

Abstract
Background
Traumatic brain edema (TBE) is caused by a specific water channel mediated by membrane aquaporins. Aquaporin-4 (AQP4) plays an especially important role in this process, but the relationship between AQP4 and TBE remains unclear. The purpose of this study was to explore expression of AQP4 in the hippocampus after traumatic brain injury (TBI), as well as the effect of brain edema on skeletal protein and its function in hippocampal neurons.
Methods
The adult male Wistar rats we divided into a sham group and a TBI group, the latter of which was further divided into 1, 3, 6, 12, 24 and 72 hours (h) and 15 days (d) post injury subgroups. A proper TBI model was established, and brain edema was assessed in each group by water content. We measured the abundance of various proteins, including hypoxia inducible factor-1α (HIF-1α), AQP4, microtubule-associated protein 2 (MAP2), tau-5 protein, phosphorylated level of TAU, synaptophysin, cyclic adenosine monophosphate response element binding protein (CREB), phosphorylated CREB and general control nonrepressed 2, in each group. Hippocampal neurons and spatial memory test were analyzed in different time points.
Results
Compared with that in the sham group, the level of AQP4 in hippocampal neurons began to significantly increase at 1 h post TBI and then decreased at 15 d post TBI. During this time frame, AQP4 level peaked at 12 and 72 h, and these peaks were closely correlated with high brain water content. HIF-1α displayed a similar trend. Conversely, levels of MAP2 began to decrease at 1 h post TBI and then increase at 15 d post TBI. In addition, the most severe brain edema in rats was found at 24 h post TBI, with neuronal loss and hippocampal dendritic spine injury. Compared to those in the sham group, rats in the TBI groups had significantly prolonged latency and significantly shortened exploration time.
Conclusions
AQP4 level was closely correlated with severity of brain edema, and abnormal levels thereof aggravated such severity after TBI.
Keywords: Aquaporin-4, Brain edema, Traumatic brain injury, Hypoxia inducible factor-1α, Microtubule-associated protein 2
Highlights.
AQP4 plays an important regulatory role in the early stage of traumatic brain injury (TBI) via.
Blocking the level of AQP4 is beneficial for attenuating brain edema, releasing the damage of hippocampus and recovering the normal levels of memory proteins in brain tissue under TBI.
AQP4 could be a checkpoint protein and potential drug target for clinical treatment of brain edema mediated by TBI in the future.
Background
The incidence and mortality of traumatic brain injury (TBI) have increased in recent years, leading TBI to become an important public-health problem worldwide [1–3]. Brain edema, one complication of brain injury, generally occurs within a few hours after TBI [4–6]. It is not only an important cause of dysfunction in brain cells, such as neurons, but also a key factor in intracranial hypertension and cerebral herniation [5]. Brain edema is the main cause of high mortality and disability in TBI patients [4], and therefore studying it has important clinical value.
As we know, traumatic brain edema (TBE) is caused by a specific water channel mediated by membrane aquaporins in cell membranes and by breakdown of the blood–brain barrier (BBB) [7–9]. Aquaporin-4 (AQP4) is a transmembrane protein that specifically regulates water balance, acting as a water channel that allows only the water molecules to pass through the cellular membrane [8, 10, 11]. It plays an important role in the processes of brain edema, stroke and other diseases [12]. One might therefore wonder whether abnormal elevation of AQP4 at the protein level could be related to severity of brain edema following TBI and whether TBE could be associated with changes in skeletal protein levels and functional deficits in hippocampal neurons. Up to now, the evidence from research on these questions has been limited.
We therefore attempted to investigate the correlation between AQP4 protein level in the hippocampus and brain edema following TBI, and the level and function of hippocampal neural skeletal protein. Using the modified Feeney method, we established a moderately closed TBI rat model, and studied the effects of TBE on the level and function of hippocampal skeletal protein in rats.
Methods
Traumatic brain injury model, grouping and technical route
Adult male Wistar rats weighing 240–310 g, were provided by the Experimental Animal Center of the Research Institute of Surgery, Daping Hospital, Army Medical University (Third Military Medical University), Chongqing, China. The corresponding animal experiments (amuwec 20 181 430) were approved on 30 June 2018, by the Research Council and Animal Use and Care Committee of Army Medical University (Third Military Medical University).
We randomly divided the animals into a sham group (n = 25) and a TBI group (n = 85). In the sham group, five rats were used for western blot, five for measurement of brain water content, five rats for immunofluorescence staining, five for Golgi staining and five for the Morris water maze (MWM) test. In the TBI group, we randomly divided 70 of the rats into seven subgroups of 10 rats each (5 for western blot, 5 for measurement of brain water content) based on time elapsed after injury: 1, 3, 6, 12, 24, 72 h and 15 d. Five more rats were used for immunofluorescence staining, and five for Golgi staining, at 24 h after TBI. The remaining five rats were used in the MWM test at 11, 13 and 15 d after TBI. In accordance with the modified Feeney method [13], we established a moderately closed TBI rat model using a small animal stereotaxic locator and a PinPoint™ craniocerebral injury impact device (Hatteras Instruments, Inc., Cary, NC, USA) to control cortical impact. The impact parameters of the bone window were as follows: diameter of the impact head, 4.0 mm; impact depth, 4.0 mm; impact velocity, 2.5 m/s; impact time, 0.85 ms. Bleeding was stopped after impact, and the bone window was closed with bone wax. The sham group received the same treatment as the TBI group except for the impact. We kept temperature strictly within the range of 22–24°C throughout the experiments. We observed the status of rats after injury, including basic vital signs, breathing, coma, movement and mortality, and recorded them in timely manner. The construction of TBI model, grouping and technical route are provided in Figure 1.
Figure 1.
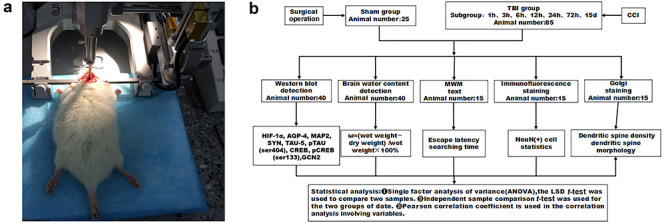
Animal model, grouping and technical schematic diagram. (a) We exposed the right side of each rat skull and subjected it to a 4-mm-diameter craniotomy using a motorized drill. The dura remained intact. The craniotomy was positioned at the center of the right parietal bone between the bregma and the lambdoid suture. This moderately controlled cortical impact model was created using a 4-mm-diameter metal tip with parameters of 4 mm below the dura, 2.5 m/s impact speed and 0.85 ms dwell time. (b) Animal grouping and technical schematic diagram. CCI Charlson Comorbidity Index, TBI traumatic brain injury, HIF-1α hypoxia inducible factor-1α, AQP4 aquaporin-4, MAP2 microtubule-associated protein 2, SYN synaptophysin, TAU-5 tau-5 protein, pTAU(ser404) phosphorylated tau protein at the ser404 site, CREB cAMP response element binding protein, pCREB(ser133) phosphorylated CREB at the ser133 site, GCN2 general control nonrepressed 2
Evaluation of brain edema
Edema was assessed directly by brain water content. In sum, rats in different groups were anesthetized with 3% pentobarbital sodium (1 milliliter per kilogram) and quickly decapitated. Then we removed the whole brain and determined its wet weight. It was dried in a drying chamber at a constant temperature of 85°C. We weighed it at 48, 72 and 96 h, taking its constant weight as the dry weight. Brain water content was calculated using the dry-wet weight method [3, 13], and expressed as a percentage of the wet weight per the Elliot formula: (wet weight-dry weight)/wet weight × 100%.
Western blot
Using western blot, we detected protein levels, including hypoxia inducible factor-1α (HIF-1α), AQP4, microtubule-associated protein 2 (MAP2), synaptophysin (SYN), phosphorylated tau protein (pTAU) at the ser404 site, TAU-5, cyclic adenosine monophosphate (cAMP) response element binding protein (CREB), phosphorylated CREB (pCREB) at the ser133 site and general control nonrepressed 2 (GCN2). The hippocampal tissues were removed from the rats and ground in a mortar containing liquid nitrogen. We then extracted total protein from tissue lysate containing protease inhibitor on ice. Concentration of total protein was determined by Coomassie Brilliant Blue protein assay. We mixed 20 μL tissue extracts normalized by protein concentration with a sodium dodecyl sulfate (SDS) sample buffer.
Samples were electrophoretically separated by 11% and 7.5% SDS-polyacrylamide gel electrophoresis and then transferred to a nitrocellulose (NC) membrane (both from Bio-Rad Laboratories, Hercules, CA, USA). Phosphate buffered saline (PBS) containing 6% skimmed milk powder (blocking buffer) was used to seal up for 3 h at room temperature (RT), and then the detected protein antibody was added. We used the following antibodies: rabbit anti-HIF-1α, rabbit anti-MAP2, rabbit anti-SYN, rabbit anti-TAU-5, rabbit anti-pTAU, rabbit anti-CREB, and rabbit anti-pCREB (all ser133; all 1:1000; Abcam Ltd, Hong Kong, China); and rabbit anti-AQP4, goat anti-GCN2 and goat anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (all 1:300; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). These antibodies were incubated with the NC membrane at RT for 1–2 h. After washing the NC membrane, we similarly incubated for 45 minutes (min) with secondary antibodies (Sigma-Aldrich, St. Louis, MO, USA) binding to horseradish peroxidase. Finally, we visualized target antigens via standard electrochemiluminescence methods (Bio-Rad, Hercules, CA, USA). In addition, we adopted GAPDH as a normalization protein in the experiment.
To quantify the relative level of target protein, we used the image analysis program (LABWORKS version 4.6) (Informer Technologies, Inc.; https://informer.com) to analyze imprinted images and compare levels of protein intensity among different groups.
Immunofluorescence staining
At 24 h after TBI, the animals received an intracardiac perfusion of cold PBS (pH 7.0) solution containing 4% polyformaldehyde. The removed brain tissue was fixed in 4% polyformaldehyde for 24 h and embedded in paraffin. We then cut 40 μm paraffin sections across the coronal plane and used immunofluorescence staining to label neuron-specific nuclear protein (NeuN). In addition, we used fluorescence microscopy for observation and photography, and we analyzed the images.
Golgi staining
For Golgi staining, we used a Rapid GolgiStain™ Kit (FD NeuroTechnologies, Columbia, MD, USA) per manufacturer’s instructions and the methods previously described by Xu and Phan [14–16]. In summary, rats were deeply anesthetized. We quickly removed the brains, rinsed them with double distilled water and immersed them in impregnation solution for 2 weeks, after which we cut them into 80 μm sections on a cryostat at −22°C and stained them for 10 min. Selected hippocampal neurons were photographed using a DFC290 camera connected to a Leica DM1000, vertical microscope (both from Leica, Wetzlar, Germany). Dendritic spine density (spines per 10 mm) and morphological changes were measured. We used three neurons from each of the five rats for statistical analysis.
Morris water maze test
Rats were randomly selected to enter the MWM from three quadrants (excluding the target quadrant) three times, until they could find the platform. On the first day, they were allowed to swim freely for 2 min. From the second day onward, they were trained three times a day at 1 h intervals. On the fourth day, the following experiments were conducted.
We performed an avoidance latency experiment, also called a positioning navigation experiment, reflecting the acquisition of spatial memory and the memory ability of reference objects, to measure the time from the rats entering the water until they climbed the hidden platform. If the rat did not find the platform within 120 seconds (s), it was led to and kept at the platform for 120 s, and the incubation period was recorded as 120 s.
A space exploration experiment, also called a transfer experiment, reflecting spatial memory retention and working memory ability, was performed as follows. The hidden platform experiment was completed on the next day. After the original platform was withdrawn, we put the rats into the water at the same entry point. Residence time was also called exploration time, and the time the rats spent passing through the quadrant of the original platform within 120 s was recorded. The above experiments were repeated at 7 d after TBI.
Statistical analysis
Data were expressed as mean ± standard deviation (SD; 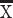 ±s). We adopted analysis of variance (ANOVA) and the least significant difference (LSD) t-test to analyze the western blot, brain water content and MWM test results. All data were analyzed using GraphPad Prism (GraphPad Software, Inc., San Diego, CA, USA), Microsoft Excel (Microsoft Corp., Redmond, WA, USA) and LABWORKS software. We used an independent-sample t-test and correlation analysis to compare average values and explore the relationship between AQP4 and brain edema. P < 0.05 was considered statistically significant.
±s). We adopted analysis of variance (ANOVA) and the least significant difference (LSD) t-test to analyze the western blot, brain water content and MWM test results. All data were analyzed using GraphPad Prism (GraphPad Software, Inc., San Diego, CA, USA), Microsoft Excel (Microsoft Corp., Redmond, WA, USA) and LABWORKS software. We used an independent-sample t-test and correlation analysis to compare average values and explore the relationship between AQP4 and brain edema. P < 0.05 was considered statistically significant.
Results
Observation of rat status after TBI
Twelve rats in the TBI group died between 7 and 24 h after TBI, the mortality rate was about 9.84%. We measured the brain water content of these dead rats and found that the ratio of brain water content between the sham group and each dead rat was ≥85.23%. None of the dead rats were included in the total number of animals used in the subsequent experiments.
AQP4 and HIF-1α levels during brain edema after TBI
Following TBI, HIF-1α level showed a tendency to increase (Figure 2a), while the level of AQP4 in the TBI group was significantly higher than that in the sham group. During the observation period of 1 h–15 d after injury, we identified two peaks in AQP4 level (Figure 2b) at the time points of 12 h and 72 h.
Figure 2.
The abnormal level of AQP4 accompanied with HIF-1α promoted the pathological process of brain edema following the TBI. (a) The level of HIF-1α in different groups under brain edema by western blot. Compared with that in the sham group, the level of HIF-1α in the TBI group increased significantly from 6 h to 72 h (p < 0.01). However, the level of HIF-1α in the TBI group decreased at 15 d after injury, although it was still higher than that in the sham group (p > 0.05). During the observation period from 1 h to 15 d after injury, the content of HIF-1α peaks at 12 h and 72 h (p < 0.01). (b) The rules of AQP4 changes were more or less in line with those of HIF-1α during the same pathological course (p < 0.01). (c) The difference of brain water content in each time points. Compared with the sham group, the brain water content at 3 h, 6 h, 12 h, 24 h, 48 h, 72 h after TBI were obviously higher (p < 0.01). Furthermore, brain edema peaks at 12 h and 72 h (p < 0.01). (d) The correlation analysis between AQP4 level and brain edema. Water content in brain tissue was positively correlated with AQP4 at hippocampal protein level, r = 0.6916, Y = 2.609*X + 75.16, p < 0.01. (e) The correlation analysis between HIF-1α and brain edema. Water content in brain tissue was positively correlated with HIF-1α at hippocampal protein level, r = 0.6939, Y = 1.998*X + 75.45, p < 0.01 (*p < 0.05; **p < 0.01). APQ4 aquaporin-4, TBI traumatic brain injury, HIF-1α hypoxia inducible factor-1α, GAPDH glyceraldehyde-3-phosphate dehydrogenase
Degree of edema was determined by mean percentage change of water content in brain tissue. One hour after TBI, brain water content began to increase, peaking at 72 h; it returned to normal in 15 d after TBI. During the observation period, two peaks of brain water content peaked twice at 12 h and 72 h (Figure 2c).
Abnormal AQP4 level was closely correlated with brain water content. The results of correlation analysis showed a positive correlation between AQP4 and brain edema (Figure 2d). Moreover, a similar correlation was shown between HIF-1α and brain edema (Figure 2e).
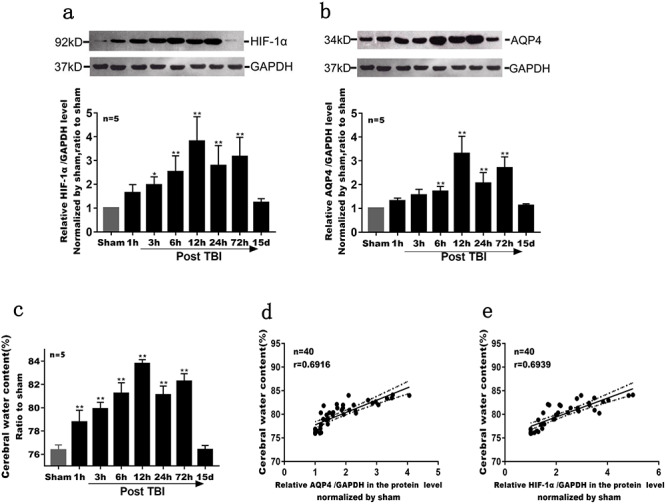
Loss of hippocampal neurons and skeletal damage
We found a significant loss of hippocampal neurons (p < 0.01) and a decrease in the number of neurons (Figure 3). The level of MAP2 in the TBI group was significantly lower than that in the sham group (p < 0.05 or p < 0.01, Figure 4a and b). Meanwhile, the level of SYN in the TBI group was also significantly decreased. The difference in these protein levels between the two groups was significant (p < 0.05 or p < 0.01, Figure 4c). In addition, compared with that in the sham group, the protein level of TAU-5, the main component of neurofibrillary tangles (NFT) [17], and the level of pTAU at the ser404 site in the TBI group gradually increased (p < 0.01, Figure 4d).
Figure 3.
Loss of hippocampal neurons post TBI as shown by NeuN fluorescence staining. After neuron was labeled by brain slices, the hippocampus was observed by confocal laser microscopy while nine visual fields (1 mm2 per visual field) were counted and mean values were taken for the t-test. (a) Immunofluorescence: NeuN (yellow), DPAI (bule), 20 × 10; (b) NeuN (+) cell statistics: the NeuN (+) cells in the dorsal hippocampus were counted and the staining results were analyzed by Image-Pro Plus 4.5 (Media Cybernetics, Silver Spring, MD, USA), p < 0.01 compared with the sham group, n = 5 (**p < 0.01). TBI traumatic brain injury, DPAI 4,6-diamidino-2-phenylindole
Figure 4.
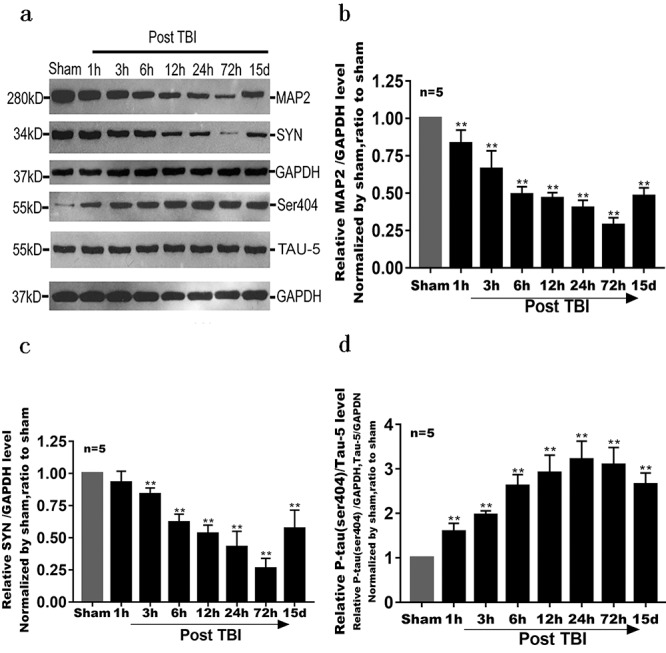
Changes in levels of several proteins (MAP2, SYN, pTAU at the ser404 site and TAU-5) after TBI. All of the above proteins were related to skeletal damage of hippocampal neurons mediated by brain edema. Detection methods were the same as for HIF-1α. (a, b) Compared with the sham group, the level of MAP2 in the TBI group continued to decrease; it recovered 15 d after injury but was still significantly lower than that in the sham group. The difference in MAP2 levels between the two groups was significant (p < 0.01). (a, c) Except at the time point of 1 h after injury (p > 0.05), SYN level differed significantly between the TBI and sham groups (p < 0.01). (a, d) Compared with the sham group, levels of pTAU at the ser404 site in the TBI group increased significantly from 1 h to 15 d (p < 0.01). However, its levels in the TBI group tended to decrease at 15 d after injury (**p < 0.01). MAP2 microtubule-associated protein 2, pTAU phosphorylated level of TAU, TBI traumatic brain injury, SYN synaptophysin, GAPDH glyceraldehyde-3-phosphate dehydrogenase
We also observed the structure of synapses (Figure 5). Compared with that in the sham group, the density of dendritic spines in the hippocampus decreased significantly at 24 h following TBI (p < 0.01). The results also showed that the density of mushroom-type (mature-type) dendritic spines were closely correlated with a decrease in spatial memory (p < 0.01). In addition, the proportion of stubby-type (disability-type) dendritic spines increased (p < 0.01), while that of thin-type (developmental) dendritic spines associated with short-term learning and memory decreased (p < 0.01).
Figure 5.
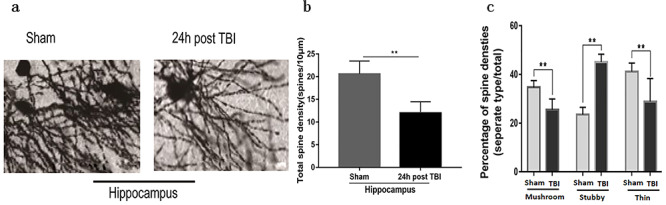
Hippocampal dendritic spine injury at 24 h after TBI. Three neurons were counted per rat, and the mean value was taken (n = 5). (a) Typical Golgi staining, 10 × 10; (b) Dendritic spine density statistics; (c) Dendritic spine morphology statistics (**p < 0.01). TBI traumatic brain injury
Neuronal dysfunction in the hippocampus
There was a significant difference in CREB and pCREB levels between the TBI and sham group (p < 0.05) (Figure 6a, b, c), which implied the weakened storage and memory ability [18]. Moreover, the level of GCN2 in the hippocampus was significantly higher in the TBI group than in the sham group (p < 0.05) (Figure 6a and d), which indicated reduced ability to transform new information into long-term memory [19].
Figure 6.
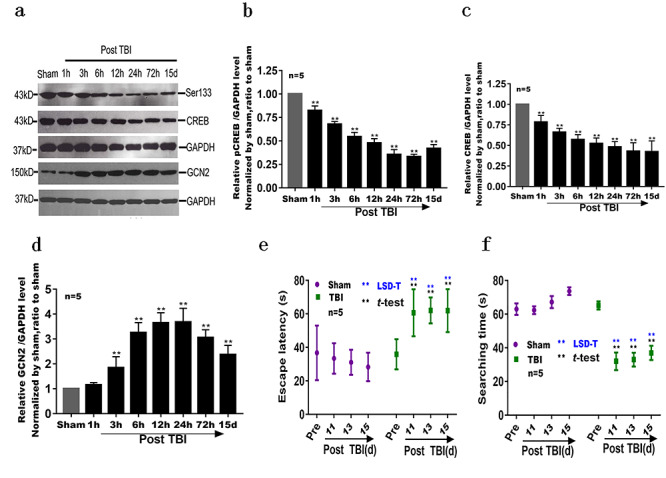
Neuronal dysfunction in hippocampus mediated by brain edema after TBI. (a, b, c) Compared with the sham group, CREB and pCREB (ser133) were at their lowest levels at 72 h post TBI (p < 0.01). (a, d) Compared with the sham group, GCN2 level increased significantly from 3 h to 15 d after TBI (p < 0.01). (e, f) Escape latency and search time in the MWM test: compared with the sham group, there were no significant differences in the results of pre-injury t-tests (p > 0.05) and significant difference in behavioral tests performed in the time points of 11, 13 and 15 d after TBI (p < 0.01) (**p < 0.01). TBI traumatic brain injury, CREB cAMP response element binding protein, d day(s), GAPDH glyceraldehyde-3-phosphate dehydrogenase, GCN2 general control nonrepressed 2
Prior to TBI, the MWM experiment showed no significant differences in spatial memory ability in rats. Following TBI, compared with that in the sham group, the latency of rats in the TBI group was significantly prolonged at 11, 13 and 15 d (p < 0.05) (Figure 6e), while exploration time in the TBI group was significantly shortened at the same time points (p < 0.01) (Figure 6f), showing decreased spatial reference memory and spatial working memory.
Discussion
At present, a moderate TBI model is most commonly used in the experimental study of TBI. After impact, 76.83% (63/82) of the rats in our study showed various degrees of neuroreflex symptoms such as conditional and unconditional reflex disappearance, coma and limb motor impairment. The mortality rate was 14.63% (12/82), which met the criteria of the moderate TBI model.
Brain edema has significant incidence and mortality rates, especially for the level of AQP4 at 24 h after TBI [20, 21]. Our experimental data supported that brain edema after brain injury was mediated by HIF-1α and AQP4. After TBI, HIF-1α level in brain tissue increased significantly at 3 h, peaked at 12 h and remained high until 72 h. AQP4 level increased at 1 h, which increased the permeability of cell membranes and destroyed the steady state and balance of the cell microenvironment. As a result, brain water content increased rapidly, exceeding 80% at 6 h. Meantime, TBE peaked at 12 h and 72 h (respectively 83.78% and 82.28% of brain tissue). The increase in brain water content was closely and positively correlated with levels of HIF-1α and AQP4, with respective correlation coefficients of 0.6939 and 0.6916. In short, following TBI, AQP4 was overexpressed and remained abnormally high, and brain edema occurred quickly and persisted. This imbalance of AQP4 contributed to persistent brain edema and subsequent damage to brain tissue.
As one of the factors in brain injury [4, 5, 22–24], brain edema can elevate intracranial pressure and increase the incidence of nerve cell injury [25] and the mortality of brain injury patients [26]. AQP4 is not only the most abundant aquaporin in the brain, but also the dominant of cerebral edema, especially vasogenic and cytotoxic edema [26, 27]. Therefore, in this study we observed dysfunction of hippocampal neurons caused by AQP4-induced brain edema. The hippocampus is the key area for spatial learning and memory, and it plays an important role in spatial navigation, goal-oriented tasks and memory storage [28]. Our results showed that neuronal loss and cerebral swelling were severe after TBI, and the level of MAP2, an important component of dendritic markers and neuronal cytoskeleton in the hippocampus, significantly decreased. The loss of neurons and the decreasing level of MAP2 suggest that TBI causes damage to dendrites and the skeletons of hippocampal neurons [29]. pTAU, which is involved in the pathological processes of many diseases, has a toxic effect on neuronal processes [17]. A large amount of pTAU produced by TBI can destroy the structure and function of normal TAU and form entanglements, which leads to the destruction of microtubules and thus affects the function of microtubules [17]. Our results showed that pTAU (ser404) significantly increased after TBI, suggesting that TAU was involved in axonal injury and cognitive dysfunction.
Moreover, as a specific marker of neuronal presynaptic terminals, SYN was confirmed to be closely related to spatial learning and memory in this study. This finding implied that the hippocampal neuronal skeleton and synaptic damage were correlated with spatial memory. In addition, the level trends of CREB, pCREB (ser133) and GCN2 also suggested that TBI impaired learning and memory in rats.
The formation of memory depends on synaptic transmission between neurons. TBI directly or indirectly leads to the destruction of neuronal skeletons and synaptic structure, hinders the formation of synapses or inhibits the regeneration of neurites (mainly axons), and blocks the functional connections between neurons leading to impairment in transmission or non-transmission of information [29–31]. This study demonstrated that the increased proportion of disabled dendritic spines appeared to be correlated with the decrease in short-term learning and memory. These results confirmed that TBI destroyed the hippocampal neuronal skeleton and synapses, and subsequently altered protein levels, which enriched the pathological basis of neurological deficits caused by TBE.
Taken together, the study showed that AQP4 played an important role after TBI (Figure 7). The abnormal level of AQP4 not only caused loss of hippocampal neurons and destroy hippocampal skeleton, but also led to hippocampal neuronal dysfunction. Furthermore, its level was closely correlated with severity of brain edema and injured memory storage in rats. Therefore, AQP4 may act as a potential drug target under TBI in the future.
Figure 7.
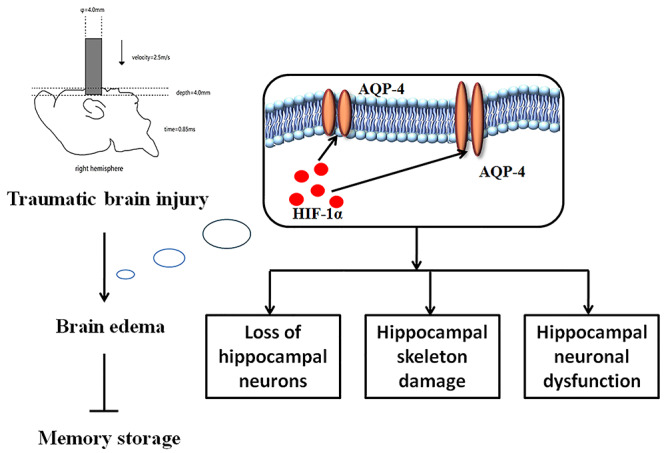
Schematic diagram for the role of AQP4 after TBI. TBI traumatic brain injury, HIF-1α hypoxia inducible factor-1, AQP4 aquaporin-4
Although our study manifested that abnormal level of AQP4 aggravated the severity of brain edema after TBI, our data did not clarify the specific relationship between AQP4 and brain edema, nor the molecular mechanism involved. So far, we cannot determine causativity in terms of these factors. Furthermore, the number of samples, type of models and methodology of experiments adopted in the study were limited. Therefore, we will need to identify the signaling pathways of AQP4 and HIF-α in further study.
Conclusions
Based on the results of this study, we concluded that HIF-1α gene was activated by ischemia and hypoxia following TBI, resulting in a sustained high level of AQP4 at the protein level and leading to sustained brain edema. TBE mediated by HIF-1α and AQP4 damaged hippocampal neurons, destroyed the skeletons of hippocampal neurons, suppressed the protein level of memory storage, and enhanced the inhibition of related protein level. This conclusion emphasizes that HIF-1α and AQP4 mediate TBE. It implies that blocking AQP4 is beneficial for attenuating brain edema, releasing damage to the hippocampus and recovering the normal levels of memory proteins in the early stage after TBI. Thus, AQP4 could be a checkpoint protein and potential drug target for clinical treatment of brain edema mediated by TBI in the future.
Abbreviations
ANOVA: analysis of variance; AQP4: aquaporin-4; cAMP: cyclic adenosine monophosphate; BBB: blood–brain barrier; CREB: cAMP response element binding protein; d: day(s); g: gram(s); GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GCN2: general control nonrepressed 2; HIF-1α: hypoxia inducible factor-1α; h: hour(s); LSD: least significant difference; MAP2: microtubule-associated protein 2; m/s: meters per second; μL: microliter; min: minutes; ms: millisecond; MWM: Morris water maze; NC: nitrocellulose; mm: millimeter; NeuN: neuron-specific nuclear protein; PBS: phosphate buffered saline; pCREB (ser133): phosphorylated CREB at the ser133 site; pTAU (P-TAU) (ser404): phosphorylated tau protein at the ser404 site; RT: room temperature; SYN: synaptophysin; TAU-5: tau-5 protein; TBE: traumatic brain edema; TBI: traumatic brain injury
Contributor Information
Ao Xiong, Department of Orthopaedics, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450042, China; State Key Laboratory of Trauma, Burns and Combined Injury, Research Institute of Surgery, Daping Hospital, Army Medical University (Third Military Medical University), Chongqing 400042, China.
Renping Xiong, State Key Laboratory of Trauma, Burns and Combined Injury, Research Institute of Surgery, Daping Hospital, Army Medical University (Third Military Medical University), Chongqing 400042, China.
Jing Yu, State Key Laboratory of Trauma, Burns and Combined Injury, Research Institute of Surgery, Daping Hospital, Army Medical University (Third Military Medical University), Chongqing 400042, China.
Yijia Liu, State Key Laboratory of Trauma, Burns and Combined Injury, Research Institute of Surgery, Daping Hospital, Army Medical University (Third Military Medical University), Chongqing 400042, China; School of Life Science and Engineering, Southwest Jiaotong University, Chengdu, Sichuan, 610031, China.
Ke Liu, State Key Laboratory of Trauma, Burns and Combined Injury, Research Institute of Surgery, Daping Hospital, Army Medical University (Third Military Medical University), Chongqing 400042, China; School of Life Science and Engineering, Southwest Jiaotong University, Chengdu, Sichuan, 610031, China.
Ge Jin, Department of Biochemistry and Molecular Biology, Basic Medical College of Zhengzhou University, Zhengzhou, Henan 450001, China.
Jianzhong Xu, Department of Orthopaedics, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450042, China.
Jun Yan, State Key Laboratory of Trauma, Burns and Combined Injury, Research Institute of Surgery, Daping Hospital, Army Medical University (Third Military Medical University), Chongqing 400042, China.
Funding
Not applicable.
Authors’ contributions
A.X. mainly performed the experiments and drafted the manuscript. R.X., J.Yu, Y.L., K.L. and G.J. participated in the study. J.X. guided the study. J.Yan performed data management and rewrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The corresponding animal experiments were approved by the Research Council and Animal Use and Care Committee of Army Medical University (Third Military Medical University).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Conflict of interest
The authors declare that no conflict of interest exists.
References
- 1. Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, et al. . Estimating the global incidence of traumatic brain injury. J Neurosurg. 2018;1:1–18. [DOI] [PubMed] [Google Scholar]
- 2. Zelinkova V, Brazinova A, Taylor MS, Rusnak M, Plancikova D, Melichova J, et al. . Location of traumatic brain injury-related deaths: epidemiological analysis of 11 European countries. Brain Inj. 2019;33:830–5. [DOI] [PubMed] [Google Scholar]
- 3. Stubbs JL, Thornton AE, Sevick JM, Silverberg ND, Barr AM, Honer WG, et al. . Traumatic brain injury in homeless and marginally housed individuals: a systematic review and meta-analysis. Lancet Public Health. 2020;5:e19–32. [DOI] [PubMed] [Google Scholar]
- 4. Shenaq M, Kassem H, Peng C, Schafer S, Ding JY, Fredrickson V, et al. . Neuronal damage and functional deficits are ameliorated by inhibition of aquaporin and HIF-1a after traumatic brain injury (TBI). J Neurol Sci. 2012;323:134–40. [DOI] [PubMed] [Google Scholar]
- 5. Chen JQ, Xia QJ. Lu H. Changes in AQP4 level and the pathology of injured cultured astrocytes after AQP4 mRNA silencing. Neuropsychiatry (London). 2017;7:426–32. [Google Scholar]
- 6. Brain Trauma Foundation, American Association of Neurological Surgeons, Congress of Neurological Surgeons . Guidelines for the management of severe traumatic brain injury. J Neurotrauma. 2007;24:S1–106. [DOI] [PubMed] [Google Scholar]
- 7. Higashida T, Kreipke CW, Rafols JA, Peng C, Schafer S, Schafer P, et al. . The role of hypoxia-inducible factor-1alpha, aquaporin-4, and matrix metalloproteinase-9 in blood–brain barrier disruption and brain edema after traumatic brain injury. J Neurosurg. 2011;114:92–101. [DOI] [PubMed] [Google Scholar]
- 8. Ho JD, Yeh R, Sandstrom A, Chorny I, Harries WE, Robbins RA, et al. . Crystal structure of human aquaporin 4 at 1.8A and its mechanism of conductance. Proc Natl Acad Sci U S A. 2009;106:7437–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Verkman AS. Aquaporins at a glance. J Cell Sci. 2011;124:2107–12. [DOI] [PubMed] [Google Scholar]
- 10. Scharfman HE, Binder DK. Aquaporin-4 water channels and synaptic plasticity in the hippocampus. Neurochem Int. 2013;63:702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Szu JI, Binder DK. The role of astrocytic aquaporin-4 in synaptic plasticity and learning and memory. Front Integr Neurosci. 2016;10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu LS, Fan YY, Ye G, Li J, Feng XP, Lin K, et al. . Curcumin alleviates brain edema by lowering AQP4 expression levels in a rat model of hypoxia-hypercapnia-induced brain damage. Exp Ther Med. 2016;11:709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury: methodology and local effects of contusions in the rats. Brain Res. 1981;211:67–77. [DOI] [PubMed] [Google Scholar]
- 14. Xu L, Yang Y, Gao L, Zhao J, Cai Y, Huang J. Protective effects of resveratrol on the inhibition of hippocampal neurogenesis induced by ethanol during early postnatal life. Biochim Biophys Acta. 2015;1852:1298–310. [DOI] [PubMed] [Google Scholar]
- 15. Phan A, Gabor CS, Favaro KJ, Kaschack S, Armstrong JN, MacLusky NJ. Low doses of 17beta-estradiol rapidly improve learning and increase hippocampal dendritic spines. Neuropsychopharmacology. 2012;37:2299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Phan A, Lancaster KE, Armstrong JN, MacLusky NJ, Choleris E. Rapid effects of estrogen receptor alpha and beta selective agonists on learning and dendritic spines in female mice. Endocrinology. 2011;152:1492–502. [DOI] [PubMed] [Google Scholar]
- 17. Zhao ZA, Ning YL, Li P, Yang N, Peng Y, Xiong RP, et al. . Widespread hyperphosphorylated tau in the working memory circuit early after cortical impact injury of brain. Behav Brain Res. 2017;323:146–53. [DOI] [PubMed] [Google Scholar]
- 18. Han JH, Kushner SA, Yiu AP, Cole CJ, Matynia A, Brown RA, et al. . Neuronal competition and selection during memory formation. Science. 2007;316:457–60. [DOI] [PubMed] [Google Scholar]
- 19. Costa-Mattioli M, Gobert D, Harding H, Herdy B, Azzi M, Bruno M, et al. . Translational control of hippocampal synaptic plasticity and memory by the eIF2alpha kinase GCN2. Nature. 2005;436:1166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Szczygielski J, Glameanu C, Müller A, Klotz M, Sippl C, Hubertus V, et al. . Changes in posttraumatic brain edema in craniectomy-selective brain hypothermia model are associated with modulation of aquaporin-4 level. Front Neurol. 2018;9:799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yao X, Uchida K, Papadopoulos MC, Zador Z, Manley GT, Verkman AS. Mildly reduced brain swelling and improved neurological outcome in aquaporin-4 knockout mice following controlled cortical impact brain injury. J Neurotrauma. 2015;32:1458–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Papadopoulos MC, Krishna S, Verkman AS. Aquaporin water channels and brain edema. Mt Sinai J Med. 2002;69:242–8. [PubMed] [Google Scholar]
- 23. Guo Q, Sayeed I, Baronne LM, Hoffman SW, Guennoun R, Stein DG. Progesterone administration modulates AQP4 expression and edema after traumatic brain injury in male rats. Exp Neurol. 2006;198:469–78. [DOI] [PubMed] [Google Scholar]
- 24. Ding JY, Kreipke CW, Speirs SL, Schafer P, Schafer S, Rafols JA. Hypoxia-inducible factor-1 alpha signaling in aquaporin upregulation after traumatic brain injury. Neurosci Lett. 2009;453:68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Griesdale DE, Honey CR. Aquaporins and brain edema. Surg Neurol. 2004;l61:418–21. [DOI] [PubMed] [Google Scholar]
- 26. Marmarou A. A review of progress in understanding the pathophysiology and treatment of brain edema. Neurosurg Focus. 2007;22:E1. [DOI] [PubMed] [Google Scholar]
- 27. Neri M, Frati A, Turillazzi E, Cantatore S, Cipolloni L, Di Paolo M, et al. . Immunohistochemical evaluation of aquaporin-4 and its correlation with CD68, IBA-1, HIF-1α, GFAP, and CD15 expressions in fatal traumatic brain injury. Int J Mol Sci. 2018;19:3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cash A, Theus MH. Mechanisms of blood–brain barrier dysfunction in traumatic brain injury. Int J Mol Sci. 2020;21:3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Peters J, Daum I, Gizewski E, Forsting M, Suchan B. Associations evoked during memory encoding recruit the context-network. Hippocampus. 2009;19:141–51. [DOI] [PubMed] [Google Scholar]
- 30. Banerjee S, Neveu P, Kosik KS. A coordinated local translational control point at the synapse involving relief from silencing and MOV10 degradation. Neuron. 2009;64:871. [DOI] [PubMed] [Google Scholar]
- 31. Brittain JM, Chen L, Wilson SM, Brustovetsky T, Gao X, Ashpole NM, et al. . Neuroprotection against traumatic brain injury by a peptide derived from the collapse in response mediator protein 2 (CRMP2). J Biol Chem. 2011;286:37778–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.