

Abstract
Chronic wounds infected with pathogens such as Staphylococcus aureus represent a worldwide health concern, especially in patients with a compromised immune system. As antimicrobial resistance has become an immense global problem, novel antibiotics are urgently needed. One strategy to overcome this threatening situation is the search for drugs targeting novel binding sites on essential and validated enzymes such as the bacterial RNA polymerase (RNAP). In this work, we describe the establishment of an in vivo wound infection model based on the pathogen S. aureus and hairless Crl:SKH1-Hrhr (SKH1) mice. The model proved to be a valuable preclinical tool to study selected RNAP inhibitors after topical application. While rifampicin showed a reduction in the loss of body weight induced by the bacteria, an acceleration of wound healing kinetics, and a reduced number of colony forming units in the wound, the ureidothiophene-2-carboxylic acid 1 was inactive under in vivo conditions, probably due to strong plasma protein binding. The cocrystal structure of compound 1 with RNAP, that we hereby also present, will be of great value for applying appropriate structural modifications to further optimize the compound, especially in terms of plasma protein binding.
Keywords: wound infection model, Staphylococcus aureus, RNA polymerase inhibitor, anti-infectives, drug development, SKH1 mice
Graphical Abstract
Skin wounds, especially chronic wounds, are causing a cycle of pain, anxiety, and reduced quality of life for the patient and are thus a worldwide healthcare issue,1 causing a considerable cost to the public health care systems and to the patients.2 A major additional problem is the fact that >90% of chronic wounds are infected with bacteria and fungi,3 which negatively influence wound healing. The Gram-positive, facultative pathogen S. aureus is a common cause of surgical-site infections (SSIs), which are a major cause of poor health outcomes, including mortality.4 As severe wound infections by S. aureus benefit from a compromised host immune system, e.g., due to HIV, chemotherapeutically-treated cancer patients,5 and diabetes,6 people suffering from these diseases are most endangered for such ailments. Furthermore, antimicrobial resistance has become a global health concern due to the rapid spread of resistant pathogens7 and the shortage of effective treatment options.8 Due to this unmet medical need, the establishment of effective treatment measures by developing novel and potent anti-infectives is urgently needed, e.g., for a prophylactic treatment of S. aureus infections to prevent SSIs.
A validated but hitherto underexploited target for the development of broad-spectrum antibiotics is the bacterial RNA polymerase (RNAP). Although this enzyme is essential for growth and survival of bacteria, rifamycins and fidaxomicin are the only RNAP inhibitors used in clinical practice to date.9–12 Rifampicin (Rif), a member of the rifamycin family, plays a fundamental role in tuberculosis treatment in combination with other first-line agents such as pyrazinamide and isoniazid.13 However, its use is compromised due to the prevalence of Rif-resistant bacterial strains.14
To overcome the problem of Rif resistance, we explored the bacterial RNAP “switch region” as an alternative target site, which is distant from the Rif binding pocket.15,16 As the switch region is not overlapping with the Rif binding site and is highly conserved among Gram-positive and Gram-negative bacteria, it is an attractive target for the development of novel broad-spectrum antibiotics.10,17,18 On the basis of a virtual screening for binders of this site, we discovered and experimentally validated a hit compound with ureidothiophene-2-carboxylic acid core structure.15 In subsequent steps, about 200 derivatives were synthesized, resulting in promising compounds with optimized RNAP inhibitory activities.15,19,20 Our subsequent investigation demonstrated, among other findings, their in vitro potency against a clinically relevant set of methicillin-resistant S. aureus (MRSA) strains, a low frequency of resistance, and the verification of the switch region as binding site.15,16 Finally, on the basis of mechanistic function similarities between the RNAP switch region and the viral non-nucleoside reverse transcriptase inhibitor (NNRTI) binding site, these compounds were further developed as dual RNAP/RT inhibitors for the treatment of MRSA/HIV coinfections.21 Here, we determined the X-ray crystal structure of the E. coli RNAP holoenzyme in complex with ureidothiophene-2-carboxylic acid 1, structurally confirming that the RNAP switch region is the binding site of this class of RNAP inhibitors.
In the present work, we established and applied a simple wound infection model in hairless Crl:SKH1-Hrhr (SKH1) mice to evaluate the potency of RNAP inhibitors against S. aureus infections under in vivo conditions. On the basis of initial experiments, we selected the mouse pathogenic laboratory strain S. aureus Newman as the bacterium of choice for our subsequent experiments. Wounds treated with Rif and a selected RNAP-inhibiting ureidothiophene-2-carboxylic acid (compound 1) were examined for wound healing and body weight kinetics of the animals. As an indicator of disease progression, we measured the acute effects of the compounds on body weight, monitored the wound size development over a period of 14 days, and determined the number of colony forming units (CFU) in the wounds on day 14.
Our model led to fruitful results based on the performance of the two RNAP inhibitors used in this study. Furthermore, we report the establishment of an efficient and easy-to-use murine infection model that allows the testing of topical drug applications that rapidly provide preclinical in vivo data, which are valuable for the design and synthesis of novel anti-infectives.
RESULTS AND DISCUSSION
Establishment of an S. aureus-Based Wound Infection Model in SKH1 Mice.
In most cases, mice other than SKH1 were used to examine the antibacterial potency of compounds under in vivo conditions.22–24 In our experiments, we decided to use hairless SKH1 mice (Crl:SKH1-Hrhr) to expedite wounding without the need to depilate the animals. The latter not only is timesaving but also avoids an impairment of the skin by secondary effects derived from the depilation procedure. Furthermore, the use of hairless mice facilitates the mounting of adhesive plaster tape on top of the wounds, which ensures a permanently moist wound environment. SKH1 mice also display a thicker surface epidermis if compared to hairy mouse strains.25 Although many strains of hairless mice are immunodeficient, no decrements in immune function have been described for SKH1 mice. To our knowledge, we are the first to utilize SKH1 mice in the context of S. aureus-infected wounds followed by treatment with novel anti-infectives and continuous monitoring of parameters, such as the wound size.26–29
To develop a system being as user-friendly as possible, we reviewed but suspended challenging and time-consuming methods such as the dorsal skin fold chamber model,26 which are not mandatory for our experimental setup. We performed punch wounds 5 mm in diameter on both flanks of each mouse, stabilized the wounds with a 5 × 1 mm silicon ring to suppress wound contraction, and sealed them directly after wounding (plus treatment) with a transparent Tegaderm film (3M, Neuss, Germany).
In order to select a suitable S. aureus strain for our experiments, we investigated the effects of JE2 (USA300),30 LS1,31 Newman,32 and SA56433 (strains commonly used in various murine S. aureus infection models) on wound closure (Figure S1). After wounding and infecting the wounds with 10 μL of exponential growth phase cells, which were washed and resuspended in phosphate-buffered saline (PBS) to an optical density (OD600) of 1.0 (~108 CFU/mL), we determined the wound size every 2 d over a period of 14 d. While the uninfected control group showed an almost complete wound closure after 10–12 d, the closure occurred significantly more slowly in the infected animals. Especially between day 6 and 14, the wound closure was more delayed in the Newman strain compared to JE2 and LS1. Although the healing process was even slower in SA564, we decided to perform subsequent experiments with S. aureus Newman (SA-N) (Figures S2 and S3: macroscopic and microscopic images of representative wounds), due to the high standard deviation seen in the SA564 group.
To investigate the impact of the infection dose on SKH1 mice, we applied different numbers of bacteria to the wounds and determined the loss of body weight 2 d after wounding/infection. While the body weight did not change in the noninfected mouse group (−0.1 ± 2.0%), we observed a concentration-dependent weight loss in the SA-N infected groups (Figure 1). While mice, whose wounds were infected with 10 μL of an OD600 0.01 culture of SA-N (~106 CFU/mL), lost 5.6 ± 2.5% of their initial body weight, higher concentrations of bacteria led to a more prominent loss of weight (i.e., 8.6 ± 2.3% at OD600 0.1, and 10.5 ± 1.9% at OD600 1.0). On the basis of this information, we chose to perform all following experiments with an OD600 of 0.1 (~107 CFU/mL). The acute effect on body weight will serve as a simple and feasible indicator, which might be valuable for the following experiments where we expected our compounds to reduce the infection burden and consequently the effect on body weight loss.
Figure 1.
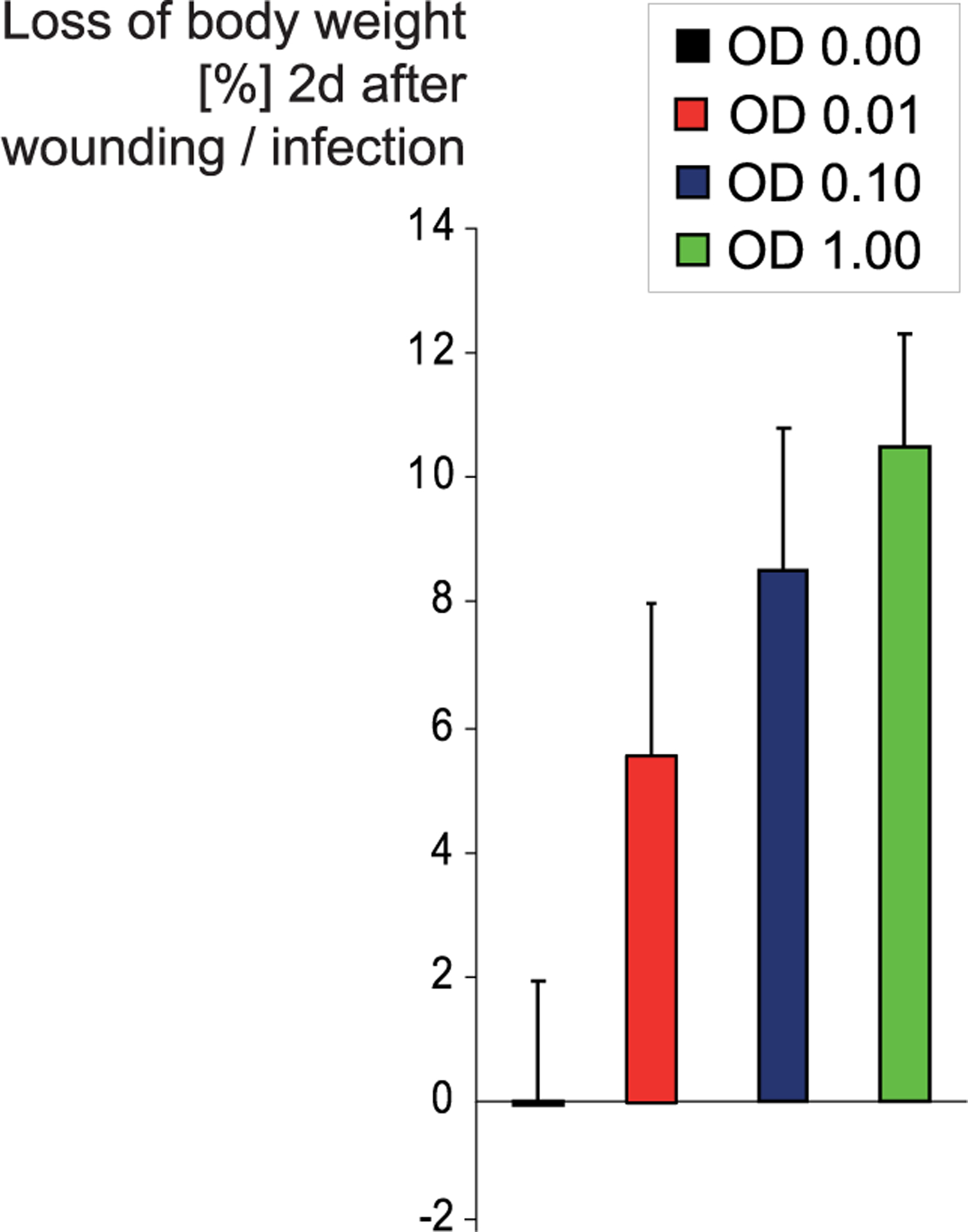
Influence of numbers of bacteria used for infection on the loss of body weight. Bacteria in the logarithmic growth phase were harvested, and their concentration was adjusted to OD600’s of 0.01, 0.10, and 1.00 (0 to ~108 CFU/mL). SKH1 mice were wounded twice dorsally with a 5 mm biopsy punch and subsequently infected with 10 μL of S. aureus Newman in the three mentioned concentrations or sham-infected (OD600 of 0.00). Both wounds of each mouse were treated identically. Two days after infection, the loss of body weight (of a total of 43 animals) compared to the weight on day 0 was determined.
Selection of RNAP Inhibitors to Be Investigated in Vivo.
The bacterial RNAP is a validated but nonetheless underexploited target. In recent years, different classes of highly promising compounds targeting this enzyme have been developed. These inhibitors range from small molecules9,11,15,19,21,34–37 to polymeric substances38 and natural products.18,39–41 Some of these compounds showed promising antibacterial activities in vitro, especially against Gram-positives, such as clinically relevant MRSA strains.16 Our wound infection model was developed to investigate the potential of such inhibitors under in vivo conditions.
Besides rifampicin42 (Figure 2), we aimed to investigate an RNAP-inhibiting small molecule. To choose the latter, we tested 200 previously described RNAP-inhibiting ureidothiophene-carboxylic acids15,19,21 (MW range: 262–562) with regard to their ability to affect the in vitro growth of SA-N. We investigated the most effective inhibitors with an IC50 of <50 μM against E. coli RNAP and an MIC of <10 μg/mL against SA-N with regard to cytotoxicity and solubility in PBS (the six most promising compounds are represented in Table S1). For our in vivo experiments, we finally chose compound 1 (Figure 2), which showed a relatively low IC50 of 14 μM against the E. coli RNAP and an MIC of 4 μg/mL against SA-N. Although the antibacterial activity is low when compared to clinically used Rif,15 it is in the range of other published RNAP-inhibiting synthetic small molecules.36,37,35 Furthermore, compound 1 displayed a low cytotoxicity (IC50 of 61 μM in HEK293 and >100 μM in NHDF cells), a solubility in PBS (including 1% DMSO) up to 100 μM, and a promising metabolic stability in human plasma and the liver S9 fraction. While the half-life (T1/2) in plasma was >60 min, compound 1 was significantly less stable (T1/2 = 9.1 ± 3.9 min) in the S9 fraction. This indicates that during a therapeutic application the inhibitor would be highly stable in the wound, while its degeneration in the liver would happen quickly, thus reducing the risk of undesired systemic side effects.
Figure 2.
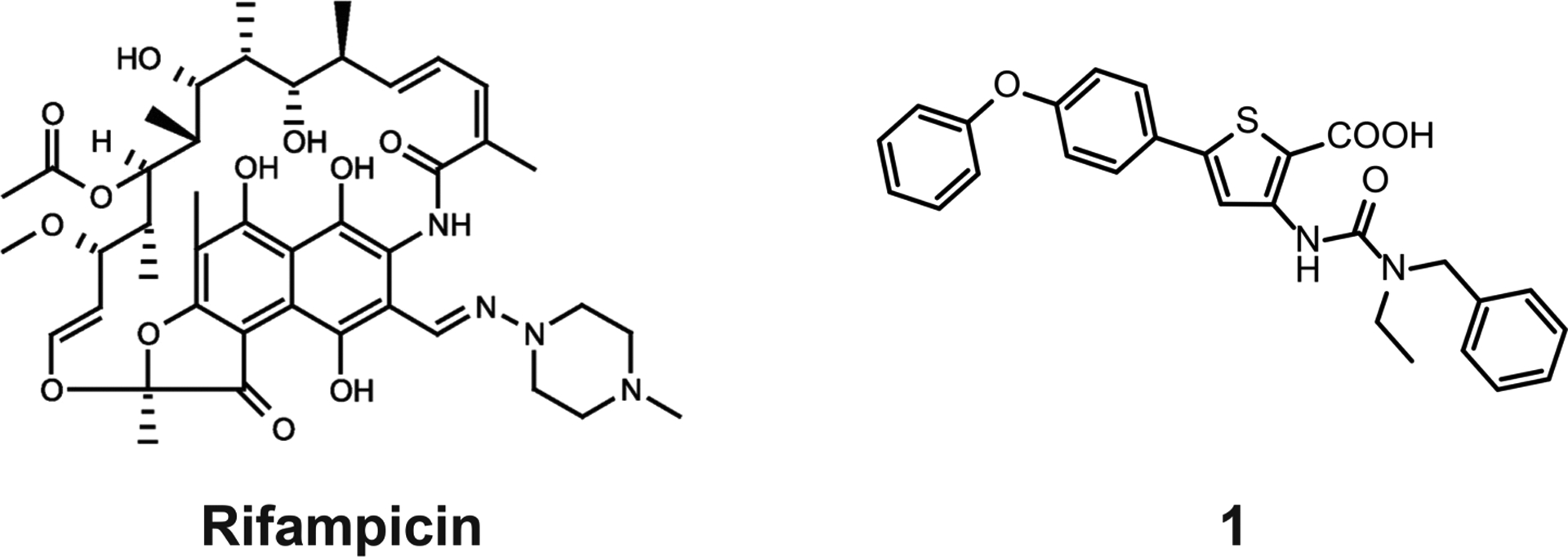
Chemical structures of rifampicin (Rif) and compound 1.
In this context, we found that compounds 1–6 lost their antibacterial efficacy after the addition of defined amounts of bovine serum albumin (BSA) to the LB growth medium (Figure S4). Even at concentrations far higher than the MIC of the six compounds (25 and 12.5 μg/mL compared to their MICs of 2–4 μg/mL), the inhibition of SA-N growth was abolished already at BSA concentrations between 0.2 and 2 mg/mL, while the potency of Rif was not influenced even at BSA concentrations of 20 mg/mL. Since considerably higher protein concentrations are generally found in exudates (>29 mg/mL),43 a significant plasma protein binding can also be expected in our experimental in vivo setup.
On the basis of these observations, we concluded that compound 1, in contrast to Rif, is unlikely to have an antibacterial effect after systemic administration, and we aimed to investigate whether this also holds true for a topical application.
Determination of the X-ray Crystal Structure of RNAP in Complex with Compound 1.
To elucidate and finally validate the mechanism of transcription inhibition by ureidothiophene-carboxylic acids and in particular of compound 1, on which we decided to focus during the following in vivo experiments, we determined the crystal structure of the E. coli σ70 RNAP holoenzyme in complex with this inhibitor (Table S2). The structure shows unambiguous density for 1 located in the RNAP switch region (Figure 3A). Due to the relatively low resolution of the structure and relatively symmetric shape of the compound, we could not determine its orientation without ambiguity. However, compound 1 with the N-benzyl-N-ethylureido moiety is on the left side, and the para-phenoxyphenyl group is on the right side, as shown in Figure 3A (yellow), fitting better than its alternative orientation (green in Figure 3A). Particularly, the placement of the para-phenoxyphenyl group on the right side covers the Fo – Fc map until its end. The orientation of compound 1 in the binding pocket is similar to one of the proposed computational docking poses.16 The binding pocket of compound 1 is a deep crescent-shaped pocket of ~22 Å in length and is largely hydrophobic (Figure 3B), which complements the hydrophobic nature of the substituents at the thiophene core including the N-benzyl-N-ethyl ureido moiety and the para-phenoxyphenyl group. The carboxyl-thiophene core is exposed to solvent and is not involved in the interaction with RNAP. The molecular surface of compound 1 is 624 Å2, and its 421 Å2 (67%) surface is involved in the RNAP binding. The contact surface area is similar to the contact areas of other switch region binding RNAP inhibitors such as myxopyronin (486 Å2) and squaramide (398 Å2),44 and these compounds accommodate at the same binding pocket (Figure 3C). Therefore, the ureidothiophene-carboxylic acid may inhibit RNAP transcription activity by preventing conformational change of the RNAP clamp required for the promoter DNA melting and/or accommodation of the template DNA into the RNAP active site cleft.17
Figure 3.
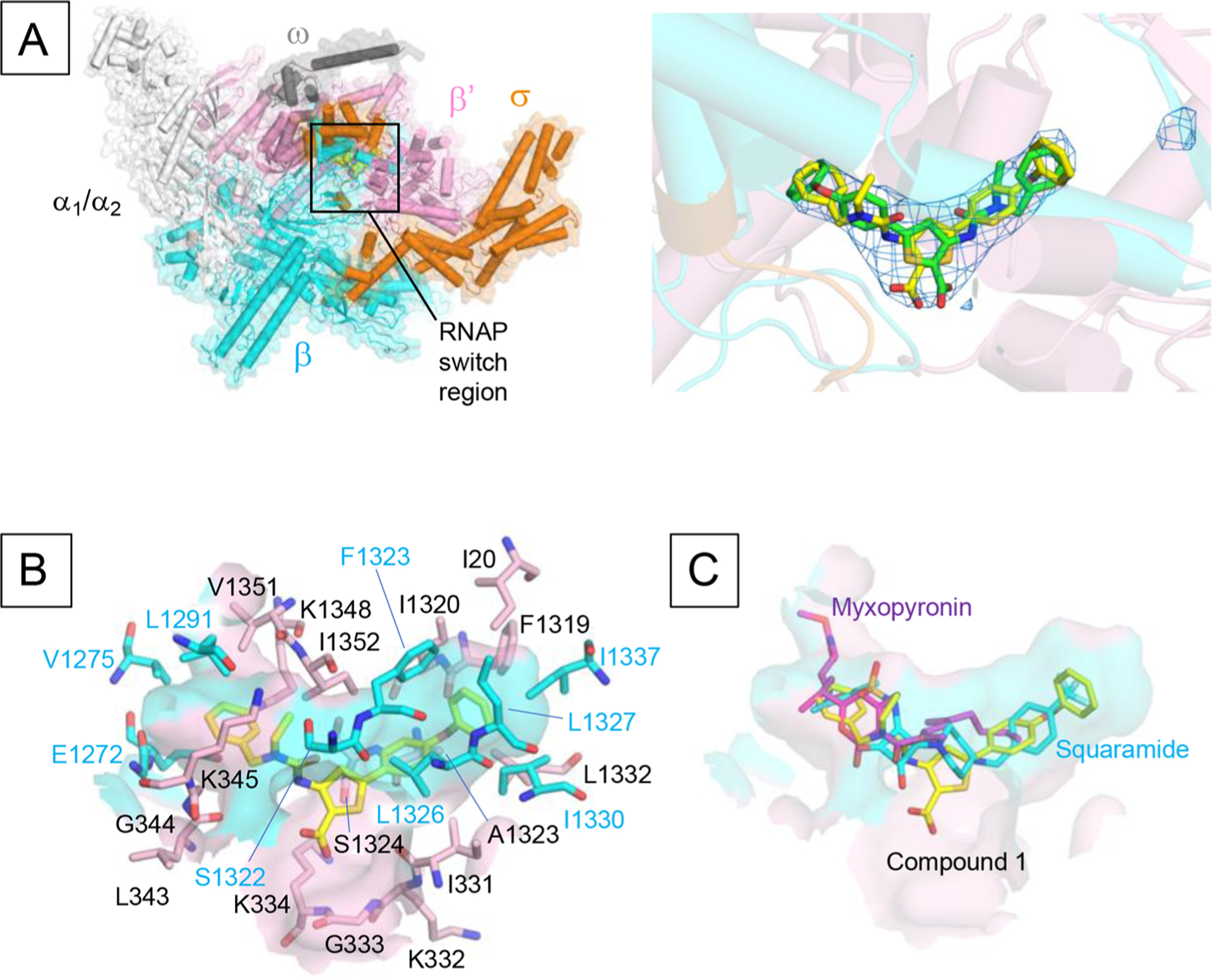
The structure of RNAP–ureidothiophene-2-carboxylic acid 1 complex. (A) Overall structure of the E. coli RNAP in complex with compound 1. RNAP is depicted as a cartoon model with a partially transparent molecular surface. The compound 1 is shown as a CPK model. The right panel shows a magnified view of the binding site of compound 1 (RNAP switch region). Compound 1 is depicted as a stick model (yellow) and one with 180° rotated orientation (green). The Fo – Fc electron density map (blue mesh) is superposed on the model. (B) Surface representation of the binding pocket of compound 1 with amino acid residues (stick models) participating in the hydrophobic interaction with compound 1 (cyan, β subunit; pink, β′ subunit). Orientation of this panel is the same as in (A). (C) Comparison of the binding of compound 1 (yellow), myxopyronin (magenta; PDB: 4YFX), and squaramide (cyan; PDB: 4FYN) to the RNAP switch region. Orientation of this panel is the same as in (A).
Anti-infective Therapy.
As the development of novel antibacterial drugs is urgently needed, we used our SKH1-based model to study the effects of inhibitor 1 and Rif on (i) the loss of body weight 2 d after wounding/infection, (ii) the wound size development over 14 d, and (iii) the number of CFUs in the wounds at the end of the experiment. Furthermore, sections of the wounds were prepared at day 8 postinfection. Rif (10 μL of 25 μg/mL in PBS) was topically administered twice on the mouse wounds: 5 min and 48 h, 3 and 48 h, or 24 and 48 h after wounding/infection. Compound 1 was also administered topically twice. Due to the BSA-mediated reduction of its antibacterial activity, we limited the treatment to one group with earlier treatment times (5 min and 48 h after wounding/infection; 10 μL of 50 μg/mL in PBS).
As the number of living bacteria in the wounds influenced the body weight (Figure 1), we measured the weight of all treated mice 2 d after wounding/infection (Figure 4). The mice with SA-N infected and PBS-treated wounds showed a body weight loss of 8.6 ± 2.3%, while the wounded but uninfected mice minimally increased their weight (−1.0 ± 1.9%). This again indicated that the wounding was per se well tolerated. In the Rif groups, the body weight loss was significantly reduced. In the groups with treatment after 3 and 48 h, the weight loss amounted to 5.4 ± 2.0%, in the group with earlier primary treatment, the loss was even more reduced (1.9 ± 1.5%). These results suggest that Rif is effective in reducing the SA-N load in skin wounds, which probably leads to an increased quality of life for the mice. The reasons why the earlier treatment was more successful might be that after 3 h, the bacteria escaped from the wound area to the surrounding tissues, that they generally increased their number by cell division (while after 5 min, the infection was not yet fully established and the bacteria were therefore more susceptible to treatment), and also the initiation of biofilm production as an intrinsic protection measure. However, when the Rif treatment was started 24 h postinfection, the SA-N infected mice lost nearly as much body weight (7.0 ± 3.1%) as the sham-treated mice. A prominent effect could unfortunately not be observed for compound 1, as the weight reduction (after treatment for 5 min and 48 h after infection) was similar to the infected control.
Figure 4.
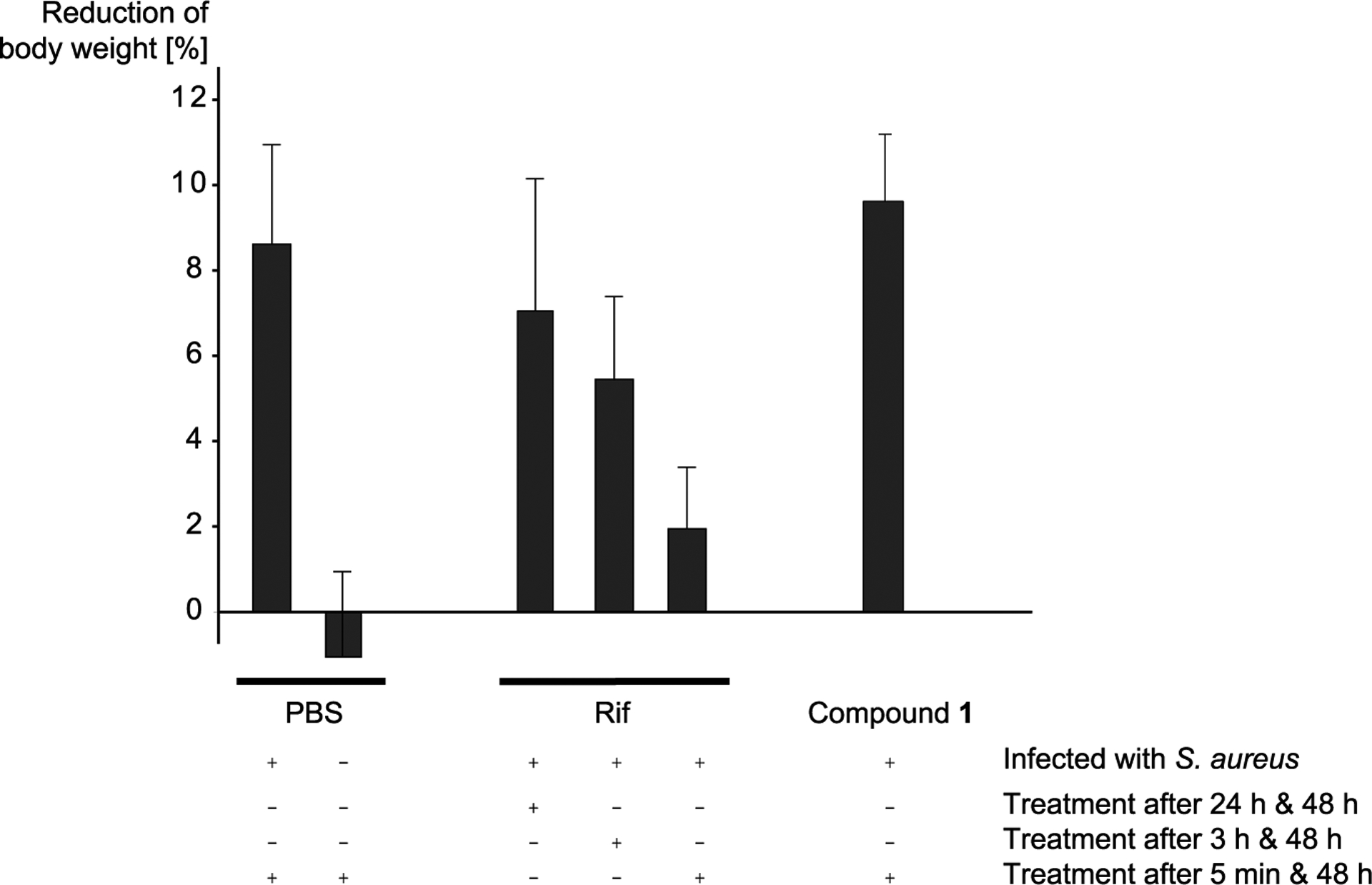
Loss of body weight. The loss of body weight (% of starting weight) 48 h after wounding/infection is shown for the six different treatment groups. Plus (+) and minus (−) signs refer to whether or not the treatment method mentioned on the right was used.
In addition to body weight, the wound area was measured every 2 d (up to day 14). As expected, the progression of wound closure was relatively slow in the infected but untreated mice (Figure 5, 6–14 d; Figure S5, 0–14 d). Even after 14 d, these wounds were not completely closed. In contrast, uninfected wounds closed much faster; e.g., on day 8, the wound area was 2.3 ± 0.7 mm2 (infected but untreated: 7.1 ± 2.6 mm2), and it was only 0.3 ± 0.4 mm2 on day 10 (vs 3.0 ± 2.4 mm2). SA-N infected wounds treated with Rif for first time 24 h postinfection displayed wound areas of 5.9 ± 2.4 mm2 on day 8, suggesting a low impact of the antibiotic on the bacterial infection. Interestingly, wound closure of both early Rif-treated groups proceeded rapidly and was comparable to the uninfected wounds. This clear effect was more prominent than expected, taking into account the results of body weight. It is possible that the systemic effects of exotoxins produced by SA-N, such as α-toxin, γ-hemolysin, and leukocidin A/B,45,46 led to these effects. If these toxins had had more time to be produced by the bacteria in the wound, as was the case in the time window of 3 h between infection and first Rif treatment, their influence would likely have a stronger effect on the well-being of the mice than on the wound healing processes. In contrast, double treatment of the wounds with compound 1 postinfection, as expected, did not lead to any change in the speed of wound healing (Figure 5).
Figure 5.
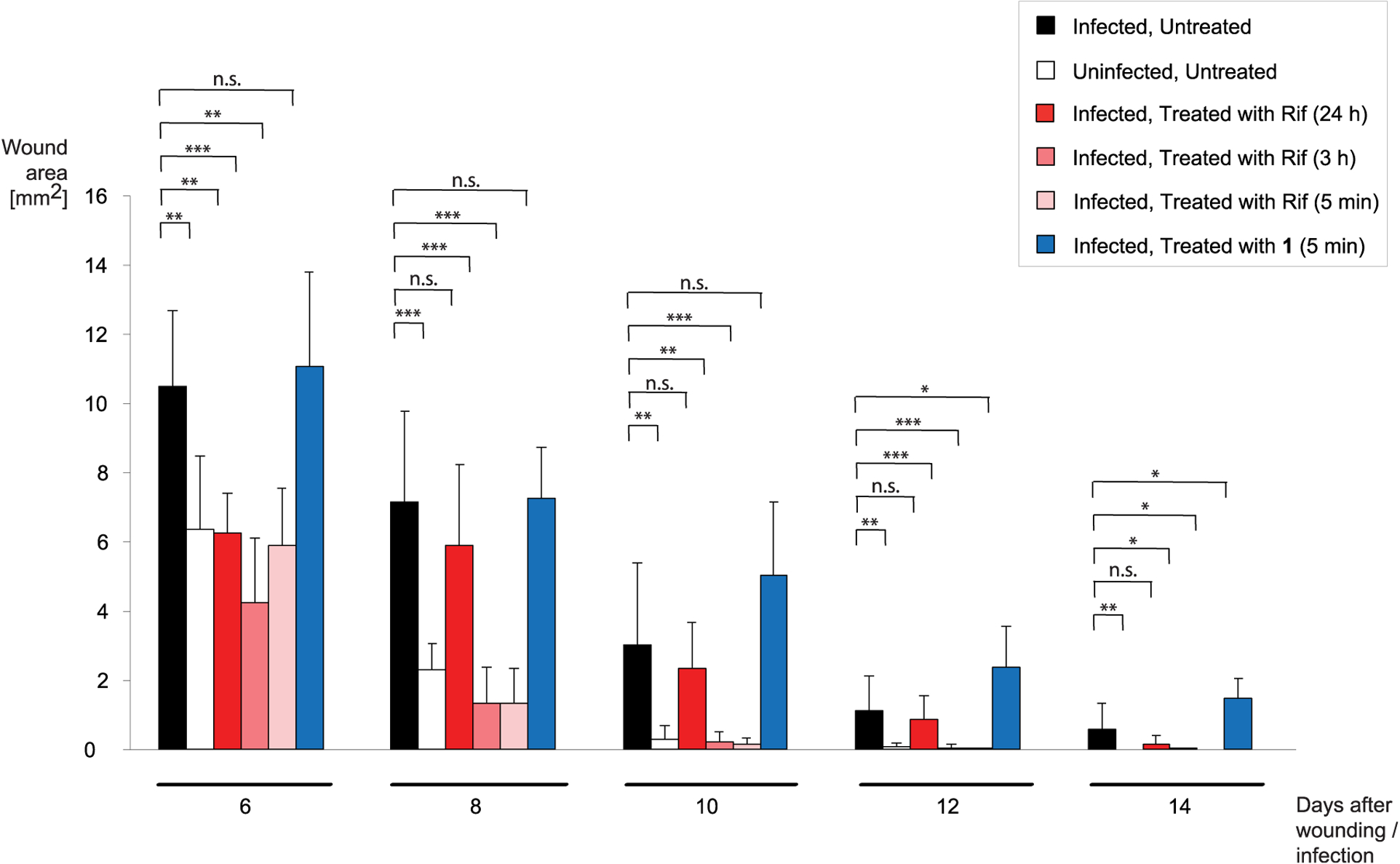
Development of wound size. The averaged wound areas (in mm2) are displayed every 2 d for the different treatment groups, between 6 and 14 d after wounding/infection. Asterisks (*) indicate a significant difference. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant (two-tailed Mann–Whitney U Test).
At the end of the experiment, the total number of CFU in the wounds was determined (Figure 6). As expected, a high number of bacteria was found in the infected and sham-treated wounds (5.5 × 105 CFU per wound on average). In comparison, the bacterial load in uninfected wounds was low (average: 95 CFU), which probably originated from the skin microbiota of the animals and/or the nonsterile environment. In line with the weight and wound healing kinetics, the average number of CFUs were also reduced in the Rif-treated groups (Figure 6); with a drastically decreasing number of CFU per wound, the earlier the timing of the first treatment was (8.6 × 105 vs 1.4 × 104 vs 1 × 101 CFU/wound).
Figure 6.
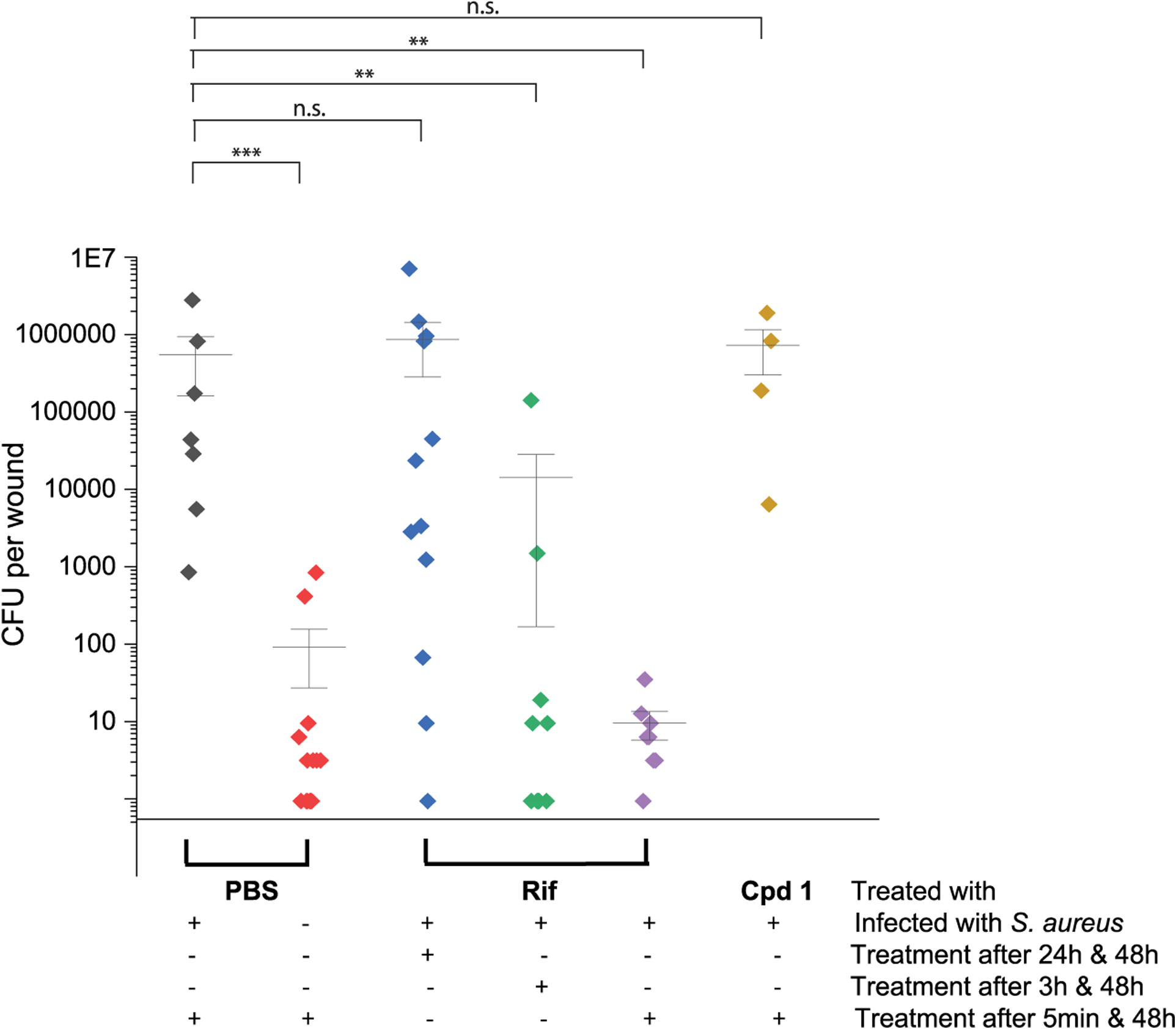
Number of CFU per wound. The total number of CFU in each wound is illustrated. Horizontal lines represent the mean value of each of the six groups; vertical lines indicate the standard deviations. Plus (+) and minus (−) signs refer to whether the treatment method mentioned on the right was applied or not. Asterisks (*) indicate a significant difference. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant (two-tailed Mann–Whitney U Test). CFUs, colony forming units.
As expected, wounds treated with compound 1 did not show a reduction in the number of CFUs.
CONCLUSIONS
In this work, we report on a powerful and easy-to-use wound infection model based on hairless SKH1 mice and the S. aureus strain Newman. As we have shown, the model enables the generation of valuable preclinical data that will be useful during the further development of novel anti-infectives. In detail, on the basis of the RNAP-inhibiting compounds Rif and compound 1, we collected three different data sets: the wounding and infection-derived body weight loss, the speed of wound healing, and the number of CFU per wound at the end of the observation period. Depending on the timing of the administration of Rif, we found that the compound showed a reduction in body weight loss induced by the bacteria, an acceleration of wound healing kinetics, and a reduced number of CFU in the wound at the end of the observation time. In contrast, compound 1 was found to be ineffective after topical administration. Our results indicate that the reason for this finding is very probably due to the strong plasma protein binding. These valuable results set the stage for subsequent structural optimization of the small molecules, which will focus not only on the in vitro enzyme inhibition and antibacterial activity but also on multiple other parameters such as the cytotoxic behavior and metabolic stability. As can be concluded from our in vivo data with compound 1, one should also focus on plasma protein binding even if a topical application is intended in order to finally generate compounds that are suitable for therapeutic application. Our new crystal structure of the E. coli σ70 RNAP holoenzyme in complex with compound 1 will be valuable for these future optimization steps.
The applicability of our animal model is illustrated by the fact that we have already successfully used it in further studies. In these, we very recently confirmed the in vivo activity of clarithromycin nanocapsules47 and compared the activity of the RNAP inhibitor sorangicin41 to a newly discovered antimicrobial natural product (manuscript in preparation).
Our work offers a model that allows for clear predictions about the efficacy of potential anti-infectives in vivo. The rapid provision of valuable preclinical data is extremely useful for the design and synthesis of such new compounds. Furthermore, our animal model provides the basis and flexibility to be used for combination therapy, e.g., by (i) combining our compounds with standard-of-care antibiotics or compounds whose activity is not directed against bacteria but, for example, against cortisol biosynthesis in skin.48 Also, (ii) the use of pathogens other than S. aureus to infect wounds may be considered, e.g., perilous wound-infecting bacteria such as Streptococcus pyogenes, Pseudomonas aeruginosa, and Clostridium perfringens.1,49
METHODS
Determination of Antibacterial Activity.
The antibacterial activity of selected compounds was determined in S. aureus Newman as described previously.34 Determination of Cytotoxicity of Compounds. HEK293 and NHDF cells (2 × 105 cells per well) were seeded in 24-well, flat-bottomed plates. Culturing of cells, incubations, and OD measurements were performed as described previously50 with slight modifications. Twenty-four hours after seeding the cells, the incubation was started by the addition of compounds in a final DMSO concentration of 1%. The cellular metabolic activity as an indicator of cell viability, proliferation, and cytotoxicity was determined after 48 h in a PHERAstar microplate reader (BMG labtech, Ortenberg, Germany). At least two independent measurements were performed for each compound.
Determination of Metabolic and Plasma Stability.
For the evaluation of combined phase I and phase II metabolic stability, the compound (1 μM) was incubated with 1 mg/mL pooled human liver S9 fraction (BD Gentest, Heidelberg, Germany), a 2 mM NADPH regenerating system, 1 mM UDPGA, 10 mM MgCl2, and 0.1 mM PAPS at 37 °C for 0, 5, 15, 30, and 60 min. The metabolic stability of testosterone (1 μM) and 7-hydroxycoumarin (1 μM) were performed in parallel to confirm the enzymatic activity of the S9 fraction. The incubation was stopped by precipitation of S9 enzymes with two volumes of cold acetonitrile containing internal standard (diphenhydramine). The stability of the compound was also determined in human pooled plasma (BioTrend, Cologne, Germany). Therefore, the compound was added to plasma at a concentration of 1 μM and incubated at 37 °C for 0, 5, 30, and 60 min. Incubation was stopped by adding four volumes of cold acetonitrile containing internal standard (diphenhydramine). Procaine was used as an activity control of metabolizing enzymes in plasma. The concentration of the remaining test compound at the different time points was analyzed by LC-MS/MS (TSQ Quantum Access MAX, Thermo Fisher, Dreieich, Germany) and used to determine half-life (T1/2).
Structure Determination of the E. coli RNAP Holoenzyme and Compound 1.
Crystals of E. coli σ70 RNAP holoenzyme were prepared as described previously.51 The holoenzyme crystals were soaked in a crystallization solution containing 30% PEG400 and 1 mM compound 1 for 12 h at 22 °C followed by flash freezing by liquid nitrogen. In addition, other compounds related to compound 1 were soaked in the RNAP crystal, but the resolution of these crystals was not enough to determine their structures. The X-ray crystallographic data sets were collected at the Macromolecular Diffraction at the Cornell High Energy Synchrotron Source (MacCHESS) F1 beamlines (Cornell University, Ithaca, NY), and the data were processed by HKL2000.52 The E. coli RNAP holoenzyme structure (PDB: 4LK1) was used as the initial model for molecular replacement followed by rigid body and positional refinements with noncrystallographic symmetry by using the program Phenix.53 The resulting maps contained additional electron densities, which allowed compound 1 not present in the initial search model to be built in using Coot.54 Final coordinate and structure factor were submitted to the PDB with the ID code listed in Table S2.
Murine S. aureus-Based Wound Infection Model.
All animal experiments were approved by the animal welfare committee Landesamt fuer Verbrauchserschutz (Saarbruecken, Germany) and were carried out in agreement with German and European legal requirements. Female SKH1 hairless mice (Crl:SKH1-Hrhr) were obtained from Charles River (Sulzfeld, Germany) and kept under specific pathogen-free conditions according to the regulations of German veterinary law.
SKH1 mice aged 8 to 12 weeks (average weight: 25.54 ± 2.34 g; we tried to keep the masses of the animals similar between the different groups) were anesthetized with an i.p. injection of 10 mg/kg body weight of xylazine hydrochloride (Bayer AG, Leverkusen, Germany) and 100 mg/kg of ketamine hydrochloride (Serumwerk Bernburg AG, Bernburg, Germany). After disinfection of the dorsal area, a sterile 5 mm biopsy punch (pfm medical, Cologne, Germany) was used to create uniform, full-thickness wounds on both dorsal sides (one wound on each side) of the mouse. To stabilize the round shape of the wounds, silicon O-rings with an external diameter of 5.5 mm (HUG Technik und Sicherheit GmbH, Ergolding, Germany) were inserted into each wound for the first 2 d after wounding. Subsequently, 10 μL of a PBS suspension with an OD600 of 0.01 to 1.0 containing washed, logarithmic growth phase SA-N cells (~1 × 104 to 1 × 106 CFU, corresponding to ~1 × 106 to 1 × 108 CFU/mL) was applied to the wound centers and allowed to drain into the tissue for 5 min. PBS without bacteria served as the control. Infected wounds were then sealed with transparent, air-permeable Tegaderm films (3M, Neuss, Germany). Treatment with 10 μL of compounds (in PBS; final DMSO concentration: 1%) was performed at different time points after infection. Body weights and wound sizes (measured with an electronic calliper in mm with two decimal digits) were determined on every second day. On this occasion, the Tegaderm film was replaced in each case. At the end of the experiment, mice were sacrificed by cervical dislocation; wound areas were aseptically excised and either prepared for histology or homogenized with a dispenser (Kinematica, Luzern, Switzerland) in PBS, and serial dilutions of the homogenates were plated on sheep blood agar plates (Becton Dickinson, Heidelberg, Germany). CFUs were counted after incubation overnight at 37 °C. The numbers of animals or wounds per group are shown in Table S3.
Paraffin Sections and Hemotoxylin and Eosin (H&E) Staining.
Immediately after cervical dislocation, wounds intended for histology were dissected and fixed overnight in 4% buffered paraformaldehyde at 4 °C, followed by dehydration and embedding of the samples in paraffin. Sections (5 μm) were prepared and mounted on slides. The H&E staining was carried out according to a described procedure.55 Microphotographs were taken with a light microscope (Nikon ECLIPSE E200, with a TrueCromeII camera).
Supplementary Material
ACKNOWLEDGMENTS
The authors thank J. Jung, I. Lang, D. Jener, and T. Wittmann for excellent technical support and J. H. Sahner for the synthesis of compounds 1-4. We thank the staff at the MacCHESS for support of X-ray crystallographic data collection. A.K.H.H. gratefully acknowledges funding from the European Research Council (ERC starting grant 757913) and the Helmholtz-Association’s Initiative and Networking Fund. This work was also supported by the National Institutes of Health (NIH) (GM087350 and GM131860 to K.S.M.).
Footnotes
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00034.
Supporting Figures S1–S5 and Tables S1–S3 showing the influence of different S. aureus strains on wound healing, pictures of an infected vs uninfected wound and corresponding paraffin sections, biological data of selected RNAP inhibitors, data collection on cocrystallization experiments, the influence of BSA on the antibacterial activity of selected compounds, the development of wound size from 0 to 14 days, and an overview of the number of animals or wounds in each experimental group; the bacterial viability assay as well as the synthesis and characterization of compounds 1–6 (PDF)
Contributor Information
Jörg Haupenthal, Department of Drug Design and Optimization, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS)–Helmholtz Centre for Infection Research (HZI), 66123 Saarbrücken, Saarland, Germany;.
Yannik Kautz, Institute of Medical Microbiology and Hygiene, Saarland University, 66421 Homburg, Saarland, Germany.
Walid A. M. Elgaher, Department of Drug Design and Optimization, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS)–Helmholtz Centre for Infection Research (HZI), 66123 Saarbrücken, Saarland, Germany;.
Linda Pätzold, Institute of Medical Microbiology and Hygiene, Saarland University, 66421 Homburg, Saarland, Germany.
Teresa Röhrig, Department of Drug Design and Optimization, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS)–Helmholtz Centre for Infection Research (HZI), 66123 Saarbrücken, Saarland, Germany.
Matthias W. Laschke, Institute for Clinical & Experimental Surgery, Saarland University, 66421 Homburg, Saarland, Germany
Thomas Tschernig, Institute of Anatomy and Cell Biology, Saarland University, 66421 Homburg, Saarland, Germany.
Anna K. H. Hirsch, Department of Drug Design and Optimization, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS)–Helmholtz Centre for Infection Research (HZI), 66123 Saarbrücken, Saarland, Germany; Department of Pharmacy, Saarland University, 66123 Saarbrücken, Saarland, Germany;.
Rolf W. Hartmann, Department of Drug Design and Optimization, Helmholtz Institute for Pharmaceutical Research Saarland (HIPS)–Helmholtz Centre for Infection Research (HZI), 66123 Saarbrücken, Saarland, Germany; Department of Pharmacy, Saarland University, 66123 Saarbrücken, Saarland, Germany;.
Markus Bischoff, Institute of Medical Microbiology and Hygiene, Saarland University, 66421 Homburg, Saarland, Germany.
REFERENCES
- (1).Kirketerp-M?ller K,Jensen PO, Fazli M, Madsen KG, Pedersen J, Moser C, Tolker-Nielsen T, Hoiby N, Givskov M, and Bjarnsholt T (2008) Distribution, organization, and ecology of bacteria in chronic wounds. J. Clin Microbiol 46, 2717–2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, and Longaker MT (2009) Human skin wounds. A major and snowballing threat to public health and the economy. Wound Repair Regen 17, 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kucera J, Sojka M, Pavlik V, Szuszkiewicz K, Velebny V, and Klein P (2014) Multispecies biofilm in an artificial wound bed–A novel model for in vitro assessment of solid antimicrobial dressings. J. Microbiol. Methods 103, 18–24. [DOI] [PubMed] [Google Scholar]
- (4).Mohamed N, Wang MY, Le Huec J-C, Liljenqvist U, Scully IL, Baber J, Begier E, Jansen KU, Gurtman A, and Anderson AS (2017) Vaccine development to prevent Staphylococcus aureus surgical-site infections. Br. J. Surg 104, e41–e54. [DOI] [PubMed] [Google Scholar]
- (5).Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Tsourdi E, Barthel A, Rietzsch H, Reichel A, and Bornstein SR (2013) Current aspects in the pathophysiology and treatment of chronic wounds in diabetes mellitus. BioMed Res. Int 2013, 385641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Matteelli A, Roggi A, and Carvalho AC (2014) Extensively drug-resistant tuberculosis. Epidemiology and management. Clin Epidemiol 6, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Conly J, and Johnston B (2005) Where are all the new antibiotics? The new antibiotic paradox. Can. J. Infect Dis Med. Microbiol 16, 159–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Artsimovitch I, and Vassylyev DG (2006) Is it easy to stop RNA polymerase? Cell Cycle 5, 399–404. [DOI] [PubMed] [Google Scholar]
- (10).Chopra I (2007) Bacterial RNA polymerase. A promising target for the discovery of new antimicrobial agents. Curr. Opin Investig Drugs 8, 600–607. [PubMed] [Google Scholar]
- (11).Villain-Guillot P, Bastide L, Gualtieri M, and Leonetti J-P (2007) Progress in targeting bacterial transcription. Drug Discovery Today 12, 200–208. [DOI] [PubMed] [Google Scholar]
- (12).Mariani R, and Maffioli SI (2009) Bacterial RNA polymerase inhibitors. An organized overview of their structure, derivatives, biological activity and current clinical development status. Curr. Med. Chem 16, 430–454. [DOI] [PubMed] [Google Scholar]
- (13).Mitchison DA (2000) Role of individual drugs in the chemotherapy of tuberculosis. Int. J. Tuberc Lung Dis 4, 796–806. [PubMed] [Google Scholar]
- (14).Mukinda FK, Theron D, van der Spuy GD, Jacobson KR, Roscher M, Streicher EM, Musekiwa A, Coetzee GJ, Victor TC, Marais BJ, Nachega JB, Warren RM, and Schaaf HS (2012) Rise in rifampicin-monoresistant tuberculosis in Western Cape, South Africa. Int. J. Tuberc Lung Dis 16, 196–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Sahner JH, Groh M, Negri M, Haupenthal J, and Hartmann RW (2013) Novel small molecule inhibitors targeting the “switch region” of bacterial RNAP. Structure-based optimization of a virtual screening hit. Eur. J. Med. Chem 65, 223–231. [DOI] [PubMed] [Google Scholar]
- (16).Fruth M, Plaza A, Hinsberger S, Sahner JH, Haupenthal J, Bischoff M, Jansen R, Muller R, and Hartmann RW (2014) Binding mode characterization of novel RNA polymerase inhibitors using a combined biochemical and NMR approach. ACS Chem. Biol 9, 2656–2663. [DOI] [PubMed] [Google Scholar]
- (17).Srivastava A, Talaue M, Liu S, Degen D, Ebright RY, Sineva E, Chakraborty A, Druzhinin SY, Chatterjee S, Mukhopadhyay J, Ebright YW, Zozula A, Shen J, Sengupta S, Niedfeldt RR, Xin C, Kaneko T, Irschik H, Jansen R, Donadio S, Connell N, and Ebright RH (2011) New target for inhibition of bacterial RNA polymerase. ‘switch region’. Curr. Opin. Microbiol 14, 532–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Mukhopadhyay J, Das K, Ismail S, Koppstein D, Jang M, Hudson B, Sarafianos S, Tuske S, Patel J, Jansen R, Irschik H, Arnold E, and Ebright RH (2008) The RNA polymerase “switch region” is a target for inhibitors. Cell 135, 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Elgaher WAM, Fruth M, Groh M, Haupenthal J, and Hartmann RW (2014) Expanding the scaffold for bacterial RNA polymerase inhibitors. Design, synthesis and structure-activity relationships of ureido-heterocyclic-carboxylic acids. RSC Adv. 4, 2177–2194. [Google Scholar]
- (20).Ruthenbeck A, Elgaher WAM, Haupenthal J, Hartmann RW, and Meier C (2017) Bacterial RNAP Inhibitors: Synthesis and Evaluation of Prodrugs of Aryl-ureidothiophene-carboxylic acids. ChemistrySelect 2, 11899–11905. [Google Scholar]
- (21).Elgaher WAM, Sharma KK, Haupenthal J, Saladini F, Pires M, Real E, Mely Y, and Hartmann RW (2016) Discovery and Structure-Based Optimization of 2-Ureidothiophene-3-carboxylic Acids as Dual Bacterial RNA Polymerase and Viral Reverse Transcriptase Inhibitors. J. Med. Chem 59, 7212–7222. [DOI] [PubMed] [Google Scholar]
- (22).Schierle CF, De la Garza M, Mustoe TA, and Galiano RD (2009) Staphylococcal biofilms impair wound healing by delaying reepithelialization in a murine cutaneous wound model. Wound Repair Regen 17, 354–359. [DOI] [PubMed] [Google Scholar]
- (23).Hurdle JG, Yendapally R, Sun D, and Lee RE (2009) Evaluation of analogs of reutericyclin as prospective candidates for treatment of staphylococcal skin infections. Antimicrob. Agents Chemother 53, 4028–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kugelberg E, Norstrom T, Petersen TK, Duvold T, Andersson DI, and Hughes D (2005) Establishment of a superficial skin infection model in mice by using Staphylococcus aureus and Streptococcus pyogenes. Antimicrob. Agents Chemother 49, 3435–3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sharma MR, Werth B, and Werth VP (2011) Animal models of acute photodamage: comparisons of anatomic, cellular and molecular responses in C57BL/6J, SKH1 and Balb/c mice. Photochem. Photobiol 87, 690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sorg H, Krueger C, and Vollmar B (2007) Intravital insights in skin wound healing using the mouse dorsal skin fold chamber. J. Anat 211, 810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Rojas I-G, Padgett DA, Sheridan JF, and Marucha PT (2002) Stress-induced susceptibility to bacterial infection during cutaneous wound healing. Brain, Behav., Immun 16, 74–84. [DOI] [PubMed] [Google Scholar]
- (28).Otto CC, Kilbourne J, and Haydel SE (2016) Natural and ion-exchanged Illite clays reduce bacterial burden and inflammation in cutaneous meticillin-resistant Staphylococcus aureus infections in mice. J. Med. Microbiol 65, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Roche ED, Renick PJ, Tetens SP, and Carson DL (2012) A model for evaluating topical antimicrobial efficacy against methicillin-resistant Staphylococcus aureus biofilms in superficial murine wounds. Antimicrob. Agents Chemother 56, 4508–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, and Bayles KW (2013) A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4, No. e00537–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bremell T, Abdelnour A, and Tarkowski A (1992) Histopathological and serological progression of experimental Staphylococcus aureus arthritis. Infect. Immun 60, 2976–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Duthie ES (1952) Variation in the antigenic composition of staphylococcal coagulase. J. Gen. Microbiol 7, 320–326. [DOI] [PubMed] [Google Scholar]
- (33).Somerville GA, Beres SB, Fitzgerald JR, DeLeo FR, Cole RL, Hoff JS, and Musser JM (2002) In vitro serial passage of Staphylococcus aureus: changes in physiology, virulence factor production, and agr nucleotide sequence. J. Bacteriol 184, 1430–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hinsberger S, Husecken K, Groh M, Negri M, Haupenthal J, and Hartmann RW (2013) Discovery of novel bacterial RNA polymerase inhibitors. Pharmacophore-based virtual screening and hit optimization. J. Med. Chem 56, 8332–8338. [DOI] [PubMed] [Google Scholar]
- (35).Ma C, Yang X, and Lewis PJ (2016) Bacterial Transcription Inhibitor of RNA Polymerase Holoenzyme Formation by Structure-Based Drug Design. From in Silico Screening to Validation. ACS Infect. Dis 2, 39–46. [DOI] [PubMed] [Google Scholar]
- (36).Artsimovitch I, Chu C, Lynch AS, and Landick R (2003) A new class of bacterial RNA polymerase inhibitor affects nucleotide addition. Science 302, 650–654. [DOI] [PubMed] [Google Scholar]
- (37).Villain-Guillot P, Gualtieri M, Bastide L, Roquet F, Martinez J, Amblard M, Pugniere M, and Leonetti J-P (2007) Structure-activity relationships of phenyl-furanyl-rhodanines as inhibitors of RNA polymerase with antibacterial activity on biofilms. J. Med. Chem 50, 4195–4204. [DOI] [PubMed] [Google Scholar]
- (38).Zhu W, Groh M, Haupenthal J, and Hartmann RW (2013) A detective story in drug discovery. Elucidation of a screening artifact reveals polymeric carboxylic acids as potent inhibitors of RNA polymerase. Chem. - Eur. J 19, 8397–8400. [DOI] [PubMed] [Google Scholar]
- (39).Haebich D, and von Nussbaum F (2009) Lost in transcription–inhibition of RNA polymerase. Angew. Chem., Int. Ed 48, 3397–3400. [DOI] [PubMed] [Google Scholar]
- (40).Maffioli SI, Zhang Y, Degen D, Carzaniga T, Del Gatto G, Serina S, Monciardini P, Mazzetti C, Guglierame P, Candiani G, Chiriac AI, Facchetti G, Kaltofen P, Sahl H-G, Dehò G, Donadio S, and Ebright RH (2017) Antibacterial Nucleoside-Analog Inhibitor of Bacterial RNA Polymerase. Cell 169, 1240–1248.E23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Irschik H, Jansen R, Gerth K, Höfle G, and Reichenbach H (1987) The sorangicins, novel and powerful inhibitors of eubacterial RNA polymerase isolated from myxobacteria. J. Antibiot 40, 7–13. [DOI] [PubMed] [Google Scholar]
- (42).Zumla A, Nahid P, and Cole ST (2013) Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discovery 12, 388–404. [DOI] [PubMed] [Google Scholar]
- (43).Heffner JE, Brown LK, and Barbieri CA (1997) Diagnostic value of tests that discriminate between exudative and transudative pleural effusions. Primary Study Investigators. Chest 111, 970–980. [DOI] [PubMed] [Google Scholar]
- (44).Molodtsov V, Fleming PR, Eyermann CJ, Ferguson AD, Foulk MA, McKinney DC, Masse CE, Buurman ET, and Murakami KS (2015) X-ray crystal structures of Escherichia coli RNA polymerase with switch region binding inhibitors enable rational design of squaramides with an improved fraction unbound to human plasma protein. J. Med. Chem 58, 3156–3171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Ma J, Gulbins E, Edwards MJ, Caldwell CC, Fraunholz M, and Becker KA (2017) Staphylococcus aureus α-Toxin Induces Inflammatory Cytokines via Lysosomal Acid Sphingomyelinase and Ceramides. Cell. Physiol. Biochem 43, 2170–2184. [DOI] [PubMed] [Google Scholar]
- (46).Dumont AL, Nygaard TK, Watkins RL, Smith A, Kozhaya L, Kreiswirth BN, Shopsin B, Unutmaz D, Voyich JM, and Torres VJ (2011) Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol. Microbiol 79, 814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Anversa Dimer F, de Souza Carvalho-Wodarz C, Goes A, Cirnski K, Herrmann J, Schmitt V, Patzold L, Abed N, De Rossi C, Bischoff M, Couvreur P, Muller R, and Lehr C-M (2020) PLGA nanocapsules improve the delivery of clarithromycin to kill intracellular Staphylococcus aureus and Mycobacterium abscessus. Nanomedicine 24, 102125. [DOI] [PubMed] [Google Scholar]
- (48).Emmerich J, van Koppen CJ, Burkhart JL, Engeli RT, Hu Q, Odermatt A, and Hartmann RW (2018) Accelerated skin wound healing by selective 11β-Hydroxylase (CYP11B1) inhibitors. Eur. J. Med. Chem 143, 591–597. [DOI] [PubMed] [Google Scholar]
- (49).Saviauk T, Kiiski JP, Nieminen MK, Tamminen NN, Roine AN, Kumpulainen PS, Hokkinen LJ, Karjalainen MT, Vuento RE, Aittoniemi JJ, Lehtimäki TJ, and Oksala NK (2018) Electronic Nose in the Detection of Wound Infection Bacteria from Bacterial Cultures. A Proof-of-Principle Study. Eur. Surg. Res 59, 1–11. [DOI] [PubMed] [Google Scholar]
- (50).Haupenthal J, Baehr C, Zeuzem S, and Piiper A (2007) RNAse A-like enzymes in serum inhibit the anti-neoplastic activity of siRNA targeting polo-like kinase 1. Int. J. Cancer 121, 206–210. [DOI] [PubMed] [Google Scholar]
- (51).Murakami KS (2013) X-ray crystal structure of Escherichia coli RNA polymerase σ70 holoenzyme. J. Biol. Chem 288, 9126–9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (53).Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Emsley P, and Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (55).Romeis B (1990) Mikroskopische Technik. J. Basic Microbiol 30, 144 DOI: 10.1002/jobm.3620300221. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.