

Abstract
Introduction:
ARID1A is commonly mutated in colorectal cancer (CRC), frequently resulting in truncation and loss of protein expression. ARID1A recruits MSH2 for mismatch-repair during DNA replication. ARID1A deficiency promotes hypermutability and immune activation in preclinical models but its role in CRC patients is being explored.
Methods:
The DNA sequencing and gene expression profiling of CRC patients were extracted from TCGA and MD Anderson Cancer Center databases, with validation utilizing external databases, and correlation between ARID1A and immunologic features. Immunohistochemistry for T-cell markers was performed on a separate cohort of patients.
Results:
28/417 MSS CRC patients (6.7%) had ARID1A mutation. Among 58 genes most commonly mutated in CRC, ARID1A mutation had the highest increase with frameshift mutation rates in MSS cases (8-fold, p<0.001). In MSS, ARID1A mutation was enriched in immune subtype (CMS1) and had a strong correlation with IFN-γ expression (Δz score +1.91, p<0.001). Compared with ARID1A wild-type, statistically significant higher expression for key checkpoint genes (e.g., PD-L1, CTLA4, and PDCD1) and genes sets (e.g., antigen presentation, cytotoxic T cell function, and immune checkpoints) was observed in mutant cases. This was validated by unsupervised differential expression of genes related to immune response and further, confirmed by higher infiltration of T-cells in IHC of tumors with ARID1A mutation (p=0.01).
Conclusion:
The immunogenicity of ARID1A mutant cases is likely due to increased level of neoantigens resulting from increased TMB and frameshift mutations. Tumors with ARID1A mutation may be more susceptible to immune therapy-based treatment strategies and should be recognized as a unique molecular subgroup in future immune therapy trials.
Keywords: ARID1A, AT-rich interactive domain 1A, colorectal cancer, immune therapy, immunotherapy, mutational burden, hypermutation
Introduction
Colorectal cancer is one of the leading causes of cancer-related death globally (1). Despite the success of conventional immunotherapy agents in various tumor types (2), these agents are effective in only a small proportion of CRC with microsatellite-instability-high (MSI-H) or mismatch-repair deficiency (dMMR) (3). MSS CRC is a heterogeneous disease (4) and one approach to develop new treatment strategies is to discover novel biomarkers identifying subsets of patients with the immunologically active microenvironment. Recently, the loss of function and mutation of the ARID1A gene have gained attention based on the newly proposed role of this protein in DNA repair (5).
AT-rich interactive domain 1A (ARID1A) is a subunit of the SWitch/Sucrose NonFermentable (SWI/SNF) chromatin remodeling complex. By hydroxylation of ATP, the SWI/SNF complex modulates the repositioning of nucleosomes and thereby regulates accessibility of chromatin to DNA transcription, replication, methylation, and repair (6). The dysregulation of this complex has been reported in cancers and among its different subunits, ARID1A is most frequently mutated (7).
Initially, the decrease in the expression of ARID1A protein and discovery and ARID1A rearrangements and deletions proposed the role of this protein as a tumor suppressor (8, 9). Later, and with the help of next-generation sequencing (NGS), somatic mutations in ARID1A were discovered in various human malignancies. Most of these heterozygous mutations are deletion or nonsense mutation and are distributed along the entire length of the gene resulting in truncation of the protein. Multiple studies have demonstrated that one only allele mutation in ARID1A gene is sufficient to result in the loss of ARID1A expression (10–13). In CRC, the somatic mutation of ARID1A is present in 6.2–9.4% of patients (14).
ARID1A has established roles in cell division and proliferation by regulating cell cycle entry and progression (15). In gynecologic cancers, restoration of wild-type ARID1A expression resulted in suppression of cell proliferation and tumor growth in mice while silencing ARID1A enhanced tumorigenicity (16). In mice model, ARID1A-deficient adenocarcinoma resembling human CRC lacks APC/β-catenin, a key gatekeeper in the regulation of gene expression (17). Existing preclinical data in gastric and biliary cancers have demonstrated similar findings supporting ARID1A as a tumor suppressor (18–20). Retrospective clinical data in CRC reveals association of ARID1A loss with late TNM stage, distant metastasis, and poor grade (21).
In addition to the functions related to cellular proliferation and gene expression, the role of this protein in genomic stability and prevention of structural aberrations in chromosomes has been proposed. One suggested mechanism shown by an in vitro study has described interaction of ARID1A with topoisomerase IIα and facilitating chromosome segregation during mitosis (22). Moreover, SWI/SNF complexes have been demonstrated to contribute to the repair of DNA double-strand breaks by promoting ATM-mediated phosphorylation of H2AX (23, 24). Also, SWI/SNF complexes have been proposed to have roles in other forms of DNA repair including nucleotide excision repair, the repair of pyrimidine dimers, and chemical-induced crosslinking of DNA (25–27). MMR deficiency and microsatellite-instability-high (MSI-H) phenotypes are associated with ARID1A mutation in various tumor types such as gastric and colorectal cancer (28–31) but it is not completely clear if the mutation is the result or the cause of MMR deficiency. A recent preclinical study has shown a reduced mismatch-repair capacity and a substantially enhanced repair capacity in ARID1A-null cells but with ARID1A expression (5). In a proteomic screen, MSH2, an important mediator in mismatch-repair, was found to be a binding partner with ARID1A. Immunoprecipitation assays further confirmed ARID1A-MSH2 interaction, which is likely mediated through the C-terminal regional of ARID1A and the N-terminal region of MSH2 (5). Also, in cell lines with intact MMR protein expression (MLH1, MSH2, and MSH6), a reduced ARID1A expression correlated with lower MMR capability, and this is regardless of ARID1A’s transcriptional regulatory role. Using orthotopic implantation of these cell lines into immunocompetent mouse models, these studies found that ARID1A deficient cell lines show MMR-defective phenotype with an increased level of infiltrating T-lymphocytes (5).
The majority of the mutations in ARID1A are non-sense or frameshift in CRC and result in truncation and functional loss of the protein (14). Despite the established role of this protein in SWI/SNF complex, the role of ARID1A mutation and its association with immune infiltration are not completely understood. Given the proposed role of this protein in DNA-mismatch repair (per mouse models), we hypothesized that ARID1A mutation in MSS CRC would lead to hypermutation and an increase in the expression of gene sets related to the immune response.
Materials and Methods
DNA sequencing, gene expression profiling, and clinical data of CRC patients from MD Anderson Cancer Center (MDACC) and The Cancer Genomic Atlas (TCGA) were used to assess the effect of ARID1A mutation.
The mRNA expression data of the TCGA CRC cohort was generated by Illumina HiSeq and GA platforms. The data was normalized, log-transformed and corrected for batch effect of the sequencing platform. In case of the MD Anderson cohort, the mRNA expression was profiled using Agilent microarrays (Agilent Technologies, Santa Clara, CA, USA). The data was preprocessed using Loess based normalization followed by background correction. Differential gene expression analysis was conducted using DESeq2 under the assumption of negative binomial distribution for the underlying gene expression count matrix and applied generalized linear model with Wald statistical test (32). Additional universal validation was performed using gene set enrichment analysis (GSEA) to examine the relation between ARID1A mutated and other hallmark gene sets (33). To analyze differentially regulated pathways and enrichment of immune signatures specifically, we used GO enrichment analysis using R package clusterProfiler, with a Bonferroni correction and p-value cutoff of 0.05 (34). We considered a gene set to be enriched when it was included in the top 100 rank in at least two subsets with a p value < 0.05, fold change greater than one and a False Discovery Rate (FDR) < 25%.
Exome-sequencing (WES) data from TCGA and MDACC was used to assess the mutational status of ARID1A in CRC. Whole-exome sequencing of MDACC cohort had been performed using HiSeq2000 system by sequencing core facility at the institution (Illumina, San Diego, CA) at a depth of at least 50x, achieving at least 80% coverage of mapping bases with at least 8x coverage and 94% of the genome being sequenced. The exome data of both cohorts were aligned to Human genome (hg19) using BWA. The variants were identified by Mutect2 after pre-processing the data in GATK pipeline (35). Variants with at least a sequencing depth of 30 and alternate alleles supported by 5% of reads were selected. Mutational status of ARID1A was defined by presence any non-silent mutation in coding region of the gene. Genes with frequent mutations in CRC were assessed for their association with the total mutational burden (TMB), frameshift mutation rate, and gene signatures of the immune response along with ARID1A.
MSI status for both TCGA and MD Anderson cohorts was determined using Immunohistochemistry (IHC) or Polymerase chain reaction (PCR) as previously described in the literature (36, 37). Additionally, we applied MSISensor (version 0.5) to identify MSI status using WES data of both cohorts. The samples were classified as MSS if MSISensor score less than 3.5 and MSI if greater than or equal to 3.5. MSISensor resulted in 100% agreement with the MSI status determined by IHC and PCR (38).
Consensus molecular subtypes (CMSs) is an established classification system in CRC; according to gene expression described in prior publication, each subtype has unique molecular and metabolic characteristics. The subtypes were defined using a large-scale analytical study interconnecting 6 CRC classification systems. The subtypes are microsatellite instability/immune (CMS1), canonical (CMS2), metabolic (CMS3), and mesenchymal (CMS4) (39). In this present study, ARID1A mutational rate was evaluated in the context of CMS subtypes of TCGA and MD Anderson cohorts.
TMB and frameshift mutation rate of MSS CRC cases were compared according to ARID1A mutational status. An external cohort was used for validation of this analysis (40). Clonality was defined as >25% of maximal allele frequency in the tumors.
Gene signatures for IFN-γ pathway and other components of immune response (Table S1) were utilized to analyze the differential RNA expression between ARID1A mutant (mt) and ARID1A wild-type (wt) cases (41, 42). In addition to ARID1A, other genes with frequent mutations in CRC (mutation frequency >5%) were assessed for their association with TMB, frameshift mutation rate, and with the expression of gene sets related to the immune response.
We also evaluated the tumor infiltration of T lymphocytes in MSS CRC according to ARID1A mutational status. Immunohistochemistry (IHC) staining was performed on FFPE tumor blocks by using the Opal fIHC Kit (PerkinElmer, Waltham, MA) as described previously (43–45). The CD3 immunofluorescence antibody for T cells was used (Dako, Carpentaria, CA). The final data were reported as number of cells/mm2.
At the end, we assessed the association of the ARID1A mt with clinical characteristics such as gender, age at the time of diagnosis, primary tumor location (right vs. left), stage, and race in MSS CRC cases. Using these variables, we performed univariate and multivariate Cox regression analyses to determine the association of ARID1A mutation with overall survival.
Statistical Analysis
The data for gene expression and the mutational burden was compared according to the ARID1A mutational status using a non-parametric (Mann-Whitney U) test. The association between ARID1A mutations and the binomial features was analyzed using χ2 test. Statistical analysis was performed using R (version 4.0.2; R Foundation for Statistical Computing, Vienna, Austria; http://www.r-project.org/) and SPSS Windows (version 24) software program (SPSS Inc, Chicago, IL, USA). All p values were 2-sided, and statistical significance was set at p<0.05. The p values for expression analyses were adjusted for multiple comparisons with a false discovery rate correction at q < 0.1.
Results
Among 502 CRC cases in MD Anderson and TCGA cohorts, 56 (11.1%) cases had a non-silent mutation in ARID1A. Among 419 patients with MSS CRC, 28 patients (6.7%) had a non-silent mutation in ARID1A. The mutation map for ARID1A gene in MSS CRC is included in supplements (Fig S1). Among 28 patients, 18 (64.2%) had inactivating mutation in ARID1A gene. Median TMB and frameshift mutation rate for all MSS CRC cases were 4.3/mb and 4.0/mb, respectively.
Non-silent mutation in the ARID1A gene was associated with an increase in TMB in MSS CRC (median mutation rate of 4.3/mb vs. 7.5/mb in wt and mt cases, respectively, p=0.045). The mutation was also associated with a higher rate of frameshift mutations in MSS CRC (median frameshift mutation rate of 4.0/mb vs. 32.0/mb in wt and mutated cases, respectively, p<.001) (Fig 1). While 41% of ARID1A mutant cases had TMB ≥ 10 mutations/Mb, only 10% of ARID1A wild-type cases had TMB ≥ 10 mutations/Mb. The findings for frameshift mutation rate and TMB were validated using MSKCC database. (p=0.002, and p<0.001, respectively).
Fig 1. TMB and frameshift mutation rate in MSS CRC according to the ARID1A mutational status.
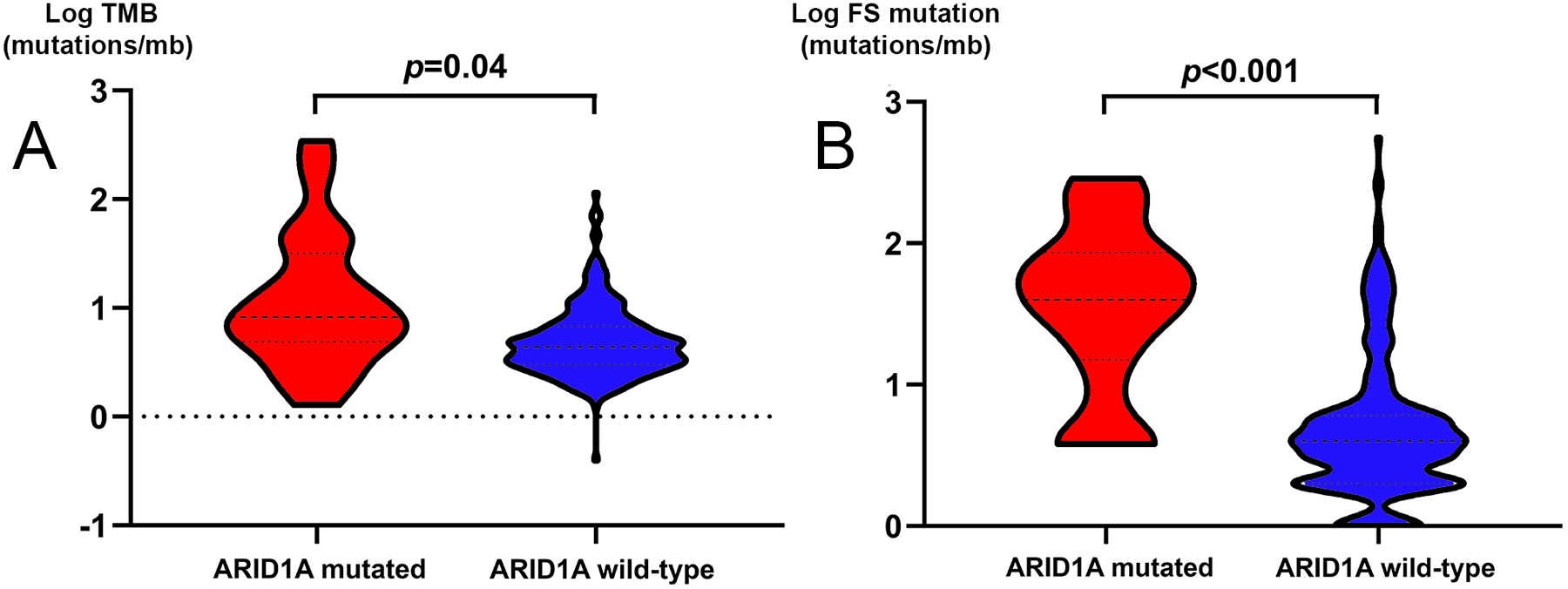
A, Violin plot of TMB in ARID1A wt and ARID1A mt in MSS CRC. B, Violin plot of frameshift mutation rate in ARID1A wt and ARID1A mt in MSS CRC.
In order to adjust for the potential confounding of high mutation rate resulting in higher number of passenger mutations in ARID1A, we conducted several additional analyses. If the ARID1A mutation was a passenger event, its frequency would correspond to the gene size. However, as shown in Figure S2, ARID1A mutation results in higher mutation rate than would be expected based on gene size alone. Second, not all ARID1A mutations are likely functional, although frameshift and nonsense mutations result in clear functional significance. Indeed, the association with increase in TMB and frameshift mutations was retained for inactivating mutation in ARID1A (p=0.008 and p=0.001, respectively) but were not observed for ARID1A missense mutations (p=0.8 and p=0.15, respectively). Third, we demonstrate that clonality impacts TMB and frameshift rates, with tumors with clonal inactivating mutations maintaining the association, while subclonal mutations do not have the same association, (p=0.001 and p<0.001, respectively). Finally, we assessed the impact of ARID1A copy number loss and found a significantly higher rate of frameshift mutations and TMB compared with those with preserved copy number (p=0.004 and p=0.016, respectively).
Next, in order to further evaluate the association of ARID1A mutation with the presence of frameshift mutation, we compared the frameshift mutation rates with mutational status of genes that are commonly mutated in MSS CRC (mutation frequency >5%). In MSS CRC, and out of the 58 genes most commonly mutated, a non-silent mutation in ARID1A had the strongest association with the frameshift mutation rate (8-fold increase for ARID1A mt cases compared to ARID1A wt, p<0.001) (Fig 2A).
Fig 2.
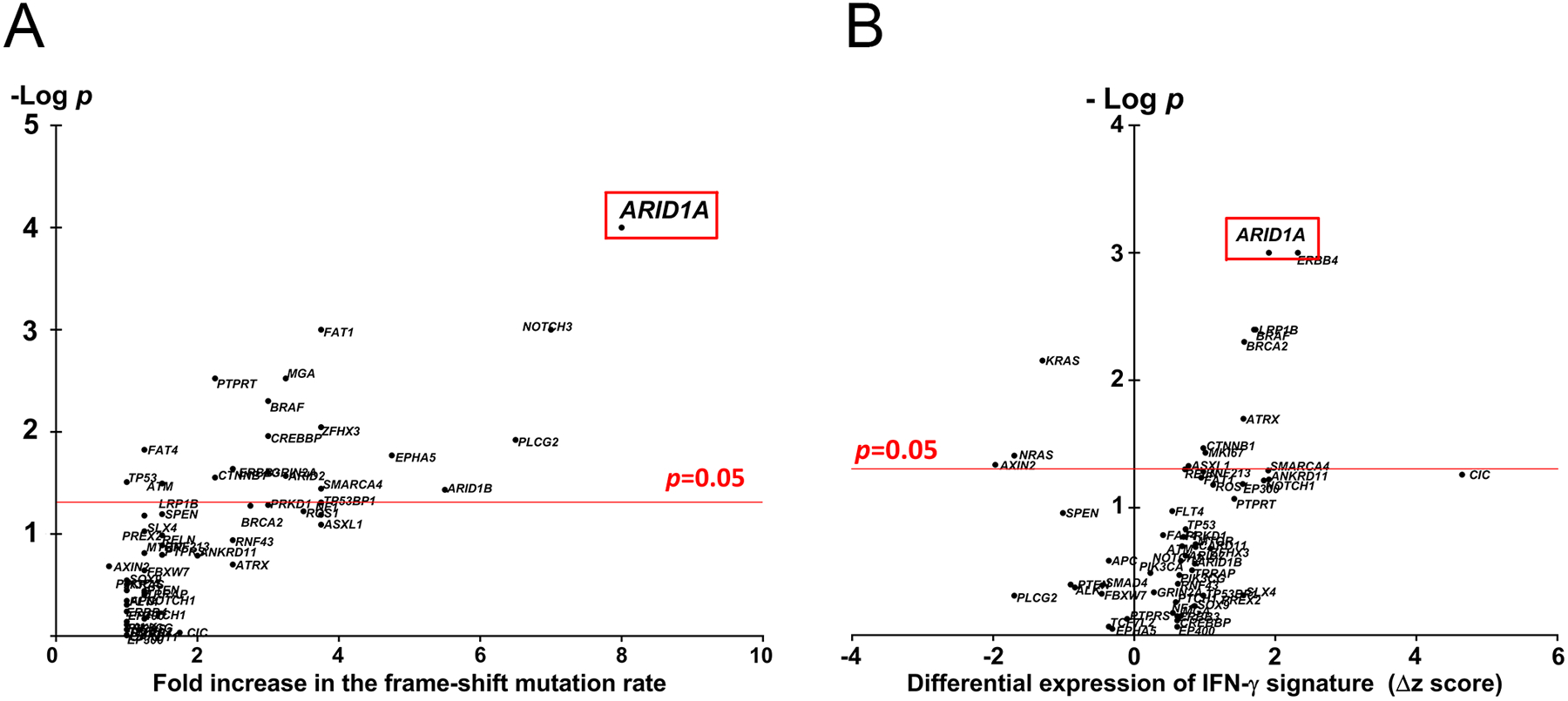
A, Association of frameshift mutation rate with the mutational status of genes commonly mutated in MSS CRC. B, Association of the differential expression of the IFN-γ pathway and mutational status of genes commonly mutated in MSS CRC.
In MSS CRC, ARID1A mutation had also a strong correlation with an increase in the expression of the IFN-γ pathway (Δz score +1.91, p=0.001) (Fig 2B).
Higher mutation rate and increase in the IFN-γ expression can be reflective of a larger gene size; however, it was noted that in comparison to other commonly mutated genes, a high increase in IFN-γ expression in ARID1A mutated cases is not due to the gene size (Fig S3).
The distribution of ARID1A mt across different molecular subtypes in all CRC cases (MSI-H and MSS) as well as MSS cases is shown in Fig 3A and Fig 3B, respectively. Out of all ARID1A mt cases, 31 (55.4%) were in CMS1. The strong enrichment of this mutation in CMS1 is due to co-occurrence with MSI-H (out of 68 MSI-H cases, 28 (40.5%) had ARID1A mutation). In MSS CRC, ARID1A mutation was still enriched in CMS1 (immune subtype) cases (7/21, 33.3%).
Fig 3. The enrichment of ARID1A mutation across different molecular subtype of CRC.
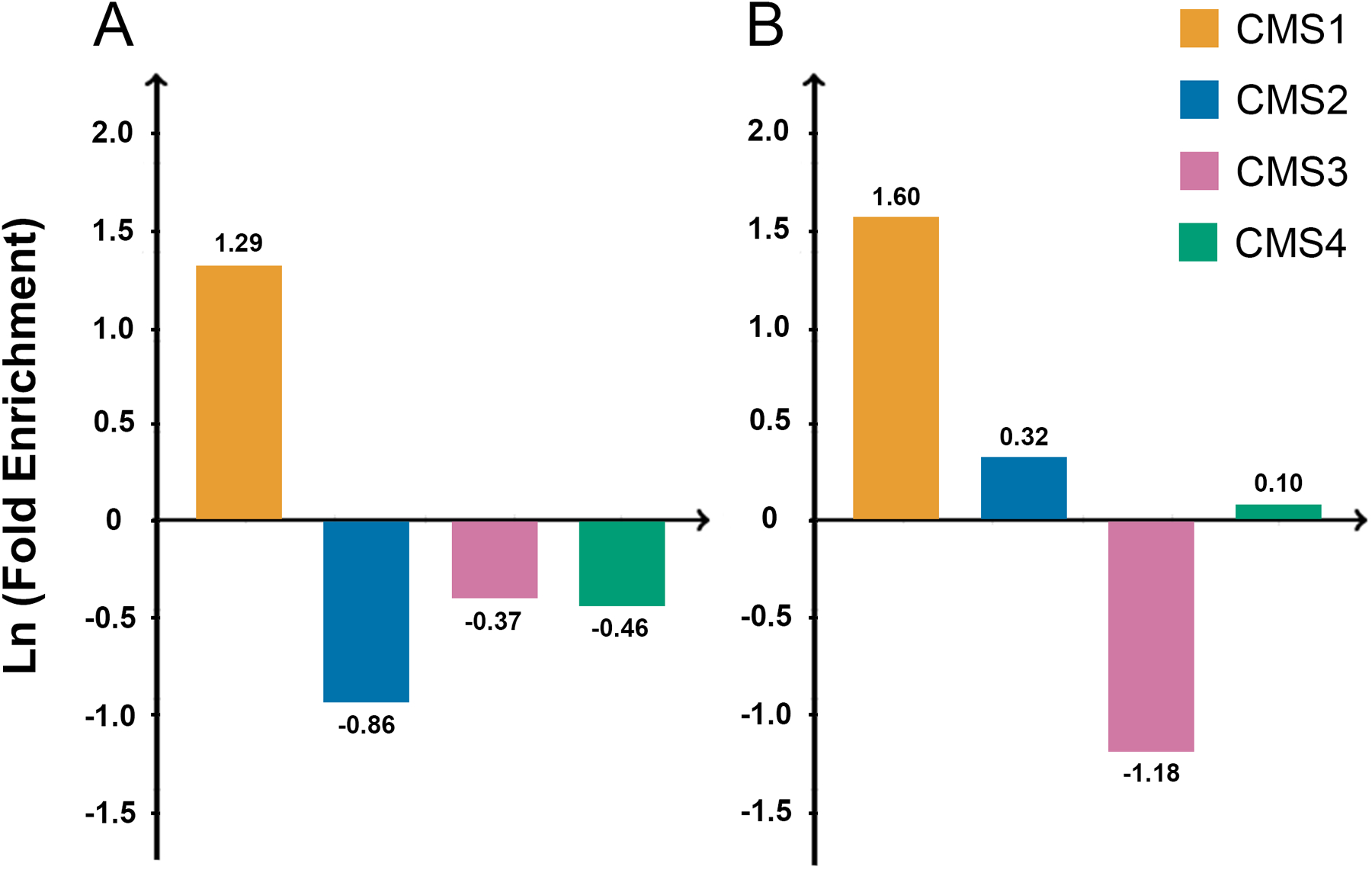
A, Fold enrichment of ARID1A mutation in each molecular subtype in all cases (MSI-H/MSS). B, Fold enrichment of ARID1A mutation in each molecular subtype in MSS CRC.
In MSS CRC, in order to further understand the association of ARID1A mutation with immune response, we looked beyond IFN-γ pathway. ARID1A mutation was associated with an increase in the expression of gene sets involved in the immune response (Fig 4A, 4B). An increase in the expression of gene sets related to NK cell, T reg, and M2 macrophage, and myeloid-derived suppressor cell (MDSC) were also observed.
Fig 4.
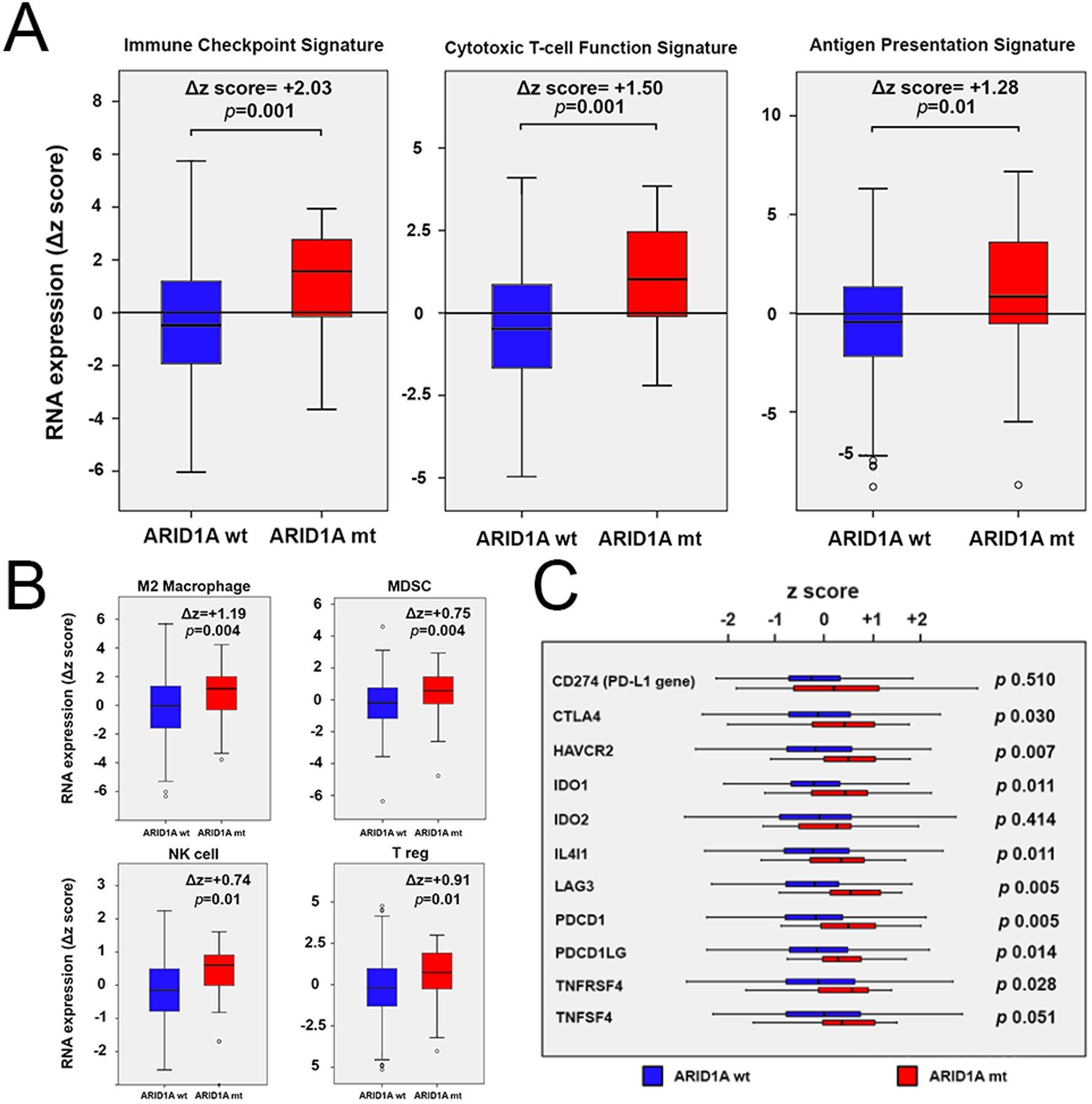
A, B, RNA expression of gene sets related to immune response in MSS CRC according to the ARID1A mutational status. C, RNA expressions of single genes involved in the immune response in MSS CRC cases according to the ARID1A mutational status.
In MSS CRC, ARID1A mutation was also associated with increased expression of immune checkpoint and key genes known to be associated with immune response (Fig 4C).
In further exploratory analyses, these hypothesis-directed finding was further validated in an unbiased differential gene expression (DEGs) analysis comparing tumors with and without ARID1A mutation. Gene set enrichment analysis demonstrated enrichment of genes involved in immune response, IFN-γ, interleukin (IL)-2, and immune response signaling (q < 0.1). The top 10 gene sets are associated with immune response signatures (Supplementary Figure S4).
In contrast to MSS, in MSI-H cases, no statistically significant difference in the expression of IFN-γ signature, frameshift mutation rate and TMB was observed between ARID1A mt and ARID1A wt cases.
In order to validate the findings seen bioinformatically, we analyzed a cohort of specimens by immunohistochemistry for CD3+ cells in cases with MSS CRC and then those with MSI-H CRC. Out of 58 samples with MSS CRC, 3 cases had ARID1A mutation. Out of 10 cases with MSI-H CRC, 5 cases had ARID1A mutation. Although limited by sample size, in comparison to ARID1A wt cases, higher intratumoral infiltration of T lymphocytes was observed in ARID1A mt samples (p=0.01) while no difference was observed in MSI-H tumors with or without ARID1A mutation (p=0.17) (Fig 5A1, 5A2, 5B, and 5C).
Fig 5.
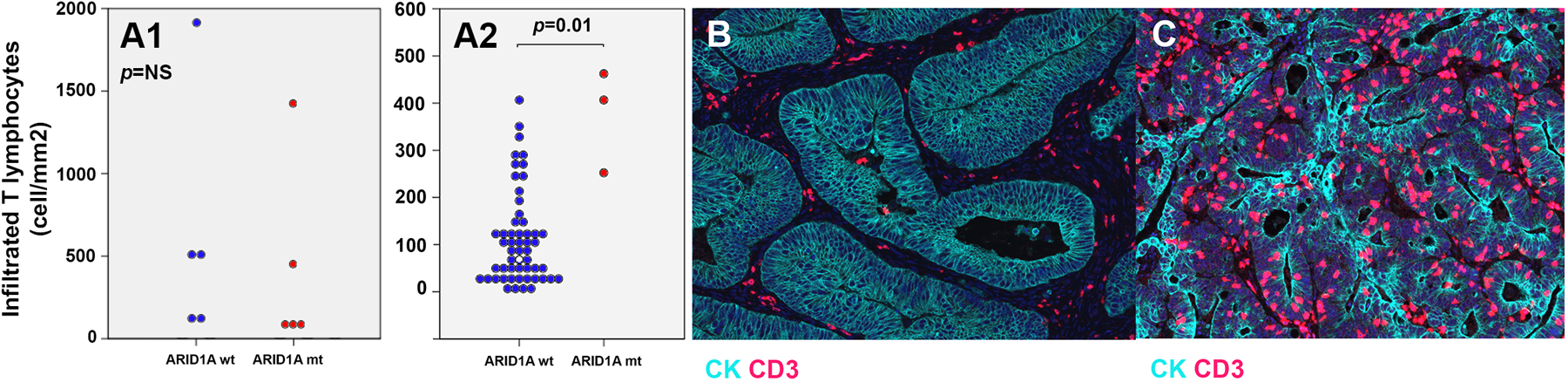
A1. Infiltration of T lymphocytes in the tumor of patients with MSI-H CRC in ARID1A mt and ARID1A wt cases. A2. Infiltration of T lymphocytes in the tumor of patients with MSS CRC in ARID1A mt and ARID1A wt cases. B, C, Higher intratumoral infiltration of T lymphocytes in an ARID1A mt MSS CRC patient (C) in comparison with that in an ARID1A wt MSS CRC case (B).
In MSS CRC, there was no associated between ARID1A mt and age at diagnosis, gender, race, primary tumor location (right vs. left), and stage at the time of diagnosis. ARID1A mutation was not associated with poor overall survival in MSS CRC patients.
Discussion
Prognosis of patients with metastatic CRC remains poor and given the heterogeneity of the disease, identifying immunologically active subsets to enhance immune response is crucial. ARID1A protein – as an important subunit of SWI/SNF complex - has been shown to contribute to cellular division, proliferation, and gene expression. The role of this protein in DNA repair, in cooperation with MMR proteins, has been revealed in preclinical models and the clinical characteristics of the loss and mutation of this protein have been investigated in retrospective studies. In this study, we discovered a strong association between ARID1A mutation and an increase in TMB and expression of genes (and gene sets) related to the immune response in MSS CRC. We also observed that in MSS CRC, compared with other commonly mutated genes, a non-silent mutation in the ARID1A gene was associated with the highest increase in the expression of IFN-γ pathway.
We also evaluated the correlation of ARID1A mutation with frameshift mutation rate (in addition to TMB) and validated these findings in a separate external cohort. The immunogenicity of ARID1A mutant cases in the MSS CRC is likely due to the increased level of neoantigens resulting from the increased TMB and frameshift mutations. Given recent FDA approval of pembrolizumab for unresectable or metastatic solid tumors with TMB≥10 mutations/Mb, further investigation of TMB in ARID1A mutant cases seems reasonable.
While TMB is a well-established biomarker that predicts a favorable response to immune therapy (46), the role of frameshift mutation rate and immune response is less defined. The immunogenicity of frameshift mutations (i.e., insertions or deletions) and its positive correlation with response to immune checkpoint blockade have been previously proposed in some tumor types (47, 48). For example, high frameshift mutation burden in renal cell carcinoma and melanoma is associated with increase in the CTL infiltration and improvement in the response to immune checkpoint inhibitors (49). The out-of-frame frameshift mutations alter the downstream DNA reading frames and therefore, could produce a higher level of neoantigens, if expressed. Hence, frameshift mutations compared with TMB (which includes all single-nucleotide variations) are felt to be more immunogenic and a better marker of response to immune-checkpoint inhibition (49). In our study, the majority of the increase in TMB was from an increase in frameshift mutation rate and thus resulting in increase in immune response.
In this study, we have shown that in MSS CRC, ARID1A mutation is correlated with higher expression of various genes and gene sets involved in the immune response. The role of ARID1A mutation in the immune microenvironment has been explored in a few studies thus far. Some studies in non-CRC cancers (e.g., in gastric cancer) have illustrated the linkage of the loss of ARID1A expression and PD-L1 expression (50, 51). A pan-cancer and a GI specific study revealed a high TMB and CD8(+) infiltrating T-cells in ARID1A altered tumors but CRC was not analyzed specifically and MSI-H cases were also included in the analysis (52, 53).
Although the recent approval of pembrolizumab for high TMB patients would suggest opportunities for treatment of these patients with ARID1A mutated MSS tumors, the overall activity of PD-1 inhibition in high-TMB MSS colorectal cancer patients is low (54). In support of this, a recent preclinical study suggests that ARID1A deficient tumors may have additional barriers to an effective immune response, including decreased expression of CXCL9, CXCL10, and an impaired IFN-γ expression in preclinical models. This was associated with a poor response to immune therapy in ARID1A deficient tumors, including reduced activity in shARID1A MC38 model with PD-L1 monoclonal antibody. These findings will be critical to integrate in applying our work to potential therapeutic strategies in the future (55).
In the present study, we also found a strong enrichment of ARID1A mutation in CMS1 CRC. This is likely due to the strong association between the mutation and MSI-H phenotype. While the correlation has also been extensively described in different tumor types, the causation is unclear. It is not completely understood if the mutation is the result of MMR deficiency or it is the cause of it (13, 15, 28, 29, 56). We further explored the role of ARID1A mutation in the MSI-H subgroup, although this was not the main objective of our study. The rate of frameshift or TMB, as well as the expressions of immune gene sets in ARID1A-mutant MSI-H were not significantly different from those in ARID1-wt MSI-H cases. This finding supports the contributory effect of ARID1A in DNA repair and reveals that a dysfunctional DNA repair state due to MMR defect is not attenuated by an intact ARID1A protein.
The limitations of our study are in part due to its retrospective nature and relatively small number of ARID1A mutant cases. The ARID1A mutation is an uncommon subgroup of CRC cases and our findings need to be validated in larger cohorts of patients. Although the strong correlation between mutation rate and neoantigen level has been shown when working with whole-exome sequencing data (57), in our study, we did not directly measure the neoantigen production in ARID1A mutant cases. While we performed an orthogonal validation of our bioinformatic findings utilizing IHC, we acknowledge that our IHC validation cohort has small number of ARID1A mutated cases and requires further integrated analyses with other immune markers and further characterization of the T-cell subsets present. While the correlation of ARID1A mutation with high mutational burden was observed in our study, the impact of this mutation on MSH2 needs additional functional evaluation.
Immune infiltration has been shown to have a reproducible prognostic impact on MSS CRC however, the molecular determinants of this have not been well described. In conclusion, we suggest that tumors with ARID1A mutation may define an immunologically active subtype of MSS colorectal. Finally, ARID1A mutant MSS CRC should be explicitly explored as a discrete subgroup in future immunotherapy trials.
Supplementary Material
Statement of Translational Relevance.
Identifying immunologically active subgroups in microsatellite-stable colorectal cancer (MSS CRC) is crucial. Recent preclinical models have proposed a role of ARID1A in DNA mismatch-repair. In this study, we demonstrate an association between ARID1A mutation, increased frameshift mutation rates, and makers of immune activation in MSS CRC patients. Intratumoral T-cell infiltration was confirmed in patient specimens, confirming a link between ARID1A mutation and an immunologically active subgroup. As only 6.7% of MSS CRC patients have ARID1A mutation, rare responses to immunotherapy in this subgroup may have been missed, and future studies enriching for this population are warranted.
Acknowledgments:
This study is supported by the UT MD Anderson Cancer Center Moon Shot Program, Support Grant No. P30CA016672 and NIH Grant No. R01CA184843 (S.K.). The results presented here are in part based upon data generated by TCGA Research Network (http://cancergenome.nih.gov/).
Footnotes
Conflict of Interest:
The authors declare that they have no conflicts of interest.
References:
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- 2.Sambi M, Bagheri L, Szewczuk MR. Current Challenges in Cancer Immunotherapy: Multimodal Approaches to Improve Efficacy and Patient Response Rates. Journal of oncology. 2019;2019:4508794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nature reviews Gastroenterology & hepatology. 2019;16(6):361–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Molinari C, Marisi G, Passardi A, Matteucci L, De Maio G, Ulivi P. Heterogeneity in Colorectal Cancer: A Challenge for Personalized Medicine? International journal of molecular sciences. 2018;19(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nature medicine. 2018;24(5):556–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annual review of biochemistry. 2009;78:273–304. [DOI] [PubMed] [Google Scholar]
- 7.Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PloS one. 2013;8(1):e55119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Nagl NG Jr., Flowers S, Zweitzig D, Dallas PB, Moran E. Expression of p270 (ARID1A), a component of human SWI/SNF complexes, in human tumors. International journal of cancer. 2004;112(4):636. [DOI] [PubMed] [Google Scholar]
- 9.Huang J, Zhao YL, Li Y, Fletcher JA, Xiao S. Genomic and functional evidence for an ARID1A tumor suppressor role. Genes, chromosomes & cancer. 2007;46(8):745–50. [DOI] [PubMed] [Google Scholar]
- 10.Pavlidou EN, Balis V. Diagnostic significance and prognostic role of the ARID1A gene in cancer outcomes (Review). World Acad Sci J. 2020;2(2):49–64. [Google Scholar]
- 11.Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer discovery. 2013;3(1):35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science (New York, NY). 2010;330(6001):228–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones S, Li M, Parsons DW, Zhang X, Wesseling J, Kristel P, et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Human mutation. 2012;33(1):100–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sarshekeh AM, Loree JM, Manyam GC, Pereira AAL, Raghav KPS, Lam M, et al. The characteristics of ARID1A mutations in colorectal cancer. Journal of Clinical Oncology. 2018;36(15_suppl):3595–. [Google Scholar]
- 15.Wu RC, Wang TL, Shih Ie M. The emerging roles of ARID1A in tumor suppression. Cancer biology & therapy. 2014;15(6):655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guan B, Wang TL, Shih Ie M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer research. 2011;71(21):6718–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathur R, Alver BH, San Roman AK, Wilson BG, Wang X, Agoston AT, et al. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nature genetics. 2017;49(2):296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nature genetics. 2012;44(5):570–4. [DOI] [PubMed] [Google Scholar]
- 19.Chan-On W, Nairismägi ML, Ong CK, Lim WK, Dima S, Pairojkul C, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nature genetics. 2013;45(12):1474–8. [DOI] [PubMed] [Google Scholar]
- 20.Wang DD, Chen YB, Pan K, Wang W, Chen SP, Chen JG, et al. Decreased expression of the ARID1A gene is associated with poor prognosis in primary gastric cancer. PloS one. 2012;7(7):e40364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei XL, Wang DS, Xi SY, Wu WJ, Chen DL, Zeng ZL, et al. Clinicopathologic and prognostic relevance of ARID1A protein loss in colorectal cancer. World journal of gastroenterology. 2014;20(48):18404–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dykhuizen EC, Hargreaves DC, Miller EL, Cui K, Korshunov A, Kool M, et al. BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature. 2013;497(7451):624–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee HS, Park JH, Kim SJ, Kwon SJ, Kwon J. A cooperative activation loop among SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. The EMBO journal. 2010;29(8):1434–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park JH, Park EJ, Lee HS, Kim SJ, Hur SK, Imbalzano AN, et al. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. The EMBO journal. 2006;25(17):3986–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong F, Fahy D, Liu H, Wang W, Smerdon MJ. Role of the mammalian SWI/SNF chromatin remodeling complex in the cellular response to UV damage. Cell cycle (Georgetown, Tex). 2008;7(8):1067–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao Q, Wang QE, Ray A, Wani G, Han C, Milum K, et al. Modulation of nucleotide excision repair by mammalian SWI/SNF chromatin-remodeling complex. The Journal of biological chemistry. 2009;284(44):30424–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kothandapani A, Gopalakrishnan K, Kahali B, Reisman D, Patrick SM. Downregulation of SWI/SNF chromatin remodeling factor subunits modulates cisplatin cytotoxicity. Experimental cell research. 2012;318(16):1973–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allo G, Bernardini MQ, Wu RC, Shih Ie M, Kalloger S, Pollett A, et al. ARID1A loss correlates with mismatch repair deficiency and intact p53 expression in high-grade endometrial carcinomas. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2014;27(2):255–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bosse T, ter Haar NT, Seeber LM, v Diest PJ, Hes FJ, Vasen HF, et al. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2013;26(11):1525–35. [DOI] [PubMed] [Google Scholar]
- 30.Wang K, Kan J, Yuen ST, Shi ST, Chu KM, Law S, et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nature genetics. 2011;43(12):1219–23. [DOI] [PubMed] [Google Scholar]
- 31.Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487(7407):330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15(12):550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(43):15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics : a journal of integrative biology. 2012;16(5):284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Current protocols in bioinformatics. 2013;43(1110):11.0.1–.0.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sepulveda AR, Hamilton SR, Allegra CJ, Grody W, Cushman-Vokoun AM, Funkhouser WK, et al. Molecular Biomarkers for the Evaluation of Colorectal Cancer: Guideline From the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology, and the American Society of Clinical Oncology. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2017;35(13):1453–86. [DOI] [PubMed] [Google Scholar]
- 37.Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer research. 1998;58(22):5248–57. [PubMed] [Google Scholar]
- 38.Niu B, Ye K, Zhang Q, Lu C, Xie M, McLellan MD, et al. MSIsensor: microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics (Oxford, England). 2014;30(7):1015–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nature medicine. 2015;21(11):1350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yaeger R, Chatila WK, Lipsyc MD, Hechtman JF, Cercek A, Sanchez-Vega F, et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer cell. 2018;33(1):125–36.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin Y, Ekmekcioglu S, Forget MA, Szekvolgyi L, Hwu P, Grimm EA, et al. Cervical Cancer Neoantigen Landscape and Immune Activity is Associated with Human Papillomavirus Master Regulators. Frontiers in immunology. 2017;8:689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. The Journal of clinical investigation. 2017;127(8):2930–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parra ER, Uraoka N, Jiang M, Cook P, Gibbons D, Forget MA, et al. Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Sci Rep. 2017;7(1):13380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stack EC, Wang C, Roman KA, Hoyt CC. Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of Tyramide signal amplification, multispectral imaging and multiplex analysis. Methods (San Diego, Calif). 2014;70(1):46–58. [DOI] [PubMed] [Google Scholar]
- 45.Parra ER, Jiang M, Solis L, Mino B, Laberiano C, Hernandez S, et al. Procedural Requirements and Recommendations for Multiplex Immunofluorescence Tyramide Signal Amplification Assays to Support Translational Oncology Studies. Cancers. 2020;12(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Annals of oncology : official journal of the European Society for Medical Oncology. 2019;30(1):44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chae YK, Viveiros P, Lopes G, Sukhadia B, Sheikh MM, Saravia D, et al. Clinical and Immunological Implications of Frameshift Mutations in Lung Cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2019;14(10):1807–17. [DOI] [PubMed] [Google Scholar]
- 48.Hanna GJ, Lizotte P, Cavanaugh M, Kuo FC, Shivdasani P, Frieden A, et al. Frameshift events predict anti-PD-1/L1 response in head and neck cancer. JCI insight. 2018;3(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. The Lancet Oncology. 2017;18(8):1009–21. [DOI] [PubMed] [Google Scholar]
- 50.Tober JM, Halske C, Behrens HM, Kruger S, Rocken C. Intratumoral heterogeneity and loss of ARID1A expression in gastric cancer correlates with increased PD-L1 expression in Western patients. Human pathology. 2019;94:98–109. [DOI] [PubMed] [Google Scholar]
- 51.Kim YB, Ahn JM, Bae WJ, Sung CO, Lee D. Functional loss of ARID1A is tightly associated with high PD-L1 expression in gastric cancer. International journal of cancer. 2019;145(4):916–26. [DOI] [PubMed] [Google Scholar]
- 52.Jiang T, Chen X, Su C, Ren S, Zhou C. Pan-cancer analysis of ARID1A Alterations as Biomarkers for Immunotherapy Outcomes. Journal of Cancer. 2020;11(4):776–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li L, Li M, Jiang Z, Wang X. ARID1A Mutations Are Associated with Increased Immune Activity in Gastrointestinal Cancer. Cells. 2019;8(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen EX, Jonker DJ, Loree JM, Kennecke HF, Berry SR, Couture F, et al. CCTG CO.26: Updated analysis and impact of plasma-detected microsatellite stability (MSS) and tumor mutation burden (TMB) in a phase II trial of durvalumab (D) plus tremelimumab (T) and best supportive care (BSC) versus BSC alone in patients (pts) with refractory metastatic colorectal carcinoma (rmCRC). Journal of Clinical Oncology. 2019;37(15_suppl):3512–. [Google Scholar]
- 55.Li J, Wang W, Zhang Y, Cieślik M, Guo J, Tan M, et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. The Journal of clinical investigation. 2020;130(5):2712–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Abe H, Maeda D, Hino R, Otake Y, Isogai M, Ushiku AS, et al. ARID1A expression loss in gastric cancer: pathway-dependent roles with and without Epstein-Barr virus infection and microsatellite instability. Virchows Archiv : an international journal of pathology. 2012;461(4):367–77. [DOI] [PubMed] [Google Scholar]
- 57.Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell reports. 2016;15(4):857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.