

Abstract
Depletion of mitochondrial copper, which shifts metabolism from respiration to glycolysis and reduces energy production, is known to be effective against cancer types that depend on oxidative phosphorylation. However, existing copper chelators are too toxic or ineffective for clinical application. Here we develop a safe, mitochondria-targeted, copper-depleting nanoparticle (CDN) and test it against triple-negative breast cancer (TNBC). We show that CDNs decrease oxygen consumption and oxidative phosphorylation, cause a metabolic switch to glycolysis, and reduce ATP production in TNBC cells. This energy deficiency, together with compromised mitochondrial membrane potential and elevated oxidative stress, results in apoptosis. CDNs should be less toxic than existing copper chelators because they favourably deprive copper in the mitochondria in cancer cells instead of systemic depletion. Indeed, we demonstrate low toxicity of CDNs in healthy mice. In three mouse models of TNBC, CDN administration inhibits tumor growth and substantially improves survival. The efficacy and safety of CDNs suggest the potential clinical relevance of this approach.
Triple-negative breast cancer (TNBC) is associated with high metastatic risk and low overall survival.1,2 TNBC is not responsive to endocrine therapy or HER2-targeted therapies, and because it is intrinsically heterogeneous, targeting a single biomarker or oncogene is often unsatisfactory. TNBC cells have a metabolic preference for importing and utilizing lipids as an energy source, likely due to their proximity to the adipose-rich mammary gland. Fatty acid oxidation is also essential in the activation of the Src pathway in TNBC cells.3 In transgenic TNBC models, pharmacologic inhibition of fatty acid oxidation blocks tumor growth and metastasis.4,5 Moreover, TNBC is less sensitive to enzyme inhibitors that target glucose transport compared to receptor-positive breast cancer types that rely more on glycolysis.6,7 Glycolysis-dependent cancer types induce fatty acid synthesis, which in turn inhibits fatty acid oxidation.8 Therefore, oxidative phosphorylation (OXPHOS) and fatty acid oxidation are attractive targets for the treatment of TNBC .
Data mining studies of breast cancer patients showed upregulation in the mitochondrial copper chaperone and co-chaperone proteins COX17 and SCO2,9,10 suggesting breast cancer cells have a higher demand for copper trafficking to the mitochondria than normal cells do. The mitochondrial copper enzyme cytochrome c oxidase (COX) is the complex IV of the electron transport chain and is responsible for energy generation and maintenance of the mitochondrial electrochemical gradient.11,12 COX is matured in the mitochondrial intermembrane space and its activity is subject to the copper supply in mitochondria.13 Limiting the availability of copper has been shown to impair the metabolism of cancer cells, especially those in high demand for copper. Inhibition of copper trafficking with inhibitors for chaperone proteins Atox 1 and CCS disrupts the bioenergetics of cancer cells.14 ATP depletion activates 5’ AMP-activated protein kinase (AMPK), which in turn increases the demand for fatty acid oxidation instead of lipogenesis.14 A similar disruption of ATP generation was observed when cancer cells in vitro were treated with tetrathiomolybdate (TM), a copper chelator for Wilson’s disease, leading to decreased oxidative phosphorylation and increased dependency on glycolysis.15,16
Here we develop a copper-depleting nanoparticle (CDN) to deplete copper in the tumor with minimal side effects to healthy tissues. CDNs combine two components: a copper-depleting moiety (CDM) and a semiconducting polymer (SPN). Mechanistically, CDN’s positive surface charge favors accumulation in mitochondria and local depletion of copper. In CDN-treated TNBC cells, mitochondrial OXPHOS activity is shut down and metabolism switches to glycolysis, with decreased ATP generation. Together with compromised mitochondrial membrane potential, decreased endogenous antioxidants, and elevated oxidative stress, TNBC cells undergo apoptosis in vitro and in vivo. Healthy cells are less susceptible owing to their lower demand for copper and lower uptake of the CDNs. Selective delivery to the tumor reduces the toxicity of CDNs for potential therapeutic applications.
Results
Design, formulation and characterization of CDN.
We formulated CDNs by incorporating a potent self-reporting copper-depleting moiety (CDM) into a semiconducting polymer nanoparticle (SPN) consisting of semiconducting polymers and phospholipid-polyethylene glycol (PEG) (Fig. 1a, Supplementary Figure 1a). The CDM is composed of N,N-Bis(2-pyridinylmethyl)-1,2-ethanediamine linked to tricarbocyanine. When copper binds, the near-infrared (NIR) fluorescence signal from tricarbocyanine is quenched, providing real-time feedback of the chelation. Two SPNs were employed to generate CDNs with different optical properties. As shown in Fig. 1b, SPN is photoacoustic at 1100 nm, and can be non-invasively tracked in vivo by photoacoustic imaging. When coupled with fluorescence signal changes from CDM, the optical signals of the system can report the amount of CDN as well as copper binding status in the organ of interest. fCDN is a fluorescence resonance energy transfer (FRET)-based formulation, where polymer acts as the FRET donor and CDM as the acceptor (Supplementary Figure 1a). fCDN detects the copper binding through fluorescence ratiometric imaging (Supplementary Figure 1d, e), which is suitable for cellular mechanism study and ex vivo quantification.
Figure 1. Design and characterization of CDN.
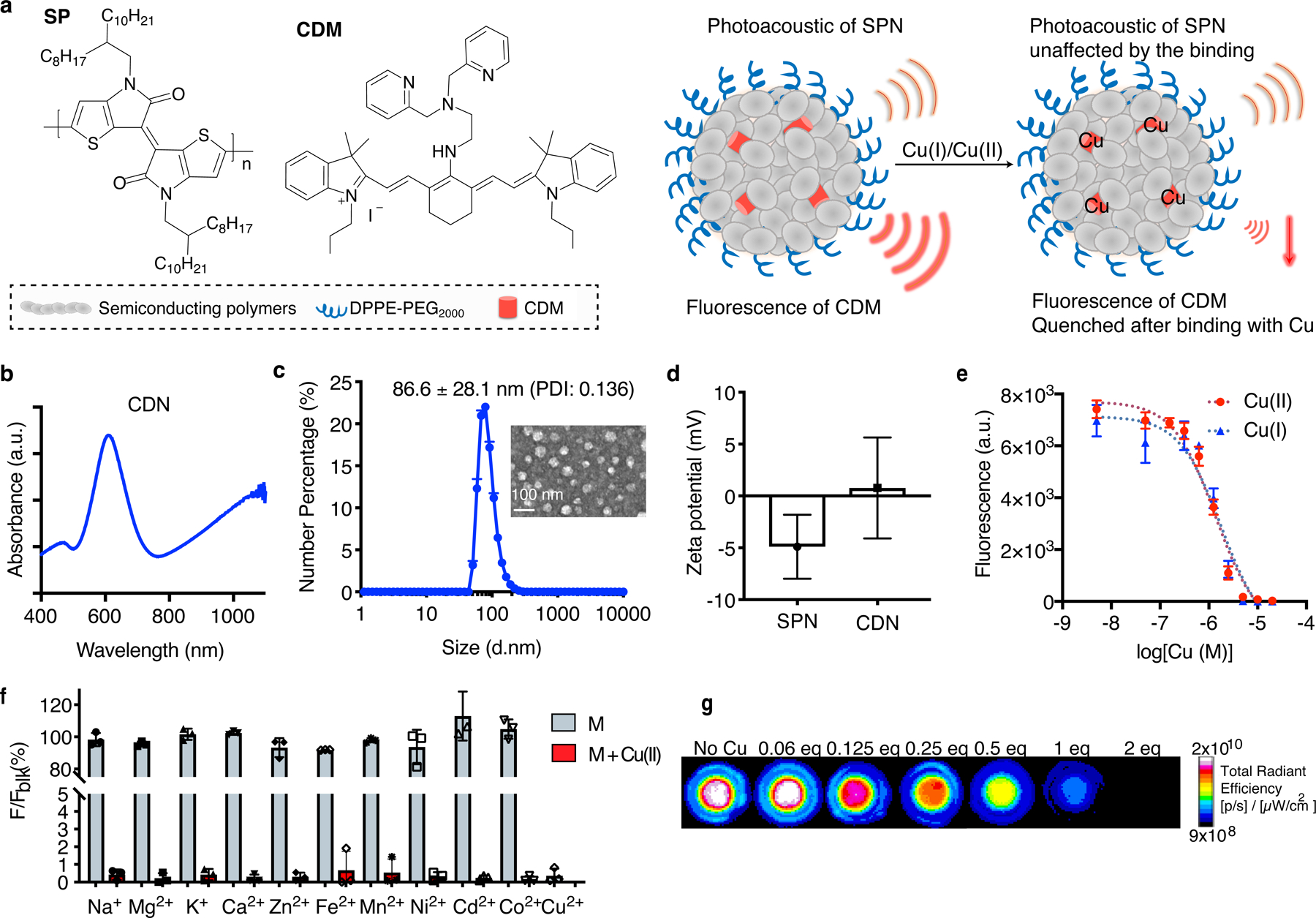
(a) Molecular components of CDN and the illustrated nanoparticle formulation. When binding with Cu(I) or Cu(II), the fluorescence of CDM is quenched but the photoacoustic signal from the semiconducting polymer remains unchanged, allowing for quantitative analysis of the chelation process. (b) UV-Vis spectrum of CDN. CDM absorption peaks at 610 nm and the polymer peaks at 1100 nm. (c) Size and morphology of CDN measured by DLS (mean ± s.e.m., n=4 independent experiments) and TEM imaging (n=2 independent experiments, 2 images acquired per experiment). Inset: TEM image of CDN with uranyl acetate staining (scale bar: 100 nm). PDI: polydisperse index. (d) Zeta potential measurement of SPN with or without CDM loading (mean ± s.d., n=5 independent samples for SPN and n=4 independent samples for CDN). (e) Fluorescence signal changes of CDN (CDM: 1 μM, Em: 760 nm) upon binding with Cu(I) or Cu(II) in buffered solution (mean ± s.d., n=3 independent samples). (f) Percentage of fluorescence signal of CDN solution (CDM: 5 μM) after mixing with various metal ions to that without metal addition (Na+: 5 mM, Mg2+: 5 mM, K+: 5 mM, Ca2+: 5 mM, Zn2+: 100 μM, Fe2+: 20 μM, Mn2+: 1 μM, Ni2+: 0.5 μM, Cd2+: 0.5 μM, Co2+: 0.5 μM) (mean ± s.d., n=3 independent samples). Concentrations of the tested metal ions are selected based on the physiological abundancy. Blue: measurement after CDN mixing with indicated physiologically abundant or trace metal ions; red: measurement after addition of extra 5 μM of Cu(II) to previous mixture of CDM and metal ions. (g) Representative fluorescence imaging of CDN mixed with different equivalents of Cu(I) in the agar phantom (n=3 independent samples). Signal intensity is normalized based on the photoacoustic signal at 1100 nm for each well.
CDNs were prepared with a nano-emulsion method as reported previously.17 The resulted CDNs were monodispersed spherical nanoparticles with a hydrodynamic size around 86.6 nm (Fig. 1c) or 81 nm (fCDN, Supplementary Figure 1c) as determined by dynamic light scattering (DLS) and transmission electron microscopy (TEM). The loading of CDM in the matrix yielded a slightly positive surface change on the nanocomplex (ζ potential of 0.77 ± 4.86 mV for CDN, 0.91 ± 4.83 mV for fCDN versus −4.89 ± 3.08 mV for SPN, Fig. 1d) that afforded the mitochondria-targeting ability of CDN. Once formulated, the CDN was stable in storage for 45 days. Minimal CDM leakage (1.38 %) was observed in serum after 24 h incubation at 37 °C (Supplementary Figure 2).
CDN binds with Cu(I) and Cu(II) promptly in a buffered solution, resulting in a decrease of CDM fluorescence at 740 nm (Fig. 1e). The dissociation constant (Kd) of CDN to Cu(I) is 1.80 ± 0.12 μM and 1.36 ± 0.07 μM to Cu(II). CDN is highly specific for copper ions and physiologically abundant metal ions were not observed to compete with copper binding when tested at the normal body content level (Fig. 1f). CDN reports the level of copper via changes in the optical signals, as demonstrated by the agar phantom imaging with IVIS Spectrum for CDM fluorescence and VisualSonics Vevo for photoacoustics for signal normalization (Fig. 1g).
CDN depletes intracellular copper and induces cell death.
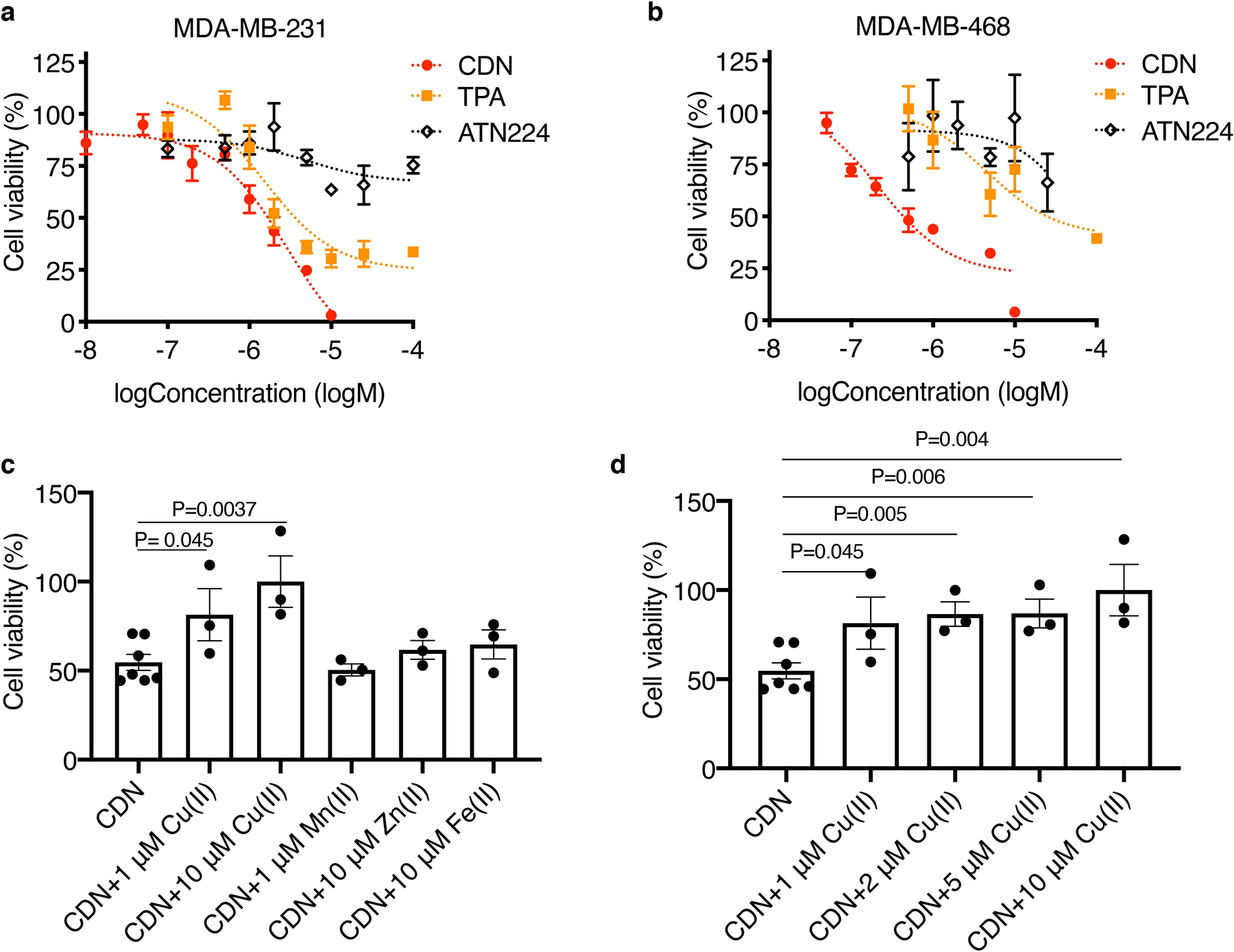
We first tested if CDN could be taken up by TNBC cells and deplete copper intracellularly. To independently measure the labile copper concentration in cells, we adopted a bioluminescent probe CCL-1 reported previously by Heffern et al,18 which is a D-luciferin analog caged by tris[(2-pyridyl)-methyl]amine (TPA) ligand. To test the cellular uptake and copper binding, we co-incubated MDA-MB-231luc or 4T1luc cells with CDN and different concentrations of copper (Fig. 2a, Supplementary Figure 3). Compared to the control group, the addition of CDN decreased the labile copper concentration as indicated by a lower bioluminescent signal from CCL-1. The fluorescence from CDM in CDN was partially quenched. Extra copper supply rescued the depleting effect, as shown by the observed increase in CCL-1 luminescence and a decrease in CDM fluorescence. Meanwhile, the copper-starved group with excess ethylenediaminetetraacetic acid (EDTA) showed the highest CDM fluorescence (123.7 ± 4.1 % of CDN group) and the lowest CCL-1 luminescence (78.6 ± 11.7 % of CDN group). With fCDN, intracellular copper binding was monitored in individual cells using confocal microscopy. Both MDA-MB-231 and MDA-MB-468 cells showed increased green/red ratios after 24 h incubation with fCDN and Cu(I) supplement in comparison to the control without Cu(I) supplement (Supplementary Figure 4). On the contrary, the EDTA supplement dramatically decreased the green/red ratio. This result again validates that CDN is able to report its own copper binding status.
Figure 2. CDN induces TNBC cell death via intracellular copper depletion.
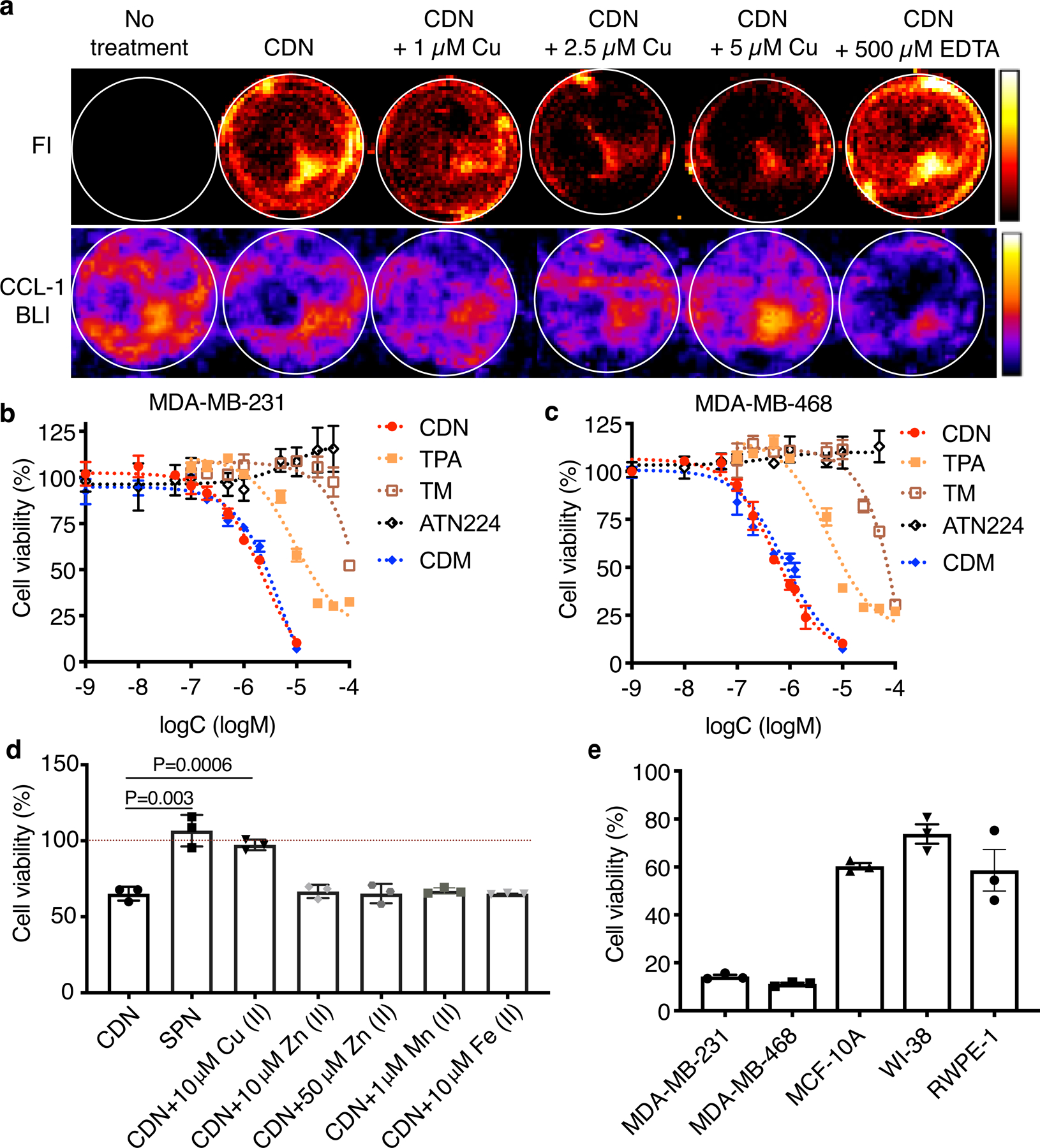
(a) Representative optical images of MDA-MB-231luc cells after 24 h incubation with CDN with or without different concentrations of copper supplements or excessive EDTA for copper deprivation (CDM: 1 μM). CDM fluorescence depicts copper binding to CDN and CCL-1 bioluminescence imaging depicts intracellular labile copper. (b) MDA-MB-231 and (c) MDA-MB-468 cell viability (percentage of cell control without treatment) after 24 h incubation with different concentrations of CDN, CDM, TPA, TM or ATN224 in complete medium with 10% FBS measured by MTS assay (mean ± s.e.m., n=3 biologically independent samples). (d) Viability of MDA-MB-231 cells after 24 h treatment with SPN or CDN with or without copper, zinc, iron or manganese addition (CDM: 1 μM, SPN: 50 μg/ml) measured by MTS assay (mean ± s.d., n=3 biologically independent samples, P value from unpaired t test, two-tailed). (e) Viability of TNBC cells (MDA-MB-231 and MDA-MB-468) and normal human cells (MCF-10A, WI-38 and RWPE-1) after 24 h treatment with CDN (CDM: 1 μM) in serum-free media (mean ± s.e.m., n=3 biologically independent samples).
Depletion of intracellular copper by CDN induced apoptosis in MDA-MB-231 and MDA-MB-468 cells, evidenced by the presence of cleaved caspase 3 after incubation (Supplementary Figure 5). CDN induced apoptosis can be effectively inhibited by BID inhibitor BI-6C9, indicating the apoptosis was associated with the release of cytochrome c or Smac from mitochondria (Supplementary Figure 5b). We hypothesized that CDN was more efficient in stopping copper trafficking than small molecular chelators due to the mitochondria targeting in cancer cells and the encapsulating effect of copper ions in the nanostructure. Three small molecular copper chelators were used to test this hypothesis: free CDM, TPA, a copper chelator that structurally resembles the chelating group in CDM without its mitochondria-targeting ability, and TM (or its choline salt ATN224). The cell viability after control agents or CDN treatment was tested with MTS assay (Fig. 2b–c) and trypan blue exclusion method (Extended Data Figure 1a–b). When treating TNBC cells in the presence of serum for 24 h, as shown in Fig. 2b, in MDA-MB-231, the IC50 of CDN is 2.31 ± 0.45 µM, in comparison to 5.48 ± 1.33 µM for CDM, 9.25 ± 2.19 µM for TPA, and over 100 µM for TM. For MDA-MB-468 cells, the IC50 of CDN is 0.53 ± 0.05 µM, 0.78 ± 0.16 µM for CDM, 5.34 ± 1.12 µM for TPA, and around 46.3 µM for TM (Fig. 2c). Lower IC50 for CDN was also observed in 4T1 cells (Supplementary Figure 6). We further validated this cytotoxic effect was due to the depletion of copper but not zinc, iron or manganese in a “metal remedy” experiment. As shown in Fig. 2d and Extended Data Figure 1c, 10 µM of copper supplement countered the cytotoxic effect of CDN; however, the addition of iron, manganese or zinc did not remediate the cytotoxicity effect. Copper ion titration study showed the restoration of the cell viability was dependent on the concentration of copper co-incubated with CDN (Extended Data Figure 1d). Most importantly, CDN is less toxic to normal healthy cells than to TNBC cells. After incubation with CDN (1 μM) in the absence of serum for 24 h, the viability was 60.30 ± 2.24 % for healthy mammary gland epithelial cell MCF-10A, 73.77 ± 6.98 % for lung fibroblast cell WI-38, and 58.59 ± 15.00 % for prostate epithelial cell RWPE-1 cell, all significantly higher than MDA-MB-231 cell (14.29 ± 1.25 %) and MDA-MB-468 cell (11.24 ± 0.88 %). It is noted that CDM became less toxic to WI-38 and RWPE-1 cells when loaded into SPN, likely due to the lower uptake of nanoparticle in the cells. No much difference was observed in MCF-10A (Supplementary Figure 7). Other than the cytotoxic effect, copper depletion also decreased the cell motility of highly invasive MDA-MB-231 cells (Supplementary Figure 8).
CDN inhibits OXPHOS in TNBC cells with a direct impact on COX activity.
We determined the subcellular localization of CDN with various organelle markers. In accordance with our hypothesis, CDN was enriched in mitochondria but less in early endosomes or endoplasmic reticulum (Supplementary Figure 9). We next investigated if CDN disrupted the mitochondrial function and metabolic pathways. We used JC-1 dye to evaluate mitochondrial integrity. MDA-MB-231 cells had strong red fluorescence (JC-1 aggregates) in the mitochondria when treated with control agents, indicating that the mitochondria remained undamaged (Fig. 3a, Supplementary Figure 10a). On the contrary, cells treated with CDN produced low red fluorescence and strong green fluorescence (JC-1 monomers), indicating that the mitochondrial membrane potential was compromised. The addition of copper prevented the mitochondrial damage and restored the red fluorescence. We further confirmed with Mitotracker staining that treatment with CDN resulted in compromised mitochondrial membrane integrity in a dose-dependent manner (Supplementary Figure 10b). As a direct result of copper depletion in the mitochondria, COX activity decreased to 49.44 ± 13.28 % as compared to control without any treatment for MDA-MB-231 cells and 30.70 ± 9.55 % as compared to control for MDA-MB-468 cells (Fig. 3b). This inhibition of COX activity by mitochondrial copper depletion is more potent than TPA or ATN224 treatment that is thought to deplete the copper pool in cytosol. CDN treatment resulted in transcriptional downregulation of COX subunits and chaperone and co-chaperone proteins that deliver copper to mitochondria. Other than COX6B2, mRNA levels of most COX units went down. COX17, COX11 and SCO2 are important for copper delivery and assembly and maturation of COX. Their mRNA levels decreased after 15 h of CDN treatment (Supplementary Figure 11).
Figure 3. CDN inhibits mitochondria complex IV.
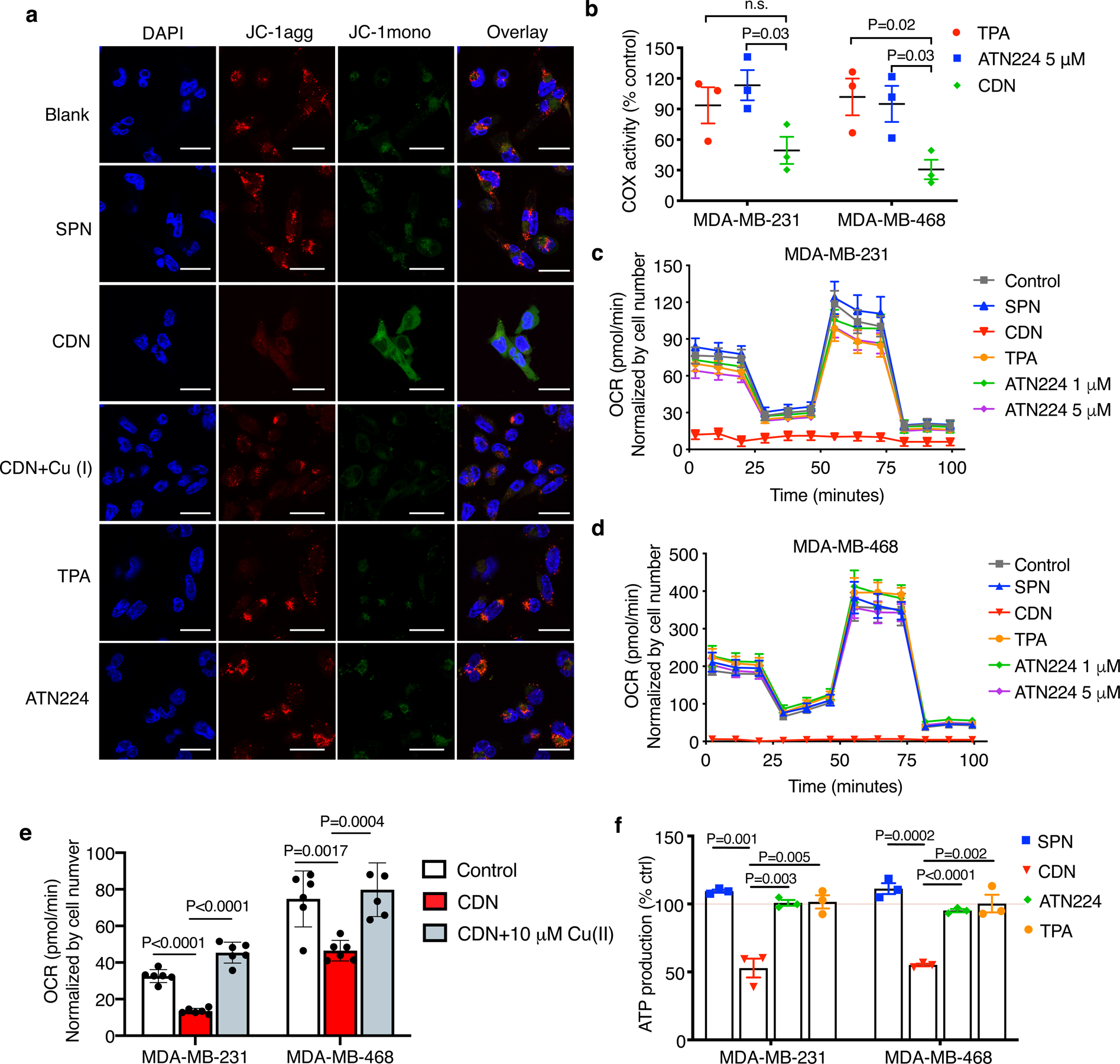
(a) Mitochondria membrane potential of MDA-MB-231 cells after 24 h incubation with CDN (CDM: 1 μM) with or without copper supplements (10 μM) or other control agents (chelator concentration: 1 μM, SPN concentration: 50 μg/ml), indicated by JC-1 staining (10 μg/ml). Red fluorescence that is emitted by aggregated JC-1 characterizes a high membrane potential while green fluorescence emitted by JC-1 monomer characterizes low membrane potential (scale bar: 50 μm, n=3 independent experiments). (b) Cytochrome c oxidase activities of MDA-MB-231 and MDA-MB-468 cells after 24 h incubation with CDN (CDM: 1 μM), TPA (1 μM) or ATN224 (5 μM) (mean ± s.e.m., n=3 biologically independent samples, P value from unpaired t test, two-tailed). Results are expressed as percentage of cell control without treatment. (c, d) CDN inhibits cellular oxygen consumption in (c) MDA-MB-231 and (d) MDA-MB-468 cells. 24 h after incubation with CDN (CDM: 1 μM) or control agents (TPA: 1 μM, ATN224: 1 μM or 5 μM, SPN: 50 μg/ml), the cellular oxygen consumption rate was measured by Seahorse analyzer. Oligomycin (1 μM) was introduced after 28 min, FCCP (1 μM) was introduced after 54 min and rotenone/antimycin A (0.5 μM) was introduced after 80 min (mean ± s.e.m., n=8 biologically independent samples). (e) Inhibition of OCR can be mitigated with the addition of copper ion. MDA-MB-231 and MDA-MB-468 cells were incubated with 1 μM of CDN for 1 h with or without copper supplement (10 μM) (mean ± s.e.m., n=6 biologically independent samples; P value from unpaired t test, two-tailed). (f) Cellular ATP level (as percentage of control with no treatment) after 24 h incubation with CDN, TPA or ATN224 (chelator concentration: 1 μM) in FBS supplemented culture medium (mean ± s.e.m., n=3 biologically independent samples; P value from unpaired t test, two-tailed).
Inhibition on the COX activity by CDN resulted in a subsequent shutdown of cellular OXPHOS. Measurement of the oxygen consumption rate (OCR) showed that treating with CDN for 24 h resulted in a complete inhibition of OCR that is as potent as well-characterized OXPHOS inhibitors (e.g., rotenone/antimycin A). In comparison, other control agents at the same or higher concentration merely impacted on the OCR in both MDA-MB-231 and MDA-MB-468 cells (Fig. 3c, d). The OCR inhibition by CDN was restored to the normal baseline level when extra copper ion was provided (Fig. 3e). An acute OCR inhibition response was also observed when incubating cells with CDN for 1 h (Extended Data Figure 2). OCR inhibition was examined at different concentrations of CDN: at a concentration higher than 1 μM, OCR was largely inhibited by CDN, and cells were not responsive to the subsequent injection of oligomycin (ATP synthase inhibitor) and carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP, uncoupling agent that collapses the proton gradient); at relatively lower concentrations, OCR response showed a mixed effect of respiratory chain component inhibition and proton leak due to mitochondria uncoupling, suggesting CDN also induced mitochondrial membrane disruption. Moreover, cellular ATP production significantly decreased in both TNBC cell lines treated with CDN (Fig. 3f, 52.8 ± 6.9 % of control for MDA-MB-231, 55.1 ± 1.0 % of control for MDA-MB-468). ATP levels in the cells treated with all control agents remained unaffected.
To further elucidate the cytotoxic mechanism of CDN, we used established OXPHOS inhibitors BAY 87–2243 and IACS-010759 as a comparison for in vitro experiments.19,20 CDN caused higher cytotoxicity in both MDA-MB-231 and MDA-MB-468 cells at 24 h after incubation comparing to the two complex I inhibitors (Extended Data Figure 3a). Cleaved caspase 3 levels after CDN treatment were also significantly higher than those after treatment of BAY 87–2243 and IACS-010759; the two inhibitors barely induced apoptosis at 24 h post-incubation (Supplementary Figure 5b). However, although CDN significantly reduced the OCR in both cell lines as compared to non-treated control, the blocking effect was not as potent as the two inhibitors, BAY 87–2243 and IACS-010759, which almost completely inhibited the oxygen consumption after 1 h incubation (Extended Data Figure 3b). All three agents were able to significantly elevate the extracellular acidification rate (ECAR) levels (Extended Data Figure 3c). Out of the three agents, IACS-010759 has the least impact on the mitochondrial membrane integrity, and BAY 87–2243 treatment resulted in partially compromised membrane integrity (Extended Data Figure 3d). This difference may contribute to the slightly higher cytotoxicity of BAY 87–2243 compared to IACS-010759. CDN treatment has proven to disrupt mitochondrial membrane in JC-1 staining and Mitotracker staining experiments (Fig. 3a, Supplementary Fig. 10). Altogether, these results indicate that CDN enabled copper depletion interfered with OXPHOS related cellular bioenergetics by directly inhibiting the COX activity and damaging the mitochondrial membrane potential.
Cell redox balance is strongly correlated with cellular copper availability as superoxide dismutase 1 (SOD1) uses copper as co-factor. We then investigated if oxidative stress induced by copper depletion contributed to cell death. Indeed, for both MDA-MB-231 and MDA-MB-468 cells, while SOD1 levels remained unaffected, SOD1 activity decreased after CDN treatment, showing a similar trend to that in treatment with a higher concentration of ATN224 (Extended Data Figure 4a, b). The superoxide level in cells was quantified with a chemiluminescent tracer 2-methyl-6-phenyl-3,7-dihydroimidazo[1,2-a]pyrazin-3-one (CLA),21 and elevated by 3-fold comparing to non-treated controls as observed in both cell lines (Extended Data Figure 4c). This elevated oxidative stress led to DNA double-strand break (marked by γH2A.X) and detectable levels of lipid peroxidation (marked by 4-HNE) with immunofluorescence staining (Extended Data Figure 4d). However, the addition of mitochondrial superoxide scavenger, MitoTEMPO, with a concentration up to 5 μM did not rescue the cells from the CDN treatment effect (Supplementary Figure 12). These suggest oxidative stress is not the direct cause of cell death but may exacerbate the cellular stress.
Inhibition of OXPHOS by CDN alters the metabolism of TNBC cells.
After treatment with CDN, the OXPHOS was largely inhibited and TNBC cells underwent a metabolic switch from OXPHOS to glycolysis. As shown in Fig. 4a, b, together with inhibited OCR, ECAR elevated in both MDA-MB-231 and MDA-MB-468 cells after 24 h treatment with CDN, but not with other control agents. The ECAR levels were not responsive to the following injection of oligomycin, suggesting cells have an increased glycolytic flux when OXPHOS is inhibited. Glucose uptake increased in both cell lines after 24 h treatment with CDN (1 μM), but not with ATN224 (5 μM) (Fig. 4c(i)). Extracellular lactate secretion significantly increased after CDN treatment (Fig. 4c(ii)), and intracellular lactate amount was comparable between CDN treated and control cells (Fig. 4d–e), suggesting increased flux to glycolysis.
Figure 4. CDN induced inhibition of mitochondrial OXPHOS changes metabolisms of TNBC cells.
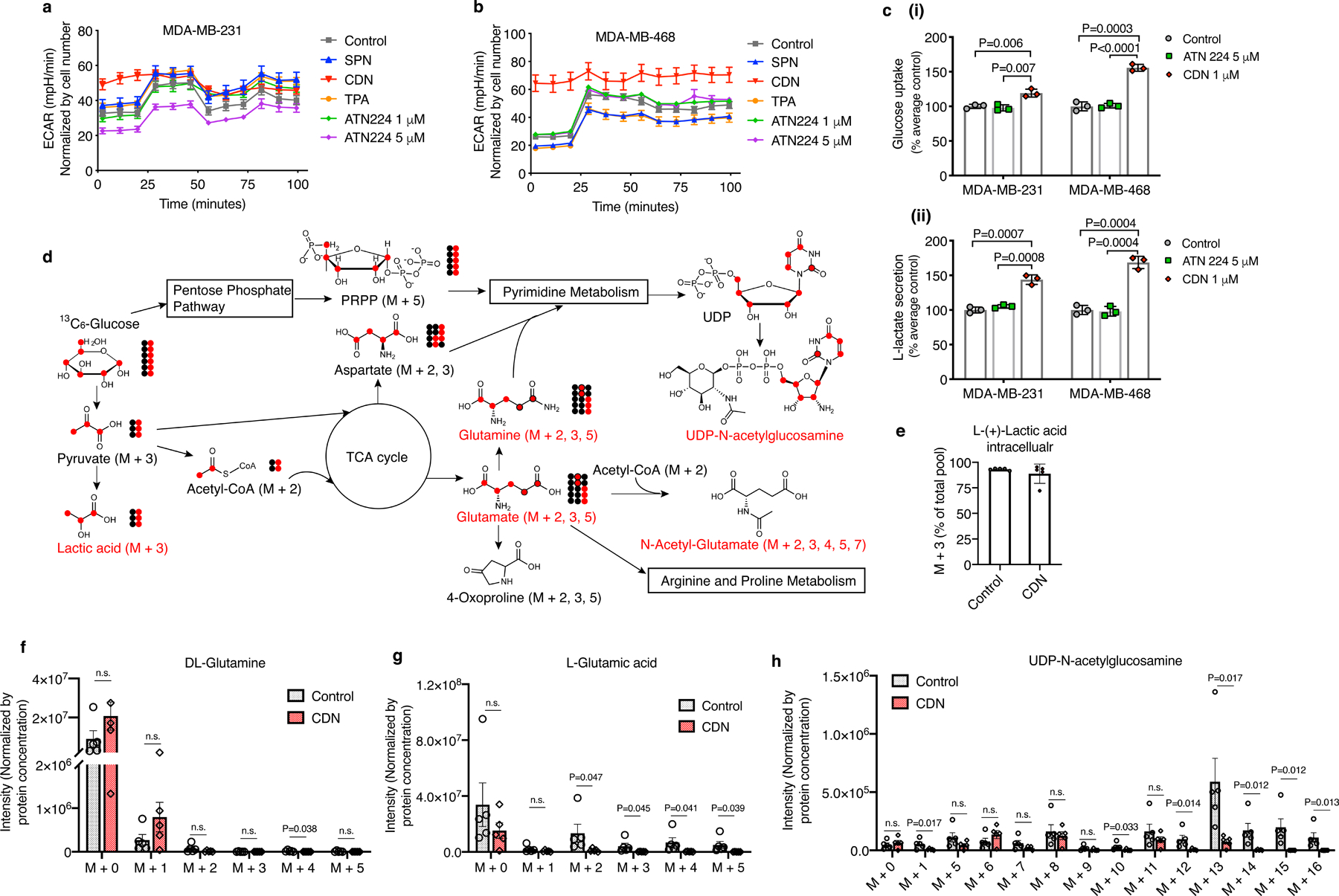
Extracellular acidification rate (ECAR) elevated in both (a) MDA-MB-231 and (b) MDA-MB-468 cells at 24 h after incubation with CDN (CDM: 1 μM). After CDN treatment, MDA-MB-468 showed minimal cellular response to the subsequent injections of oligomycin (1 μM, introduced after 28 min), FCCP (1 μM, introduced after 54 min) and rotenone/antimycin A (0.5 μM, introduced after 80 min). MDA-MB-231 exhibited no response to oligomycin and rotenone/antimycin A. Unlike CDN, other control agent treatment (TPA: 1 μM, ATN224: 1 μM or 5 μM, SPN: 50 μg/ml) slightly decreased or did not impact the ECAR levels (mean ± s.e.m., n=8 biologically independent samples). (c) (i) glucose uptake and (ii) lactate secreted into the medium over 24 h incubation with ATN224 (5 μM) or CDN (CDM: 1 μM) (mean ± s.e.m., n=3 biologically independent samples, P value from unpaired t test, two-tailed). Results are expressed as percentage of cell control without treatment. (d) Illustration of glucose metabolism pathways affected by CDN treatment. (e) Percentage of M+3 lactic acid over the total pool. Intensities of isotopologues of metabolites, including, (f) glutamine, (g) glutamate, (h) UDP-N-acetylglucosamine, produced from 13C6-labeled glucose in vitro. Metabolites with isotopologues shown are written in red. Other important intermediate metabolites are written in black. Non-labeled 12C are shown as black-filled circles while labeled 13C are shown as red-filled circles. Raw intensities are normalized by protein concentration. Data are shown as mean ± s.e.m. (n=5 biologically independent samples, P value from unpaired t test, two-tailed).
Stable isotope-tracing experiments using 13C6-glucose confirmed that CDN incubation led to a reduction of amino acid synthesis activity (Fig. 4d, f, g, Supplementary Figure 13). CDN treatment also caused changes in the hexosamine pathway as evidenced by a significant decrease in UDP- N-acetylglucosamine (Fig. 4h). Stable isotope-tracing 13C515N2-glutamine also found significantly decreased glutamine contribution to amino acid biosynthesis upon CDN treatment (Extended Data Figure 5). And glutamine did not serve as an alternative compensatory mechanism (Extended Data Figure 6). Notably CDN impaired cellular redox homeostasis beyond superoxide production and SOD activity. In CDN treated cells, endogenous antioxidants such as glutathione and its product cysteinylglycine levels were significantly decreased (Extended Data Figure 5). This decrease in the antioxidant production might further exacerbate the cellular oxidative stress. Taken together, CDN treatment significantly decreased the utilization of glucose and glutamine for amino acid and hexosamine synthesis and redox homeostasis, which all contributed to the potency of CDN treatment.
In vivo imaging of copper depletion and disruption of mitochondrial potential in the tumor by CDN.
To study the copper depleting dynamics in vivo, we longitudinally monitored CCL-1 bioluminescence, CDM fluorescence, and SPN photoacoustic signals in MDA-MB-231luc tumors after CDN administration. The labile copper level depicted by CCL-1 showed a continued decrease till day 3, and slowly resumed to the original level at day 7 after a single injection of 1.35 mg/kg CDN (Fig. 5a, Supplementary Figure 14). While the nanoparticles were retained in the tumor region for as long as 10 days (Fig. 5b, Supplementary Figure 15), CDM binding to copper was saturated around day 5, which explains the slow restoration of copper levels in tumors (Fig. 5b). This data suggests that CDN can accumulate in the tumor region, deplete copper locally and maintain a low copper level in the tumor for up to five days after a single injection (1.35 mg/kg).
Figure 5. CDN depletes copper efficiently in vivo.
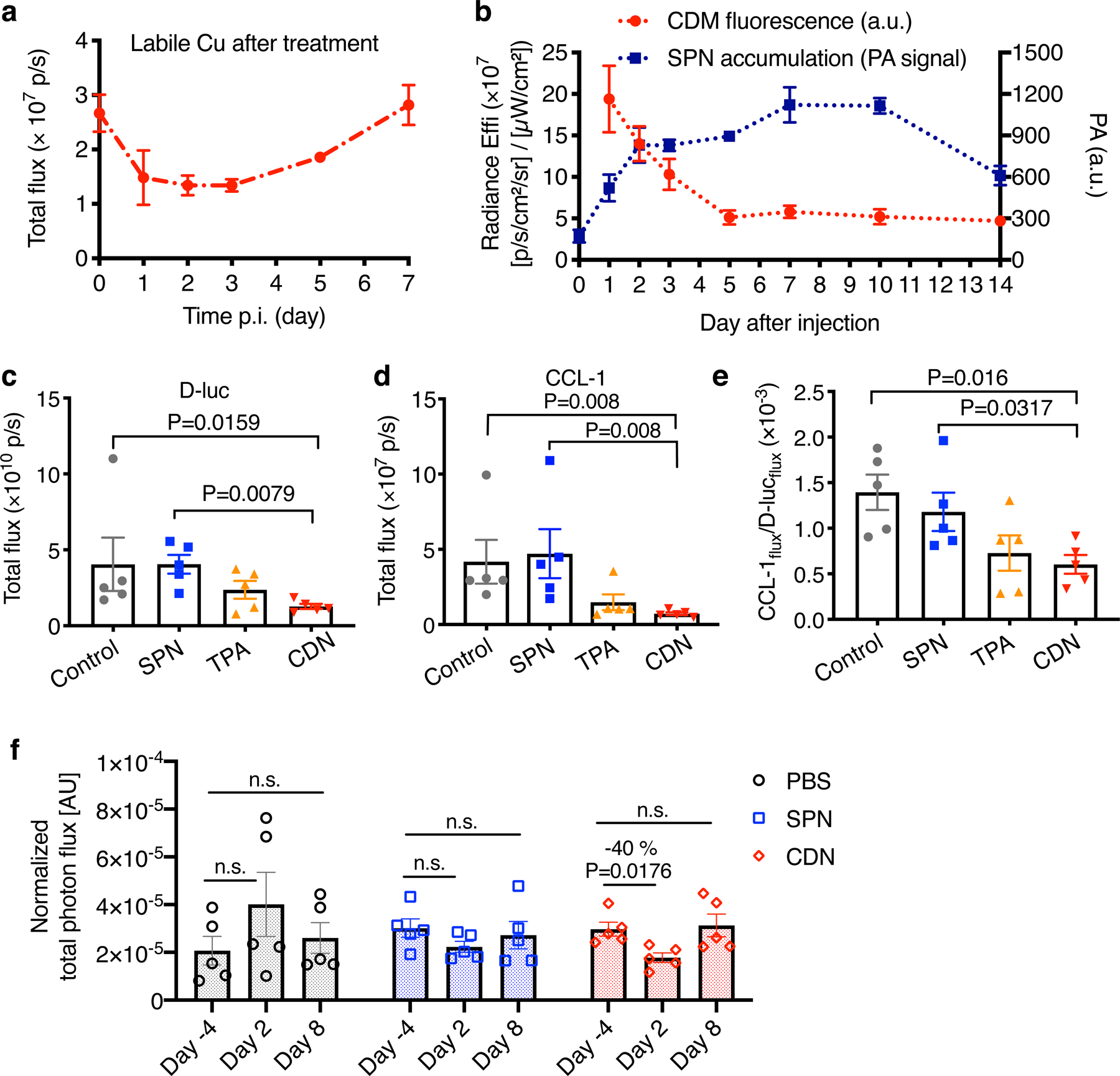
(a) Labile copper concentration detected by CCL-1 bioluminescence imaging after i.v. injection of CDN (1.35 mg/kg) in MDA-MB-231luc tumor bearing mice (mean ± s.e.m., n=3 independent animals). (b) Longitudinal monitoring of nanocomplex delivery and copper binding via optical imaging of CDN after i.v. injection of CDN. Photoacoustic signal in the tumor region (blue) reflects the accumulation and retention of CDN while fluorescence signal reports the copper binding to CDM (red). (mean ± s.d., n=3 independent animals) (c) D-luciferin bioluminescence imaging and (d) CCL-1 imaging of mice receiving SPN, TPA or CDN treatment (chelator dose: 1.35 mg/kg). MDA-MB-231luc tumor bearing mice were i.v. administered with the indicated agents every three days with a total of 5 doses. Images were acquired at day 25 after the first injection (mean ± s.e.m., n=5 independent animals, P value from unpaired t test, two-tailed). (e) The labile copper levels in the tumor region for different treatment groups were quantified as CCL-1 to D-luciferin flux ratio (mean ± s.e.m., n=5 independent animals; P value from unpaired t test, two-tailed). (f) In vivo mitochondrial membrane potential measured by MAL3 bioluminescence imaging. MDA-MB-231luc tumor bearing mice were imaged before and after injection with PBS, SPN or CDN. The total photon flux of MAL3 bioluminescence signals was normalized by D-luciferin bioluminescence signal (mean ± s.e.m., n=5 independent animals, P value from unpaired t test, two-tailed).
We proceeded to investigate if strategic administration of CDN led to effective copper depletion and inhibition of tumor growth in MDA-MB-231luc orthotopic breast cancer bearing mice (n = 5 each group). For the treatment groups, mice received i.v. injection of CDN or TPA (at the same chelator concentration of 1.35 mg/kg) every three days. On day 25 from the first treatment, mice were subjected to D-luciferin and CCL-1 bioluminescence imaging (Supplementary Figure 16). For both D-luciferin and CCL-1, the total photon flux values of the CDN group were significantly lower than the blank control and SPN control (Fig. 5c, d). To normalize the cancer cell number effect on the CCL-1 imaging result, we calculated the total flux ratio of CCL-1 to D-luciferin (Fig. 5e). Both TPA and CDN successfully depleted the copper in the tumor region compared to the control group (TPA: P = 0.04, CDN: P = 0.016). However, CDN exhibited higher depletion efficiency and better treatment efficacy than TPA.
We then investigated the mitochondrial membrane potential after CDN treatment in vivo using mitochondria-activatable luciferin 3 (MAL3)22 probe (Supplementary Figure 17). The mitochondrial membrane potential significantly decreased at 2 days after CDN treatment (P=0.0176, n=5), then restored to pre-treatment level when imaged at day 8 after treatment (Fig. 5f). In comparison, PBS or SPN treatment did not cause a significant change in the mitochondrial membrane potential. This result was consistent with our in vitro finding that CDN treatment compromised mitochondrial membrane potential, which contributed to the apoptosis of cancer cells.
CDN serves as an effective, safe drug for TNBC.
A potential side effect of systemic copper depletion is that it may result in overall copper deficiency and induce hematological disorders. We explored the biodistribution and copper depletion of the CDN in tumors and various healthy organs with fCDN. At 24 h after i.v. injection of fCDN, mice were sacrificed, and the tumor and normal organs including contralateral mammary fat pad, heart, lung, liver, spleen, adrenals, kidneys, stomach, small intestine, big intestine, ovary, and uterus, were collected and imaged with an IVIS imager. As shown in Extended Data Figure 7a, the tumor showed significantly higher SPN fluorescence signal (green color) than any other healthy organs whereas the CDM fluorescence at 740 nm (red color) was weak compared to that of the liver. Quantitatively (Extended Data Figure 7b–d), the tumor showed the highest fluorescence radiance ratio of 40.5 ± 7.3, reflecting high accumulation and effective copper binding in the tumor. The liver also showed a relatively high amount of CDN as indicated by the CDM fluorescence, but the Em540/Em740 ratio was only 2.5 ± 0.9 for the liver. This indicated that although CDNs were cleared through the hepatobiliary route, they did not deprive the liver of copper ions. Distinct distribution patterns inside the tumor and liver tissues have been observed with microscopic imaging (Supplementary Figure 18). CDN signals in the liver appeared punctuated and showed a good correlation between green and red signals (low copper binding). In the tumor, the green signals were relatively homogenously distributed and red signals were rather dim (due to high copper binding). CDN likely resided in the Kupffer cells in the liver instead of hepatocytes. Other healthy major organs showed minimal fluorescence signals in both channels, indicating fairly low uptakes in normal tissues.
We further evaluated the potential cumulative toxicity originated from repetitive injections and acute toxicity. For the cumulative toxicity, healthy Balb/c mice received weekly intravenous administration of CDN. During the course of the treatment, no weight loss or behavioral abnormality was observed. After seven injections of CDN, mice were sacrificed for hematological and pathological analysis. No blood count suppression was observed in the treated group as shown in Extended Data Figure 8a i–vii and no difference was observed in the liver panel comparing to the saline control group (Extended Data Figure 8a viii–x). No pathological changes were observed in major organs including the hearts, lungs, livers, kidneys, and spleens (Extended Data Figure 8b). For the acute toxicity study, healthy mice were i.v. injected with 100 mg/kg CDN or saline and monitored for two weeks. We found that high dose injection did not cause either body weight loss or behavioral abnormality in the following two weeks, nor hematological or pathological changes comparing to the control group (Extended Data Figure 9).
The therapeutic effect of CDN for TNBC was first evaluated in a long-term survival study in MDA-MB-231 breast cancer model. For the treatment groups, nude mice bearing MDA-MB-231 orthotopic tumors received 7 doses of intravenous injections of TPA, CDM or CDN (chelator concentration of 1.35 mg/kg, n=12 each group) or daily oral gavage of ATN224 (0.7 mg/kg/day till day 42, n=12). Since the copper level in the tumor resumed to the original level at day 7 after a single injection (Fig. 5a), a weekly dosing strategy was adopted to explore if a lower overall dosing was applicable (Fig. 6a). As shown in Fig. 6b, c, tumor growth in the saline group and SPN group was rapid, and the median survival was 25.5 days and 27 days, respectively. The ATN224 treatment group showed moderate therapeutic efficacy with a median survival of 32 days. The TPA and CDM groups showed similar therapeutic efficacy (median survival: 33.5 days for CDM and 35 days for TPA). The CDN treatment significantly inhibited tumor progression: 50 % of the treated mice survived over 68 days after the first injection, and the tumor volumes of the surviving mice remained fairly small except for one mouse (mean ± s.e.m. 99 ± 29 mm3, n=5). This promising therapeutic efficacy of CDN for primary TNBC tumors was also observed in mouse models bearing MDA-MB-468 and 4T1 tumors (Fig. 6d, Supplementary Figure 19). Metabolomic analysis of the tumors further confirmed that CDN treatment truly led to elevated glycolysis, evidenced by significantly higher levels of glucose (Fig. 6e), glycolysis end product lactate (Fig. 6f), and alanine, the product of pyruvate via transaminase reactions (Fig. 6g).
Figure 6. CDN inhibits TNBC tumor growth.
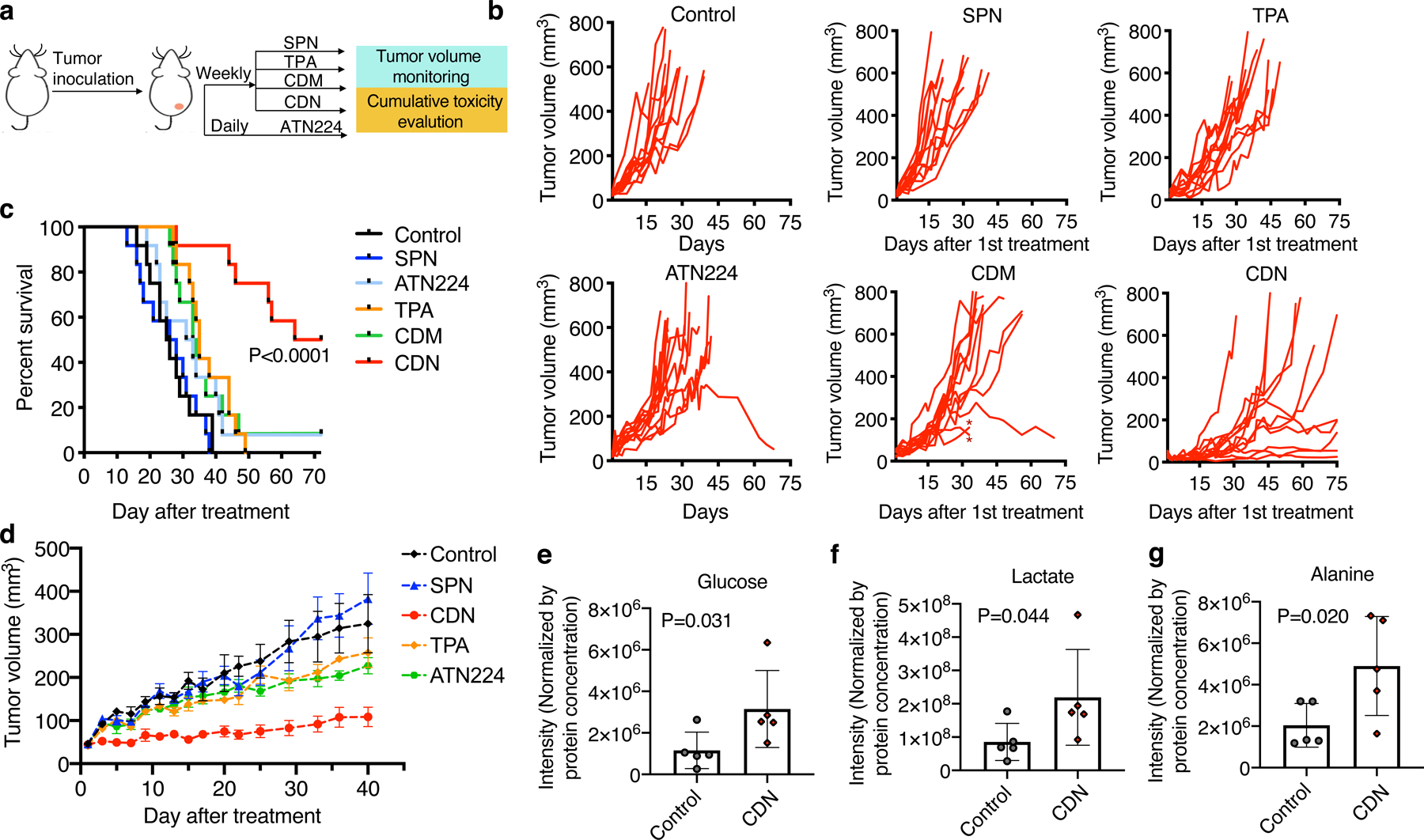
(a) Treatment strategy for long-term survival study with MDA-MB-231 tumor bearing mice. Six control and treatment groups were included in the study: control, SPN, TPA, ATN224, CDM and CDN. Mice received SPN, TPA, CDM or CDN (chelator dose: 1.35 mg/kg) via i.v. injection weekly and ATN224 via oral gavage daily (0.7 mg/kg/day). (b) Tumor growth curve of each individual mouse in different treatment groups (n=12 independent animals). In the CDM treatment group, * indicates individuals that received early euthanasia under the instruction of veterinarian due to skin rashes. (c) Survival curve of different treatment groups up to 70 days after the initial treatment. (d) Tumor growth curve of MDA-MB-468 tumor bearing mice during CDN and control agent treatment (mean ± s.e.m., n=5 independent animals). At day 40 after initial treatment, tumor burden in CDN treatment group was significantly smaller than that in the non-treated control (P=0.016), SPN group (P=0.003), TPA group (P=0.004) and ATN224 group (P=0.004) (P value from unpaired t test, two-tailed). Three days after the 7th dose, mice were sacrificed, and tumors were subject to metabolite analysis. (e) Glucose, (f) lactate and (g) alanine levels were determined (mean ± s.d., n=5 independent animals, P value from unpaired t test, two-tailed).
Discussion
Although many cancers undergo glycolysis under aerobic conditions,23,24 there is an increasing understanding that some cancer cell types rely on mitochondrial OXPHOS. Cancer types including breast cancer,3,25 prostate cancer,26 metastatic ovarian cancer,27,28 and glioblastoma29 have been found to import lipid from adjacent adipocytes and employ fatty acid oxidation as their primary energy source. Patient tumor immunohistochemistry staining has also shown that TNBC has the highest percentage of negative staining of pyruvate kinase M2 isoform (PKM2),30 the enzyme catalyzing the final step in glycolysis and generating ATP, suggesting glycolysis is not considered indispensable for TNBC. In this work we show targeting mitochondria OXOPHOS by CDN inhibits TNBC in vitro and in vivo with high efficacy. CDN also decreased the viability of some ER-positive breast cancer cell line (such as HCC1428) (Extended Data Figure 10a) and prostate cancer cell lines (Extended Data Figure 10b) in vitro, with low toxicity to normal prostate epithelial cells (Fig. 2e).
Existing drugs targeting oxidative metabolism have several limitations. First, they have modest efficacy and inadequate accumulation. For example, biguanides, such as metformin, which is clinically approved for the treatment of diabetes and metabolic disorders, inhibits complex I and disrupts mitochondrial OXPHOS,31 but its cellular IC50 is at hundreds of micromolar, too high to be useful as potent cytotoxic agent.32 Second, off-target toxicity is a concern; potent electron transport chain inhibitors, such as rotenone, exhibit very high off-target neurotoxicity.33 Finally, the pharmacokinetic profiles of some of these drugs, such as oligomycin,20 are unacceptable.
Our design and treatment strategy overcome these obstacles with a dual-targeting mechanism to improve the potency of CDNs and decrease their potential toxicity to normal cells. Cancer cell mitochondria typically feature a hyperpolarized membrane potential.34–36 To take advantage of this high negative transmembrane potential, we coupled a positively charged fluorescent moiety to direct the chelator specifically to cancer cell mitochondria. Moreover, instead of directly inhibiting the electron transfer complex, we chose to decrease the availability of an enzyme co-factor. This will largely spare healthy cells in low need of copper supply (their copper concentration is lower than the Kd of the chelator), which further decreases the potential off-site toxicity. The delivery of CDM via the polymeric matrix structure has been proven to improve the efficiency and in vivo pharmacokinetics of CDM. With the favorable prolonged tumor retention time and imaging-enabled treatment stratification, a much lower dose is needed.
Glycolysis is negatively regulated by OXPHOS.37 Mitochondrial oxidation suppression releases the inhibition of glycolysis, which turns out to be the Achilles heel for OXPHOS-dependent cancers. In response to mitochondrial OXPHOS inhibition by CDN, TNBC cells increased glycolytic activity, evidenced by ECAR elevation and increased uptake of glucose and production of lactate in vitro and in vivo. This switch to glycolysis is a compensatory metabolic flux due to the absence of mitochondrial OXPHOS that would normally utilize the carbons from pyruvate and acetyl Co-A. However, this increased glycolysis flux was not able to counter ATP deficiency and oxidative stress. Reduced TCA activity also limited the carbon source for amino acid and nucleotide synthesis. Of note, analysis of the mRNA sequencing results revealed that lipid metabolism was likely altered in response to copper depletion. TNBC typically shows upregulation of genes encoding enzymes for fatty acid oxidation and downregulation of genes encoding enzymes for fatty acid synthesis in primary human tumors.8 After CDN treatment, MDA-MB-231 cells showed upregulation of many genes that encode activators of fatty acid, cholesterol and triglyceride synthesis including ACLY, FASN, SCAP, SREBF1, SREBF2 (Supplementary Figure 20). Enhanced fatty acid synthesis may further exacerbate the energy deficiency as increased malonyl-CoA inhibits CPT1 and suppresses fatty acid transport into the mitochondria for oxidation.38,39
Copper enzymes including COX40 or phosphodiesterase 3B41 have been found excessive and essential for cancer cell metabolism, and various others identified prerequisite during all stages of cancer development involving uncontrolled cancer cell proliferation, dysregulated metabolism, invasion and migration to distant sites.15,42–47 Despite their recognized importance, successful attempts to treat primary or metastatic cancer with copper chelation are rather limited.48 Conventional copper chelator TM and ATN224 are reported to be anti-angiogenesis agents.49,50 The cytotoxicity of TM and ATN224 to TNBC cells is not potent as shown in our results and other studies (IC50 of over 100 μM).51 And their therapeutic effect in primary tumor treatment is not satisfactory for many types of cancer.52–54 The most successful case so far is seen in a Phase II clinical trial with TM as a chemo-preventive method in prolonging the recurrence-free window of patients with no trace of tumor by the time of the treatment.55–57 Moreover, both molecules deplete copper systemically, and serious concerns have been raised that it may result in overall copper deficiency and induce hematological disorder and neurotoxicity. Functioning differently than these conventional copper chelators, CDN induced mitochondrial dysfunction and altered the metabolism of TNBC cells. Copper depletion by CDN resulted in a combination of energy and nutrient deficiency, as well as elevated oxidative stress and mitochondrial membrane rupture, which all contributed to the apoptosis of TNBC cells. The design and application of CDN may open new avenues for copper-depleting cancer intervention.
Methods
Chemicals.
All chemicals were obtained from Sigma-Aldrich unless otherwise stated. 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (ammonium salt)(DPPE-PEG2000) was purchased from Avanti Polar Lipids. fSP (Poly[(9,9-dioctyl-2,7-divinylenefluorenylene)-alt-{2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylene}]) was purchased from Luminescence Technology Corp. 2-Methyl-6-phenyl-3,7-dihydroimidazo[1,2-a]pyrazin-3-one (CLA) was purchased from TCI America. JC-1 dye was purchased from Invitrogen. ATN224 was purchased from Cayman Chemical Company. XenoLight™ D-Luciferin was purchased from Perkin Elmer. TRIzol® reagent was purchase from Thermo Fisher Scientific. Ultrapure water was purchased from Invitrogen. 1× PBS, 1× D-PBS, and 1× HBSS were purchased from Gibco. Sterile 0.9 % saline solution was purchased from TEKnova. CCL-1 was synthesized with the procedures reported previously.58
General information for compound synthesis and characterization.
Compound synthesis routes and characterizations are detailed in Supplementary Notes. The nuclear magnetic resonance (NMR) spectra were taken on Bruker 400 MHz magnetic resonance spectrometer. Chemical shifts are reported as δ in units of p.p.m. relative to TMS (δ 0,s); multiplicities are reported as follows: s (singlet), d (doublet), m (multiplet) or br (broadened); coupling constants are reported as a J value in hertz (Hz); the number of protons (n) for a given resonance is indicated nH, and based on the spectral integration values. Electrospray ionization mass spectrometry (ESI MS) spectra were collected at the Mass Spectrometry Facility of Stanford University. Elemental analysis was performed on a FlashEA1112 Elementar Analysis Instrument. Gel permeation chromatography (GPC) analysis was conducted on a Waters 2414 system with polystyrene (PS) as standard and chloroform as eluent. The UV-vis-NIR absorption spectra were recorded on an Agilent spectrophotometer.
CDN formulation and characterization.
CDN and fCDN were formulated with a nano-emulsion method reported previously.17 Briefly, 1 ml tetrahydrofuran (THF) solution of SP or (Poly[(9,9-dioctyl-2,7-divinylenefluorenylene)-alt-{2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylene}]) (fSP, 0.125 mg), DPPE-PEG2000 (2.5 mg) and CDM (2.5 μg) was rapidly injected into distilled deionized water (9 ml) under continuous sonication with a microtip-equipped probe sonicator (Branson, W-150) at a power output of 6 watts RMS for 2 min. After sonication THF was evaporated at 45 °C under nitrogen atmosphere. The aqueous solution was filtered through a polyvinylidene difluoride (PVDF) syringe driven filter (0.22 μm) (Millipore) to remove large nanoparticles. Then the solution was washed three times with HEPES buffer (10 mM, pH 7.4) with a 30 K centrifugal filter unit (Millipore) under centrifugation at 3,500 rpm for 8 min at 4 °C. After ultrafiltration, the final concentration of CDM in CDN was 132 μM (total nanoparticle matrix concentration was 6.5 mg/ml). CDN solution was stored in dark at 4 °C. The sizes of CDN and fCDN were determined by dynamic light scattering (Malvern ZetaSizer Nano S) and presented as number percentage and intensity percentage with polydisperse index. The transmission electron microscopy (TEM) images were obtained on a transmission JEM 1230 electron microscope with an accelerating voltage of 200 kV. Zeta potential measurements were conducted on the Malvern ZetaSizer Nano S with folded capillary cells. Fluorescence spectra were recorded on a wavelength-calibrated FluoroMax-3 fluorometer (Horiba Jobin Yvon).
Fluorescence response of CDN to copper and other ions.
To measure the fluorescence signal changes of CDN upon mixing with Cu+ and Cu2+, different equivalents of copper ions were added to CDN solution (CDM: 1 μM), and the fluorescence emission spectra were read on a 96-well plate reader (Tecan). For fCDN, the fluorescence spectra were measured upon mixing with different equivalents of Cu2+ with an excitation wavelength at 453 nm. The FRET ratio was calculated as the ratio between emission at 540 nm and 740 nm. To test the specificity of CDN, CDN solution (CDM: 5 μM) was first mixed with physiologically relevant metal ions, including Na+ (5 mM), Mg2+ (5 mM), K+ (5 mM), Ca2+ (5 mM), Zn2+ (100 μM), Fe2+ (20 μM), Mn2+ (1 μM), Ni2+ (0.5 μM), Cd2+ (0.5 μM), Co2+ (0.5 μM). Concentrations of the tested metal ions are selected based on the physiological abundancy. The fluorescence emissions of CDM after mixing with metal ions were measured (Ex: 610 nm, Em: 740 nm). Then extra 5 μM of Cu2+ was added to each group of mixture to determine the CDM fluorescence change. For the phantom imaging, agar phantom, CDN solutions with different equivalents of Cu+ to CDM were added to warm agar solution (1 %, 20 μl, 40 °C) and immediately added to pre-formulated agar phantom well. Each well was sealed with another layer of agar. The fluorescence image was acquired with IVIS Spectrum (Ex: 640 nm, Em: 740 nm), and the photoacoustic signal of each well was acquired with LAZR VisualSonics (1064 nm).
Serum stability of CDN.
fCDN (4 μl) was added to 396 μl of mouse serum (Sigma Aldrich) without or with 500 μM EDTA for labile copper removal and incubated for 24 h at 37 °C. The FRET ratio between 540 nm and 740 nm (Ex: 453 nm) and CDM fluorescence (Ex: 610 nm, Em: 740 nm) prior to or after incubation were measured with a plate reader (Tecan). To determine the CDM leakage in serum over 24 h, CDN in serum after incubation was washed with an ultracentrifugation unit (Amicon Ultra-0.5 ml). The CDM amount in the elute was determined by fluorescence measurement. To determine the effect of CDN on serum copper, CDN (CDM: 2 μM or 4 μM) was added to serum and incubated for 24 h at 37 °C. The ceruloplasmin (Cp) activity in the serum was determined by Ceruloplasmin Colorimetric Activity Kit (Invitrogen, EIACPLC) under the sample preparation guideline and assay protocol provided. Results were presented as ratio percentage to the Cp activity (mU/ml) before incubation.
Cell culture.
MDA-MB-231, MDA-MB-468, BT-20, 4T1, WI-38, MCF-10A, RWPE-1, PC-3, 22Rv1, HCC1428, MCF7, and T47D were purchased from ATCC. MDA-MB-231luc cells were purchased from Promega. 4T1luc cell line was a kind gift from Dr. Edward Graves’ Lab. Cells were cultured in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C unless otherwise stated. MDA-MB-231, MDA-MB-231luc, MDA-MB-468, 4T1, 4T1luc, 22Rv1, and HCC1428 cells were cultured in RPMI-1640 medium (Gibco, Catalog No. 22400089) with 10 % FBS and 1 % pen/strep. MCF7 cells were cultured in ATCC-formulated Eagle’s Minimum Essential Medium (ATCC, Catalog No. 30–2003) supplemented with 0.01 mg/ml human recombinant insulin (Gibco, Catalog No. 12585–014), 10 % FBS and 1 % pen/strep. T47D cells were cultured in RPMI-1640 medium (Gibco, Catalog No. 22400089) supplemented with 0.2 U/ml human recombinant insulin (Gibco, Catalog No. 12585–014), 10 % FBS and 1 % pen/strep. BT-20 and WI-38 cells were cultured in ATCC-formulated Eagle’s Minimum Essential Medium (ATCC, Catalog No. 30–2003) with 10 % FBS and 1 % pen/strep. PC-3 cells were cultured in ATCC-formulated F-12K medium (ATCC, Catalog No. 30–2004) with 10 % FBS and 1 % pen/strep. MCF-10A cells were cultured in MEBM base medium with MEGM kit supplement (without the addition of gentamycin-amphotericin B mix, Lonza/Clonetics, Catalog No. CC-3150), 100 ng/ml cholera toxin (Sigma Aldrich, Catalog No. C8052) and 1 % pen/strep. RWPE-1 cells were cultured in Keratinocyte Serum Free medium (Gibco, Catalog No. 17005042) with K-SFM supplement, including EGF and BPE. Mycoplasma test was carried out routinely to detect contamination.
In vitro copper depletion.
The copper level after CDN or control agent treatment was measured by CCL-1 luminescence imaging or ratiometric fluorescence microscopic imaging of fCDN. For CCL-1 imaging, MDA-MB-231luc or 4T1luc cells were seeded in 6-well plates (106 per well) 24 h prior to the treatment and cultured in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C. For MDA-MB-231luc cells, CDN (CDM: 1 μM) with or without extra copper ion supplements at concentrations indicated in the figure legend or 500 μM of EDTA was added to each well (fresh medium with 10 % FBS) and incubated for 24 h. For 4T1luc cells, CDN, SPN, ATN224, or TPA (chelator concentration: 1 μM, SPN: 50 μg/ml) was added and continued incubation for 24 h. Cells were washed three times with PBS, and 1 ml of CCL-1 in D-PBS (50 μM) was added to each well. Cells were immediately imaged by IVIS Spectrum (Perkin Elmer) for 45 min. The integration images of luminescent radiance were presented. For ratiometric fluorescence imaging, MDA-MB-231 or MDA-MB-468 cells were seeded (2×104 per well) in Lab-Tek II chamber (NUNC™) 24 h prior to the experiment and cultured in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C. After 24 h incubation with fCDN or with supplements of either 10 μM of Cu2+ or 500 μM of EDTA, cells were washed three times with PBS and changed to fresh medium containing 10 μg/mL Hoechst 33342. For live cell confocal microscopic imaging (Zeiss LSM 710), 488 nm excitation wavelength was used and fluorescence signals at 525 ± 30 nm were collected as the green channel (fSP) and 630–733 nm were collected as the red channel (CDM). Images were processed as Green/Red ratios profiled by Image J and presented in pseudo-colors.
Cytotoxicity.
For MTS assay, TNBC and normal cells were seeded into 96-well plates 24 h prior to the test and cultured in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C. Cells were then incubated with CDN or control agents with the concentrations indicated in the corresponding figure legends in complete medium or base medium without FBS supplement. 24 h after incubation, cells were washed with PBS 3 times and changed to fresh medium. The viability of the cells after treatment was measured using the MTS assay (CellTiter 96, Promega, Catalog No. G3580). The viability was presented as the percentage of control. (n=3 per condition for viability) For trypan blue exclusion test, TNBC cells were seeded into 6-well plates 24 h prior to the test and cultured in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C. Cells were then incubated with CDN or control agents with the concentrations indicated in the corresponding figure legends in complete medium. 24 h after incubation, cells were washed with PBS for 3 times and harvested with a scraper. The cell suspension was then mixed with 0.4 % trypan blue dye (1:1, Thermo Fisher Scientific, Catalog No.15250061) and incubated for less than three minutes at room temperature. The viability of the cells was measured with Countess II (Life Technology). The viability was presented as the percentage of control (n=3 per condition for viability).
Cell invasion study.
Cell suspension was added into the upper chambers of the pre-hydrated 24-well plate Matrigel invasion chamber (Corning, Catalog No. 08–774-122) and allowed to grow for 24 h in complete medium with or without CDN or control agent treatment. After incubation, the non-invading cells were removed with cotton swab scrubbing, and the invaded cells were stained with Diff-Quik™. Cell numbers were counted with a microscope with at least 15 views per well. For MDA-MB-231 cells, n=3 biologically independent samples for blank, SPN, CDN with copper, TPA, and ATN224 group; n=5 biologically independent samples for CDN group. For MDA-MB-468 cells, n=4 biologically independent samples for blank and SPN group, and n=5 biologically independent samples for CDN group.
Western Blots.
Cells were treated with CDN or control agents with the concentration indicated in the corresponding figure legends. Then cells were trypsinized, and cell lysates were prepared with RIPA buffer (Thermo, Catalog No. 89900). The protein concentrations were quantified by Pierce BCA assay (Thermo Scientific, Catalog No. 23250). Cleaved caspase-3 antibody was purchased from Cell Signaling Technologies (Asp175, #9661). SOD1 antibody was purchased from Santa Cruz Biotechnologies (sc-101523).
In vitro mitochondria membrane potential imaging.
Mitochondria membrane potential was characterized by JC-1 staining (Thermo Fisher, T3168). Briefly, cells were seeded (2×104 per well) in Lab-Tek II chamber (NUNC™) 24 h prior to the treatment and cultured in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C. After 24 h incubation with CDN (with or without 10 μM of copper supplements) or control agents, cells were washed three times with PBS and incubated with JC-1 (working concentration of 10 μg/ml) in warm DPBS containing 10 μg/ml DAPI for 10 min. Then cells were washed three times with PBS and imaged with a confocal microscope (Zeiss LSM 710). For JC-1 monomers, 488 nm excitation wavelength was used and fluorescence signals at 525 ± 15 nm were collected as the green channel. For JC-1 aggregates, 546 nm excitation wavelength was used and fluorescence signals at 600 ± 15 nm were collected as the red channel.
Subcellular localization.
Subcellular localization of CDN was determined by analyzing the colocalization of CDM signal with subcellular organelles labeled by commercially available trackers. Briefly, cells were seeded (2×104 per well) in Lab-Tek II chamber (NUNC™) and cultured in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C overnight prior to the experiments. For mitochondria, cells were co-incubated with Mitotracker (Thermo Fisher, M7514) at a working concentration of 200 nM and CDN (CDM: 1 μM) for 30 min. For endoplasmic reticulum, cells were pre-incubated with CDN (CDM: 0.1 μM) for 4 h. Cells were then washed with HBSS for 3 times and stained with ER-tracker Green (Thermo Fisher, E34251) for 30 min. For the early endosome, cells were pre-incubated with CellLightTM Early Endosomes-GFP (Thermo Fisher, C10586) overnight, then incubated with CDN for 1 h prior to imaging. Nucleus staining was done in all experiments with Hoechst 33342 (10 μg/ml, Thermo Fisher, Catalog No. 62249). The confocal microscopic imaging settings: 488 nm excitation / 525 ± 15 nm emission for the green channel and 633 nm excitation/ 650–730 nm emission for the red channel. The correlation was drawn out of three different views of cells imaged with 20× lens, approximately 150 cells per view. Pearson R-value was calculated using ImageJ with Costes threshold regression.
COX activity assay.
Cytochrome c oxidase activity was measured by Cytochrome c oxidase assay kit (abcam, ab239711). Briefly, 2×107 cells were incubated with CDN or control agents at concentrations indicated in the figure legend. 24 h after incubation, cells were collected and washed three times with cold PBS. The cell pallets were processed with Mitochondria Isolation Kit for Cultured Cells (Thermo Fisher, Catalog No. 89874) to obtain mitochondrial extraction. After protein quantification, 5 μg of total purified mitochondrial protein was used for COX activity measurement. The activity of the enzyme was determined by measuring the oxidation of reduced cytochrome c as an absorbance decrease at 550 nm. The rate of the enzyme reaction was calculated in the linear range. (n=3 for each treatment condition)
Oxygen consumption rate and extracellular acidification rate measurement.
OCR and ECAR were measured by Seahorse Bioscience instrument (XF96, Agilent). Briefly, on the day following MDA-MB-231 or MDA-MB-468 cell seeding, cells were incubated with CDN or control agents for 24 h (chelator concentration: 1 μM). On the day of the measurement, cells were washed three times with D-PBS, changed into fresh medium without phenol red and bicarbonate (Seahorse base medium, Catalog No. 103336–100) and equilibrated for 30 min in a 37 °C incubator lacking CO2. Oxygen concentration and extracellular acidification in media was measured at basal conditions and after sequential addition of oligomycin (1 μM, introduced after 28 min), FCCP (1 μM, introduced after 54 min), and rotenone/antimycin A (0.5 μM, introduced after 80 min) (n=8 per condition for OCR and ECAR for each cell line). For acute OCR inhibition titration assay and copper remedy assay, on the day following cell seeding, cells were incubated with CDN at various CDM concentrations for 1 h or co-incubated with 1 μM of Cu(II). Cells were then changed into Seahorse base medium and equilibrated for 30 min in a 37 °C incubator lacking CO2. Oxygen concentration and extracellular acidification in media was measured at basal conditions and after sequential addition of oligomycin (1 μM, introduced after 28 min), FCCP (1 μM, introduced after 54 min) and rotenone/antimycin A (0.5 μM, introduced after 80 min) (n=6 per condition for OCR and ECAR for each cell line).
Cellular ATP measurement.
ATP levels after CDN or control agent treatment were determined by Adenosine 5’-triphosphate Bioluminescent Somatic Cell Assay Kit (Sigma-Aldrich, FLASC). Briefly, 106 cells were seeded in 6-well plate 24 h prior to the treatment. Other than the control group, cells were incubated with CDN, SPN, TPA or ATN224 at concentrations indicated in the figure legends for 24 h in complete medium or medium without FBS supplement. For ROS scavenger experiments, various concentrations of MitoTEMPO was added to the medium for co-incubation. Cells were then trypsinized and resuspended in ultrapure water for assay. The luminescence signals were measured by Turner Biosystems 20/20n luminometer. The ATP concentration of each group was determined based on the ATP standard assay (n=3 per condition ATP production measurement for each cell line).
Glucose uptake and lactic acid secretion.
Glucose uptakes from the culture medium and lactic acid secretion were measured by assay kits according to the manufacturer’s protocols. Briefly, cells were seeded in 6-well plates (106 per well) 24 h prior to the treatment and cultured in a humidified atmosphere of 5 % CO2 and 95 % air at 37 °C. Then cells were incubated with 2 mL of fresh medium containing CDN or control agents for 24 h. Same volume of medium in wells without cell seeding was used as baseline (same culturing conditions). The glucose level in the medium was measured by Glucose Colorimetric Assay Kit (Cayman Chemical, 10009582), and calculated as the decrease in medium glucose level after incubation. The lactic acid secretion into the medium was measured by Glycolysis Cell-Based Assay Kit (Cayman Chemical, 600450), and calculated as the increase of lactate in the medium after incubation. (n=3 per condition for glucose uptake and lactate secretion, respectively)
In vitro labeling using 13C6-glucose or 13C5, 15N2-glutamine.
MDA-MB-231 cells were plated into 10 cm dishes and cultured at 37 °C with 5 % CO2 prior to treatment. On the day of treatment, for the 13C6-glucose labeled groups, normal media was replaced with media containing 2 g/l labeled 13C6-glucose (Sigma, Catalog No. 660663) instead of 12C6-glucose. For the13C5, 15N2-glutamine labeled groups, normal media was replaced with media containing 0.3 g/l labeled 13C515N2-glutamine (Sigma, Catalog No. 607983) instead of 12C5,14N2-glutamine. Half of the dishes of cells grown in non-labeled control media, half of the dishes of cells grown in 13C6-glucose labeled media, and half of the dishes of cells grown in 13C5, 15N2-glutamine labeled media were treated with CDN (CDM: 1 μM), while the other half were treated with control (n=5 per group). Cells were harvested 24 hours after treatment.
Metabolomics analysis of labeled in vitro samples.
The cells were subjected to metabolite extraction using our established protocol as described before.59–61 The lyophilized aqueous phase metabolite samples were re-suspended in 50 % (vol/vol) acetonitrile diluted with mass-spec-grade water, while the dried organic phase metabolite samples were re-suspended in 2: 1 (vol: vol) of chloroform: methanol. Metabolomics data from the samples were acquired using a Thermo Scientific Q Exactive Orbitrap Mass Spectrometer Plus with a Vanquish UPLC system at Johns Hopkins Metabolomics Facility overseen by Dr. Anne Le. The Vanquish UPLC auto-sampler systems were used to uptake 2 μl of each sample and were maintained at 4 °C. Reverse-phase chromatography using 0.1 % formic acid in mass spec-grade water as mobile aqueous phase and 0.1 % formic acid in 98 % acetonitrile as the mobile organic phase was employed. The total runtime for each sample was 13 minutes. A Discovery® HS F5 HPLC Column with 3 μm particle size and 15 cm × 2.1 mm L × I.D. (Sigma) and a compatible guard column (Sigma) were used and were maintained at 35 °C. Data were analyzed using Thermo Fisher Scientific Compound discoverer®, Xcalibur®, and TraceFinder® softwares. The raw intensities were normalized based on protein concentration and cell weight of each sample to get the final normalized intensities.
Metabolomics analysis of in vivo samples.
MDA-MB-468 tumors in control and CDN treatment groups were subjected to metabolite extraction as described in the in vitro section above (n=5 for each group). The lyophilized samples were re-suspended in 50 % (vol/vol) acetonitrile diluted with mass-spec-grade water. Metabolomics data were acquired in the same way as described above in the Metabolomics Analysis for Labeled in Vitro Samples section. Data were analyzed using Thermo Fisher Scientific Compound discoverer®, Xcalibur®, and TraceFinder® softwares. The raw intensities were normalized based on protein concentration and tumor weight of each sample to get the final normalized intensities.
mRNA sequencing.
107 MDA-MB-231 cells in 75 cm2 flasks were incubated with CDN (CDM: 1 μM) for 15 h or 24 h. Then non-treated control and CDN treated cells were collected and resuspended in Trizol (Life Technology). The RNA extraction, sample quality control, library preparation and sequencing (6G raw data/sample) were performed by Novogene Corporation Inc. RNAseq data analysis was carried out by Stanford Bioinformatics Service Center. For the data analysis, each condition had average 25.5 million 150 bp long paired-end reads. Fastqc (version 0.11.2) was used for sequencing quality assessment (Multiqc (version 1.5) was used to aggregate results into a single report). Reads were then aligned to GRCh38(hg38) genome using STAR version 2.5.3a with splice junctions being defined in GTF file (obtained from GRCh38). An average of 89 % of reads was aligned to the reference genome. Expression at gene level was determined by calculating reads per kilo base per million aligned reads (FPKM) as well as raw count using RSEM (version 1.2.30). Analysis methods are detailed in Supplementary Notes.
SOD activity and oxidative stress.
SOD activity measurement: 1.5×106 cells in 25 cm2 flasks were incubated with CDN or control agents for 24 h. After treatment, cells were gently washed twice with D-PBS, followed by trypsinization and centrifugation at 1000 rcf for 10 min at 4 °C. To the pellet was added 500 μl of cold HEPES buffer and sonicated with probe sonicator for 10 s in ice bath (6 RWS, twice). After centrifuged at 1500 rcf for 5 min at 4 °C, the supernatant was removed for assay and stored at −80 °C before the test. For sample measurement, 100 μl of each sample was centrifuged at 10,000 rcf for 15 min at 4 °C. The supernatant was collected as cytosolic content and measured for the protein concentration with BCA assay (Thermo Scientific, Catalog No. 23250). The sample SOD activity was measured with Superoxide Dismutase Assay Kit (Cayman Chemical, 706002) according to the provided protocol (n=3 per condition).
Superoxide measurement: the superoxide level after CDN or control agent treatment was measured by a chemiluminescent sensor for superoxide. Briefly, after 24 h pretreatment with CDN or control agents, cell suspension (106 cells/mL in PBS) was collected, mixed with 200 μl of 2-methyl-6-phenyl-3,7-dihydroimidazo[1,2-a]pyrazin-3-one (CLA, TCI America, Catalog No. A5307) in HBSS solution (working concentration of 3.6 μg/ml) and incubated at 37 °C for 3 min. The chemiluminescence signals were measured by Turner Biosystems 20/20n luminometer (n=3 per condition).
DNA damage and lipid peroxidation caused by oxidative stress: the DNA damage and lipid peroxidation in cells after CDN or control agent treatment for 24 h were determined by immunofluorescent staining. DNA damage was detected by anti-γH2A.X (phosphor S139) antibody (abcam, ab11174), and lipid peroxidation was detected by anti-4 hydroxynonenal antibody (abcam, ab46545).
Animal models.
All animal experiments except for the MAL3 imaging were carried out at Stanford University in strict compliance with the Guidelines for the Care and Use of Research Animals established by the Stanford University Institutional Animal Care and Use Committee. The MAL3 imaging experiment was performed in the École Polytechnique Fédérale de Lausanne (EPFL). Animal use was approved by Ethical Committee of Canton Vaud, Switzerland (License VD3316c). All mice used in the animal experiments were 6–8-week female mice purchased from Charles River. For animal models used in labile copper bioluminescence imaging experiment, 2×106 of MDA-MB-231luc cells were inoculated into the 4th mammary fat pad of NSG™ mice to generate orthotopic models. For animal models used in MAL3 bioluminescence imaging experiment, 3×106 of MDA-MB-231luc cells were inoculated into the 4th mammary fat pad of swiss nu/nu mice to generate orthotopic models. For treatment efficacy and survival study, 5×105 4T1 cells were inoculated into the 4th mammary fat pad of Blab/c mice or 2×106 of MDA-MB-231 or MDA-MB-468 cells were inoculated into the 4th mammary fat pad of nude mice to generate orthotopic models. Animals were maintained daily by the Stanford Veterinary Service Center (VSC) or EPFL veterinary care for the routine care including housing, feeding, sanitation, animal health care for non-research related illnesses, and disease surveillance.
Copper depleting dynamics and tumor retention of CDN in vivo.
For the copper depleting dynamics study, the labile copper levels in tumor were determined by CCL-1 imaging. MDA-MB-231luc tumor-bearing mice were intraperitoneally injected with freshly prepared CCL-1 in D-PBS solution (6 mg/kg) before and at each time point after CDN injection (i.v., CDM: 1.35 mg/kg). 30 min after CCL-1 administration, mice were imaged with IVIS Spectrum for luminescence signal (n=3). For the tumor retention and copper depletion monitoring study, MDA-MB-231 tumor-bearing mice were intravenously injected with CDN (CDM: 1.35 mg/kg). The fluorescence signal from CDM and photoacoustic signal from SPN were acquired before and at day 1, 2, 3, 5, 7, 10, and 14 after CDN administration. The CDM fluorescence was acquired with IVIS Spectrum with an excitation at 640 nm and an emission filter at 740 nm. The photoacoustic signal from SPN was acquired with LAZR VisualSonics. Three-dimensional photoacoustic and ultrasound images were recorded. Laser pulses at the wavelength of 1064 nm were routed directly from the pump laser (Q switch Nd:YAG laser) of an optical parametric oscillator (OPO) system. The laser pulses were coupled to an optical fiber bundle that was integrated on the ultrasound transducer. The averaged output fluence of the laser was adjusted by neutral density (ND) filters to 60 ± 3 mJ cm−2 (7 ns). To record photoacoustic and ultrasound images, we used an ultrasound/photoacoustic micro-imaging system (LAZR, VisualSonics, Inc.) with a 40 MHz array ultrasound transducer (LZ550, VisualSonics, Inc.). A volume of 14 mm × 15 mm × 15 mm was mechanically scanned with a step size of 63 μm. Given the imaging parameters, each 3D scan required 240 frames.
Mitochondrial membrane potential imaging in vivo.
30 swiss nu/nu (female, 8 weeks old) mice were injected with suspension of MDA-MB-231luc cells (3×106 cells per mouse). When half of the animals developed tumors of about 150–300 mm3 (this reflects approximately 50 % take rate for this cell line), mice were split into 3 groups (5 mice per group) and imaged according to the scheme shown on Supplementary Figure 17. All the mice were imaged with ΔΨm-specific bioluminescent MAL3 probe to assess the basal level of ΔΨm in the tumors 4 days prior to CDN or control agent administration. MAL3 probe consists of 2 reagents that result in light production proportional to the level of ΔΨm (TPP-CL2 and azido-TPP1).22 In a typical ΔΨm imaging experiment, mice received i.v. injection of 100 µl of 700 µM TPP-CL2 solution in the vehicle comprising 0.1% fatty acid-free BSA in PBS. 20 hours later, the animals received i.p. injection of azido-TPP1 solution (100 µl, 7 mM in PBS) followed by continuous imaging during 50 min using IVIS Spectrum (PerkinElmer, USA) using following settings: exposure 3 min, binning 16, aperture F1, field of view D. To take into account heterogeneity of tumor sizes, all the mice received subsequent i.p. injection of large dose of luciferin potassium salt solution in PBS (100 µL, 33 mM) and imaged for another 10 minutes using the following settings: exposure auto, binning 8, aperture F1, field of view D.
Next, the mice were allowed to rest for 3 days to assure clearance of the imaging probes. On Day 0 of the experiment, mice in each group received i.v. injection of 150 µL of either PBS, SPN solution, or CDN (CDM: 1.35 mg/kg) solution. ΔΨm in tumor xenografts was monitored using MAL3 probe as described above on Day 2 and 8 of the experiment (Supplementary Figure 17c). Similar to Day −4, the mice were imaged with luciferin right after being imaged with MAL3 probe to account for differences in tumor size.
The total photon flux from each mouse was quantified by calculating the area under the kinetic curve resulting after MAL3 and luciferin injections. The total photon flux from MAL3 probe was normalized to the total photon flux resulting from the injection of luciferin for each animal to take into account differences in tumor size. Resulting normalized values on Day −4 (before injection of nanoparticles and corresponding controls), Day 2 (2 days after the injection of reagents), and Day 8 (8 days after the reagent injection) for each group were plotted.
Tissue distribution.
To determine the ex vivo tissue distribution of CDN, MDA-MB-231 tumor-bearing mice (n=6) were intravenously injected with fCDN (CDM: 1.35 mg/kg). At 24 h after injection, mice were whole-body imaged for CDM and fSPN fluorescence and then sacrificed to collect major organs for ex vivo imaging, including tumor, mammary fat pad, heart, lung, liver, spleen, adrenal, kidney, stomach, small intestine, large intestine, ovary, and uterus. The excitation wavelength is 500 nm, the emission wavelength for green fluorescence from fSPN is 540 nm, and for red fluorescence from CDM is 740 nm. After fluorescence imaging, tumor and liver tissues were embedded in OCT gel and the frozen tissue blocks were sliced for microscopic imaging. Confocal microscopic imaging settings for tissue slices were excitation wavelength at 488 nm, emission wavelengths from 500 to 560 nm for green fluorescence, and 610 to 733 nm for red fluorescence.
Therapeutic effect of CDN.
(1) Copper level after treatment: MDA-MB-231luc tumor-bearing mice were randomly divided into four groups (n=5 per group): control, SPN (same SPN concentration with CDN), TPA (1.35 mg/kg), and CDN (CDM: 1.35 mg/kg). For the treatment group, mice were intravenously injected with CDN or control agents every three days, five doses in total. Tumor volume was continuously monitored over the treatment plan. At day 25 after initial treatment, mice were subjected to D-luciferin bioluminescence imaging (150 mg/kg) and CCL-1 luminescence imaging (6 mg/kg). (2) Long-term survival study: MDA-MB-231 tumor bearing-mice were randomly divided into six groups (n=12 per group): control, SPN, TPA, ATN224, CDM, and CDN group. TPA, CDM, and CDN groups received an intravenous injection of the corresponding agent at the same chelator concentration of 1.35 mg/kg weekly for 7 doses in total. SPN group received an intravenous injection of the same amount of SPN as in CDN group weekly for 7 doses in total. ATN224 group received a daily oral gavage of ATN224 at a dose of 0.7 mg/kg/day for 42 days at maximum. The treatment plan was initiated when the average tumor size reached 50 mm3 and the endpoint of the survival study was the primary tumor volume over 500 mm3 and/or other signs of early euthanasia identified by the VSC veterinarian. The tumor volumes were continuously monitored over the treatment plan. (3) Therapeutic efficacy in 4T1 model: 4T1 tumor-bearing mice were randomly divided into three groups (n=5 per group): control, TPA, and CDN group. TPA and CDN groups received an intravenous injection of the corresponding agent at the same chelator concentration of 1.35 mg/kg weekly for 5 doses in total. The treatment plan was initiated when the average tumor size reached 50 mm3 and the endpoint was the primary tumor volume over 500 mm3. All surviving animals were euthanized at day 32 after the initial treatment to compare the tumor sizes after treatment. (4) Therapeutic efficacy in MDA-MB-468 model and in vivo metabolomics study: MDA-MB-468 tumor-bearing mice were randomly divided into five groups (n=5 per group): control, SPN, TPA, ATN224, and CDN group. TPA and CDN groups received an intravenous injection of the corresponding agent at the same chelator concentration of 1.35 mg/kg weekly for 7 doses in total. SPN group received an intravenous injection of the same amount of SPN as in CDN group weekly for 7 doses in total. ATN224 group received a daily oral gavage of ATN224 at a dose of 0.7 mg/kg/day for 42 days at maximum. The treatment plan was initiated when the average tumor size reached 50 mm3. Early endpoint of the study was determined based on the sign of early euthanasia identified by the VSC veterinarian. The tumor volumes were continuously monitored over the treatment plan. 3 days after the last injection, animals were sacrificed. Tumors were collected, weighed, and snap-frozen for metabolite extraction and analysis.
Acute toxicity and cumulative toxicity of CDN.
For the acute toxicity study, healthy female Balb/c mice were i.v. injected with saline or CDN (CDM: 100 mg/kg) and monitored for two weeks (n=3 per group). Mice were observed for behavioral changes, and body weight was monitored over two weeks before sacrificed for hematological analysis, Mammalian Liver Profile tests, and tissue histology analysis. Tissues to examine included heart, lungs, liver, kidneys, and spleen. Hematology and pathological analyses were performed by Stanford Veterinary Service Center. For the cumulative toxicity study, healthy female Balb/c mice were i.v. injected with saline or CDN (CDM: 1.35 mg/kg) weekly for a total of 7 doses (n=5 per group). Mice were observed for behavioral changes, and body weight was monitored over the treatment course. Mice then were sacrificed at day 45 after initial treatment for hematological analysis, Mammalian Liver Profile tests, and tissue histology analysis. Tissues to examine included heart, lungs, liver, kidneys, and spleen.
Statistical Analysis.
Unpaired t test was performed to compare two groups (two-tailed). Kruskal Wallis tests were performed for multiple group comparisons and Wilcoxon rank sum test was conducted for post hoc pair comparisons. For survival study, Log rank tests were conducted for multiple groups in overall survival.
Extended Data
Extended Figure 1. Cytotoxicity of CDN measured with trypan blue exclusion method.
Cell viability of (a) MDA-MB-231 and (b) MDA-MB-468 cells after treatment with various concentrations of CDN, TPA or ATN224 (mean ± s.e.m., n=3 independent samples). (c) Metal remedy experiment measuring the cell viability of MDA-MB-231 cells after 24 h treatment with CDN (CDM: 1 μM) with (n=3 independent samples) or without (n=6 independent samples) various metal ion supplement (mean ± s.e.m., P values from unpaired t test, two-tailed). (d) Titration study showing the remedy effect of various concentrations of copper ions on MDA-MB-231 cell viability measured by trypan blue method (mean ± s.e.m., n=6 independent samples for CDN group, n=3 independent samples for copper addition group, P values from unpaired t test, two-tailed).
Extended Figure 2. Acute OCR response of MDA-MB-231 cells after 1 h treatment with CDN.
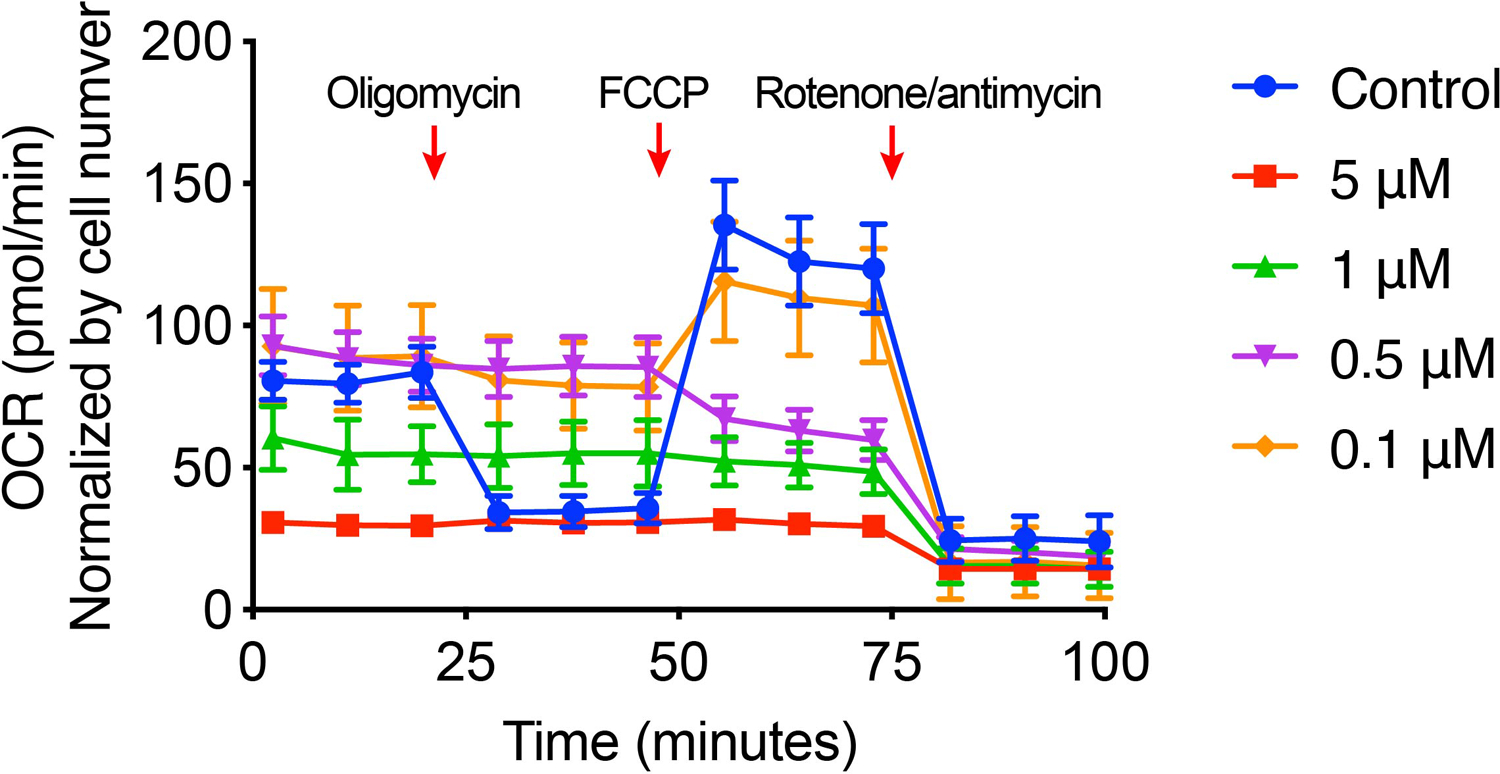
OCR was measured with a serial injection of oligomycin (1 μM), FCCP (1 μM) and rotenone and antimycin A (0.5 μM) (mean ± s.e.m., n=6 biologically independent samples).
Extended Figure 3. Comparison on the cytotoxicity, OCR inhibition and mitochondria membrane potential damaging effect between CDN and established complex I inhibitors.
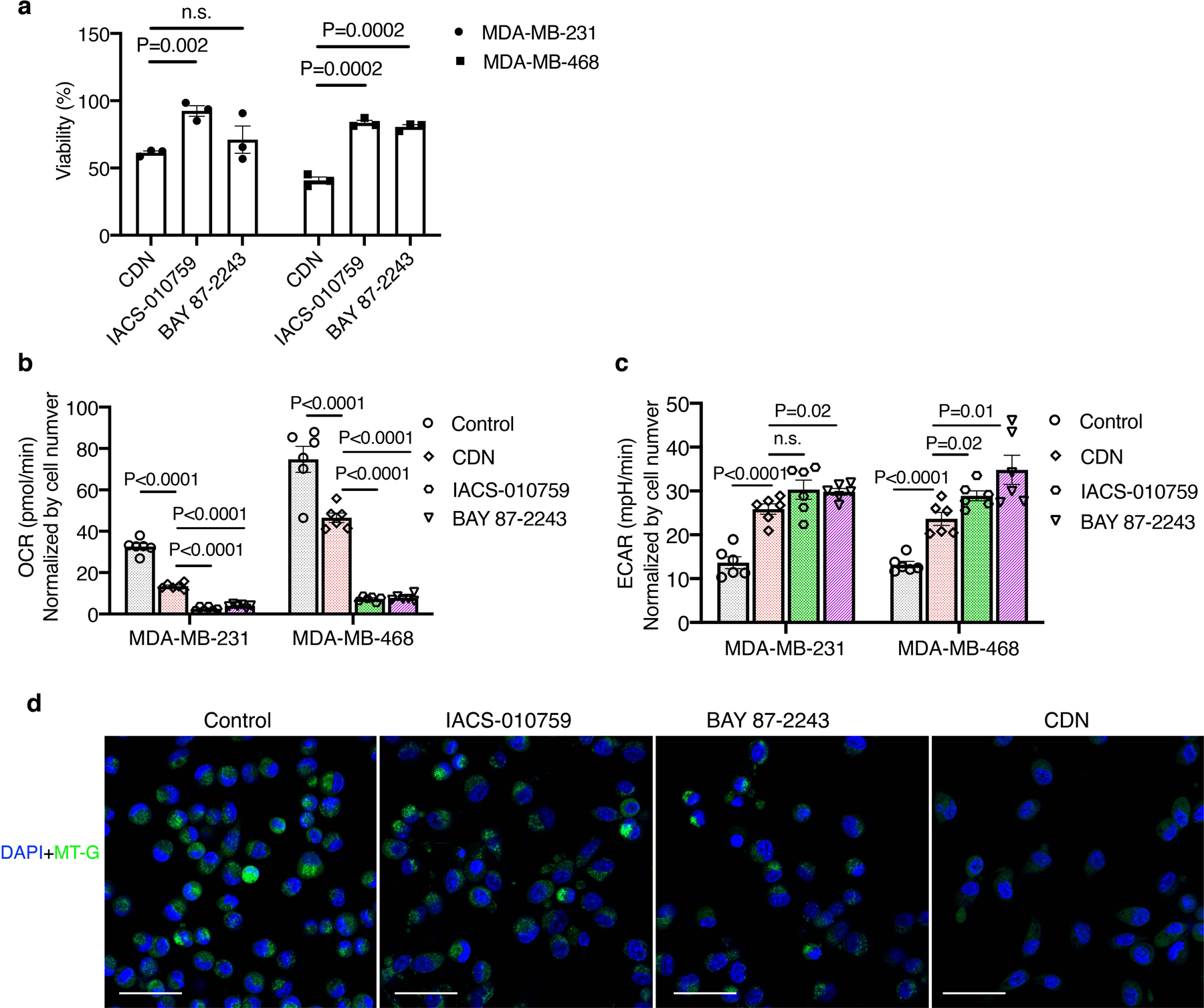
MDA-MB-231 or MDA-MB-468 cells were incubated with 1 μM of CDN, IACS-010759 or BAY 87–2243. (a) Cell viability after 24 h of treatment was measured by MTS assay (mean ± s.e.m., n=3 biologically independent samples, P values from unpaired t test, two-tailed). (b) OCR and (c) ECAR were determined via Seahorse assay at 1 h after incubation. The results were normalized by cell number (mean ± s.e.m., n=6 biologically independent samples, P values from unpaired t test, two-tailed)). (d) Representative confocal microscopy images of cells stained with MitoTracker Green (MT-G, green) and DAPI (blue) after 24 h treatment. (scale bar: 50 μm). Three experiments were repeated independently with similar results.
Extended Figure 4. CDN-enabled copper depletion elevated cellular oxidative stress.
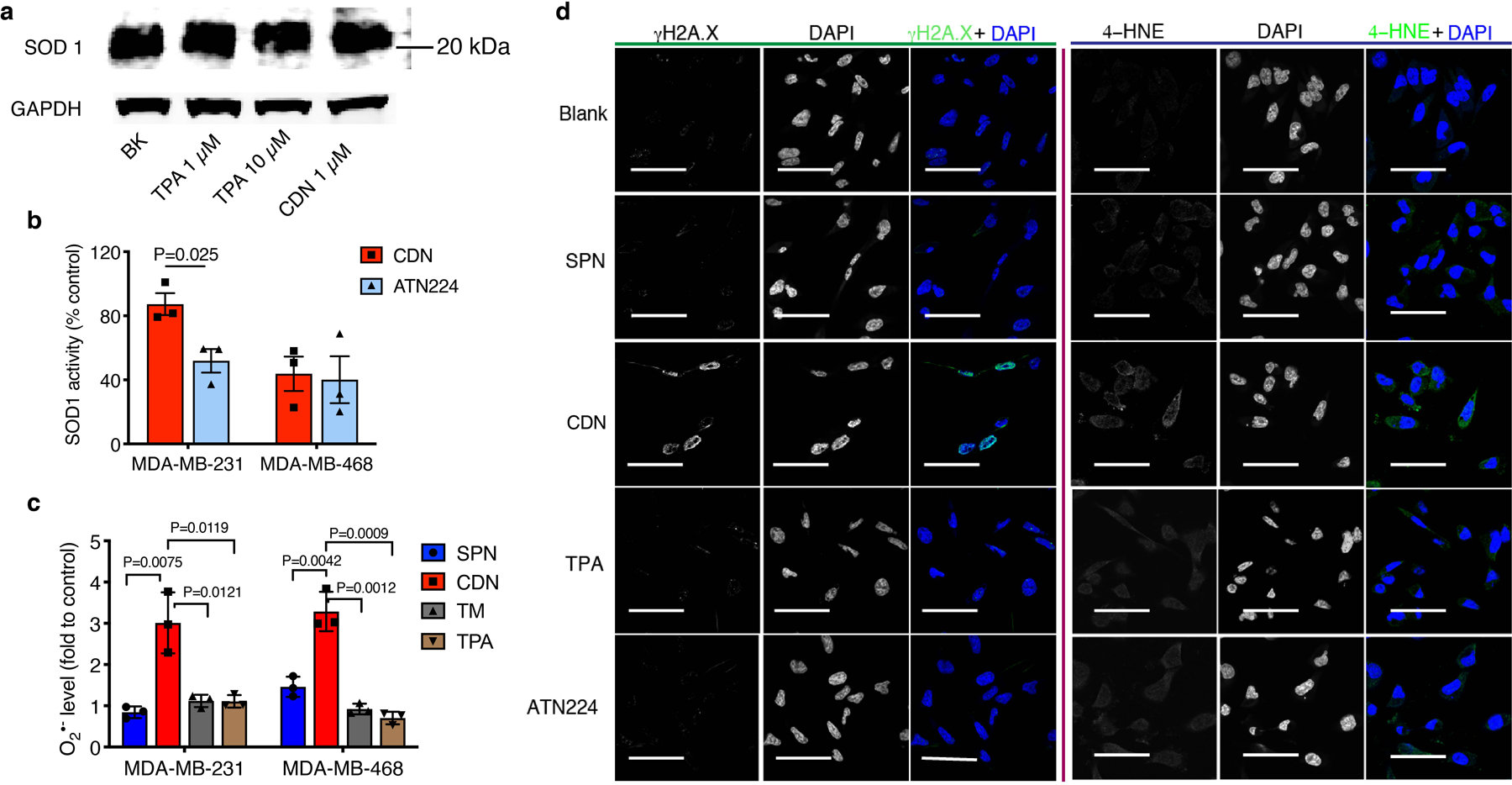
(a) SOD1 levels of MDA-MB-231 after treatment with TPA and CDN at concentrations indicated by western blot analysis. Two experiments were repeated independently with similar results. (b) SOD1 activity of TNBC cells treated with CDN (1 μM) or ATN224 (5 μM) (mean ± s.e.m., n=3 biologically independent samples, P>0.05 for MDA-MB-468, P value from unpaired t test, two-tailed). (c) Cellular superoxide level measured by the chemiluminescence signal of CLA after treatment of SPN (50 μg/ml), CDN treatment (CDM: 1 μM, SPN: 50 μg/ml), TM (1 μM) and TPA (1 μM), shown as a ratio of treated to non-treated control (mean ± s.e.m., n=3 biologically independent samples, P values from unpaired t test, two-tailed). (d) Immunofluorescence staining of γH2AX (left) and 4-hydroxynonenal (4-HNE, right) on MDA-MB 231 cells at 24 h after incubation with SPN (50 μg/ml), CDN (CDM: 1 μM, SPN: 50 μg/ml), TPA (1 μM) or ATN224 (1 μM). (Scale bar=50 μm). Three experiments were repeated independently with similar results.
Extended Figure 5. Intensities of isotopologues of metabolites (glutamine, cysteinyl glycine, glutathione (reduced), 4-oxoproline, hydroxyproline) produced from 13C515N2-labeled glutamine in vitro.
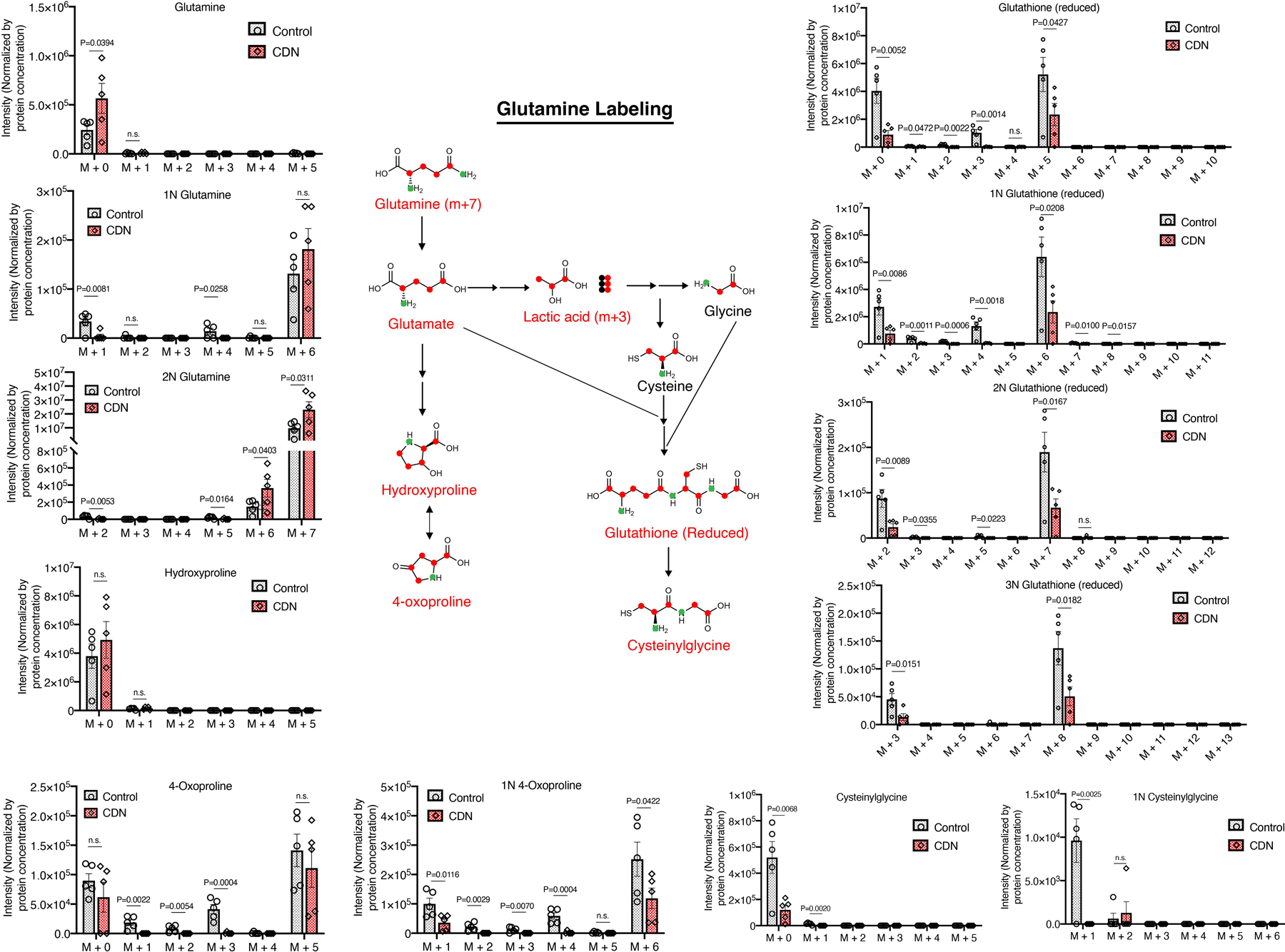
Illustration of the corresponding pathways through which these metabolites are produced is also shown. Metabolites with isotopologues shown are written in red. Other important intermediate metabolites are written in black. Non-labeled 12C and 14N are shown as black-filled circles while labeled 13C are shown as red-filled circles and labeled 15N are shown as green-filled circles. Raw intensities are normalized by the protein concentration. Data are shown as mean ± s.e.m. (n = 5 for biologically independent samples, P values from unpaired t test, two-tailed).
Extended Figure 6. Intensities of isotopologues of metabolites (lactic acid, glutamate, asparagine, and taurine) produced from 13C515N2-labeled glutamine in vitro and illustration of the corresponding pathways through which these metabolites are produced.
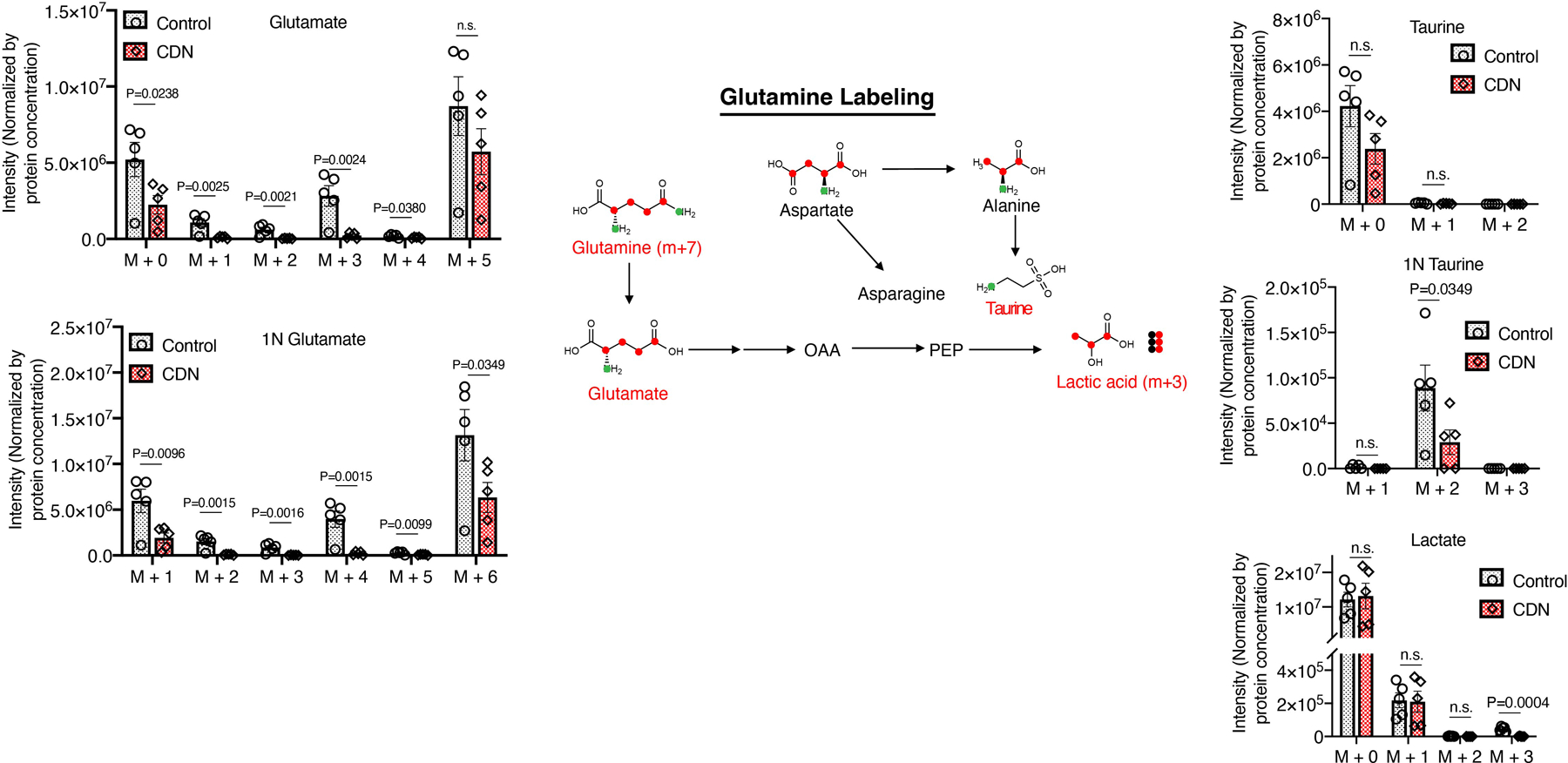
Metabolites with isotopologues shown are written in red. Other important intermediate metabolites are written in black. Non-labeled 12C and 14N are shown as black-filled circles while labeled 13C are shown as red-filled circles and labeled 15N are shown as green-filled circles. Raw intensities are normalized by both the protein concentration. Data are shown as mean ± s.e.m. (n = 5 for biologically independent samples, P values from unpaired t test, two-tailed).
Extended Figure 7. Ex vivo imaging of MDA-MB-231 tumor bearing mice administered with fCDN.
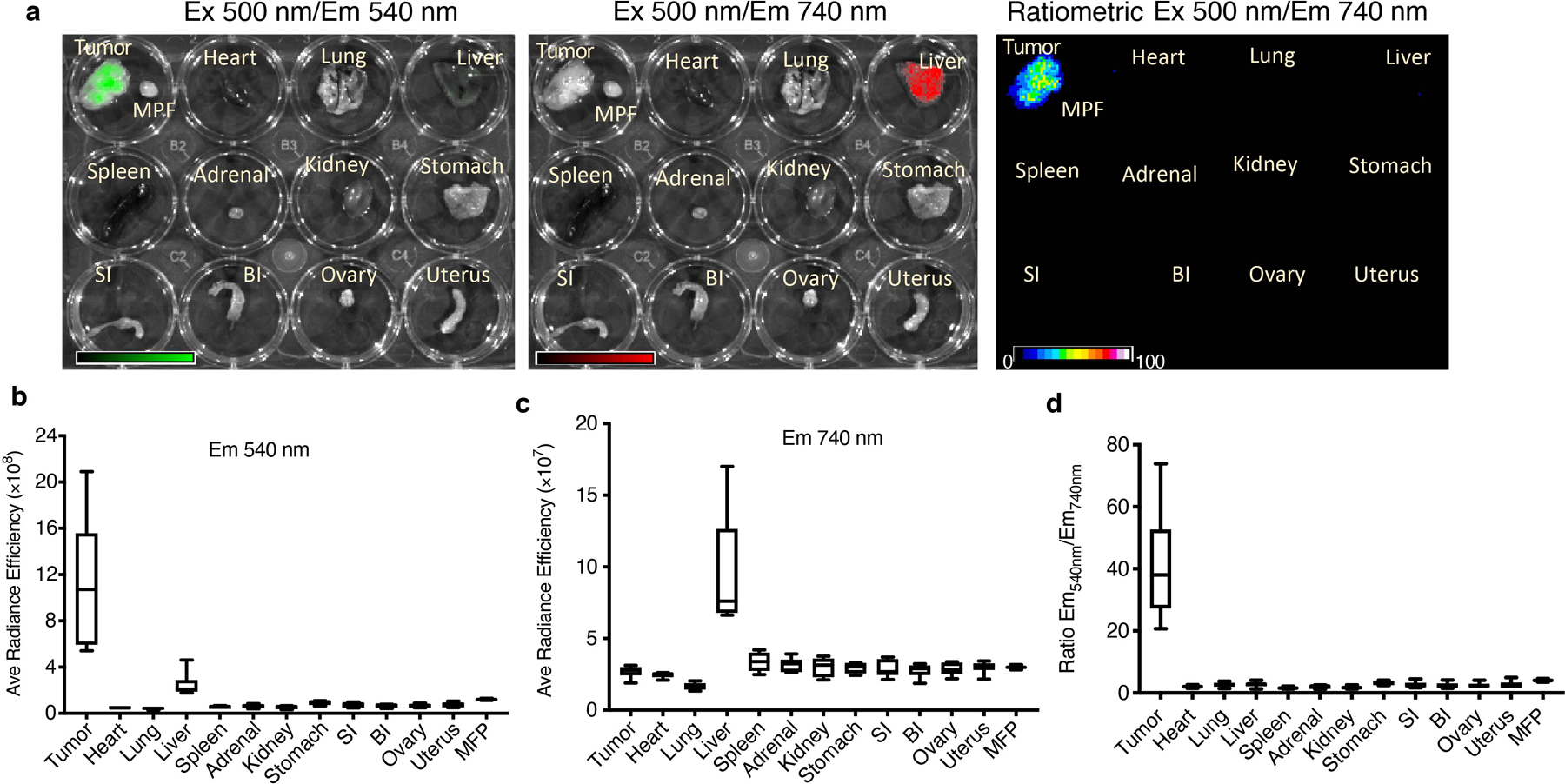
Nude mice bearing orthotopic MDA-MB-231 tumors were injected i.v. with fCDN (CDM dose: 1.35 mg/kg, n=6 independent animals). At 24 h after injection, mice were sacrificed, and major organs were collected and imaged with IVIS. (a). Representative images of ex vivo imaging emitted at 540 nm and 740 nm (excited at 500 nm). The ratiometric fluorescence graphs were shown as Em540/Em740. Average fluorescence efficiency from all organs was quantified at the (b) 540 nm emission (from the SPN group) and (c) 740 nm emission (from the CDM group). (d) The ratio of fluorescence emission between 540 nm and 740 nm. For all the boxplots: center line, median; box limits, first and third quartiles; whiskers, min to max values.
Extended Figure 8. Cumulative toxicity profile for CDN.
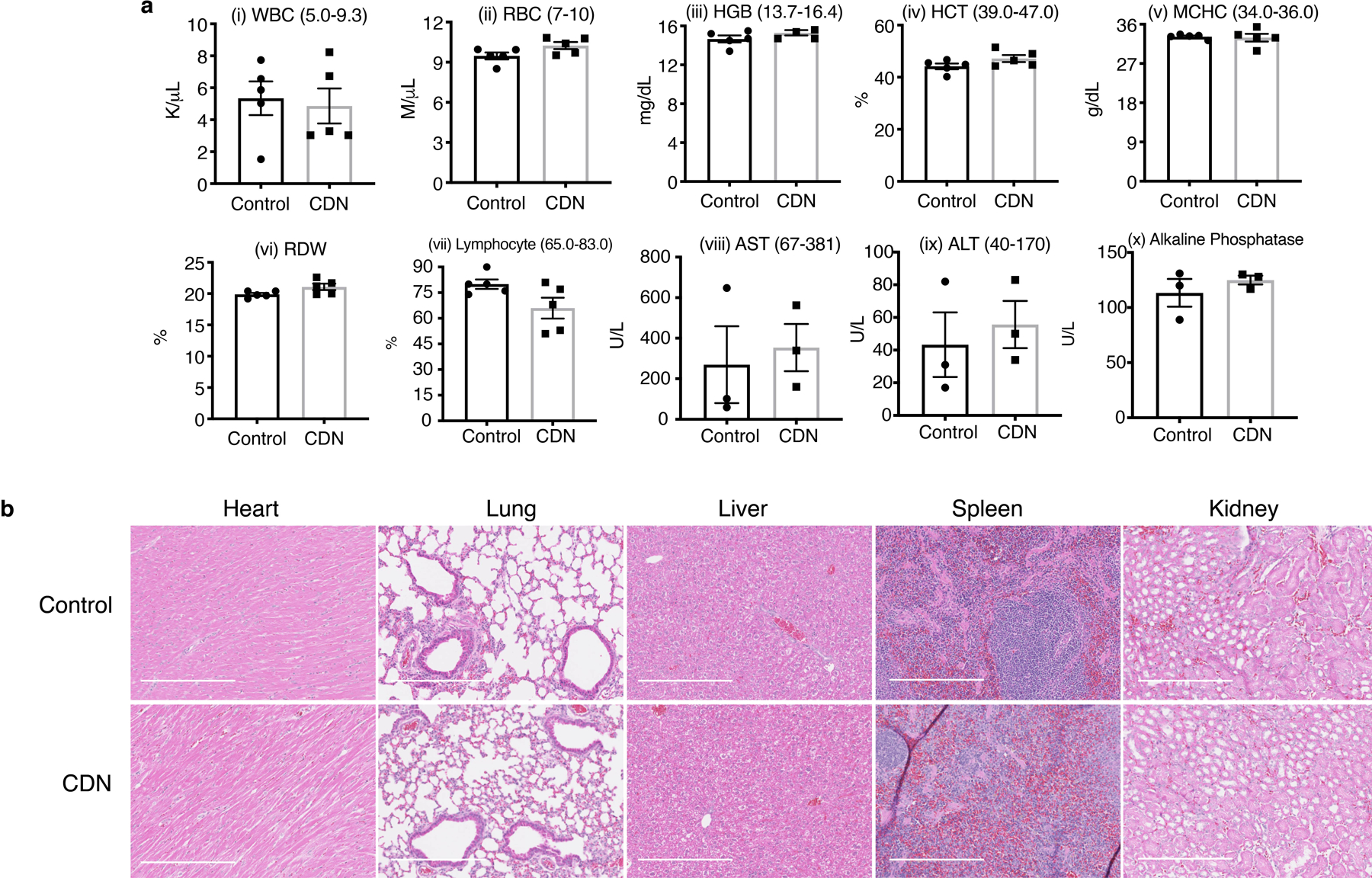
(a) Hematological (i-vii) and liver panel (viii-x) analysis of mice after receiving either saline or CDN (CDM dose: 1.35 mg/kg, intravenous administration weekly, 7 doses in total). (mean ± s.e.m., n=5 independent animals for blood test, n=3 independent animals for liver panel test). (b) Representative haematoxylin and eosin (H&E) staining of normal tissue slice from mice treated with saline or treatment strategy shown in Figure 6a for CDN (scale bar: 50 μm). Slides from 5 independent animals were imaged and showed similar results.
Extended Figure 9. Acute toxicity profile for CDN.
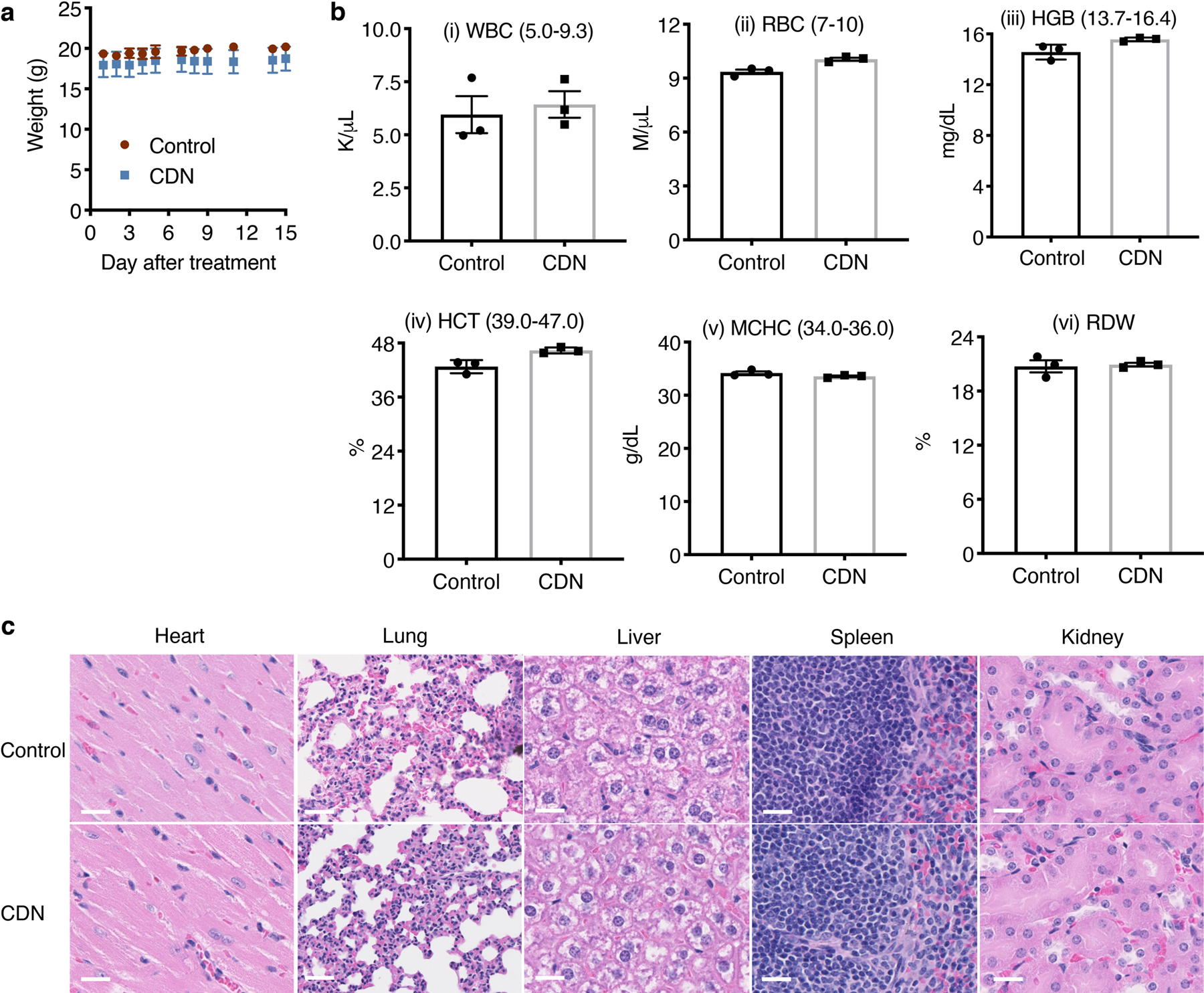
(a) Body weight changes of mice after i.v. injection of saline or single large dose of CDN (CDM: 100 mg/kg, n=3 independent animals). (b) Blood test parameters of treated mice comparing to control mice (mean ± s.e.m., n=3 independent animals). (c) representative H&E stained tissue slices of indicated organs from CDN treated and control mice (scale bar: 20 μm). Slides from 3 independent animals were imaged and showed similar results.
Extended Figure 10. Cytotoxicity of CDN to receptor-positive breast cancer and prostate cancer cells.
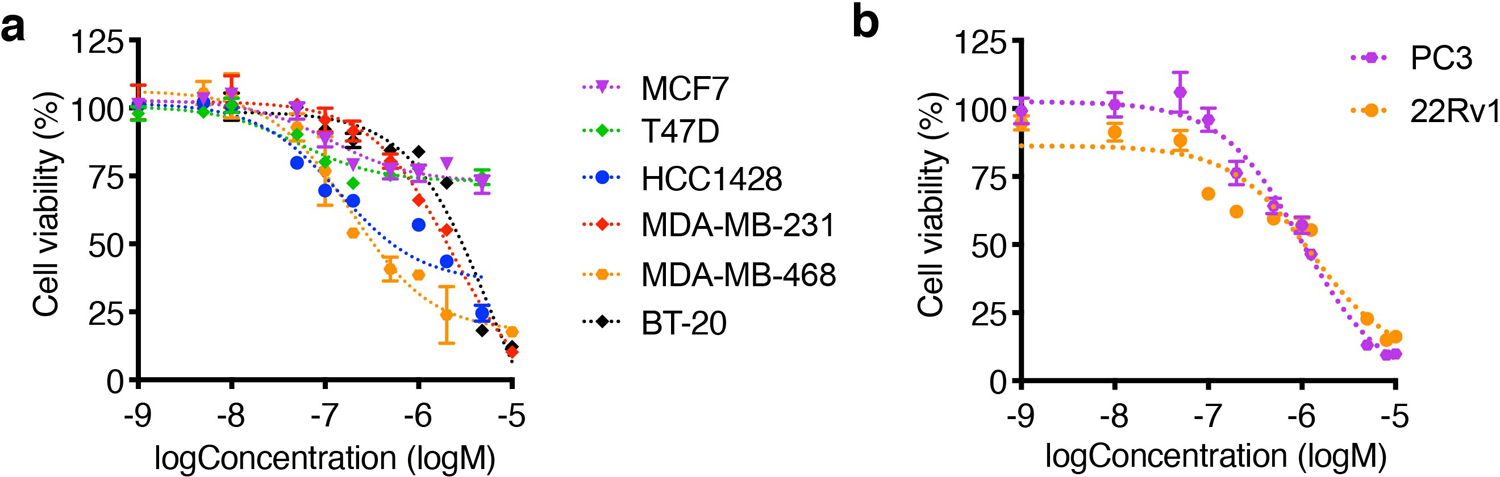
(a) viability of receptor-positive breast cancer cell lines (HCC1428, MCF7, T47D) and TNBC cells (MDA-MB-231, MDA-MB-468, BT-20) after 24 h of treatment with CDN measured by MTS assay (mean ± s.e.m., n=3 biologically independent samples). (b) Prostate cancer cell lines, PC3 and 22Rv1, also responded to CDN treatment. Viability after 24 h of CDN treatment was presented as percentage of cell control without treatment (mean ± s.e.m., n=3 biologically independent samples).
Supplementary Material
Acknowledgements
L.C. acknowledges the support by the Office of the Assistant Secretary of Defense for Health Affairs through the Breast Cancer Research Program (BCRP) under Award No. (W81XWH-18–1-0591). A.M.G. and M.C. acknowledges the support by the Stanford Cancer Translational Nanotechnology Training (TNT) T32 training grant funded by the National Cancer Institute (T32 CA196585). This work was also supported by the US National Institutes of Health National Cancer Institute (NCI) grant R01CA243033 (to J.R.), R35CA197713 (to A.M.G.) and the Shared Instrument Grant (1S10OD025226–01) funded by the NIH. We acknowledge the use of the Mass Spectrometry Facility, Department of Chemistry NMR Facility, SCi3 Core Facility, the Neuroscience Microscopy Service Facility (NIH NS069375), Cell Sciences Imaging Facility, Animal Histology Services and Diagnostic Lab at Veterinary Service Center and the Genetics Bioinformatics Service Center at Stanford University. We thank Dr. Andrew Olson for his expertise with tissue preparation and imaging by confocal microscopy, and Drs. Jarrett Rosenburg and Tie Liang for their assistance in biostatistical analysis.
Footnotes
Competing interest statement
Stanford University is seeking to file a patent application covering part of the information contained in the paper.
Data availability
All important data generated or analyzed during this study are included in this article and its supplementary information files. Additional data that support the findings of the study are available from the corresponding author upon reasonable request. RNA sequencing data are available from NCBI Sequence Read Archive (SRA) under accession number PRJNA587318.
Code availability
Custom code described in this work for the RNA sequencing data analysis is available for academic research upon request from the authors.
References
- 1.Foulkes WD, Smith IE & Reis-Filho JS Triple-negative breast cancer. N. Engl. J. Med 363, 1938–1948 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Bianchini G, Balko JM, Mayer IA, Sanders ME & Gianni L Triple-negative breast cancer: challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol 13, 674–690 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park JHH et al. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep 14, 2154–2165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Camarda R et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med 22, 427–432 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Weverwijk A et al. Metabolic adaptability in metastatic breast cancer by AKR1B10-dependent balancing of glycolysis and fatty acid oxidation. Nat. Commun 10, 2698 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srirangam A et al. Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer. Clin. Cancer Res 12, 1883–1896 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koppenol WH, Bounds PL & Dang CV Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Gouw AM et al. The MYC oncogene cooperates with sterol-regulated element-binding protein to regulate lipogenesis essential for neoplastic growth. Cell Metab 30, 556–572 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagaraja GM et al. Gene expression signatures and biomarkers of noninvasive and invasive breast cancer cells: comprehensive profiles by representational difference analysis, microarrays and proteomics. Oncogene 25, 2328–2338 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Blockhuys S et al. Defining the human copper proteome and analysis of its expression variation in cancers. Metallomics 9, 112–123 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Hargreaves IP et al. Inhibition of mitochondrial complex IV leads to secondary loss complex II–III activity: Implications for the pathogenesis and treatment of mitochondrial encephalomyopathies. Mitochondrion 7, 284–287 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Li Y et al. An assembled complex IV maintains the stability and activity of complex I in mammalian mitochondria. J. Biol. Chem 282, 17557–17562 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Carr HS & Winge DR Assembly of cytochrome c oxidase within the mitochondrion. Acc. Chem. Res 36, 309–316 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Wang J et al. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat. Chem 7, 968–979 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishida S, Andreux P, Poitry-Yamate C, Auwerx J & Hanahan D Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci 110, 19507–19512 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim KK et al. Tetrathiomolybdate inhibits mitochondrial complex IV and mediates degradation of hypoxia-inducible factor-1α in cancer cells. Sci. Rep 5, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pu K et al. Semiconducting polymer nanoparticles as photoacoustic molecular imaging probes in living mice. Nat. Nanotechnol 9, 233–239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Palma G et al. Metallic elements in pulmonary biopsies from lung cancer and control subjects. Acta Biomed 79 Suppl 1, 43–51 (2008). [PubMed] [Google Scholar]
- 19.Ellinghaus P et al. BAY 87–2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med 2, 611–624 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molina JR et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med 24, 1036–1046 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Yasuta N, Takenaka N & Takemura T Mechanism of photosensitized chemiluminescence of 2-methyl-6-phenylimidazo[1,2-a]pyrazin-3(7 H )-one (CLA) in aqueous solution. Chem. Lett 28, 451–452 (1999). [Google Scholar]
- 22.Bazhin AA et al. A bioluminescent probe for longitudinal monitoring of mitochondrial membrane potential. Nat. Chem. Biol (2020). doi: 10.1038/s41589-020-0602-1 [DOI] [PubMed]
- 23.Weinberg SE & Chandel NS Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol 11, 9–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vyas S, Zaganjor E & Haigis MC Mitochondria and cancer. Cell 166, 555–566 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoy AJ, Balaban S & Saunders DN Adipocyte-tumor cell metabolic crosstalk in breast cancer. Trends Mol. Med 23, 381–392 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Liu Y Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis 9, 230–234 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Nieman KM et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med 17, 1498–1503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schild T, Low V, Blenis J & Gomes AP Unique metabolic adaptations dictate distal organ-specific metastatic colonization. Cancer Cell 33, 347–354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duman C et al. Acyl-CoA-binding protein drives glioblastoma tumorigenesis by sustaining fatty acid oxidation. Cell Metab 30, 274–289 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Israelsen WJ et al. PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155, 397–409 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bridges HR, Jones AJY, Pollak MN & Hirst J Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J 462, 475–487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ariaans G, Jalving M, Vries E. G. E. de & Jong S de. Anti-tumor effects of everolimus and metformin are complementary and glucose-dependent in breast cancer cells. BMC Cancer 17, 232:1–13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sanchez M, Gastaldi L, Remedi M, Cáceres A & Landa C Rotenone-Induced Toxicity is Mediated by Rho-GTPases in Hippocampal Neurons. Toxicol. Sci 104, 352–361 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Modica-Napolitano, Josephine S, Aprille JR Delocalized lipophilic cations selectively target the mitochondria of carcinoma cells. Adv. Drug Deliv. Rev 49, 63–70 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Heerdt BG, Houston MA & Augenlicht LH The intrinsic mitochondrial membrane potential of colonic carcinoma cells is linked to the probability of tumor progression. Cancer Res 65, 9861–9867 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Shi Y et al. Gboxin is an oxidative phosphorylation inhibitor that targets glioblastoma. Nature 567, 341–346 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Krebs HA The Pasteur effect and the relations between respiration and fermentation. Essays Biochem 8, 1–34 (1972). [PubMed] [Google Scholar]
- 38.McGarry JD, Mannaerts GP & Foster DW A possible role for malonyl-CoA in the regulation of hepatic fatty acid oxidation and ketogenesis. J. Clin. Invest 60, 265–270 (1977). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGarry JD, Leatherman GF & Foster DW Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J. Biol. Chem 253, 4128–4136 (1978). [PubMed] [Google Scholar]
- 40.Ackerman CM & Chang CJ Copper signaling in the brain and beyond. J. Biol. Chem 293, 4628–4635 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krishnamoorthy L et al. Copper regulates cyclic-AMP-dependent lipolysis. Nat. Chem. Biol 12, 586–592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Turski ML & Thiele DJ New Roles for Copper Metabolism in Cell Proliferation, Signaling, and Disease. J. Biol. Chem 284, 717–721 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blockhuys S & Wittung-Stafshede P Roles of copper-binding proteins in breast cancer. Int. J. Mol. Sci 18, 871 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Denoyer D, Masaldan S, La Fontaine S & Cater MA Targeting copper in cancer therapy: ‘Copper That Cancer’. Metallomics 7, 1459–76 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Grubman A & White AR Copper as a key regulator of cell signalling pathways. Expert Rev. Mol. Med 16, e11 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Matson Dzebo M, Ariöz C & Wittung-Stafshede P Extended functional repertoire for human copper chaperones. Biomol. Concepts 7, 29–39 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Shao S et al. A non-cytotoxic dendrimer with innate and potent anticancer and anti-metastatic activities. Nat. Biomed. Eng 1, 745–757 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Pan Q et al. Copper deficiency induced by tetrathiomolybdate suppresses tumor growth and angiogenesis. Cancer Res 62, 4854–4859 (2002). [PubMed] [Google Scholar]
- 50.Doñate F et al. Identification of biomarkers for the antiangiogenic and antitumour activity of the superoxide dismutase 1 (SOD1) inhibitor tetrathiomolybdate (ATN-224). Br. J. Cancer 98, 776–783 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chisholm CL et al. Ammonium tetrathiomolybdate treatment targets the copper transporter ATP7A and enhances sensitivity of breast cancer to cisplatin. Oncotarget 7, 84439–84452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin J et al. A non-comparative randomized phase II study of 2 doses of ATN-224, a copper/zinc superoxide dismutase inhibitor, in patients with biochemically recurrent hormone-naïve prostate cancer. Urol. Oncol. Semin. Orig. Investig 31, 581–588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Redman BG et al. Phase II trial of tetrathiomolybdate in patients with advanced kidney cancer. Clin. Cancer Res 9, 1666–1672 (2003). [PubMed] [Google Scholar]
- 54.Schneider BJ et al. Pre-operative chemoradiation followed by post-operative adjuvant therapy with tetrathiomolybdate, a novel copper chelator, for patients with resectable esophageal cancer. Invest. New Drugs 31, 435–442 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garber K Targeting copper to treat breast cancer. Science 349, 128–129 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Chan N et al. Influencing the tumor microenvironment: a phase II study of copper depletion using tetrathiomolybdate in patients with breast cancer at high risk for recurrence and in preclinical models of lung metastases. Clin. Cancer Res 23, 666–676 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Sahota S et al. A phase II study of copper-depletion using tetrathiomolybdate (TM) in patients (pts) with breast cancer (BC) at high risk for recurrence: Updated results. J. Clin. Oncol 35, 2557–2557 (2017). [Google Scholar]
- 58.Heffern MC et al. In vivo bioluminescence imaging reveals copper deficiency in a murine model of nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci 113, 14219–14224 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nguyen T et al. Uncovering the role of N-acetyl-aspartyl-glutamate as a glutamate reservoir in cancer. Cell Reports 27, 491–501 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Udupa S et al. Upregulation of the glutaminase II pathway contributes to glutamate production upon glutaminase 1 inhibition in pancreatic cancer. Proteomics 19, 1800451 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elgogary A et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Natl. Acad. Sci 113 (36), E5328–5336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.