

Abstract
Influenza A virus (IAV) and SARS‐CoV‐2 (COVID‐19) cause pandemic infections where cytokine storm syndrome and lung inflammation lead to high mortality. Given the high social and economic cost of respiratory viruses, there is an urgent need to understand how the airways defend against virus infection. Here we use mice lacking the WD and linker domains of ATG16L1 to demonstrate that ATG16L1‐dependent targeting of LC3 to single‐membrane, non‐autophagosome compartments – referred to as non‐canonical autophagy – protects mice from lethal IAV infection. Mice with systemic loss of non‐canonical autophagy are exquisitely sensitive to low‐pathogenicity IAV where extensive viral replication throughout the lungs, coupled with cytokine amplification mediated by plasmacytoid dendritic cells, leads to fulminant pneumonia, lung inflammation and high mortality. IAV was controlled within epithelial barriers where non‐canonical autophagy reduced IAV fusion with endosomes and activation of interferon signalling. Conditional mouse models and ex vivo analysis showed that protection against IAV infection of lung was independent of phagocytes and other leucocytes. This establishes non‐canonical autophagy in airway epithelial cells as a novel innate defence that restricts IAV infection and lethal inflammation at respiratory surfaces.
Keywords: ATG16L1 WD Domain, cytokine storm, influenza, intrinsic defence, non‐canonical autophagy
Subject Categories: Autophagy & Cell Death; Microbiology, Virology & Host Pathogen Interaction
Deletion of WD domain mediating non‐canonical functions of ATG16L1 sensitizes mice to cytokine storm and lethal pneumonia upon influenza infection.
Introduction
Influenza A virus (IAV) is a respiratory pathogen of major global public health concern (Yamayoshi & Kawaoka, 2019). As with SARS‐CoV‐2, animal reservoirs of IAV can contribute to zoonotic infection leading to pandemics with a high incidence of viral pneumonia, morbidity and mortality. IAV infects airway and alveolar epithelium and damage results from a combination of the intrinsic pathogenicity of individual virus strains as well as the strength and timing of the host innate/inflammatory responses. Optimal cytokine levels protect from IAV replication and disease but excessive cytokine production and inflammation worsens the severity of lung injury (Davidson et al, 2014; Iwasaki & Pillai, 2014; Teijaro et al, 2014; Herold et al, 2015; Ramos & Fernandez‐Sesma, 2015). Even though infection of the lower respiratory tract can result in inflammation, flooding of alveolar spaces, acute respiratory distress syndrome and respiratory failure, the factors that control IAV replication at epithelial surfaces and limit lethal lung inflammation remain largely unknown.
The transport of viruses to lysosomes for degradation provides an important barrier against infection. Transport to lysosomes can be enhanced by non‐canonical autophagy pathways which conjugate autophagy marker protein LC3 to endo‐lysosome compartments to increase lysosome fusion. In phagocytes, LC3‐associated phagocytosis (LAP) conjugates LC3 to phagosomes and enhances phagosome maturation (Sanjuan et al, 2007; Delgado et al, 2008; Martinez et al, 2015; Lamprinaki et al, 2017; Fletcher et al, 2018). In non‐phagocytic cells, LC3 is conjugated to endo‐lysosome compartments during the uptake of particulate material such as apoptotic cells and aggregated β‐amyloid, and following membrane damage during pathogen entry or osmotic imbalance induced by lysosomotropic drugs (Florey et al, 2011, 2015; Roberts et al, 2013; Tan et al, 2018; Heckmann et al, 2019). It is known from in vitro studies that LC3 can be recruited to endo‐lysosome compartments during the uptake of pathogens, but the roles played by non‐canonical autophagy during viral infection in vivo are largely unknown.
A role for non‐canonical autophagy in host defence has been implied from in vitro studies of LAP in phagocytes infected with free living microbes with a tropism for macrophages such as bacteria (Listeria monocytogenes (Gluschko et al, 2018), Legionella dumoffii (Hubber et al, 2017)), protozoa (Leishmania major) and fungi (Aspergillus fumigatus (Akoumianaki et al, 2016, Kyrmizi et al, 2018, Matte et al, 2016)). It is also known that IAV induces non‐canonical autophagy during infection of cells in culture (Fletcher et al, 2018); however, the role played by non‐canonical autophagy in controlling IAV infection and lung inflammation in vivo is currently unknown. It is not known, for example, whether non‐canonical autophagy is important in the control of IAV infection by epithelial cells at sites of infection or whether it plays a predominant role within phagocytes and antigen‐presenting cells during development of an immune response. Herein we use mice with specific loss of non‐canonical autophagy to determine the role played by non‐canonical autophagy in host defence against IAV infection of the respiratory tract. The mice (δWD) lack the WD and linker domains of ATG16L1 that are required for conjugation of LC3 to endo‐lysosome membranes (Rai et al, 2019) but express the N‐terminal ATG5‐binding domain and the coiled coil domain (CCD) and linker residues up to glutamate at position 230 (E230) of ATG16L1 that are required for WIPI2 binding and autophagy (Dooley et al, 2014). Importantly, the δWD mice grow normally and maintain tissue homeostasis (Rai et al, 2019), and unlike mice with LysMcre‐mediated deletion of autophagy genes from myeloid cells (Lu et al, 2016), or disruption of ATG16L1 through loss of the CCD (Saitoh et al, 2008), the δWD mice do not have a pro‐inflammatory phenotype.
We show that loss of non‐canonical autophagy from all tissues renders mice highly sensitive to low‐pathogenicity murine‐adapted IAV (A/X‐31) leading to extensive viral replication throughout the lungs, cytokine dysregulation and high mortality typically seen after infection with highly pathogenic IAV. Conditional mouse models and ex vivo analysis showed that protection against IAV infection of lung was independent of phagocytes and other leucocytes, and that infection was controlled within epithelial barriers where non‐canonical autophagy slowed fusion of IAV with endosomes and reduced interferon signalling. This establishes non‐canonical autophagy pathways in airway epithelial cells as a novel innate defence mechanism that restricts IAV infection at respiratory surfaces.
Results
Mice with systemic loss of the WD and linker domains of ATG16L1 are highly sensitive to IAV infection
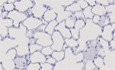
Panels A and B of Fig EV1 show the rationale for removing the WD and linker domains from ATG16L1 to generate mice (δWD) with a specific loss of non‐canonical autophagy (E230 mice described in Rai et al, 2019). The consequences of loss of the WD and linker domains of ATG16L1 on conventional autophagy and non‐canonical autophagy were confirmed using cell lines taken from controls and δWD mice. Figure 1A shows that mouse embryo fibroblasts (MEFs) from littermate control mice recruited LC3 to small puncta indicative of autophagosomes when they were starved in HBSS to induce conventional autophagy and recruited LC3 to large endo‐lysosomal vacuoles when non‐canonical autophagy was induced by chloroquine or monensin. HBSS induced LC3 puncta in MEFs from δWD mice, but the MEFs were unable to recruit LC3 to large vacuoles induced by chloroquine or monensin. LC3 recruitment was quantified by imaging LC3‐positive puncta and vacuoles. The graphs (Fig 1B) show that numbers of autophagosomes induced by HBSS were the same in each cell type but MEFs from δWD were unable to recruit LC3 to large vacuoles when incubated with chloroquine or monensin. The results indicated a selective loss of non‐canonical autophagy. LC3 recruitment was quantified by Western blot to detect LC3II, the lipidated form of LC3 that binds membranes. Figure 1C shows that control MEFs expressed the full‐length α and β forms of ATG16L1 at 70 kDa and generated increased levels of LC3II following starvation in HBSS or incubation with monensin or chloroquine. MEFs from δWD mice expressed a truncated ATG16L1 at 30 kDa (Fig 1C). The LC3II signal in δWD cells increased after starvation to levels that were similar to starvation controls. LC3II also increased in δWD MEFs incubated with monensin and chloroquine. Monensin and chloroquine raise lysosomal pH, and the consequent inhibition of proteolysis slows the efflux of amino acids from the lysosome. This in turn inhibits the Ragulator‐Rag:MTORC1 complex and induces autophagy. Previous work (Fletcher et al, 2018) has shown that monensin activates conventional autophagy and at the same time raised lysosomal pH slows fusion of autophagosomes with lysosomes. This explains the accumulation of small LC3 puncta and increased LC3II observed in δWD cells incubated with monensin or chloroquine. Quantification of Western blots (Fig 1D) showed that there was no significant difference between LC3II signals in control and δWD MEFs after starvation suggesting that autophagy was equally active in the two cell types. The LC3II signal in δWD MEFs incubated with monensin or chloroquine was however lower than controls but not significantly different. Studies in phagocytic cells have shown that non‐canonical autophagy/LAP is downstream of Rubicon and PHOX:NOX2 ROS signalling (Martinez et al, 2015). Addition of diphenyliodonium (DPI), an inhibitor of NOX2, to cells incubated with monensin or chloroquine inhibited recruitment of LC3 to large vacuoles (Figure EV1C) indicating that WD domain‐dependent non‐canonical autophagy is also downstream of ROS signalling in non‐phagocytic cells. Microscopy and line profile analysis were used to determine whether recruitment of LC3 to phagosomes in bone marrow‐derived macrophages (BMDM) induced by Zymosan also required the WD domain of ATG16L1. Figure 1E‐H shows that LC3 was recruited to phagosomes in control BMDM, but was not recruited to phagosomes in BMDM from δWD mice. Taken together, the results show that the δWD mice have specific loss of non‐canonical autophagy in both myeloid and non‐myeloid cells.
Figure EV1. Deletion of the WD domain of ATG16L1 causes selective loss of non‐canonical autophagy.
- The WD domain is not required for canonical/conventional autophagy. Conventional autophagy is activated by the initiation complex containing ULK1, FIP200 and the PI3 kinase VPS34 (i) which responds to starvation. VPS34 phosphorylates lipids in membranes to generate sites for WIPI2 binding (ii). WIPI2 binds the coiled coil domain (CCD) and short stretch of linker of ATG16L1 (ii) leading to recruitment of ATG16L1 and the LC3 conjugation machinery (ATG16l1:ATG5‐ATG12, ATG7, ATG3) (iii). This results in conjugation of LC3 to PE in the surface of the autophagosome (iv) to promote expansion and fusion with lysosomes.
- The WD domain is required for non‐canonical autophagy. During non‐canonical autophagy, signalling from the lumen of the endosome or phagosome recruits UVRAG, Rubicon and VPS34 (i). VPS34 phosphorylates lipids in endo‐lysosome membranes to generate sites for binding the PHOX:NOX2 complex (ii) which is stabilised by Rubicon to generate reactive oxygen species (ROS). ROS induces binding of the WD domain of ATG16L1 to endo‐lysosome membranes (iii) leading to recruitment of the LC3 conjugation machinery (ATG16l1:ATG5‐ATG12, ATG7, ATG3) (iii) and conjugation of LC3 to PE (iv).
- ROS is required for recruitment of LC3 to vacuoles in non‐phagocytic cells. MEFs from dWD mice or littermate controls were incubated with chloroquine or monensin in the presence or absence of DPI, an inhibitor of NOX2 as indicated. Fixed and permeabilised cells were immunostained for LC3. Recruitment of LC3 to vacuoles was determined by fluorescence microscopy and expressed as numbers of LC3‐positive vacuoles per cell (n = 10). Recruitment of LC3 required the WD domain of ATG16L1 and was inhibited by DPI. Data from 10 cells are shown, and bars represent the mean ± SD and were compared by Student’s t‐test (****P < 0.001).
Figure 1. Deletion of the WD and linker domains of ATG16L1 leads to selective loss of non‐canonical autophagy.

-
AMEFs from δWD and littermate control mice were incubated with HBSS, chloroquine or monensin for 2 h as indicated. Cells were fixed and permeabilised and stained for LC3. Arrows indicate LC3 vacuoles where a ring of LC3 signal surrounds vacuoles ranging between 2 and 8 μm diameter.
-
BNumbers of fluorescent LC3 puncta and LC3 vacuoles in each cell were quantified by fluorescence microscopy. LC3 puncta were identified using spot function software to locate puncta ranging from 0.5 to 1.0 μm diameter. LC3 vacuoles were identified by eye as rings of fluorescence ranging between 2 and 8 μm diameter. Data from 10 cells are shown, and bars represent the mean ± SD and were compared by Student’s t‐test (*P < 0.05, ****P < 0.0001).
-
C–EMEFs from δWD and littermate control mice were incubated with HBSS, chloroquine or monensin for 2 h as indicated and cell lysates analysed by Western blot for ATG16L1, δWD and LC3 as indicated. Control MEFs express α and β isoforms of ATG16L1 at 70 kDa, and MEFs from δWD mice express a truncated ATG16L1 at 30kDa. (D) shows the level of conversion of LC3 to LC3II estimated by densitometry from a mean (±SD) of three replicate blots. (E) shows fluorescence images of phagosomes following engulfment of Zymosan (red) by bone marrow‐derived macrophages (BMDM) from control and δWD mice. White line indicates track used for line profile analysis to compare the LC3 signal on the limiting membrane of the phagosome with the centre.
-
FShows examples of line profile analysis
-
G, H(G) shows the percentage of LC3‐positive phagosomes per cell, and (H) shows line profile analysis of data from 10 cells, and bars represent the mean ± SD and were compared by Student’s t‐test (****P < 0.0001).
IAV enters airway and lung epithelial cells by endocytosis, and in tissue culture IAV induces non‐canonical autophagy leading to ATG16L1‐WD domain‐dependent conjugation of LC3 to the plasma membrane and peri‐nuclear structures (Fletcher et al, 2018). To test whether non‐canonical autophagy has a host defence function in vivo, δWD mice were infected with IAV. We used a low‐pathogenicity murine‐adapted IAV (A/X31) that does not normally lead to extensive viral replication throughout the lungs, or cause the cytokine storm syndrome and death typically seen after infection with highly pathogenic viral strains. The results (Fig 2) showed that δWD mice became moribund and showed severe signs of clinical illness (rapid breathing, piloerection). They also displayed rapid weight loss compared to littermate controls (Fig 2A) and had increased mortality with survivors recovering more slowly from infection (Fig 2B). Virus titres in the lungs of both mice increased with time (Fig 2C) and increased weight loss in δWD mice was associated with an approx. log increase in lung virus titre at 5 days post‐infection (d.p.i). Furthermore, histopathology and immunohistochemistry (IH) analysis of lungs from δWD mice showed fulminant viral pneumonia with large numbers of IAV‐positive cells (Fig 2D). Lungs from control and δWD mice did not show signs of inflammation before infection (Fig EV2).
Figure 2. Systemic loss of non‐canonical autophagy increases susceptibility to IAV infection.

-
A–DLittermate control and δWD mice were challenged intranasally with IAV strain X31 (103 pfu). (A) Mice were monitored for weight loss at indicated time points. (n = 8). Data represent the mean value ± SEM. Comparisons were made using a repeated‐measures two‐way ANOVA (Bonferroni post‐test (**P < 0.01, ***P < 0.001)). (B) Survival was assessed at indicated time points (n = 15). Comparisons were made using log‐rank (Mantel–Cox) test ***P < 0.001. (C) IAV titre in lungs was determined by plaque assay at 5 d.p.i. (n = 6). Data for individual animals are shown, and bars represent the mean ± SD. Mann–Whitney U‐test was used to determine significance (**P < 0.01. (D) The presence of IAV antigen was assessed by IH at 7 d.p.i. (representative images from n = 6).
Figure EV2. Histological analysis of littermate control and δWD mice.
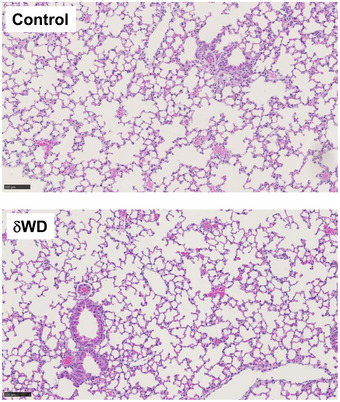
Formalin‐fixed, paraffin‐embedded lung tissue from uninfected littermate control and δWD mice was stained with H&E. Both groups of mice were similar, displaying no histological abnormalities (representative images from n = 6). Scale bars represent 100 µM.
Non‐canonical autophagy controls lung inflammation after IAV infection
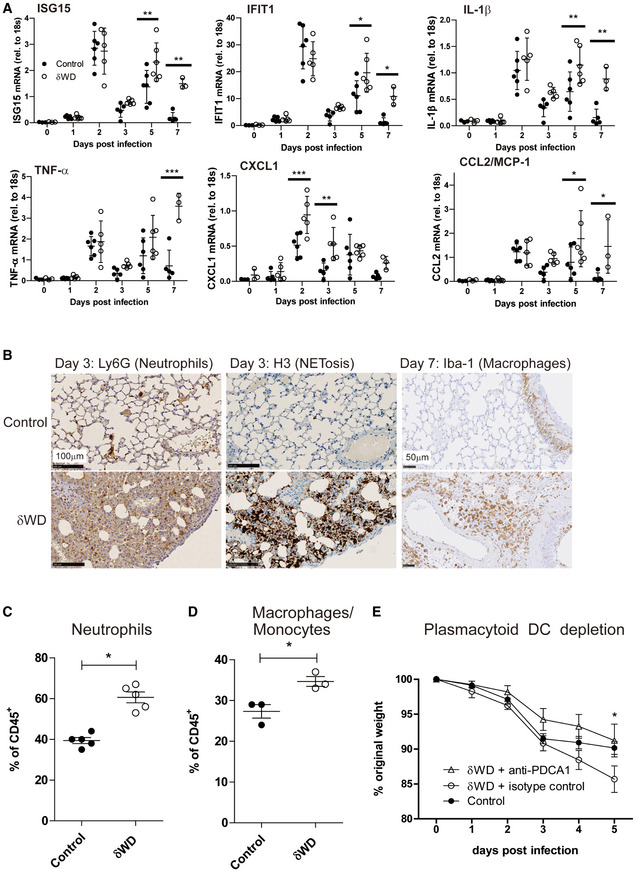
Innate protection against IAV is provided by type 1 (α, β) and III (λ) interferon (IFN) with severe IAV infection causing excessive airway inflammation and pulmonary changes attributable in part to IFNαβ and TNF‐α (Szretter et al, 2007; Davidson et al, 2014). Measurement of cytokine expression at 2 d.p.i showed that IAV induced a transient increase in transcripts for interferon‐stimulated genes (ISGs), ISG15 and IFIT1 (Iwasaki & Pillai, 2014) and pro‐inflammatory cytokines (IL‐1β, TNF‐α and CCL2 [MCP‐1]) in the lungs of both control and δWD mice (Fig 3A). This increase in cytokine expression was resolved by 3 d.p.i. before a second wave of increased cytokine expression at 5 d.p.i. This second wave of cytokine expression was resolved by 7 d.p.i in control mice, but δWD mice showed sustained increases in ISG15, IFIT1, IL‐1β, TNF‐α and CCL2 transcripts, coincident with exacerbated weight loss. At 3 d.p.i, lungs of δWD mice showed increased expression of neutrophil chemotaxis factor CXCL1 mRNA (Fig 3A), coincident with increased neutrophil infiltration of airways and parenchyma, and extensive neutrophil extracellular traps (NETs) as a consequence of neutrophil degeneration as shown by IH (Fig 3B and Appendix Fig S1). Increased neutrophil infiltration of airways in δWD mice at 2 d.p.i. was confirmed and quantified using flow cytometric analysis of bronchoalveolar lavage (BAL; Fig 3C). At 5–7 d.p.i., increased expression of CCL2 mRNA in δWD mice was coincident with extensive macrophage/monocyte infiltration into lung parenchyma observed by IH (Fig 3B and Appendix Fig S2) which was not seen in controls or the lungs of δWD mice before infection with virus (EV2). This increased macrophage/monocyte infiltration in δWD mice was confirmed and quantified using flow cytometric analysis of single‐cell suspensions from lung tissue (Fig 3D). It is known that, in severe IAV infection, a cytokine storm occurs that is amplified by plasmacytoid dendritic cells (pDCs) (Davidson et al, 2014). pDCs detect virus‐infected cells and produce large amounts of cytokines, in particular IFNαβ, that in severe infections can enhance disease. In these cases, depletion of pDCs can decrease morbidity (Davidson et al, 2014). Depletion of pDCs in IAV‐infected δWD mice using anti‐PDCA‐1 led to markedly decreased weight loss as compared with isotype control‐treated mice and that was similar to that seen in littermate controls (Fig 3E). This indicates that excessive cytokine production amplified by pDCs is responsible for the increased morbidity seen in the δWD mice.
Figure 3. Systemic loss of non‐canonical autophagy leads to extensive lung inflammation and damage.
-
A–ELittermate control and δWD mice (n = 5) were challenged with IAV X31 (103 pfu). (A) At the indicated time points, cytokine mRNA transcripts in lung tissue (n = 5) were evaluated by qPCR. Data for individual animals are shown, and bars represent the mean ± SD and were compared by 2‐way ANOVA with Bonferroni post‐tests (*P < 0.05, **P < 0.01, ***P < 0.001). (B) Representative lung sections from animals (n = 6) taken at 3 d.p.i. were stained by IH for neutrophils (Ly6G) or neutrophil extracellular traps (NET; anti‐H3). Sections at 7 d.p.i. were stained for macrophages (Iba‐1). Further micrographs are shown in Appendix Figs S1 and S2. (C) BAL (n = 5) was taken at 2 d.p.i and evaluated by flow cytometry, with pre‐gating on CD45+. The percentage of neutrophils (CD11b+, Ly6G+) cells is shown ± SEM and was compared using Mann–Whitney U‐test (*P < 0.05). (D) Single‐cell suspensions were prepared from lungs taken at 5 d.p.i and evaluated by flow cytometry, with pre‐gating on CD45+. The percentage of macrophage/monocytes (CD11b+, F4/80+) cells is shown ± SEM and was compared using Mann–Whitney U‐test (*P < 0.05). (E) δWD mice were treated with either anti‐PCDA‐1 (to deplete plasmacytoid DC) or an isotype‐matched control. Littermate control mice were used as comparator. Weight loss was measured at the indicated days p.i. (n = 5). Comparisons were made using a repeated‐measures two‐way ANOVA (Bonferroni post‐test, *P < 0.05).
Thus, mice with systemic loss of non‐canonical autophagy failed to control lung virus replication and inflammation, leading to increased cytokine production, morbidity and mortality.
Systemic loss of the WD and linker domains of ATG16L1 does not lead to gross changes in inflammatory threshold or immunological homeostasis
Macrophages cultured from embryonic livers from mice with loss of the coiled coil domain of ATG16L1 are unable to activate canonical autophagy and secrete high levels of IL1‐β (Saitoh et al, 2008). Similarly, LysMcre‐mediated deletion of genes essential for conventional autophagy (e.g. Atg5, Atg7, Atg14, Atg16L1, FIP200) in mice leads to raised pro‐inflammatory cytokine expression in the lung. This has been reported to increase resistance to IAV infection (Lu et al, 2016), and this was also observed in mice used in our study (Appendix Fig S3) where LysMcre‐mediated loss of Atg16L1 resulted in weight loss similar to controls (Appendix Fig S3A) and reduced overall virus titre (Appendix Fig S3B). This led us to test the possibility that the δWD mutation to ATG16L1 could also increase IL‐1β secretion and cause the increased inflammation observed during IAV infection. This was tested by incubating BMDM with LPS and purine receptor agonist, BzATP (Appendix Fig S4A), or by challenging mice with LPS (Appendix Fig S4B). Mice with a complete loss of ATG16L1 in myeloid cells (Atg16L1fl/fl‐lysMcre) showed threefold increases in IL‐1β in serum and increased secretion IL‐1β from BMDM in vitro. In contrast IL‐1β secretion in δWD mice did not differ significantly from littermate controls (Appendix Fig S4A and B). This was consistent with lack of elevated cytokines in lungs prior to infection (see day 0 in Fig 3A), and our previous work shows that serum levels of IL‐1β, IL‐12p70, IL‐13 and TNF‐α in δWD mice are the same as in littermate controls at 8‐12 and 20‐24 weeks (Rai et al, 2019). The exaggerated inflammatory response to IAV in δWD mice did not therefore result from a raised pro‐inflammatory threshold or dysregulated IL‐1β responses in the lung. Also, the frequencies of T cells, B cells and macrophages were similar in δWD mice to littermate controls (Appendix Fig S5). These data suggest that the exaggerated responses of δWD mice to IAV do not occur because the mice have a raised inflammatory threshold or abnormal immunological homeostasis.
Non‐canonical autophagy limits IAV infection independently of phagocytic cells
The link between non‐canonical autophagy, TLR signalling, NADPH oxidase activation and ROS production (Sanjuan et al, 2007; Delgado et al, 2008; Martinez et al, 2015) provides phagocytes with a powerful mechanism to limit infections in vivo. To test whether wild‐type bone marrow‐derived cells could protect susceptible δWD mice from lethal IAV infection, we generated radiation chimaeras (Fig EV3). When challenged with IAV, δWD mice reconstituted with either wild‐type or δWD bone marrow remained highly sensitive to IAV (Fig 4A and B) with body weight reduced by up to 25% and decreased survival by 5 d.p.i. As seen for δWD mice, weight loss was associated with a 10‐fold increase in lung viral titre (Fig 4C), fulminant pneumonia and inflammatory infiltration into the lung (Fig 4D). This increased susceptibility to IAV was not observed for control mice reconstituted with wild‐type bone marrow, showing that non‐canonical autophagy pathways in phagocytes and other leucocytes from control mice were not able to protect δWD mice against lethal IAV infection. In a reciprocal experiment (Fig 5), mice expressing Cre recombinase in myeloid cells (LysMcre) were used to generate mice (called δWDphag), where the truncated Atg16L1δWD gene was restricted to phagocytic cells (Appendix Fig S6). In these mice, non‐canonical autophagy was absent in cultured phagocytes (BMDM) but it was present in skin fibroblasts (Appendix Fig S6E). After infection with IAV, δWDphag mice showed comparable weight loss and virus titres to those seen in littermate control mice (Fig 5A and B). Likewise, the raised IL‐1β levels (Fig 5C) and profuse macrophage and neutrophil lung infiltration observed in δWD mice were absent (Fig EV4) and similar to littermate controls. The ability of the WD and linker domains of ATG16L1 to protect epithelial cells against IAV infection was tested ex vivo to further exclude any contribution from recruited leucocytes. Virus titres in precision‐cut lung slices (Fig 5D) increased over 3 days and similar to the kinetics seen in vivo titres from δWD mice rose to 10‐fold greater than controls. Thus, the sensitivity of δWD mice to IAV was not due to the loss of non‐canonical autophagy from myeloid cells, making it likely that non‐canonical autophagy mediated by the WD and linker domains of ATG16L1 protects against lethal IAV infection in non‐myeloid tissue.
Figure EV3. Confirmation of bone marrow transplant radiation chimaerism.

- Strategy for making bone marrow chimaeras.
- Chimaerism was confirmed 12 weeks post‐transplant in spleen cells by flow cytometric analysis of congenic markers on leucocytes (CD45.1, CD45.2). Flow plot shows representative plot from one C57BL/6 WT (CD45.1) bone marrow → δWD (CD45.2) recipient chimaera and one C57BL/6 WT (CD45.1) bone marrow → littermate control (CD45.2) recipient chimaera. All animals were > 95% chimaeric.
Figure 4. Bone marrow‐derived lymphoid tissue cannot reverse sensitivity to IAV infection.
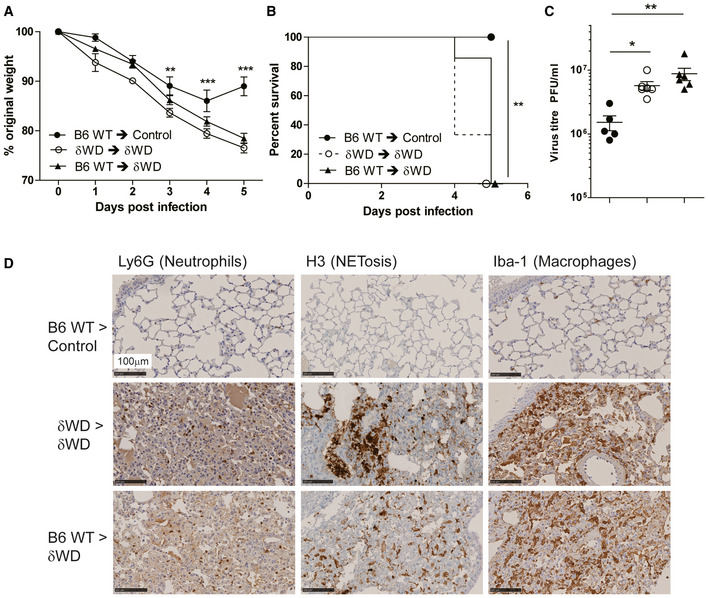
-
A–DBone marrow from wild‐type (Atg16L1+/+) was used to reconstitute irradiated littermate control mice (B6 WT → control [●]) or δWD mice (B6 WT → δWD [○]). Bone marrow from δWD mice was used to reconstitute irradiated δWD mice (δWD → δWD [▲]). After 12 weeks, mice (n = 5 per group) were challenged with IAV X31 (103 pfu). (A) Mice were monitored for weight loss at indicated time points. Data represent the mean value ± SEM. Comparisons were made using a repeated‐measures two‐way ANOVA (Bonferroni post‐test, **P < 0.01, ***P < 0.001). (B) Survival was assessed at indicated time points. Comparisons were made using log‐rank (Mantel–Cox) test **P < 0.01). (C) IAV titre in lungs was determined by plaque assay at 5 d.p.i. (n = 6). Data for individual animals are shown using symbols described in (A) and (B), and bars represent the mean ± SD. A one‐way ANOVA with Tukey’s post hoc analysis was used to determine significance (*P < 0.05, **P < 0.01). (D) Lungs taken at 5 d.p.i. were analysed for neutrophils (Ly6G), neutrophil extracellular traps (NET; anti‐H3) and macrophages (Iba‐1).
Figure 5. Loss of non‐canonical autophagy from phagocytes does not increase sensitivity to IAV infection.

-
A–CδWDphag mice lack non‐canonical autophagy in myeloid (LysMcre) cells (for construction see Appendix Fig S6A). Offspring negative for LysMcre were used as littermate controls. Mice (n = 6 per group) were challenged intranasally with IAV X31 (103 pfu). (A) Mice were monitored for weight loss at indicated time points. Data represent the mean value ± SEM. Comparisons were made using a repeated‐measures two‐way ANOVA (Bonferroni post‐test). (B) IAV titre in lungs was determined by plaque assay at 5 d.p.i. (n = 6). Data for individual animals are shown, and bars represent the mean ± SD. Mann–Whitney U‐test was used to determine significance. (C) IL‐1β mRNA transcripts in lung at 5 d.p.i. were determined by qPCR. Mann–Whitney U‐test was used to determine significance (**P < 0.01).
-
DPrecision‐cut lung slices from control and δWD mice were infected with IAV. Virus titres were determined at indicated time points. Comparisons were made using two‐way ANOVA with Bonferroni post‐tests (**P < 0.01).
Figure EV4. Mice deficient in non‐canonical autophagy/LAP in phagocytes control IAV infection and do not show increased lung inflammation.
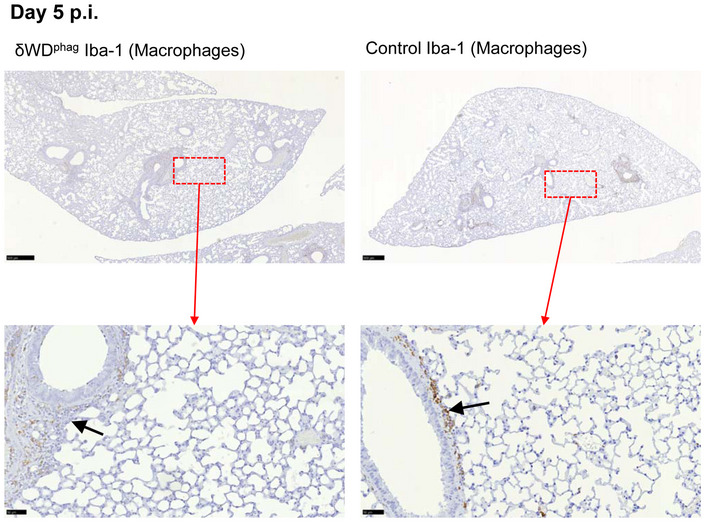
δWDphag mice that are deficient in non‐canonical autophagy in phagocytes and littermate control mice were infected i.n. with 103 pfu IAV X31. Lung tissues were harvested at 5 d p.i. Macrophages were detected by IH using anti‐Iba‐1, visualised with DAB and counterstained with haematoxylin. Scale bars represent 500 µm (upper panels) and 50 µm (lower panels). Micrographs of representative areas from lungs of six mice are shown. Both δWDphag and control mice show little inflammatory response in the parenchyma and mild, macrophage‐rich peri‐bronchiolar infiltration (black arrows).
Non‐canonical autophagy slows IAV fusion with endosomes and reduces interferon signalling
IAV enters cells by receptor‐mediated endocytosis where acidification of late endosomes results in fusion with the endosomal membrane and delivery of viral ribonuclear proteins (RNPs) into the cytoplasm (Wharton et al, 1994; Skehel & Wiley, 2000). RNPs are then imported into the nucleus for genome replication (Boulo et al, 2007). Figure 6A shows that the generation of infectious virus was greater in δWD MEFs compared to control. IAV binding was analysed using fluorescent virus, and Fig 6B shows that binding was similar between control and δWD MEFs suggesting that subsequent steps of endocytosis and viral fusion may be increased in δWD MEFs. The possibility that the WD domain of ATG16L1 affected replication of IAV independently of endocytosis was tested using an acid bypass assay. IAV was bound to cells at 4° and warmed to 37°C for 2 min at pH 6.8 (control) or pH 5 to induce direct fusion with the plasma membrane. When early stages of replication were assessed by staining for nuclear protein, there was no difference between δWD MEFs and control (Fig 6C). This made it unlikely that the WD domain has a direct role in facilitating IAV replication. The effect of non‐canonical autophagy on IAV entry was tested using fluorescence de‐quenching assay where the envelope of purified IAV was labelled with green (DiOC18) and red (R18) lipophilic dyes. Individual fusion events in cells estimated by automated confocal microscopy (Fig 6D) show that the number of fusion events per cell was increased after 60 min at 37°C in δWD MEFs compared to control. Similarly, FACS analysis (see also Fig EV5) of the percentage of cells with de‐quenched signal showed greater fusion in δWD MEFs (60%) compared to controls (40%) at 30 min and this increased at 60 min (73% versus 56% for controls; Fig 6E), as did mean fluorescence intensity (Fig 6F). Endocytosed viruses were also estimated by automated confocal microscopy (Fig 6 G and H) of permeabilised cells and again there was a significant increase in endocytosis of IAV in δWD MEFs at 30 min. Taken together, the results showed that the WD domain of ATG16L1 slowed fusion of IAV with endosome membranes. The recognition of viral RNA by interferon sensors following delivery of RNPs into the cytoplasm was used as a second assay for IAV entry. MEFs from δWD mice showed between three‐ and fivefold increases in expression of IFN responsive genes, ISG15 and IFIT1 (Fig 6I and J), and this was also observed in the lung in vivo (Fig 3A). Taken together, the results demonstrate for the first time that the WD and linker domains of ATG16L1 allow non‐canonical autophagy to provide a novel innate defence mechanism against lethal IAV infection within the epithelial barrier in vivo.
Figure 6. Non‐canonical autophagy reduces virus replication, endosome fusion and interferon signalling.
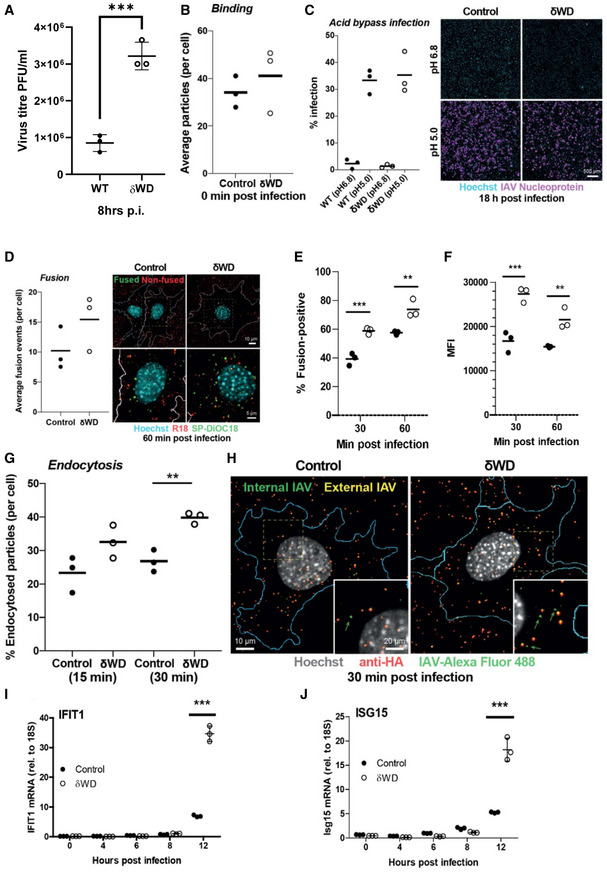
-
AThe WD domain of ATG16L1 reduces IAV replication in MEFs. Cells were infected with IAV, and virus titres were estimated at 8 h post‐infection by plaque assay. Graphs show three individual experiments with a bar (± SD) at the mean and were compared using two‐way ANOVA with Bonferroni post‐tests ***P < 0.001).
-
BIAV binding is similar between control and δWD MEFs. Cells were bound with IAV X31 labelled with AF488 on ice for 1 h and fixed immediately. The cells were stained with anti‐HA (H3SKE, red) without permeabilisation to visualise extracellular IAV particles and counterstained with Hoechst (nuclei) and WGA‐AF647 to label the cell membrane. Cells were imaged by automated confocal microscopy using a 40x objective, and the maximum intensity projection images were analysed to quantify the number of bound IAV particles per cell. The results and means (horizontal line) of n = 3 independent experiments (50‐100 cells per experiment) are shown.
-
CIAV infection following acid bypass is comparable between control and δWD MEFs. IAV was bound on ice for 1 h and warmed at either neutral pH (6.8) or low pH (pH 5.0) for 2 min, followed by incubation in STOP medium containing NH4Cl for 18 h. Cells were fixed and stained for NP and Hoechst, imaged by automated confocal microscopy and maximum intensity projection images analysed to quantify viral infectivity. The results from N = 3 independent experiments (5,000‐10,000 cells quantified per experiment), and the means are shown. Scale bar; 500 µm.
-
DIAV fusion with endosomes is increased in δWD MEFs. Cells were infected with dual‐labelled (R18/SP‐DiOC18) IAV for 1 h, fixed and counterstained with Hoechst (nuclei blue) and WGA‐AF647 (cell perimeter in white). Cells were imaged by automated confocal microscopy, and the number of fusion events per cell (represented by number of SP‐DiOC18 puncta) was quantified. Fused and non‐fused viral particles are shown as green and red spots, respectively. The results and means (horizontal lines) of n = 3 independent experiments (30‐60 cells per experiment) are shown. Representative cells overlayed with the cell boundary segmented from the WGA staining are shown on the right.
-
E, FNumbers of fusion‐positive cells are increased for δWD MEFs. MEFs from δWD (white circles) or littermate control mice (black circles) were incubated with dual‐labelled (SP‐DiOC18/R18) IAV at 4°C for 45 min and warmed to 37°C for 30 ad 60 min. Cells were harvested by trypsinisation, fixed in PFA and analysed by flow cytometry to determine to determine percentage of cells positive for fusion (E) and median fluorescence intensity (MFI) of de‐quenched SP‐DiOC18 signa (F). Graphs show individual replicates (n = 3) with a horizontal line at the mean and were compared using two‐way ANOVA with Bonferroni post‐tests **P < 0.01, ***P < 0.001). pH‐dependent fusion was assessed by adding bafilomycin A1 to the infection assay (Fig EV5).
-
G, HEndocytosis of IAV increases in δWD MEFs. MEFs from δWD or littermate control mice were incubated with Alexa Fluor 488‐labelled IAV at 4°C for 1hr and warmed to 37°C for 15. Cells were fixed and permeabilised and stained with antibody against HA (red). The blue line indicates the plasma membrane of the cell. Green arrows indicate examples of viruses only labelled with green fluorescence. Double‐labelled virus particles (green‐red) represent non‐endocytosed particles bound at the cell surface, whereas green particles represent endocytosed particles. The results and means of n = 3 independent experiments (50‐100 cells per experiment) are shown. Student’s t‐test; P < 0.01. The dotted square in the top row panels is enlarged in the bottom row. Scale bars; 10 µm (top row) and 5 µm (bottom row).
-
I, JInterferon signalling increases in δWD MEFs. MEFs from δWD or littermate control mice were infected with IAV. At the indicated time points, mRNA transcripts were evaluated by qPCR for IFIT1 (I) and ISG15 (J). Data from 3 replicates are shown, and bars represent the mean ± SD and were compared by two‐way ANOVA with Bonferroni post‐tests (***P < 0.001)
Figure EV5. Non‐canonical autophagy reduces endosome fusion in vitro .

-
AAfter labelling, cells were warmed to 37°C for increasing times, harvested by trypsinisation, fixed in PFA and analysed by flow cytometry. Representative plots (from n = 3) showing de‐quenched SP‐DiOC18. Numbers in the gates show percentage of cells positive for fusion as determined by comparison to uninfected cells. Graphical representation of all data is shown in Fig 6E and F.
-
B–DFluorescent signal in cells infected with SP‐DiOC18‐labelled IAV is pH‐dependent fusion. pH‐dependent fusion was assessed by adding bafilomycin A1 (BAF) to some wells during labelling and then warmed to 37°C for 90 min. (B). Representative plots (n = 3) showing de‐quenched SP‐DiOC18. Numbers in the gates show percentage of cells positive for fusion as determined by comparison to uninfected cells. (C) Percentage of cells positive for fusion. (D) Median fluorescence intensity of de‐quenched SP‐DiOC18 signal. (C) and (D) show individual replicates (n = 3) with a bar at the mean and were compared using Mann–Whitney U‐test (*P < 0.05).
Discussion
Respiratory viruses such as influenza A virus (IAV) and SARS‐CoV‐2 can move from animal reservoirs to create human pandemics with high morbidity and mortality. The danger posed by pandemic spread of respiratory viruses underlines an urgent need to understand how the airways defend against viral infection. In this study, we have analysed the role played by non‐canonical autophagy in defending the respiratory tract against infection by IAV in vivo. Mice with systemic loss of non‐canonical autophagy (δWD) showed profound sensitivity to infection by a low‐pathogenicity murine‐adapted IAV (A/X31) leading to extensive viral replication throughout the lungs, dysregulated cytokine production and fulminant pneumonia leading to high mortality and death usually seen after infection with virulent strains (Belser et al, 2011). These signs mirror the cytokine storms and mortality seen in humans infected with highly pathogenic strains of IAV such as the 1918 “Spanish” Influenza (Belser et al, 2011).
The observation that bone marrow transfers from wild‐type mice were unable to protect δWD mice from IAV suggested that protection against IAV infection in vivo was independent of leucocytes and did not require non‐canonical autophagy in leucocyte populations (e.g. macrophages, dendritic cells, neutrophils, granulocytes, lymphocytes). In a reciprocal experiment, the linker and WD domains of ATG16L1 were deleted specifically from myeloid cells. These mice, which lack non‐canonical autophagy in phagocytic cells (LAP), but maintain non‐canonical autophagy in other tissues, failed to show increased sensitivity to IAV infection. Thus, protection against severe IAV‐associated disease in the respiratory tract of the host relies heavily on non‐canonical autophagy in non‐leucocyte populations.
Activation of non‐canonical autophagy in phagocytic cells leads to LC3‐associated phagocytosis (LAP) where TLR signalling and reactive oxygen species (ROS) recruit LC3 to phagosomes. A lack of involvement of non‐canonical autophagy/LAP in protection against IAV disease in vivo was surprising because the activation of LAP in phagocytic cells such as macrophages, dendritic cells and neutrophils would provide a powerful means of recognising and controlling microbial infection in vivo. In vitro studies show that activation of acid sphingomyelinase by Listeria monocytogenes (Gluschko et al, 2018) and subsequent ROS production by NOX2 recruit LC3 to phagosomes. Similarly, activation of TLR2 and NOX2 by Legionella dumoffii in vitro signals ULK1‐independent translocation of LC3 to single‐membraned vacuoles containing Legionella (Hubber et al, 2017). In both cases, LC3 promotes fusion with lysosomes. The observation that virulence factors such as the GP63 metalloprotease of Leishmania major and melanin of Aspergillus fumigatus prevent recruitment of NOX2 to phagosomes to prevent LAP (Akoumianaki et al, 2016; Matte et al, 2016; Kyrmizi et al, 2018) further suggests that non‐canonical autophagy in phagocytes should provide a defence against infection. One reason for the discrepancy may be that the studies cited above have focused on in vitro experiments using microbes with a tropism for macrophages, rather than in vivo studies where pathogens encounter epithelial barriers.
Intranasal infection of mice with IAV results in rapid infection of principally airway and pulmonary epithelial cells (Akram et al, 2018). The results of in vivo challenge of radiation chimaeras and δWDphag mice strongly suggest that non‐canonical autophagy in the epithelium rather than leucocytes is responsible for restricting IAV infection. This was supported by ex vivo experiments where virus titres and interferon responses were five‐ to 10‐fold greater in precision‐cut lung slices and MEFs from δWD mice. Furthermore, loss of non‐canonical autophagy increased fusion of IAV envelope with endosomes and increased activation of interferon signalling pathways. Both assays suggest that non‐canonical autophagy reduces IAV entry and delivery of viral RNA to the cytoplasm. This would explain reduced interferon signalling, and at the same time the delayed escape of IAV into the cytoplasm would increase the transfer of endocytosed virus to lysosomes for degradation. The precise mechanisms employed by non‐canonical autophagy to reduce virus entry from endosomes are unknown. This may involve recruitment of LC3 to endosomes by TMEM59 (Boada‐Romero et al, 2016) to increase fusion with lysosomes, or by maintaining membrane repair during virus entry, as observed for bacteria such as S. Typhimurium and Listeria monocytogenes (Kreibich et al, 2015; Tan et al, 2018). A p22phox‐NOX2 pathway that recruits LC3 to vacuoles containing S. Typhimurium in epithelial cells (Huang et al, 2009) may also be activated during IAV entry and hamper lethal infection.
δWD mice infected with IAV appeared to be unable to resolve inflammatory responses resulting in sustained expression of pro‐inflammatory cytokines, morbidity and a striking lung changes characterised by profuse migration of neutrophils into the airway at day 3 followed by macrophages on day 7. pDCs detect IAV‐infected cells and produce large amounts of cytokines, in particular IFNαβ, that in severe infections can enhance disease (Davidson et al, 2014). The fact that morbidity in δWD mice could be decreased by depleting pDCs indicates that excessive cytokine production, amplified by pDCs, was a major factor. This is not due to a lack of non‐canonical autophagy/LAP in pDC as bone marrow chimaeras of δWD mice with wild‐type leucocytes have the same phenotype as δWD mice. IAV is recognised by endosomal TLR3 in respiratory epithelial cells and RIG‐I detects virus replicating in the cytosol leading to activation of IRF3 and NFkB with subsequent induction of interferon, ISG and pro‐inflammatory cytokine production (Iwasaki & Pillai, 2014). Increased inflammation may result directly from increased virus in the lungs, but the increased fusion of IAV envelope with endosomes in δWD mice may increase delivery of viral RNA to the cytoplasm resulting in the sustained pro‐inflammatory cytokine signalling. A similar pro‐inflammatory phenotype resulting from decreased trafficking of inflammatory cargoes is observed following disruption of non‐canonical autophagy by LysMcre‐mediated loss of Rubicon from macrophages or microglia (Martinez et al, 2016; Heckmann et al, 2019). Studies also show that the WD domain of ATG16L1 can modulate endocytosis of cytokine receptors (Serramito‐Gómez et al, 2020). Interaction of the WD domain with the IL10 receptor (IL10‐R), for example, promotes formation of endosomes containing IL‐10/IL‐10 receptor complexes leading to an enhanced anti‐inflammatory signalling, that would be lost in δWD mice. Impaired recycling of Toll‐like receptor 4, CD36 and the β‐amyloid receptor TREM2 is also observed in microglia lacking the WD domain of ATG16L1 leading to neuroinflammation (Heckmann et al, 2020). Inhibition of receptor recycling results from slowed return of receptors to the plasma membrane, rather than increased endocytosis (Heckmann et al, 2019). This makes it unlikely that the increased fusion of IAV with endosomes we see in δWD cells results from upregulation of endocytosis following loss of the ATG16L1 WD domain.
We have dissected the roles played by conventional autophagy and non‐canonical autophagy in vivo by removing the linker and WD domain from ATG16L1 to prevent conjugation of LC3 to single‐membraned endo‐lysosome compartments (Rai et al, 2019). Quantitative analysis of conventional autophagy by fluorescence microscopy of LC3 puncta (Fig 1B) and Western blot of LC3II (Fig 1C&D) did not reveal a loss of canonical autophagy in cells from δWD mice compared to controls. Nevertheless, it is not possible to exclude the possibility that removal of the WD and linker domains of ATG16L1 has a minor effect on canonical autophagy that might affect infection “in vivo”. There are however examples where the WD domain of ATG16L1 has roles during infection that are separate from conventional autophagy, and this may be true also for control of IAV. The WD domain of ATG16L1 maintains membrane repair during Listeria infection independently of conventional autophagy (Tan et al, 2018). The Salmonella T3SS effector protein SopF reduces recruitment of LC3 to vacuoles containing S. Typhimurium by inhibiting the interaction between the WD domain of ATG16L1 and the vacuolar ATPase recruited to sites of vacuole damage (Xu et al, 2019). This promotes growth and virulence of S. Typhimurium, but is independent of FIP200, an essential component of conventional autophagy. An alternative approach to studying non‐canonical autophagy “in vivo” has been to target pathways upstream of LC3 conjugation where deletion of Rubicon produces a selective block in LAP (Martinez et al, 2015; Heckmann et al, 2019). Rubicon stabilises the PHOX:NOX2 complex (Yang et al, 2009) allowing reactive oxygen species (ROS) to induce binding of ATG16L1 to endo‐lysosome membranes (Martinez et al, 2015). Mouse models relying on loss of Rubicon show defects in the clearance of bacterial and fungal pathogens and apoptotic cells (Martinez et al, 2015; Martinez et al, 2016), but have not yet been studied in the context of viral infection. Furthermore, disruption of Rubicon leads to upregulation of IL‐1β, IL6 and TNF‐α secretion, and the mice fail to gain weight and develop an autoimmune disease that resembles systemic lupus erythematosus (Martinez et al, 2016; Heckmann et al, 2017). This exaggerated inflammation might make it difficult to predict if any altered responses to infection observed in Rubicon‐/‐ mice, particularly lung inflammation, resulted directly from loss of non‐canonical autophagy, or from upstream changes in cytokine regulation caused by loss of Rubicon.
Several non‐canonical pathways leading to recruitment of LC3 to endo‐lysosomal compartments, rather than phagosomes, are beginning to emerge. Non‐canonical autophagy in microglia facilitates endocytosis of β‐amyloid and TLR receptors to reduce β‐amyloid deposition and inflammation in mouse models of Alzheimer’s disease (Heckmann et al, 2019). This may involve interaction between the WD domain and TMEM59 which is required for β‐amyloid glycosylation (Ullrich et al, 2010). Lysosomotropic drugs, which stimulate direct recruitment of LC3 to endosomes, create pH and osmotic changes that may mimic the consequences of viral infections that perturb endosome membranes or deliver viroporins to endo‐lysosome compartments. It will be interesting to see if the WD and linker domains of ATG16L1 limit infection by other microbes at epithelial barriers in vivo, particularly infection of the respiratory tract by SARS‐CoV‐2. This may be true for picornaviruses where LC3 is recruited to enlarged endosomes during entry of foot‐and‐mouth disease virus (Berryman et al, 2012) and following LC3 accumulation on megaphagosomes in pancreatic acinar cells during coxsackievirus B3 infection (Kemball et al, 2010). In the specific cases of IAV and SARS‐CoV‐2, non‐canonical autophagy at epithelial barriers is likely important for innate control of new pathogenic strains, where acquired immunity from previous infection may be absent or less effective. It will be valuable to assess whether human allelic variants of ATG16L1 confer altered resistance/susceptibility to respiratory infections such as IAV and whether drug‐based manipulation of non‐canonical autophagy can increase resistance at the respiratory epithelial barrier.
Materials and Methods
Cell culture and virus
Influenza virus A/HKx31 (X31, H3N2) was propagated in the allantoic cavity of 9‐day‐old embryonated chicken eggs at 35°C for 72 h. Titres were determined by plaque assay using MDCK cells with an Avicel overlay.
Mice
All experiments were performed in accordance with UK Home Office guidelines and under the UK Animals (Scientific procedures) Act1986.
The generation of δWD mice (Atg16L1 δWD/δWD) has been described previously (Rai et al, 2019) where they are called “E230” mice to distinguish them from E226 mice described in the same paper which lack both canonical and conventional autophagy. Generation of δWDphag and Atg16L1 fl/fl‐LysMCre mice is described in detail in Appendix Fig S7. Comparisons were made using age‐ and sex‐matched littermate control mice for each individual genotype. Generation and breeding of mice was approved by the University of East Anglia Animal Welfare and Ethical Review Body and performed under UK Home Office Project License 70/8232.
Influenza infection studies were performed at the University of Liverpool, approved by the University of Liverpool Animal Welfare and Ethical Review Body and performed under UK Home Office Project License 70/8599. Studies used 2‐ to 3‐month‐old male and female mice that had been back‐crossed to C57BL/6J. Mice were maintained under specific pathogen‐free barrier conditions in individually ventilated cages (Greenline GM500, Tecniplast) at a temperature of 22°C (± 2°C), humidity 55% (± 10%), light/dark cycle 12/12 h (7 am to 7 pm), food CRM(P) and RO or filtered water ad lib. Colonies were screened using the Charles River surveillance plus PRIA health screening profile every 3 months to ensure SPF status.
For IAV infection, animals were randomly assigned into multiple cohorts, anaesthetised lightly by the i.m. route with 150 mg/kg ketamine (Ketavet, Zoetis UK Ltd) and separate cohorts inoculated intranasally with 103 PFU IAV strain X31 in 50 µl sterile PBS. Mice were infected between 9 and 11 AM. Animals were sacrificed at variable time points after infection by cervical dislocation. Tissues were removed immediately for downstream processing. Sample sizes of n = 6 were used as determined using power calculations and previous experience of experimental infection with these viruses. For survival analysis, a humane endpoint was determined using a scoring matrix that included excessive (>20%) weight loss.
To specifically deplete plasmacytoid dendritic cells (pDCs), mice were treated with anti‐PDCA‐1 (Cambridge Bioscience) or IgG2b isotype‐matched control, using a dose of 500 mg per 200 ml via the i.p. route on day 1 of infection with IAV and every 48 h thereafter (Davidson et al, 2014).
Generation and analysis of radiation chimaeras
The general strategy is shown in Fig EV3. Mice were subjected to whole body irradiation with 11 Gy in two doses 4 h apart using a 137Cs source in a rotating closed chamber. Bone marrow was collected from male wild‐type C57BL/6‐Ly5.1 (B6.SJL‐PtprcaPepcb/BoyCrl; Atg16L1+/+) mice that are congenic for the CD45.1 allele or from δWD mice (that are congenic for CD45.2). The C57BL/6 CD45.1 marrows were used to enable confirmation of chimaerism by FACS analysis of bone marrow‐derived cells as littermate control and δWD mice are CD45.2 (Fig EV3B). The femur and tibia of the donor mouse was collected and sterilised for 2 min in 70% ethanol. The ends of the bones were removed, and PBS was used to flush out the bone marrow through a 40 μm cell sieve. Red blood cell lysis was performed using 0.83% ammonium chloride, and the cells were washed twice in PBS and resuspended at a concentration of 107 cells/ml. T‐cell depletion was performed prior to transfusion by using a commercial mouse hematopoietic progenitor cell isolation kit (EasySep, STEMCELL™ Technologies, #19856).
After depletion, 106 donor bone marrow cells were injected into each irradiated mouse by tail vein injection 3 h following irradiation. Mice were then allowed to recover for 12 weeks with daily monitoring of mouse weights and general condition for at least the first two weeks to monitor for any severe radiation sickness or illness due to being immunocompromised.
For chimaerism analysis, approximately 106 spleen cells were analysed by flow cytometry using fluorochrome‐conjugated monoclonal antibodies specific for CD45.1 (clone A20 eBioscience) and CD45.2 (clone 104 eBioscience). As shown in Fig EV3B, in the groups where CD45.1 marrow was transplanted, all mice were > 95% chimaeric.
Flow cytometric analysis of cells
Bronchoalveolar lavage (BAL) fluid was obtained by lavage of mice via the trachea using 1 ml ice‐cold RPMI containing 5% FCS. For lung tissue, single‐cell suspensions were made from minced lung and subjected to collagenase and DNase I digestion and then treated with ACK buffer to remove red blood cells. In both cases, approximately 106 cells were incubated in 100 µl of Fc block (clone 2.4G2, BD Biosciences) diluted in PBS, 2% FCS (PBS‐FCS) for 15 min at 4°C prior to the addition of fluorochrome‐conjugated monoclonal antibodies and incubation for 30 min at 4°C in the dark. Cells were then washed in PBS‐FCS, fixed in 4% paraformaldehyde in PBS for 15 min at 20°C prior to analysis on a MACSQuant Analyzer 10 (Miltenyi Biotec UK). Data were analysed using FlowJo (FlowJo, LLC). Antibodies used included CD45, Ly6G, CD11c, CD11b and F4/80 (all eBioscience). Neutrophil populations in BAL were identified as CD45+, CD11c‐, CD11b+ and Ly6G+. Macrophage/monocyte populations in lung tissue were identified as CD45+, CD11c‐, CD11b+ and F4/80+.
Histology, immunohistochemistry
Tissues were fixed in 4% buffered paraformaldehyde (PFA; pH7.4) for 24 h and routinely paraffin wax embedded. Consecutive sections (3‐5 µm) were either stained with haematoxylin and eosin (HE) or used for immunohistochemistry (IH).
IH was performed to detect influenza antigens and to identify neutrophils and neutrophil extracellular traps (NETs) and macrophages using the horseradish peroxidase (HRP) and the avidin–biotin complex (ABC) method. The following primary antibodies were applied: goat anti‐IAV (Meridian Life Sciences Inc., B65141G), rat anti‐mouse Ly6G (clone 1A8, BioLegend; neutrophil marker), rabbit anti‐Iba‐1 (antigen: AIF1; Wako Chemicals; microglia/macrophage specific marker) and rabbit anti‐histone H3 (citrulline R2 + R8 + R17; Abcam; NET marker). Briefly, after deparaffinization, sections underwent antigen retrieval in citrate buffer (pH 6.0, 20 min at 98°C) followed by blocking of endogenous peroxidase (peroxidase block, S2023, Dako) for 10 min at room temperature (RT). Slides were then incubated with the primary antibodies (diluted in dilution buffer, Dako) for a) Iba‐1 (60 min at RT), followed by a 30 min incubation at room temperature with the secondary antibody (Envision mouse and rabbit, respectively, Dako) in an autostainer (Dako), and b) Ly6G (60 min at RT), followed by rabbit anti‐rat IgG and the ABC kit (both 30 min at RT; Ventana). Staining for histone H3 was undertaken with an autostainer (Discovery XT, Ventana), using citrate buffer, dilution buffer and detection kits provided by the manufacturer. The antibody reaction was visualised with 3,3'‐diaminobenzidine, and sections were counterstained with haematoxylin.
Statistical analysis
Data were analysed using the Prism package (version 5.04 GraphPad Software). P values were set at 95% confidence interval. A repeated‐measures two‐way ANOVA (Bonferroni post‐test) was used for time‐courses of weight loss; two‐way ANOVA (Bonferroni post‐test) was used for other time‐courses; log‐rank (Mantel–Cox) test was used for survival curves; one‐way ANOVA (Tukey’s post hoc) was used to compare three or more groups side‐by‐side; Mann–Whitney U‐test was used to compare two groups. Numbers of replicates are shown in the individual figure legends. All differences not specifically stated to be significant were not significant (P > 0.05). For all figures, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Primary cell culture
Mouse embryonic fibroblasts (MEFs) were generated by serial passage of cells taken from mice at embryonic day 13.5 and cultured in DMEM (Thermo Fisher Scientific, 11570586) with 10% FCS. Bone marrow‐derived macrophages (BMDMs) were generated from femur and tibia flushed with RPMI‐1640 (Sigma, R8758). Macrophages were generated from adherent cells in RPMI‐1640 containing 10% FCS and M‐CSF (Peprotech, 315‐02) (30 ng/ml) for 6 d. Macrophage populations were quantified by FACS using antibodies against CD16/CD32, F4/80 and CD11b (BioLegend, 101320, 123107).
Precision‐cut lung slices
Infection of ex vivo lung slices was used to examine the responses of lungs without any contribution from recruited leucocytes, which could not be present. Mouse lungs were inflated with 2% low melting point agarose in HBSS and then sliced into 300 µm sections using a vibrating microtome. They were then cultured overnight in DMEM/F12 medium (Thermo Fisher 21331020) prior to infection with IAV.
IAV binding and endocytosis assay
IAV X31 was labelled with Alexa Fluor 488 (Thermo Fisher Scientific) as described (Hoffmann et al, 2018). IAV entry assays were performed as previously described (Banerjee et al, 2013; Banerjee et al, 2014; Miyake et al, 2019). Sub‐confluent monolayers of MEFs cultured on optical 96‐well plates (Greiner 655090) in DMEM 10% FBS were incubated with the labelled virus on ice for 1 h in infection medium (DMEM, 50 mM HEPES pH 6.8, 0.2% BSA) and warmed for 0 (for binding experiments) 15 and 30 min at 37°C by addition of warmed infection medium and transfer of the plates to 37°C, followed by fixation with 4% paraformaldehyde in PBS. Fixed cells were blocked for 1 h with 1% BSA in PBS before incubation with a mouse anti‐haemagglutinin monoclonal antibody (H3SKE) for 1h at RT, followed by a 30‐min incubation with a second goat anti‐mouse‐Alexa Fluor 594 antibody (Thermo Fisher Scientific, A11005), together with Hoechst 33342 (Thermo Fisher Scientific) and Wheat Germ agglutinin labelled with Alexa Fluor 647 (WGA‐AF647, Thermo Fisher Scientific, W32466). Image acquisition was performed with a Yokogawa CQ1 spinning disc confocal microscope, using a 40x air objective with an NA of 0.95, in 4‐channel mode. Image analysis was performed using the Cell Path Finder version 3.04.03.02 (Yokogawa Electric Corporation). Briefly, images were subjected to segmentation analysis, wherein overlap between red (H3SKE/goat anti‐mouse‐AF594) and green (X31‐AF488) signals was quantified within the boundaries of a segmented cell outline generated by the same programme, thus allowing the quantification of endocytosed virus per cell. Signals colocalising with the nucleus (Hoechst) were omitted. Double‐labelled virus particles (Green‐Red) represent non‐endocytosed particles bound at the cell surface, whereas green particles represent endocytosed particles. Within cell outlines, the number of green (endocytosed) particles normalised against the total number of particles (surface‐bound and endocytosed, Green‐Red + Green) gives the fraction of endocytosed particles.
IAV endosome fusion assessed in cell populations by FACS
The envelope of purified IAV (0.1 mg protein mL‐1) was labelled using an ethanol solution containing 33 µM 3,3’‐dioctadecyloxacarbocyanine (DIOC18) and 67 µM octadecyl rhodamine B (R18). Aggregated virus was removed by a 0.22 µm filter (Millipore). Sub‐confluent monolayers of MEFs cultured in DMEM (50 mM HEPES, 0.2% BSA) were incubated with the labelled virus at 40°C for 45 min and warmed to 37°C for increasing times. Cells were harvested by trypsinisation and fixed in 4% PFA for 20 min. Cell pellets (2500 rpm, 4 min) were resuspended in 100 µL FACS buffer (1xPBS 1%BSA) and analysed by FACs using a NovoCyte Flow cytometer FlowJo software. pH independent fusion was assessed by adding bafilomycin A1 to the infection assay.
IAV fusion assessed in single cells by microscopy
X31/R18 double‐labelled X31 was prepared as herein described. For the fusion experiment, MEF cells were incubated in binding media for 1h on ice and then warmed for 1h at 37°C by addition of warm binding media and transfer of the plates to 37°C, followed by fixation with 4% paraformaldehyde in PBS. The cells were then labelled with Hoechst and WGA‐AF647 as herein described. Image acquisition was performed with a Yokogawa CQ1 spinning disc confocal microscope, using a 40x air objective with an NA of 0.95, in 4‐channel mode. Image analysis was performed using the Cell Path Finder version 3.04.03.02 (Yokogawa Electric Corporation).
IAV acid bypass assay
Sub‐confluent monolayers of MEFs cultured on optical 96‐well plates (Greiner 655090) in DMEM 10% FBS were incubated with IAV X31 on ice for 1 h in infection medium (DMEM, 50 mM HEPES pH 6.8, 0.2% BSA) and warmed for 2 min at 37°C by addition of warm medium (pH 6.8) or warm medium adjusted to low pH (pH 5.0) by citrate buffer, on a metal block immersed in a 37°C water bath. Another warm metal block was placed on top of the plate. The medium was replaced with STOP medium (DMEM, 50mM HEPES pH7.4, 20 mM NH4Cl) to block viral entry through the endocytic route, and the plate was transferred to 37°C and incubated for 18 h, fixed with 4% paraformaldehyde in PBS. Fixed cells were blocked for 1 h with 1% BSA in PBS before incubation with a mouse anti‐nucleoprotein monoclonal antibody (HB‐65, ATCC) for 1h at RT, followed by a 30‐min incubation with a second goat anti‐mouse‐Alexa Fluor 647 antibody (Invitrogen, A21235), together with Hoechst 33342 (Thermo Fisher Scientific). Image acquisition was performed with a Yokogawa CQ1 spinning disc confocal microscope, using a 10x air objective with an NA of 0.40, in 2‐channel mode. Image analysis was performed using the Cell Path Finder version 3.04.03.02 (Yokogawa Electric Corporation).
Image acquisition by spinning disc confocal microscopy
Image acquisition was performed with a Yokogawa CQ1 spinning disc confocal microscope, using a 10x air objective with an NA of 0.40, or a 40x air objective with an NA of 0.95. Imaging was performed with up to four excitation laser lines (405/488/561/640nms) with spinning disc confocal. For 10x objective, images were acquired with 5 z‐stacks to cover 40 µm; for 40x objective, images were acquired with 20 z‐stacks to cover 30 µm. Maximum intensity projected images were used for image analysis using the Cell Path Finder version 3.04.03.02 (Yokogawa Electric Corporation).
Autophagy and non‐canonical autophagy
Autophagy was activated by incubating MEF cells in Hanks balanced salt solution (HBSS) (Thermo Fisher, 11550456) for 2 h at 37°C. Non‐canonical autophagy was stimulated in MEFs with monensin (Sigma‐Aldrich, M5273) or chloroquine (Sigma‐Aldrich, C6628) with a final concentration of 100 µM for 2 h. Numbers of fluorescent LC3 puncta were quantified by fluorescence microscopy using spot function software (IMARIS package [BITPLANE sScientific Software]) to locate puncta ranging from 0.5–1.0 μm diameter. LC3 vacuoles were identified by eye as rings of fluorescence ranging between 2 and 8 μm diameter. Non‐canonical autophagy/LAP was assessed in BMDMs by incubation with Zymosan A (Alexa Fluor 594‐labelled; Thermo Fisher Z23374). Phagosomes containing Zymosan were observed by eye using immunofluorescence microscopy. Line profile analysis was applied across the centre of the phagosome using ImageJ. The extent of recruitment of LC3 to the phagosome was calculated by subtracting the LC3 pixel intensity at the centre of the phagosome from intensity observed at the perimeter. Conjugation of LC3 to PE was also assessed by Western blot from the ratio of LC3II.
qPCR for cytokine transcription
Lung lobes were snap‐frozen and homogenised using a TissueLyser (Qiagen). Tissue culture cells were washed twice using PBS. Total RNA was extracted by TRIzol–chloroform (Thermo Fisher 15596018) and purified by RNeasy MinElute Cleanup Kit (Qiagen 74204). RNA was analysed by qPCR using SYBR Green/7500 (Thermo Fisher S7563) Standard Real‐Time PCR System (Applied Biosystems, Grand Island, NY) and primer sets as detailed in Table S1. Relative amounts of mRNA expression were normalised to 18S rRNA (Table 1).
Table 1.
Primer sequences for mRNAs analysed by RT–qPCR
| Target | Catalogue number a |
|---|---|
| ISG15 | QT00322749 |
| IFIT1 | QT01161286 |
| IL‐1β | QT01048355 |
| TNF‐α | QT00104006 |
| CXCL1 | QT00115647 |
| CCL2/MCP‐1 | QT00167832 |
| 18S ribosomal RNA | QT02448075 |
Catalogue numbers refer to validated QuantiTect primer sets (Qiagen).
Western blotting
Cells were lysed using M‐PER reagent (Thermo Fisher 78501) with complete protease inhibitor cocktail (Sigma, 04693159001) and clarified by centrifugation. Extracted proteins (20 µg) were separated on a precast 4–12% gradient SDS–PAGE gels (Expedeon, NBT41212), transferred to immobilon PVDF (Millipore, IPFL00010) and probed using antibodies for ATG16L1 (MBL M150‐3), LC3A/B (Cell Signaling 41085) and actin (Sigma, A5441). Primary antibodies were detected using IRDye‐labelled secondary antibodies (LI‐COR biosciences, 926‐32211, 926‐68020) and visualised by Odyssey infrared system (LI‐COR).
Fluorescence imaging
Cells were fixed in ice‐cold methanol, and non‐specific binding was blocked using 5% goat serum plus 0.3% Triton X‐100 in PBS followed by incubating with anti LC3A/B (Cell Signaling 4108) or anti‐ATG16L1 (MBL M150‐3). Cells were washed and then incubated with anti‐rabbit‐Alexa 488 (Thermo Fisher 10729174). After washing, cells were counterstained with 4’, 6 diamidino‐2‐phenylindole (DAPI) (Thermo Fisher Scientific, 10116287) and mounted with Fluoromount‐G (Cambridge Bioscience). Cells were imaged on a Zeiss Imager M2 Apotome microscope with a 63x, 1.4 NA oil‐immersion objective.
Author contributions
TW, JPS, PPP, SRC and JLC conceived the experiments. Mouse strains were generated by UM and genotyped by MJ, SR and WZ. Immunological homeostasis was assessed by WZ, AM and AZ. Animal infections were carried out by JPS, YW, WZ and PS and histology and immunohistology by AK. IAV fusion was analysed by YY with DB and BB. Downstream analysis was performed by YW, WZ, PS, TP and PPP. In vitro analysis was performed by BB, TP, YY and PPP. The manuscript was drafted by TW, JPS, YB, PPP, RAT and UM and edited and approved by all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
This work was funded in part by Biotechnology and Biological Sciences Research Council (BBSRC) grant BB/R00904X/1 to JPS, JLC SRC, PPP, UM and TW, and through BBSRC Institute Strategic Programme Gut Microbes and Health BB/R012490/1: BBS/E/F/000PR10353, BBS/E/F/000PR10355. H3SKE was a kind gift of John Skehel. We thank Alina Rozanova with help on IAV Alexa Fluor labelling and IAV endocytosis assay set up, Mahomi Suzuki and Toshiaki Endo of Yokogawa Electric Corporation for help with image analysis.
The EMBO Journal (2021) 40: e105543.
Contributor Information
James P Stewart, Email: j.p.stewart@liv.ac.uk.
Thomas Wileman, Email: t.wileman@uea.ac.uk.
Data availability
This study includes no data deposited in external repositories.
References
- Akoumianaki T, Kyrmizi I, Valsecchi I, Gresnigt MS, Samonis G, Drakos E, Boumpas D, Muszkieta L, Prevost MC, Kontoyiannis DP et al (2016) Aspergillus Cell Wall Melanin Blocks LC3‐Associated Phagocytosis to Promote Pathogenicity. Cell Host Microbe 19: 79–90 [DOI] [PubMed] [Google Scholar]
- Akram KM, Moyo NA, Leeming GH, Bingle L, Jasim S, Hussain S, Schorlemmer A, Kipar A, Digard P, Tripp RA et al (2018) An innate defense peptide BPIFA1/SPLUNC1 restricts influenza A virus infection. Mucosal Immunol 11: 71–81 [DOI] [PubMed] [Google Scholar]
- Banerjee I, Miyake Y, Nobs SP, Schneider C, Horvath P, Kopf M, Matthias P, Helenius A, Yamauchi Y (2014) Influenza A virus uses the aggresome processing machinery for host cell entry. Science 346: 473–477 [DOI] [PubMed] [Google Scholar]
- Banerjee I, Yamauchi Y, Helenius A, Horvath P (2013) High‐content analysis of sequential events during the early phase of influenza A virus infection. PLoS One 8: e68450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belser JA, Gustin KM, Maines TR, Blau DM, Zaki SR, Katz JM, Tumpey TM (2011) Pathogenesis and transmission of triple‐reassortant swine H1N1 influenza viruses isolated before the 2009 H1N1 pandemic. J Virol 85: 1563–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berryman S, Brooks E, Burman A, Hawes P, Roberts R, Netherton C, Monaghan P, Whelband M, Cottam E, Elazar Z et al (2012) Foot‐and‐mouth disease virus induces autophagosomes during cell entry via a class III phosphatidylinositol 3‐kinase‐independent pathway. J Virol 86: 12940–12953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boada‐Romero E, Serramito‐Gomez I, Sacristan MP, Boone DL, Xavier RJ, Pimentel‐Muinos FX (2016) The T300A Crohn's disease risk polymorphism impairs function of the WD40 domain of ATG16L1. Nat Commun 7: 11821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulo S, Akarsu H, Ruigrok RW, Baudin F (2007) Nuclear traffic of influenza virus proteins and ribonucleoprotein complexes. Virus Res 124: 12–21 [DOI] [PubMed] [Google Scholar]
- Davidson S, Crotta S, McCabe TM, Wack A (2014) Pathogenic potential of interferon alphabeta in acute influenza infection. Nat Commun 5: 3864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V (2008) Toll‐like receptors control autophagy. EMBO J 27: 1110–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA (2014) WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12‐5‐16L1. Mol Cell 55: 238–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher K, Ulferts R, Jacquin E, Veith T, Gammoh N, Arasteh JM, Mayer U, Carding SR, Wileman T, Beale R et al (2018) The WD40 domain of ATG16L1 is required for its non‐canonical role in lipidation of LC3 at single membranes. EMBO J 37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, Gammoh N, Kim SE, Jiang X, Overholtzer M (2015) V‐ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 11: 88–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M (2011) Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol 13: 1335–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluschko A, Herb M, Wiegmann K, Krut O, Neiss WF, Utermohlen O, Kronke M, Schramm M (2018) The beta2 Integrin Mac‐1 Induces Protective LC3‐Associated Phagocytosis of Listeria monocytogenes. Cell Host Microbe 23: 324–337 [DOI] [PubMed] [Google Scholar]
- Heckmann BL, Boada‐Romero E, Cunha LD, Magne J, Green DR (2017) LC3‐Associated Phagocytosis and Inflammation. J Mol Biol 429: 3561–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann BL, Teubner BJW, Tummers B, Boada‐Romero E, Harris L, Yang M, Guy CS, Zakharenko SS, Green DR (2019) LC3‐associated endocytosis facilitates beta‐amyloid clearance and mitigates neurodegeneration in murine Alzheimer's disease. Cell 178: 536–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann BL, Teubner BJW, Boada‐Romero E, Tummers B, Guy C, Fitzgerald P, Mayer U, Carding S, Zakharenko SS, Wileman T et al (2020) Noncanonical function of an autophagy protein prevents spontaneous Alzheimer's disease. Sci Adv 6: eabb9036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold S, Becker C, Ridge KM, Budinger GR (2015) Influenza virus‐induced lung injury: pathogenesis and implications for treatment. Eur Respir J 45: 1463–1478 [DOI] [PubMed] [Google Scholar]
- Hoffmann AB, Mazelier M, Leger P, Lozach PY (2018) Deciphering virus entry with fluorescently labeled viral particles. Methods Mol Biol 1836: 159–183 [DOI] [PubMed] [Google Scholar]
- Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, Glogauer M, Grinstein S, Brumell JH (2009) Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci U S A 106: 6226–6231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubber A, Kubori T, Coban C, Matsuzawa T, Ogawa M, Kawabata T, Yoshimori T, Nagai H (2017) Bacterial secretion system skews the fate of Legionella‐containing vacuoles towards LC3‐associated phagocytosis. Sci Rep 7: 44795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Pillai PS (2014) Innate immunity to influenza virus infection. Nat Rev Immunol 14: 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemball CC, Alirezaei M, Flynn CT, Wood MR, Harkins S, Kiosses WB, Whitton JL (2010) Coxsackievirus infection induces autophagy‐like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J Virol 84: 12110–12124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreibich S, Emmenlauer M, Fredlund J, Ramo P, Munz C, Dehio C, Enninga J, Hardt WD (2015) Autophagy proteins promote repair of endosomal membranes damaged by the salmonella type three secretion system 1. Cell Host Microbe 18: 527–537 [DOI] [PubMed] [Google Scholar]
- Kyrmizi I, Ferreira H, Carvalho A, Figueroa JAL, Zarmpas P, Cunha C, Akoumianaki T, Stylianou K, Deepe GS Jr, Samonis G et al (2018) Calcium sequestration by fungal melanin inhibits calcium‐calmodulin signalling to prevent LC3‐associated phagocytosis. Nat Microbiol 3: 791–803 [DOI] [PubMed] [Google Scholar]
- Lamprinaki D, Beasy G, Zhekova A, Wittmann A, James S, Dicks J, Iwakura Y, Saijo S, Wang X, Chow CW et al (2017) LC3‐Associated phagocytosis is required for dendritic cell inflammatory cytokine response to gut commensal yeast Saccharomyces cerevisiae . Front Immunol 8: 1397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Q, Yokoyama CC, Williams JW, Baldridge MT, Jin X, DesRochers B, Bricker T, Wilen CB, Bagaitkar J, Loginicheva E et al (2016) Homeostatic control of innate lung inflammation by Vici syndrome gene Epg5 and additional autophagy genes promotes influenza pathogenesis. Cell Host Microbe 19: 102–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, Li QZ, Yan M, Janke L, Guy C et al (2016) Noncanonical autophagy inhibits the autoinflammatory, lupus‐like response to dying cells. Nature 533: 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J et al (2015) Molecular characterization of LC3‐associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol 17: 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Matte C, Casgrain PA, Seguin O, Moradin N, Hong WJ, Descoteaux A (2016) Leishmania major promastigotes evade LC3‐associated phagocytosis through the action of GP63. PLoS Pathog 12: e1005690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y, Keusch JJ, Decamps L, Ho‐Xuan H, Iketani S, Gut H, Kutay U, Helenius A, Yamauchi Y (2019) Influenza virus uses transportin 1 for vRNP debundling during cell entry. Nat Microbiol 4: 578–586 [DOI] [PubMed] [Google Scholar]
- Rai S, Arasteh M, Jefferson M, Pearson T, Wang Y, Zhang W, Bicsak B, Divekar D, Powell PP, Naumann R et al (2019) The ATG5‐binding and coiled coil domains of ATG16L1 maintain autophagy and tissue homeostasis in mice independently of the WD domain required for LC3‐associated phagocytosis. Autophagy 15: 599–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos I, Fernandez‐Sesma A (2015) Modulating the innate immune response to influenza A virus: potential therapeutic use of anti‐inflammatory drugs. Front Immunol 6: 361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts R, Al‐Jamal WT, Whelband M, Thomas P, Jefferson M, van den Bossche J, Powell PP, Kostarelos K, Wileman T (2013) Autophagy and formation of tubulovesicular autophagosomes provide a barrier against nonviral gene delivery. Autophagy 9: 667–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M et al (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1beta production. Nature 456: 264–268 [DOI] [PubMed] [Google Scholar]
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S et al (2007) Toll‐like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450: 1253–1257 [DOI] [PubMed] [Google Scholar]
- Serramito‐Gómez I, Boada‐Romero E, Villamuera R, Fernández‐Cabrera A, Cedillo JL, Martín‐Regalado A, Carding S, Mayer U, Powell PP, García‐Higuera I, Pimentel‐Muiños FX (2020) Regulation of cytokine signalling through direct interaction between cytokine receptors and the ATG16L1 WD40 domain. Nat Commun 11: 5919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel JJ, Wiley DC (2000) Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem 69: 531–569 [DOI] [PubMed] [Google Scholar]
- Szretter KJ, Gangappa S, Lu X, Smith C, Shieh WJ, Zaki SR, Sambhara S, Tumpey TM, Katz JM (2007) Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol 81: 2736–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JMJ, Mellouk N, Osborne SE, Ammendolia DA, Dyer DN, Li R, Brunen D, van Rijn JM, Huang J, Czuczman MA et al (2018) An ATG16L1‐dependent pathway promotes plasma membrane repair and limits Listeria monocytogenes cell‐to‐cell spread. Nat Microbiol 3: 1472–1485 [DOI] [PubMed] [Google Scholar]
- Teijaro JR, Walsh KB, Rice S, Rosen H, Oldstone MB (2014) Mapping the innate signaling cascade essential for cytokine storm during influenza virus infection. Proc Natl Acad Sci U S A 111: 3799–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich S, Munch A, Neumann S, Kremmer E, Tatzelt J, Lichtenthaler SF (2010) The novel membrane protein TMEM59 modulates complex glycosylation, cell surface expression, and secretion of the amyloid precursor protein. J Biol Chem 285: 20664–20674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton SA, Belshe RB, Skehel JJ, Hay AJ (1994) Role of virion M2 protein in influenza virus uncoating: specific reduction in the rate of membrane fusion between virus and liposomes by amantadine. J Gen Virol 75(Pt 4): 945–948 [DOI] [PubMed] [Google Scholar]
- Xu Y, Zhou P, Cheng S, Lu Q, Nowak K, Hopp AK, Li L, Shi X, Zhou Z, Gao W et al (2019) A Bacterial Effector Reveals the V‐ATPase‐ATG16L1 Axis that Initiates Xenophagy. Cell 178: 552–566. [DOI] [PubMed] [Google Scholar]
- Yamayoshi S, Kawaoka Y (2019) Current and future influenza vaccines. Nat Med 25: 212–220 [DOI] [PubMed] [Google Scholar]
- Yang CS, Shin DM, Kim KH, Lee ZW, Lee CH, Park SG, Bae YS, Jo EK (2009) NADPH oxidase 2 interaction with TLR2 is required for efficient innate immune responses to mycobacteria via cathelicidin expression. J Immunol 182: 3696–3705 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
This study includes no data deposited in external repositories.