

Abstract
We seek to manipulate gene function here through CRISPR-Cas9 editing of cis-regulatory sequences, rather than the more typical mutation of coding regions. This approach would minimize secondary effects of cellular responses to nonsense mediated decay pathways or to mutant protein products by premature stops. This strategy also allows for reducing gene activity in cases where a complete gene knockout would result in lethality, and it can be applied to the rapid identification of key regulatory sites essential for gene expression. We test this strategy here with genes of known function as a proof of concept, and then applied it to examine the upstream genomic region of the germline gene Nanos2 in the sea urchin, Strongylocentrotus purpuratus. We first used CRISPR-Cas9 to target established genomic cis-regulatory regions of the skeletogenic cell transcription factor, Alx1, and the TGF-β signaling ligand, Nodal, which produce obvious developmental defects when altered in sea urchin embryos. Importantly, mutation of cis-activator sites (Alx1) and cis-repressor sites (Nodal) result in the predicted decreased and increased transcriptional output, respectively. Upon identification of efficient gRNAs by genomic mutations, we then used the same validated gRNAs to target a deadCas9-VP64 transcriptional activator to increase Nodal transcription directly. Finally, we paired these new methodologies with a more traditional, GFP reporter construct approach to further our understanding of the transcriptional regulation of Nanos2, a key gene required for germ cell identity in S. purpuratus. With a series of reporter assays, upstream Cas9-promoter targeted mutagenesis, coupled with qPCR and in situ RNA hybridization, we concluded that the promoter of Nanos2 drives strong mRNA expression in the sea urchin embryo, indicating that it’s primordial germ cell (PGC)-specific restriction may rely instead on post-transcriptional regulation. Overall, we present a proof-of-principle tool-kit of Cas9-mediated manipulations of promoter regions that should be applicable in most cells and embryos for which CRISPR-Cas9 is employed.
Introduction
The mechanisms of cell fate determination are typically interrogated by their gene regulatory networks (GRNs), as they summarize analyses of pathways governing embryonic development (Davidson et al., 2002). A GRN is constructed from gene perturbation experiments to identify the function of transcription factors or signaling ligands which control cell fate decisions in an embryo (Davidson et al., 2002; Peter and Davidson, 2015; Saudemont et al., 2010). Currently, CRISPR-Cas9 genome editing technology is a preferred approach to test and to construct GRNs. Typically, Cas9 and its associated guide RNA (gRNA) are used to mutate coding exons of the gene of interest. This approach is overall extremely successful in a wide variety of vertebrate and invertebrate animal (Gandhi et al., 2018; Lin and Su, 2016; Moreno-Mateos et al., 2015). However, it was recently demonstrated that nonsense mediated decay of a mutant transcript can affect gene expression and cell cycle progression secondarily and yield partial protein derivatives of unknown impact (Anderson et al., 2017; Tuladhar et al., 2019). To obviate these concerns, and to add an additional level of understanding in transcriptional control, we sought to test the efficacy of altering transcription by targeting non-coding, cis-regulatory regions flanking the gene of interest (Williams et al., 2018). To first assess the efficacy of CRISPR-Cas9 mutation on predicted cis-regulatory regions, we assayed two well characterized genes, Aristaless-like Homeobox 1 (Alx1) and the TGF-β family ligand, Nodal. Alx1 is a transcription factor that directs skeletogenesis in the embryo of the sea urchin S. purpuratus (Damle and Davidson, 2011; Ettensohn et al., 2003). Nodal is a TGF-β signaling ligand responsible for Left/Right and Dorsal-Ventral axis specification essential for the bilateral asymmetry in embryos (Aihara and Amemiya, 1999; Aihara and Amemiya, 2001; Duboc et al., 2004; Duboc et al., 2005; Flowers et al., 2004; Nam et al., 2007; Su, 2014). Importantly, Nodal signaling is also needed for the spatial restriction of germ cell factors in echinoderm embryos (Fresques and Wessel, 2018; Luo and Su, 2012). Both Alx1 and Nodal have previously been integrated into a gene regulatory network (GRN) that is well established (Damle and Davidson, 2011; Longabaugh et al., 2005; Range et al., 2007; Saudemont et al., 2010), and offer both activation and repression elements, making them tractable for initial testing of CRISPR-Cas9-mediated mutagenesis of cis-regulatory elements (Damle and Davidson, 2011; Nam et al., 2007). Since Alx1 and Nodal each regulate development through distinct pathways, and when mutated, yield unique and robust phenotypes, we reasoned that they were excellent targets for testing the efficacy of Cas9 cis-regulatory element mutagenesis (Duboc et al., 2005; Ettensohn et al., 2003). In the case of Alx1 expression, we targeted cis-elements essential for Alx1 transcription. In contrast, for Nodal expression, we targeted a predicted repressive site in the Nodal promoter and induced de-repression of Nodal gene activity.
We then applied this technology to test modification of germline gene function by targeting predicted cis-regulatory regions upstream of Nanos2. Nanos2, the sea urchin ortholog of the PGC-specific RNA binding protein Nanos, is a potent translational repressor of PGCs. In collaboration with Pumilio, Nanos protein binds specific sequences in the 3’UTR of mRNAs to enable translational repression, and/or transcript degradation (Parisi and Lin, 2000; Weidmann and Goldstrohm, 2012; Weidmann et al., 2016). The result of Nanos2 directed translational repression and degradation is the repression of somatic cell gene expression (Irish et al., 1989; Lai et al., 2012), formation of a developmental gradient (Kobayashi et al., 1996), and induction of quiescence within PGCs (Oulhen et al., 2017). Each of these Nanos dependent functions are essential for PGC identity and maintenance in all species tested.
Earlier work revealed that Nanos2 is essential for PGC maintenance in the sea urchin, S. purpuratus, and that its mRNA and protein both accumulate specifically within PGCs (Juliano et al., 2010; Oulhen et al., 2013; Wessel et al., 2014). Remarkably, Nanos2 overexpression in the sea urchin embryo has not been possible because of highly selective post-transcriptional and post-translational mechanisms (Campanale et al., 2019; Oulhen and Wessel, 2016b; Oulhen et al., 2013). We tested here genomic sequences of interest that contribute to its expression using conventional GFP-reporter assays as well as newly developed Cas9-mediated cis-element mutagenesis.
Materials and Methods
Animal Care, Embryo Injection and Culturing
Adult S. purpuratus animals were supplied by Pete Halmay of Pt. Loma Marine Invertebrate Lab (Lakeside, CA, email: peterhalmay@gmail.com). Animals are housed in 35ppt artificial seawater at 16 °C. Zygotes are injected via capillary needle, pressurized using an Eppendorf femtojet for injection of all constructs (Yaguchi, 2019). Embryos were cultured in filtered seawater collected from Woods Hole, Massachusetts, in Corning Nunc® IVF dishes at 16° C in a benchtop incubator. At designated stages of development, 50 embryos were collected for qPCR assays, several hundred for RNA in situ hybridization, and ten of each group for genotyping and PCR analysis for mutation efficiencies.
Analyses of Sequence Conservation
Binding Site Prediction.
For Alx1 and Nodal cis-regulatory analyses, minimum upstream regulatory sequences needed to drive specific expression were referenced from previous publications (Damle and Davidson, 2011; Nam et al., 2007). For Nanos2 cis-regulatory analyses, predicted transcription factor binding sites were chosen based on previous data from CHIP-qPCR analyses (Oulhen et al., 2019). All binding site motif predictions for Alx1, Nodal, and Nanos2, were performed using TFsiteSCAN software with parameters for validated amphibian transcription factor binding site data (http://www.ifti.org/cgi-bin/ifti/Tfsitescan.pl). All sequences were .FASTA files accessed from Echinobase.org, S. purpuratus genome version 3.1 (http://www.echinobase.org/)(Cameron et al., 2009).
Analysis of Sequence Conservation.
Conservation of echinoderm sequences in upstream regions was performed using MussaGL software (http://mussa.caltech.edu/mussa/wiki/MussaglBuild), accessed from Caltech.edu (Haeussler and Joly, 2011). MUSSA (multiple species sequence analysis) utilizes sliding windows to analyze conserved sequences between two sequences of DNA. For Nanos2 promoter analysis, alignments between S. purpuratus and H. pulcherrimus were conducted, as well as MussaGL analysis of the three different Nanos genes (Nanos1, Nanos2 and Nanos3) with genomic sequences from S. purpuratus. All genomic sequences were obtained from EchinoBase.org, S.purpuratus genome version 3.1(http://www.echinobase.org/Echinobase/jbrowse/).
CRISPR-Cas9 targeting genomic sites
gRNA design and synthesis.
gRNAs were designed using CRISPRscan (https://www.crisprscan.org/) software, and only those with no predicted off-target binding sites in the genome were selected for use in this study (Moreno-Mateos et al., 2015). For Alx1 and Nodal, the promoter region had been established in previous publications (Damle and Davidson, 2011; Nam et al., 2007). For Nanos2, an upstream region was selected from the 2.5kb of upstream genomic sequence. Genomic sequences upstream of genes Nodal, Alx1 and Nanos2 were retrieved from Echinobase.org, using the S.purpuratus genome version 3.1 (http://www.echinobase.org/Echinobase/). CRISPRscan guide RNAs were ordered as synthetic oligos from Eurofins.com with an accompanying tailing sequence described in CRISPRscan.org (Moreno-Mateos et al., 2015). DNA templates with a T7 promoter were generated using Gotaq Green Master Mix (Promega). gRNAs were synthesized following the Megashortscript T7 transcription kit from ThermoFisher and purified using Qiagen RNA column reagents (microRNA cleanup kit, Qiagen). Cas9 mRNA (1ug) and a pooled mixture of promoter-targeting gRNAs (2ug) were combined in a 1:2 ratio by mass and diluted with nuclease-free H2O and 10,000 MW dextran conjugated to Alexa Fluor 488 (Thermofisher) to a final concentration of 500ng/uL for injection into fertilized zygotes of S. purpuratus, (n=200 for each injection experiment) using parameters described (Yaguchi, 2019).
Overexpression of dCas9-VP64.
For use in echinoderm embryos, a synthetic FLAG VP64dCas9 in an expression plasmid was generated using Gibson assembly. For the fusion protein, the dCas9 sequence contained in the pcDNA-dCas9-VP64 Plasmid #47107 from Addgene (Addgene.com) was used as a template. The plasmid backbone for cloning was PCS2-GFP with an SP6 transcription site, and poly-A signal for mRNA expression in echinoderm embryos. For these experiments, mRNA of the dCas9VP64 fusion was injected in either a 1:2 ratio to gRNAs, or a 1:5 ratio by mass. Each of these experiments is denoted in subsequent analysis as “1:2” or “1:5.” The intact Cas9 mRNA and gRNA used in previous experiments was included as a positive control for Cas9/gRNA functionality.
Analyses of Genomic Mutations
Mutation identification, and gRNA efficiency calculations.
To determine efficiency of Cas9 mutation with each gRNA, individual embryos were collected in 200uL PCR tubes. Mutant and control embryos were collected in separate tubes and lysed in 12uL QuickExtract DNA extraction buffer from Lucigen.com. Individual lysates were run in a standard PCR cycle, using platinum Taq reagents from ThermoFisher. 10ng of purified PCR products were cloned into the PGEMT-EZ vector and transformed into XL1 E. coli for subsequent blue-white selection. From each single embryo, sequences representative of 7–10 colonies were analyzed to determine mutation efficiencies for each gRNA from DNA clones (Figure 1).
Figure 1. CRISPR Cas9 Promoter Analysis Workflow.
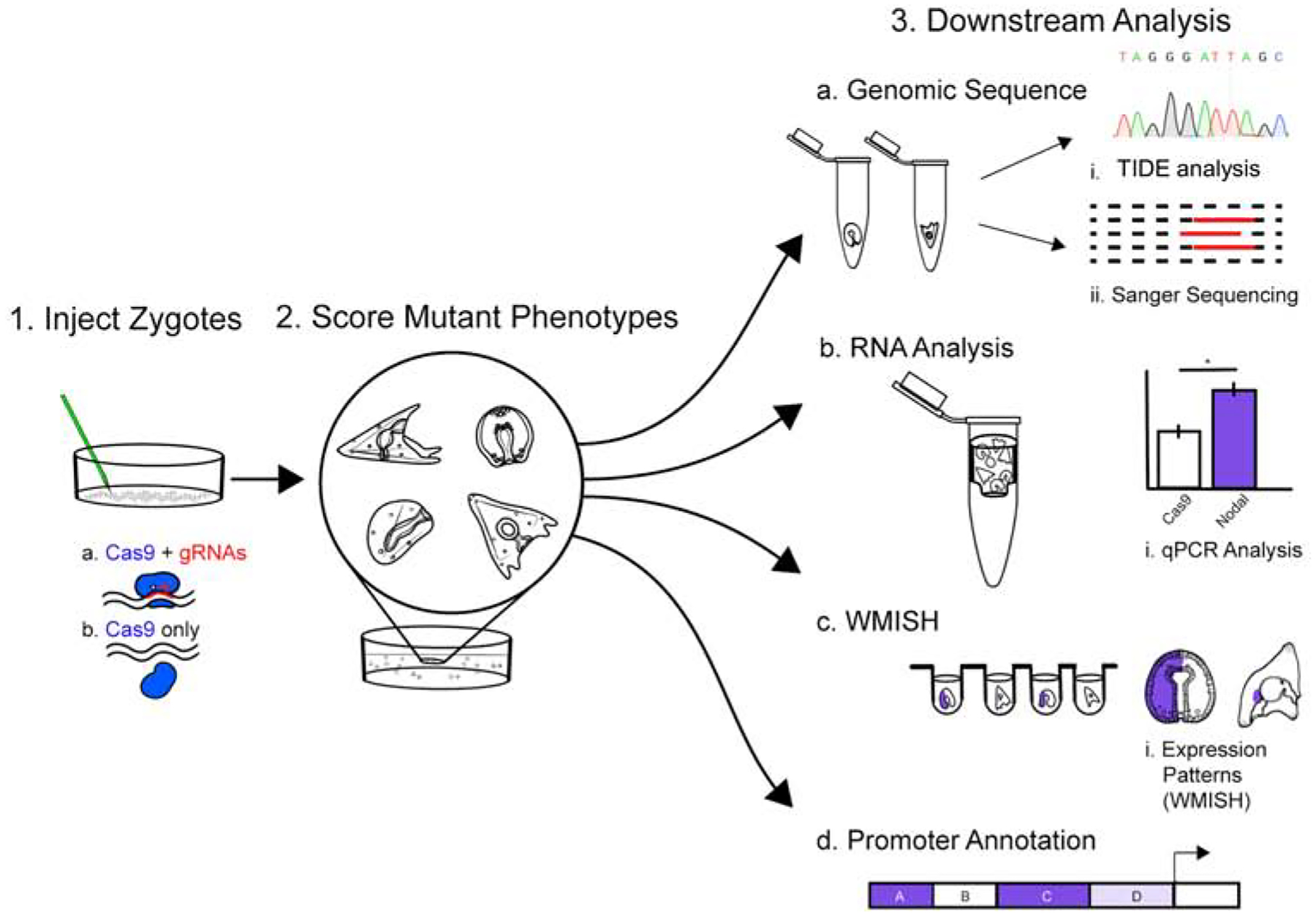
Step 1. Cas9 mRNA +/− gRNA is injected into individual zygotes of S.purpuratus. Cas9 mRNA alone serves as injection and vehicle control. Step 2. Injected embryos at desired developmental stages are collected and the phenotypes are scored. Step 3. Downstream analysis. a) Mutations are assessed from individual embryos (N=7) from each group, per each experiment, by TIDE analysis and Sanger sequencing of genomic PCR products; b) N = 50 embryos from each group are collected for qPCR analysis of transcriptional changes relative to Cas9 only controls. c) N = 100 embryos are collected and fixed for imaging, or whole-mount RNA in situ hybridization (WMISH) analysis. d) A map of the promoter and cis-regulatory regions is then constructed from these data.
TIDE analysis.
Single embryo PCR products were isolated using standard column DNA purification reagents and were Sanger sequenced as a population with the primers used to generate the PCR product. These sequences represented a mosaic of the mutant embryo. For TIDE analysis, spectral data (.ab1) files were downloaded and organized into Cas9 only (control spectra), and Cas9+gRNA spectra for three embryos selected at random (experimental). For visual analysis of mutations, spectra were aligned in SnapGene software (Figure 1). For quantification of mutations, the first gRNA sequence within the amplicon was entered into the TIDE web app (https://tide.nki.nl/) with an alignment region set to maximum boundaries, and p-value threshold at <0.001 for indel detection (Brinkman and van Steensel, 2019). For quantification of spectral decay following the gRNA cut site, the same parameters were used (Brinkman et al., 2014).
Expression Quantification by qPCR
For each cis-element analysis, the qPCR results represent the average expression level over 50 injected embryos. For CRISPR-Cas9 mutagenesis, 10,000 MW dextran conjugated to Alexa Fluor 488 (Thermofisher) is used as an injection indicator, and healthy embryos (N=50) were selected by green fluorescence. RNA was prepared using Qiagen RNeasy micro kit reagents (cat. 74004), and cDNA synthesized using the MAXIMA cDNA synthesis kit from ThermoFisher (cat.K1671). qPCR was performed in the Applied Biosystems 7300 real-time system, using SYBR green/ROX 2X Mastermix from ThermoFisher (cat. vFERK0222). All qPCR experiments are aliquoted in 96-well plates, with triplicate reactions for each sample. qPCR reactions are analyzed using the AB sequence detection software (version1.4) and Delta-Delta CT analysis for expression of each gene is performed relative to Cas9 injected control samples. Fold change is relative to a ubiquitin control primer, and significance is calculated using two-tailed t test. All FC values and p-values are reported using PRISM software (Figure 1).
Imaging and Image Analysis
Expression, Localization, Intensity, RNA in situ hybridization.
Whole-mount in situ hybridization of embryos is performed using previously published protocols (Arenas-Mena et al., 1998). Probes were generated with primers against the cDNA for the respective gene, with an added T7 promoter site and targeted an amplicon of 800–1000 bp. The PCR product was purified, and probes were synthesized using the Roche digoxygenin labelling kit (cat. 11277073910). A sense probe was generated as a negative control. RNA hybridization was developed using an anti-Dig alkaline phosphatase-conjugated-Fab fragment, Sigma (cat. 11093274910). Antibody staining imaged on an inverted light microscope using NBT/BCIP for chromogenic WMISH. Probe signal was quantified using min/max, mean grey values through imageJ software (Figure 1).
Immunofluorescence.
Immunofluorescence of Alx1 and Nodal promoter mutant embryos was performed using previously published protocols. First, injected embryos at desired timepoints were collected and fixed in either ice-cold methanol, or 4% PFA in seawater. Primary antibodies used were Anti Phospho-Smad 1/5/8, Rabbit monoclonal (Cell signaling, cat# 9516), and EctoV (marker of oral ectoderm) mouse monoclonal, a generous gift of Dr. David R. McClay (Duke University. Durham, North Carolina. dmcclay@duke.edu). Antibodies to Vasa were used as previously described (Voronina et al., 2008). Secondary anti-rabbit 488 (Invitrogen, cat#A32731) and anti-mouse 594 (Invitrogen, cat#A32742) were used to visualize protein localization. All images were taken on a Nikon CSU-W1 confocal microscope. Localization and intensity of signal was quantified using imageJ software.
EctoV Immunofluorescence.
48hpf Nodal promoter mutant, and Cas9 only injected embryos were fixed in ice-cold methanol for 10 minutes, and immunolabeled as described above. EctoV signal was visualized using anti-mouse 594 secondary (Invitrogen, cat#A32742). Images taken with Nikon CSU-W1 confocal microscope were saved as .TIFF image files for import into ImageJ software. Thirteen images each of Nodal mutant and Cas9 only embryos in the same orientation (presumptive oral-aboral axis visible) were quantified. First, visible ectoderm was measured as a perimeter (arbitrary units, relative to scalebar). Then, the visible ectodermal surface with EctoV+ signal (magenta, over threshold defined from secondary only control image) was measured as a second perimeter value. These two values were then used to create an “EctoV+” percentage value for each embryo. An average percentage of the EctoV+ surface for all embryos in each group are reported as a table, as well as the individual EctoV+ surface normalized to embryo size as individual points on a Scatter plot. P-values were determined by a standard two-tailed t-test on the mean percent of each group.
Nanos2 GFP reporter injections
S. purpuratus or H. pulcherrimus Nanos2-promoter GFP constructs were made as described (Yajima et al., 2010). Sm50 reporter construct was used as a positive control (Makabe et al., 1995). Briefly, primers were designed within the desired upstream or downstream region of Nanos2 using sequences retrieved from Echinobase.org (http://www.echinobase.org/Echinobase/) to PCR-amplify the fragment of interest. For HpNanos2, PCR-based genome walking was also performed to identify the upstream and downstream sequences (Triglia et al., 1988). Each fragment was then cloned into the pGreenlantern1 vector (GIBCO, BRL) to construct GFP-fusions. These constructs were injected as described previously (Yajima et al., 2007). Approximately, 6 pl of 6 ng/μl GFP reporter construct linearized with SacI and 20 ng/μl genomic DNA as a carrier were injected into fertilized eggs (McMahon et al., 1985). Expression of GFP was detected and analyzed at the desired developmental stage under the fluorescence microscope (Zeiss, Axioplan).
Results
Mutations in the basal promoter of Alx1 transcription produce skeleton defects
Aristaless-Like Homeobox 1 (Alx1) is a transcription factor that is broadly conserved across diverse phyla (Ettensohn et al., 2003; Khor and Ettensohn, 2017). In sea urchins, Alx1 is a potent inducer of skeletogenic mesenchyme and has well established upstream cis-regulatory sites required for its transcription (Damle and Davidson, 2011; Ettensohn et al., 2003). Four gRNAs were designed, targeting known regions of function near the transcription start site. Two gRNAs were generated targeting the first 50 bp of the basal promoter region, one targeting −183 bp, which has an established HesC binding site, and the fourth targeting −279 bp from the TSS as it defined the boundary of the minimum essential upstream region required for Alx1 transcription (Figure2S). Cas9 mRNA was mixed with each of the gRNAs in a 1:2 ratio by mass, and the workflow was followed as described in Figure 1. Across four independent matings, a significant 2–3 fold decrease in Alx1 mRNA was observed relative to Cas9 only injected controls by qPCR assay of n=50 embryos (Figure 2A). Embryos with a mild phenotype often had one defective spicule, sometimes confined to only one side of the embryo (Figure 2D, arrowhead). In other embryos with a “mild” phenotype, skeletal elements were visible, but abnormal; they displayed a ratio ≤ 3.7 to 1 to the dorsoventral connecting rod, in contrast to the average 5:1 ratio observed in Cas9 only control embryos (Figure S1).
Figure 2. Mutations in the promoter of Alx1 yield skeletal defects.
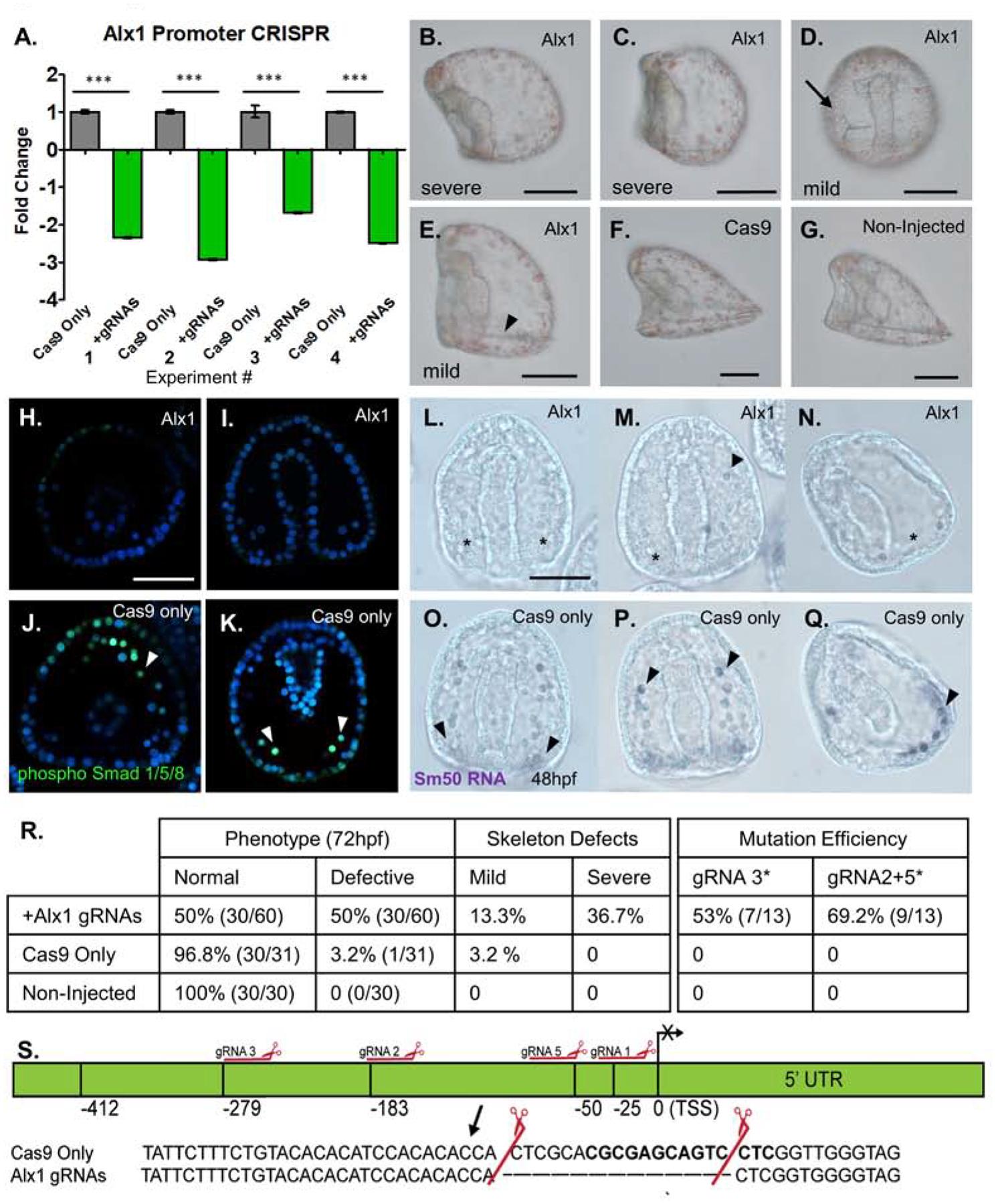
Proof-of concept analysis using cis-regulatory regions of a known promoter for the skeletogenic transcription factor, Alx1. Cas9 mRNA mixed with four gRNAs targeting the promoter of Alx1 were injected into zygotes. Cas9 mRNA only served as the injection control.
A: qPCR of Alx1 mRNA following the Alx1 promoter mutation. Each bar represents n=50 injected embryos collected at 18hpf (blastula) stage. Alx1 expression was normalized to ubiquitin mRNA. A significant and consistent 2–3 fold decrease of Alx1 transcription was observed across four independent matings.
B–G: Representative phenotypes from Alx1 promoter mutation embryos at 72hpf, all embryos are shown in a lateral orientation, except for embryo D. (Scalebar = 60μm)
B,C: Embryos with no detectable skeleton at 72hpf were scored as having a “severe” phenotype.
D: An embryo viewed from the ventral aspect shows a unilateral spicule on the right side (arrow) and is scored as “mild”.
E: Alx1 promoter mutant embryos with skeletal elements, but with defects (arrowhead) in both length and formation of the post-oral and body rods at this stage of development (72hpf) considered “mild”.
F: Cas9 only injected control with normal skeleton, 72hpf.
G: Non-injected age matched control with normal skeleton, 72hpf.
H, I: phosphorylated Smad 1/5/8 antibody marks active pSmad+ nuclei in the cells of the PMC ring at 48hpf. Alx1 promoter mutants have reduced PMCs in the ring, and a lack of Smad+ PMCs. Embryos are viewed along the Dorsal/Ventral axis.
J, K: Normal PMCs in Cas9 only injected control embryo at 48hpf. Cas9 injected control embryos have between 13–20 pSmad 1/5/8+ PMC cells visible in the PMC ring, see arrowheads.
L: Chromogenic WMISH of SM50 mRNA in Alx1 promoter mutant embryo at 48hpf. Sm50 staining is not present in the mutant embryos, indicating an absence of PMCs expressing Sm50, which is a skeletogenic factor transcribed by Alx1 (asterisks).
M: RNA WMISH of Alx1 promoter mutant embryo with unilateral single visible Sm50 expressing PMC (arrowhead).
N: Lateral view of 48hpf Alx1 promoter mutant embryo with no Sm50 expression in the PMC ring. Note the region with a complete absence of PMCs (asterisk).
O,P: Cas9 only control embryos at 48hpf showing normal expression of Sm50 in skeletogenic PMCs, (arrowheads). Note abundance of Sm50 signal surrounding the base of the archenteron.
Q. Lateral view of Cas9 only embryo at 48hpf showing normal PMC ring with Sm50 expressing cells (arrowhead).
R. Table of phenotypes and mutation efficiencies from Alx1 promoter mutagenesis experiments. General defects are summarized in the left two columns, while specific skeletogenesis effects are quantified in the right columns. The second table summarizes the overall mutation efficiency observed when targeting the Alx1 promoter. The mutations produced by gRNA3 did not result in skeletogenic defects are show, as well as gRNAs 2+5, which cut as a pair and produced severe skeletogenic phenotypes. Mutation efficiency is assessed by single embryo genotypic analysis.
S. Promoter map constructed for Alx1 using CRISPR-Cas9 mutagenesis data. Mutation by gRNAs 2+5 are shown as an inset. This mutation 183 bp upstream of the transcription start site corresponded to the 2–3 fold decrease in the level of Alx1 mRNA via qPCR, and was observed in embryos with no skeleton (severe) phenotype.
Using an antibody against nuclear phosphorylated Smad1/5/8 (pSmad1/5/8), PMC number and distribution was assessed by immunofluorescence (Lapraz et al., 2009). Alx1 promoter knockout embryos displayed reduced pSmad+ PMCs when viewed both anteriorly, and from the ventral surface (Figure2 H, I) relative to Cas9 only embryos when viewed in the same orientations (Figure 2J, K). Further, to quantify defects in PMC skeletogenic mesenchyme fate, late gastrula (48hpf) embryos were collected, just before spicule elaboration. The PMC-specific transcript, spicule matrix protein 50 (Sm50) was assayed by in situ RNA-hybridization, as it is a marker of skeletogenic mesenchyme fate, downstream of Alx1 (Cheers and Ettensohn, 2005). An antisense RNA probe against sea urchin Sm50 was visualized using chromogenic WMISH, and a distinct lack of Sm50+ PMCs is apparent at 48hpf, relative to Cas9 only injected control embryos from the same mating (Figure 2L–Q).
All of the phenotypic analyses of Alx1 mutant embryos and gRNA efficiencies are summarized in tabular form (Figure 2R), with a breakdown of the mild and severe phenotypes observed, which correspond to the representative embryo images in Figure 2B–G. Sequence analysis of genomic DNA (Supplement Figure 1) revealed consistent mutations that correlated to both decrease in transcription in qPCR at 18hpf (Figure 2R, S), and the severe phenotype of no skeletogenesis at 72hpf (Figure 2B–G). The causal mutation which resulted in severe skeletogenic defects was induced by gRNAs 2 and 5, cutting as a pair. From Sanger sequencing data of individual embryos, integrated with the qPCR data, an Alx1 promoter map was constructed showing the causal mutation associated with severe skeletogenic defects (Figure 2S). Mutation within the targeted basal promoter site resulted in an absence of skeleton and this phenotype was observed across 4 independent experiments. Thus, we concluded that mutation of cis-regulatory sites needed for transcription can produce robust phenotypes in the sea urchin embryo.
Mutation of a repressor site induces overexpression of Nodal
Nodal is responsible for the establishment of the Left-Right embryonic axis, as well as Dorsal-Ventral polarization of the late gastrula (48hpf) and prism (72hpf) stage sea urchin embryo (Duboc et al., 2004; Duboc et al., 2005; Flowers et al., 2004; Yaguchi et al., 2010). Previous results also implicated Nodal in restriction of germline gene expression (Fresques and Wessel, 2018; Luo and Su, 2012) as well as left-side restriction of germline genes such as Nanos2. Previously, the upstream region of Nodal was interrogated for regulation of Nodal by use of a reporter construct, which found both cis-activator and repressor sites (Nam et al., 2007). Our goal was to target discrete sites within this promoter region of the genome through Cas9 mutagenesis, and to assess associated phenotypes, if any (Duboc et al., 2005; Flowers et al., 2004; Range et al., 2007).
For targeting the region upstream of Nodal, three gRNAs flanking a repressive Hes/ETS-1 binding site (predicted from TFsiteSCAN software), one at the core promoter −76 bp from the TSS, and three more within the bounds of the previously established promoter region used in the previously published reporter assays were selected (Figure 3H). Cas9 mRNA was mixed with seven gRNAs as described in Figure 1. Among n=50 embryos, significant upregulation of Nodal transcript between 2- and 8- fold was observed in three independent experiments via qPCR (Figure 3A). Importantly, across three independent matings, we observed a range in severity of the Nodal overexpression following Hes/ETS-1 site knockout.
Figure 3. Mutation of a repressive HES/ets-1 motif in the Nodal promoter yields increased Nodal transcription.
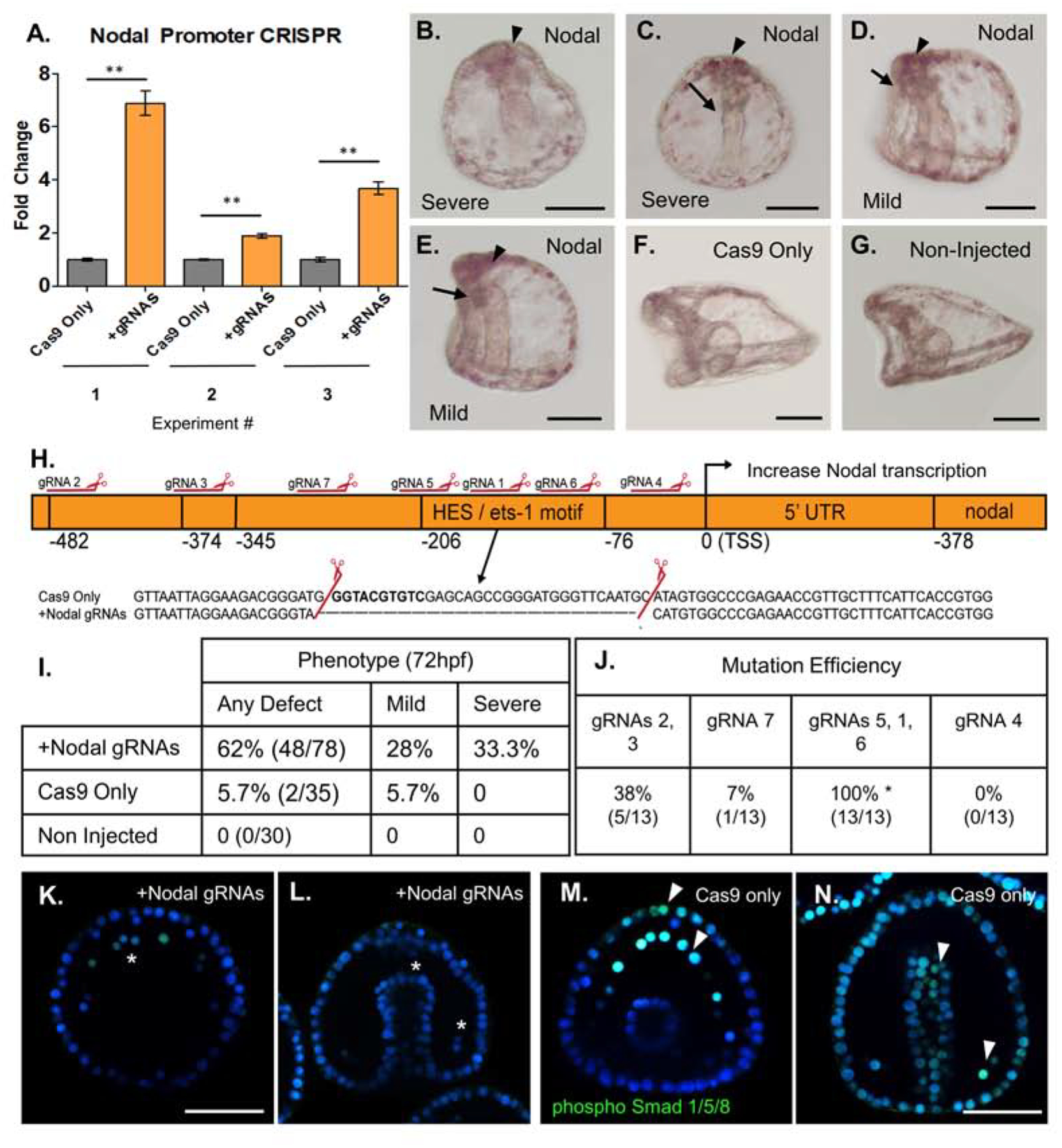
Cas9 mRNA mixed with gRNAs targeting the Nodal promoter were injected into zygotes to test sites important for Nodal expression. Cas9 mRNA-only served as an injection control and results shown are embryos from three separate matings.
A: qPCR (n=50 embryos per experiment) from each of three independent experiments demonstrated a significant (2- to 7- fold) increase in Nodal mRNA relative to Cas9 only injected control groups at 18hpf. Nodal expression was normalized to ubiquitin mRNA.
B–G: Phenotypic analysis of Nodal promoter KO embryos at 72hpf (scale bar = 60uM).
B: Severe Nodal promoter KO phenotype, formation of mouth not observed, absent dorsal-ventral flattening.. Note apical clustering of pigment cells (arrowhead).
C: Severe polarity defect at 72hpf. Gut is straight along Dorsal-Ventral axis, with no formation of mouth (arrow). Note apical clustering of pigment cells (arrowhead).
D. Mild phenotype Nodal promoter mutant embryo has normal flattening of oral and anal surfaces of the embryo, but formation of mouth is defective or absent (arrow). Apical clustering of pigment cells observed (arrowhead).
E: Mild phenotype, L/R polarity is intact, but gut formation is defective (arrow).
F: Cas9-only injected control embryo 72hpf (lateral view) shows normal gut development and flattening of oral surface and extended aboral ectoderm.
G: Age-matched control embryo 72hpf (lateral view) with normal oral-aboral patterning, gut structures, and development.
H. Nodal promoter map with mutation and repressive HES/ets-1 motif highlighted. Mutations identified through single embryo PCR genotyping demonstrated a mutation within this region, corresponding to cleavage by gRNAs 5,1 and 6. The causal mutation corresponding to the observed 2–8 fold Nodal overexpression is highlighted in the inset.
I. Summary of Nodal promoter mutant phenotypes at 72hpf. All defects are summarized in the left column, with a breakdown of mild (gut defect, oral-aboral rounding) and severe (three or more body plan defects) phenotypes in two columns.
J. Summary of gRNA mutation efficiencies at each site, as quantified from single embryo genotyping. gRNAs are grouped by their location within the promoter region. Some gRNAs produce no mutations, while others generate rather large deletions when active in pairs or a group of three. The causal deletion of the repressor HES/ets-1 site was produced by gRNAs1, 5, and 6 with 100% efficiency in sequenced individual embryos with a severe phenotype.
K. Nodal overexpressing embryo with nuclear localization of phosphorylated Smad 1/5/8 (green). pSmad 1/5/8 marks BMP2/4 responsive cells, including PMCs (asterisks). Nodal signaling is inhibitory to BMP 2/4 signaling, thus in Nodal overexpression mutants, only five pSmad1/5/8+ cells are visible in the PMC ring at 48hpf. (Scale bar = 60uM).
L. Nodal overexpressing embryos viewed dorsally demonstrate defective gut elongation, as well as an absence of pSmad 1/5/8+ nuclear signal. pSmad 1/5/8 is normally observed in response to BMP2/4, but is diminished in Nodal overexpressing mutants at 48hpf. (wildtype PMCs shown with arrowhead).
M. Phosphorylated Smad 1/5/8 nuclear signal in a Cas9 only injected embryo, showing thirteen PMCs with strong pSmad 1/5/8+ nuclear signal, and significant signal seen also in the anterior-most ectoderm (arrowheads).
N. Cas9 only control embryos viewed dorsally showing normal gut development at 48hpf, as well as phosphorylated Smad 1/5/8 nuclear signal in the BMP2/4 responsive cells (PMCs shown with arrowhead, and broadly in the ectoderm).
Phenotypic analysis revealed a diversity of mutant phenotypes, with gut and dorsal-ventral axis defects ranging from mild to severe when compared to non-injected and Cas9 only injected control embryos from the same experiment (Figure 3B–G). All resulting phenotypic effects of Nodal promoter mutation observed are summarized in tabular form (Figure 3I), divided by severity of the Nodal overexpression phenotype. Sequencing of amplicons from individual embryos with severe phenotypes identified the corresponding mutation within the Hes/ETS-1 motif. This mutation was produced by site mutation of gRNAs 1,5, and 6, as a group. The causal mutation observed in the Hes/ETS-1 motif is shown inset to the map of the Nodal promoter, with resultant Nodal overexpression at the TSS (Figure 3H). Efficiencies of mutation at each site in the promoter are summarized in tabular form in Figure 3J. Importantly, the Nodal overexpression phenotypes observed included a significant loss of pSmad1/5/8 signal in 48hpf gastrula stage embryos, viewed across both anterior-posterior and dorsal-ventral axes (Figure3K–N). This is consistent with a previous publication that reported Nodal signaling as antagonistic to BMP2/4 dependent nuclear pSmad1/5/8, in multiple cell types at 48hpf (Luo and Su, 2012).
Finally, Nodal overexpression by deletion of an Hes/ets-1 site led to a loss of oral-aboral patterning at 48hpf and 72hpf. To quantify this phenotype, a marker of oral ectoderm, EctoV was visualized using immunofluorescence (Figure 4A–D) (Coffman and McClay, 1990; Flowers et al., 2004; Hardin et al., 1992). EctoV signal was not confined to the flattened, ventral side of developing Nodal promoter mutant embryos, as is normally observed (Angerer et al., 2001; Hardin et al., 1992). In the Nodal de-repressed mutants, EctoV was instead broadly expressed across most of the ectodermal surface at 48hpf (Figure 4A–D). This result was found to be significant across (N=13; Nodal promoter mutant and Cas9 control) embryos at 48hpf. Interestingly, another distinct phenotype resulting from Nodal de-repression was a clustering of pigment cells in the apical-most region of the embryo. To quantify this phenotype, embryos were divided into quadrants delineating four asymmetric regions visible at the 48hpf gastrula stage. When quantified, a significant over-enrichment of pigment cells within Quadrant 1, the apical-anterior-most ectodermal surface of the embryo was observed, when compared to Cas9 only injected controls (Supplemental Figure 2).
Figure 4. Nodal over-expression promoter mutants show expanded EctoV localization.
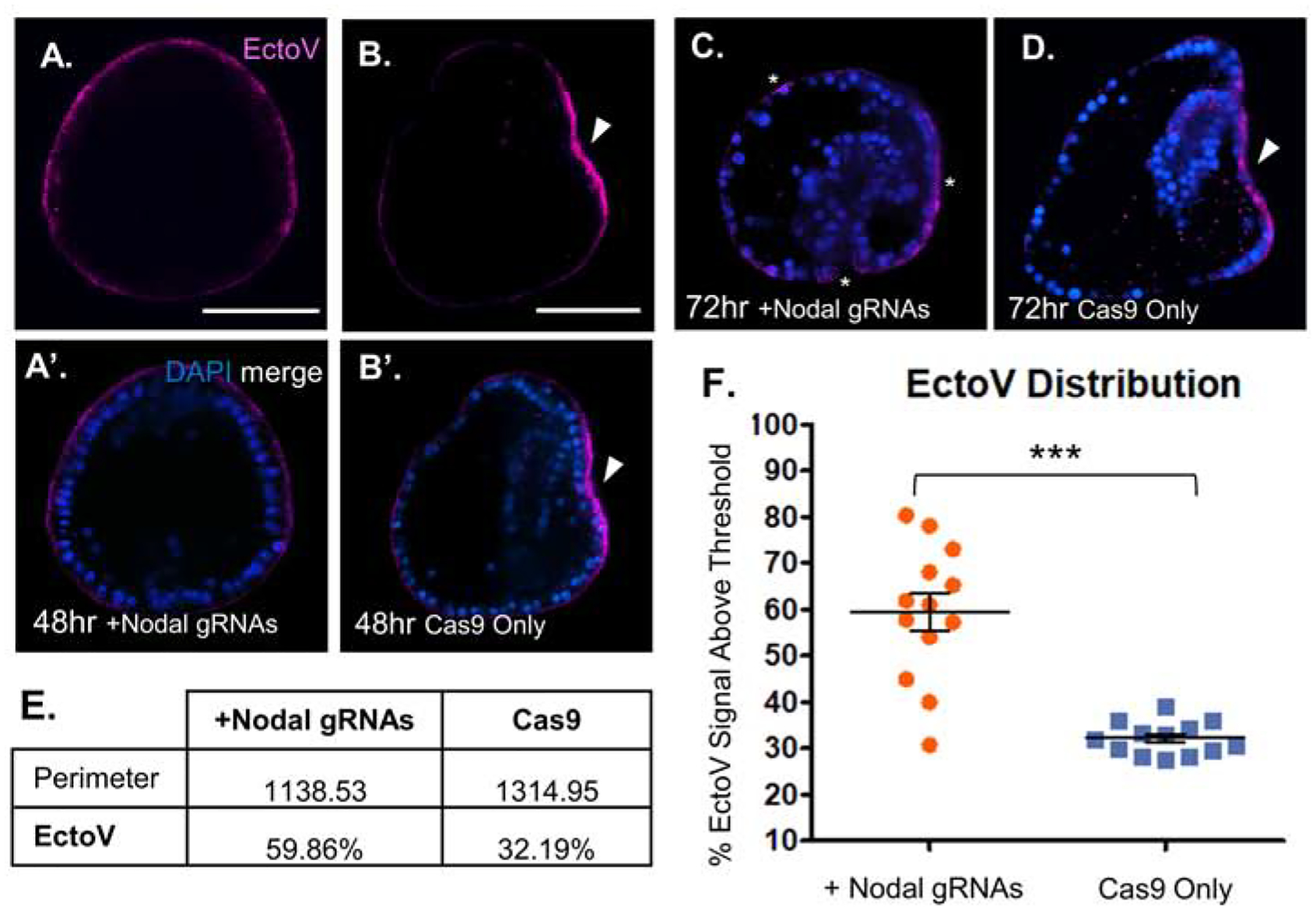
A, A’. Overexpression of Nodal from a promoter mutant embryo at 48hpf reveals ubiquitous EctoV surface expression (magenta). Merge with DAPI shown below, Scalebar = 60μm.
B, B’. Cas9 only control embryo viewed laterally shows strongly localized EctoV (magenta) expression on the flattened oral surface (arrowhead). Merge with DAPI shown below, Scalebar = 60μm.
C. Nodal overexpressing promoter mutant embryo at 72hpf viewed laterally showing persistent EctoV expression broadly along all surfaces of the embryo (asterisks). Note defective gut elongation, as well as absence of oral-aboral flattening.
D. Cas9 only control embryo at 72hpf viewed laterally showing tightly restricted EctoV expression on the flattened surface of the oral ectoderm (arrowhead).
E. EctoV localization is not restricted to the oral surface in Nodal promoter mutant embryos. The perimeter of each embryo, representing visible ectodermal surface (relative units), and the percentage of that surface with positive EctoV signal are summarized for the Nodal promoter mutants and Cas9 only control embryos at 48hpf. N=13 each, Nodal mutant and Cas9 only injected embryos were imaged, and compared from the same orientation.
F. Scatterplot of EctoV signal distribution from individual embryos imaged at 48hpf. EctoV expression was quantified as a percentage of magenta signal above threshold (background fluorescence) across the ectodermal surface of the embryo at 48hpf. Nodal promoter mutant (orange) embryos had a significant (p = 3.97E–05) expansion of EctoV signal across the surface ectoderm compared to Cas9 only control (blue) embryos.
Overexpression of Nodal by VP64-dCas9
We leveraged the result of defined gRNA sites of known efficiency upstream of the Nodal locus to test the ability to overexpress endogenous Nodal mRNA using the transcriptional activator VP64, fused to a nonfunctional (dead)-Cas9 (Konermann et al., 2015). We used the same promoter-targeting mixture of gRNAs plus dCas9-VP64 mRNA (Figure 5A). With this experiment, we sought to test whether dCas9-VP64 targeted to predetermined regions upstream of the Nodal 5’UTR could induce an increase in gene transcription. We assessed the impact of dCas9-VP64 on Nodal overexpression by measuring Nodal mRNA and known downstream targets of Nodal (Duboc et al., 2008; Duboc and Lepage, 2008; Fresques and Wessel, 2018). Two independent experiments were conducted, testing both a 2:1 mass ratio and a 5:1 mass ratio of gRNA/ dCas9-VP64 mRNA for efficacy. In both experiments, Nodal was significantly overexpressed using dCas9-VP64 relative to control (Cas9 mRNA) injected embryos by qPCR (Figure 5B). To assay Nodal overexpression indirectly, its direct downstream target, Lefty, was assayed, and found to be significantly upregulated relative to controls in the 1:2 mass ratio experiment (Figure 5C). Finally, the germline genes Nanos2 and Vasa were assayed in response to dCas9-VP64 induced Nodal overexpression at 18hpf. Nanos2 was significantly overexpressed 2.5-fold relative to controls, while Vasa expression was increased only 1.4-fold (Figure 5D). Overexpression of Lefty, and the significant effects on germline gene expression were only observed in the dCas9:VP64 mixed in a 1:2 ratio with gRNAs targeting the Nodal promoter. These results demonstrate that using experimentally validated gRNAs, a dCas9-VP64 construct can be used successfully to activate cis-regulatory regions for a gene of interest, providing additional strategies to control gene transcription in the sea urchin embryo, and give insight into what mass ratio is needed for efficient transcriptional manipulation by dCas9 in the sea urchin.
Figure 5. A VP64-dCas9 fusion protein induces overexpression of Nodal.
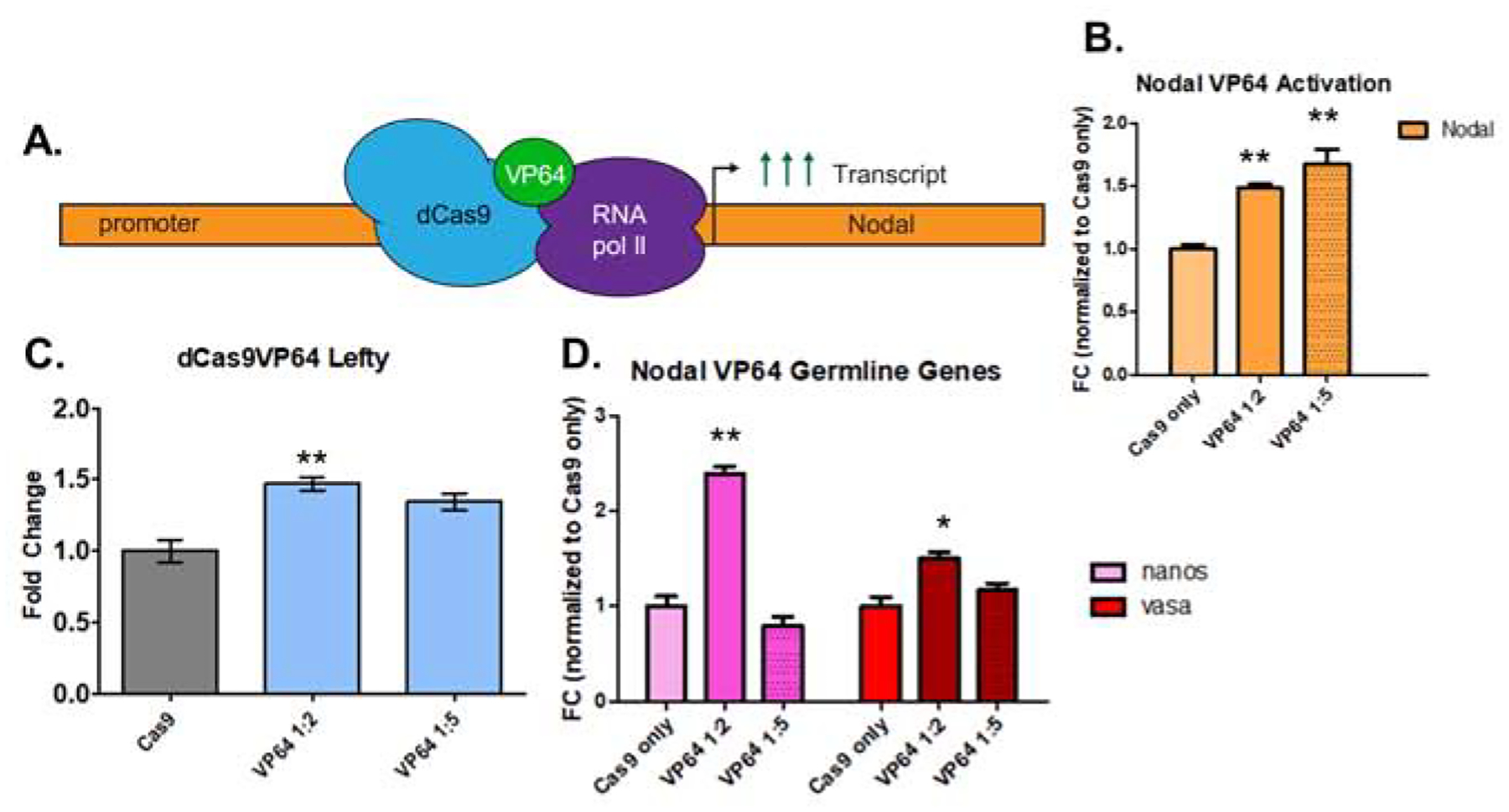
Identifying efficient gRNA targeting in the previous experiments enabled us to test a dCas9-VP64 construct for targeting and activation of Nodal from the defined promoter site. We assessed the effect on Nodal transcription by analyzing the direct downstream target, Lefty, as well as two germline genes: Nanos2 and Vasa.
A. schematic of dCas9-VP64 expression of the Nodal gene. Seven small gRNAs used for the cis-regulatory analysis were used in the overexpression experiment.
B. qPCR analysis of dCas9-VP64 induced nodal overexpression. Two experiments, one in which a 1:2 mass ratio of dCas9-VP64 mRNA to gRNA was used, and another with a 1:5 mass ratio (500ng of Cas9-Vp64 mRNA is used for injection mixes). In both experiments, (n=50 embryos) a statistically significant 1.6-fold upregulation of Nodal relative to Cas9-only injected controls was observed.
C: dCas9-VP64 induced nodal overexpression resulted in an increase of Lefty, a single, direct downstream target of nodal activation in the 1:2 mass ratio injected group.
D. qPCR analysis of the germline genes Nanos2 and Vasa (DDX4) in response to Nodal overexpression. At 18hpf, an increase of the germline genes Vasa and Nanos2 was observed in response to Nodal overexpression.
The Nanos2 promoter drives broad expression of a GFP reporter in sea urchin embryos
We next sought to identify potential cis-regulatory regions driving Nanos2 transcription. First, to dissect functionality of a noncoding region upstream of Nanos2, we made four reporter constructs containing between 1–3 kb of Sp-Nanos2 upstream sequence, each of which was fused to a GFP reporter (Figure 6A). Each construct was injected into zygotes of S.purpuratus, and the GFP expression in the resultant embryos was classified as either within the small micromere (Smms) derived cells alone, or ectopic (in non-Smm lineages). Smms give rise to the PGCs, and large micromeres (Lmm) give rise to the primary mesenchyme cells (PMCs), which form skeleton. Thus, endogenous Nanos2 expression is restricted to the Smms, and its mRNA is not observed in the PMCs nor other cell types via in situ hybridization (Juliano et al., 2006). These reporter constructs seldom produced GFP expression within only the PGCs (less than 4% of the embryos had PGC-specific GFP). The most common expression pattern observed in all four experiments was GFP within PMCs as well as PGCs and in varied cells of the ectoderm or endoderm (Figure 6B). Embryos with varying GFP reporter activity in all cells were quantified for each construct (figure 6D–F). Expression outside of the Smms appeared to be independent of the length of upstream sequence fused to GFP, as no significant difference in GFP expression localization was observed between the four constructs (Figure 6B). In gastrulae, the vast majority of GFP expression driven by upstream Nanos2 DNA was not restricted to the Smms (Figure 6G, H). As a control for these experiments, we tested a known, tightly cell-specific GFP reporter; the previously validated Sm50 promoter GFP fusion construct (Makabe et al., 1995). In these experiments, SM50-GFP reporter activity was observed strictly within the PMCs (Figure 6I–K), a result in stark contrast to Nanos2-GFP expression.
Figure 6. Genomic regions upstream of Nanos2 drive ectopic expression of a GFP reporter.
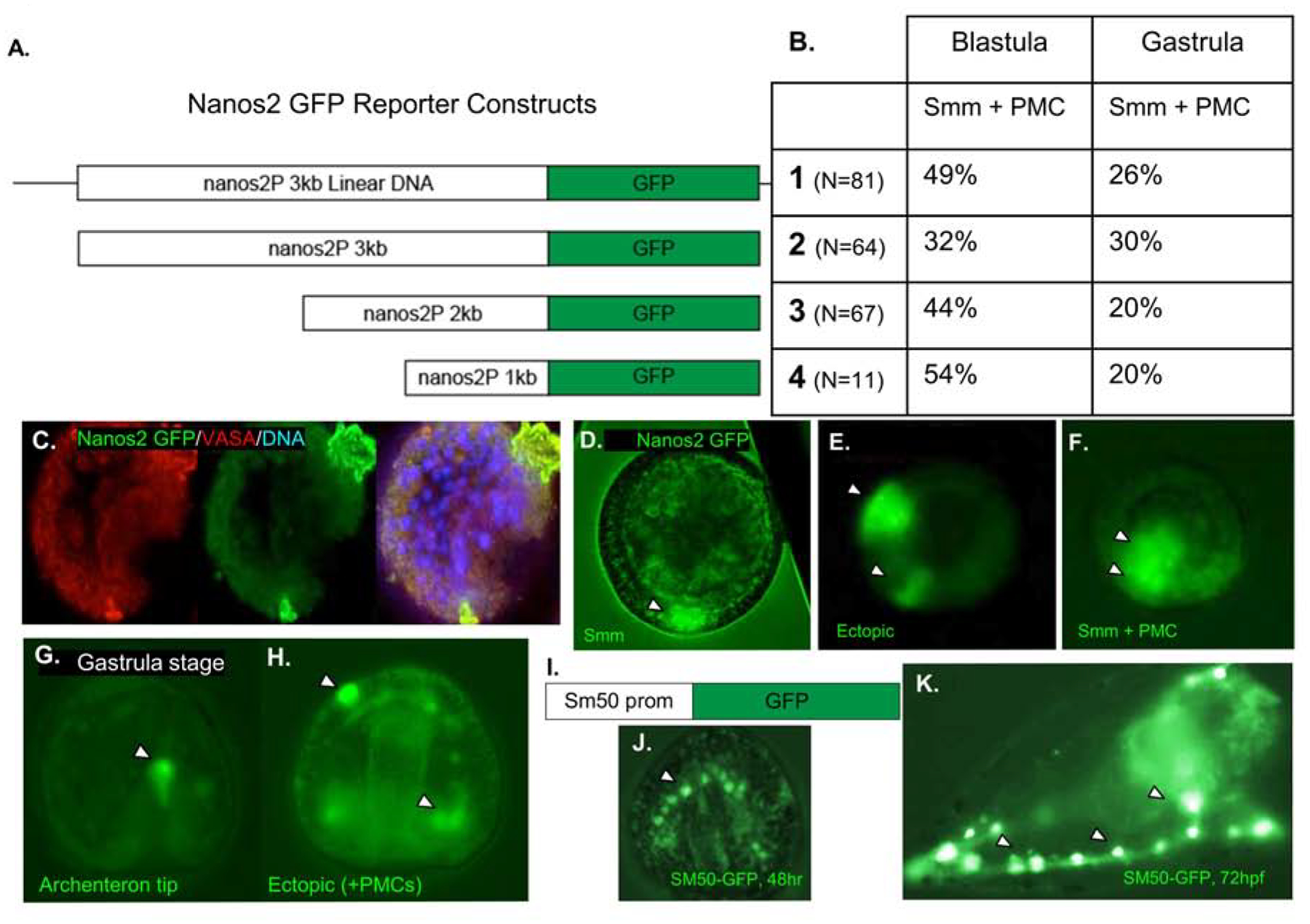
A. Schematic of four Nanos2 promoter GFP fusion constructs that were used in four independent experiments. The first construct is the entire linearized plasmid, while the following three are PCR products from the same construct, producing smaller fragments of the upstream region fused to a GFP reporter.
B. Quantification of Nanos2 promoter GFP reporter expression within the small micromeres (Smms/PGCs) and primary mesenchyme cells (PMCs) as shown in Figure C–H. A negligible number of embryos (<4%, each trial) demonstrated PGC specific GFP localization (shown in C and D). All other embryos demonstrated ectopic expression, and representatives are shown in E–H. No significant difference was seen in expression localization depending on the length of reporter construct injected, and all blastula stage embryos quantified had between 32–54% of embryos with GFP expression localized within PMC + Smms and 40–60% ectopic expression.
C. Representative embryo with Nanos2 promoter driven GFP expression in Smms, counterstained with anti-Vasa antibodies.
D. Representative embryo imaged with Nanos2 promoter driven GFP reporter expressed within the Small micromeres (Smm).
E. 18hpf embryo with GFP expression within the presumptive ectoderm (arrowheads).
F. 18hpf embryo with ectopic GFP expression, in both Smms and PMCs (arrowheads).
G. Representative 48hpf gastrula with GFP reporter in the tip of the archenteron (arrowhead).
H. Representative 48hpf gastrula embryo with Nanos2 promoter driven GFP reporter expressed in multiple different cell types (arrowheads).
I. As a positive control, DNA constructs driving GFP reporter expression by the Sm50 promoter were tested in these experiments
J. At 48hpf, Gastrula stage embryo with GFP expression restricted solely to the PMCs (arrowhead).
K. At 72hpf, Sm50 promoter driven GFP expression is only observed in the PMCs (arrowheads).
Importantly, these Sp-Nanos2 upstream regions drove similar broad expression patterns even in another sea urchin species, Lytechinus variegatus (Lv) (Figure S3), suggesting that these cis-elements might be conserved, and function similarly in both species. Additionally, for both the 3kb linearized GFP reporter construct, and the 1kb PCR amplified reporter construct, only 3.5% of Lv embryos demonstrated PGC-specific expression. The Nanos2 upstream region of a related sea urchin species, Hemicentrotus pulcherrimus (Hp) was also back-tested in S. purpuratus embryos. For this experiment, a 6.3kb upstream region of Hp-Nanos2 was used as it was shown to be highly conserved with the Sp-Nanos2 upstream region via MussaGL analysis (Figure S3). GFP was again predominantly observed ectopic to PGCs independent of the length of the upstream region cloned, or presence/absence of UTRs (FigureS3F). Together, these data indicate that the region upstream of Sp-Nanos2 is conserved within related sea urchin species (Figure S3), and while capable of driving similar expression patterns in cross-species injection experiments, it is not capable of driving PGC-specific expression of a GFP reporter in either S.purpuratus or L.variegatus embryos. Rather, these data indicate the upstream sequence alone is not sufficient to drive Nanos2 PGC-specific expression. Further, the conservation of this upstream sequence, at most, may be responsible for a relatively broad expression pattern in blastula stage embryos (18–24hpf).
Discrete cis-regulatory elements upstream of Nanos2 are needed for its transcription in sea urchin embryos
Following the Nanos2 GFP-reporter experiments, we sought to test directly in genomic DNA if any specific regions within the upstream sequence might influence Nanos2 transcription. We used the same Cas9-promoter mutagenesis approach (Figure 1), and designed gRNAs to assay whether we could alter transcription of Nanos2 by deleting predicted sites in vivo. We targeted putative Forkhead-box and Tcf/Lef binding sites, as these have been shown to influence Nanos2 transcription (Figure S4). In total, 9 gRNAs were tested for upstream deletions that might lead to Nanos2 transcriptional perturbation, and those responsible for the two essential mutations are reported (Figure 7A). Mutagenesis of putative transcription sites were tested in experiments from three independent matings. The mutations were first identified by single embryo PCR, and TIDE analysis (Figure 1; Figure S5). Two arrowheads denote the gRNAs responsible for the two most common mutation sites identified via TIDE analysis, and Sanger sequencing (Figure 7A). The first arrowhead denotes the first detected mutation, identified via TIDE analysis, −1700 bp upstream of the TSS, corresponding to the cut site of gRNA 61 (Figure 7D). The second arrowhead denotes the mutation of the FoxY binding site identified via sequencing of genomic DNA from single embryos selected at random from experiments with decreased Nanos2 expression at the 18hpf blastula stage. The overall mutation efficiencies for both mutations identified via sequencing of individual embryos selected at random are summarized in Figure 7B. In three independent experiments, Nanos2 mRNA decreased 2-fold relative to Cas9-only controls (Figure 7C). Sanger sequencing of genomic PCR fragments isolated from individual embryos identified deletions between gRNA 4 and gRNA 33, which corresponded to a significant downregulation of Nanos2 at 18hpf blastula stage. Importantly, this deletion was within a predicted FoxY binding site −1000 bp upstream of the Nanos2 TSS, that was also a site of open chromatin at both 18hpf, and 24hpf as implicated by ATAC-seq data (Figure 7G; Figure S4). In each experiment, deletions at the first gRNA site −1700 bp upstream were observed (Figure S5), as well as a deletion that ranged from 12 bp to 75 bp, which removed a predicted FoxY binding site (Figure 7G). None of these mutations disrupted the spatial restriction of Nanos2 expression, but they did decrease mRNA signal observed via in situ RNA hybridization (Figure 7E, F). Collectively, these results, paired with the GFP-reporter constructs,suggest that the ~2kb of Nanos2 upstream sequence tested is necessary for driving Nanos2transcription, but does not provide PGC-specific spatial restriction. We hypothesize that, consistent with our previous findings, the spatial regulation of Nanos2 transcript is more likely a product of post-transcriptional mechanisms in this embryo (Oulhen et al., 2013; Oulhen 2016).
Figure 7. Mutations in the Nanos2 upstream sequences define essential regions for Nanos2 transcription.
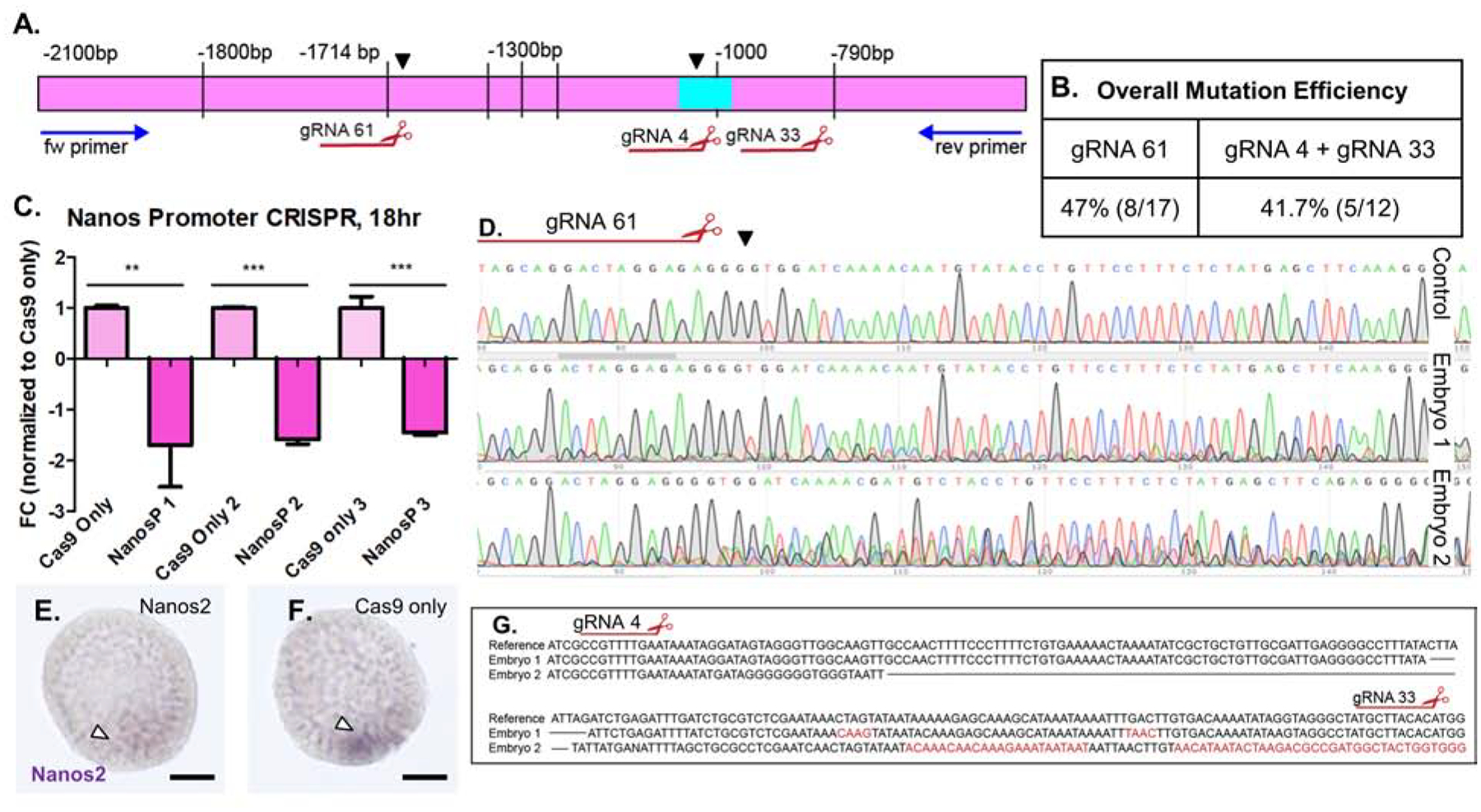
Injection of 9 Nanos2 sgRNAs targeting upstream sequences of the Nanos2 gene, along with Cas9 mRNA, generated deletions within a region −1.8kb-800bp upstream of the TSS of Nanos2.
A. Blue arrows represent primers used for sequencing this region and mutations causing transcriptional changes. Black arrowheads denote characterized mutations. Large deletion (aqua box) corresponding to decrease in Nanos2 expression over three independent experiments.
B. Table summarizing overall mutation efficiencies corresponding to the observed decreases in Nanos2 transcription in qPCR data. gRNA 61 produced a mutation identified in 3/5 TIDE sequencing analyses, as well as 5/12 individual embryo genotyping experiments. gRNA 4 and gRNA 33 together mutated a predicted FOXY binding site −1000bp upstream of the TSS observed in 5/12 single embryo genotyping analyses. These mutations are from individual embryos selected at random, not by phenotype.
C. qPCR assay from three independent experiments targeting the Nanos2 upstream region. In each independent experiment, a significant decrease in Nanos2 transcript (1.7–1.9-fold decrease) was observed at the 18hpf blastula stage, when Nanos2 expression is at its highest levels. Single embryo genotyping and sequencing from each of these experiments confirmed mutagenesis within the presumptive promoter region as highlighted by arrowheads in A.
D. TIDE analysis of PCR products from three embryos (two shown) reveals mosaicism following the cut site of gRNA 61, the mosaic pattern of sequencing and peak mismatch continued to decay until −790 bp (see also Supplemental Figure 5).
E. WMISH using Nanos2 dig labelled RNA probe in a representative embryo from the Nanos2 promoter targeted (cas9 + gRNA) injected group. Arrowhead denotes faint Nanos2 signal in Smms. (Scalebar = 20μM).
F. WMISH using Nanos2 dig labelled RNA probe in a representative embryo from the control (Cas9 mRNA only) injected group, note intense purple signal in a small cluster of Smms at the base of blastula (arrowhead).
G. Sanger sequencing from two embryos reveal a large deletion between gRNA 4 and gRNA 33. These embryos represent mutations corresponding to the qPCR (Figure 7C).
Discussion
Building a GRN by identifying cis-regulatory sequences for a gene of interest using reporter assays can be a time-consuming process (Damle and Davidson, 2011; Makabe et al., 1995). In this study, we instead tested a strategy of promoter mutagenesis to determine if Cas9 mutation of predicted regulatory sequences may be used for the identification of cis-regulatory sites. With the simplicity in time and cost of gRNA synthesis, one can test many sites predicted for transcriptional regulation, and rapidly validate mutations of these sites simply by genotyping individual sea urchin embryos (Oulhen and Wessel, 2016a). While large segments of DNA may be important to test, for which reporter gene constructs may be ideal (Makabe et al., 1995; Revilla-i-Domingo et al., 2004), the Cas9-promoter approach paired with TIDE and qPCR, is rapid and efficient for validation. Here, we demonstrated Cas9 promoter analyses of the Alx1 upstream HesC site, the Nodal predicted HES/ets-1 repressor site, and the Nanos2 predicted FoxY binding site, which all produced unique phenotypes and implications for genomic cis-regulation. For further transcriptional analyses, we also demonstrated dCas9-VP64 transcriptional activation for overexpression of endogenous Nodal, and its downstream effector, Lefty (Duboc et al., 2008). In this case, a dead Cas9 enzyme is used to target gene activity, instead of mutating a region of DNA (Konermann et al., 2015). Such application opens a strategy of transiently activating specific gene expression, or with use of a repressor domain linked to deadCas9,repressing gene activity during early embryogenesis, with assurance that later development may proceed with a wild-type genome. Overall, we found a highly efficient method for mutating cis-regulatory regions of interest to alter gene transcription by either reducing expression through mutation of cis-activation sites (Alx1, Nanos2) or by increasing expression through mutation of cis-repressive sites (Nodal). This approach opens the possibility of rapidly annotating smaller mutations of cis-elements in a gene, especially now, with high fidelity Cas9-PRIME (Anzalone et al., 2019). In comparison to current approaches using reporter gene constructs, morpholino-based knockdown, or mRNA overexpression strategies, this Cas9-promoter targeting approach relies only on the endogenous transcriptional activity, in the context of the genome anatomy, and not on random integration of multiple copies of exogenous DNA. The resultant phenotypes also are independent of artifactual misexpression of signaling molecules, which result by injection of mRNAs into zygotes that may have dramatic, but non-physiological outcomes. The transcriptional effects reported here are instead the result of manipulating endogenous gene activity. The Cas9-promoter approach also allows for more rapid and higher fidelity tests of gene regulatory mechanisms than classical reporter-construct based approaches. These approaches are applicable for embryos of diverse species.
We used a multimodal approach here to assess functionality of upstream noncoding regions of Nanos2 by mutating several predicted cis-regulatory sites, as well as supplementing these analyses with GFP-reporter construct injections in multiple species of sea urchin. These data, demonstrate that based upon the upstream sequences alone, Nanos2 is readily transcribed in several cell lineages in the 18hpf and 24hpf embryo. Our data suggest that the noncoding genomic sequences we tested here are functional, needed to drive Nanos2 expression, but not necessarily in a PGC- restricted pattern. Instead, taken together with previous findings, we hypothesize that the spatial specificity of Nanos2 mRNA and protein accumulation in this embryo likely relies more on post-transcriptional and post-translational selectivity. Mechanisms exist to restrict Nanos2 accumulation within PGCs via selective mRNA degradation outside of the germline directed by a 3’UTR GNARLE sequence, and post-translationally by a combination of stability and lability motifs within the protein itself (Oulhen and Wessel, 2014, 2016b). When integrating this new cis-regulatory data with our previous findings, we conclude that Nanos2 expression relies on a strong, promoter-driven expression of Nanos2 in multiple cell types, paired with high-fidelity, post-transcriptional degradation to result in the highly restricted PGC-specific expression.
In the future, Cas9-targeted cis-element testing is a tractable, and robust method for studying native gene transcription. However, while the Cas9-promoter method does yield rapid mutagenesis results, and is fine-tunable to predicted sites, a caveat is that the targeted region must contain a PAM sequence, “NGG,” for Cas9 enzyme cleavage of DNA (Moreno-Mateos et al., 2015). This restriction limits the approach from targeting regions that are often AT-rich. Additionally, a high-efficiency gRNA with no off-target sites, a suitable “NGG,” and functionality in-vivo is not guaranteed for every predicted cis-regulatory site of importance (Moreno-Mateos et al., 2015). To increase the fidelity of this approach in the future, further tests will employ alternative gene editing enzymes with differing targeting capabilities. We plan to establish genome-editing technologies in the sea urchin using the enzyme Cpf1, which instead utilizes a “TTN” motif for cutting, and is thus better suited to editing AT-rich regions (Safari et al., 2019). Finally, future studies may now also include CRISPR-Cas9 PRIME editing system, which induces a more specific, targeted nucleotide change (Anzalone et al., 2019) for high-stringency, single nucleotide, cis-element testing.
Supplementary Material
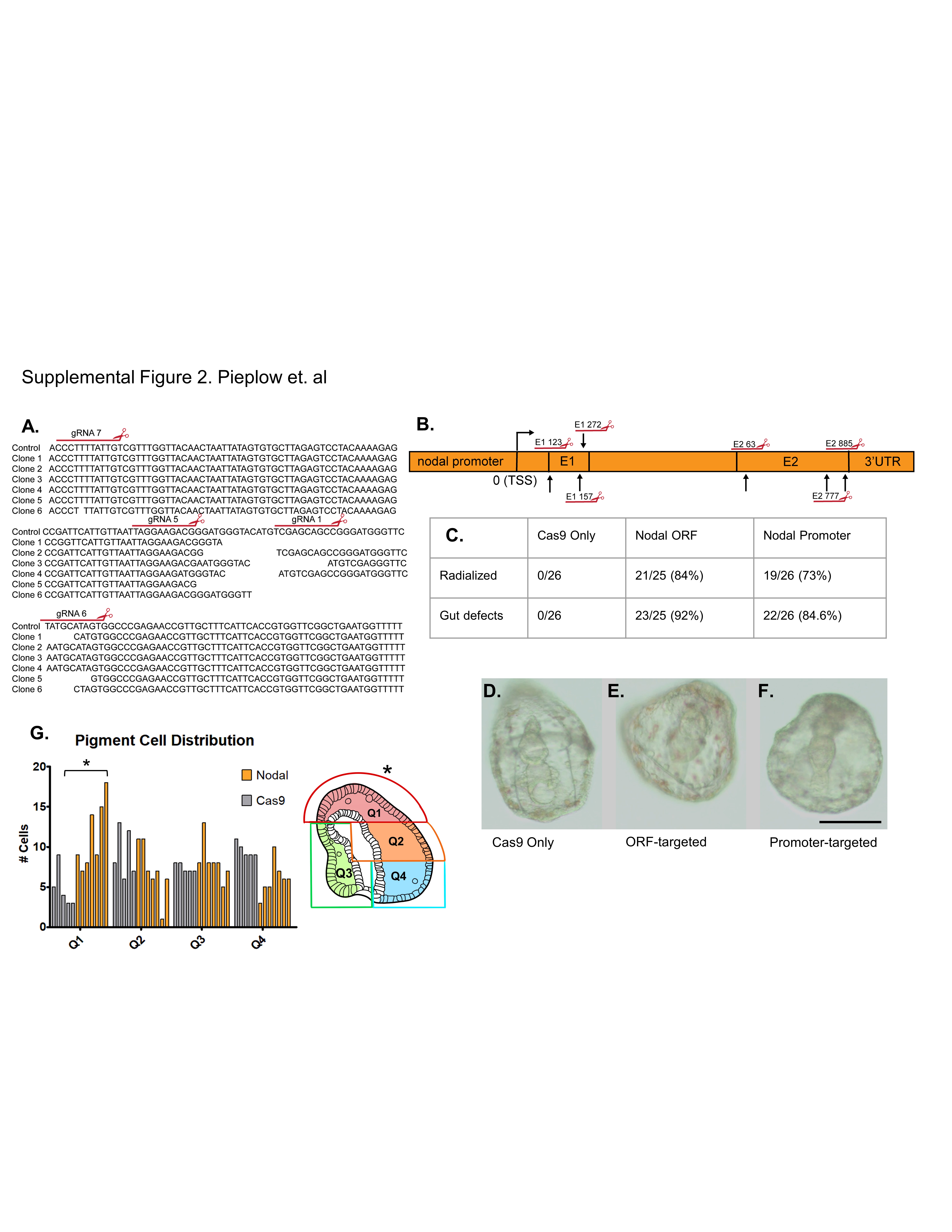
Supplemental Figure 2. Mutagenesis of the repressor site in the Nodal promoter is highly efficient, and the phenotype is distinct from a Nodal exon mutant.
A. Clonal analysis demonstrates high efficiency of promoter mutation associated with Nodal overexpression (Figure 2).
B. Schematic of Exon-Targeting gRNAs used to compare to Nodal promoter targeting mutant to Nodal ORF mutant phenotypes. All six Exon-targeting gRNAs are from a previous publication (Lin and Su, 2016).
C. Table of phenotypes associated with mutagenesis of the promoter, or within the open reading frame (ORF) of Nodal reveal differences in mutant phenotypes.
D. Cas9 only injected control at 72hpf, showing normal developmental features.
E. Representative Nodal Exon (open reading frame; ORF) -targeted embryo at 72hpf demonstrating a loss of Dorsal-Ventral patterning, mild gut defects, and skeletogenic defects. Note that the distribution of pigmented cells is unremarkable compared to that of the control shown in C.
F. Representative Nodal promoter-targeted embryo at 72hpf demonstrates a distinct phenotype, which involves unique gut features, radialization with apical (“pinching up”) defects, as well as pigment cell clumping on the apical ectoderm, unlike exon-targeted embryos.
G. Histogram of pigment cell distribution in Nodal promoter mutant (orange) and Cas9 only injected controls (grey). Embryos at 48hpf were divided into four quadrants based on asymmetrical features for pigment cell quantification. Note density of pigment cells observed in the apical ectoderm, denoted as Q1, is significantly higher in the Nodal promoter mutant embryos (p val=0.009).
{kind=link}
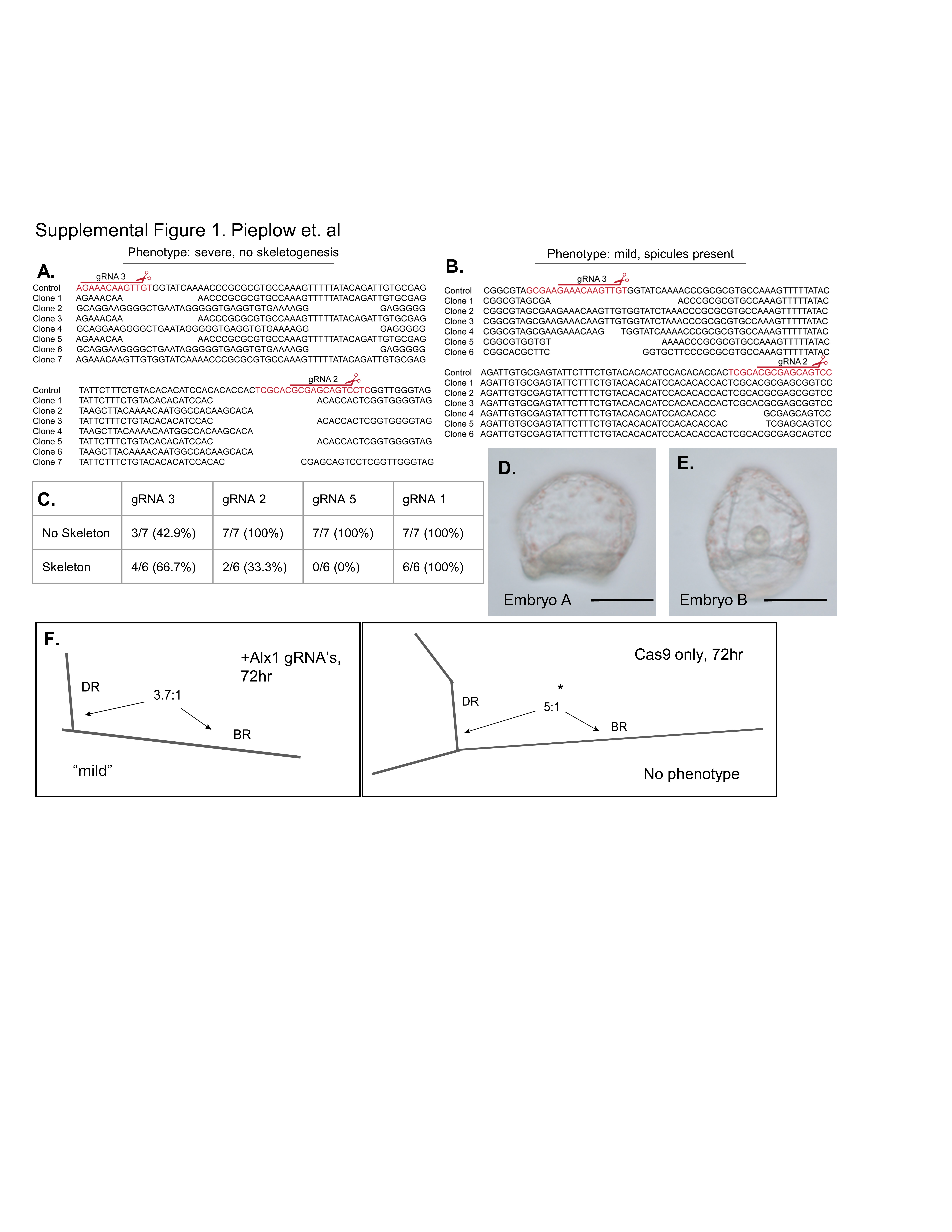
Supplemental Figure 1. Site-specific mutations upstream of Alx1 influence severity of skeletogenic defects
A. Clonal analysis of sequences obtained via a single-embryo PCR of a representative Alx1 promoter mutated embryo with a strong phenotype (no skeleton). The clonal analysis reveals that there is a deletion in 7/7 embryos at the site between sgRNA3 and sgRNA2, not observed in embryos with a mild phenotype. This mutation was associated with the strong phenotype of no skeletogenesis.
B. A clonal analysis of sequences obtained from a single-embryo PCR of an Alx1 promoter mutated embryo with a mild phenotype (defective, but present skeletogenesis)
C. Table with the mutation efficiency for each individual gRNA for an embryo collected with a mild phenotype, and for one collected with a severe phenotype. Table denotes the efficiencies of mutation at each gRNA site, and how they relate to penetrance of skeletogenic phenotype (severe, mild).
D. Image of respective 72hpf embryo from PCR in A.
E. Image of 72hpf embryo from PCR in B.
F. Schematic of control embryo skeleton, and defective skeletogenesis from Alx1 promoter mutant embryos considered a “mild” phenotype. The average DR (dorsoventral connecting rod), and BR (body rod) ratio was 5:1 in Cas9 only embryos. Mild phenotype embryos had a ratio of BR-DR length of 3.7:1 or less. (With stunted or absent post oral rods).
{kind=link}
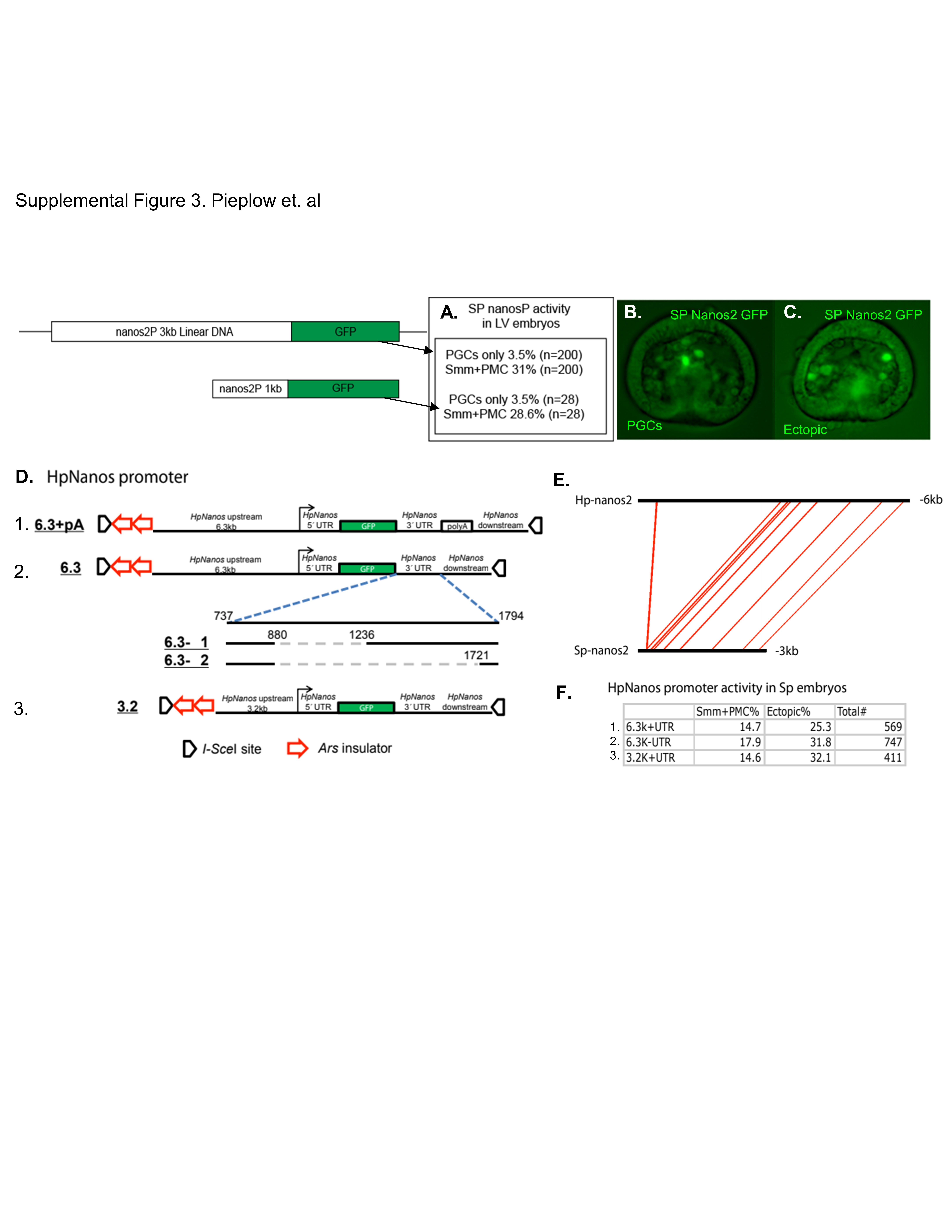
Supplemental Figure 3. Conservation of the Nanos2 upstream sequences and broad Nanos2 expression
To test the conservation of transcriptional promiscuity observed for the GFP reporter constructs, two of the expression constructs of Sp Nanos2 promoter were selected and used for injection into fertilized eggs of related species, Lytechinus variegatus (Lv).
A. Table outlines GFP expression observed in Lv embryos, corresponding to each construct (see number at left). Constructs correspond to the 3kb linearized plasmid (1.) and 1kb PCR fragment (4.) used in reporter injections from Figure 5.
B. Representative 24hpf gastrula stage embryo with Nanos2 promoter GFP expression only observed in PGCs (3.5% of embryos).
C. Representative 24hpf gastrula stage embryo with the predominantly observed pattern of GFP expression in multiple cell types, Smm and PMCs (30%), marked as “Smm +PMC” in accompanying table (Figure 6A).
D. GFP reporter expression cassette with insert of 6.3kb of the Hemicentrotus pulcherrimus (Hp) Nanos2 promoter driving a reporter: GFP open reading frame (ORF). Construct 6.3+pA has a polyA tail flanking GFP ORF. Construct 6.3 has 6.3Kb promoter of Hp Nanos2, with or without Hp Nanos2 3’UTRS flanking the GFP sequence, (see inset). 3.2 cassette contains half, or 3.2kb of Hp Nanos2 promoter driving GFP.
E. MussaGL genomic alignment of Hp Nanos2 promoter (6.3kb) with Sp Nanos2 promoter (3kb). Red lines are conserved sequences within the expression. Due to the high level of conservation, 6.3 kb of the Hp promoter was selected for expression. Alignments were performed using MUSSAGL software.
F. Table quantifying Hp Nanos2 driven GFP expression localization following injection of three cassettes outlined in (A). Cassettes were injected into fertilized eggs of a related species, S. purpuratus, and phenotypes were quantified (n=500 injected per construct).
{kind=link}
Supplemental Figure 4. Nanos2 promoter region has many distinct sites required for normal expression
A schematic of the Nanos2 promoter, gRNAs used in Figure 7 promoter knockouts are shown, with predicted binding sites and transcription factor motifs used for targeting the gRNA design. Knockouts resulting in significant loss of Nanos2 transcription corresponded to mutations of a region followed by multiple Tcf/Lef sites and the loss of the FOXY binding site 2 (arrowhead). Below: ATAC-seq data from Echinobase (Echinobase.org) is included below the schematic to denote regions of open chromatin upstream of Nanos2 at relevant developmental timepoints. A summary of intensities at each site across the promoter is shown in green.
{kind=link}
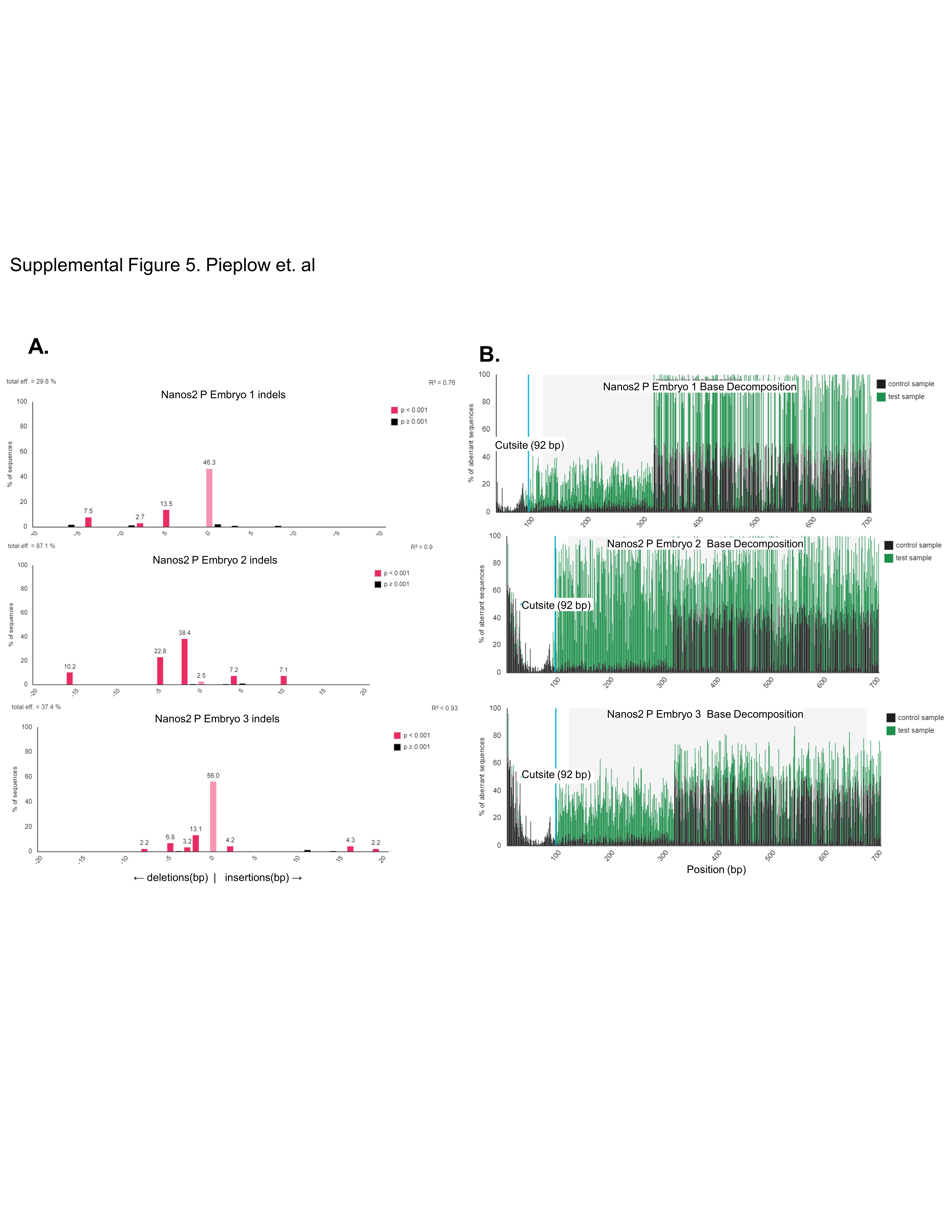
Supplemental Figure 5. Methodology of TIDE analysis and quantitation.
A. TIDE analysis quantification of indels from Nanos2 upstream region CRISPR experiments. Graph represents quantitation of deletions observed from spectral data of 3 individual embryos. Percentage of sequence affected by indels on x-axis, and size of indel at each site on the x axis, note that the scale ranges from insertions to deletions.
B. Base decay quantitation by TIDE analysis. Following a cut at gRNA 61 site, base decomposition is quantified relative to a Cas9 only control embryo. Spectral decay or mismatch when aligned to control sequence indicates that the injected embryo was mosaic and mutant upstream of Nanos2 following a cut −1714kb.
{kind=link}
We seek to test gene function by CRISPR-Cas9 editing of cis-regulatory sequences
Targeting cis-regulatory sites enables unique control over endogenous transcriptional levels
We optimize this approach using the well annotated test genes, Alx1 and Nodal.
Specific accumulation of Nanos2 in the germ line appears largely post-transcriptional
Acknowledgements:
The authors are grateful for, and acknowledge the support for this work from the National Science Foundation (1923445 to GMW) and the National Institutes of Health (9RO1GM125071 and 1R01GM132222 to GMW and 1P20GM119943 to NO. We also acknowledge the Ethiopian Biotechnology Institute of Addis Ababa University for travel support of MD. The EctoV (marker of oral ectoderm) mouse monoclonal antibody was a generous gift of Dr. David R. McClay (Duke University. Durham, North Carolina. dmcclay@duke.edu). A.P. also acknowledges valuable discussions with Cheng-Yi Chen about Nanos and germline gene transcription (Stowers Medical Institute, KS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aihara M, Amemiya S, 1999. Inversion of left–right asymmetry in the formation of the adult rudiment in sea urchin larvae: removal of a part of embryos at the gastrula stage. Zygote 8, S82–S83. [PubMed] [Google Scholar]
- Aihara M, Amemiya S, 2001. Left-right positioning of the adult rudiment in sea urchin larvae is directed by the right side. Development 128, 4935–4948. [DOI] [PubMed] [Google Scholar]
- Anderson JL, Mulligan TS, Shen M-C, Wang H, Scahill CM, Tan FJ, Du SJ, Busch-Nentwich EM, Farber SA, 2017. mRNA processing in mutant zebrafish lines generated by chemical and CRISPR-mediated mutagenesis produces unexpected transcripts that escape nonsense-mediated decay. PLOS Genetics 13, e1007105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer LM, Oleksyn DW, Levine AM, Li X, Klein WH, Angerer RC, 2001. Sea urchin goosecoid function links fate specification along the animal-vegetal and oral-aboral embryonic axes. Development 128, 4393–4404. [DOI] [PubMed] [Google Scholar]
- Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, Chen PJ, Wilson C, Newby GA, Raguram A, Liu DR, 2019. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas-Mena C, Martinez P, Cameron RA, Davidson EH, 1998. Expression of the Hox gene complex in the indirect development of a sea urchin. Proceedings of the National Academy of Sciences 95, 13062–13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, van Steensel B, 2014. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research 42, e168–e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, van Steensel B, 2019. Rapid Quantitative Evaluation of CRISPR Genome Editing by TIDE and TIDER. Methods Mol Biol 1961, 29–44. [DOI] [PubMed] [Google Scholar]
- Cameron RA, Samanta M, Yuan A, He D, Davidson E, 2009. SpBase: the sea urchin genome database and web site. Nucleic Acids Res 37, D750–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanale JP, Hamdoun A, Wessel GM, Su YH, Oulhen N, 2019. Methods to label, isolate, and image sea urchin small micromeres, the primordial germ cells (PGCs). Methods Cell Biol 150, 269–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheers MS, Ettensohn CA, 2005. P16 is an essential regulator of skeletogenesis in the sea urchin embryo. Dev Biol 283, 384–396. [DOI] [PubMed] [Google Scholar]
- Coffman JA, McClay DR, 1990. A hyaline layer protein that becomes localized to the oral ectoderm and foregut of sea urchin embryos. Dev Biol 140, 93–104. [DOI] [PubMed] [Google Scholar]
- Damle S, Davidson EH, 2011. Precise cis-regulatory control of spatial and temporal expression of the alx-1 gene in the skeletogenic lineage of s. purpuratus. Developmental Biology 357, 505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, Otim O, Brown CT, Livi CB, Lee PY, Revilla R, Rust AG, Pan Z, Schilstra MJ, Clarke PJ, Arnone MI, Rowen L, Cameron RA, McClay DR, Hood L, Bolouri H, 2002. A genomic regulatory network for development. Science 295, 1669–1678. [DOI] [PubMed] [Google Scholar]
- Duboc V, Lapraz F, Besnardeau L, Lepage T, 2008. Lefty acts as an essential modulator of Nodal activity during sea urchin oral-aboral axis formation. Dev Biol 320, 49–59. [DOI] [PubMed] [Google Scholar]
- Duboc V, Lepage T, 2008. A conserved role for the nodal signaling pathway in the establishment of dorso-ventral and left-right axes in deuterostomes. J Exp Zool B Mol Dev Evol 310, 41–53. [DOI] [PubMed] [Google Scholar]
- Duboc V, Rottinger E, Besnardeau L, Lepage T, 2004. Nodal and BMP2/4 signaling organizes the oral-aboral axis of the sea urchin embryo. Dev Cell 6, 397–410. [DOI] [PubMed] [Google Scholar]
- Duboc V, Rottinger E, Lapraz F, Besnardeau L, Lepage T, 2005. Left-right asymmetry in the sea urchin embryo is regulated by nodal signaling on the right side. Dev Cell 9, 147–158. [DOI] [PubMed] [Google Scholar]
- Ettensohn CA, Illies MR, Oliveri P, De Jong DL, 2003. Alx1, a member of the Cart1/Alx3/Alx4 subfamily of Paired-class homeodomain proteins, is an essential component of the gene network controlling skeletogenic fate specification in the sea urchin embryo. Development 130, 2917–2928. [DOI] [PubMed] [Google Scholar]
- Ewen-Campen B, Schwager EE, Extavour CG, 2010. The molecular machinery of germ line specification. Mol Reprod Dev 77, 3–18. [DOI] [PubMed] [Google Scholar]
- Extavour CG, Akam M, 2003. Mechanisms of germ cell specification across the metazoans: epigenesis and preformation. Development 130, 5869–5884. [DOI] [PubMed] [Google Scholar]
- Flowers VL, Courteau GR, Poustka AJ, Weng W, Venuti JM, 2004. Nodal/activin signaling establishes oral-aboral polarity in the early sea urchin embryo. Dev Dyn 231, 727–740. [DOI] [PubMed] [Google Scholar]
- Fresques TM, Wessel GM, 2018. Nodal induces sequential restriction of germ cell factors during primordial germ cell specification. Development 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Razy-Krajka F, Christiaen L, Stolfi A, 2018. CRISPR Knockouts in Ciona Embryos. Adv Exp Med Biol 1029, 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Joly JS, 2011. When needles look like hay: how to find tissue-specific enhancers in model organism genomes. Developmental biology 350, 239–254. [DOI] [PubMed] [Google Scholar]
- Hardin J, Coffman JA, Black SD, McClay DR, 1992. Commitment along the dorsoventral axis of the sea urchin embryo is altered in response to NiCl2. Development 116, 671–685. [DOI] [PubMed] [Google Scholar]
- Irish V, Lehmann R, Akam M, 1989. The Drosophila posterior-group gene nanos functions by repressing hunchback activity. Nature 338, 646–648. [DOI] [PubMed] [Google Scholar]
- Juliano CE, Voronina E, Stack C, Aldrich M, Cameron AR, Wessel GM, 2006. Germ line determinants are not localized early in sea urchin development, but do accumulate in the small micromere lineage. Developmental Biology 300, 406–415. [DOI] [PubMed] [Google Scholar]
- Juliano CE, Yajima M, Wessel GM, 2010. Nanos functions to maintain the fate of the small micromere lineage in the sea urchin embryo. Developmental biology 337, 220–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor JM, Ettensohn CA, 2017. Functional divergence of paralogous transcription factors supported the evolution of biomineralization in echinoderms. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Yamada M, Asaoka M, Kitamura T, 1996. Essential role of the posterior morphogen nanos for germline development in Drosophila. Nature 380, 708–711. [DOI] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F, 2015. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai F, Singh A, King ML, 2012. Xenopus Nanos1 is required to prevent endoderm gene expression and apoptosis in primordial germ cells. Development 139, 1476–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapraz F, Besnardeau L, Lepage T, 2009. Patterning of the dorsal-ventral axis in echinoderms: insights into the evolution of the BMP-chordin signaling network. PLoS Biol 7, e1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko PF, Ashburner M, 1988. The product of the Drosophila gene vasa is very similar to eukaryotic initiation factor-4A. Nature 335, 611–617. [DOI] [PubMed] [Google Scholar]
- Lasko PF, Ashburner M, 1990. Posterior localization of vasa protein correlates with, but is not sufficient for, pole cell development. Genes Dev 4, 905–921. [DOI] [PubMed] [Google Scholar]
- Lin C-Y, Su Y-H, 2016. Genome editing in sea urchin embryos by using a CRISPR/Cas9 system. Developmental Biology 409, 420–428. [DOI] [PubMed] [Google Scholar]
- Lin H, Spradling AC, 1997. A novel group of pumilio mutations affects the asymmetric division of germline stem cells in the Drosophila ovary. Development 124, 2463–2476. [DOI] [PubMed] [Google Scholar]
- Longabaugh WJ, Davidson EH, Bolouri H, 2005. Computational representation of developmental genetic regulatory networks. Dev Biol 283, 1–16. [DOI] [PubMed] [Google Scholar]
- Luo Y-J, Su Y-H, 2012. Opposing Nodal and BMP Signals Regulate Left–Right Asymmetry in the Sea Urchin Larva. PLOS Biology 10, e1001402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnúsdóttir E, Azim Surani M, 2014. How to make a primordial germ cell. Development 141, 245–252. [DOI] [PubMed] [Google Scholar]
- Makabe KW, Kirchhamer CV, Britten RJ, Davidson EH, 1995. Cis-regulatory control of the SM50 gene, an early marker of skeletogenic lineage specification in the sea urchin embryo. Development 121, 1957–1970. [DOI] [PubMed] [Google Scholar]
- McLaren A, 1981. Germ cells and soma : a new look at an old problem. Yale University Press, New Haven. [Google Scholar]
- McMahon AP, Flytzanis CN, Hough-Evans BR, Katula KS, Britten RJ, Davidson EH, 1985. Introduction of cloned DNA into sea urchin egg cytoplasm: replication and persistence during embryogenesis. Dev Biol 108, 420–430. [DOI] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Vejnar CE, Beaudoin J-D, Fernandez JP, Mis EK, Khokha MK, Giraldez AJ, 2015. CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nature Methods 12, 982–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J, Su Y-H, Lee PY, Robertson AJ, Coffman JA, Davidson EH, 2007. Cis-regulatory control of the nodal gene, initiator of the sea urchin oral ectoderm gene network. Developmental biology 306, 860–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulhen N, Swartz SZ, Laird J, Mascaro A, Wessel GM, 2017. Transient translational quiescence in primordial germ cells. Development 144, 1201–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulhen N, Swartz SZ, Wang L, Wikramanayake A, Wessel GM, 2019. Distinct transcriptional regulation of Nanos2 in the germ line and soma by the Wnt and delta/notch pathways. Developmental Biology 452, 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulhen N, Wessel G, 2016a. Albinism as a visual, in vivo guide for CRISPR/Cas9 functionality in the sea urchin embryo. Molecular reproduction and development 83, 1046–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulhen N, Wessel GM, 2014. Every which way—nanos gene regulation in echinoderms. genesis 52, 279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulhen N, Wessel GM, 2016b. Differential Nanos 2 protein stability results in selective germ cell accumulation in the sea urchin. Developmental Biology 418, 146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oulhen N, Yoshida T, Yajima M, Song J, Sakuma T, Sakamoto N, Yamamoto T, Wessel G, 2013. The 3’UTR of Nanos2 directs enrichment in the germ cell lineage of the sea urchin. Developmental biology 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Lin H, 2000. Translational repression: a duet of Nanos and Pumilio. Curr Biol 10, R81–83. [DOI] [PubMed] [Google Scholar]
- Peter IS, Davidson EH, 2015. Genomic control process : development and evolution. Academic Press is an imprint of Elsevier, London, UK; San Diego, CA, USA. [Google Scholar]
- Range R, Lapraz F, Quirin M, Marro S, Besnardeau L, Lepage T, 2007. Cis-regulatory analysis of nodal and maternal control of dorsal-ventral axis formation by Univin, a TGF-beta related to Vg1. Development 134, 3649–3664. [DOI] [PubMed] [Google Scholar]
- Revilla-i-Domingo R, Minokawa T, Davidson EH, 2004. R11: a cis-regulatory node of the sea urchin embryo gene network that controls early expression of SpDelta in micromeres. Developmental Biology 274, 438–451. [DOI] [PubMed] [Google Scholar]
- Safari F, Zare K, Negahdaripour M, Barekati-Mowahed M, Ghasemi Y, 2019. CRISPR Cpf1 proteins: structure, function and implications for genome editing. Cell & Bioscience 9, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudemont A, Haillot E, Mekpoh F, Bessodes N, Quirin M, Lapraz F, Duboc V, Rottinger E, Range R, Oisel A, Besnardeau L, Wincker P, Lepage T, 2010. Ancestral regulatory circuits governing ectoderm patterning downstream of Nodal and BMP2/4 revealed by gene regulatory network analysis in an echinoderm. PLoS Genet 6, e1001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y-H, 2014. Telling left from right: Left-right asymmetric controls in sea urchins. genesis 52, 269–278. [DOI] [PubMed] [Google Scholar]
- Suh N, Blelloch R, 2011. Small RNAs in early mammalian development: from gametes to gastrulation. Development 138, 1653–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triglia T, Peterson MG, Kemp DJ, 1988. A procedure for in vitro amplification of DNA segments that lie outside the boundaries of known sequences. Nucleic Acids Res 16, 8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuladhar R, Yeu Y, Tyler Piazza J, Tan Z, Rene Clemenceau J, Wu X, Barrett Q, Herbert J, Mathews DH, Kim J, Hyun Hwang T, Lum L, 2019. CRISPR-Cas9-based mutagenesis frequently provokes on-target mRNA misregulation. Nature Communications 10, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronina E, Lopez M, Juliano CE, Gustafson E, Song JL, Extavour C, George S, Oliveri P, McClay D, Wessel G, 2008. Vasa protein expression is restricted to the small micromeres of the sea urchin, but is inducible in other lineages early in development. Dev Biol 314, 276–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidmann CA, Goldstrohm AC, 2012. Drosophila Pumilio protein contains multiple autonomous repression domains that regulate mRNAs independently of Nanos and brain tumor. Mol Cell Biol 32, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidmann CA, Qiu C, Arvola RM, Lou TF, Killingsworth J, Campbell ZT, Tanaka Hall TM, Goldstrohm AC, 2016. Drosophila Nanos acts as a molecular clamp that modulates the RNA-binding and repression activities of Pumilio. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel GM, Brayboy L, Fresques T, Gustafson EA, Oulhen N, Ramos I, Reich A, Swartz SZ, Yajima M, Zazueta V, 2014. The biology of the germ line in echinoderms. Mol Reprod Dev 81, 679–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel GM, Morita S, Oulhen N, 2020. Somatic cell conversion to a germ cell lineage - a violation or a revelation? Journal of Experimental Zoology [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RM, Senanayake U, Artibani M, Taylor G, Wells D, Ahmed AA, Sauka-Spengler T, 2018. Genome and epigenome engineering CRISPR toolkit for in vivo modulation of cis-regulatory interactions and gene expression in the chicken embryo. Development 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie C, 1999. Germ cells. Cell 96, 165–174. [DOI] [PubMed] [Google Scholar]
- Yaguchi J, 2019. Microinjection methods for sea urchin eggs and blastomeres, Methods in Cell Biology. Elsevier, pp. 173–188. [DOI] [PubMed] [Google Scholar]
- Yaguchi S, Yaguchi J, Angerer RC, Angerer LM, Burke RD, 2010. TGFbeta signaling positions the ciliary band and patterns neurons in the sea urchin embryo. Dev Biol 347, 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima M, Kiyomoto M, Akasaka K, 2007. Ars insulator protects transgenes from long-term silencing in sea urchin larva. Development Genes and Evolution 217, 331–336. [DOI] [PubMed] [Google Scholar]
- Yajima M, Umeda R, Fuchikami T, Kataoka M, Sakamoto N, Yamamoto T, Akasaka K, 2010. Implication of HpEts in gene regulatory networks responsible for specification of sea urchin skeletogenic primary mesenchyme cells. Zoological Science 27, 638–646. [DOI] [PubMed] [Google Scholar]
- Yajima M, Wessel GM, 2011. Small micromeres contribute to the germline in the sea urchin. Development 138, 237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 2. Mutagenesis of the repressor site in the Nodal promoter is highly efficient, and the phenotype is distinct from a Nodal exon mutant.
A. Clonal analysis demonstrates high efficiency of promoter mutation associated with Nodal overexpression (Figure 2).
B. Schematic of Exon-Targeting gRNAs used to compare to Nodal promoter targeting mutant to Nodal ORF mutant phenotypes. All six Exon-targeting gRNAs are from a previous publication (Lin and Su, 2016).
C. Table of phenotypes associated with mutagenesis of the promoter, or within the open reading frame (ORF) of Nodal reveal differences in mutant phenotypes.
D. Cas9 only injected control at 72hpf, showing normal developmental features.
E. Representative Nodal Exon (open reading frame; ORF) -targeted embryo at 72hpf demonstrating a loss of Dorsal-Ventral patterning, mild gut defects, and skeletogenic defects. Note that the distribution of pigmented cells is unremarkable compared to that of the control shown in C.
F. Representative Nodal promoter-targeted embryo at 72hpf demonstrates a distinct phenotype, which involves unique gut features, radialization with apical (“pinching up”) defects, as well as pigment cell clumping on the apical ectoderm, unlike exon-targeted embryos.
G. Histogram of pigment cell distribution in Nodal promoter mutant (orange) and Cas9 only injected controls (grey). Embryos at 48hpf were divided into four quadrants based on asymmetrical features for pigment cell quantification. Note density of pigment cells observed in the apical ectoderm, denoted as Q1, is significantly higher in the Nodal promoter mutant embryos (p val=0.009).
Supplemental Figure 1. Site-specific mutations upstream of Alx1 influence severity of skeletogenic defects
A. Clonal analysis of sequences obtained via a single-embryo PCR of a representative Alx1 promoter mutated embryo with a strong phenotype (no skeleton). The clonal analysis reveals that there is a deletion in 7/7 embryos at the site between sgRNA3 and sgRNA2, not observed in embryos with a mild phenotype. This mutation was associated with the strong phenotype of no skeletogenesis.
B. A clonal analysis of sequences obtained from a single-embryo PCR of an Alx1 promoter mutated embryo with a mild phenotype (defective, but present skeletogenesis)
C. Table with the mutation efficiency for each individual gRNA for an embryo collected with a mild phenotype, and for one collected with a severe phenotype. Table denotes the efficiencies of mutation at each gRNA site, and how they relate to penetrance of skeletogenic phenotype (severe, mild).
D. Image of respective 72hpf embryo from PCR in A.
E. Image of 72hpf embryo from PCR in B.
F. Schematic of control embryo skeleton, and defective skeletogenesis from Alx1 promoter mutant embryos considered a “mild” phenotype. The average DR (dorsoventral connecting rod), and BR (body rod) ratio was 5:1 in Cas9 only embryos. Mild phenotype embryos had a ratio of BR-DR length of 3.7:1 or less. (With stunted or absent post oral rods).
Supplemental Figure 3. Conservation of the Nanos2 upstream sequences and broad Nanos2 expression
To test the conservation of transcriptional promiscuity observed for the GFP reporter constructs, two of the expression constructs of Sp Nanos2 promoter were selected and used for injection into fertilized eggs of related species, Lytechinus variegatus (Lv).
A. Table outlines GFP expression observed in Lv embryos, corresponding to each construct (see number at left). Constructs correspond to the 3kb linearized plasmid (1.) and 1kb PCR fragment (4.) used in reporter injections from Figure 5.
B. Representative 24hpf gastrula stage embryo with Nanos2 promoter GFP expression only observed in PGCs (3.5% of embryos).
C. Representative 24hpf gastrula stage embryo with the predominantly observed pattern of GFP expression in multiple cell types, Smm and PMCs (30%), marked as “Smm +PMC” in accompanying table (Figure 6A).
D. GFP reporter expression cassette with insert of 6.3kb of the Hemicentrotus pulcherrimus (Hp) Nanos2 promoter driving a reporter: GFP open reading frame (ORF). Construct 6.3+pA has a polyA tail flanking GFP ORF. Construct 6.3 has 6.3Kb promoter of Hp Nanos2, with or without Hp Nanos2 3’UTRS flanking the GFP sequence, (see inset). 3.2 cassette contains half, or 3.2kb of Hp Nanos2 promoter driving GFP.
E. MussaGL genomic alignment of Hp Nanos2 promoter (6.3kb) with Sp Nanos2 promoter (3kb). Red lines are conserved sequences within the expression. Due to the high level of conservation, 6.3 kb of the Hp promoter was selected for expression. Alignments were performed using MUSSAGL software.
F. Table quantifying Hp Nanos2 driven GFP expression localization following injection of three cassettes outlined in (A). Cassettes were injected into fertilized eggs of a related species, S. purpuratus, and phenotypes were quantified (n=500 injected per construct).
Supplemental Figure 4. Nanos2 promoter region has many distinct sites required for normal expression
A schematic of the Nanos2 promoter, gRNAs used in Figure 7 promoter knockouts are shown, with predicted binding sites and transcription factor motifs used for targeting the gRNA design. Knockouts resulting in significant loss of Nanos2 transcription corresponded to mutations of a region followed by multiple Tcf/Lef sites and the loss of the FOXY binding site 2 (arrowhead). Below: ATAC-seq data from Echinobase (Echinobase.org) is included below the schematic to denote regions of open chromatin upstream of Nanos2 at relevant developmental timepoints. A summary of intensities at each site across the promoter is shown in green.
Supplemental Figure 5. Methodology of TIDE analysis and quantitation.
A. TIDE analysis quantification of indels from Nanos2 upstream region CRISPR experiments. Graph represents quantitation of deletions observed from spectral data of 3 individual embryos. Percentage of sequence affected by indels on x-axis, and size of indel at each site on the x axis, note that the scale ranges from insertions to deletions.
B. Base decay quantitation by TIDE analysis. Following a cut at gRNA 61 site, base decomposition is quantified relative to a Cas9 only control embryo. Spectral decay or mismatch when aligned to control sequence indicates that the injected embryo was mosaic and mutant upstream of Nanos2 following a cut −1714kb.