

Abstract
The popularity of botanical and other purported medicinal natural products (NPs) continues to grow, especially among patients with chronic illnesses and patients managed on complex prescription drug regimens. With few exceptions, the risk of a given NP to precipitate a clinically significant pharmacokinetic NP-drug interaction (NPDI) remains understudied or unknown. Application of static or dynamic mathematical models to predict and/or simulate NPDIs can provide critical information about the potential clinical significance of these complex interactions. However, methods used to conduct such predictions or simulations are highly variable. Additionally, published reports using mathematical models to interrogate NPDIs are not always sufficiently detailed to ensure reproducibility. Consequently, guidelines are needed to inform the conduct and reporting of these modeling efforts. This recommended approach from the Center of Excellence for Natural Product Drug Interaction Research describes a systematic method for using mathematical models to interpret the interaction risk of NPs as precipitants of potential clinically significant pharmacokinetic NPDIs. A framework for developing and applying pharmacokinetic NPDI models is presented with the aim of promoting accuracy, reproducibility, and generalizability in the literature.
Significance Statement
Many natural products (NPs) contain phytoconstituents that can increase or decrease systemic or tissue exposure to, and potentially the efficacy of, a pharmaceutical drug; however, no regulatory agency guidelines exist to assist in predicting the risk of these complex interactions. This recommended approach from a multi-institutional consortium designated by National Institutes of Health as the Center of Excellence for Natural Product Drug Interaction Research provides a framework for modeling pharmacokinetic NP-drug interactions.
I. Introduction: Application of Static and Dynamic Models to Natural Products
Static and dynamic [i.e., physiologically-based pharmacokinetic (PBPK)] models are mainstay tools during drug development. For applications such as estimating dissolution and bioavailability, triaging early-phase new chemical entities (NCEs) with suboptimal pharmacokinetic characteristics (e.g., high clearance or low oral bioavailability), or predicting drug-drug interactions (DDIs), PBPK models can be used to design and occasionally replace clinical studies (Sager et al., 2015). Botanical dietary supplements and other purported medicinal natural products (NPs) often contain phytoconstituents that can precipitate clinically significant pharmacokinetic and potential pharmacodynamic NP-drug interactions (NPDIs) with conventional medications (both approved prescription and nonprescription) (Grimstein and Huang, 2018; Johnson et al., 2018; Paine et al., 2018). NPs can also contain misidentified plants or toxic chemical constituents introduced through suboptimal harvesting, production, and/or manufacturing practices (van Breemen et al., 2008). Induction or inhibition of cytochrome P450 (CYP) 3A by St. John’s wort or grapefruit juice, respectively, are textbook examples of NPDIs that can increase or decrease the systemic exposure to CYP3A object drugs (Bailey et al., 1998; Henderson et al., 2002).
As with DDIs, NPDIs can perturb object drug systemic exposure to subtherapeutic or supratherapeutic concentrations, which in turn can lead to altered therapeutic response to the drug. However, mathematical modeling of NPDIs has not kept pace with that of DDIs. Unlike DDIs, to date, NPDI prediction is not driven by guidance documents from regulatory agencies, including the US Food and Drug Administration (FDA), European Medicines Agency, and the Pharmaceuticals and Medical Devices Agency. Silence on this challenging topic may have arisen from the intricacies of NPDI modeling and simulation, which require special attention to the phytochemical complexity of NPs, inconsistencies in formulations, differences in botanical taxonomy and nomenclature, and the paucity of human pharmacokinetic data for most commercially available NPs.
Despite the absence of guidance documents, static and PBPK models for estimating changes in object-drug systemic exposure have been developed (Zhou et al., 2005; Brantley et al., 2013; Ainslie et al., 2014; Brantley et al., 2014b; Gufford et al., 2015a; Tian et al., 2018; Adiwidjaja et al., 2019, 2020b). That NPDI models continue to be developed in the absence of regulatory guidance underscores the timeliness and importance of NPDI modeling and simulation and the need for resources and guidelines to support this research effort.
Compared with DDIs, NPDIs remain uniquely difficult to predict because of several key factors that preclude accurate in vitro-to-in vivo extrapolation: 1) the inherently complex and variable composition of phytoconstituents among marketed products of presumably the same NP, 2) identification of all possible constituents that contribute to NPDIs, 3) the often relatively sparse human pharmacokinetic information about precipitant (“perpetrator”) NP constituents, and 3) potentially complex and varying interactions between the precipitants (e.g., synergy between constituents, inhibition by one constituent, and induction by another) due to the variable composition of precipitants in the same NP (Grimstein and Huang, 2018; Paine et al., 2018; Sorkin et al., 2020). The limited plasma exposure data for most commercially available NPs as well as the general absence of physicochemical data for their major phytoconstituents are perhaps the greatest impediments to developing robust PBPK models in this field. Indeed, the FDA recognizes these deficiencies as “technical challenges in determining standard pharmacokinetic measurements (https://www.fda.gov/media/93113/download).” This recommended approach lays a framework for selection of robust in vitro data, appropriate model parameterization and verification, and clear communication of model characteristics in the literature with the aim of promoting accuracy, reproducibility, and generalizability of pharmacokinetic NPDI models.
Recognizing that NPDIs are a pressing but understudied public health risk, the National Center for Complementary and Integrative Health established the Center of Excellence for Natural Product Drug Interaction Research (NaPDI Center), which is tasked with developing recommended approaches to guide researchers on the conduct of rigorous NPDI studies (Paine et al., 2018). The NaPDI Center has released recommended approaches for selecting and prioritizing NPs as potential precipitants of NPDIs and for sourcing and characterizing NPs for research studies (Johnson et al., 2018; Kellogg et al., 2019). This recommended approach summarizes existing challenges and potential solutions related to mathematical modeling of pharmacokinetic NPDIs with the goal of facilitating more rapid and systematic identification of clinically significant NPDIs.
II. Generating and Selecting Data for Static and Physiologically Based Pharmacokinetic Models
A. Identification of Precipitant Phytoconstituents
For many commercial NPs, precipitant phytoconstituent(s) (i.e., inducers and inhibitors of drug metabolizing enzymes and transporters) may not have been identified. These situations merit judicious sourcing and characterization of the crude NP followed by identification and quantification of precipitant constituents. One of the NaPDI Center’s recommended approaches details pivotal considerations for sourcing and characterizing NPs for both in vitro and in vivo studies involving an NP (Kellogg et al., 2019). These considerations mirror those put forth by the FDA for ensuring therapeutic consistency and quality control during botanical drug development (https://www.fda.gov/media/93113/download) and by National Center for Complementary and Integrative Health for promoting consistency in grant applications and research reporting (https://nccih.nih.gov/research/policies/naturalproduct.htm#requestedpi).
Identifying phytoconstituents as precipitants of pharmacokinetic NPDIs is a complex and variable process, which typically includes a screening and/or experimental approach involving human-derived in vitro systems expressing relevant drug metabolizing enzymes and/or transporters. Experimental approaches include iterative fractionation and screening of crude extracts, during which an NP is partitioned into aqueous and organic phases and separated chromatographically into discrete pools of phytochemicals. These fractions are subsequently tested for bioactivity (induction or inhibition) across a predefined array of concentrations against a panel of drug metabolizing enzymes and transporters. Such biochemometric analysis or bioactivity-directed fractionation allows the bioactive fraction(s) to be refined and rescreened iteratively, progressively isolating fractions containing relatively purified mixtures of bioactive constituents or highly purified individual constituents (Kim et al., 2011; Kellogg et al., 2016; Rivera-Chávez et al., 2017a,b, 2019a,b; Amrine et al., 2018; Britton et al., 2018; Caesar et al., 2018; Tian et al., 2018; El-Elimat et al., 2019; Paguigan et al., 2019).
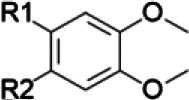
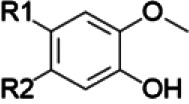
If the NP constituents are known and corresponding chemical structures are available, structure-activity comparisons may be used to anticipate the likelihood of NPDIs based solely on the presence of certain functional groups in individual constituent structures (Johnson et al., 2018) (Table 1). For example, methylenedioxyphenyl groups are well known structural alerts for potential time-dependent inhibition of the cytochrome P450 enzymes that involve stable heme coordination, whereas catechol groups or α,β-unsaturated aldehydes and ketones are structural alerts for time-dependent inhibition of cytochrome P450 enzymes that produce reactive intermediates and covalent protein adduction (Johnson et al., 2018).
TABLE 1.
Structural alerts for constituents in select natural products
Reprinted with permission from the American Society for Pharmacology and Experimental Therapeutics from Johnson et al. (2018).
| Constituent(s)/Natural Product | Structural Alert | Alert Substructure |
|---|---|---|
| Flavonoids, phenylpropanoids/Echinacea glycyrrhizin, glycyrrhizinic acid/licorice | Catechols | 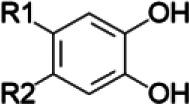 |
| Isoquinoline alkaloids/goldenseal terpenoids/cinnamon curcuminoids/turmeric | Masked catechol |
 , , 
|
| Isoquinoline alkaloids/goldenseal shizandrins/Schisandra spp. Gomisins/Schisandra spp. | Methylenedioxyphenyl | 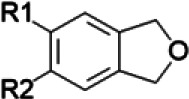 |
| Cycloartenol/black cohosh | Subterminal olefin | 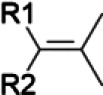 |
| Polyacetylenes/Echinacea | Terminal and subterminal acetylenes |
 , , 
|
| Terpenoids/cinnamon diallyl disulfides and trisulfides/garlic | Terminal olefin | 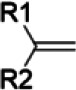 |
| Cinnamaldehyde/cinnamon | α,β-Unsaturated aldehyde | 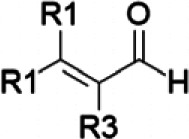 |
| Curcuminoids/turmeric | α,β-Unsaturated ketone | 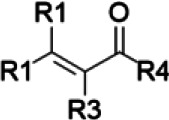 |
B. Obtaining Existing Data to Populate Static and Physiologically-Based Pharmacokinetic Models with Requisite Parameters
1. Collecting Physicochemical Data
Several open-source and/or commercial screening libraries exist specifically for the purpose of collating physicochemical characteristics of NPs (Gao et al., 2008; Valli et al., 2013; Mirza et al., 2015; Xie et al., 2015; Chen et al., 2018; Pilón-Jiménez et al., 2019). These databases are designed primarily to facilitate in silico identification of NCEs and to obtain experimentally determined characteristics, including structure, pKa, logarithm of octanol:water partition ratio, stereochemistry, and possible mechanisms of action. Additionally, the CHEMFATE data base curates available physicochemical data for many chemical entities (https://cfpub.epa.gov/si/si_public_record_Report.cfm?Lab=&dirEntryID=2897).
For constituents whose physicochemical characteristics have not been determined experimentally, structure-based prediction of chemical properties can be made provided that the molecular structure is known. Structure-based prediction of phase partitioning has shown excellent coefficients of determination with direct measurement (r2 = 0.51–0.91) (Eros et al., 2002; An et al., 2014; National Research Council, 2014), although performance is less accurate for phosphorus- and halogen-containing chemical entities (An et al., 2014). Similarly, pKa can be predicted using a variety of computational tools (Voutchkova et al., 2012). The intestinal effective permeability and absorption rate constant (ka) can be predicted from basic molecular attributes (polar surface area, phase partitioning, and hydrogen-bond donors), showing relatively high predictive performance with experimental Fa (fraction of the oral dose absorbed into the intestinal wall) values (r2 > 0.70) (Winiwarter et al., 1998; Linnankoski et al., 2006). When an NP is formulated as a capsule or tablet, solubility and dissolution may be limiting factors for absorption. Alternatively, a conservative estimate of 100% Fa may be used to predict the highest degree of exposure to precipitant constituents.
In contrast to physicochemical factors, computational prediction of factors influencing distribution (e.g., plasma protein and tissue binding) remains less developed (Poulin, 2015a). Previous studies that estimated the extent of plasma protein binding using sigmoidal functions of logarithm of octanol:water partition ratio showed high predictive performance compared with direct measurement (r2 = 0.79), whereas others have proposed simulating unbound drug concentrations in tissue compartments (Yamazaki and Kanaoka, 2004; Poulin, 2015b). Experimental methods for measuring the extent of plasma protein binding or fraction unbound (fu) rely on long-established techniques for separating bound and unbound drug (Rowland, 1980). Until further research validates novel methods for simulating protein binding behavior of NP constituents, determining fu experimentally is recommended based on data generated by the NaPDI Center (Nguyen et al., 2019). In brief, fu for multiple NP constituents (n = 14–17) in human liver microsomes (HLMs) and plasma was generated in silico using two modeling and simulation platforms (www.certara.com, v17; Simcyp and www.simulations-plus.com/software/gastroplus, v9.6; GastroPlus) and compared with experimentally determined values. Experimental fu was recovered via equilibrium dialysis using a 96-well device as described (Zamek-Gliszczynski et al., 2011). In silico–generated values ranged from 0.48 to 1.00 and from 0.01 to 0.75 in HLMs and plasma, respectively. Average (±S.D. of at least three determinations) experimental fu ranged from 0.052 ± 0.008 to 1.21 ± 0.09 for HLMs and from 0.013 ± 0.003 to 0.95 ± 0.20 for plasma. The ratio of in silico–generated fu values to experimental fu values was assessed for low, moderate, and high binding constituents (Fig. 1). Experimental fu for plasma proteins was generally lower than that for HLMs, which was consistent with values generated in silico. Both modeling and simulation platforms consistently predicted fu values for low binding constituents to within 30% of experimental values, suggesting that in silico–generated values are reasonable estimates for low binding constituents. However, predictive performance diminished for moderate and high binding constituents. Continued comparisons of in silico–generated and experimental fu values for additional NP constituents will form a database that can be used to develop predictive models of fu as described for pharmaceutical drugs (Obach, 1999; Lombardo et al., 2018).
Fig. 1.

Variability in the geometric mean of in-silico-to-observed fu ratios for high binding (low fu, denoted by experimental fu ≤ 20%), moderate binding (moderate fu, denoted by 20% < experimental fu < 80%), and low binding (high fu, denoted by experimental fu ≥ 80%) natural product constituents in human liver microsomes and plasma. Error bars denote 90% confidence intervals. Closed diamonds denote values generated by GastroPlus, whereas open diamonds denote values generated by Simcyp. Natural product constituents evaluated are 4-methylumbelliferone, 7-hydroxymitragynine, berberine, bergamottin, hydrastine, hydrastinine, isosilybin A, isosilybin B, isosilychristin, mitraciliatine, mitragynine, paynantheine, silybin A, silybin B, silychristin, silydianin, and speciogynine.
2. Generating Requisite Model Parameters from In Vitro Experiments
In general, based on the morphologic, transcriptomic, and proteomic differences among animal, immortalized human, and primary human tissues, the latter are the preferred experimental systems for characterizing xenobiotic metabolism and transport (Baillie and Rettie, 2011; Kauffman et al., 2013; Sawant-Basak et al., 2018). For these reasons, FDA guidance documents and the International Transporter Consortium recommend conducting in vitro DDI studies using human-derived systems or systems modified to express human drug-metabolizing enzymes and/or transporters (Brouwer et al., 2013; Chu et al., 2018; Evers et al., 2018; Zamek-Gliszczynski et al., 2018; FDA, 2020). These systems include recombinant enzymes, human subcellular tissue fractions (e.g., microsomes, cytosol), cell lines expressing human transporters, and intact human cell systems (i.e., hepatocytes, enterocytes). Recommended panels of enzymes and transporters against which potential NP precipitant constituents should be screened have been proposed in a previous NaPDI Center recommended approach (Johnson et al., 2018) (Table 2). In addition to the systems proposed in the earlier recommended approach, kidney- and intestine-derived microsomes and cell lines should be considered because of the well known interorgan differences in enzyme and transporter expression and function (Loretz et al., 2020).
TABLE 2.
Recommended enzymes, transporters, and experimental systems for screening natural products for inhibition and/or induction
Adapted with permission from the American Society for Pharmacology and Experimental Therapeutics from Johnson et al. (2018).
| Cytochrome P450 enzymes | ||
|---|---|---|
| Essential: CYP1A2, CYP2B6, CYP2C19, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A | ||
| Experimental System | Inhibition | Induction |
| Recombinant enzymes | As needed | NA |
| Human liver microsomes | a | NA |
| Human hepatocytes | As needed | a |
| Human intestinal microsomes | As needed | NA |
| Human intestinal cells | As needed | As neededb |
| Human kidney microsomes | As needed | NA |
| Human kidney cells | As needed | As neededb |
| Other cell lines | As needed | NA |
| UGTs | ||
|---|---|---|
| Essential: UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A8, UGT1A9, UGT1A10, UGT2B7, UGT2B10, UGT2B15 | ||
| Experimental System | Inhibition | Induction |
| Recombinant enzymes | As needed | NA |
| Human liver microsomes | a | NA |
| Human hepatocytes | a | a |
| Human intestinal microsomes | As needed | NA |
| Human intestinal cells | As needed | As neededb |
| Human kidney microsomes | As needed | NA |
| Human kidney cells | As needed | As neededb |
| Other cell lines | As needed | NA |
| Other Enzymes that May Be Considered | ||
|---|---|---|
| hCE1, hCE2, SULT1A1, SULT1A3, SULT1B1, SULT1E1, SULT2A1 | ||
| Experimental System | Inhibition | Induction |
| Recombinant enzymes | a | NA |
| Human liver microsomes | a | NA |
| Human hepatocytes | As needed | a |
| Human intestinal microsomes | As needed | NA |
| Human intestinal cells | As neededb | As neededb |
| Human kidney microsomes | As neededb | NA |
| Human kidney cells | As neededb | As neededb |
| Transporters | ||
|---|---|---|
| Essential: BCRP, BSEP, MATE1, MATE2-K, MRP2, MRP3, NTCP, OATP1B1, OATP1B3, OATP2B1, OAT, OCT, P-gp | ||
| Experimental System | Inhibition | Induction |
| Transfected cell lines (single, double) | a | NA |
| Human intestinal cells | As neededb | As neededb |
| Human kidney cells | As neededb | As neededb |
| Human hepatocytes | a | a |
| Membrane vesicles | a | NA |
BCRP, breast cancer resistance protein; BSEP, bile salt export pump; hCE, human carboxylesterase; MATE, multidrug and toxin extrusion protein; MRP, multidrug resistance–associated protein; NA, not applicable; NTCP, sodium taurocholate–cotransporting polypeptide; OAT, organic anion transporter; OATP, organic anion–transporting polypeptide; OCT, organic cation transporter; P-gp, P-glycoprotein; SULT, sulfotransferase.
Essential system. Inhibition studies in hepatocytes may involve multiple transporters.
These models represent an emerging field and will be refined with time. Expression levels of enzymes and transporters in these models are lower than those in vivo (Speer et al., 2019; Chapron et al., 2020; Kasendra et al., 2020).
Generally, NPDI prediction models are designed for the purpose of evaluating NPs as inhibitors or inducers of drug metabolizing enzymes and transporters rather than predicting exposure to NPs. Because the dose(s) of, and thus exposure to, NP constituents are difficult to standardize, these models provide only a conservative estimate of the magnitude of an interaction to confirm or rule out potential NPDIs. Thus, the goal of in vitro experiments is to generate robust parameters related to activation and induction (e.g., EC50, maximum inductive effect) or inhibition (e.g., IC50, reversible Ki, time-dependent KI, kinact) behavior as well as parameters related to clearance (e.g., t1/2, clearance). The following recommendations pertain to selecting concentration ranges for such experiments.
Prior to isolation of individual NP precipitant constituents, in vitro testing with crude fractions derived during bioactivity-directed fractionation is recommended. Concentrations used for initial bioactivity screening may vary because of differences in extraction methods and assay methodology. Based on the NaPDI Center investigators’ collective experience, relatively higher concentrations of the extracts may be needed to identify potential pharmacokinetic interactions mediated by UDP-glucuronosyltransferases (UGTs) compared with the CYPs. For example, cranberry extracts/fractions at 5 and 50 µg/ml showed concentration-dependent inhibition of intestinal CYP3A activity (Kim et al., 2011), and silymarin at 5 and 50 µg/ml showed similar percentage inhibition toward CYP3A and UGT activities (Brantley et al., 2013; Gufford et al., 2014), whereas higher concentrations (20, 60, and 180 µg/ml) were needed to produce concentration-dependent inhibition of UGTs by green tea extracts/fractions (Tian et al., 2018). These concentration ranges can be used to test for reversible inhibition as well as time-dependent CYP inhibition (e.g., based on structural alerts). NADPH or another relevant cofactor (e.g., UDP glucuronic acid) and substrate, respectively, should be used to initiate these reactions.
When testing isolated bioactive constituents, the concentration range should span a pharmacologically relevant concentration of individual constituents (i.e., maximum unbound plasma concentration) and a 10-fold higher concentration. If human plasma concentrations of a given constituent are not available, simulated unbound gut concentrations, simulated unbound hepatic portal venous inlet concentrations, and concentrations approaching constituent solubility can provide initial estimates of the concentrations to be tested (Tian et al., 2018; Cox et al., 2019). Three concentrations of constituents (e.g., 1, 10, and 100 µM) are recommended during initial screening to assess potential concentration-dependent alteration in enzyme/transporter activity. Depending on the results, this concentration range can be adjusted accordingly or used to guide determination of induction (e.g., EC50), reversible inhibition (e.g., IC50, Ki), and/or time-dependent inhibition (e.g., IC50 shift, KI, kinact) potency.
III. Applying or Developing Static and Physiologically Based Pharmacokinetic Models
There are two major categories of modeling strategies that are applicable to different pharmacokinetic NPDI scenarios. Static models refer to those that generate the estimated change in a pharmacokinetic endpoint of the object drug (typically AUC) in the presence of a single concentration of one or more NP constituents. Unless the NP is administered to steady state as an intravenous infusion, the plasma (or gut) concentration of the constituent causing the NPDI will change with time. Dynamic models, such as PBPK models, are capable of incorporating these changing concentrations to predict NPDIs. Such models are used with increasing frequency in the academic, regulatory, and commercial sectors to characterize and simulate DDIs. Both techniques have been used successfully to predict NPDIs involving curcumin and constituents of St. John’s wort and milk thistle (Table 3). Publications using PBPK modeling have proliferated approximately 4-fold since 2011, and the FDA has released 24 rule-making and guidance documents on this topic (Kola and Landis, 2004; Tan et al., 2018).
TABLE 3.
Examples of natural product–drug interactions predicted using static and PBPK models
| Natural Product | Object Drug(s) | Biochemical Target(s) | Model Type | Change in Object-Drug AUC or R2 | Reference(s) | |||
|---|---|---|---|---|---|---|---|---|
| Common Name | Latin Name | Precipitant Constituent(s) | Predicted | Observed | ||||
| Cannabis, marijuana | Cannabis sativa L. | CBD, THC | Phenacetin, diclofenac, omeprazole, dextromethorphan, testosterone | CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A | Static | CBD: >5-fold ↑ for CYP2C19 and CYP3A substrates; THC: >5-fold ↑ for CYP2C9 substrates | NR | Bansal et al. (2020) |
| Cinnamon | Cinnamomum spp. | trans-Cinnamic aldehyde | Letrozole, nicotine | CYP2A6 | Static | 1.1- to 3.6-fold ↑ | NR | Chan et al. (2016) |
| trans-Cinnamic aldehyde, 2-methoxycinnamaldehyde | ∼4- to 5-fold ↑ | Espiritu et al. (2020) | ||||||
| Curcumin (as a solid lipid nanoparticle) | Curcuma longa L. | Curcumin, curcumin glucuronide | Imatinib, bosutinib, paclitaxel | CYP2C8, CYP3A4 | PBPK | ≤1.10-Fold ↑ for imatinib and bosutinib | NR | Adiwidjaja et al. (2020a) |
| Goldenseal | Hydrastis canadensis L. | Berberine, (-)-β-hydrastine, hydrastinine | Dextromethorphan, midazolam, diclofenac | CYP2C9, CYP2D6, CYP3A | Static | R2 = 1.00, 7.90, and 1.26 for berberine and CYP2C9, CYP2D6, and CYP3A4, respectively; R2 = 8.94, ∼4, and 17.8 for (-)-β-hydrastine and CYP2C9, CYP2D6, and CYP3A4, respectively | NR for dextromethorphana 1.62-fold ↑ for midazolam after 2 wk of goldenseal administration (1323 mg three times daily) NR for diclofenac | Gurley et al., (2008); Guo et al. (2012); McDonald et al. (2020) |
| Grapefruit juice | Citrus × paradisi Macfad. | 6′,7′-Dihydroxy-bergamottin | Loperamide | CYP3A | Static | 1.6-Fold ↑ | 1.7-fold ↑ | Ainslie et al. (2014) |
| Green tea | Camellia sinensis (L.) Kuntze | ECG, EGCG | Raloxifene | UGTs | Static | 6.1- and 1.3-fold ↑, respectively, based on estimated concentrations in intestinal lumen and enterocyte | 30% ↓ | Tian et al. (2018b); Judson et al. (2020) |
| Kratom | Mitragyna speciosa (Korth.) Havil. | Mitragynine | Diclofenac, dextromethorphan, midazolam | CYP2C9, CYP2D6, CYP3A | Static | 1.1- and 5.7-fold ↑ for dextromethorphan and midazolam, respectively | NR | Tanna et al. (2020) |
| St. John’s wort | Hypericum perforatum L. | Hyperforinb | Alprazolam, carbamazepine, docetaxel, ethinyl estradiol, imatinib, midazolam, tacrolimus, verapamil, zolpidem, ibuprofen, tolbutamide, S-warfarin, clopidogrel, omeprazole | CYP2C19, CYP2C9, CYP3A | PBPK | 2%–79% ↓ for all substrates except clopidogrel (1.31-fold ↑) and S-warfarin (1.06-fold ↑). | Close agreement with observed changes (18%–23% difference) | Adiwidjaja et al. (2019) |
| Silibininb | Silybum marianum (L.) gaertn. | Silybin A, silybin B | Midazolam, warfarin | CYP3A, CYP2C9 | PBPK | 1.05- and 1.04-fold ↑ for midazolam and S-warfarin, respectively | 1.09 and 1.13-fold ↑ for midazolam and S-warfarin, respectively | Brantley et al. (2014b) |
| Silymarin | Silybum marianum (L.) gaertn. | Silybin A, silybin B, isosilybin A, isosilybin B, silychristin | Midazolam | CYP3A | Static | 1.75-fold ↑ | NR | Brantley et al. (2013) |
| Silibinin | Silybum marianum (L.) gaertn. | Silybin A, silybin B | Raloxifene | UGTs | PBPK | Up to 1.3-fold ↑ | 1.09-fold ↑ | Gufford et al. (2015a) |
| Silibinin, silymarin | NA, Silybum marianum (L.) gaertn. | Silybin A, silybin B | Raloxifene | UGT1A1, UGT1A8, UGT1A10 | Static | 4- to 5-fold ↑ by silibinin and silymarin, respectively | 1.09-fold ↑ | Gufford et al. (2015a,b) |
CBD, cannabidiol; CYP, cytochrome P450; EGCG, epigallocatechin gallate; NA, not applicable; NR, not reported; R2, intrinsic clearance ratio (without:with a time-dependent inhibitor); THC, tetrahydrocannabinol; UGT, UDP-glucuronosyltransferase.
9-Fold ↑ in urinary dextromethorphan:dextrorphan after 2 wk of goldenseal administration (300 mg three times daily).
Semipurified extract of milk thistle containing silybin A and silybin B in approximately an equimolar ratio.
Selection of a static model to predict NPDI risk is a conservative approach. If the NP is a potent inhibitor that results in maximum inhibition of the enzyme/transporter at all plasma or gut concentrations of the NP constituent, then the static and PBPK models will yield identical predictions. Static models that estimate fold changes in object drug AUC have been used to predict pharmacokinetic NPDIs (Zhou et al., 2004, 2005; Brantley et al., 2013; Ainslie et al., 2014; Gufford et al., 2015b; Tian et al., 2018; Bansal et al., 2020; Espiritu et al., 2020; McDonald et al., 2020). In contrast, PBPK models incorporate systems of differential equations to predict the time course of plasma concentrations of both object drug and precipitant NP constituent(s) using an array of in vitro data and a sequence of physiologic compartments (e.g., intestine and liver) in which distribution of the object drug/NP constituent is governed by blood flow, protein binding, and influx and efflux processes, and elimination is governed by blood flow, protein binding, and the intrinsic clearance of metabolic or excretory processes.
A. Developing Pharmacologically Based Pharmacokinetic Models for Natural Product–Drug Interaction Prediction
Few PBPK models for estimating the extent of NPDIs have been reported, although PBPK modeling strategies have been used successfully to predict drug interactions involving silibinin (Brantley et al., 2014b; Gufford et al., 2015a), Schisandra sphenanthera (Adiwidjaja et al., 2020b), and St. John’s wort (Adiwidjaja et al., 2019). Historically, PBPK modeling was a niche skill that involved solving systems of differential equations, often with manually coded programs. The general structure of a PBPK model is illustrated conceptually (Fig. 2).
Fig. 2.

General structure of a physiologically-based pharmacokinetic model designed to evaluate a natural product–drug interaction. Intravenous administration is rarely, if ever, used for natural products; rather, common routes include oral consumption and inhalation. The number of tissue compartments is variable, but N compartments can be included in a full physiologically-based pharmacokinetic model. Input and output blood flow rates (Q) describe constituent passage between the arterial and venous circulation.
Strategies for developing PBPK models depend on the available data and can be bottom-up, top-down, or middle-out. Various platforms have been used to construct bottom-up concentration-time prediction models, and differential equation–solving applications have proven to be useful tools for developing PBPK models (Allen, 1990; Lu et al., 2016). When some in vivo data are available, a middle-out approach that integrates existing in vivo and in vitro data can be used to refine uncertain or unknown parameters in the PBPK model; the advantage of this approach is that the model is informed by limited in vivo data (Tsamandouras et al., 2015). Finally, when complete clinical pharmacokinetic data are available, top-down models can be constructed to estimate organ exposures, although these models usually require the assumption of homogenous distribution.
Each modeling strategy requires assumptions (e.g., the expression and abundance of tissue-specific enzymes and transporters). Tutorials and reviews for building these models are available (Sager et al., 2015; Kuepfer et al., 2016). Thus, the scope of this recommended approach is to tailor these recommendations for building PBPK models for NPs and NPDIs.
B. Natural Product Dose Selection
As mentioned earlier, dose estimation is difficult for NPs. Currently, no database exists to collate information on the relative proportions of individual constituents in commercially available NPs. In addition, estimating average consumer NP doses is difficult because NP formulations vary widely between manufacturers, lots, and batches, and NP standardization is relatively nonexistent (Brantley et al., 2014a; Paine et al., 2018). For NPs administered as an aqueous solution (e.g., flavonoids in grapefruit juice), the dose can be approximated as the quantity of constituent in the volume of a glass of juice (e.g., 250 ml) (Johnson et al., 2017). The lack of standardized NP doses necessitates a sensitivity analysis with varying doses to predict the magnitude range for an NPDI.
C. Modeling Using Commercial Applications
Commercially available software platforms are designed to require minimal input from the end user and typically run full PBPK models that operate on systems of differential equations governing dissolution, solubility, absorption, distribution, metabolism, and excretion. An advantage of these platforms is the ability to simulate populations with large intersubject variation (e.g., by Monte Carlo methods) in these determinants of xenobiotic disposition. Additionally, effects of age, sex, race, and physiologic conditions, such as disease and pregnancy, on xenobiotic disposition can be simulated using commercial software.
Because manual entry of physiologic model parameters and equations is not required, end users may run simulations without changing input parameters. At minimum, the default software settings should be carefully evaluated, and all input values and settings should be reported. Commercial applications typically include a library of default object drugs. These drugs should be carefully evaluated to ensure that the correct object drugs are selected according to published guidelines (Fuhr et al., 2019).
IV. Building Physiologically Based Pharmacokinetic Models De Novo for NPDIs
Unlike PBPK models developed using commercial software, PBPK models developed de novo provide full control over model characteristics. Design considerations are described below.
A. Compartments and Parameterization
The degree of complexity used in a PBPK model can vary from minimal (e.g., a three-compartment model) to high (e.g., a model with many physiologic compartments) (Sager et al., 2015). A full PBPK model can produce concentration-versus-time estimates in many physiologic compartments, potentially providing greater insight into the mechanism of an NPDI. However, the potential increase in accuracy from a more compartmentalized model can be achieved only if the necessary physiologic parameters (blood flow, organ composition) and NP physicochemical parameters (e.g., tissue partition coefficient, pKa) are available. Complicated dissolution and absorption models may improve model performance but can be implemented only if the necessary physicochemical and in vitro data are available.
B. Verification
PBPK models can be built manually as systems of differential equations or generated using machine-learning approaches. Regardless of the approach, a separate verification data set should be used for final assessment of model prediction accuracy. Acceptable prediction accuracy should be specified before conducting PBPK modeling and simulation.
C. Error Checking
To avoid physiology-related errors while parameterizing models, checkpoints should be used to ensure physiologic relevance (e.g., the sum of blood flows should be equivalent to the expected cardiac output scaled for a human of certain age and sex). Sources of these reference values may include curated databases, such as those maintained by the US Environmental Protection Agency for PBPK modeling (https://cfpub.epa.gov/ncea/risk/recordisplay.cfm?deid=204443). Evaluating models in alternate programming languages and/or with independent datasets provides an additional layer of model verification and quality assurance. When possible, comparing a de novo model to that developed using a commercial program may provide insight into critical differences in predicted pharmacokinetic endpoints (Gufford et al., 2015a).
D. Reporting
Reproduction of a PBPK model is impossible without comprehensive reporting of model characteristics. Ideally, the complete code for a custom PBPK model should be published or made available for purposes of reproduction (Sager et al., 2015). Likewise, all inputs for a PBPK model developed using commercial software should be provided. Ensuring the availability of the relevant information is incumbent on both the editors and reviewers of relevant journals.
V. Using Static and Physiologically Based Pharmacokinetic Models to Prioritize Natural Product–Drug Interaction Risk
The NaPDI Center posits that NPDIs should be evaluated with at least the same level of rigor as that mandated for DDIs (FDA, 2020). Thus, a sequential set of decision trees are proposed to guide decision-making (Fig. 3).
Fig. 3.

Decision tree for the development of PBPK models of natural product–drug interactions. Selection of a modeling strategy depends on the available data. If data about the induction and inhibition behavior of the natural product constituent(s) are not available in the literature, these data can be gathered from in vitro experiments. If the predicted concentrations of the constituent(s) in either the gut or the plasma exceed the cutoffs [Table 4 and FDA and European Medicines Agency (EMA) guidance], different types of modeling are warranted. Cmax,u, maximum unbound concentration; Emax, maximum inductive effect; kdeg, degradation rate constant; KI, inhibitor concentration at one-half maximum inactivation rate; kobs, inactivation rate constant (observed).
A. Initial Assessment of Natural Product–Drug Interaction Risk
Investment of time and computing resources into development of complex PBPK models is not necessary for every NP constituent. Rather, simple initial assessments should be conducted to determine which constituent(s) may merit modeling studies.
For rapid triage of multiple NP constituents, predicted physicochemical properties can be used to populate commercial modeling software programs that include the target tissue as a compartment. Estimated concentrations at the tissue site(s) of interest can then be compared with reported inductive or inhibitory concentrations from in vitro experiments. If the predicted maximum unbound plasma concentration of the NP constituent(s) is within 10% of (FDA, 2020) or exceeds the in vitro unbound inductive or inhibitory concentration (e.g., unbound concentration at half maximum inductive effect, unbound IC50, Ki,u), then PBPK modeling of the NPDI is warranted. Alternatively, if the target drug metabolizing enzyme or transporter is pharmacologically important in the gut (e.g., CYP3A or organic anion–transporting polypeptide 2B1) (Won et al., 2010; 2012) and the gut tissue/luminal concentration estimated by the modeling approach is near or exceeds the unbound inductive or inhibitory concentration, then static and PBPK models should be used to predict the likelihood and magnitude of an NPDI (FDA, 2020). A decision process for developing PBPK models of NPDIs is presented (Fig. 3; Table 4).
TABLE 4.
Gut-specific cutoffs or criteria for natural product–drug interactions
Cutoffs or criteria used in decision tree for physiologically-based pharmacokinetic modeling of natural product–drug interactions depicted in Fig. 3. For additional information or for cutoff values related to other organ systems, refer to (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf; FDA, 2020).
| Transporter Inhibition | ||
|---|---|---|
| P-gp and BCRP | (Dose/250 ml)/Ki,u (or IC50,u) ≥ 10 | |
| Enzyme Inhibition | ||
| Reversible Inhibition |
Time-Dependent Inhibition |
|
| CYP3A | (Dose/250 ml)/Ki,u ≥ 10 | kobs/kdeg ≥ 10, wherein kobs = (kinact ⋅ Dose/250 ml)/(KI,u + Dose/250 ml) |
| CYP Inductiona | ||
| 1. Concentration-dependent increase in mRNA expression of a CYP | ||
| 2. ≥ 2-Fold increase of CYP mRNA expression relative to vehicle control at expected gut drug concentrations | ||
| 3. Increase ≥20% of the positive control response | ||
BCRP, breast cancer resistance protein; CYP, cytochrome P450; IC50,u, unbound IC50; kdeg, degradation rate constant; Ki,u, unbound reversible inhibition constant; kobs, inactivation rate constant (observed); P-gp, P-glycoprotein.
Must satisfy all three criteria to qualify as a CYP inducer. Criteria are based on those recommended for hepatic CYP induction (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf; FDA, 2020).
VI. Future Research
As for DDIs, if a clinically significant pharmacokinetic NPDI is suspected, the interaction merits advancement to a clinical study. The design of such a study is critical and will be addressed in a separate recommended approach from the NaPDI Center.
A. Natural Product–Drug Interactions within the Gastrointestinal Tract
Precision in modeling NPDIs mediated by drug metabolizing enzymes and transporters expressed in the intestine is governed primarily by the difficulty in predicting intracellular unbound concentrations of absorbed and effluxed NP constituents. Because intestinal epithelial cells polarize into an apical (brush border) and a basolateral domain, intestinal transporters show orientation-related expression. Thus, the extent of an NPDI mediated by an intestinal transporter should be driven by the local intracellular (for efflux transporters) or extracellular (for uptake transporters) concentration of the NP constituent at the membrane (apical or basolateral) where the transporter is expressed (Fig. 4). These concentrations may be reasonably approximated by the concentration of an NP constituent in the intestinal lumen. For example, for uptake transporters expressed on the apical membrane, unbound intestinal lumen concentrations would be the driving force. Further complicating these calculations is the unstirred water layer covering the apical membrane of enterocytes, which effectively constitutes an aqueous barrier to absorption both in vitro and in vivo (Korjamo et al., 2008; Wood et al., 2018). For an intracellular enzyme or efflux transporter expressed on the basolateral membrane of enterocytes, the intracellular unbound concentration would be more relevant, with the intestinal lumen concentration serving as a driver of intracellular concentration during the absorptive phase.
Fig. 4.

Illustration of intestinal cell polarization and the relative orientations of uptake and efflux transporters.
Another area of future research for PBPK modeling of NPDIs relates to the impact of the gut microbiota on plasma and target tissue exposure to object drugs. Gut microbiota can contribute to prodrug activation (e.g., the sulphanilamide-generating prodrugs prontosil and neoprontosil and the 5-aminosalisylic acid–generating prodrugs sulfasalazine, balsazide, and osalazine) (Wilson and Nicholson, 2017). Additionally, the gut flora directly execute a number of drug metabolic reactions, including decarboxylation, demethylation, hydrolysis, and dehydration (Wilson and Nicholson, 2017; Clarke et al., 2019). There is also emerging evidence that the secretory gut flora metabolome can alter drug metabolizing enzyme and transporter expression in the gut and liver (Fu and Cui, 2017; Nichols et al., 2019) and the drug molecules on which they act. Thus, there may be NPDIs mediated by gut microbiota. The contribution of the gut flora to NPDIs is a largely untapped area of future research.
B. Natural Product Metabolites
Currently, for NCEs, evaluation of a metabolite as a substrate and inducer/inhibitor of drug metabolizing enzymes and transporters is warranted if a metabolite is 1) less polar and exhibits at least 25% of the AUC compared with the parent or 2) more polar and has equal or greater AUC compared with the parent (FDA, 2020).
For NP phytoconstituents, metabolite data are often not available, raising concerns about the risk of unidentified NPDIs. NP phytoconstituents can undergo significant first-pass metabolism in the gut and liver, generating quantitatively major circulating products with uncharacterized effects on pharmacokinetic processes, as well as reactive metabolites that inactivate the enzymes that produce them. The recent development of the biochemometric approach discussed above may identify NP constituent metabolites that are precipitants of NPDIs. However, such examples have yet to be reported.
C. Systems Biology
Another logical step for improving the understanding of pharmacokinetic NPDIs is to integrate systems biology models with PBPK models. One systems biology tool potentially helpful to NPDI research is the virtual metabolic human database (Noronha et al., 2019). This recently developed database connects human metabolism with genetics, human-associated microbial metabolism, nutrition, and diseases. The use of -omics tools and the virtual human metabolic database have yet to be explored for NPDIs but may eventually offer unique mechanistic insight that can contribute to PBPK modeling.
VII. Conclusions
The application of static and PBPK models to potential NPDIs may allow rapid and systematic assessment of NPDI risk. Given the breadth and popularity of the NP consumer market, the lack of strict regulation on NPs with high NPDI risk, and the cost and time associated with conducting clinical studies, mathematical modeling provides a plausible method for mitigating the public health risk of NPDIs. Widespread adoption of systematic approaches to NPDI model development and application will facilitate the identification and investigation of NPDIs and promote the dissemination of critical NPDI information to researchers, clinicians, and patients.
Acknowledgments
The authors thank Nicholas Oberlies and Preston Manwill (University of North Carolina Greensboro, Greensboro, NC) for confirming the Latin names of the natural products listed in Table 3. M.F.P. dedicates this article to David P. Paine.
Abbreviations
- AUC
area under the concentration-versus-time curve
- DDI
drug-drug interaction
- Fa
fraction of oral dose absorbed into the intestinal wall
- FDA
US Food and Drug Administration
- fu
fraction unbound
- HLM
human liver microsome
- KI
inhibitor concentration at half maximum inactivation rate
- Ki
reversible inhibition constant
- Ki,u
unbound reversible inhibition constant
- kinact
maximum inactivation rate constant
- NaPDI Center
Center of Excellence for Natural Product Drug Interaction Research
- NCE
new chemical entity
- NP
natural product
- NPDI
NP-drug interaction
- PBPK
physiologically-based pharmacokinetic
- UGT
UDP-glucuronosyltransferase
Authorship Contributions
Performed data analysis: Cox, Tian, Paine.
Wrote or contributed to the writing of the manuscript: Cox, Tian, Clarke, Rettie, Unadkat, Thummel, McCune, Paine.
Footnotes
This work was supported by National Institutes of Health National Center for Complimentary and Integrative Health and Office of Dietary Supplements [Grant U54-AT008909] and in part by National Institute on Drug Abuse [Grant P01-DA032507]. E.J.C. was formerly a postdoctoral research associate with the Center of Excellence for Natural Product Drug Interaction Research (NaPDI Center) and is now a paid medical writer for the NaPDI Center.
The authors have no financial conflicts of interest to disclose.
References
- Adiwidjaja J, Boddy AV, McLachlan AJ (2019) Physiologically based pharmacokinetic modelling of hyperforin to predict drug interactions with St John’s wort. Clin Pharmacokinet 58:911–926. [DOI] [PubMed] [Google Scholar]
- Adiwidjaja J, Boddy AV, McLachlan AJ (2020a) Physiologically-based pharmacokinetic predictions of the effect of curcumin on metabolism of imatinib and bosutinib: in vitro and in vivo disconnect. Pharm Res 37:128. [DOI] [PubMed] [Google Scholar]
- Adiwidjaja J, Boddy AV, McLachlan AJ (2020b) Potential for pharmacokinetic interactions between Schisandra sphenanthera and bosutinib, but not imatinib: in vitro metabolism study combined with a physiologically-based pharmacokinetic modelling approach. Br J Clin Pharmacol 86:2080–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainslie GR, Wolf KK, Li Y, Connolly EA, Scarlett YV, Hull JH, Paine MF (2014) Assessment of a candidate marker constituent predictive of a dietary substance-drug interaction: case study with grapefruit juice and CYP3A4 drug substrates. J Pharmacol Exp Ther 351:576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen GD (1990) MODFIT: a pharmacokinetics computer program. Biopharm Drug Dispos 11:477–498. [DOI] [PubMed] [Google Scholar]
- Amrine CSM, Raja HA, Darveaux BA, Pearce CJ, Oberlies NH (2018) Media studies to enhance the production of verticillins facilitated by in situ chemical analysis. J Ind Microbiol Biotechnol 45:1053–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An N, Van Der Mei F, Voutchkova-Kostal A (2014) Global model for octanol-water partition coefficients from proton nuclear magnetic resonance spectra. Mol Inform 33:286–292. [DOI] [PubMed] [Google Scholar]
- Bailey DG, Malcolm J, Arnold O, Spence JD (1998) Grapefruit juice-drug interactions. Br J Clin Pharmacol 46:101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillie TA, Rettie AE (2011) Role of biotransformation in drug-induced toxicity: influence of intra- and inter-species differences in drug metabolism. Drug Metab Pharmacokinet 26:15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal S, Maharao N, Paine MF, Unadkat JD (2020) Predicting the potential for cannabinoids to precipitate pharmacokinetic drug interactions via reversible inhibition or inactivation of major cytochromes P450. Drug Metab Dispos 48:1008–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantley SJ, Argikar AA, Lin YS, Nagar S, Paine MF (2014a) Herb-drug interactions: challenges and opportunities for improved predictions. Drug Metab Dispos 42:301–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantley SJ, Graf TN, Oberlies NH, Paine MF (2013) A systematic approach to evaluate herb-drug interaction mechanisms: investigation of milk thistle extracts and eight isolated constituents as CYP3A inhibitors. Drug Metab Dispos 41:1662–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brantley SJ, Gufford BT, Dua R, Fediuk DJ, Graf TN, Scarlett YV, Frederick KS, Fisher MB, Oberlies NH, Paine MF (2014b) Physiologically based pharmacokinetic modeling framework for quantitative prediction of an herb-drug interaction. CPT Pharmacometrics Syst Pharmacol 3:e107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton ER, Kellogg JJ, Kvalheim OM, Cech NB (2018) Biochemometrics to identify synergists and additives from botanical Medicines: a case study with hydrastis canadensis (goldenseal). J Nat Prod 81:484–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer KL, Keppler D, Hoffmaster KA, Bow DA, Cheng Y, Lai Y, Palm JE, Stieger B, Evers R, International Transporter Consortium (2013) In vitro methods to support transporter evaluation in drug discovery and development. Clin Pharmacol Ther 94:95–112. [DOI] [PubMed] [Google Scholar]
- Caesar LK, Kellogg JJ, Kvalheim OM, Cech RA, Cech NB (2018) Integration of biochemometrics and molecular networking to identify antimicrobials in angelica keiskei. Planta Med 84:721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan J, Oshiro T, Thomas S, Higa A, Black S, Todorovic A, Elbarbry F, Harrelson JP (2016) Inactivation of CYP2A6 by the dietary phenylpropanoid trans-cinnamic aldehyde (cinnamaldehyde) and estimation of interactions with nicotine and letrozole. Drug Metab Dispos 44:534–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapron A, Chapron BD, Hailey DW, Chang SY, Imaoka T, Thummel KE, Kelly E, Himmelfarb J, Shen D, Yeung CK (2020) An improved vascularized, dual-channel microphysiological system facilitates modeling of proximal tubular solute secretion. ACS Pharmacol Transl Sci 3:496–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Garcia de Lomana M, Friedrich NO, Kirchmair J (2018) Characterization of the chemical space of known and readily obtainable natural products. J Chem Inf Model 58:1518–1532. [DOI] [PubMed] [Google Scholar]
- Chu X, Liao M, Shen H, Yoshida K, Zur AA, Arya V, Galetin A, Giacomini KM, Hanna I, Kusuhara H, et al. International Transporter Consortium (2018) Clinical probes and endogenous biomarkers as substrates for transporter drug-drug interaction evaluation: perspectives from the international transporter consortium. Clin Pharmacol Ther 104:836–864. [DOI] [PubMed] [Google Scholar]
- Clarke G, Sandhu KV, Griffin BT, Dinan TG, Cryan JF, Hyland NP (2019) Gut reactions: breaking down xenobiotic-microbiome interactions. Pharmacol Rev 71:198–224. [DOI] [PubMed] [Google Scholar]
- Cox EJ, Maharao N, Patilea-Vrana G, Unadkat JD, Rettie AE, McCune JS, Paine MF (2019) A marijuana-drug interaction primer: precipitants, pharmacology, and pharmacokinetics. Pharmacol Ther 201:25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Elimat T, Raja HA, Ayers S, Kurina SJ, Burdette JE, Mattes Z, Sabatelle R, Bacon JW, Colby AH, Grinstaff MW, et al. (2019) Meroterpenoids from neosetophoma sp.: a dioxa[4.3.3]propellane ring system, potent cytotoxicity, and prolific expression. Org Lett 21:529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eros D, Kövesdi I, Orfi L, Takács-Novák K, Acsády G, Kéri G (2002) Reliability of logP predictions based on calculated molecular descriptors: a critical review. Curr Med Chem 9:1819–1829. [DOI] [PubMed] [Google Scholar]
- Espiritu MJ, Chen J, Yadav J, Larkin M, Pelletier RD, Chan JM, Gc JB, Natesan S, Harrelson JP (2020) Mechanisms of herb-drug interactions involving cinnamon and CYP2A6: focus on time-dependent inhibition by cinnamaldehyde and 2-methoxycinnamaldehyde. Drug Metab Dispos 48:1028–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers R, Piquette-Miller M, Polli JW, Russel FGM, Sprowl JA, Tohyama K, Ware JA, de Wildt SN, Xie W, Brouwer KLR, International Transporter Consortium (2018) Disease-associated changes in rug transporters may impact the pharmacokinetics and/or toxicity of drugs: a white paper from the International Transporter Consortium. Clin Pharmacol Ther 104:900–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA (2020) In Vitro Drug Interaction Studies - Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry, U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER), Silver Spring, Maryland.
- Fu ZD, Cui JY (2017) Remote sensing between liver and intestine: importance of microbial metabolites. Curr Pharmacol Rep 3:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhr U, Hsin CH, Li X, Jabrane W, Sörgel F (2019) Assessment of pharmacokinetic drug-drug interactions in humans: in vivo probe substrates for drug metabolism and drug transport revisited. Annu Rev Pharmacol Toxicol 59:507–536. [DOI] [PubMed] [Google Scholar]
- Gao Z, Li H, Zhang H, Liu X, Kang L, Luo X, Zhu W, Chen K, Wang X, Jiang H (2008) PDTD: a web-accessible protein database for drug target identification. BMC Bioinformatics 9:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimstein M, Huang SM (2018) A regulatory science viewpoint on botanical-drug interactions. Yao Wu Shi Pin Fen Xi 26:S12–S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gufford BT, Barr JT, González-Pérez V, Layton ME, White JR Jr, Oberlies NH, Paine MF (2015a) Quantitative prediction and clinical evaluation of an unexplored herb-drug interaction mechanism in healthy volunteers. CPT Pharmacometrics Syst Pharmacol 4:701–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gufford BT, Chen G, Lazarus P, Graf TN, Oberlies NH, Paine MF (2014) Identification of diet-derived constituents as potent inhibitors of intestinal glucuronidation. Drug Metab Dispos 42:1675–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gufford BT, Chen G, Vergara AG, Lazarus P, Oberlies NH, Paine MF (2015b) Milk thistle constituents inhibit raloxifene intestinal glucuronidation: a potential clinically relevant natural product-drug interaction. Drug Metab Dispos 43:1353–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Chen Y, Tan ZR, Klaassen CD, Zhou HH (2012) Repeated administration of berberine inhibits cytochromes P450 in humans. Eur J Clin Pharmacol 68:213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley BJ, Swain A, Hubbard MA, Hartsfield F, Thaden J, Williams DK, Gentry WB, Tong Y (2008) Supplementation with goldenseal (Hydrastis canadensis), but not kava kava (Piper methysticum), inhibits human CYP3A activity in vivo. Clin Pharmacol Ther 83:61–69. [DOI] [PubMed] [Google Scholar]
- Henderson L, Yue QY, Bergquist C, Gerden B, Arlett P (2002) St John’s wort (Hypericum perforatum): drug interactions and clinical outcomes. Br J Clin Pharmacol 54:349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EJ, González-Peréz V, Tian DD, Lin YS, Unadkat JD, Rettie AE, Shen DD, McCune JS, Paine MF (2018) Selection of priority natural products for evaluation as potential precipitants of natural product-drug interactions: a NaPDI center recommended approach. Drug Metab Dispos 46:1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson EJ, Won CS, Köck K, Paine MF (2017) Prioritizing pharmacokinetic drug interaction precipitants in natural products: application to OATP inhibitors in grapefruit juice. Biopharm Drug Dispos 38:251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson S, Paine MF, Kirby T, Tanna R, Gallagher E, Matula-Péntek A, Horváth M, Layton M, White J, Thummel K, et al. (2020) Influence of transporters and the gut microbiota on the pharmacokinetic green tea-raloxifene interaction. Clin Pharmacol Ther 107:S71. [Google Scholar]
- Kasendra M, Luc R, Yin J, Manatakis DV, Kulkarni G, Lucchesi C, Sliz J, Apostolou A, Sunuwar L, Obrigewitch J, et al. (2020) Duodenum intestine-chip for preclinical drug assessment in a human relevant model. eLife 9:e50135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman AL, Gyurdieva AV, Mabus JR, Ferguson C, Yan Z, Hornby PJ (2013) Alternative functional in vitro models of human intestinal epithelia. Front Pharmacol 4:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg JJ, Paine MF, McCune JS, Oberlies NH, Cech NB (2019) Selection and characterization of botanical natural products for research studies: a NaPDI center recommended approach. Nat Prod Rep 36:1196–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg JJ, Todd DA, Egan JM, Raja HA, Oberlies NH, Kvalheim OM, Cech NB (2016) Biochemometrics for natural products research: comparison of data analysis approaches and application to identification of bioactive compounds. J Nat Prod 79:376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Sy-Cordero A, Graf TN, Brantley SJ, Paine MF, Oberlies NH (2011) Isolation and identification of intestinal CYP3A inhibitors from cranberry (Vaccinium macrocarpon) using human intestinal microsomes. Planta Med 77:265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola I, Landis J (2004) Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov 3:711–715. [DOI] [PubMed] [Google Scholar]
- Korjamo T, Heikkinen AT, Waltari P, Mönkkönen J (2008) The asymmetry of the unstirred water layer in permeability experiments. Pharm Res 25:1714–1722. [DOI] [PubMed] [Google Scholar]
- Kuepfer L, Niederalt C, Wendl T, Schlender JF, Willmann S, Lippert J, Block M, Eissing T, Teutonico D (2016) Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacometrics Syst Pharmacol 5:516–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnankoski J, Mäkelä JM, Ranta VP, Urtti A, Yliperttula M (2006) Computational prediction of oral drug absorption based on absorption rate constants in humans. J Med Chem 49:3674–3681. [DOI] [PubMed] [Google Scholar]
- Lombardo F, Berellini G, Obach RS (2018) Trend analysis of a database of intravenous pharmacokinetic parameters in humans for 1352 drug compounds. Drug Metab Dispos 46:1466–1477. [DOI] [PubMed] [Google Scholar]
- Loretz C, Ho MD, Alam N, Mitchell W, Li AP (2020) Application of cryopreserved human intestinal mucosa and cryopreserved human enterocytes in the evaluation of herb-drug interactions: evaluation of CYP3A inhibitory potential of grapefruit juice and commercial formulations of twenty-nine herbal supplements. Drug Metab Dispos 48:1084–1091. [DOI] [PubMed] [Google Scholar]
- Lu J, Goldsmith MR, Grulke CM, Chang DT, Brooks RD, Leonard JA, Phillips MB, Hypes ED, Fair MJ, Tornero-Velez R, et al. (2016) Developing a physiologically-based pharmacokinetic model knowledgebase in support of provisional model construction. PLOS Comput Biol 12:e1004495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald MG, Tian DD, Thummel KE, Paine MF, Rettie AE (2020) Modulation of major human liver microsomal cytochromes P450 by component alkaloids of goldenseal: time-dependent inhibition and allosteric effects. Drug Metab Dispos 48:1018–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirza SB, Bokhari H, Fatmi MQ (2015) Exploring natural products from the biodiversity of Pakistan for computational drug discovery studies: collection, optimization, design and development of a chemical database (ChemDP). Curr Comput Aided Drug Des 11:102–109. [DOI] [PubMed] [Google Scholar]
- Nguyen J, Tian D, Tanna R, Paine MF (2019) “Assessing the predictive performance of in silico generated binding parameters for various natural product constituents,” in 12th International ISSX (International Society for the Study of Xenobiotics) Meeting; 2019 July 30; Portland, Oregon. [Google Scholar]
- Nichols RG, Peters JM, Patterson AD (2019) Interplay between the host, the human microbiome, and drug metabolism. Hum Genomics 13:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noronha A, Modamio J, Jarosz Y, Guerard E, Sompairac N, Preciat G, Daníelsdóttir AD, Krecke M, Merten D, Haraldsdóttir HS, et al. (2019) The Virtual Metabolic Human database: integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res 47:D614–D624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council (2014) A Framework to Guide Selection of Chemical Alternatives, National Academies Press, Washington (DC). [PubMed] [Google Scholar]
- Obach RS (1999) Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos 27:1350–1359. [PubMed] [Google Scholar]
- Paguigan ND, Rivera-Chávez J, Stempin JJ, Augustinović M, Noras AI, Raja HA, Todd DA, Triplett KD, Day C, Figueroa M, et al. (2019) Prenylated diresorcinols inhibit bacterial quorum sensing. J Nat Prod 82:550–558. [DOI] [PubMed] [Google Scholar]
- Paine MF, Shen DD, McCune JS (2018) Recommended approaches for pharmacokinetic natural product-drug interaction research: a NaPDI center commentary. Drug Metab Dispos 46:1041–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilón-Jiménez BA, Saldívar-González FI, Díaz-Eufracio BI, Medina-Franco JL (2019) BIOFACQUIM: a Mexican compound database of natural products. Biomolecules 9:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin P (2015a) Drug distribution to human tissues: prediction and examination of the basic assumption in In Vivo pharmacokinetics-pharmacodynamics (PK/PD) research. J Pharm Sci 104:2110–2118. [DOI] [PubMed] [Google Scholar]
- Poulin P (2015b) A paradigm shift in pharmacokinetic-pharmacodynamic (PKPD) modeling: rule of thumb for estimating free drug level in tissue compared with plasma to guide drug design. J Pharm Sci 104:2359–2368. [DOI] [PubMed] [Google Scholar]
- Rivera-Chávez J, Caesar L, Garcia-Salazar JJ, Raja HA, Cech NB, Pearce CJ, Oberlies NH (2019a) Mycopyranone: a 8,8′-binaphthopyranone with potent anti-MRSA activity from the fungus Phialemoniopsis sp. Tetrahedron Lett 60:594–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Chávez J, El-Elimat T, Gallagher JM, Graf TN, Fournier J, Panigrahi GK, Deep G, Bunch RL, Raja HA, Oberlies NH (2019b) Delitpyrones: α-pyrone derivatives from a freshwater delitschia sp. Planta Med 85:62–71. [DOI] [PubMed] [Google Scholar]
- Rivera-Chávez J, Raja HA, Graf TN, Burdette JE, Pearce CJ, Oberlies NH (2017a) Biosynthesis of fluorinated peptaibols using a site-directed building block incorporation approach. J Nat Prod 80:1883–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Chávez J, Raja HA, Graf TN, Gallagher JM, Metri P, Xue D, Pearce CJ, Oberlies NH (2017b) Prealamethicin F50 and related peptaibols from Trichoderma arundinaceum: validation of their authenticity via in situ chemical analysis. RSC Adv 7:45733–45751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland M (1980) Plasma protein binding and therapeutic drug monitoring. Ther Drug Monit 2:29–37. [DOI] [PubMed] [Google Scholar]
- Sager JE, Yu J, Ragueneau-Majlessi I, Isoherranen N (2015) Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos 43:1823–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawant-Basak A, Rodrigues AD, Lech M, Doyonnas R, Kasaian M, Prasad B, Tsamandouras N (2018) Physiologically relevant, humanized intestinal systems to study metabolism and transport of small molecule therapeutics. Drug Metab Dispos 46:1581–1587. [DOI] [PubMed] [Google Scholar]
- Sorkin BC, Kuszak AJ, Bloss G, Fukagawa NK, Hoffman FA, Jafari M, Barrett B, Brown PN, Bushman FD, Casper SJ, et al. (2020) Improving natural product research translation: from source to clinical trial. FASEB J 34:41–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speer JE, Gunasekara DB, Wang Y, Fallon JK, Attayek PJ, Smith PC, Sims CE, Allbritton NL (2019) Molecular transport through primary human small intestinal monolayers by culture on a collagen scaffold with a gradient of chemical cross-linking. J Biol Eng 13:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan YM, Worley RR, Leonard JA, Fisher JW (2018) Challenges associated with applying physiologically based pharmacokinetic modeling for public health decision-making. Toxicol Sci 162:341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanna RS, Tian D, Cech NB, Oberlies NH, Rettie AE, Thummel KE, Paine MF (2020) Refined prediction of pharmacokinetic kratom-drug interactions: time-dependent inhibition considerations. J Pharmacol Exp Ther 376:64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian DD, Kellogg JJ, Okut N, Oberlies NH, Cech NB, Shen DD, McCune JS, Paine MF (2018) Identification of intestinal UDP-glucuronosyltransferase inhibitors in green tea (Camellia sinensis) using a biochemometric approach: application to raloxifene as a test drug via in vitro to in vivo extrapolation. Drug Metab Dispos 46:552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsamandouras N, Rostami-Hodjegan A, Aarons L (2015) Combining the ‘bottom up’ and ‘top down’ approaches in pharmacokinetic modelling: fitting PBPK models to observed clinical data. Br J Clin Pharmacol 79:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valli M, dos Santos RN, Figueira LD, Nakajima CH, Castro-Gamboa I, Andricopulo AD, Bolzani VS (2013) Development of a natural products database from the biodiversity of Brazil. J Nat Prod 76:439–444. [DOI] [PubMed] [Google Scholar]
- van Breemen RB, Fong HH, Farnsworth NR (2008) Ensuring the safety of botanical dietary supplements. Am J Clin Nutr 87:509S–513S. [DOI] [PubMed] [Google Scholar]
- Voutchkova A, Kostal J, Anastas P (2012) Property-based approaches to design rules for reduced toxicity, in Handbook of Green Chemistry, Part 9 Designing Safer Chemicals pp 349–373, Wiley, New York. [Google Scholar]
- Wilson ID, Nicholson JK (2017) Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res 179:204–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winiwarter S, Bonham NM, Ax F, Hallberg A, Lennernäs H, Karlén A (1998) Correlation of human jejunal permeability (in vivo) of drugs with experimentally and theoretically derived parameters. A multivariate data analysis approach. J Med Chem 41:4939–4949. [DOI] [PubMed] [Google Scholar]
- Won CS, Oberlies NH, Paine MF (2010) Influence of dietary substances on intestinal drug metabolism and transport. Curr Drug Metab 11:778–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won CS, Oberlies NH, Paine MF (2012) Mechanisms underlying food-drug interactions: inhibition of intestinal metabolism and transport. Pharmacol Ther 136:186–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood FL, Houston JB, Hallifax D (2018) Importance of the unstirred water layer and hepatocyte membrane integrity In Vitro for quantification of intrinsic metabolic clearance. Drug Metab Dispos 46:268–278. [DOI] [PubMed] [Google Scholar]
- Xie T, Song S, Li S, Ouyang L, Xia L, Huang J (2015) Review of natural product databases. Cell Prolif 48:398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki K, Kanaoka M (2004) Computational prediction of the plasma protein-binding percent of diverse pharmaceutical compounds. J Pharm Sci 93:1480–1494. [DOI] [PubMed] [Google Scholar]
- Zamek-Gliszczynski MJ, Ruterbories KJ, Ajamie RT, Wickremsinhe ER, Pothuri L, Rao MV, Basavanakatti VN, Pinjari J, Ramanathan VK, Chaudhary AK (2011) Validation of 96-well equilibrium dialysis with non-radiolabeled drug for definitive measurement of protein binding and application to clinical development of highly-bound drugs. J Pharm Sci 100:2498–2507. [DOI] [PubMed] [Google Scholar]
- Zamek-Gliszczynski MJ, Taub ME, Chothe PP, Chu X, Giacomini KM, Kim RB, Ray AS, Stocker SL, Unadkat JD, Wittwer MB, et al. International Transporter Consortium (2018) Transporters in drug development: 2018 ITC recommendations for transporters of emerging clinical importance. Clin Pharmacol Ther 104:890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Chan E, Li SC, Huang M, Chen X, Li X, Zhang Q, Paxton JW (2004) Predicting pharmacokinetic herb-drug interactions. Drug Metabol Drug Interact 20:143–158. [DOI] [PubMed] [Google Scholar]
- Zhou S, Huang M, Xu A, Yang H, Duan W, Paxton JW (2005) Prediction of herb-drug metabolic interactions: a simulation study. Phytother Res 19:464–471. [DOI] [PubMed] [Google Scholar]