sufficient amount of microglia from the neocortex for immunoblotting. In addition, Mef2C is a transcription factor that translocates to the nucleus upon activation (Dong et al., 2017). Immunofluorescent assays for Mef2C could provide us with critical data for a correlation of Mef2C expression with its nuclear translocation to reflect Mef2C functional status in microglia. To this end, Mef2C was labeled using its specific antibody. Microglia were determined by the co-staining of ionized calcium binding adaptor molecule 1 (Iba-1), a specific microglial marker (Ohsawa et al., 2004) and nuclei were visualized by the staining of 4′,6-diamidino-2-phenylindole (DAPI). In comparison with their nonTg control counterparts, microglia from 5×FAD mice at 3–4 months old only demonstrated a marginal decrease in Mef2C levels (Fig. 1A & E). This slight decrease of total Mef2C levels devoid of statistical significance persisted in 5×FAD mice at 9–10 months old (Fig. 1A & E); however, in comparison with their age- and gender-matched nonTg littermates, 5×FAD mice exhibited remarkably decreased levels of nuclear Mef2C (Fig. 1B & E), suggesting microglial Mef2C deactivation in early stages of AD pathology. Importantly, such changes in microglial Mef2C observed in young 5×FAD mice were exacerbated with age (Fig. 1B & E), implicating declining microglial Mef2C deregulation with the development of brain amyloidopathy. As a result, the distribution of Mef2C in the cytosol was increased in the microglia of young 5×FAD mice (Fig. 1C & E) with a decreased ratio of nuclear Mef2C to cytoplasmic Mef2C (Fig. 1D). Increased proliferation and the shift of microglial morphology from a spider-like shape to a de-ramified pattern are pathological features of AD-related microgliosis and are indicative of inflammatory microglial activation (Franco-Bocanegra et al., 2019). In line with Mef2C deregulation, microglia in 5×FAD mice exhibited increased density (Supplementary Fig. 2A & B), decreased convex hull area (Supplementary Fig. 2C & F), lessened number of nodes (Supplementary Fig. 2D & F), as well as reduced total process length (Supplementary Fig. 2E & F), suggesting microglial proliferation and activation. A prominent microglial phenotype that occurs early in AD is increased phagocytosis of synaptic content, which constitutes a critical mechanism of synapse elimination in this neurological disorder (Hong et al., 2016; Rajendran and Paolicelli, 2018). Concurring with microglial morphological changes, an increased engulfment of postsynaptic content (PSD95) was observed in microglia from 5×FAD mice, indicating enhanced synapse elimination by microglia in AD-brain-related conditions (Supplementary Fig. 2G&H). Put together, these results suggest that impaired microglial Mef2C function in 5×FAD mice comprises an early microglial phenotype in Aβ-rich milieus.
Fig. 1.

Decreased microglial Mef2C nuclear translocation in 5×FAD mice brain. A. Measurement of total Mef2C expression in microglia from 3–4–month– and 9–10–month–old nonTg and 5×FAD mice. Two-way ANOVA followed by Bonferroni post hoc analysis. 3–4 months: nonTg, n = 6; 5×FAD, n = 5. 9–10 months: nonTg, n = 6; 5×FAD, n = 6. B&C. Analysis of nuclear (B) and cytoplasmic (C) expression of Mef2C in microglia from 3–4–month– and 9–10–month–old nonTg and 5×FAD mice. Two-way ANOVA followed by Bonferroni post hoc analysis. 3–4 months: nonTg, n = 6; 5×FAD, n = 5. 9–10 months: nonTg, n = 6; 5×FAD, n = 6. D. Calculation of nuclear/cytoplasmic ratio of Mef2C expression in microglia from 3–4–month– and 9–10–month–old nonTg and 5×FAD mice. Two-way ANOVA followed by Bonferroni post hoc analysis. 3–4 months: nonTg, n = 6; 5×FAD, n = 5. 9–10 months: nonTg, n = 6; 5×FAD, n = 6. E. Representative confocal microscopy images of quantification in (A-D). Iba1 was used to visualize the microglia. Nuclei were stained with DAPI. Scale bar = 10 μm.
3.2. Early microglial IFN-I response in 5×FAD mice
Previous studies suggest that IFN-I signaling negatively regulates Mef2C in microglia in aging mice (Deczkowska et al., 2017). If this relationship in aging brains was to be applied to an AD-relevant setting, we would expect to see an early activation of IFN-I signaling in microglia from young 5×FAD mice. To this end, we collected microglia from the whole neocortex of nonTg mice along with their 5×FAD littermates at 3–4 months old using fluorescence-activated cell sorting (FACS) (Nugent et al., 2020), and examined their transcriptional profile through mRNA sequencing (RNAseq). In comparison to their nonTg counterparts, the microglia of 5×FAD mice had decreased expression of 298 genes and increased expression of 364 genes (P < 0.05) (Fig. 2A. & Supplementary database). Metascape enrichment analysis (http://metascape.org/gp/index.html#/main/step1) showed that regulated genes were most related to “immune system process,” “metabolic process,” and “response to stimuli” pathways (Fig. 2B). Among the differentially-expressed genes, a total of 149 genes were identified as interferon-stimulated genes (ISGs) by definition of Interferome 2.01 (http://www.interferome.org/interferome/search/showSearch.jspx) (Fig. 2C & D). Most of them are associated with IFN-I signaling, which suggests that IFN-I signaling is an essential regulation pathway in microglia for 5×FAD mice (Roy et al., 2020). Although the source of IFN-I in 5×FAD brains remains to be determined, the results indicate that activated IFN-I signaling is an early microglial phenotype concurring with Mef2C deregulation in 5×FAD mice.
Fig. 2.

Early activation of IFN-I signaling in microglia from 5×FAD mice. A. Volcano plot showing differences in gene expression in purified microglia from 3–4–month–old 5×FAD mice with respect to nonTg littermates. Unpaired two-way Student’s t-test. Significance is plotted against fold change. Up- or down- regulated genes are highlighted in red and green, respectively, with adjusted significance of P < 0.05. n = 3 per group. B. Top 20 significantly enriched Gene Ontology (GO) terms from genes differentially expressed in purified microglia from 3–4–month–old 5×FAD mice compared to nonTg littermates. The significance of the observed GO enrichment was estimated by the P value plotted on a log10 scale. n = 3 per group. C. Heat map showing clustering of upregulated and downregulated interferon stimulated genes (ISGs) expressed in purified microglia from 3–4–month–old nonTg and 5×FAD mice. The major upregulated and downregulated genes were listed. D&E. Venn diagram showing upregulated (D) and downregulated (E) interferon stimulated genes (ISGs) induced by Type I and/or Type II interferons in purified microglia from 3–4–month–old 5×FAD mice compared to nonTg littermates.
3.3. Involvement of IFN-I signaling in microglial activation in soluble Aβ-rich milieus
In comparison with Aβ plaques, soluble Aβ in its oligomeric forms plays a central role in AD pathogenesis (Walsh and Selkoe, 2007) and drives early microglial activation (Hong et al., 2016). The intensive microglial IFN responses, as observed in our transcriptomics study on young 5×FAD mice with limited parenchymal Aβ plaques, further implicates an effect of oligomeric Aβ. To determine the involvement of IFN signaling in Aβ-mediated microglial changes, we removed circulating IFN-I and IFN-I producing cells in nonTg mice at 3–4 months old through transcardial perfusion, and prepared acute organotypic brain slices for incubation with soluble oligomeric Aβ42 at 1 μM for up to 6 h. After the treatment, brain slices were subjected to immunostaining assays for morphological features of microglial activation. As early as after 2 h incubation time with oligomeric Aβ42, microglia showed remarkable morphological changes demonstrated by decreased convex hull area (Fig. 3A & D), lowered number of nodes (Fig. 3B & D), as well as reduced total process length (Fig. 3C & D), implicating a quick microglial response to soluble Aβ toxicity. Such histopathological changes of microglia were exacerbated with increased incubation time (data not shown). In agreement with microglial morphological changes, a substantial increase in microglial engulfed PSD95 content was observed in oligomeric Aβ42-exposed brain slices (Fig. 3E & F), indicating enhanced synapse elimination through microglial phagocytosis in soluble Aβ-rich milieus. Importantly, oligomeric Aβ42-induced changes in microglial morphology (Fig. 3A–D), and engulfment of PSD95 (Fig. 3E & F) were markedly mitigated by the blockade of IFN-I signaling via a pretreatment of antibodies against IFN-I receptor (IFNAR). Collectively, these results indicate a fast response of microglia to soluble Aβ insult and suggest a role of IFN-I signaling in Aβ-mediated microglial phenotype.
Fig. 3.

Involvement of IFN-I signaling in microglial activation in soluble Aβ-rich milieus. A, B & C. Effect of Aβ (1 μM, 2 h) treatment on microglia convex hull area (A), number of nodes (B), and total processes length (C) from acute organotypic brain slices in the presence or absence of a 20 min pretreatment of α-IFNAR (10 μg/ml). The brain slices were prepared from 3–month–old nonTg mice. Two-way ANOVA followed by Bonferroni post hoc analysis. n = 18–24 microglia. D. Representative confocal microscopy images of quantification in (A-C). Iba1 was used to visualize microglia. Nuclei were stained with DAPI. Scale bar = 10 μm. E. Effect of Aβ (1 μM, 2 h) treatment on microglial engulfment of synapses from acute organotypic brain slices in the presence or absence of a 20 min pretreatment of α-IFNAR (10 μg/ml). The brain slices were prepared from 3–month–old nonTg mice. Two-way ANOVA followed by Bonferroni post hoc analysis. n = 27–34 microglia. F. Representative confocal microscopy images of microglial engulfment of synapses. Iba1 (red) and PSD95 (green) were used to visualize microglia and synapses respectively. The overlapped staining of microglia and PSD95 indicates synapses engulfment. Scale bar = 10 μm.
3.4. Ameliorated microglial Mef2C nuclear translocation by the blockade of IFN-I signaling in soluble Aβ-rich milieus
Loss of Mef2C is suggested to render sensitivity to stimulus-induced microglial activation (Deczkowska et al., 2017). In view of the prompt Aβ effect in stimulating IFN signaling-related microglial activation and the negative impact of IFN signaling on microglial Mef2C regulation (Deczkowska et al., 2017), we treated organotypic brain slices from nonTg mice at 3–4 months old with 1 μM oligomeric Aβ42 for 2 h to establish a direct link between soluble Aβ toxicity and microglial Mef2C deregulation. After the treatment, brain slices were used for immunostaining for Mef2C, microglia and the nuclei. Analysis of the data showed that oligomeric Aβ42 negatively affected microglial Mef2C nuclear translocation demonstrated by decreased nuclear Mef2C (Fig. 4A & E), augmented cytoplasmic Mef2C (Fig. 4B & E), and a decreased ratio of nuclear Mef2C to cytoplasmic Mef2C (Fig. 4C) with no significant change in the levels of total Mef2C in microglia (Fig. 4D & E). The results corroborate our findings in 5×FAD mice. To determine the cause-effect relationship between IFN response and microglial Mef2C deregulation in this Aβ context, we preincubated the organotypic brain slices with anti-IFNAR antibody and continued the experiments. Anti-IFNAR antibody alone substantially enhanced Mef2C nuclear translocation (Fig. 4A & E), resulting in an increase in the ratio of nuclear Mef2C to cytoplasmic Mef2C (Fig. 4C & E). Those impacts vanished microglial Mef2C changes in response to oligomeric Aβ42 (Fig. 4A–E). Intriguingly, the application of anti-IFNAR antibody surprisingly upregulated the expression of Mef2C in regardless of the presence of absence of oligomeric Aβ42 (Fig. 4D & E), indicating a positive regulation of Mef2C by blocking IFN-I signaling. These findings support the involvement of IFN-I response in Aβ-mediated microglial Mef2C deregulation.
Fig. 4.

Reduced microglia Mef2C nuclear translocation in soluble Aβ-rich milieus is IFN-I dependent. A, B & C. Effect of Aβ (1 μM, 2 h) treatment on nuclear Mef2C (A), cytoplasmic Mef2C (B), and nuclear/cytoplasmic ratio of Mef2C (C) in microglia of acute organotypic brain slices in the presence or absence of a 20 min pretreatment of α-IFNAR (10 μg/ml). The brain slices were prepared from 3–month–old nonTg mice. Two-way ANOVA followed by Bonferroni post hoc analysis. n = 18–24 microglia. D. Effect of Aβ (1 μM, 2 h) treatment on total Mef2C expression in microglia of acute organotypic brain slices in the presence or absence of a 20 min pretreatment of α-IFNAR (10 μg/ml). The brain slices were prepared from 3–month–old nonTg mice. Two-way ANOVA followed by Bonferroni post hoc analysis. n = 18–24 microglia. E. Representative confocal microscopy images of quantification in (A-D). Iba1 was used to visualize the microglia. Nuclei were stained with DAPI. Scale bar = 10 μm.
4. Discussion
With the increased recognition of neuroinflammation as an early and critical pathological feature of AD, microglial mechanisms are accentuated as a crucial supplement to the current neurocentric view of the pathogenesis of this neurodegenerative disorder (Hansen et al., 2018). Known as the major component of innate immunity in the central nervous system (CNS), microglia are responsive to a variety of inflammatory mediators including cytokines and chemokines, fatty acid metabolites, and Aβ in AD-relevant pathological settings (Riedel et al., 2016; Smith et al., 2012; Vehmas et al., 2003). Enhanced microglial activity limits Aβ plaque buildup in AD brains and favors the clearance of Aβ (Mandrekar et al., 2009; Mawuenyega et al., 2010; Zhao et al., 2017). However, the pro-inflammatory activation of microglia decreases binding and degradation of Aβ in microglia (Hickman et al., 2008), and paradoxically promotes Aβ plaque formation (Venegas and Heneka, 2019). Importantly, synapse pruning via the phagocytosis of activated microglia is a critical pathway for early synapse loss in AD-relation conditions (Hong et al., 2016; Rajendran and Paolicelli, 2018). In addition to the many AD genetic risks clustered in innate immunity related to microglial function (Sierksma et al., 2020), the contribution of microglia to the development of AD-related brain pathologies is further supported by the ameliorated brain amyloidopathy in microglia-depleted 5×FAD mice (Spangenberg et al., 2019). Although the deleterious role of pro-inflammatory microglia is well-appreciated in AD pathogenesis, the detailed mechanisms that drive microglial activation, especially at the prodromal or early stage of AD remain unresolved. In our current study, we have determined that elicited IFN responses and suppressed Mef2C activation are prominent microglial changes at the early stage of AD pathology in 5×FAD mice. The concurrence of Mef2C deactivation and microglial activation in 5×FAD mice further implicates an association of Mef2C deregulation and microglial phenotypes in AD-relative settings, which furnishes our knowledge on the pathways of oligomeric Aβ-potentiated microglial activation. Moreover, the aging-like changes of microglial Mef2C in AD-related conditions adds novel evidence to foster our understanding of the mechanisms underlying the transition of pathological aging to AD.
Mef2C is a transcription factor with poorly defined functions. In addition to its epigenetic regulation of osteocytes (Kramer et al., 2012; Velazquez-Cruz et al., 2014), cardiomyocytes (Liu and Tang, 2019), and myocytes (Anderson et al., 2015), Mef2C has been implicated in the regulation of innate and adaptive immune cells with myeloid origin including microglia (Ben-Yehuda et al., 2020; Brown et al., 2018; Fu et al., 2006; Kong et al., 2016; Zuurbier et al., 2014). Mef2C-deficient microglia exhibit increased sensitivity to inflammatory mediator-induced pro-inflammatory changes underpins the importance of Mef2C in microglial biology (Deczkowska et al., 2017). Despite the physiological role of Mef2C in microglial function in developmental brains (Ben-Yehuda et al., 2020), the involvement of microglial Mef2C deregulation in CNS diseases is largely unclear until a recent study attributed pro-inflammatory microglial responses to Mef2C instability in aging brains (Deczkowska et al., 2017). In agreement with the many overlapped aspects of microglial phenotypes between brain aging and AD (Mosher and Wyss-Coray, 2014), we have determined suppressed microglial Mef2C from activation in AD-related conditions demonstrated by decreased Mef2C nuclear translocation in 5×FAD mice as well as in Aβ-treated organotypic brain slices. In view of the concurrence of Mef2C changes and microglial activation in 5×FAD mice and the close association between Mef2C and microglial response in the aging context, we thus postulate an Mef2C-associated mechanism of inflammatory microglial activation in Aβ-rich milieus. Indeed, in our current experimental setting, we cannot establish the cause-effect relationship between Mef2C deregulation and the development of pro-inflammatory microglial activation in AD-related conditions, but the results from this proof-of-concept study forms groundwork for our further investigation into this topic.
Another critical issue related to Mef2C is the influence of IFN-I. Deczkowska and colleagues found that microglial Mef2C nuclear translocation is negatively regulated by IFNβ alongside an IFNβ-related downregulation of Mef2C transcripts (Deczkowska et al., 2017). Such a finding with increased ISGs in microglia from aging brains indicate the role of IFN-I signaling in shaping microglial profiles during aging (Deczkowska et al., 2017). Noteworthy, the involvement of IFN-I signaling in AD pathogenesis has received increasing recognition. Elevated IFN-I response and its role in promoting microglial activation have been consistently determined in several Aβ-based mouse models including, 5×FAD (Roy et al., 2020), APP/PS1 (Minter et al., 2016; Moore et al., 2020), and J20 lines (Mesquita et al., 2015). Moreover, genetic depletion of IFN-I alpha receptor-1 (IFNAR1) in APP/PS1 mice inhibits neuroinflammatory responses, ameliorates microgliosis, and improves mouse spatial navigation without affecting brain Aβ burden (Minter et al., 2016; Moore et al., 2020). Our transcriptional data of increased ISGs in microglia from young 5×FAD mice and the preventive effects of antibodies against IFNAR on Aβ-induced synapse loss due to microglial engulfment corroborate these reports, adding credit to an IFN-I hypothesis of microgliopathy and neuroinflammation in AD. However, the emerging roles of IFN-I in AD pathogenesis has raised a critical question about the source of IFN-I in AD brains. Type I interferons are pleiotropic cytokines that are involved in antiviral immunity and autoimmunity (Pestka et al., 2004). Although many cell types, especially the myeloid cells and dendritic cell precursors, can produce IFN-I following viral stimulation (Fitzgerald-Bocarsly and Feng, 2007), the provenance of IFN-I in immune-privileged sites, such as the brain, is still under intensive investigation. Previous studies have determined IFN-I production in microglia, astrocytes, and neurons after viral challenge (Delhaye et al., 2006). In addition, circulating IFN-I species like IFNα can trespass the blood-brain barrier (BBB) and exert immune-modulating influence in the brain (Aw et al., 2020). However, the source of brain IFN-I in chronic neurodegenerative disorders is still poorly understood. Although choroid plexus epithelial cells (CPEs) have been implicated to be a critical IFN-I provisioner in aging and AD brains (Baruch et al., 2013; Gorle and Vandenbroucke, 2019), microglia in contact with Aβ plaques are a recently highlighted IFN-I producer in AD brains (Roy et al., 2020). Given the myeloid origin of microglial cells and capability of microglia to produce cytokines and chemokines, we cannot fully rule out the possibility of IFN-I production in microglia not in direct contact with Aβ plaques. Because Mef2C response to IFN-I challenge may confer vulnerability to Aβ-induced microglial activation, IFN-I signaling and Mef2C deregulation-promoted microglial activation may reinforce each other, resulting in a feeding-forward vicious cycle. Disregarding the source of IFN-I in AD brains, microglia, along with astrocytes and neurons, are target cells of brain interferons (Roy et al., 2020); and microglia are more responsive to IFN-I challenge (Li et al., 2018). Therefore, the interferon responses may serve as a critical factor for the decision of microglial fate in AD-relevant pathological settings.
Lastly, in addition to suppressed translocation to microglial nuclei, Mef2C expression is decreased at the transcription level in mice at 22 months old (Deczkowska et al., 2017). The downregulation of Mef2C mRNA copies in microglia was not observed with 5×FAD mice in this study. This disparity in Mef2C transcription may reflect an age effect. Interestingly, although we failed to see any negative association of IFN signaling and microglial Mef2C transcription in Aβ-rich environments, the administration of anti-IFNAR antibody upregulated Mef2C expression in microglia, which seems to suggest the function of IFN-I signaling in restraining Mef2C from overactivation in physiology. Therefore, the depression of Mef2C in aging and AD brains may represent an effect of IFN-I overdose. Of note, decreased Mef2C mRNA levels in peripheral leucocytes were previously reported in Japanese patients with late-onset AD and were correlated with the severity of their cognitive impairment (Sao et al., 2018). Although we cannot ignore the potential impact of peripheral circulating IFN-I on this cohort due to the lack of related information, such an AD-related change in Mef2C may suggest a relationship of Mef2C deregulation with AD at systems levels, supporting a crosstalk between peripheral immune system with AD brain changes. Moreover, Mef2C polymorphism has been linked with increased susceptibility to sporadic AD (Lambert et al., 2013; Tang et al., 2016). Therefore, further studies on the role of Mef2C in the development of AD will render us profound insight into the contribution of microglia and, in a broader spectrum, the adaptive and innate immunity to AD pathogenesis. Noteworthy, Mef2C is implicated in the regulation of excitatory neurons and genetic depletion of Mef2C leads to changes in neurons towards inhibitory synaptic transmission (Harrington et al., 2016). In current study, we primarily focused on Mef2C status in microglia. Whether Mef2C is affected in AD neurons will be examined in our future investigation. Nevertheless, our current proof-of-concept study suggests a potential role of Mef2C as a mediator for IFN-I response-related microglial activation in AD-related conditions. The identification of the common change of Mef2C in aging and AD constitutes a novel mechanistic link between the two conditions, which will foster us a better understanding of the mechanisms driving the transition of pathological aging to this devastating neurodegenerative disorder. The most parsimonious interpretation of our results is that the concurrence of Mef2C deactivation and IFN-I response constitutes a microglial signature in Aβ-rich milieus, which lays the groundwork for our future research on an IFN signaling- and Mef2C-related mechanism of AD pathogenesis.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2021.105272.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgement
This work was supported by research funding from NIH (R01AG053588, and R01AG059753), Alzheimer’s Association (AARG-16–442863), and BrightFocus Foundation (A20201159S). Nazia A. Hafeez helped with data analysis. Tienju Wang helped with proofreading.
Footnotes
Declaration of Competing Interest
The authors declare no conflict of interest.
References
- Anderson CM, et al. , 2015. Myocyte enhancer factor 2C function in skeletal muscle is required for normal growth and glucose metabolism in mice. Skelet. Muscle 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh M, et al. , 2019. Exercise reverses behavioral and synaptic abnormalities after maternal inflammation. Cell Rep. 27 (2817–2825), e5. [DOI] [PubMed] [Google Scholar]
- Aw E, et al. , 2020. Microglial responses to peripheral type 1 interferon. J. Neuroinflammation 17, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruch K, et al. , 2013. CNS-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc. Natl. Acad. Sci. U. S. A 110, 2264–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck SJ, et al. , 2016. Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat. Commun. 7, 11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yehuda H, et al. , 2020. Maternal Type-I interferon signaling adversely affects the microglia and the behavior of the offspring accompanied by increased sensitivity to stress. Mol. Psychiatry 25, 1050–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordt EA, et al. , 2020. Isolation of microglia from mouse or human tissue. STAR Protoc 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown FC, et al. , 2018. MEF2C phosphorylation is required for chemotherapy resistance in acute myeloid leukemia. Cancer Discov 8, 478–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna M, Butovsky O, 2017. Microglia function in the central nervous system during health and neurodegeneration. Annu. Rev. Immunol 35, 441–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings J, 2019. The role of biomarkers in Alzheimer’s disease drug development. Adv. Exp. Med. Biol 1118, 29–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak F, et al. , 2016. Recent developments in understanding brain aging: implications for Alzheimer’s disease and vascular cognitive impairment. J. Gerontol. A Biol. Sci. Med. Sci 71, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska A, et al. , 2017. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat. Commun 8, 717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhaye S, et al. , 2006. Neurons produce type I interferon during viral encephalitis. Proc. Natl. Acad. Sci. U. S. A 103, 7835–7840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, et al. , 2017. Myocyte enhancer factor 2C and its directly-interacting proteins: a review. Prog. Biophys. Mol. Biol 126, 22–30. [DOI] [PubMed] [Google Scholar]
- Dubois B, et al. , 2010. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 9, 1118–1127. [DOI] [PubMed] [Google Scholar]
- Eimer WA, Vassar R, 2013. Neuron loss in the 5×FAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol. Neurodegener 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald-Bocarsly P, Feng D, 2007. The role of type I interferon production by dendritic cells in host defense. Biochimie 89, 843–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco-Bocanegra DK, et al. , 2019. Microglial motility in Alzheimer’s disease and after Abeta42 immunotherapy: a human post-mortem study. Acta Neuropathol Commun 7, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, et al. , 2006. MEF2C mediates the activation induced cell death (AICD) of macrophages. Cell Res 16, 559–565. [DOI] [PubMed] [Google Scholar]
- Gorle N, Vandenbroucke RE, 2019. Interferons: a molecular switch between damage and repair in ageing and Alzheimer’s disease. Mech. Ageing Dev 183, 111148. [DOI] [PubMed] [Google Scholar]
- Hansen DV, et al. , 2018. Microglia in Alzheimer’s disease. J. Cell Biol 217, 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AJ, et al. , 2016. MEF2C regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, et al. , 2015. Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, et al. , 2008. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci 28, 8354–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, et al. , 2016. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong NR, et al. , 2016. MEF2C and EBF1 co-regulate B cell-specific transcription. PLoS Genet 12, e1005845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongsui R, et al. , 2014. Quantitative assessment of microglial morphology and density reveals remarkable consistency in the distribution and morphology of cells within the healthy prefrontal cortex of the rat. J. Neuroinflammation 11, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer I, et al. , 2012. Mef2c deletion in osteocytes results in increased bone mass. J. Bone Miner. Res 27, 360–373. [DOI] [PubMed] [Google Scholar]
- Lambert JC, et al. , 2013. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45, 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Kirsch MA, et al. , 2015. Type I interferonopathies–an expanding disease spectrum of immunodysregulation. Semin. Immunopathol 37, 349–357. [DOI] [PubMed] [Google Scholar]
- Li W, et al. , 2018. Microglia have a more extensive and divergent response to interferon-alpha compared with astrocytes. Glia 66, 2058–2078. [DOI] [PubMed] [Google Scholar]
- Liu CF, Tang WHW, 2019. Epigenetics in cardiac hypertrophy and heart failure. JACC Basic Transl. Sci 4, 976–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik M, et al. , 2015. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener 10, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrekar S, et al. , 2009. Microglia mediate the clearance of soluble Abeta through fluid phase macropinocytosis. J. Neurosci 29, 4252–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mawuenyega KG, et al. , 2010. Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330, 1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesquita SD, et al. , 2015. The choroid plexus transcriptome reveals changes in type I and II interferon responses in a mouse model of Alzheimer’s disease. Brain Behav. Immun 49, 280–292. [DOI] [PubMed] [Google Scholar]
- Minter MR, et al. , 2016. Deletion of the type-1 interferon receptor in APPSWE/PS1DeltaE9 mice preserves cognitive function and alters glial phenotype. Acta Neuropathol. Commun 4, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore Z, et al. , 2020. Abrogation of type-I interferon signalling alters the microglial response to Abeta1–42. Sci. Rep 10, 3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosher KI, Wyss-Coray T, 2014. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem. Pharmacol. 88, 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent AA, et al. , 2020. TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105 (837–854), e9. [DOI] [PubMed] [Google Scholar]
- Ohsawa K, et al. , 2004. Microglia/macrophage-specific protein Iba1 binds to fimbrin and enhances its actin-bundling activity. J. Neurochem 88, 844–856. [DOI] [PubMed] [Google Scholar]
- Pathan M, et al. , 2015. FunRich: an open access standalone functional enrichment and interaction network analysis tool. Proteomics 15, 2597–2601. [DOI] [PubMed] [Google Scholar]
- Perea JR, et al. , 2018. The role of microglia in the spread of tau: relevance for Tauopathies. Front. Cell. Neurosci 12, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestka S, et al. , 2004. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev 202, 8–32. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM, 2010. Alzheimer’s disease. N Engl J Med 362, 329–344. [DOI] [PubMed] [Google Scholar]
- Rajendran L, Paolicelli RC, 2018. Microglia-mediated synapse loss in Alzheimer’s disease. J. Neurosci 38, 2911–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro LF, et al. , 2014. Ghrelin triggers the synaptic incorporation of AMPA receptors in the hippocampus. Proc. Natl. Acad. Sci. U. S. A 111, E149–E158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel BC, et al. , 2016. Age, APOE and sex: triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 160, 134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy ER, et al. , 2020. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J. Clin. Invest. 130, 1912–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusinova I, et al. , 2013. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res 41, D1040–D1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sao T, et al. , 2018. MEF2C mRNA expression and cognitive function in Japanese patients with Alzheimer’s disease. Psychiatry Clin. Neurosci 72, 160–167. [DOI] [PubMed] [Google Scholar]
- Sierksma A, et al. , 2020. Translating genetic risk of Alzheimer’s disease into mechanistic insight and drug targets. Science 370, 61–66. [DOI] [PubMed] [Google Scholar]
- Sims R, et al. , 2017. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat. Genet 49, 1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, et al. , 2012. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 87, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg E, et al. , 2019. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun 10, 3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K, et al. , 2016. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun 7, 11295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH, 2011. Brain aging, Alzheimer’s disease, and mitochondria. Biochim. Biophys. Acta 1812, 1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SS, et al. , 2016. MEF2C rs190982 polymorphism with late-onset Alzheimer’s disease in Han Chinese: a replication study and meta-analyses. Oncotarget 7, 39136–39142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, Takaoka A, 2001. A weak signal for strong responses: interferon-alpha/beta revisited. Nat. Rev. Mol. Cell Biol 2, 378–386. [DOI] [PubMed] [Google Scholar]
- Tian J, et al. , 2019. Disrupted hippocampal growth hormone secretagogue receptor 1alpha interaction with dopamine receptor D1 plays a role in Alzheimer’s disease. Sci. Transl. Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Platas SG, et al. , 2014. Morphometric characterization of microglial phenotypes in human cerebral cortex. J. Neuroinflammation 11, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vehmas AK, et al. , 2003. Immune reactive cells in senile plaques and cognitive decline in Alzheimer’s disease. Neurobiol. Aging 24, 321–331. [DOI] [PubMed] [Google Scholar]
- Velazquez-Cruz R, et al. , 2014. Analysis of association of MEF2C, SOST and JAG1 genes with bone mineral density in Mexican-Mestizo postmenopausal women. BMC Musculoskelet. Disord 15, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venegas C, Heneka MT, 2019. Inflammasome-mediated innate immunity in Alzheimer’s disease. FASEB J 33, 13075–13084. [DOI] [PubMed] [Google Scholar]
- Venegas C, et al. , 2017. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 552, 355–361. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ, 2007. A beta oligomers - a decade of discovery. J. Neurochem 101, 1172–1184. [DOI] [PubMed] [Google Scholar]
- Yegambaram M, et al. , 2015. Role of environmental contaminants in the etiology of Alzheimer’s disease: a review. Curr. Alzheimer Res 12, 116–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, et al. , 2017. Microglia limit the expansion of beta-amyloid plaques in a mouse model of Alzheimer’s disease. Mol. Neurodegener 12, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, et al. , 2019. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuurbier L, et al. , 2014. Immature MEF2C-dysregulated T-cell leukemia patients have an early T-cell precursor acute lymphoblastic leukemia gene signature and typically have non-rearranged T-cell receptors. Haematologica 99, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.