

Abstract
Cancer incidences are rising globally. Therefore, in order to prevent and treat cancer, understanding cancer pathology is crucial. Tumors reprogram their metabolic phenotype to meet their needs for bioenergy, biosynthesis, and redox control. Alteration of the metabolic pathway has been proposed as the hallmark of cancer and explains the distinction between normal and cancer cells concerning nutrient utilization. Changes in the metabolism of nutrients such as glucose, amino acid, and fatty acid are associated with cancer risk. Luckily, this can be controlled with lifestyle modifications. Improvements in lifestyle behaviors to reduce cancer risks include a healthy diet, calorie restriction, and regular physical activity. This review begins with the understandings of metabolic reprogramming in cancer. Then, there will be evidence on the correlation between lifestyle factors and altered nutrient metabolism suggesting an application of lifestyle intervention for cancer risk reduction.
Keywords: Lifestyle, Cancer, Metabolism, Nutrient, Oncogene
INTRODUCTION
Cancer is a group of diseases defined as the uncontrollable proliferation and invasion of abnormal cells. The normal cells transform into cancer cells through a multistep process including alterations in both molecular signature and metabolic phenotype. The final stage of the invasion process called “metastasis” potentially attributes to a fatal outcome.
The incidences of cancer morbidity and mortality are expected to increase rapidly over the next decades. Global Cancer Statistics (GLOBOCAN) estimated 18.1 million new cancer cases and 9.6 million cancer deaths in 2018 [1,2], marking cancer as the second major cause of death. America alone had more than 16.9 million people alive with at least one experience of cancer as of January 1 of 2019 [3]. Advances in cancer research; however, cannot overcome challenges in cancer treatment since cancer cells evolve to acquire resistance to the therapy and recur as secondary cancer which in most cases, is more malignant and aggressive [4]. The risk factors for cancer are strongly associated with the consequences of population growth and social development [5]. The evolution of society is indispensably linked with lifestyle changes that increase metabolic diseases such as diabetes and obesity, metabolic disorder, and even cancer [6,7]. Nutrients such as red meat or processed meat can increase risks of colorectal cancer whereas calcium, fiber, milk, and whole-grain lower cancer risks. This can be explained via the improvement of immune responsiveness, inflammation, and over-nutrient which are risk factors for colorectal cancer development [8]. Different from classical opinion, novel cancer trait also includes deregulating cellular energetics, which indicates the important contribution of cellular metabolism in cancer development [9]. Indeed, different from their normal counterparts, cancer cells display a diversity of metabolic reprogramming which is controlled by not only intrinsic genetic mutations but also the tumor microenvironment. This metabolic adaptation eventually supports cancer cells with energy in the form of ATP as well as building blocks to maintain biosynthesis capacity and the balance of redox status [10]. This review will discuss the importance of changes in molecular signatures and metabolic phenotype in understanding cancer pathology and the importance of lifestyle factors on the reprogramming nutrient metabolism, strongly suggesting lifestyle modification for cancer prevention and treatment.
MOLECULAR BIOLOGY OF CANCER
Centuries of cancer research has proven that cancer is a disease involving dynamic changes in the genome. Cancer development is associated with the mutations generating oncogene with a gain of function and tumor suppressor gene with loss of function. Evidence indicates that tumorigenesis is the result of a process driving the transformation of normal human cells into a highly malignant and invasive tumor, which is in parallel with the instability of the genome. It further suggests that the development of cancer is reflected by the alterations of six manifestations including sufficient growth signals, resistance to antigrowth signal, uncontrollable replicative capability, sustained angiogenesis, tissue invasion, and metastasis (Fig. 1) [11].
Fig. 1.

Alteration of molecular biology during cancer development.
A growth signal is required for both normal cells and cancer cells to move from a quiescent state into an active proliferative state. These signals are transmitted into the cells via transmembrane receptors. Normal cells are unable to proliferate without those stimulatory signals while many oncogenes can mimic growth factors to induce cancer cell proliferation. One typical example is the automatically active Ras signaling cascade in 25% of human tumors which provides the release of a flux of mitogenic signals into cells without touching their upstream regulators [12].
Similar to the constitutive active transduction of growth signal, cancer cells also show insensitivity to antiproliferative signals. Those signals are also received by transmembrane cell surface receptors to inhibit cell proliferation by keeping cells into the quiescent (Go) state or induce specific differentiation-associated traits. Some types of cancer cells are known to evade these antiproliferative signals to prosper. For example, they try to disrupt the retinoblastoma protein (Rb) signaling to liberate the E2F transcription factor, and eventually continue the cell cycle [13].
The mechanism that cancer cells can expand uncontrollably is determined not only by the rate of cell proliferation but also by the resistance mechanism to apoptosis, a kind of programmed cell death. Apoptosis is triggered by multiple physiologic signals which lead to membrane disrupted and broken-down cellular skeletons. While normal cells show normal apoptotic programs in all cell types throughout the body, cancer cells exert the loss of proapoptotic regulators for example via the mutation of the p53 tumor suppressor gene [14].
Besides proliferation and apoptosis regulation, telomere maintenance is an important characteristic to distinguish normal cells from cancer cells. Cancer cells can upregulate the expression of the telomerase enzyme which adds hexanucleotide repeats onto the ends of telomeric DNA [15]. This leads to telomere maintenance for limitless replication of cancer cells.
Angiogenesis is also essential for cancer progression. Indeed, oxygen and nutrients are important for cell function and survival. Therefore, the vasculature is extremely required for tumors to grow. The tumor can activate the angiogenic process by changing the regulation between angiogenic inducer and inhibitor. For example, they increased the expression of Vascular endothelial growth factor (VEGF) or basic fibroblast growth factor to induce the development of endothelial cells leading to the generation of blood vessels [16].
Tissue invasion and metastasis are the last stages of cancer progression. It indicates the process that primary tumors move out and invade adjacent tissues, which eventually travels to a distant site to generate new cancer colonies. This is also called metastasis which is responsible for cancer death. One typical example is related to the changes in integrin expression which can support the invasion of cancer cells. Cancer cells can shift the expression of integrin used in normal cells to other integrins (e.g., α3β1 and αVβ3) which can degrade stromal components produced by extracellular proteases leading to favor metastasis [17,18].
REPROGRAMMING NUTRIENT METABOLISM IN CANCER
Different from classical opinions, novel cancer trait also includes deregulating cellular energetics as a new hallmark of cancer [9]. Nutrients are required for cell survival since it provides elements for energy generation and biosynthesis. Nutrient balance is well regulated to maintain cell homeostasis and cell proliferation. Therefore, any abnormality in nutrient metabolism including glucose, amino acid, and lipid metabolism can lead to a variety of metabolic diseases including cancer.
Nutrients are required for cell survival since they provide elements for energy generation and biosynthesis. By breaking down into simple components after food digestion, nutrients can enter the bloodstream for a further metabolic process by various types of cells. Nutrient balance is well regulated to maintain cell homeostasis and cell proliferation. Therefore, any abnormality in cellular metabolism can lead to a variety of metabolic diseases including cancer (Fig. 2).
Fig. 2.

Metabolic reprogramming in cancer cells. Different from their normal counterparts, oncogenes can regulate cancer cells to reprogram their metabolic phenotype to satisfy their demand of bioenergy, biosynthesis and redox control.
The discovery of the Warburg Effect [19] has emphasized the role of glucose metabolism in cancer development. This phenomenon is known as aerobic glycolysis which is different from normal cells that mostly prefer glycolysis in the absence of oxygen. Warburg effect accompanies the faster incorporation of carbon into biomass which facilitates rapid cell divisions. Moreover, by eliminating oxidative phosphorylation which is the main source of reactive oxygen species (ROS), it protects cells from excess oxidative stress during proliferation [20]. While most of the cells require aerobic glycolysis for survival, some cancer cells also show a dependency on glutamine, which is the most abundant amino acid in the blood and muscle [21,22]. Many cancer cells express high levels of glutamine transporters to support their high demand for glutamine [23]. After entering into the cells via transporters, glutamine is metabolized into glutamate and then α-ketoglutarate which continues the TCA cycle for energy production and macromolecule biosynthesis [21]. Moreover, glutamine can supply nitrogen sources for other non-essential amino acids and nucleotide synthesis and provide glutamate for glutathione which is important to control intracellular ROS level [24]. In some tumors with defective mitochondria, glutamine can be utilized by the cancer cells via reductive carboxylation rather than oxidative metabolism which glutamine-derived citrate can provide acetyl-coenzyme A for both lipid synthesis and the intermediates needed for the remaining TCA cycle metabolites [25]. Besides glucose and glutamine metabolism, lipid metabolism is also altered in cancer cells. While normal cells usually use exogenous fatty acid, cancer cells are capable of uptake and de novo synthesize fatty acid by upregulating important enzymes. Fatty acids not only provide an efficient source for ATP production but also serve as the important signaling molecules or contribute to membrane components [25]. Excess fatty acids can be stored in the form of lipid droplets which cancer cells can exploit for energy requirement under nutrient starvation or metabolic stress [26]. Overall, cancer cells can reprogram their metabolism in the most effective methods to help their survival against harsh tumor environments such as lack of nutrients and oxygen.
As the central regulator for metabolism, mitochondria are the keys to tumor development by their ability to generate ATP and building blocks as well as their capacity to produce ROS and regulate apoptosis. Nuclear DNA and mitochondrial DNA are both essential for mitochondria assembly. Mutations of mitochondrial genes were reported in many cancer cell lines which highlights how alterations of mitochondria lead to metabolic reprogramming [27]. For instance, germline mutations in mitochondrial complex II succinate dehydrogenase were observed in patients with paragangliomas and phaeochromocytomas [28] or somatic missense mutations in isocitrate dehydrogenase can induce metabolic changes that promote tumor growth [29]. Therefore, mitochondrial dysfunction is associated with malignant transformation which contributes to tumorigenesis [30].
Cancer is driven by the alterations in oncogenes or tumor suppressor genes such as the amplification of growth factor receptors or inactivation of p53 as an important tumor suppressor [31]. Although many mechanisms have been identified, the role of oncogenes in shaping metabolic phenotype only got attention recently. For instance, c-myc is a well- known oncogene regulating key enzymes in glycolysis which support the Warburg Effect [32]. Also, Kras mutation reprograms glutamine metabolism by upregulating aspartate transaminase and suppressing glutamate dehydrogenase in pancreatic cancer [33]. This evidence for oncogene-driven metabolic regulation provides underlying mechanisms that can be further exploited for cancer therapy development.
IMPLICATION FOR CANCER TREATMENT AND PREVENTION
Metabolic reprogramming in cancer cells is a promising target in cancer treatment since the dependency of cancer cells on specific metabolic pathways makes those cells susceptible to metabolic perturbation (Fig. 3). Indeed, a glycolysis inhibitor 2-deoxyglucose (2-DG) is a well-known anticancer treatment in most cancer cells [34]. Statins, known as cholesterol synthesis inhibitors, are reported to suppress RAS and growth factor receptor signaling pathways both of which are important for tumor growth. Metabolite depletion is also employed for targeting cancer such as recombinant arginine deiminase and arginase I which decreases arginine level or L-asparaginase which is effective for hematopoietic tumors [35-38]. Metformin, a type 2 diabetes drug, can inhibit tumor growth alone or in combination with other therapies which is also under clinical trials [39]. Not only providing important targets for cancer treatment but understanding cancer metabolism is also required for diet control in cancer patients. Indeed, nutrients such as red meat or processed meat augment can increase the risks of colorectal cancer whereas calcium, fiber, milk, and whole-grain lower cancer risks. This can be explained via the improvement of immune responsiveness, inflammation, and over-nutrient which are risk factors for colorectal cancer development [8]. The ketogenic diet (KD) which contains high fat, low carbohydrate, and adequate protein is based on the Warburg effect. As most cancer cells are dependent on glycolysis for survival, the limitation of carbohydrate can deplete ATP source in cancer cells while normal cells can utilize ketone bodies for survival. Besides, KD also decreases blood glucose level as well as insulin production which is important for cancer growth. Indeed, several clinical trials have shown the effect of KD on suppressing tumor growth in some types of cancer such as glioblastoma, prostate, colon, pancreatic, and lung cancer which proposed the role of KD as adjuvant therapy in cancer treatment [40,41].
Fig. 3.
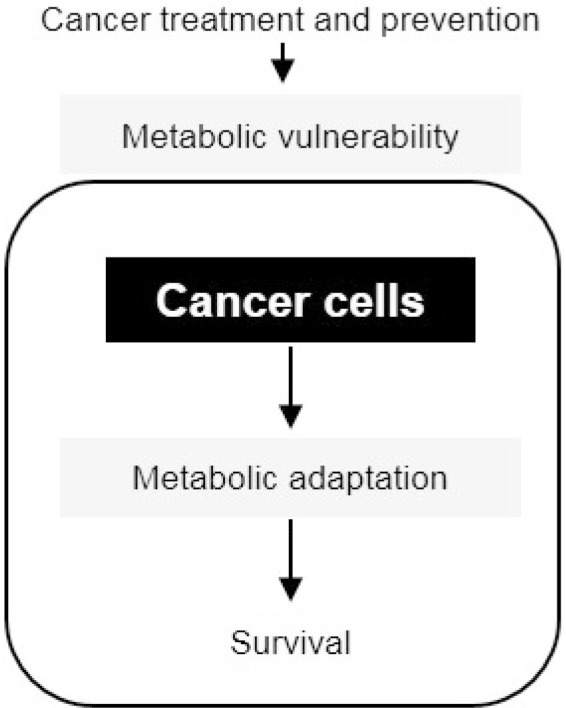
Metabolic vulnerability as the promising therapeutic target in cancer treatment and prevention.
MODIFICATION OF LIFESTYLE BEHAVIORS FOR CANCER RISK REDUCTION
Understanding the relationship between nutrient metabolism and cancer risk is required for the application of lifestyle medicine in cancer patients (Fig. 4). A meta-analysis including three studies has shown that a post-diagnostic low- fat diet can reduce the risk of breast cancer recurrence and improve breast cancer survival [42]. Since overweight and obesity have a direct linkage to breast cancer development, it is essential to establish an intensive modification of lifestyle behaviors including regular exercise and healthy diets rich in fruit, vegetables, whole grains, and low in red meat and saturated fat [43]. Since cancer cells require more nutrients to support their uncontrollable proliferation, calorie restriction (CR) has been suggested to reduce the risk of cancer. CR would mean chronic reduction of energy intake by about 30% without malnutrition occurring. CR can extend lifespan and reduce age-related diseases including cancer [44]. A systemic review reported that CR displayed a 75.5% reduction in tumor incidence with a variety of tumor models [45]. Chronic CR is challenging to apply to cancer patients, it is more suitable to employ intermittent CR, CR mimetic drugs, or other dietary regimens. Intermittent CR has been suggested to increase the effect of chemotherapy and radiation therapy in cancer patients [46,47] which indicates a potential adjuvant therapy of intermittent CR. CR mimetics with the application of pharmacological agents such as metformin, a biguanide used in type 2 diabetes, or resveratrol, a polyphenolic compound found in grapes, berries, and red wine are believed to sensitize cancer cells to conventional anti-cancer therapies [48,49]. An alternative dietary regimen such as a ketogenic diet is also helpful in combination therapies for cancer patients. The ketogenic diet (KD) which contains high fat, low carbohydrate, and adequate protein can decrease blood glucose level as well as insulin production which is important for cancer growth. Indeed, several clinical trials have shown the effect of KD on suppressing tumor growth in some types of cancer such as glioblastoma, prostate, colon, pancreatic, and lung cancer which proposed the role of KD as adjuvant therapy in cancer treatment [40,41]. In addition to diet control, physical activity is known to improve cancer risk in most cancer types [50]. Physical activity can increase or maintain muscle mass and decrease fat mass. It is suggested that physical activity reduced 25-30% the risk of breast cancer [51]. Therefore, it is important to promote the individual's motivation to regularly exercise such as jogging, walking, or exercising at the gym.
Fig. 4.

Lifestyle behavior modification for cancer prevention.
Smoking is also known as the risk of many types of cancers such as lung, oral, pharyngeal, laryngeal, esophageal, bladder, kidney, and pancreatic cancer. Supporting evidence also implicates tobacco as a risk factor for other cancers, including the cancer of the colon, stomach, and cervix, and leukemia. Prevention of smoking is the best way to prevent cancer; however, currently smoking cancer patients are expected to quit smoking also for risk of other diseases. People who quit smoking live longer than continuing smokers. For example, people who discontinue smoking before age 50 have as much as half the risk of dying in the next 15 years than people who continue to smoke. Also, the reduction in the risk of death compared with that for people who continue smoking can start shortly after quitting and lasts for at least 10 to 15 years. Education plays an important role in the cessation of tobacco use. Furthermore, the maintenance of a non-smoking lifestyle is critical to prevent the risks of multiple types of cancer [52-55].
CONCLUSION
The pathology of cancer involves the alterations of molecular biology and metabolic characteristics. Different from classical opinions which focus on the dysregulation of signaling pathways, metabolic reprogramming has now become the notable hallmark of cancer [9]. Many progress has been made to elucidate the mechanism, biological influences, and clinical application associated with cancer metabolism. This review has summarized opinions about common themes related to cancer pathology. First, alterations of both molecular features and nutrient metabolism including glucose, glutamine, and fatty acid metabolism can facilitate tumor growth. Second, metabolic reprogramming is associated with not only dysfunctional mitochondria but also the activation of oncogenes. Finally, the diversity of metabolic features in cancer cells suggests that the modification of lifestyle behaviors can be beneficial for cancer prevention and treatment. Taken together, understanding cancer development would provide insights to target cancer, which is one of the deadliest diseases.
REFERENCES
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018 doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Saito H, Kilpatrick C, Pittet D. The 2018 World Health Organization SAVE LIVES: Clean Your Hands Campaign targets sepsis in health care. Intensive Care Med. 2018;44:499–501. doi: 10.1007/s00134-018-5097-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30. doi: 10.3322/caac.21590. [DOI] [PubMed] [Google Scholar]
- 4.Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina- Pinelo S, Paz-Ares L. Current Challenges in Cancer Treatment. Clin Ther. 2016;38:1551–66. doi: 10.1016/j.clinthera.2016.03.026. [DOI] [PubMed] [Google Scholar]
- 5.Omran AR. he epidemiologic transition: a theory of the epidemiology of population change. 1971. Milbank Q. 2005;83:731–57. doi: 10.1111/j.1468-0009.2005.00398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiderpass E. Lifestyle and cancer risk. J Prev Med Public Health. 2010;43:459–71. doi: 10.3961/jpmph.2010.43.6.459. [DOI] [PubMed] [Google Scholar]
- 7.Shigeta H, Shigeta M, Nakazawa A, Nakamura N, Yoshikawa T. Lifestyle, obesity, and insulin resistance. Diabetes Care. 2001;24:608. doi: 10.2337/diacare.24.3.608. [DOI] [PubMed] [Google Scholar]
- 8.Song M, Garrett WS, Chan AT. Nutrients, foods, and colorectal cancer prevention. Gastroenterology. 2015;148:1244–60.:e16. doi: 10.1053/j.gastro.2014.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 10.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 11.Gutschner T, Diederichs S. The hallmarks of cancer: a long non-coding RNA point of view. RNA Biol. 2012;9:703–19. doi: 10.4161/rna.20481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Medema RH, de Vries-Smits AM, van der Zon GC, Maassen JA, Bos JL. Ras activation by insulin and epidermal growth factor through enhanced exchange of guanine nucleotides on p21ras. Mol Cell Biol. 1993;13:155–62. doi: 10.1128/MCB.13.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–30. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 14.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–31. doi: 10.1016/S0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 15.Bryan TM, Cech TR. Telomerase and the maintenance of chromosome ends. Curr Opin Cell Biol. 1999;11:318–24. doi: 10.1016/S0955-0674(99)80043-X. [DOI] [PubMed] [Google Scholar]
- 16.Veikkola T, Alitalo K. VEGFs, receptors and angiogenesis. Semin Cancer Biol. 1999;9:211–20. doi: 10.1006/scbi.1998.0091. [DOI] [PubMed] [Google Scholar]
- 17.Varner JA, Cheresh DA. Integrins and cancer. Curr Opin Cell Biol. 1996;8:724–30. doi: 10.1016/S0955-0674(96)80115-3. [DOI] [PubMed] [Google Scholar]
- 18.Lukashev ME, Werb Z. ECM signalling: orchestrating cell behaviour and misbehaviour. Trends Cell Biol. 1998;8:437–41. doi: 10.1016/S0962-8924(98)01362-2. [DOI] [PubMed] [Google Scholar]
- 19.Warburg O. On the origin of cancer cells. Science. 1956;123:309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 20.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–33. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wise DR, Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci. 2010;35:427–33. doi: 10.1016/j.tibs.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Windmueller HG, Spaeth AE. Uptake and metabolism of plasma glutamine by the small intestine. J Biol Chem. 1974;249:5070–9. doi: 10.1016/S0021-9258(19)42329-6. [DOI] [PubMed] [Google Scholar]
- 23.Mohamed A, Deng X, Khuri FR, Owonikoko TK. Altered glutamine metabolism and therapeutic opportunities for lung cancer. Clinical Lung Cancer. 2014;15:7–15. doi: 10.1016/j.cllc.2013.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lushchak VI. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids. 2012;2012:736837. doi: 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481:385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tirinato L, Pagliari F, Limongi T, Marini M, Falqui A, Seco J, Candeloro P, Liberale C, Di Fabrizio E. An Overview of Lipid Droplets in Cancer and Cancer Stem Cells. Stem Cells International. 2017;2017:1656053. doi: 10.1155/2017/1656053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brandon M, Baldi P, Wallace DC. Mitochondrial mutations in cancer. Oncogene. 2006;25:4647–62. doi: 10.1038/sj.onc.1209607. [DOI] [PubMed] [Google Scholar]
- 28.Bardella C, Pollard PJ, Tomlinson I. SDH mutations in cancer. Biochim Biophys Acta. 2011;1807:1432–43. doi: 10.1016/j.bbabio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Ichimura K. Molecular pathogenesis of IDH mutations in gliomas. Brain Tumor Pathol. 2012;29:131–9. doi: 10.1007/s10014-012-0090-4. [DOI] [PubMed] [Google Scholar]
- 30.Porporato PE, Filigheddu N, Pedro JMB, Kroemer G, Galluzzi L. Mitochondrial metabolism and cancer. Cell Res. 2018;28:265–80. doi: 10.1038/cr.2017.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Croce CM. Oncogenes and cancer. N Engl J Med. 2008;358:502–11. doi: 10.1056/NEJMra072367. [DOI] [PubMed] [Google Scholar]
- 32.Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res. 2012;18:5546–53. doi: 10.1158/1078-0432.CCR-12-0977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, Kang Y, Fleming JB, Bardeesy N, Asara JM, Haigis MC, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–5. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633–46. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 35.Vlodavsky I, Mohsen M, Lider O, Svahn CM, Ekre HP, Vigoda M, Ishai-Michaeli R, Peretz T. Inhibition of tumor metastasis by heparanase inhibiting species of heparin. Invasion metastasis. 1994;14:290–302. [PubMed] [Google Scholar]
- 36.Ott PA, Carvajal RD, Pandit-Taskar N, Jungbluth AA, Hoffman EW, Wu BW, Bomalaski JS, Venhaus R, Pan L, Old LJ, Pavlick AC, Wolchok JD. Phase I/II study of pegylated arginine deiminase (ADI-PEG 20) in patients with advanced melanoma. Invest New Drugs. 2013;31:425–34. doi: 10.1007/s10637-012-9862-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yau T, Cheng PN, Chan P, Chan W, Chen L, Yuen J, Pang R, Fan ST, Poon RT. A phase 1 dose-escalating study of pegylated recombinant human arginase 1 (Peg-rhArg1) in patients with advanced hepatocellular carcinoma. Invest New Drugs. 2013;31:99–107. doi: 10.1007/s10637-012-9807-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agrawal NR, Bukowski RM, Rybicki LA, Kurtzberg J, Cohen LJ, Hussein MA. A Phase I-II trial of polyethylene glycol-conjugated L-asparaginase in patients with multiple myeloma. Cancer. 2003;98:94–9. doi: 10.1002/cncr.11480. [DOI] [PubMed] [Google Scholar]
- 39.Morales DR, Morris AD. Metformin in cancer treatment and prevention. Annu Rev Med. 2015;66:17–29. doi: 10.1146/annurev-med-062613-093128. [DOI] [PubMed] [Google Scholar]
- 40.Klement RJ. Beneficial effects of ketogenic diets for cancer patients: a realist review with focus on evidence and confirmation. Med Oncol. 2017;34:132. doi: 10.1007/s12032-017-0991-5. [DOI] [PubMed] [Google Scholar]
- 41.Weber DD, Aminazdeh-Gohari S, Kofler B. Ketogenic diet in cancer therapy. Aging. 2018;10:164–5. doi: 10.18632/aging.101382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xing MY, Xu SZ, Shen P. Effect of low-fat diet on breast cancer survival: a meta-analysis. Asian Pac J Cancer Prev. 2014;15:1141–4. doi: 10.7314/APJCP.2014.15.3.1141. [DOI] [PubMed] [Google Scholar]
- 43.Taha Z, Eltom SE. The Role of Diet and Lifestyle in Women with Breast Cancer: An Update Review of Related Research in the Middle East. BioRes Open Access. 2018;7:73–80. doi: 10.1089/biores.2018.0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hursting SD, Dunlap SM, Ford NA, Hursting MJ, Lashinger LM. Calorie restriction and cancer prevention: a mechanistic perspective. Cancer Metab. 2013;1:10. doi: 10.1186/2049-3002-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lv M, Zhu X, Wang H, Wang F, Guan W. Roles of caloric restriction, ketogenic diet and intermittent fasting during initiation, progression and metastasis of cancer in animal models: a systematic review and meta- analysis. PloS One. 2014;9:e115147. doi: 10.1371/journal.pone.0115147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Safdie FM, Dorff T, Quinn D, Fontana L, Wei M, Lee C, Cohen P, Longo VD. Fasting and cancer treatment in humans: a case series report. Aging. 2009;1:988–1007. doi: 10.18632/aging.100114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Raffaghello L, Safdie F, Bianchi G, Dorff T, Fontana L, Longo VD. Fasting and differential chemotherapy protection in patients. Cell Cycle. 2010;9:4474–6. doi: 10.4161/cc.9.22.13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Linden MA, Lopez KT, Fletcher JA, Morris EM, Meers GM, Siddique S, Laughlin MH, Sowers JR, Thyfault JP, Ibdah JA, Rector RS. Combining metformin therapy with caloric restriction for the management of type 2 diabetes and nonalcoholic fatty liver disease in obese rats. Appl Physiol Nutr Metab. 2015;40:1038–47. doi: 10.1139/apnm-2015-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gupta SC, Kannappan R, Reuter S, Kim JH, Aggarwal BB. Chemosensitization of tumors by resveratrol. Ann N Y Acad Sci. 2011;1215:150–60. doi: 10.1111/j.1749-6632.2010.05852.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brown JC, Winters-Stone K, Lee A, Schmitz KH. Cancer, physical activity, and exercise. Compr Physiol. 2012;2:2775–809. doi: 10.1002/cphy.c120005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Friedenreich CM, Cust AE. Physical activity and breast cancer risk: impact of timing, type and dose of activity and population subgroup effects. Br J Sports Med. 2008;42:636–47. doi: 10.1136/bjsm.2006.029132. [DOI] [PubMed] [Google Scholar]
- 52.Kispert S, McHowat J. Recent insights into cigarette smoking as a lifestyle risk factor for breast cancer. Breast Cancer. 2017;9:127–32. doi: 10.2147/BCTT.S129746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jacob L, Freyn M, Kalder M, Dinas K, Kostev K. Impact of tobacco smoking on the risk of developing 25 different cancers in the UK: a retrospective study of 422,010 patients followed for up to 30 years. Oncotarge. 2018;9:17420–9. doi: 10.18632/oncotarget.24724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Alshammari FD, Ahmed HG, Alshammari D, Alharbi AM, Alsaedi AS, Elasbaly A. Population insight of the relationship between lifestyle and cancer: A population- based survey. AIMS Public Health. 2019;6:34–48. doi: 10.3934/publichealth.2019.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Novello AC. Surgeon General's report on the health benefits of smoking cessation. Public Health Rep. 1990;105:545–8. [PMC free article] [PubMed] [Google Scholar]