

Abstract
Diabetes-induced coronary endothelial cell (CEC) dysfunction contributes to diabetic heart diseases. Angiotensin II (Ang II), a vasoactive hormone, is upregulated in diabetes, and is reported to increase oxidative stress in CECs. 4-hydroxy-2-nonenal (4HNE), a key lipid peroxidation product, causes cellular dysfunction by forming adducts with proteins. By detoxifying 4HNE, aldehyde dehydrogenase (ALDH) 2 reduces 4HNE mediated proteotoxicity and confers cytoprotection. Thus, we hypothesize that ALDH2 improves Ang II-mediated defective CEC angiogenesis by decreasing 4HNE-mediated cytotoxicity. To test our hypothesis, we treated the cultured mouse CECs (MCECs) with Ang II (0.1, 1 and 10 μM) for 2, 4 and 6 hours. Next, we treated MCECs with Alda-1 (10 μM), an ALDH2 activator or disulfiram (2.5 μM)/ALDH2 siRNA (1.25 nM), the ALDH2 inhibitors, or blockers of angiotensin II type-1 and 2 receptors i.e. Losartan and PD0123319 respectively before challenging MCECs with 10 μM Ang II. We found that 10 μM Ang II decreased tube formation in MCECs with in vitro angiogenesis assay (P < .0005 vs control). 10 μM Ang II downregulated the levels of vascular endothelial growth factor receptor 1 (VEGFR1) (p < .005 for mRNA and P < .05 for protein) and VEGFR2 (p < .05 for mRNA and P < .005 for protein) as well as upregulated the levels of angiotensin II type-2 receptor (AT2R) (p < .05 for mRNA and P < .005 for protein) and 4HNE-adducts (P < .05 for protein) in cultured MCECs, compared to controls. ALDH2 inhibition with disulfiram/ALDH2 siRNA exacerbated 10 μM Ang II-induced decrease in coronary angiogenesis (P < .005) by decreasing the levels of VEGFR1 (P < .005 for mRNA and P < .05 for protein) and VEGFR2 (P < .05 for both mRNA and protein) and increasing the levels of AT2R (P < .05 for both mRNA and protein) and 4HNE-adducts (P < .05 for protein) relative to Ang II alone. AT2R inhibition per se improved angiogenesis in MCECs. Additionally, enhancing ALDH2 activity with Alda 1 rescued Ang II-induced decrease in angiogenesis by increasing the levels of VEGFR1, VEGFR2 and decreasing the levels of AT2R. In summary, ALDH2 can be an important target in reducing 4HNE-induced proteotoxicity and improving angiogenesis in MCECs. Finally, we conclude ALDH2 activation can be a therapeutic strategy to improve coronary angiogenesis to ameliorate cardiometabolic diseases.
Keywords: Aldehyde dehydrogenase 2, 4-hydroxy-2-nonenal, Angiotensin II, angiogenesis, cardiometabolic diseases, disulfiram, Alda 1, VEGFR1, VEGFR2
1. INTRODUCTION
Transport of nutrients, oxygen, hormones, drugs and immune cells to the target organs is carried out through blood vessels [1]. Endothelial cell (EC) line of the blood vessel plays an indispensable role in the transport. EC provides an interface between blood plasma and the tissues associated with blood vessels [1]. So, it can be surmised that any structural or functional defect to the vascular ECs can cause several diseases associated with the cardiovascular system. Modulation of cell signaling in vascular ECs can lead to excessive or inadequate angiogenesis and consequently trigger the progression of diseases including cancer, skin diseases, age-related blindness, diabetic ulcers, cardiovascular diseases and stroke [2].
Generally, the term “angiogenesis or neovascularization” means the development of new blood vessels in both healthy and diseased states. However, in the strict sense, the definition of angiogenesis is the process of vessel sprouting from pre-existing ones [3]. In normal physiology, the process of angiogenesis is controlled by an intricate balance between pro-angiogenic and anti-angiogenic factors. When the balance leans toward the pro-angiogenic factors, it increases blood vessel formation which is observed in malignant tumors [1]. On the other hand, when the balance leans toward anti-angiogenic factors, it decreases blood vessel formation that is observed in diabetic heart. In diabetic milieu, oxidative stress, inflammation, metabolic changes and neurohormonal disturbances are key contributors for the pathogenesis.
All the key contributors can lead to increase in angiotensin II (Ang II), an important bioactive peptide from renin-angiotensin-aldosterone system, plays a fundamental role in the cardiovascular system [4, 5]. Ang II was primarily recognized as a vasoconstrictor, but several follow-up studies revealed that it also contributes to cellular growth, differentiation and migration, apoptosis, extracellular matrix formation, and inflammation. Ang II-induced cellular growth and differentiation are associated with the upregulation of different types of growth factors including vascular endothelial growth factor (VEGF), platelet-derived growth factor, epidermal growth factor, transforming growth factor β, insulin-like growth factor 1, platelet activator factor and fibroblast growth factor [6]. Ang II also causes EC dysfunction through the upregulation of mitochondrial reactive oxygen species (ROS) production as well as contributes to the development and progression of atherosclerosis by augmenting oxidative stress [7]. Oxidative stress-induced ROS formation causes an increased generation of 4-hydroxy-2-nonenal (4HNE), a cytotoxic aldehyde [8]. 4HNE can alter protein function and gene regulation through the formation of adducts with these macromolecules [9]. 4HNE adducted proteins are recycled by proteasomes [10]. However, if 4HNE levels are extremely high in the cells, then the proteins in proteasomal machinery also undergo 4HNE adduction and become dysfunctional, thus leading to proteotoxicity as the oxidized and damaged proteins were not recycled by proteasome, resulting in cellular dysfunction and ultimately tissue damage [11]. Thus, 4HNE is one of the upstream signaling molecules of proteotoxicity. According to Ohki et al. 4HNE levels were increased significantly in the skeletal muscle of Ang II-infused mice compared to untreated mice [12]. It is not only ROS-mediated lipid peroxidation that increases 4HNE even Ang II also increases 4HNE levels and contribute to the cellular dysfunction in cardiovascular diseases [13, 14].
In mammals, the action of Ang II is mediated by the activation of angiotensin II type 1 receptor (AT1R) and angiotensin II type 2 receptor (AT2R), both of which are G-protein coupled receptors. Impaired activation of Ang II and its receptors are associated with several cardiovascular diseases including hypertension, atherosclerosis, heart failure and restenosis after angioplasty [5]. However, the role of Ang II and its receptors in coronary angiogenesis is not well known. Ang II-induced ROS formation in ECs is mainly triggered by the activation of AT1R [14, 15]. A study in mammals reported that vascular proliferation in tumor and non-tumor models is associated with the expression and activation of AT1R [16]. Some other studies in breast cancer patients revealed that the activation of the AT1 receptor triggers the modulation of angiogenesis, cell proliferation, migration, and inflammation [16]. Contrary to these findings, another study exhibited that Ang II-induced activation of AT2R promotes apoptosis and inhibits angiogenesis in human bladder cancer [17]. Thus, the modulation of AT1R and AT2R is critical in understanding the cellular process including angiogenesis. Therefore, in this study, we plan to use AT1 receptor antagonist and AT2 receptor antagonists to determine their effects.
Aldehyde dehydrogenase (ALDH) 2 is a key mitochondrial enzyme that converts toxic aldehydes into non-reactive acids [18]. ALDH2 is cardioprotective by detoxifying 4HNE [19]. Pharmacologically activating ALDH2 has been shown to protect the heart against ischemia / reperfusion injury and cardiac remodeling [20, 21]. The pleiotropic roles of ALDH2 has been emerging [22]. However, the role of ALDH2 in the modulation of coronary angiogenesis has not been studied well yet. A recent study reported that deficiency of ALDH2 impairs angiogenesis by inhibiting hypoxia-inducible factor-1α / VEGF signaling cascade in ECs [21]. We have identified that lower ALDH2 activity can potentiate 4HNE-induced mitochondrial dysfunction in cultured cardiomyocytes [23] and impaired tube formation in cultured mouse coronary ECs [24]. Based on the notion, for the first time, we propose to determine the effect of ALDH2 modulation in Ang II-mediated dysregulation of coronary angiogenesis, in vitro.
2. METHODS
2.1. Mammalian cell culture
Mouse coronary EC (MCEC) line was obtained from the Cedarlane (#CLU510). The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (GE Life Sciences) supplemented with 10% fetal bovine serum (FBS) (v/v) and 1% (v/v) penicillin / streptomycin (P / S). The cells were grown in 100-mm plates and maintained at 37 °C with a continuous supply of 95% air / 5% CO2. The medium was changed every 48 hours intervals. For each passage, the cells were washed with PBS and incubated at 37°C with .05% trypsin / EDTA (GE life Sciences). The cells after fourth passage were grown in new DMEM supplemented with 0.2% FBS (low serum) and 1% P / S. After 24 hours of low serum treatment the cells were counted with a hemocytometer. Once the concentrations of MCECs were determined, the cells were seeded in Matrigel coated 96-well plates for tube formation assay and in 100‐mm plates for extraction of mRNA and protein followed by an hour of incubation at 37°C with a continuous supply of 95% air / 5% CO2. Then, the cells were treated with vehicle or disulfiram (DSF) (2.5 μM), an ALDH2 inhibitor / Alda 1 (10 μM), an ALDH2 activator / Losartan (1 μM), an AT1R inhibitor / PD0123319 (1 μM), an AT2R inhibitor for an hour (Fig. 1). After DSF / Losartan / PD0123319 treatment, the cells were treated with different doses of Ang II (0.1, 1 and 10 μM) (and respective vehicles) for 2 and 4 hours (Fig. 1). We performed at least four replications. Microscopic pictures of the tube formation assay were taken at 2 and 4 hours after Ang II treatment. Cell viability assay and collection of cell lysates for mRNA and protein extraction were also done in the same time frame.
Fig. 1.
Scheme depicting the treatment protocol.
2.2. siRNA-mediated transfection
siRNA-mediated transfection of MCECs was performed according to the manufacturers protocol. Briefly, 2 × 105 MCECs were seeded in a well of a 6-wells plate containing DMEM supplemented with 10% FBS. The MCECs were treated with ALDH2 siRNA (1.25 nM) (#sc-60148, SCBT) and control siRNA (1.25 nM) (#sc-37007, SCBT) at 60% confluency and subsequently incubated in a CO2 incubator for 32 h. SiRNA transfected MCECs were further used for tube formation assay and mRNA/protein extraction.
2.3. Tube formation assay
Preparation of Matrigel coated cell culture plates were already discussed in our previously published paper [24]. Briefly, based on the number of required wells, 75-μl growth factor reduced Matrigel (Corning) were taken into each well of a 96 well plate. Then, the plates were immediately incubated at 37°C in a cell culture incubator for 30 minutes in order to make the Matrigel coat to solidify. 2 × 104 MCECs were re-suspended in 50-μl DMEM supplemented with 10% FBS and 1% P / S and subsequently pipetted in Matrigel coated individual wells. MCECs were treated as in Fig. 1. Images of tube formation on Matrigel were captured under a 10x phase contrast microscope (Olympus IX81). Around 4 random images were captured from each well. The number of circles formed by the MCECs under each high-power field (HPF) were indicating the blood vessel formations which were counted manually (unbiased) using ImageJ software. This quantification of tube formation under HPF was compiled on excel spreadsheets for further statistical analysis.
2.4. Cell viability assay
The viability of MCECs was determined with trypan blue kit (ThermoFisher Scientific) according to the manufacturer protocol.
2.5. mRNA extraction and measurement of mRNA concentrations
Total mRNAs from MCECs were carried out with the RNeasy Mini kit (Qiagen) according to the manufacturers protocol. RNA concentrations were measured with Qubit RNA BR assay kit (Life Technologies) and Qubit 3.0 fluorometer (Life Technologies) according to the manufacturer protocol.
2.6. Real-time qPCR
Real-time quantitative PCR (qPCR) was performed using iTaq universal SYBR Green one-step kit and a CFX96 Real-time system (Bio-Rad). 25 ng of mRNA with a reaction volume of 10 μl were used for the qPCR reaction. The primer sets (mouse-specific) used for the qPCR are listed on the table (Table 1). The qPCR data were analyzed using the MS-Excel spreadsheets. The relative expression of target mRNAs was calculated as fold change using the [25]. Fold changes of the target mRNAs were normalized with the mouse ribosomal protein L27 (RpL27), a housekeeping gene.
Table 1.
Primer sequences for RT-qPCR reaction.
| Genes | Forward primer (5’−3’) | Reverse primer (5’−3’) |
|---|---|---|
| RpL27 | GCTCGTGCTGCTAATAAAGC | GTTTCATGAACTTGCCCATC |
| VEGFR1 | CGGAAG GAAGACAGCTCATC | CTTCACGCGACAGGTGTAGA |
| VEGFR2 | GGCGGTGGTGACAGTATCTT | TCTCCGGCAAGCTCAAT |
| AT2R | GTTCCCCTTGTTTGGTGTAT | CATCTTCAGGACTTGGTCAC |
2.7. ALDH2 activity assay
ALDH2 activity was measured according to the protocol described elsewhere [26]. Briefly, MCECs were grown in 150 mm plates and treated with DMSO (vehicle), Ang II, DSF and Alda1 for 2 h. Cellular protein was extracted from cultured MCECs using tissue protein extraction reagent (ThermoFisher Scientific) containing protease/phosphatase inhibitors. Protein concentrations of the samples were measured using the Bradford protein assay protocol with the aid of a microplate reader. 100 μg of total cellular protein from each sample was used for this assay. Freshly made 50 mM sodium pyrophosphate solution as a buffer, 2.5 mM NAD+ solution as a cofactor and 10 mM acetaldehyde as a substrate were used. Enzymatic activity of ALDH2 from cell lysate was determined spectrophotometrically by using the reductive reaction of NAD+ to NADH at λ340 nm wavelength at 37 °C temperature with the aid of a microplate reader.
2.8. Western immunoblotting
4HNE, VEGFR1, VEGFR2 and AT2 protein levels were evaluated using Western immunoblot (WB) assay. In brief, after treatment, cellular protein was extracted from cultured MCECs using tissue protein extraction reagent (ThermoFisher Scientific) containing protease/phosphatase inhibitors. Whole cell lysates were homogenized using a tissue homogenizer. Total protein concentration was determined with the Bradford protein assay. Specific protein bands were separated using SDS-PAGE and the proteins were then transferred to nitrocellulose membranes. The membranes were blocked using 5% bovine serum albumin (BSA) and subsequently incubated with 4HNE mouse mAb (#MAB3249, R&D Systems), ALDH2 mouse mAb (# MA5–17029, Invitrogen), VEGFR1 mouse mAb (#NB600–1004, NB), VEGFR2 (D5B1) rabbit mAb (#9698, CST), AT2 (EPR3876) rabbit mAb (#ab92445, AB) and β-Actin (8H10D10) Mouse mAb (#3700, CST) primary antibodies at a concentration of 1:1000 overnight in a cold refrigerator (4 °C). Depend on the sources of primary antibodies the membrane bound antibodies were incubated with anti-rabbit/anti-mouse horseradish peroxidase (HRP)-coupled secondary antibodies (1:1000) for 1 h at room temperature. Immunolabeling was detected using ECL detection reagents according to the manufacturer’s protocol. The images of the protein bands were taken with FluorChem E imaging system. Intensity of scanned WB images were analyzed with image J software. β-actin protein was used as a loading control to normalize the proteins of interest.
2.9. Statistical analysis
Calculation of means, standard error of the mean (SEM) and the generation of bar diagrams were carried out with excel spreadsheets. The statistical significances were determined with paired and unpaired t-test using GraphPad Prism (GraphPad Software).
3. RESULTS
3.1. Ang II decreases angiogenesis
MCEC lines were treated with different doses of Ang II (0.1, 1 & 10 μM). 0.1 μM Ang II-treated MCECs did not show any significant change in circle formation relative to the control after 2 hours (Fig. 2A, 2B & 2M). However, after 4 and 6 hours, 0.1 μM Ang II-treated MCECs showed significant decrease in circle formation relative to the control (p < .005 for 4 hours and p < .0005 for 6 hours) (Fig. 2E, 2F, 2I, 2J & 2N). 1 μM Ang II-treated MCECs significantly decreased circle formation relative to the control after all time points (p < .05 after 2 hours and p < .0005 both after 4 & 6 hours) (Fig. 2C, 2G, 2K, 2M – 2O). 10 μM Ang II-treated MCECs significantly decreased circle formation after 2, 4 and 6 hours (p < .0005 vs control after 2 & 4 hours, p < .0001 vs control after 6 hours, p < .0005 vs 0.1 μM Ang II after 2 hours, p < .005 vs 0.1 μM Ang II both after 4 & 6 hours, p < .005 vs 1 μM Ang II after 2 hours and p < .05 vs 1 μM Ang II after 6 hours) (Fig. 2A – 2D, 2E, 2F, 2H, 2I – 2L & 2M – 2O).
Fig. 2. Dose dependent effect of Ang II in MCECs angiogenesis.
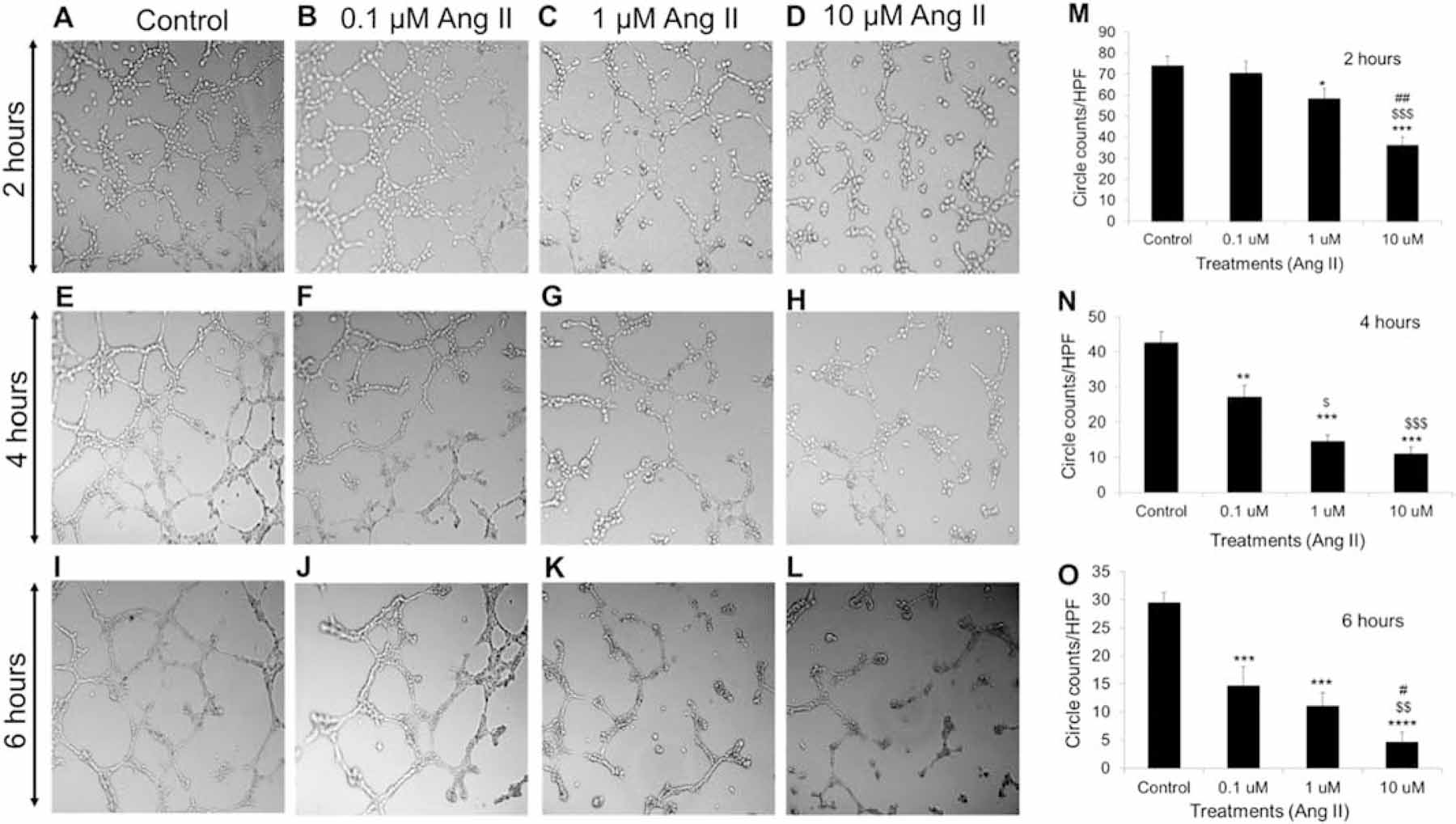
(A) – (D) Representative micrographs of tube formation by control, 0.1, 1 and 10 μM Ang II treated MCECs on matrigel after 2 hours respectively. Magnification: 10x. (E) – (H) Representative micrographs of tube formation by control, 0.1, 1 and 10 μM Ang II treated MCECs on matrigel after 4 hours respectively. Magnification: 10x. (I) – (L) Representative micrographs of tube formation by control, 0.1, 1 and 10 μM Ang II treated MCECs on matrigel after 6 hours respectively. Magnification: 10x. (M) Quantitative data of circles with different treatments stated in A through D. (N) Quantitative data of circles with different treatments stated in E through H. (O) Quantitative data of circles with different treatments stated in I through L. n=8 for each group. Each bar represents mean ± SEM. *P < .05 vs control, **P < .005 vs control, ***P < .0005 vs control, ****P < .0001 vs control, $p < .05 vs 0.1 μM Ang II, $ $p < .005 vs .1 μM Ang II, $ $ $p < .0005 vs 0.1 μM Ang II, #p < .05 vs 1 μM Ang II and ##p < .005 vs 1 μM Ang II. HPF = high power field.
3.2. Pharmacological Inhibition of ALDH2 activity exacerbates Ang II-induced decreases in angiogenesis by impairing VEGFR1, VEGFR2 and AT2R expression which was rescued by ALDH2 activation.
To study the effect of ALDH2 inhibition and activation, we used DSF (2.5 μM) and Alda 1 (10 μM) respectively (Fig. 3). MCECs separately treated with 10 μM Ang II and 2.5 μM DSF significantly reduced tube formation compared with control after two hours (P < .005) (Fig. 3A – 3D & 3H). DSF pretreatment before challenging the MCECs with Ang II caused further reduction in circle formation compared with individual effects of Ang II and DSF after two hours (P < .0005 vs control, P < .005 vs both Ang II and DSF alone) (Fig. 3A – 3D, 3F & 3H). However, individual treatment with 10 μM Alda 1 significantly increased circle formation relative to the control, Ang II and DSF alone (P < .05 vs control, P < .0005 vs both Ang II and DSF alone) (Fig. 3A – 3E & 3H). Alda 1 pretreatment before Ang II challenge rescued the Ang II mediated decrease in tube formation which is significantly higher than the individual effect of Ang II and DSF as well as the combined effects of Ang II and DSF in tube formation (P < .005 vs both Ang II and DSF and P < .0005 vs DSF + Ang II) (Fig. 3C, 3D, 3F, 3G & 3H).
Fig. 3. Role of pharmacological inhibition of ALDH2 activity in Ang II-mediated angiogenesis and mRNA/protein levels of 4HNE-protein adducts, VEGFR1, VEGFR2 and AT2R in MCECs.
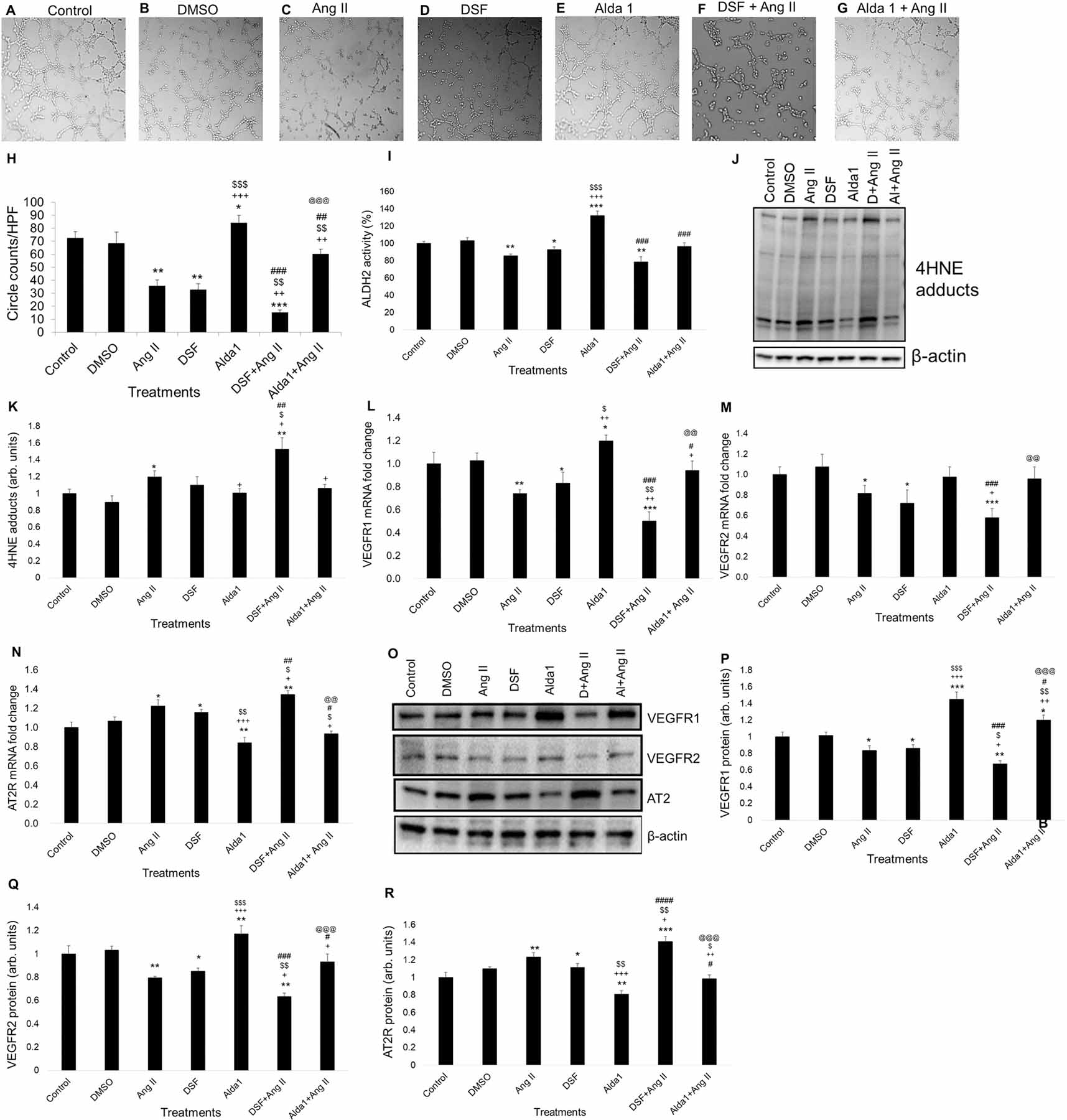
(A) - (G) Representative micrographs of tube formation on Matrigel after 1 h of DSF (2.5 μM) /Alda1 (10 μM) treatments followed by 2 h of Ang II (10 μM) treatment. Magnification: 10x. A to E show tube formation with control, DMSO (vehicle), Ang II (10 μM), DSF (2.5 μM) and Alda 1 (10 μM)-treated MCECs. F shows tube formation with DSF pretreatment followed by Ang II treatment. G shows tube formation with Alda 1 pretreatment followed by Ang II treatment. (H) Quantitative data of circles with different treatments stated in A through G. (I) Quantitative data of ALDH2 activity in MCECs after 1 h of DSF (2.5 μM) /Alda1 (10 μM) treatments following 2 h of Ang II (10 μM) treatment. (J) Representative WB band image of 4HNE protein adducts after 1 h of DSF (2.5 μM) /Alda1 (10 μM) treatments following 2 h of Ang II (10 μM) treatment. (K) Quantification of WB data in panel J. (L) Representative qPCR data of VEGFR1 mRNA expression after 2 h. (M) Representative qPCR data of VEGFR2 mRNA expression after 2 h. (N) Representative qPCR data of AT2R mRNA expression after 2 h. (O) Representative Western blot (WB) band images of VEGFR1, VEGFR2, AT2R and β-actin proteins in MCECs after 1 h of DSF (2.5 μM) /Alda1 (10 μM) treatments followed by 2 h of Ang II (10 μM) treatment. (P) Quantification of WB data of VEGFR1 protein levels stated in panel O. (Q) Quantification of WB data of VEGFR2 protein levels stated in panel O. (R) Quantification of WB data of AT2R protein levels stated in panel O. n=6 for each group. Each bar represents mean ± SEM. *p < .05 vs control, **p < .005 vs control, ***p < .0005 vs control, +p < .05 vs Ang II, ++p < .005 vs Ang II, +++p < .0005 vs Ang II, $p < .05 vs DSF, $ $p < .005 vs DSF, $ $ $p < .0005 vs DSF, #p < .05 vs Alda 1, ##p < .005 vs Alda 1, ###p < .0005 vs Alda 1, ####p < .0001 vs Alda 1, @@p < .005 vs DSF+Ang II and @@@p < .0005 vs DSF+Ang II. HPF = high power field, Ang II = Angiotensin II and DSF = disulfiram.
VEGFR1 activation in EC was shown to upregulate peripheral angiogenesis [27]. Treatment with 10 μM Ang II caused a significant reduction in VEGFR1 mRNA (P < .005 vs control) and protein (P < .05) levels compared with control in cultured MCECs after 2 hours (Fig. 3L, 3O & 3P). Similarly, treatment with DSF alone also caused a significant reduction in VEGFR1 mRNA (P < .05 vs control) and protein (P < .05) levels relative to control after 2 hours as well (Fig. 3L, 3O & 3P). DSF pretreatment enhanced Ang II-induced decrease in VEGFR1 mRNA (P < .0005 vs control and P < .005 vs both Ang II and DSF alone) and protein (P < .005 vs control, P < .05 vs both Ang II and DSF alone) levels compared with Ang II and DSF alone in MCECs (Fig. 3L, 3O & 3P). However, treatment with 10 μM Alda 1 significantly increased VEGFR1 mRNA (P < .05 vs control, P < .005 vs Ang II & P < .05 vs DSF) and protein (P < .0005 vs control, P < .0005 vs Ang II & P < .0005 vs DSF) levels compared to control, Ang II and DSF alone (Fig. 3L, 3O & 3P). Alda 1 pretreatment before challenging the MCECs with Ang II rescued the Ang II mediated decrease in VEGFR1 levels which is significantly higher than the individual effects of Ang II and DSF as well as the combined effect of Ang II and DSF in VEGFR1 mRNA (P < .05 vs Ang II and P < .005 vs DSF + Ang II) and protein (P < .005 vs both Ang II and DSF alone and P < .005 vs DSF + Ang II) levels (Fig. 3L, 3O & 3P).
Activation of VEGFR2 in cardiac endothelial cell contributes coronary angiogenesis in mouse [28]. Treatment with 10 μM Ang II caused a significant reduction in VEGFR2 mRNA (P < .05 vs control) and protein (P < .005) levels compared with control in cultured MCECs after 2 hours (Fig. 3M, 3O & 3Q). Similarly, treatment with DSF alone also caused a significant reduction in VEGFR2 mRNA (P < .05 vs control) and protein (P < .05) levels relative to control after 2 hours as well (Fig. 3M, 3O & 3Q). DSF pretreatment enhanced Ang II-induced decreases in VEGFR2 mRNA (P < .0005 vs control and P < .05 vs Ang II) and protein (P < .005 vs control, P < .05 vs Ang II and P < .005 vs DSF) levels compared with Ang II and DSF alone in MCECs (Fig. 3M, 3O & 3Q). However, treatment with 10 μM Alda 1 significantly increased VEGFR2 protein (P < .005 vs control and P < .0005 vs both Ang II & DSF alone) levels compared to control, Ang II and DSF alone (Fig. 3O & 3Q). Alda 1 pretreatment before challenging the MCECs with Ang II rescued the Ang II mediated decrease in VEGFR2 levels which is significantly higher than the individual effects of Ang II as well as the combined effect of Ang II and DSF in VEGFR2 protein (P < .05 vs Ang II and P < .0005 vs DSF + Ang II) levels (Fig. 3O & 3Q).
Ang II-induced inhibition of CEC proliferation is mediated via AT2R [29]. We found that treatment with Ang II significantly increased AT2R mRNA (P < .05) and protein (P < .005) levels compared with control in cultured MCECs after 2 hours (Fig. 3N, 3O & 3R). Likewise, treatment with DSF alone also caused a significant increase in AT2R mRNA (P < .05) and protein (P < .05) levels compared with control after 2 hours as well (Fig. 3N, 3O & 3R). DSF pretreatment enhanced Ang II-induced increase in AT2R mRNA (P < .005 vs control and P < .05 vs both Ang II and DSF alone) and protein (P < .0005 vs control, P < .05 vs Ang II and P < .005 vs DSF) levels compared with Ang II and DSF alone in MCECs (Fig. 3N, 3O & 3R). However, Alda 1 treatment significantly decreased AT2R mRNA (P < .005 vs control, P < .0005 vs Ang II and P < .005 vs DSF) and protein (P < .005 vs control, P < .0005 vs Ang II and P < .005 vs DSF) levels relative to control, Ang II and DSF alone (Fig. 3N, 3O & 3R). Alda 1 pretreatment before challenging the MCECs with Ang II rescued the Ang II mediated increase in AT2R levels which is significantly lower than the individual effects of Ang II and DSF as well as the combined effect of Ang II and DSF in AT2R mRNA (P < .05 vs both Ang II and DSF alone and P < .005 vs DSF + Ang II) and protein (P < .005 vs Ang II, P < .05 vs DSF and P < .0005 vs DSF + Ang II) levels (Fig. 3N, 3O & 3R).
3.3. Increase in 4HNE adduct levels due to the decrease in ALDH2 activity with DSF and ALDH2 siRNA treatment
To confirm whether Ang II-induced decrease in MCEC angiogenesis is due to decreased ALDH2 activity and subsequent elevation of 4HNE-protein adducts in MCECs, we performed ALDH2 activity assay and WB using 4HNE protein adducts antibodies respectively. Our data showed that treatment with 10 μM Ang II significantly increased 4HNE-protein adducts (P < .05) in MCECs after 2 hours (Fig. 3J & 3K). DSF pretreatment exacerbated Ang II-mediated increase in 4HNE adduct levels compared to control, Ang II and DSF alone (P < .005 vs control and P < .05 vs Ang II and P < .05 vs DSF) in cultured MCECs (Fig. 3J & 3K). However, treatment with Alda 1 decreases Ang II-mediated increase in 4HNE adduct (P < .05 vs Ang II) levels in cultured MCECs (Fig. 3J & 3K). Alda 1 pretreatment decreased the Ang II mediated increase in 4HNE adduct levels (P < .05 vs Ang II) in cultured MCECs (Fig. 3J & 3K).
3.4. Genetic inhibition of ALDH2 activity by ALDH2 siRNA exacerbates Ang II-induced decrease in angiogenesis by impairing VEGFR1, VEGFR2 and AT2R levels which is rescued after ALDH2 activation
To reconfirm the role of ALDH2 inhibition in the impairment of VEGFR1, VEGFR2 and AT2 levels and subsequent decrease in MCEC angiogenesis, we inhibited the activity of ALDH2 with ALDH2 siRNA (1.25 nM) (Fig. 4). Our data showed that 10 μM Ang II and ALDH2 siRNA significantly reduced tube formation compared with control after two hours in MCECs (P < .05) (Fig. 4A, 4C, 4E & 4J). ALDH2 siRNA transfection prior to Ang II challenge caused further reduction in circle formation compared with individual effects of Ang II after two hours (P < .005 vs control and P < .05 vs Ang II) (Fig. 4A, 4C, 4H & 4J). However, 10 μM Alda 1 alone significantly increased circle formation relative to the control, Ang II and ALDH2 siRNA alone (P < .005 vs control, P < .0005 vs both Ang II and ALDH2 siRNA alone) (Fig. 4A, 4C, 4E, 4F & 4J). Alda 1 pretreatment before Ang II challenge rescued the Ang II mediated decrease in tube formation which is significantly higher than the individual effect of Ang II and ALDH2 siRNA as well as the combined effects of Ang II and ALDH2 siRNA in tube formation (P < .005 vs Ang II, P < .0005 vs ALDH2 siRNA and P < .0005 vs ALDH2 siRNA + Ang II) (Fig. 4C, 4E, 4H, 4I & 4J).
Fig. 4. Role of genetic inhibition of ALDH2 activity in Ang II-mediated angiogenesis and mRNA/protein expression of VEGFR1, VEGFR2 and AT2R in MCECs.
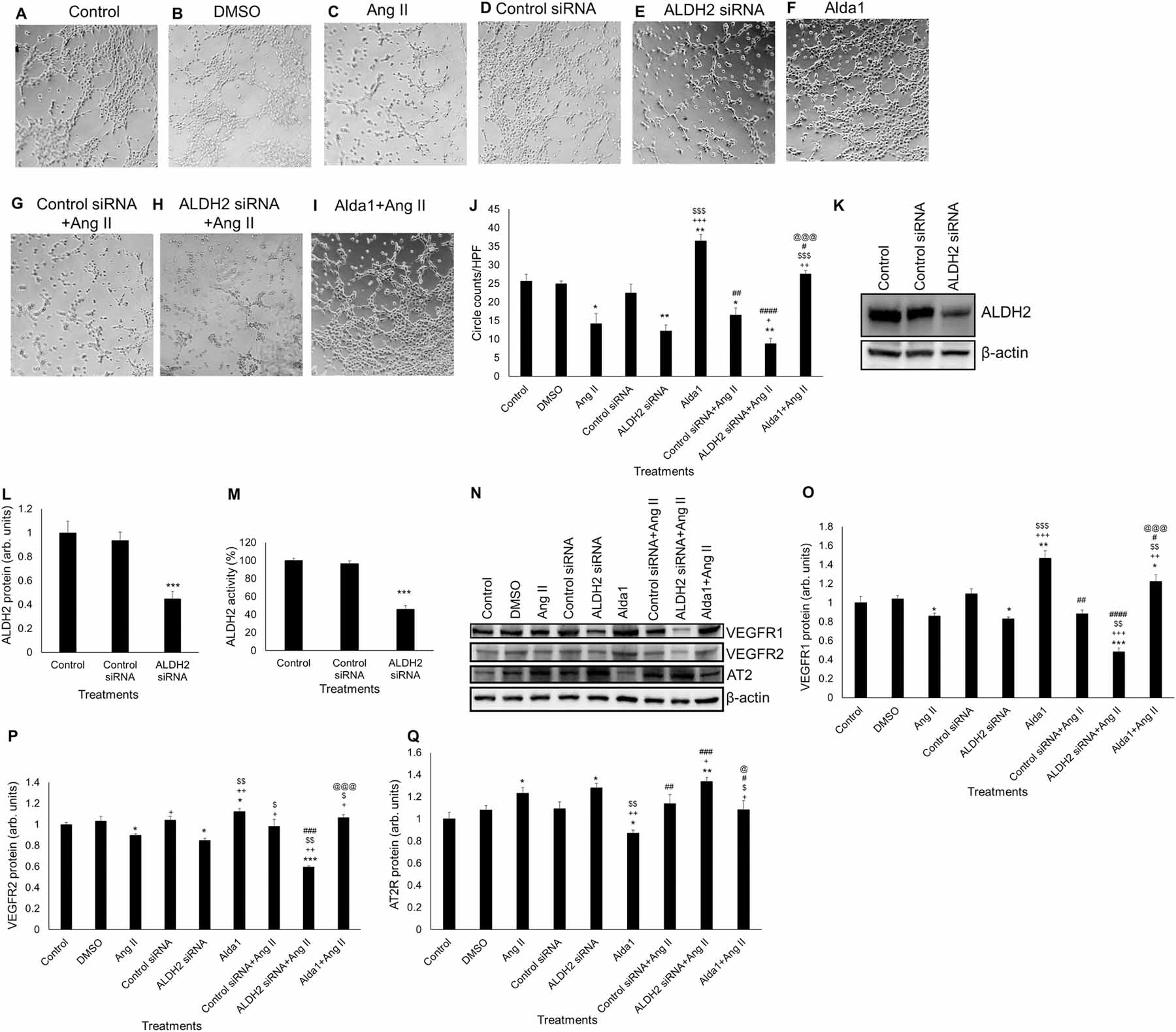
(A) - (I) Representative micrographs of tube formation on Matrigel after 32 h of siRNA (1.25 nM)/1 h of Alda1 (10 μM) pretreatments followed by 2 h of Ang II (10 μM) treatment. Magnification: 10x. A to F show tube formation with control, DMSO (vehicle), Ang II (10 μM), control siRNA (1.25 nM), ALDH2 siRNA (1.25 nM) and Alda 1 (10 μM)-treated MCECs. G shows tube formation with control siRNA pretreatment followed by Ang II treatment. H shows tube formation with ALDH2 siRNA pretreatment followed by Ang II treatment. I shows tube formation with Alda 1 pretreatment followed by Ang II treatment. (J) Quantitative data of circles with different treatments stated in A through I. (K) Representative WB band image of ALDH2 siRNA (1.25 nM) treated MCECs after 32 h. (L) Quantification of WB data in panel K. (M) Quantitative data of ALDH2 siRNA activity in MCECs after 32 h of siRNA (1.25 nM) treatment. (N) Representative Western blot (WB) band images of VEGFR1, VEGFR2, AT2R and β-actin proteins in MCECs after 32 h of siRNA (1.25 nM)/1 h of Alda1 (10 μM) pretreatments followed by 2 h of Ang II (10 μM) treatment. (O) Quantification of WB data of VEGFR1 protein levels stated in panel N. (P) Quantification of WB data of VEGFR2 protein levels stated in panel N. (Q) Quantification of WB data of AT2R protein levels stated in panel N. n=6 for each group. Each bar represents mean ± SEM. *p < .05 vs control, **p < .005 vs control, ***p < .0005 vs control, +p < .05 vs Ang II, ++p < .005 vs Ang II, +++p < .0005 vs Ang II, $p < .05 vs DSF, $ $p < .005 vs DSF, $ $ $p < .0005 vs DSF, #p < .05 vs Alda 1, ##p < .005 vs Alda 1, ###p < .0005 vs Alda 1, ####p < .0001 vs Alda 1, @p < .05 vs DSF+Ang II and @@@p < .0005 vs DSF+Ang II. HPF = high power field, Ang II = Angiotensin II and DSF = disulfiram.
To confirm whether ALDH2 siRNA treatment reduces the levels as well as the activity of ALDH2, we performed ALDH2 WB and ALDH2 activity assay respectively. The data showed that treatment with ALDH2 siRNA for 32 hours significantly decreased the ALDH2 protein levels (P < .0005) compared with controls in cultured MCECs (Fig. 4K–4M).
Treatment with 10 μM Ang II caused a significant reduction in VEGFR1 protein levels (P < .05) compared to control in cultured MCECs after 2 hours (Fig. 4N & 4O). Similarly, treatment with ALDH2 siRNA alone also caused a significant reduction in VEGFR1 protein levels (P < .05) relative to control after 2 hours as well (Fig. 4N & 4O). ALDH2 siRNA pretreatment enhanced Ang II-induced decrease in VEGFR1 protein (P < .0005 vs control, P < .0005 vs Ang II and P < .005 vs ALDH2 siRNA) levels compared with Ang II and ALDH2 siRNA alone in MCECs (Fig. 4N & 4O). On the other hand, treatment with 10 μM Alda 1 significantly increased VEGFR1 protein levels compared to control, Ang II and ALDH2 siRNA alone (P < .005 vs control, P < .0005 vs Ang II, P < .0005 vs ALDH2 siRNA) (Fig. 4N & 4O). Alda 1 pretreatment rescued the Ang II mediated decrease in VEGFR1 levels which is significantly higher than the individual effects of Ang II and ALDH2 siRNA as well as the combined effect of Ang II and ALDH2 siRNA (P < .05 vs control, P < .005 vs Ang II, P < .005 vs ALDH2 siRNA and P < .0005 vs ALDH2 siRNA + Ang II) (Fig. 4N & 4O).
Treatment with 10 μM Ang II caused a significant reduction in VEGFR2 protein levels compared with control (P < .05) in cultured MCECs after 2 hours (Fig. 4N & 4P). Similarly, treatment with ALDH2 siRNA alone also caused a significant reduction in VEGFR2 protein levels relative to control (P < .05) after 2 hours as well (Fig. 4N & 4P). ALDH2 siRNA pretreatment enhanced Ang II-induced decrease in VEGFR2 protein levels compared with Ang II and ALDH2 siRNA alone in MCECs (P < .0005 vs control, P < .005 vs both Ang II and ALDH2 siRNA alone) (Fig. 4N & 4P). However, treatment with 10 μM Alda 1 significantly increased VEGFR2 protein levels compared to control, Ang II and ALDH2 siRNA alone (P < .05 vs control and P < .005 vs both Ang II ALDH2 siRNA alone) (Fig. 4N & 4P). Alda 1 pretreatment prior to Ang-II challenge rescued the Ang II mediated decrease in VEGFR2 levels in MCECs which is significantly higher than the individual effects of Ang II and ALDH2 siRNA as well as the combined effect of Ang II and ALDH2 siRNA in VEGFR2 protein (P < .05 vs both Ang II and ALDH2 siRNA alone and P < .0005 vs ALDH2 siRNA + Ang II) (Fig. 4N & 4P).
Our data also showed that treatment with Ang II significantly increased AT2R protein (P < .05) levels compared with control in cultured MCECs after 2 hours (Fig. 4N & 4Q). Likewise, treatment with ALDH2 siRNA alone also caused a significant increase in AT2R protein (P < .05) levels compared with control after 2 hours as well (Fig. 4N & 4Q). ALDH2 siRNA pretreatment enhanced Ang II-induced increase in AT2R protein levels compared with Ang II alone (P < .005 vs control and P < .05 vs Ang II) in MCECs (Fig. 4N & 4Q). In contrast, Alda 1 treatment significantly decreased AT2R protein levels relative to control, Ang II and ALDH2 siRNA alone (P < .05 vs control and P < .005 vs both Ang II ALDH2 siRNA alone) (Fig. 4N & 4Q). Alda 1 pretreatment prior to Ang II rescued the Ang II mediated increase in AT2R levels which is significantly lower than the individual effects of Ang II and ALDH2 siRNA as well as the combined effect of Ang II and ALDH2 siRNA in AT2R protein levels (P < .05 vs both Ang II and ALDH2 siRNA and P < .05 vs ALDH2 siRNA + Ang II) (Fig. 4N & 4Q).
3.5. Ang II, disulfiram and Alda 1 did not decrease the viability of mouse coronary ECs
10 μM Ang II did not show any significant effect in MCEC viability after 2 hours (Fig. 5). Additionally, treatment with 2.5 μM DSF and 10 μM Alda 1 alone or in combination with Ang II did not alter MCECs viability (Fig. 5).
Fig. 5. Viability test for Ang II treated MCECs.
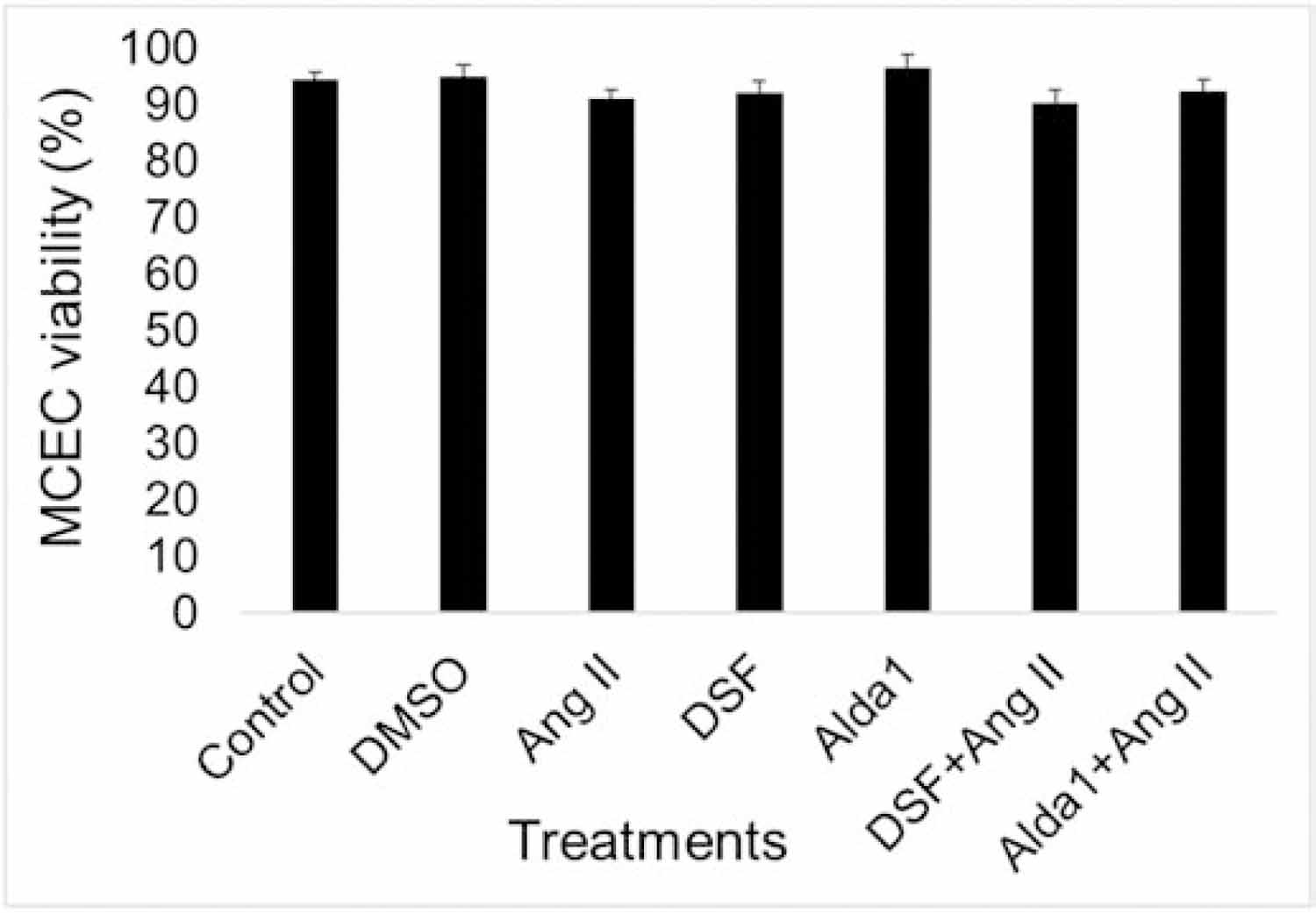
Trypan blue assay for Ang II (10 μM) treated MCECs after 2 hours. All data were represented as means ± SEM, n = 6.
3.6. Blockade of angiotensin II type 2 receptor attenuates Ang II-induced decrease in coronary angiogenesis via rescuing impaired expression of VEGFR1, VEGFR2 and AT2R
Treating MCECs with Losartan before Ang II challenge did not alter tube formation significantly compared to Ang II alone (Fig. 6A – 6C & 6H). However, PD0123319 pretreatment prior to Ang II challenge rescued Ang II mediated decrease in tube formation (Fig. 6A, 6B & 6F). Pretreatments with Losartan and PD0123319 before Ang II challenge also rescued Ang II mediated decrease in tube formation in MCECs (Fig. 6B, 6G & 6H).
Fig. 6. Role of Ang II receptors inhibition in angiogenesis and mRNA/protein expression of VEGFR1, VEGFR2 and AT2R in MCECs.
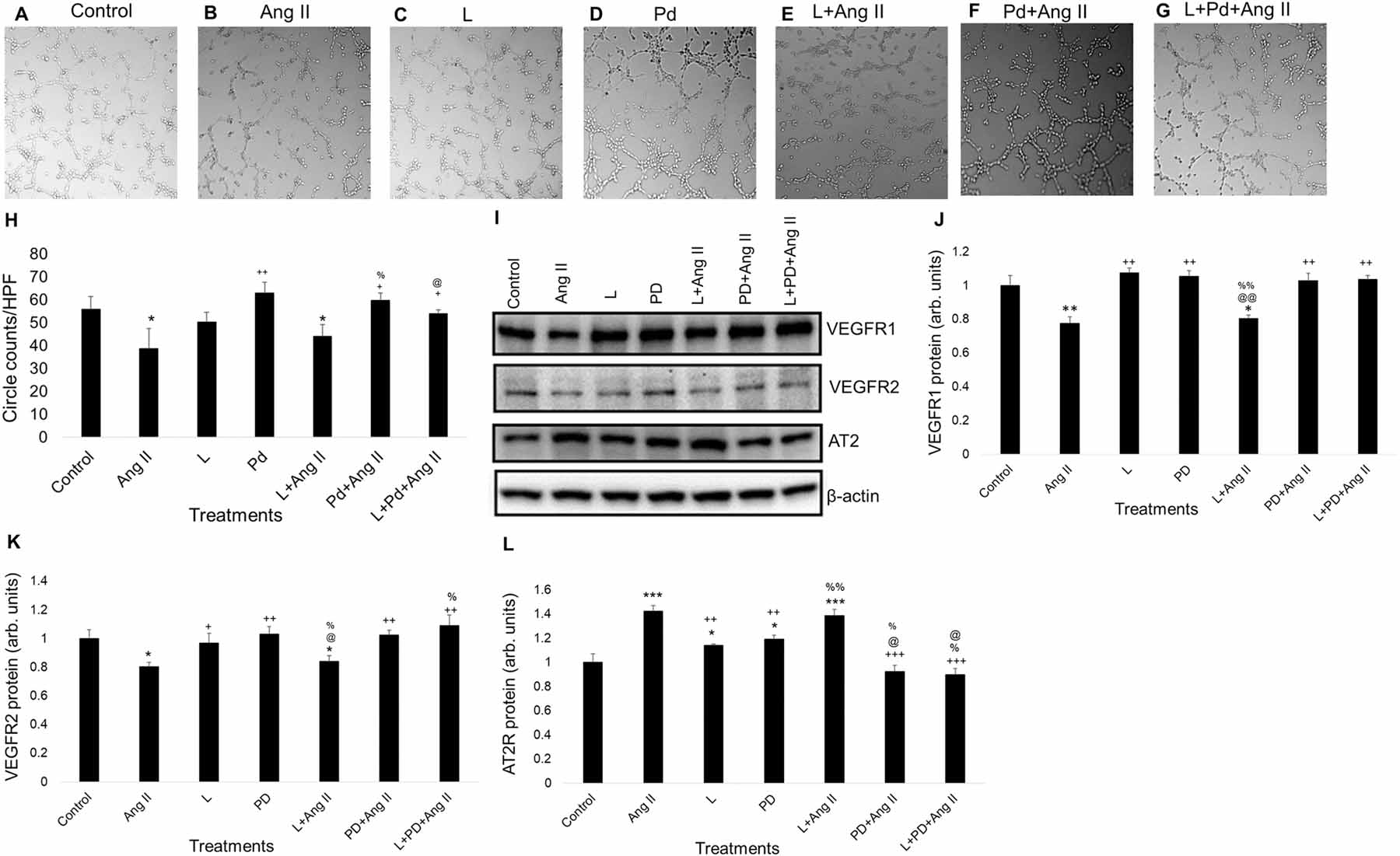
(A)-(G) Representative micrographs (PCM) of tube formation on Matrigel with losartan/PD0123319 pretreatment followed by Ang II treatment for 2 h. Magnification: 10x. A to D show tube formation with control, Ang II (10 μM), losartan (1 μM) and PD0123319 (1 μM)-treated MCECs. E shows tube formation with losartan pretreatment followed by Ang II treatment for 2 h. F shows tube formation with PD0123319 pretreatment followed by Ang II treatment for 2 h. G shows tube formation with losartan and PD0123319 pretreatment followed by Ang II treatment for 2 h. (H) Quantitative data of circles with different treatments stated in A through G. (I) Representative WB band images of VEGFR1, VEGFR2, AT2R and β-actin proteins in MCECs with losartan/PD0123319 pretreatment followed by Ang II treatment for 2 h. (J) Quantification of WB data of VEGFR1 protein levels stated in panel I. (K) Quantification of WB data of VEGFR2 protein levels stated in panel I. (L) Quantification of WB data of AT2R protein levels stated in panel I. n=6 for each group. Each bar represents mean ± SEM. *p < .05 vs control, **p < .005 vs control, and ***p < .005 vs control, +p < .05 vs Ang II, ++p < .005 vs Ang II, +++p < .0005 vs Ang II, %p < .05 vs L, %%p < .005 vs L, @p < .05 vs PD and @@p < .005 vs PD. HPF = high power field, L = Losartan and PD/Pd = PD0123319.
Losartan pretreatment prior to Ang II challenge did not alter VEGFR1 protein levels compared to Ang II alone (Fig. 6I & 6J). However, PD0123319 pretreatment prior to Ang II challenge rescued Ang II mediated decrease in VEGFR1 protein levels (Fig. 6I & 6J). Pretreatments with Losartan and PD0123319 before Ang II challenge rescued Ang II mediated decrease in VEGFR1 protein levels in MCECs (Fig. 6I & 6J).
Pretreatment with Losartan prior to Ang II challenge did not alter VEGFR2 protein levels compared to Ang II alone (Fig. 6I & 6K). PD0123319 pretreatment to MCECs prior to Ang II challenge rescued Ang II mediated decrease in VEGFR2 protein (Fig. 6I & 6K). Pretreatments with Losartan and PD0123319 prior to Ang II challenge also rescued Ang II mediated decrease in VEGFR2 protein levels in MCECs (Fig. 6I & 6K).
Pretreatment with Losartan before Ang II challenge did not alter AT2R protein levels compared to Ang II alone (Fig. 6I & 6L). However, pretreatment with PD0123319 before Ang II challenge rescued Ang II mediated increase in AT2R mRNA and protein levels (Fig. 5J, 5K & 5N). Pretreatments with Losartan and PD0123319 before Ang II challenge also rescued Ang II mediated increase in AT2R protein levels in MCECs (Fig. 6I & 6L).
3.7. Effect of ALDH2 activity on Losartan or PD0123319 mediated inhibition of Ang II receptors.
We found that treating MCECs with DSF and Losartan or PD0123319 showed significant decrease in tube formation compared with controls after 2 hours (P < .0005) (Fig. 7A – 7D & 7K). Likewise, treating MCECs with Alda 1 prior to Losartan or PD0123319 treatment for 2 hours showed significant increase in tube formation compared with control (P < .05) (Fig. 7A, 7B, 7E, 7F & 7K). Additionally, pretreating MCECs with DSF and Losartan following treatment with Ang II significantly decreased tube formation compared with control after 2 hours (Fig. 7A, 7G & 7K). Although, there is no significant change in tube formation compared with the combined effect of DSF and Ang II (P < .0001 vs control) (Fig. 3F, 3H, 7A, 7G & 7K). However, pretreating MCECs with DSF and PD0123319 following treatment with Ang II significantly increased tube formation compared with the combined effects of DSF+ Losartan, and Ang II. However, there is no significant change in tube formation compared with the combined effect of DSF and PD0123319 (P < .05 vs DSF + L + Ang II) (Fig. 7D, 7H & 7K). Additionally, pretreating MCECs with Alda 1 and Losartan prior to Ang II significantly decreased tube formation compared with control after 2 hours (Fig. 7A, 7I & 7K). There is no significant change in tube formation compared with the combined effect of DSF and Ang II (P < .0001 vs control) (Fig. 3G, 3H, 7A, 7I & 7K). However, pretreating MCECs with Alda 1 and PD0123319 prior to Ang II challenge significantly increased tube formation (P < .005) compared with the combined effect of Alda 1, Losartan and Ang II (Fig. 7I, 7J & 7K). However, there is no significant change in tube formation compared with the combined effect of Alda 1 and PD0123319 (P < .005 vs DSF + L + Ang II) (Fig. 7F, 7J & 7K).
Fig. 7. Role of ALDH2 and Ang II receptors in the alterations of angiogenesis and mRNA/protein expression of VEGFR1, VEGFR2 and AT2R in MCECs.
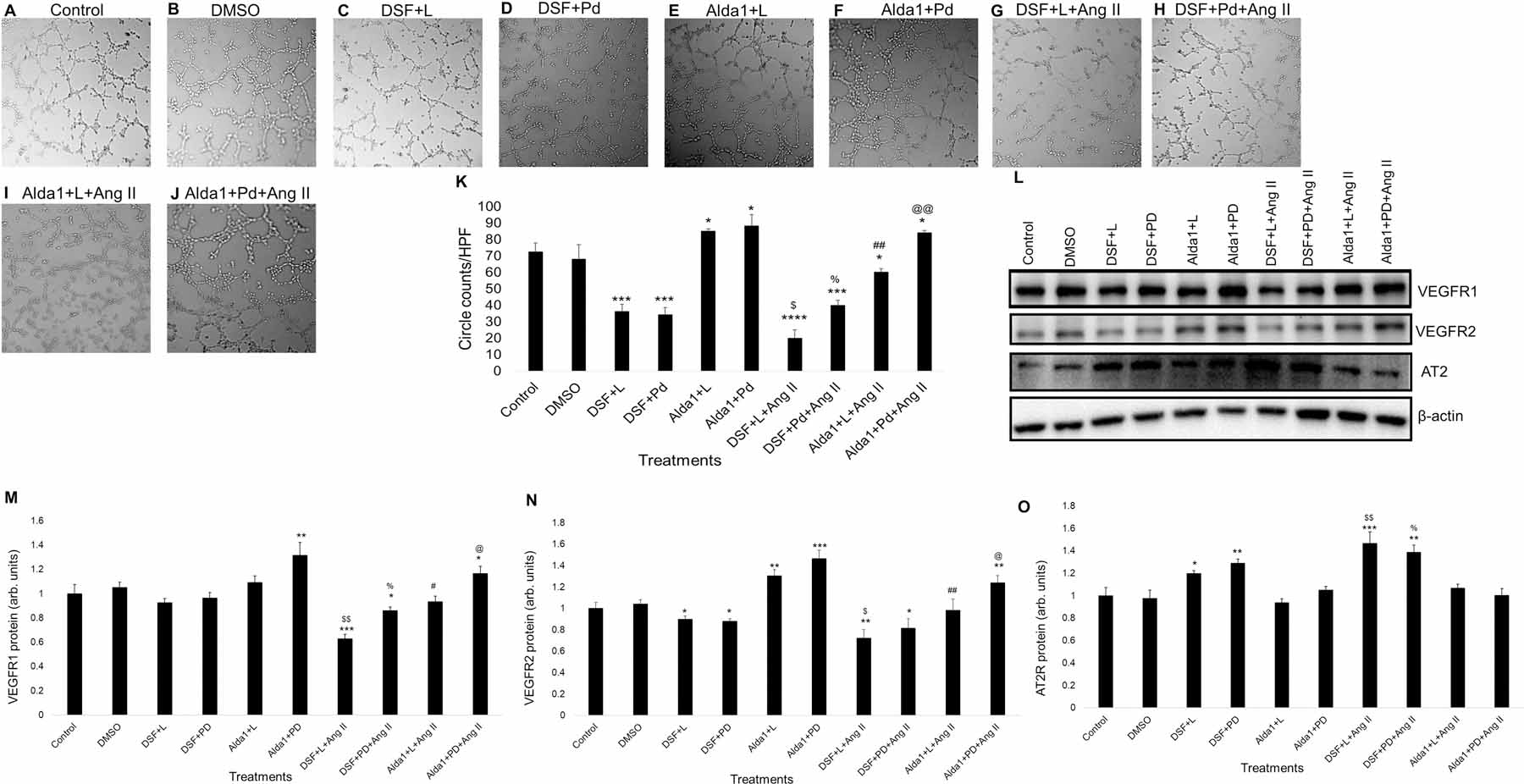
(A)-(J) Representative micrographs of tube formation on Matrigel with DSF/Alda1/Losartan / PD0123319 pretreatment followed by Ang II treatment for 2 h. Magnification: 10x. A and B show tube formation with control and DMSO (vehicle)-treated MCECs respectively. C shows tube formation with DSF (2.5 μM) pretreatment followed by Losartan (1 μM) treatment. D shows tube formation with DSF (2.5 μM) pretreatment followed by PD0123319 (1 μM) treatment. E shows tube formation with Alda 1 (10 μM) pretreatment followed by Losartan (1 μM) treatment. F shows tube formation with Alda 1 (10 μM) pretreatment followed by PD0123319 (1 μM) treatment. G shows tube formation with DSF (2.5 μM) and Losartan (1μM) pretreatment followed by Ang II (10 μM) treatment. H shows tube formation with DSF (2.5 μM) and PD0123319 (1μM) pretreatment followed by Ang II (10 μM) treatment. I shows tube formation with Alda 1 (10 μM) and Losartan (1μM) pretreatment followed by Ang II (10 μM) treatment. J shows tube formation with Alda 1 (10 μM) and PD0123319 (1μM) pretreatment followed by Ang II (10 μM) treatment. (K) Quantitative data of circles with different treatments stated in A through J. (L) Representative WB band images of VEGFR1, VEGFR2, AT2R and β-actin proteins in MCECs with DSF/Alda1/Losartan/ PD0123319 pretreatment followed by Ang II treatment for 2 h. (M) Quantification of WB data of VEGFR1 protein levels stated in panel L. (N) Quantification of WB data of VEGFR2 protein levels stated in panel L. (O) Quantification of WB data of AT2R protein levels stated in panel L. n=6 for each group. Each bar represents mean ± SEM. *p < .05 vs control, **p < .005 vs control, ***p < .0005 vs control, ****p < .0001 vs control, $p < .05 vs DSF + L, $ $p < .005 vs DSF + L, %p < .05 vs DSF + L + Ang II, %%p < .005 vs DSF + L + Ang II, #p < .05 vs Alda 1 + L, ##p < .005 vs Alda 1 + L, @p < .05 vs Alda 1 + L + Ang II and @@p < .005 vs Alda 1 + L + Ang II. HPF = high power field, DSF = disulfiram, L = losartan and PD/Pd = PD0123319.
Treating MCECs with DSF and then Losartan or PD0123319 did not show any significant change in VEGFR1 protein levels compared with DSF alone (Fig. 3O, 3P, 7L & 7M). Similarly, treating MCECs with Alda 1 and then Losartan or PD0123319 for 2 hours did not show any significant change in protein levels compared with Alda 1 alone (Fig. 3O, 3P, 7L & 7M). Additionally, MCECs pretreated with DSF and then Losartan prior to Ang II challenge significantly decreased VEGFR1 protein (P < .0005) levels compared with control (Fig. 7L & 7M). Even though, there is no significant change in protein levels compared with the combined effect of DSF and Ang II (Fig. 3O, 3P, 7L & 7M). However, MCECs pretreated with DSF and then PD0123319 prior to Ang II significantly increased VEGFR1 protein levels compared with the combined effects of DSF, Losartan and Ang II after 2 hours (P < .05) (Fig. 7L & 7M). Even though, there is no significant change in VEGFR1 protein levels compared with the combined effects of DSF and PD0123319 (Fig. 7L & 7M). Additionally, MCECs pretreated with Alda 1 and then Losartan prior to Ang II challenge significantly decreased VEGFR1 protein (P< .05) levels compared with the combined effect of Alda 1 and then Losartan (Fig. 7L & 7M). However, MCECs pretreated with Alda 1 and then PD0123319 prior to Ang II challenge significantly increased VEGFR1 protein levels (P < .05) compared with the combined effects of Alda 1, Losartan and Ang II (Fig. 7L & 7M). There is no significant change in VEGFR1 protein levels compared with the combined effect of Alda 1 and PD0123319 (Fig. 7L & 7M).
Challenging MCECs with DSF and Losartan or PD0123319 did not show any significant change in VEGFR2 protein levels compared with DSF alone (Fig. 3O, 3Q, 7L & 7N). Even though it showed significant decrease in VEGFR2 protein levels compared with control (P < .05) (Fig. 7L & 7N). Similarly, challenging MCECs with Alda 1 and then Losartan or PD0123319 for 2 hours did not show any significant change in VEGFR2 protein levels compared with Alda 1 alone (Fig. 3O, 3Q, 7L & 7N), even though it showed significant increase in VEGFR2 protein (P < .005 for Alda 1+Losartan and P < .0005 for Alda 1+ PD0123319) levels compared with control (Fig. 7L & 7N). Additionally, MCECs pretreated with DSF and then Losartan prior to Ang II treatment significantly decreased VEGFR2 protein (P < .005) levels compared with control (Fig. 7L & 7N) but not in comparison with the combined effects of DSF and Ang II (Fig. 3O, 3Q, 7L & 7N). DSF and then PD0123319 pretreatments before Ang II treatment significantly decreased VEGFR2 protein levels (P < .05) compared with control (Fig. 7L & 7N), but not in comparison to the combined effects of DSF and PD0123319 (Fig. 7L & 7N). Additionally, pretreating MCECs with Alda 1 and then Losartan before Ang II challenge significantly decreased VEGFR2 protein (P< .005) levels compared to the combined effect of Alda 1 and Losartan (Fig. 7L & 7N). However, pretreating MCECs with Alda 1 and then PD0123319 following treatment with Ang II for 2 hours significantly increased VEGFR2 protein (P < .05) levels compared with the combined effects of Alda 1, Losartan and Ang II but no significant change compared to the combined effects of Alda 1 and PD0123319 (Fig. 7L & 7N).
DSF and then Losartan / PD0123319 pretreatment to MCECs did not show any significant change in AT2R protein levels compared to DSF alone (Fig. 3O, 3R, 7L & 7O). Similarly, pretreating MCECs with Alda 1 and Losartan / PD0123319 did not show any significant change in AT2R protein levels compared to Alda 1 alone (Fig. 3O, 3R, 7L & 7O). Additionally, pretreating MCECs with DSF and then Losartan bprior to Ang II treatment significantly increased AT2R protein (P < .0005) levels compared with control (Fig. 7L & 7O). but no significant change compared with the combined effect of DSF and Ang II (Fig. 3O, 3R, 7L & 7O). Additionally, pretreating MCECs with Alda 1 and then Losartan prior to treating with Ang II did not show significant change in AT2R protein levels compared with the combined effects of Alda 1 and Losartan (Fig. 7L & 7O).
4. DISCUSSION
This study showed that Ang II dose-dependently decreased angiogenesis which has been evident from circle counts indicating reduced blood vessel formation as evaluated by tube formation assay.
Inhibition of ALDH2 activity pharmacologically with DSF or genetically with the use of ALDH2 siRNA decreased angiogenesis compared to control, similar to that of Ang II-mediated decrease. Additionally, pretreatment with DSF or ALDH2 siRNA exacerbated Ang II-mediated decrease in angiogenesis. On the other hand, activation of ALDH2 by Alda 1 pretreatment increased angiogenesis compared with control. Alda 1 also rescued Ang II-mediated decrease in angiogenesis.
We wanted to confirm whether the decrease in angiogenesis with tube formation assay is due to Ang II and / or DSF-induced cell death. However, the cell viability assay results confirmed that treatment of MCECs with Ang II / DSF alone or the combination of both did not alter cell viability.
To determine the involvement of specific Ang II receptors in Ang II-induced angiogenesis, we pharmacologically inhibited AT1R with Losartan and AT2R with PD0123319. We found that inhibition of AT1R did not alter Ang II mediated decrease in MCECs angiogenesis. However, inhibition of AT2R attenuated Ang II-mediated decrease in angiogenesis. Though Ang II is implicated in altering endothelial cell function including angiogenesis [30], the exact molecular signaling mechanism of Ang II-mediated coronary angiogenesis is unclear. Based on the pharmacological study, it appears AT2R is involved in the CEC proliferation (ref 29) in vitro. Thus, first we checked the transcript and protein expression levels of AT2R. Additionally, AT2R inhibition attenuated ang II-induced impairment of the levels of signaling proteins required for angiogenesis such as VEGFR1 and VEGFR2, but not with AT1R inhibition.
Paquin-Veillette et al, showed that deletion of AT2R downregulates SHP-1 activity followed by the restoration of VEGF actions that leads to increased angiogenesis in diabetic mice [31]. In contrast, deficiency of AT2R was shown to be associated with impaired production of VEGF and thereafter decreases angiogenesis in tumor malignancy [32]. This may be due to the difference between normal and tumor angiogenesis.
In this study we found that inhibition of ALDH2 activity with DSF or ALDH2 siRNA exacerbated Ang II-mediated increase in antiangiogenic AT2R transcript/protein levels. Additionally, we found that activation of ALDH2 with Alda 1 attenuated Ang II-mediated increase in AT2R transcript/protein levels. Our study also showed that inhibition of ALDH2 activity with DSF or ALDH2 siRNA potentiated Ang II-mediated increase in 4HNE-protein adduct levels in MCECs. Based on this finding and combined pharmacological interventions with AT2R inhibitor and ALDH2 modulators, we postulate that 4HNE may be involved in the activation of antiangiogenic AT2R activation. It is currently unknown how 4HNE elicits this effect whether by increasing the transcription or decrease the protein degradation by affecting proteasomal function. However, tt is reported that 4HNE can cause proteotoxicity and affect cardiovascular function [33].
There are contradictory findings about the role of VEGFR1, an important transmembrane tyrosine kinase receptor for VEGF, in coronary angiogenesis. Few studies showed that activation of VEGFR1 with VEGF, activates cyclic guanosine monophosphate-dependent protein kinase-1 signaling pathways followed by the formation of new blood vessels and regression of hypertrophy [34, 35]. However, some other studies showed that ablation of VEGFR1 increased angiogenesis in mice [36, 37]. From our current study, we found that inhibition of ALDH2 activity with DSF or ALDH2 siRNA exacerbated Ang II-mediated decrease in proangiogenic VEGFR1 levels. This study also showed that activation of ALDH2 with Alda 1 rescued Ang II-mediated decrease in VEGFR1 levels. In this study, we did not attempt to investigate the downstream signaling cascades of VEGFR1 in vascular ECs which are not well characterized yet.
Activation of VEGFR2 with VEGF, is a prominent signaling pathway in ECs that involved in EC migration, proliferation, survival and sprouting of new blood vessels, the required stages of angiogenesis [38]. In our previous studies we also showed that the proangiogenic factor, VEGFR2 which is involved in MCECs migration as well as angiogenesis is downregulated with the inhibition of ALDH2 activity by DSF [24, 26]. From our current study, we found that inhibition of ALDH2 activity with DSF or ALDH2 siRNA exacerbated Ang II-mediated decrease in proangiogenic VEGFR2 levels. Additionally, activation of ALDH2 with Alda 1 attenuated Ang II-mediated decrease in VEGFR2 levels. VEGFR2 mediates phosphorylation of AKT and which in turn phosphorylates endothelial nitric oxide synthase (eNOS) and thus releases nitric oxide (NO). Then, NO activates soluble guanylyl cyclase (sGC) which releases cGMP and then cGMP activates protein kinase G (PKG) to cause of migration of ECs. Parallelly, PKG can activate extracellular signal regulated kinase (ERK) ½ to cause EC proliferation via Ras-Raf-MEK-pathway [39]. EC proliferation and migration are important stages of coronary angiogenesis; however, they were not individually investigated in this study, which is a limitation. Furthermore, changes in those downstream pathways of VEGFR2 signaling where eNOS acts as a central molecule were not examined as well, which is another key limitation. Finally, reaffirmation of our current findings in human ECs could have been increased the translational potential of this study which is also a limitation.
In conclusion, we state that Ang II decreases MCEC angiogenesis through the activation of AT2R followed by the downregulation of VEGFR1 and VEGFR2 expression and upregulation of AT2R expression in cultured MCECs. We further demonstrated that activation of ALDH2 activity can rescue the coronary angiogenesis by attenuating the inhibitory effect of Ang II through upregulation of VEGFR1 and VEGFR2 and downregulation of AT2R expression in cultured MCECs. Whereas, inhibition of ALDH2 activity can exacerbate the inhibitory effect of Ang II in angiogenesis through downregulation of VEGFR1 and VEGFR2 and upregulation of AT2R expression in cultured MCECs. Since Ang II levels are increased in cardiovascular tissues in patients with chronic metabolic diseases such as diabetes, hypertension and metabolic syndromes [40–42]. ALDH2 can be used as an important therapeutic target to alleviate defective coronary angiogenesis in chronic pathological states such as cardiomyopathy or heart failure with preserved ejection fraction in diabetes.
Highlights.
Angiotensin II-induced decrease in coronary angiogenesis is mediated via angiotensin type 2 receptor.
Inhibition of ALDH2 activity exacerbates angiotensin II-induced decrease in coronary angiogenesis.
Increasing ALDH2 activity attenuates angiotensin II-induced decrease in coronary angiogenesis.
Angiotensin II-induced decrease in coronary angiogenesis is associated with VEGFR1 and VEGFR2 downregulation.
Acknowledgments:
Funding Source: This work was partially supported by NHLBI-NIH grant 1R01HL139877–01A1 and an internal grant from Henry Ford Health System A10249 (SSP) and Departmental Discretionary Fellowship from Department of Physiology, Wayne State University (BR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability: All relevant data are within the paper. All data are fully available without restriction.
Declaration of conflicting interests:
The Author(s) declare(s) that there is no conflict of interest.
Disclosures:
None
REFERENCES
- 1.Potente M, Gerhardt H, and Carmeliet P, Basic and therapeutic aspects of angiogenesis. Cell, 2011. 146(6): p. 873–87. [DOI] [PubMed] [Google Scholar]
- 2.Timar J, et al. , Angiogenesis-dependent diseases and angiogenesis therapy. Pathol Oncol Res, 2001. 7(2): p. 85–94. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P and Jain RK, Molecular mechanisms and clinical applications of angiogenesis. Nature, 2011. 473(7347): p. 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carbajo-Lozoya J, et al. , Angiotensin II modulates VEGF-driven angiogenesis by opposing effects of type 1 and type 2 receptor stimulation in the microvascular endothelium. Cell Signal, 2012. 24(6): p. 1261–9. [DOI] [PubMed] [Google Scholar]
- 5.Nakashima H, et al. , Angiotensin II regulates vascular and endothelial dysfunction: recent topics of Angiotensin II type-1 receptor signaling in the vasculature. Curr Vasc Pharmacol, 2006. 4(1): p. 67–78. [DOI] [PubMed] [Google Scholar]
- 6.Escobar E, et al. , Angiotensin II, cell proliferation and angiogenesis regulator: biologic and therapeutic implications in cancer. Curr Vasc Pharmacol, 2004. 2(4): p. 385–99. [DOI] [PubMed] [Google Scholar]
- 7.Bian F, et al. , Reactive oxygen species mediate angiotensin II-induced transcytosis of low-density lipoprotein across endothelial cells. Int J Mol Med, 2017. 39(3): p. 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mihalas BP, et al. , The lipid peroxidation product 4-hydroxynonenal contributes to oxidative stress-mediated deterioration of the ageing oocyte. Sci Rep, 2017. 7(1): p. 6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhong H and Yin H, Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox Biol, 2015. 4: p. 193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H and Forman HJ, Signaling by 4-hydroxy-2-nonenal: Exposure protocols, target selectivity and degradation. Arch Biochem Biophys, 2017. 617: p. 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mihalas BP, et al. , Oxidative damage in naturally aged mouse oocytes is exacerbated by dysregulation of proteasomal activity. J Biol Chem, 2018. 293(49): p. 18944–18964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohki K, et al. , Angiotensin II Type 1 Receptor-associated Protein Inhibits Angiotensin II-induced Insulin Resistance with Suppression of Oxidative Stress in Skeletal Muscle Tissue. Sci Rep, 2018. 8(1): p. 2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minas JN, et al. , Angiotensin and mineralocorticoid receptor antagonism attenuates cardiac oxidative stress in angiotensin II-infused rats. Clin Exp Pharmacol Physiol, 2015. 42(11): p. 1178–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee SH, et al. , Angiotensin II-Induced Oxidative Stress in Human Endothelial Cells: Modification of Cellular Molecules through Lipid Peroxidation. Chem Res Toxicol, 2019. 32(7): p. 1412–1422. [DOI] [PubMed] [Google Scholar]
- 15.Suzuki H, et al. , Current understanding of the mechanism and role of ROS in angiotensin II signal transduction. Curr Pharm Biotechnol, 2006. 7(2): p. 81–6. [DOI] [PubMed] [Google Scholar]
- 16.Arrieta O, et al. , Association between AT1 and AT2 angiotensin II receptor expression with cell proliferation and angiogenesis in operable breast cancer. Tumour Biol, 2015. 36(7): p. 5627–34. [DOI] [PubMed] [Google Scholar]
- 17.Pei N, et al. , Angiotensin II type 2 receptor promotes apoptosis and inhibits angiogenesis in bladder cancer. J Exp Clin Cancer Res, 2017. 36(1): p. 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen CH, Sun L, and Mochly-Rosen D, Mitochondrial aldehyde dehydrogenase and cardiac diseases. Cardiovasc Res, 2010. 88(1): p. 51–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nene A, et al. , Aldehyde dehydrogenase 2 activation and coevolution of its epsilonPKC-mediated phosphorylation sites. J Biomed Sci, 2017. 24(1): p. 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen CH, et al. , Targeting aldehyde dehydrogenase 2: new therapeutic opportunities. Physiol Rev, 2014. 94(1): p. 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X, et al. , Mitochondrial Aldehyde Dehydrogenase 2 Regulates Revascularization in Chronic Ischemia: Potential Impact on the Development of Coronary Collateral Circulation. Arterioscler Thromb Vasc Biol, 2015. 35(10): p. 2196–206. [DOI] [PubMed] [Google Scholar]
- 22.Pang J, et al. , Targeting acetaldehyde dehydrogenase 2 (ALDH2) in heart failure-Recent insights and perspectives. Biochim Biophys Acta Mol Basis Dis, 2017. 1863(8): p. 1933–1941. [DOI] [PubMed] [Google Scholar]
- 23.Pan G, et al. , ALDH2 Inhibition Potentiates High Glucose Stress-Induced Injury in Cultured Cardiomyocytes. J Diabetes Res, 2016. 2016: p. 1390861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roy B and Palaniyandi SS, Aldehyde dehydrogenase 2 inhibition potentiates 4-hydroxy-2-nonenal induced decrease in angiogenesis of coronary endothelial cells. Cell Biochem Funct, 2020. 38(3): p. 290–299. [DOI] [PubMed] [Google Scholar]
- 25.Livak KJ and Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 2001. 25(4): p. 402–8. [DOI] [PubMed] [Google Scholar]
- 26.Roy B, Sundar K, and Palaniyandi SS, 4-hydroxy-2-nonenal decreases coronary endothelial cell migration: Potentiation by aldehyde dehydrogenase 2 inhibition. Vascul Pharmacol, 2020. 131: p. 106762. [DOI] [PubMed] [Google Scholar]
- 27.Ganta VC, et al. , VEGF165b Modulates Endothelial VEGFR1-STAT3 Signaling Pathway and Angiogenesis in Human and Experimental Peripheral Arterial Disease. Circ Res, 2017. 120(2): p. 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kivela R, et al. , Endothelial Cells Regulate Physiological Cardiomyocyte Growth via VEGFR2-Mediated Paracrine Signaling. Circulation, 2019. 139(22): p. 2570–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stoll M, et al. , The angiotensin AT2-receptor mediates inhibition of cell proliferation in coronary endothelial cells. J Clin Invest, 1995. 95(2): p. 651–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dang H, et al. , Atorvastatin reverses the dysfunction of human umbilical vein endothelial cells induced by angiotensin II. Exp Ther Med, 2018. 16(6): p. 5286–5297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paquin-Veillette J, et al. , Deletion of AT2 Receptor Prevents SHP-1-Induced VEGF Inhibition and Improves Blood Flow Reperfusion in Diabetic Ischemic Hindlimb. Arterioscler Thromb Vasc Biol, 2017. 37(12): p. 2291–2300. [DOI] [PubMed] [Google Scholar]
- 32.Clere N, et al. , Deficiency or blockade of angiotensin II type 2 receptor delays tumorigenesis by inhibiting malignant cell proliferation and angiogenesis. Int J Cancer, 2010. 127(10): p. 2279–91. [DOI] [PubMed] [Google Scholar]
- 33.Wu B, et al. , Aldehyde dehydrogenase 2 activation in aged heart improves the autophagy by reducing the carbonyl modification on SIRT1. Oncotarget, 2016. 7(3): p. 2175–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gogiraju R, Bochenek ML, and Schafer K, Angiogenic Endothelial Cell Signaling in Cardiac Hypertrophy and Heart Failure. Front Cardiovasc Med, 2019. 6: p. 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hou J and Kang YJ, Regression of pathological cardiac hypertrophy: signaling pathways and therapeutic targets. Pharmacol Ther, 2012. 135(3): p. 337–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ho VC and Fong GH, Vasculogenesis and Angiogenesis in VEGF Receptor-1 Deficient Mice. Methods Mol Biol, 2015. 1332: p. 161–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Z and Zhou B, Accelerated coronary angiogenesis by vegfr1-knockout endocardial cells. PLoS One, 2013. 8(7): p. e70570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abhinand CS, et al. , VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J Cell Commun Signal, 2016. 10(4): p. 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bir SC, et al. , Emerging role of PKA/eNOS pathway in therapeutic angiogenesis for ischaemic tissue diseases. Cardiovasc Res, 2012. 95(1): p. 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singh VP, et al. , Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes, 2008. 57(12): p. 3297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yokota R, et al. , Intra-Renal Angiotensin Levels Are Increased in High-Fructose Fed Rats in the Extracorporeal Renal Perfusion Model. Front Physiol, 2018. 9: p. 1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lavrentyev EN, Estes AM, and Malik KU, Mechanism of high glucose induced angiotensin II production in rat vascular smooth muscle cells. Circ Res, 2007. 101(5): p. 455–64. [DOI] [PubMed] [Google Scholar]