

Abstract
Methylation of carbon‐5 of cytosines (m5C) is a post‐transcriptional nucleotide modification of RNA found in all kingdoms of life. While individual m5C‐methyltransferases have been studied, the impact of the global cytosine‐5 methylome on development, homeostasis and stress remains unknown. Here, using Caenorhabditis elegans, we generated the first organism devoid of m5C in RNA, demonstrating that this modification is non‐essential. Using this genetic tool, we determine the localisation and enzymatic specificity of m5C sites in the RNome in vivo. We find that NSUN‐4 acts as a dual rRNA and tRNA methyltransferase in C. elegans mitochondria. In agreement with leucine and proline being the most frequently methylated tRNA isoacceptors, loss of m5C impacts the decoding of some triplets of these two amino acids, leading to reduced translation efficiency. Upon heat stress, m5C loss leads to ribosome stalling at UUG triplets, the only codon translated by an m5C34‐modified tRNA. This leads to reduced translation efficiency of UUG‐rich transcripts and impaired fertility, suggesting a role of m5C tRNA wobble methylation in the adaptation to higher temperatures.
Keywords: 5‐methylcytosine, Caenorhabditis elegans, NSUN, RNA modifications, translation efficiency
Subject Categories: RNA Biology
Analysis of worm mutants devoid of m5C RNA modifications uncovers their contribution to efficient Leu/Pro codon translation, and requirement for fertility‐supporting Leu‐UUG decoding at higher temperatures.

Introduction
The methylation of carbon‐5 of cytosines (m5C) in RNA is a conserved modification in biological systems. m5C has been detected in tRNAs, rRNAs, mRNAs and non‐coding RNAs and is catalysed by m5C RNA‐methyltransferases that utilise SAM as a methyl donor (Liu & Santi, 2000; Boccaletto et al, 2017). In humans, RNA m5C formation is catalysed by the tRNA aspartic acid MTase 1 (TRDMT1/DNMT2), and by seven proteins of the NOP2/Sun domain family (NSUN1‐7) (García‐Vílchez et al, 2019). Pathogenic mutations in humans have been mapped to several genes involved in the m5C pathway (Abbasi‐Moheb et al, 2012; Khan et al, 2012; Martinez et al, 2012; Khosronezhad et al, 2015; Komara et al, 2015; Ren et al, 2015; Haag et al, 2016; Nakano et al, 2016; Van Haute et al, 2016). Despite its conservation and clear relevance, the functions and molecular interactions of the RNA m5C methylome remain largely unknown. Here, we use Caenorhabditis elegans as a model to study the genetic requirements and molecular functions of m5C modification and its methyltransferases in vivo.
m5C has been implicated in a variety of molecular roles. Among the most highly modified methyltransferase targets are tRNAs and rRNAs, the core components of the translation machinery. tRNAs are methylated by NSUN2, NSUN3, NSUN6 and DNMT2, while rRNAs are methylated by NSUN1, NSUN4 and NSUN5 (García‐Vílchez et al, 2019).
In some tRNAs, m5C protects from degradation. Loss of NSUN2‐mediated tRNA methylation has been shown to promote cleavage by angiogenin and accumulation of tRNA fragments that interfere with the translation machinery (Tuorto et al, 2012; Blanco et al, 2014, 2016; Flores et al, 2016). Similarly, DNMT2‐mediated methylation was found important for protection of tRNAs from stress‐induced cleavage in Drosophila and mice (Schaefer et al, 2010; Tuorto et al, 2012, 2015). NSUN6‐mediated methylation of tRNAs Cys and Thr promotes a slight enhancement of tRNA thermal stability (Haag et al, 2015; Li et al, 2018).
In other tRNAs, m5C modulates translational fidelity. DNMT2 has been shown to facilitate charging of tRNA Asp and discrimination between Asp and Glu near‐cognate codons, thus controlling the synthesis of Asp‐rich sequences and promoting translational fidelity (Tuorto et al, 2015; Shanmugam et al, 2015). NSUN3 methylates exclusively mitochondrial tRNA Met‐CAU at the wobble position, which is further modified into f5C by the dioxygenase ALKBH1, facilitating the recognition of AUA and AUG codons as methionine in the mitochondria (Takemoto et al, 2009; Van Haute et al, 2016; Nakano et al, 2016; Haag et al, 2016). Lack of f5C affects mitochondrial translation rates in human fibroblasts (Van Haute et al, 2016).
The rRNA methyltransferase NSUN1 (yeast nop2) methylates C2870 in yeast 25S rRNA. While NSUN1 is an essential gene in all organisms studied thus far, it remains unclear whether this is dependent on its catalytic activity (Sharma et al, 2013). Similarly, NSUN4 acts in complex with MTERF4 for assembly of small and large mitochondrial ribosome subunits; however, m5C catalysis does not seem to be essential (Metodiev et al, 2014). NSUN5 has been shown to methylate position C2278 in yeast 25S rRNA (Sharma et al, 2013; Schosserer et al, 2015). Loss of NSUN5‐mediated methylation induces conformational changes in the ribosome and modulation of translational fidelity, favouring the recruitment of stress‐responsive mRNAs into polysomes and promoting lifespan enhancement in yeast, flies and nematodes. In mice, Nsun5 knockout causes reduced body weight and reduced protein synthesis in several tissues (Schosserer et al, 2015; Heissenberger et al, 2019).
It remains less clear whether mRNAs are specific targets of m5C‐methyltransferases and if m5C is functional in mRNA. Several methods have been used to investigate the presence and function of m5C in coding transcripts and two m5C‐binding proteins have been identified so far (Yang et al, 2017; Chen et al, 2019). However, there is a lack of consensus on the abundance, distribution and relevance of this mark in mRNAs, as the number and identity of putative mRNA m5C sites varies widely between studies (Squires et al, 2012; Zhang et al, 2012; Tang et al, 2015; David et al, 2017; Legrand et al, 2017; Li et al, 2017; Yang et al, 2017; Huang et al, 2019).
Although previous studies have explored the roles of individual m5C methyltransferases, none have established a systematic dissection of these enzymes as a class, of their specificity, or of their potential molecular and genetic interactions, in any organism. Many questions remain on how the m5C methylome sustains development and normal physiology. In this work, we generated the first mutant animal devoid of any detectable levels of cytosine C5 methylation in RNA, demonstrating that m5C is a non‐essential modification under standard conditions. We then used this mutant strain as a genetic tool to map m5C sites onto RNA in vivo and determined their impact on translation, development, physiology and stress.
Results
m5C and its derivatives are non‐essential RNA modifications in C. elegans
To identify putative m5C RNA methyltransferases in C. elegans, we performed a BLAST analysis and found that the open reading frames W07E6.1, Y48G8AL.5, Y39G10AR.21 and Y53F4B.4 are likely homologues of the human genes NSUN1, NSUN2, NSUN4 and NSUN5, respectively (Fig 1A). Knockdown of these genes through RNAi by feeding revealed that nsun‐1 is an essential gene, as 100% of the animals that had this gene silenced from the first larval stage onwards developed into sterile adults (Fig 1B and C). We could not identify homologous genes of NSUN3, NSUN6, NSUN7 or DNMT2.
Figure 1. m5C and its derivatives are non‐essential RNA modifications in C. elegans .

-
APhylogenetic relationship among human and putative nematode NSUN proteins. Unrooted phylogenetic tree of NSUN homologues in Homo sapiens and C. elegans using entire protein sequence. Phylogenetic tree reconstructed using the maximum likelihood method implemented in the PhyML program (v3.0).
-
B, CKnock‐down of nsun genes through RNAi by feeding. Representative images of wild‐type adult animals after silencing of nsun genes via RNAi by feeding. Widefield DIC images are 10× magnification (B). Percentage of fertile adults after gene silencing by RNAi (C). n = 2 independent experiments, 3 biological replicates each.
-
DMutant alleles used in this study. CRISPR‐Cas9 strategy for creation and screening of catalytically inactive alleles of nsun‐1 (mj473), nsun‐2 (mj458) and nsun‐4 (mj457). A homologous recombination template bearing a point mutation to convert the catalytic cysteine into alanine while creating a restriction site for HaeIII was co‐injected. For the study of nsun‐5, a 928 bp deletion allele (tm3898) was used. Image not to scale.
-
E, FMass spectrometry quantification of m5C (D) and hm5Cm (E) levels in total RNA from nsun mutants. RNA was extracted from populations of L1 animals synchronised by starvation, digested to nucleosides and analysed via LC‐MS. n = 3 independent biological replicates. n.d. = not detected.
-
GFold change in total RNA modification levels upon loss of m5C. Fold changes were calculated by dividing the peak area ratio of noNSUN samples by the one of wild‐type samples. n = 3 independent biological replicates. Multiple t‐tests.
Data information: In (C, E, F, G), data are presented as mean ± SEM. In (C), a representative plot of two independent experiments is shown.
m5C RNA methyltransferases utilise two conserved cysteine residues for the methyl group transfer, one of which (TC‐Cys) is required for the covalent adduct formation, and the other (PC‐Cys) for the release of the substrate following m5C catalysis (King & Redman, 2002). Using CRISPR‐Cas9, we introduced mutations converting the TC‐Cys into alanine in nsun‐1 (mj473), nsun‐2 (mj458) and nsun‐4 (mj457) (Fig 1D, and Appendix Fig S1A and B). These mutants, as well as a previously reported knockout mutant of nsun‐5 (tm3898), are viable and produce viable progeny, suggesting that the individual activity of m5C methyltransferases is not essential for the viability of C. elegans. In addition, these results suggest that the essential role played by NSUN‐1 in fertility (Fig 1B and C) is independent of the catalytic functions of this protein.
To investigate epistatic interactions among the nsun genes in C. elegans, we performed genetic crosses between the individual mutants and produced a quadruple mutant in which all nsun genes are predicted to be inactive. This strain was viable and fertile and was termed noNSUN. To confirm whether the introduced mutations resulted in catalytic inactivation, and to rule out the existence of additional unknown m5C RNA methyltransferases, we performed mass spectrometry analyses in total RNA from the mutant strains and confirmed that m5C is no longer detectable in this genetic background (Fig 1E; lower limit of detection ~ 0.3 ng/ml, average amount detected in wild type samples 477 ng/ml). We additionally quantified the m5C metabolic derivative 2′‐O‐methyl‐5‐hydroxymethylcytosine (hm5Cm) (Huber et al, 2017) and found that this modification is not present either in nsun‐2 or noNSUN mutants (Fig 1F). We therefore conclude that m5C and its derivatives are not essential for C. elegans viability under laboratory conditions. Furthermore, we showed that NSUN‐2 is the main source of m5C (88% of total) and that hm5Cm sites exclusively derive from NSUN‐2 targets in C. elegans.
It has been proposed that some RNA modifications may act in a combinatorial manner, providing compensatory effects to each other (Hopper & Phizicky, 2003). This prompted us to investigate whether complete loss of m5C would significantly interfere with the levels of other RNA modifications. We performed a mass spectrometry analysis to quantify 15 different modifications in total RNA and found no significant differences between wild‐type and noNSUN samples (Fig 1G). Taken together, our data establish the noNSUN strain as a highly specific genetic tool for the study of m5C distribution and function in vivo.
The m5C methylome of C. elegans
Schosserer et al demonstrated that position C2381 of 26S rRNA is methylated at carbon‐5 by NSUN‐5 in C. elegans, being involved in lifespan modulation (Schosserer et al, 2015). Nevertheless, the m5C methylome of this organism remained to be determined. We therefore used the noNSUN strain as a negative control for whole‐transcriptome bisulphite sequencing (WTBS) analysis (Legrand et al, 2017), aiming to determine the localisation of m5C sites in C. elegans RNA at single‐nucleotide resolution.
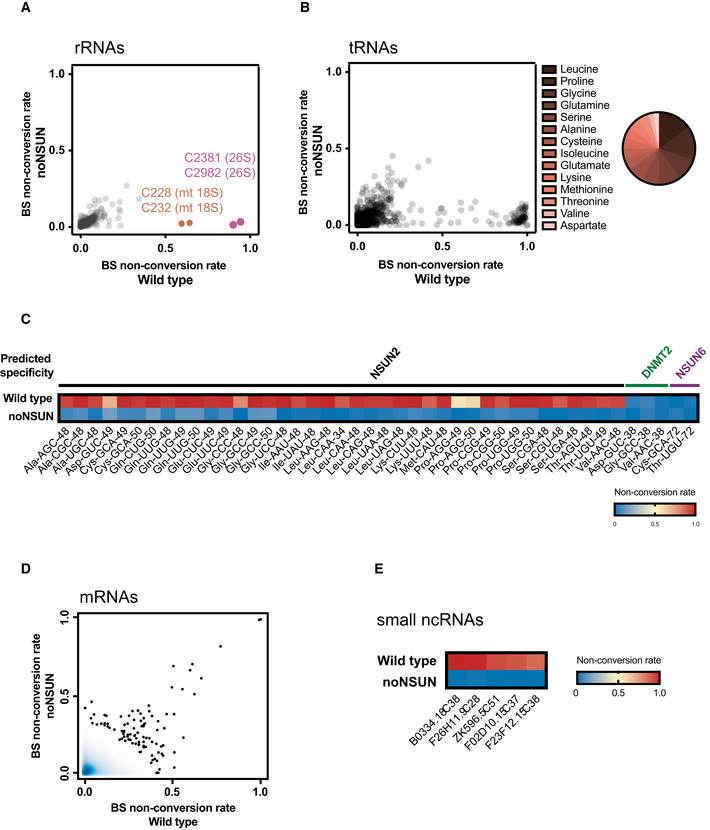
We identified C5 methylation at positions C2982 and C2381 of 26S cytoplasmic rRNA and positions C628 and C632 of 18S mitochondrial rRNA (Fig 2A). Using alignment to rRNA of different organisms, we found that position C2982 is a conserved NSUN1 target (Sharma et al, 2013), which has also been recently reported in C. elegans (Heissenberger et al, 2020). In addition, C2381 has previously been reported as a conserved NSUN5 target (Sharma et al, 2013; Schosserer et al, 2015). We further confirmed the specificity of these sites using a targeted bisulphite sequencing (BS‐seq) approach in individual mutants (Fig EV1A and B). Interestingly, other groups had previously identified adjacent modified sites in mt‐rRNA in mice; however, the methylation of only one of the positions was shown to be dependent on NSUN4 activity, while the other was interpreted as a 4‐methylcytosine site (Metodiev et al, 2014). In the case of C. elegans, both positions are NSUN‐dependent (Fig 2A).
Figure 2. The m5C methylome of C. elegans .
-
A, BSite‐specific methylation analysis by whole‐transcriptome bisulphite sequencing. Scatter plots show individual cytosines and their respective non‐conversion rates in rRNAs (A) and tRNAs (B) of wild‐type and noNSUN strains; pie chart showing most frequently methylated tRNA isoacceptors.
-
CHeat map showing non‐conversion rates of tRNA positions methylated in stoichiometry higher than 50% and of tRNA positions predicted to be targets of DNMT2 and NSUN6.
-
DSite‐specific methylation analysis by whole‐transcriptome bisulphite sequencing. Scatter plot shows density of cytosines and their respective non‐conversion rates in mRNAs of wild‐type and noNSUN strains.
-
EHeat map showing non‐conversion rates of small non‐coding RNA positions methylated in stoichiometry higher than 50%.
Data information: In (A–E), n = 3 independent biological replicates.
Figure EV1. Related to Fig 2. Enzymatic specificity of NSUN proteins in C. elegans .

-
A–CDetermination of enzymatic specificity of 26S rRNA C2381 (A), 26S rRNA C2982 (B) and tRNA Leu‐CAA C34 and C48 (C) methylation by targeted bisulphite‐sequencing. Each column represents one cytosine in the sequence of interest; each line represents one clone sequenced. Enzymatic specificity of C2381 and C2982 in C. elegans has been independently demonstrated in other publications (Schosserer et al, 2015; Heissenberger et al, 2020).
We found 40 positions to be methylated in stoichiometry higher than 50% in tRNAs, the majority of which being detected in leucine and proline isoacceptors (Fig 2B and C). As anticipated for NSUN2 targeting, modified positions are found in the variable loop region (positions 48, 49, 50), with cytoplasmic tRNA Leu‐CAA carrying an additional modification at the wobble position (C34) (Blanco et al, 2014; Burgess et al, 2015) (Fig 2C). Using targeted BS‐seq, we demonstrated that NSUN‐2 is indeed responsible for both C34 and C48 methylation in tRNA‐Leu (Fig EV1C). In agreement with the lack of homologous genes of DNMT2 and NSUN6 in C. elegans, no methylation was found on conserved RNA targets of these enzymes (Goll et al, 2006; Schaefer et al, 2010; Long et al, 2016; Fig 2C).
Contrasting with what was observed for tRNAs and rRNAs, non‐conversion of mRNA sites in C. elegans is rare and occurs at much lower stoichiometry. Lowering the non‐conversion threshold from 50 to 25–40%, we detected 188 positions that remained unconverted after bisulphite treatment exclusively in wild‐type samples, i.e. putative m5C sites (Fig 2D, x‐axis). Using the same thresholds to probe for likely artefacts revealed 88 positions that remained unconverted exclusively in noNSUN samples, i.e. non‐conversion was only 46% less frequent (Fig 2D, y‐axis). It is also noteworthy that positions remaining reproducibly highly unconverted equally in wild‐type and noNSUN samples are more frequent in mRNAs, when compared to tRNAs and rRNAs (Fig 2A, B and D). In summary, we found no evidence of a widespread distribution of m5C in coding transcripts. To investigate whether the presence of common characteristics could support a subset of the aforementioned 188 positions as bona fide methylated sites, we performed gene ontology, motif search, genomic localisation and secondary structure analyses on these transcripts and sites; however, no significant shared features were found. While our data do not completely rule out the existence of m5C methylation in mRNAs, it demonstrates that this mark cannot be detected in high stoichiometry in C. elegans, as observed in tRNAs and rRNAs.
Finally, we attempted to identify m5C sites in small RNAs in our data set. Given that the fractionation used in this protocol aimed to enrich for tRNAs (60–80 nt), we could not detect microRNA reads in abundance for confident analysis. Nevertheless, we found high NSUN‐dependent non‐conversion rates (> 80%) in five non‐coding RNAs (approximately 60 nt long) previously identified in C. elegans (Lu et al, 2011; Xiao et al, 2012) (Fig 2E). Secondary structure predictions suggest that the methylated sites are often found on the base of a stem‐loop, reminiscent of tRNA variable loops (Fig EV2). Further experiments will be required to determine the functionality of these m5C sites.
Figure EV2. Related to Fig 2. Predicted secondary structures of m5C‐methylated ncRNAs.

Red dot indicates the methylated position. Structures predicted by the Predict a Secondary Structure Web Server (David Mathews Lab, University of Rochester) as the lowest free energy structures generated using default data.
NSUN‐4 is a multisite‐specific tRNA/rRNA‐methyltransferase in the mitochondria of C. elegans
Interestingly, we found the mitochondrial tRNA Met‐CAU to be methylated at a very high rate (94.7%) (Fig 3A). Supporting our finding, previous articles also indicated the detection of this modified site in Ascaris suum and C. elegans (Watanabe et al, 1994; Nakano et al, 2016). This was unexpected, as previous reports have shown that this position is methylated by NSUN3 (Haag et al, 2016; Nakano et al, 2016; Van Haute et al, 2016). As C. elegans does not have an NSUN3 homologue, this implies that mitochondrial tRNAs can be modified by alternative enzymes. A BLAST analysis of the human NSUN3 methyltransferase domain against the C. elegans proteome showed higher similarity to NSUN‐4 (30% identity), followed by NSUN‐2 (26% identity). Moreover, we observed that, among human NSUN genes, NSUN3 and NSUN4 share the highest percentage of similarity (Fig 3B). Using a targeted BS‐seq approach, we probed the methylation status of position C34 in mitochondrial tRNA Met‐CAU from wild‐type, nsun‐2 and nsun‐4 strains. Our results indicate that NSUN‐4 is responsible for the catalysis of m5C in this position in C. elegans (Fig 3C).
Figure 3. NSUN‐4 is a dual tRNA/rRNA methyltransferase in C. elegans .

-
ARNA bisulphite sequencing map for mitochondrial tRNA Met‐CAU in wild‐type (top) and noNSUN (bottom) strains. Each row represents one sequence read, and each column one cytosine.
-
BPer cent identity matrix of human NSUN proteins according to the Clustal Omega multiple alignment tool.
-
C, DTargeted bisulphite‐sequencing heat map showing non‐conversion rates of cytosines in mitochondrial tRNA Met‐CAU (C) and mitochondrial 18S rRNA (D). Each row represents one genetic strain analysed, and each column represents one cytosine.
-
ETreefam phylogenetic tree based on sequence conservation of NSUN3 proteins in different model organisms. Bootstrap values are indicated on branches.
Data information: In (A), a representative map of the replicates is shown, n = 3 independent biological replicates. In (C, D), the average of two experiments is plotted, n = 2 independent biological replicates, 10 clones sequenced per strain, per replicate. Similar effects were observed in all replicates analysed.
NSUN‐4 is the only mitochondrial rRNA m5C methyltransferase identified to date. To confirm that the previously reported role of NSUN4 is also conserved in C. elegans, we performed targeted BS‐seq in 18S mitochondrial rRNA and found that methylation of positions C628 and C632, as well as C631, is mediated by NSUN‐4 (Fig 3D). Notably, methylation of position C631 was also detected by WTBS, however in reduced stoichiometry (23.5% in wild type vs. 0.04% in noNSUN). Taken together, our results show that NSUN‐4 is a multisite‐specific tRNA/rRNA mitochondrial methyltransferase in C. elegans.
To investigate when the divergence of NSUN3 arose in evolution, a phylogenetic analysis of NSUN3 was performed using Treefam and, given the high sequence similarity, NSUN3 and NSUN4 sequences were automatically included in the generated cladogram. While Drosophila and C. elegans only have NSUN4, vertebrate model organisms as basal as zebrafish have both NSUN3 and NSUN4 (Fig 3E). A more expanded version of the tree indicates the presence of NSUN4, but not NSUN3, in sea lampreys (http://www.treefam.org/family/TF321304#tabview=tab1), suggesting that NSUN3 diverged from NSUN4 in vertebrates.
Loss of m5C leads to temperature‐sensitive reproductive phenotypes
The individual or collective introduction of mutations in nsun genes failed to induce noticeable abnormal phenotypes. We therefore performed a more extensive characterisation of the mutant strains using a live imaging‐based phenotypic analysis (Akay et al, 2019). As a proxy for reduced fitness, we chose to analyse the number of viable progeny and occurrence of developmental delay (growth rate, as measured by body length). In comparison with wild‐type animals, we observed a delay in all mutant strains, which persists throughout development and into adulthood. This difference is greater in noNSUN animals, especially as this strain transitions from L4 stage to young adulthood (Fig 4A). When comparing mutants’ sizes at young adult stage at 20°C, noNSUN animals are, on average, five times smaller than wild type (Fig 4B). While this difference reflects a developmental delay, noNSUN animals remain 20% smaller even when they reach adulthood themselves (Fig 4B). In addition, the noNSUN strain shows a 25% reduction in brood size, which is comparable to what is observed in nsun‐1 and nsun‐5 individual mutants (Fig 4C).
Figure 4. Loss of m5C leads to a temperature‐sensitive reproductive phenotype.

-
ABody length of individual nsun mutants throughout development (n = 44, 7, 7, 7, 8, 8) in ~ 4 h windows. L1‐L4 refers to the larval stages, YA and Ad to young adult and adult, respectively.
-
BSize of mutant nsun strains at young adult and egg‐laying stages. Approximately 100 synchronised young adults of each strain were measured. Images were processed using custom algorithms to recognise C. elegans and measure their cross‐sectional area.
-
C, DViable progeny counts of wild‐type and nsun mutant strains at 20°C (C) and of wild‐type and noNSUN strains at 20°C and 25°C (D). Automatic counting was done using a MATLAB script which processed plate images in real‐time.
Data information: In (A), error bars indicate the 95% confidence interval of the median. In (C), data are presented as mean ± SEM, one‐way ANOVA. In (D), data are presented as mean ± SEM, unpaired two‐tailed Student’s t‐test.
Caenorhabditis elegans stocks can be well maintained between 16 and 25°C, being most typically kept at 20°C. To gain insights into how the loss of m5C impacts development under different environmental conditions, wild‐type and noNSUN animals were cultured at 25°C for three generations and subjected to automated measurements. As shown in Fig 4D, the reproductive phenotype previously observed in the noNSUN strain (Fig 4C) is significantly aggravated at this temperature. This suggests that the phenotypes arising from loss of m5C are temperature‐sensitive, pointing towards an involvement of this modification in the adaptation to environmental changes.
Loss of m5C impacts translation efficiency of leucine and proline codons
To explore the impact of temperature stress in the absence of m5C while avoiding the confounding effect introduced by differences in brood size, we performed further experiments using an acute heat shock treatment. To investigate whether the observed phenotypes are linked to abnormalities in protein translation rates, we quantified the polysomal fraction in wild‐type and noNSUN adult animals subject to heat shock at 27°C for 4 h and found no significant differences (Fig 5A, Appendix Fig S2).
Figure 5. Loss of m5C impacts translation efficiency of leucine and proline codons.

- Fraction of polysomal ribosomes quantified from polysome profiles in the wild‐type and noNSUN strains subject to a 4 h heat shock at 27°C. ns = non‐significant.
- Heat map showing P‐site codon occupancy according to the colour scale at 20 and 27°C. Red and blue refer to enhanced and reduced codon occupancy, respectively, in the noNSUN strain relative to wild type. Leucine and proline codons are marked in red.
- Ribosome‐protected fragment (RPF) counts in each sample plotted along ife‐1 and pat‐10 CDS. Vertical grey lines indicate UUG codons.
- Translation efficiency of UUG‐enriched, leucine‐enriched, proline‐enriched and random genes in each sample. A gene was considered enriched in a certain codon when the proportion of this codon in the gene was at least 3‐fold higher than the proportion of the same codon across the transcriptome.
Data information: In (A–E), n = 3 biological replicates. In (A), data are presented as mean ± SEM, unpaired two‐tailed Student’s t‐test. In (D), box plots show the median (central band) and IQR (boxes) ± 1.5 × IQR (whiskers), Welch’s t‐test, P‐value < 0.05.
To gain insights into transcriptional and translational differences resulting from the loss of m5C, we performed transcriptomic and ribosome profiling analyses. The latter allows the quantification of active translation by deep‐sequencing of the mRNA fragments that are protected from nuclease digestion by the presence of ribosomes (ribosome‐protected fragments—RPFs). RPFs showed the expected 3 nt periodicity along the coding domain sequences of mRNA, with the majority of reads in frame (Appendix Fig S3A and B). Furthermore, RNA‐seq and Ribo‐seq counts of genes showed high correlation, and variation in the gene counts could be attributed to the difference in samples analysed (Appendix Fig S3C–E). Loss of m5C did not greatly impact the nature of the heat stress response, as most differentially transcribed and translated genes upon heat stimulus showed agreement, or very subtle differences between wild‐type and noNSUN strains (Appendix Fig S4). We found that differentially transcribed genes upon loss of m5C are mainly involved in cuticle development (Fig EV3A), while differentially translated genes are enriched in components of the cuticle and ribosomes, as well as RNA‐binding proteins (Fig EV3B).
Figure EV3. Related to Fig 5. Differentially transcribed and translated genes upon loss of m5C at different temperatures.

-
A, BHeat maps and gene ontology enrichment (biological process) analysis for the comparison between wild‐type and noNSUN samples. Panel (A) shows RNA‐seq (scaled normalised expression) and panel (B) shows Ribo‐seq (scaled normalised RPFs). Sets of significant non‐redundant GO terms are clustered according to semantic similarity; size indicates the frequency of the GO term in the underlying database WT = wild type; n = 3 biological replicates.
We then evaluated genome‐wide codon occupancy during translation elongation in both temperatures and found that loss of m5C leads to increased ribosome occupancy at leucine and proline codons. Upon heat shock, Leu‐UUG codons showed the highest ribosome density observed in the noNSUN strain, suggesting that translation of this codon is slowed during heat stress in the absence of m5C (Figs 5B and EV4A). We investigated this phenomenon more closely in different transcripts and found that ribosome stalling at UUG codons seems to be context‐dependent, as it only occurs in a small subset of UUG codons (Figs 5C and EV4B). Interestingly, as shown in our WTBS analysis, leucine and proline are the most frequently methylated tRNA isoacceptors in C. elegans (Fig 2B). In addition, tRNA Leu‐CAA, responsible for decoding of UUG codons, is the only cytoplasmic tRNA bearing an m5C‐modified wobble position (Fig 2C).
Figure EV4. Related to Fig 5. Codon occupancy analyses.

- Fold change of P‐site codon occupancy in noNSUN over wild‐type samples at 20 and 27°C. P‐values for the fold change occupancy of each codon are indicated in a heat map below the graph, where asterisks indicate statistical significance. n = 3 biological replicates.
- Representative examples of UUG codon occupancy in different affected genes. Ribosome‐protected fragment counts (RPF) plotted along the gene’s CDS. Vertical grey lines indicate UUG codons. n = 3 biological replicates.
Data information: In (A), P < 0.05, t‐test.
As a downstream consequence of ribosome stalling, we found translation efficiency of UUG‐, leucine‐ and proline‐rich genes to be significantly reduced in the noNSUN strain. While this effect can be observed in both temperatures in leucine‐rich transcripts and at 20°C in proline‐rich ones, it occurs in a heat shock‐dependent manner in UUG‐rich transcripts, suggesting an involvement of m5C wobble methylation in the adaptation to heat stress (Fig 5D). Finally, we found that translation efficiency is further reduced as transcripts get more enriched in the affected codons (Fig EV5).
Figure EV5. Related to Fig 5. Translation efficiency of leucine and proline‐enriched transcripts.

Translation efficiency of genes with increasing enrichment for leucine (top) or proline codons (bottom). Box plots show the median (central band) and IQR (boxes) ± 1.5 × IQR (whiskers). n = 3 biological replicates.
Discussion
Chemical modifications of RNA occur in organisms from all kingdoms of life and are often highly conserved throughout evolution, as is the case of the methylation of carbon‐5 in cytosines (Huber et al, 2015; Boccaletto et al, 2017). Despite that, there is a growing body of evidence showing that several RNA modifications are individually not required for development under controlled conditions (O’Connor et al, 2018, reviewed in Sharma & Lafontaine, 2015 and Hopper & Phizicky, 2003). Our results reignite a recurrent question in the epitranscriptomics field: why are so many of these chemical marks extensively conserved throughout evolution and, yet, organisms often present subtle phenotypes in their absence? Ribonucleoside modifications occur in an overwhelming diversity and, in some cases, might (i) exert subtle molecular effects, (ii) act in a combinatorial or redundant manner with other modifications or (iii) be the result of relaxed enzymatic specificity (Phizicky & Alfonzo, 2010; Jackman & Alfonzo, 2013).
While the absence of m5C in RNA did not give rise to overt phenotypes under standard laboratory conditions, a more detailed analysis of the mutants revealed developmental and fertility defects. Previous studies have shown that levels of several RNA modifications, including m5C, are responsive and can react dynamically to a wide range of environmental challenges, such as toxicants, starvation and heat shock, thus potentially supporting organismal adaptation (Chan et al, 2010; van Delft et al, 2017). In agreement with this idea, we observed a temperature‐dependent aggravation of reproductive phenotypes in m5C‐deficient C. elegans. In nature, where the environmental conditions vary greatly, such genotypes would likely be selected against in a wild population.
Our results suggest an RNA methylation‐independent essential role for NSUN‐1 in germline development. Consistently, Nop2p/Nol1/NSUN1 has been shown to be an essential gene in yeast, mice and Arabidopsis (De Beus et al, 1994; Sharma et al, 2013; Burgess et al, 2015; Kosi et al, 2015). In Saccharomyces cerevisiae, both depletion and catalytic mutation of Nop2p lead to lower levels of 60S ribosomal subunits, supporting the idea that reduced methylation affects rRNA processing and translation (Hong et al, 1997; Honget al, 2001; Sharma et al, 2013). In contrast, Bourgeois et al (2015) reported that loss of Nop2p‐mediated m5C had no effect on ribosome synthesis and phenotype. A similar phenomenon has been observed for NSUN4 in mice, as well as for Dim1 and Trmt12 in yeast, and their human homologues DIMT1L and WBSCR22, where the presence of the enzyme, rather than its catalytic activity, is required for viability (Lafontaine et al, 1995; Metodiev et al, 2014; Zorbas et al, 2015). It has been proposed that the essential binding of certain rRNA methyltransferases represents a quality control step in ribosome biogenesis, committing rRNA to methylation during the maturation process (Lafontaine et al, 1998).
Taking advantage of the noNSUN strain as a tool to increase the confidence of WTBS analysis, we produced the first comprehensive list of m5C sites throughout C. elegans transcriptome. Using a targeted approach, we showed that NSUN‐4 has both rRNA and tRNA targeting capabilities in the mitochondria. It has been suggested that binding of MTERF4 and NSUN4 in a complex is responsible for targeting the methyltransferase to rRNA in the mitochondria (Spåhr et al, 2012; Yakubovskaya et al, 2012; Metodiev et al, 2014). Nevertheless, genetic evidence suggests that NSUN4 methylates rRNA independently of MTERF4 in mice (Metodiev et al, 2014). C. elegans has an MTERF4 homologue (K11D2.5), and most residues involved in the interaction with NSUN4 appear conserved, suggesting that a similar interaction with this co‐factor could occur (Spåhr et al, 2012).
In humans, NSUN3‐mediated methylation at position 34 of mitochondrial tRNA Met‐CAU is further modified by the dioxygenase ALKBH1 to form f5C (Haag et al, 2016; Nakano et al, 2016). Previous studies explored differential methods for the detection of f5C, indicating that 35–100% of tRNA Met‐CAU molecules are f5C‐modified, while the whole population is at least m5C‐modified (Haag et al, 2016; Van Haute et al, 2016; Kawarada et al, 2017). As f5C reacts as an unmodified cytosine upon sodium bisulphite treatment, it was surprising to detect high levels of non‐conversion (95%) in our study. Nakano et al (2016) have used DNA probes in reciprocal circulating chromatography followed by mass spectrometry to demonstrate high stoichiometry of f5C in this site in C. elegans RNA. In addition, an ALKBH1 homologue (Y51H7C.5) has recently been discovered and implicated in mitochondrial protein biogenesis in the nematode (Wagner et al, 2019). These results support the existence of a f5C pathway in C. elegans. However, as we used a sequencing‐based method that does not discriminate between precursors or mature tRNAs, it is possible that our method detects mainly primary transcripts or precursor molecules, which have not been oxidised by ALKBH1.
Using the noNSUN strain as a negative control, we investigated the presence of m5C in coding transcripts. Several reports have shown that m5C is a common mRNA modification (Squires et al, 2012; Amort et al, 2017; David et al, 2017; Yang et al, 2017). However, results derived from BS‐seq can be influenced by several factors, such as incomplete deamination, protection due to secondary structures, presence of other modifications, protein binding and sequencing errors, among others (summarised in Legrand et al, 2017). Given these technical drawbacks, the noNSUN strain represented an unprecedentedly stringent negative control, which allowed for exclusive detection of highly specific methylation. Despite the detection of positions with 20–30% NSUN‐dependent non‐conversion, we detected a similar number of positions with NSUN‐independent non‐conversion at these rates, which we interpret as false positives. This poses a statistical challenge on the interpretation of such non‐converted positions as methylated. Our main conclusion, therefore, is that the data do not provide evidence for widespread or high stoichiometry m5C methylation of coding transcripts in C. elegans. This is in agreement with earlier work using chromatography (Desrosiers et al, 1974; Adams & Cory, 1975; Salditt‐Georgieff et al, 1976) and other reports that have detected very few or no m5C sites in eukaryotes by BS‐seq (Edelheit et al, 2013; Khoddami & Cairns, 2013; Legrand et al, 2017; Khoddami et al, 2019).
Using ribosome profiling, we investigated the genome‐wide effect of loss of m5C in translational speed and efficiency and found leucine and proline translation to be affected. The strongest effect by far was observed in a heat shock‐dependent manner in UUG codons, which rely on the only tRNA modified at the wobble position—tRNA Leu‐CAA. Chan et al (2012) found that m5C level specifically at position 34 of tRNA Leu‐CAA is upregulated upon oxidative stress in yeast. The presence of this modification was shown to enhance translation efficiency of a UUG‐rich luciferase reporter construct, as trm4Δ (NSUN2 homologue mutant) cells showed significantly lower levels of reporter activity, especially under oxidative stress. The biological relevance of these findings was linked to an abnormally high frequency of UUG codons in transcripts of specific ribosomal protein paralogues (Chan et al, 2012).
In summary, m5C supports C. elegans fitness at higher temperatures and enhances the translational efficiency of leucine and proline codons in physiology and stress. Our work highlights a specific role of cytosine C5 methylation in facilitating translation of leucine UUG codons upon heat shock, suggesting that m5C tRNA wobble methylation is involved in the adaptation to heat stress.
Materials and Methods
Genetics
Caenorhabditis elegans strains were grown and maintained as described in Brenner (Brenner, 1974). The strains were kept at 20°C, unless otherwise indicated. HB101 strain Escherichia coli was used as food source (Caenorhabditis Genetics Center, University of Minnesota, Twin Cities, MN, USA). Bristol N2 was used as the wild type strain.
Gene silencing by RNAi
Empty vector, nsun‐1 (W07E6.1), nsun‐2 (Y48G8AL.5), nsun‐4 (Y39G10AR.21) and nsun‐5 (Y53F4B.4) bacterial feeding clones were kindly provided by Prof. Julie Ahringer’s laboratory (Kamath & Ahringer, 2003). Single colonies were inoculated in LB‐Ampicillin 100 μg/ml and cultured for 8 h at 37°C. Bacterial cultures were seeded onto 50 mm NGM agar plates containing 1 mM IPTG and 25 μg/ml Carbenicillin at a volume of 200 μl of bacterial culture per plate, and left to dry for 48 h. Fifty synchronised L1 larvae were placed onto RNAi plates and left to grow until adult stage. Adults were scored for fertility (presence of embryos in the germline).
CRISPR‐Cas9 gene editing
CRISPR‐Cas9 gene editing was performed as in Paix et al (2015). Briefly, injection mixes were prepared in 5 mM Tris pH 7.5 as follows: 20 μg of tracrRNA (Dharmacon), 3.2 μg of dpy‐10 crRNA (Dharmacon), 200 ng of dpy‐10 homologous recombination template (Sigma Aldrich), 8 μg of target gene gRNA (Dharmacon), 1.65 μg of homologous recombination template (Sigma), up to a volume of 11.5 μl. The mix was added to 10 μg of Cas9 (Dharmacon) to a final volume of 15 μl and incubated at 37°C for 15 min. For the creation of nsun catalytic mutants, a homologous recombination template bearing a point mutation to convert the catalytic cysteine into alanine while creating a restriction site for HaeIII was co‐injected. Following incubation, the mix was immediately micro‐injected into the germline of N2 young adults. After injection, animals were left to recover in M9 medium, then transferred to individual plates and left to recover overnight at 20°C. Successful injections led to the hatch of dumpy and roller animals. From positive plates, 96 animals were individualised for self‐fertilisation and genotyped for the relevant alleles. Same process was performed with F2s, until a homozygous population was isolated. Each strain was backcrossed at least three times with the wild type strain.
RNA extraction (mass spectrometry and WTBS)
The strains of interest (N2 and noNSUN) were grown in 90 mm plates until gravid adult stage, washed three times with M9 and pelleted by centrifugation at 775 g for 2 min. Gravid adults were resuspended in 4 ml of bleaching solution (final concentration 177 mM NaOH, 177 mM NaOCl solution—free chlorine 4–5%) and vortexed vigorously for 7 min. Recovered embryos were washed four times to remove any traces of bleach and left to hatch in ml of M9 for 24 h at 20°C in a rotating wheel. Synchronised L1 starved larvae were used for RNA extraction. Independent triplicates were obtained from three different generations.
Nematodes were washed thoroughly in M9 to remove bacterial residue and pelleted in RNAse‐free tubes at 775 g for 2 min. 500–1,000 μl of TRIsure (Bioline) and 100 μl of zirconia beads were added, and the samples were subjected to three cycles of 4000 g with 20 s breaks on Precellys to crack open the animals. 100 μl of chloroform was added to the tubes, which were then shaken vigorously for 15 s and incubated at room temperature for 3 min. Samples were centrifuged at 12,000 g for 15 min at 4°C, and the aqueous phase of the mixture was carefully recovered and transferred to a fresh RNAse‐free tube. RNA was precipitated with 500 μl of cold isopropanol at room temperature for 10 min and then centrifuged at 12,000 g for 15 min at 4°C. The supernatant was carefully removed, the pellet was washed and vortexed with 1 ml of 75% ethanol and centrifuged at 7,500 g for 5 min at 4°C. RNA pellet was air‐dried, dissolved in the appropriate volume of DEPC‐treated water and the concentration, and 260/280 and 260/230 ratios were measured by Nanodrop. RNA integrity was evaluated in the Agilent 2200 Tapestation system.
RNA mass spectrometry
Up to 10 μg of RNA was digested by adding 1 μl digestion enzyme mix per well in a digestion buffer (4 mM Tris–HCl pH 8, 5 mM MgCl2, 20 mM NaCl) in a total volume of up to 100 μl. The digestion enzyme mix was made by mixing benzonase (250 U/μl, Sigma Aldrich), phosphodiesterase I from Crotalus adamanteus venom (10 mU/μl, Sigma Aldrich) and Antarctic phosphatase (5 U/μl, NEB) in a ratio of 1:10:20. The reaction was incubated overnight at 37°C. The following day, an equal volume of 13C, 15N‐labelled uridine (internal control, previously dephosphorylated; Sigma Aldrich) in 0.1% formic acid was added to each reaction and this was subsequently prepared for LC‐MS‐MS by filtration through 30 kDa molecular weight cut‐off filters (Sigma).
Samples were resolved using a Thermo Scientific U3000 UPLC system on a gradient of 2–98% (0.1% formic acid/acetonitrile) through an Acquity 100 × 2.1 mm C‐18 HSS T3 column and analysed on a QExactive‐HF Orbitrap High Resolution Mass Spectrometer (Thermo Fisher Scientific, IQLAAEGAAPFALGMBFZ) in positive full‐scan mode, and the results were deconvoluted using the accompanying Xcalibur Software. Nucleosides of interest were identified by both retention times and accurate masses, compared with purified standards and quantified accordingly.
Whole transcriptome bisulphite sequencing
Bisulphite sequencing experiments were performed as previously described in Legrand et al (2017). RNA was fractionated into < 200 nt and > 200 nt using a modified mirVana miRNA isolation kit (AM1560) protocol. Briefly, 50 μg of RNA in a volume of 80 μl were mixed with 400 μl of mirVana lysis/binding buffer and 48 μl of mirVana homogenate buffer and incubated for 5 min at room temperature. Next, 1/3 volume (176 μl) of 100% ethanol was added and thoroughly mixed by inversion, and the mixture was incubated for 20 min at room temperature. After addition of 0.8 μg of Glycoblue, the samples were spun down at 2,500 g for 8 min at 21°C for precipitation of long RNAs. The supernatant containing the short fraction was transferred to a fresh tube, and the RNA pellet was washed in 1 ml of cold 75% ethanol before centrifugation at maximum speed for at least 20 min at 4°C. The pellet was finally air‐dried and resuspended in DEPC‐treated water. For short fraction RNA precipitation, 800 μl of isopropanol was added to the supernatant and the mixture was incubated at −80°C for at least 20 min. Next, 20 μg of Glycoblue was added and the mixture was spun down at maximum speed for at least 20 min at 4°C. The pellet was washed with cold 70% ethanol and air‐dried before resuspension in DEPC‐treated water. Depletion of ribosomal RNA was performed on the short fractions and on half of the long fractions using a Ribo‐zero rRNA removal kit (Illumina), according to the supplier’s instructions. The other half of long fractions was processed as Ribo + samples. RNA was stored at −80°C until the moment of use.
The long fractions (with and without rRNA depletion) were further processed with the NEBNext Magnesium RNA Fragmentation Module (NEB), as described in the manual. 3 min of fragmentation at 94°C has been established to lead to a peak at approximately 250 nt, appropriate for the final 100 bp paired‐end sequencing. The fragmented RNA was precipitated using ethanol with 20 μg GlycoBlue at −80°C for at least 10 min.
Samples were treated with TURBO DNase (Ambion) in a final volume of 20 μl, according to the manufacturer’s instructions. DNase‐treated samples were bisulphite‐converted using an EZ RNA Methylation Kit (Zymo Research), following the manufacturer’s manual. As a final step before library preparation, a stepwise RNA end repair was carried out using T4 polynucleotide kinase (TaKaRa). A 3′‐dephosphorylation and 5′‐phosphorylation reaction was performed using T4 PNK enzyme (TaKaRa). The enzyme was removed by phenol‐chloroform purification. Library preparation was done using a NEBNext Small RNA Library Prep Set, according to the manufacturer’s protocol. cDNA was amplified with 12 cycles of PCR and purified using the QIAquick PCR Purification Kit (Qiagen). The libraries were size‐selected on a 6% polyacrylamide gel. Compatible barcodes were selected, and samples were pooled in equimolar ratios on multiple lanes in an Illumina HiSeq 2000 platform. A 100 bp paired‐end sequencing approach was used.
Bioinformatics, statistical analyses and methylation calling were performed as described in Legrand et al (2017), utilising the BisRNA software. Adapters were removed from sequenced reads using Cutadapt version 1.8.1 (with options: ‐‐error‐rate = 0.1 ‐‐times = 2 ‐‐overlap = 1 and adapter sequences AGATCGGAAGAGCACACGTCT and GATCGTCGGACTGTAGAACTCTGAAC for forward and reverse reads, respectively (Martin, 2011). Reads were further trimmed of bases with phred quality score < 30 on 5′ and 3′ ends, and reads shorter than 25 nucleotides were discarded (Trimmomatic version 0.36) (Bolger, Lohse, & Usadel, 2014). Reads were aligned uniquely using Bsmap (version 2.87, options: ‐s 12 ‐v 0.03 ‐g 0 ‐w 1000 ‐S 0 ‐p 1 ‐V 1 ‐I 1 ‐n 0 ‐r 2 ‐u ‐m 15 ‐x 1000) (Xi & Li, 2009). Reference sequences were downloaded from Gtrnadb (version ce10), Ensembl (release 90, version WBcel235) and Arb‐Silva (Chan & Lowe, 2016; Kersey et al, 2016; Lee et al, 2018; Quast et al, 2012). End sequence “CCA” was appended to tRNA if missing. Bisulphite‐identical sequences, where only C>T point differences were present, were merged, keeping the C polymorphism. Similarly to Legrand et al (2017), tRNA sequences were further summarised to the most exhaustive yet unambiguous set of sequences, using sequence similarity matrix from Clustal Omega (Sievers et al, 2011). Methylation calling was performed as described in Legrand et al (2017), utilising the BisRNA software. Methylation frequency was calculated as the proportion of cytosines with coverage higher than 10 in three wild‐type and noNSUN replicates and bisulphite non‐conversion ratio higher than 0.1. The measure for reproducibility was the standard error. Deamination rates were calculated as the count of converted cytosines divided by the sum of converted and non‐converted cytosines. This calculation was carried out on nuclear and mitochondrial rRNA. Known methylation sites in rRNA were removed from the calculations. WTBS raw data have been deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE144822.
Targeted bisulphite sequencing
1 μg of total RNA was bisulphite‐modified with the EZ RNA Methylation Kit (Zymo Research). Briefly, samples were first treated with DNase I for 30 min at 37°C in 20 μl volume. The DNAse reaction was stopped and immediately applied to the EZ RNA Methylation Kit (Zymo Research) according to manufacturer’s instructions. Converted RNAs were eluted in 12 μl of distilled water.
Reverse transcription was performed with the purified RNA, and adaptors were added to the amplicons using reverse oligonucleotides designed for the bisulphite‐converted sequences of interest and SuperScript III reverse transcriptase (Invitrogen). cDNA was cleaned from any residual RNA with an RNase H treatment at 37°C for 20 min and then used for PCR amplification and adaptor addition using forward oligonucleotides. Low annealing temperature (58°C) was used to overcome high A‐T content after bisulphite treatment. 4 μl of PCR product was used for ligation and transformation into TOP10 competent cells using the Zero Blunt TOPO PCR Cloning Kit (Invitrogen) according to the manufacturer’s instructions. Following overnight culture, 24 colonies were individually lysed and used for PCR amplification using M13 primers, in order to confirm the presence of the insert at the correct size by DNA electrophoresis. The remaining PCR product (10 clones per condition) was used for Sanger sequencing using T3 primers (Genewiz).
Automated phenotypical characterisation
Viable progeny
Viable progeny refers to the number of progeny able to reach at least the L4 stage within ~ 4 days. Measurements were completed over three 24‐h intervals. First, eggs were prepared by synchronisation via coordinated egg laying. When these animals had grown to the L4 stage, single animals were transferred to fresh plates (day 0). For 3 days, each day (days 1–3), each animal was transferred to a new plate, while the eggs were left on the old plate and allowed to hatch and grow for ~ 3 days, after which, the number of animals on each of these plates was counted (Hodgkin & Barnes, 1991) using a custom animal counting program utilising short video recordings. Animals were agitated by tapping each plate four times; after this, 15 frames were imaged at 1 Hz and the maximum projection was used as a background image. Animals were then detected by movement using the difference in the image between each frame and this background image and counted this way for ten additional frames. The final count was returned as the mode of these counts. This system was tested on plates with fixed numbers of animals and was accurate to within 5%, comparable to human precision. Total viable progeny was reported then as the sum for 3 days. Data are censored for animals that crawled off of plates (Akay et al, 2019).
Single worm growth curves
Populations of Caenorhabditis elegans were synchronised by coordinated egg laying. Single eggs were transferred to individual wells of a multi‐well NGM plate solidified with Gelrite (Sigma). Each well was inoculated with 1 µl of OD 20 E. coli HB101 bacteria (~ 18 million) and imaged periodically using a camera mounted to a computer controlled XY plotter (EleksMaker, Jiangsu, China) which moved the camera between different wells. Images were captured every ~ 11 min for ~ 75 h. Image processing was done in real‐time using custom MATLAB scripts, storing both properties of objects identified as C. elegans, and subimages of regions around detected objects. Body length was calculated using a custom MATLAB (Mathworks, Natick, MA) algorithm, and all other properties were measured using the regionprops function. Growth curves were aligned to egg‐hatching time, which was manually determined for each animal.
Polysome profiling
Synchronised populations of the strains of interest (N2 and noNSUN) were grown until adult stage (3 days) in 140 mm NGM agar plates seeded with concentrated E. coli HB101 cultures at 20°C. Next, the animals were harvested from the plates, transferred to liquid cultures in S‐medium supplemented with E. coli HB101 and incubated at 20°C or 27°C for 4 h in a shaking incubator at 200 rpm before harvesting. Sample preparation for polysome profiling was adapted from Arnold et al (2014). The animals were harvested, washed 3× in cold M9 buffer supplemented with 1 mM cycloheximide and once in lysis buffer (20 mM Tris pH 8.5, 140 mM KCl, 1.5 mM MgCl2, 0.5% Nonidet P40, 2% PTE (polyoxyethylene‐10‐tridecylether), 1% DOC (sodium deoxycholate monohydrate), 1 mM DTT, 1 mM cycloheximide). The animals were pelleted, and as much liquid as possible was removed before the samples were frozen as droplets in liquid nitrogen, using a Pasteur pipette. Frozen droplets were transferred to metallic capsules and cryogenically ground for 25 s in a mixer (Retsch MM 400 Mixer Mill). The resulting frozen powder was stored at −80°C until the moment of use.
Approximately 250 μl of frozen powder was added to 600 μl of lysis buffer and mixed by gentle rotation for 5 min at 4°C. The samples were centrifuged at 10,000 g for 7.5 min, the supernatant was transferred to fresh tubes and the RNA concentration was quantified by Nanodrop. For ribosome footprinting, 400 μl of lysate was treated with 4 μl of DNase I (1 U/μl, Thermo Scientific) and 8 μl of RNase I (100 U/μl, Ambion) for 45 min at room temperature with gentle shaking. 20 μl of RNasin ribonuclease inhibitor (40 U/μl, Promega) was added to quench the reaction when appropriate. The tubes were immediately put on ice, and 220 μl of lysate was loaded into 17.5–50% sucrose gradients and ultracentrifuged for 2.5 h at 165,000 gmax, 4°C in a Beckman SW60 rotor. In parallel, undigested samples used for polysome profiling were equally loaded into sucrose gradients under the same conditions. Gradient fractions were eluted with an ISCO UA‐6 gradient fractionator while the absorbance at 254 nm was continuously monitored. The fraction of polysomes engaged in translation was calculated as the area under the polysomal part of the curve divided by the area below the entire curve.
Ribosome profiling
Sucrose gradient fractions were collected in tubes containing 300 μl of 1 M Tris–HCl pH 7.5, 5 M NaCl, 0.5 M EDTA, 10% SDS, 42% urea and then mixed by vortexing with 300 μl of Phenol‐Chloroform‐Isoamylalcohol (PCL, 24:25:1). Fractions corresponding to 80S monosomes were heated for 10 min at 65°C and centrifuged at 16,000 g for 20 min at room temperature. The upper aqueous phase was transferred to a fresh tube and mixed well with 600 μl of isopropanol and 1 μl of Glycoblue for precipitation overnight at −80°C. RNA was pelleted by centrifugation at 16,000 g for 20 min at 4°C and washed in 800 μl of cold ethanol. After supernatant removal, the pellet was left to dry for 1–2 min and then dissolved in 60 μl of RNase‐free water. RNA concentration and quality were measured by Nanodrop and Tapestation (2200 Agilent R6K), respectively.
A dephosphorylation reaction was performed by adding 7.5 μl T4 polynucleotide kinase (PNK) 10× buffer, 1.5 μl ATP, 1.5 μl RNase OUT, 1.5 μl T4 PNK (TaKaRa) and 3 μl RNase‐free water up to a final volume of 75 μl and incubated for 1.5 h at 37°C. The enzyme was removed by acid‐phenol extraction and the RNA was precipitated with 1/10 volume 3 M sodium acetate pH 5.2, 2.5× volume 100% cold ethanol and 1 μl Glycoblue overnight at −80°C. Pelleted RNA was dissolved in RNase‐free water. For footprint fragments purification, RNA was denaturated for 3 min at 70°C and loaded into a 15% polyacrylamide TBE‐urea gel alongside a small RNA marker. Gel was run for 1 h at 150 V and stained for 10 min with 1:10,000 SYBR Gold in 0.5× TBE. The gel was visualised under UV light, and the region between the 20 nt and 30 nt marks (28–32 nt) was excised with a sterile scalpel. The gel band was crushed into small pieces and incubated in 300 μl of 0.3 M RNase‐free NaCl solution with 2 μl of RNase OUT overnight at 4°C on an Intelli‐Mixer (Elmi). The gel slurry was transferred to a 0.45 μm NanoSep MF Tube (Pall Lifesciences) and centrifuged at maximum speed for 5 min at 4°C. After overnight precipitation with 30 μl of 3 M sodium acetate pH 5.2, 1 μl Glycoblue and 800 μl 100% ethanol, the RNA was dissolved in RNase‐free water.
Libraries were prepared using a NEB NEXT Small RNA Library Prep Set for Illumina (Multiplex compatible) E7330 Kit, following the manufacturer’s instructions. cDNA libraries were purified according to the manual, followed by a QIAQuick PCR Purification Kit and a 6% polyacrylamide gel, where a band of 150 bp (120 bp adapter +28–32 footprint fragments) was excised. Gel extraction was performed as described above for footprint fragments purification. Libraries were sequenced at the Genomics and Proteomics Core Facility of the German Cancer Research Centre (DKFZ), Heidelberg.
Raw reads were assessed for quality using FastQC and Trimmed for low quality bases and adapter sequences using Trimmomatic (version 0.39, parameters—ILLUMINACLIP:2:30:10 SLIDINGWINDOW:4:20 MINLEN:20) (Bolger et al, 2014). SortMERNA was used to remove any rRNA sequences (Kopylova et al, 2012). Remaining reads were uniquely aligned to the C. elegans (WBCel235) reference genome using HISAT2 (version 2.1.0) (Kim et al, 2015). The longest transcript was chosen for each gene from the WBCel235 reference genome and the CDS for these transcripts was extracted. Reads were length stratified and checked for periodicity, only read lengths showing periodicity over the three frames were retained for further analysis (26 bp −30 bp). Reads aligned to the genome were shifted 12 bp from the 5′‐end towards the 3′‐end (Ingolia et al, 2009). Any reads aligned to the first 10 codons of each gene were then removed, and the remaining reads with a 5′ end aligning to a CDS were kept for further analysis (Lecanda et al, 2016).
Bulk codon occupancy in the P‐Site for each codon was calculated as the number of shifted RPFs assigned to the first nucleotide of the codon. This value was then normalised by the frequency of the counts for the same codon in the +1, +2 and +3 codons relative to the A‐Site (Stadler & Fire, 2011). Fold changes were then computed as the normalised bulk codon occupancies for noNSUN/ wild type. Ribosome occupancy for gene in a sample was calculated as the number of shifted in frame RPFs aligned to the CDS of the gene (not including the first 10 codons). These values were inputted into DESeq2 (Love et al, 2014). Translation efficiency was calculated by dividing the ribosome occupancy of each gene (disregarding the first 10 codons) by the mRNA abundance of the same gene. Ribo‐seq raw data have been deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE146256.
RNA sequencing
Input RNA was extracted from aliquots from the samples used for polysome profiling and ribosome footprinting. 100 μl of chloroform were added to the tubes, which were then shaken vigorously for 15 s and incubated at room temperature for 3 min. Samples were centrifuged at 12,000 × g for 15 min at 4°C, and the aqueous phase of the mixture was carefully recovered and transferred to a fresh RNAse‐free tube. RNA was precipitated with 500 μl of cold isopropanol at room temperature for 10 min and then centrifuged at 12,000 g for 15 min at 4°C. The supernatant was carefully removed, the pellet was washed and vortexed with 1 ml of 75% ethanol and centrifuged at 7,500 g for 5 min at 4°C. RNA pellet was air‐dried, dissolved in the appropriate volume of DEPC‐treated water and the concentration, 260/280 and 260/230 ratios were measured by Nanodrop. RNA integrity was evaluated in the Agilent 2200 Tapestation system. RNA was depleted of DNA with a TURBO DNA‐free kit (Invitrogen), according to the manufacturer’s instructions. Libraries were prepared with 750 ng of starting material using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina, following rRNA depletion using a NEBNext rRNA Depletion Kit (Human/Mouse/Rat) (NEB).
Raw reads were assessed for quality using FastQC (Andrews, 2010) and Trimmed for low quality bases and adapter sequences using Trimmomatic (version 0.39, parameters—ILLUMINACLIP:2:30:10 SLIDINGWINDOW:4:20 MINLEN:25) (Bolger et al, 2014). SortMERNA (Kopylova et al, 2012) was used to remove any reads matching rRNA sequences. Remaining reads were aligned to the C. elegans reference genome (WBCel235) using HISAT2 (version 2.1.0, default parameters) (Kim et al, 2015). Read alignments were then counted using HTSeq‐count (Anders et al, 2015) and gene counts inputted into DESeq2 (Love et al, 2014). RNA‐seq raw data have been deposited in the Gene Expression Omnibus (GEO) database under the accession number GSE146256.
Author contributions
Conceptualisation, ICN and EAM; Investigation, ICN, FT, DJ, AGH, FB, AK and AA; Formal analysis, DJ, CL and JP; Writing—Original Draft, ICN; Writing—Review and Editing, ICN, FT, AA, FL and EAM; Supervision: MH, FL and EAM; Funding Acquisition, MH, FL and EAM.
Conflict of interest
E.A.M. is a co‐founder and director of Storm Therapeutics, Cambridge, UK. A.H. is an employee of Storm Therapeutics, Cambridge, UK.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We would like to thank the Gurdon Institute Media Kitchen for their support providing reagents and media. We thank Kay Harnish for the Gurdon Institute Sequencing Facility management. We thank Julie Ahringer’s laboratory for kindly sharing RNAi bacterial clones. We thank the National Bioresource Project (Tokyo, Japan) for providing the nsun‐5 deletion allele. We thank Claudia Flandoli for illustrating our working model. We thank Archana Yerra for manuscript editing. We are grateful to the Miska Laboratory members, especially Kin Man Suen, Eyal Maori, Ragini Medhi and Grégoire Vernaz, for helpful discussions and advice. We are grateful to Miranda Landgraf for her constant support and to Marc Ridyard for laboratory management and maintenance of our nematode collection. This research was supported by a Wellcome Trust Senior Investigator award (104640/Z/14/Z) and Cancer Research UK award (C13474/A27826) to E.A.M ; Conselho Nacional de Desenvolvimento Científico e Tecnológico doctorate scholarship (CNPq, Brazil—205589/2014‐6) to I.C.N.; Deutsche Forschungsgemeinschaft (SPP1784) to F.L. The Miska Laboratory was also supported by core funding from the Wellcome Trust (092096/Z/10/Z, 203144/Z/16/Z) and Cancer Research UK (C6946/A24843).
The EMBO Journal (2021) 40: e105496.
Data availability
BS‐seq: Gene Expression Omnibus (GEO) GSE144822 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144822).
RNA‐seq: Gene Expression Omnibus (GEO) GSE146256 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE146256)
Ribo‐seq: Gene Expression Omnibus (GEO) GSE146256 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE146256)
References
- Abbasi‐Moheb L, Mertel S, Gonsior M, Nouri‐Vahid L, Kahrizi K, Cirak S, Wieczorek D, Motazacker MM, Esmaeeli‐Nieh S, Cremer K et al (2012) Mutations in NSUN2 cause autosomal‐ recessive intellectual disability. Am J Hum Genet 90: 847–855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JM, Cory S (1975) Modified nucleosides and bizarre 5′‐termini in mouse myeloma mRNA. Nature 255: 28–33 [DOI] [PubMed] [Google Scholar]
- Akay A, Jordan D, Navarro IC, Wrzesinski T, Ponting CP, Miska EA, Haerty W (2019) Identification of functional long non‐coding RNAs in C. elegans . BMC Biol 17: 14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amort T, Rieder D, Wille A, Khokhlova‐Cubberley D, Riml C, Trixl L, Jia X‐Y, Micura R, Lusser A (2017) Distinct 5‐methylcytosine profiles in poly(A) RNA from mouse embryonic stem cells and brain. Genome Biol 18: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W (2015) HTSeq‐A Python framework to work with high‐throughput sequencing data. Bioinformatics 31: 166–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. Retrieved from http://www.bioinformatics.babraham.ac.uk/projects/fastqc [Google Scholar]
- Arnold A, Rahman MM, Lee MC, Muehlhaeusser S, Katic I, Gaidatzis D, Hess D, Scheckel C, Wright JE, Stetak A et al (2014) Functional characterization of C. elegans Y‐box‐binding proteins reveals tissue‐specific functions and a critical role in the formation of polysomes. Nucleic Acids Res 42: 13353–13369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Beus E, Brockenbrough JS, Hong B, Aris JP (1994) Yeast NOP2 encodes an essential nucleolar protein with homology to a human proliferation marker. J Cell Biol 127: 1799–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco S, Dietmann S, Flores JV, Hussain S, Kutter C, Humphreys P, Lukk M, Lombard P, Treps L, Popis M et al (2014) Aberrant methylation of tRNAs links cellular stress to neuro‐developmental disorders. EMBO J 33: 1–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J, Sajini A, Tanna H, Cortés‐Garrido R, Gkatza N et al (2016) Stem cell function and stress response are controlled by protein synthesis. Nature 534: 335–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccaletto P, Machnicka MA, Purta E, Piątkowski P, Bagiński B, Wirecki TK, de Crécy‐Lagard V, Ross R, Limbach PA, Kotter A et al (2017) MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res 46: 303–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgeois G, Ney M, Gaspar I, Aigueperse C, Schaefer M, Kellner S, Helm M, Motorin Y (2015) Eukaryotic rRNA modification by yeast 5‐methylcytosine‐methyltransferases and human proliferation‐associated antigen p120. PLoS One 10: e0133321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess AL, David R, Searle IR (2015) Conservation of tRNA and rRNA 5‐methylcytosine in the kingdom Plantae. BMC Plant Biol 15: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PP, Lowe TM (2016) GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res 44: D184–D189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CTY, Dyavaiah M, DeMott MS, Taghizadeh K, Dedon PC, Begley TJ (2010) A quantitative systems approach reveals dynamic control of tRNA modifications during cellular stress. PLoS Genet 6: e1001247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CTY, Pang YLJ, Deng W, Babu IR, Dyavaiah M, Begley TJ, Dedon PC (2012) Reprogramming of tRNA modifications controls the oxidative stress response by codon‐biased translation of proteins. Nat Commun 3: 937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Li A, Sun B‐F, Yang Y, Han Y‐N, Yuan X, Chen R‐X, Wei W‐S, Liu Y, Gao C‐C et al (2019) 5‐methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat Cell Biol 21: 978–990 [DOI] [PubMed] [Google Scholar]
- David R, Burgess A, Parker B, Li J, Pulsford K, Sibbritt T, Preiss T, Searle IR (2017) Transcriptome‐wide Mapping of RNA 5‐Methylcytosine in Arabidopsis mRNAs and non‐coding RNAs. Plant Cell 29: 445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Delft P, Akay A, Huber SM, Bueschl C, Rudolph KLM, Di Domenico T, Schuhmacher R, Miska EA, Balasubramanian S (2017) The profile and dynamics of RNA modifications in animals. ChemBioChem 18: 979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desrosiers R, Friderici K, Rottman F (1974) Identification of methylated nucleosides in messenger RNA from novikoff hepatoma cells. Proc Natl Acad Sci USA 71: 3971–3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelheit S, Schwartz S, Mumbach MR, Wurtzel O, Sorek R (2013) Transcriptome‐wide mapping of 5‐methylcytidine RNA modifications in bacteria, archaea, and yeast reveals m5C within archaeal mRNAs. PLoS Genet 9: e1003602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores JV, Cordero‐Espinoza L, Oeztuerk‐Winder F, Andersson‐Rolf A, Selmi T, Blanco S, Tailor J, Dietmann S, Frye M (2016) Cytosine‐5 RNA methylation regulates neural stem cell differentiation and motility. Stem Cell Reports 8: 112–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Vílchez R, Sevilla A, Blanco S (2019) Post‐transcriptional regulation by cytosine‐5 methylation of RNA. Biochim Biophys Acta Gene Regul Mech 1862: 240–252 [DOI] [PubMed] [Google Scholar]
- Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh C, Zhang X, Golic KG, Jacobsen SE, Bestor TH (2006) Methylation of tRNA Asp by the DNA methyltransferase homolog Dnmt2. Science 311: 395–398 [DOI] [PubMed] [Google Scholar]
- Haag S, Warda AS, Kretschmer J, Günnigmann MA, Höbartner C, Bohnsack MT (2015) NSUN6 is a human RNA methyltransferase that catalyzes formation of m5C72 in specific tRNAs. RNA 21: 1532–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag S, Sloan KE, Ranjan N, Warda AS, Kretschmer J, Blessing C, Hübner B, Seikowski J, Dennerlein S, Rehling P et al (2016) NSUN3 and ABH1 modify the wobble position of mt‐tRNAMet to expand codon recognition in mitochondrial translation. EMBO J 35: 2104–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heissenberger C, Liendl L, Nagelreiter F, Gonskikh Y, Yang G, Stelzer EM, Krammer TL, Micutkova L, Vogt S, Kreil DP et al (2019) Loss of the ribosomal RNA methyltransferase NSUN5 impairs global protein synthesis and normal growth. Nucleic Acids Res 47: 11807–11825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heissenberger C, Rollins JA, Krammer TL, Nagelreiter F, Stocker I, Wacheul L, Shpylovyi A, Tav K, Snow S, Grillari J Rogers AN, Lafontaine DL et al (2020) The ribosomal RNA m5C methyltransferase NSUN‐1 modulates healthspan and oogenesis in Caenorhabditis elegans . eLife 9: e56205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin J, Barnes TM (1991) More is not better: Brood size and population growth in a self‐fertilizing nematode. Proc R Soc B Biol Sci 246: 19–24 [DOI] [PubMed] [Google Scholar]
- Hong B, Brockenbrough JS, Wu P, Aris JP (1997) Nop2p is required for pre‐rRNA processing and 60S ribosome subunit synthesis in yeast. Mol Cell Biol 17: 378–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong B, Wu K, Brockenbrough JS, Wu P, Aris JP (2001) Temperature sensitive nop2 alleles defective in synthesis of 25S rRNA and large ribosomal subunits in Saccharomyces cerevisiae . Nucleic Acids Res 29: 2927–2937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper AK, Phizicky EM (2003) tRNA transfers to the limelight. Genes Dev 17: 162–180 [DOI] [PubMed] [Google Scholar]
- Huang T, Chen W, Liu J, Gu N, Zhang R (2019) Genome‐wide identification of mRNA 5‐methylcytosine in mammals. Nat Struct Mol Biol 26: 380–388 [DOI] [PubMed] [Google Scholar]
- Huber SM, van Delft P, Mendil L, Bachman M, Smollett K, Werner F, Miska EA, Balasubramanian S (2015) Formation and abundance of 5‐hydroxymethylcytosine in RNA. ChemBioChem 23: 752–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber SM, Van Delft P, Tanpure A, Miska EA, Balasubramanian S (2017) 2′‐O‐methyl‐5‐hydroxymethylcytidine: A second oxidative derivative of 5‐methylcytidine in RNA. J Am Chem Soc 139: 1766–1769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS (2009) Genome‐wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman JE, Alfonzo JD (2013) Transfer RNA modifications: nature’s combinatorial chemistry playground. Wiley Interdiscip Rev RNA 4: 35–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Ahringer J (2003) Genome‐wide RNAi screening in Caenorhabditis elegans . Methods 30: 313–321 [DOI] [PubMed] [Google Scholar]
- Kawarada L, Suzuki T, Ohira T, Hirata S, Miyauchi K, Suzuki T (2017) ALKBH1 is an RNA dioxygenase responsible for cytoplasmic and mitochondrial tRNA modifications. Nucleic Acids Res 45: 7401–7415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersey PJ, Allen JE, Armean I, Boddu S, Bolt BJ, Carvalho‐Silva D, Christensen M, Davis P, Falin LJ, Grabmueller C, et al (2016) Ensembl Genomes 2016: more genomes, more complexity. Nucleic Acids Res 44: D574–D580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Rafiq MA, Noor A, Hussain S, Flores JV, Rupp V, Vincent AK, Malli R, Ali G, Khan FS et al (2012) Mutation in NSUN2, which encodes an RNA methyltransferase, causes autosomal‐recessive intellectual disability. Am J Hum Genet 90: 856–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoddami V, Cairns BR (2013) Identification of direct targets and modified bases of RNA cytosine methyltransferases. Nat Biotechnol 31: 458–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoddami V, Yerra A, Mosbruger TL, Fleming AM, Burrows CJ, Cairns BR (2019) Transcriptome‐wide profiling of multiple RNA modifications simultaneously at single‐base resolution. Proc Natl Acad Sci USA 116: 6784–6789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosronezhad N, Hosseinzadeh Colagar A, Mortazavi SM (2015) The Nsun7 (A11337)‐deletion mutation, causes reduction of its protein rate and associated with sperm motility defect in infertile men. J Assist Reprod Genet 32: 807–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL (2015) HISAT: A fast spliced aligner with low memory requirements. Nat Methods 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MY, Redman KL (2002) RNA methyltransferases utilize two cysteine residues in the formation of 5‐methylcytosine. Biochemistry 41: 11218–11225 [DOI] [PubMed] [Google Scholar]
- Komara M, Al‐Shamsi AM, Ben‐Salem S, Ali BR, Al‐Gazali L (2015) A novel single‐nucleotide deletion (c.1020delA) in NSUN2 causes intellectual disability in an emirati child. J Mol Neurosci 57: 393–399 [DOI] [PubMed] [Google Scholar]
- Kopylova E, Noé L, Touzet H (2012) SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28: 3211–3217 [DOI] [PubMed] [Google Scholar]
- Kosi N, Alić I, Kolačević M, Vrsaljko N, Jovanov Milošević N, Sobol M, Philimonenko A, Hozák P, Gajović S, Pochet R et al (2015) Nop2 is expressed during proliferation of neural stem cells and in adult mouse and human brain. Brain Res 1597: 65–76 [DOI] [PubMed] [Google Scholar]
- Lafontaine D, Vandenhaute J, Tollervey D (1995) The 18s rRNA dimethvlase D i m l ~ is required for pre‐ribo ~ omal RNA processing in yeast. Genes Dev 9: 2470–2481 [DOI] [PubMed] [Google Scholar]
- Lafontaine DL, Preiss T, Tollervey D (1998) Yeast 18S rRNA dimethylase Dim1p: a quality control mechanism in ribosome synthesis? Mol Cell Biol 18: 2360–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecanda A, Nilges BS, Sharma P, Nedialkova DD, Schwarz J, Vaquerizas JM, Leidel SA (2016) Dual randomization of oligonucleotides to reduce the bias in ribosome‐profiling libraries. Methods 107: 89–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RYN, Howe KL, Harris TW, Arnaboldi V, Cain S, Chan J, Chen WJ, Davis P, Gao S, Grove C, et al (2018) WormBase 2017: molting into a new stage. Nucleic Acids Res 46: D869–D874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legrand C, Tuorto F, Hartmann M, Liebers R, Jacob D, Helm M, Lyko F (2017) Statistically robust methylation calling for wholetranscriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs. Genome Res 27: 1589–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Li X, Tang H, Jiang B, Dou Y, Gorospe M, Wang W (2017) NSUN2‐mediated m5C methylation and METTL3/METTL14‐mediated m6A methylation cooperatively enhance p21 translation. J Cell Biochem 118: 2587–2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Li H, Long T, Dong H, Wang E‐D, Liu R‐J (2018) Archaeal NSUN6 catalyzes m5C72 modification on a wide‐range of specific tRNAs. Nucleic Acids Res 28: 2041–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Santi DV (2000) m5C RNA and m5C DNA methyl transferases use different cysteine residues as catalysts. Proc Natl Acad Sci USA 97: 8263–8265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long T, Li J, Li H, Zhou M, Zhou XL, Liu RJ, Wang ED (2016) Sequence‐specific and shape‐selective RNA recognition by the human RNA 5‐methylcytosine methyltransferase NSun. J Biol Chem 291: 24293–24303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu ZJ, Yip KY, Wang G, Shou C, Hillier LDW, Khurana E, Agarwal A, Auerbach R, Rozowsky J, Cheng C et al (2011) Prediction and characterization of noncoding RNAs in C. elegans by integrating conservation, secondary structure, and high‐throughput sequencing and array data. Genome Res 21: 276–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal 17: 10 [Google Scholar]
- Martinez FJ, Lee JH, Lee JE, Blanco S, Nickerson E, Gabrie S, Frye M, Al‐Gazali L, Gleeson JG (2012) Whole exome sequencing identifies a splicing mutation in NSUN2 as a cause of a Dubowitz‐like syndrome. J Med Genet 49: 380–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metodiev MD, Spåhr H, Loguercio Polosa P, Meharg C, Becker C, Altmueller J, Habermann B, Larsson NG, Ruzzenente B (2014) NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet 10: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano S, Suzuki T, Kawarada L, Iwata H, Asano K, Suzuki T (2016) NSUN3 methylase initiates 5‐formylcytidine biogenesis in human mitochondrial tRNAMet. Nat Chem Biol 12: 546–551 [DOI] [PubMed] [Google Scholar]
- O’Connor M, Leppik M, Remme J (2018) Pseudouridine‐free Escherichia coli ribosomes. J Bacteriol 200: e00540–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paix A, Folkmann A, Rasoloson D, Seydoux G (2015) High efficiency, homology‐directed genome editing in Caenorhabditis elegans using CRISPR‐Cas9 ribonucleoprotein complexes. Genetics 201: 47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phizicky EM, Alfonzo JD (2010) Do all modifications benefit all tRNAs? FEBS Lett 584: 265–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2012) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 41: D590–D596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren HY, Zhong R, Ding XP, Chen ZY, Jing YL (2015) Investigation of polymorphisms in exon7 of the NSUN7 gene among Chinese Han men with asthenospermia. Genet Mol Res 14: 9261–9268 [DOI] [PubMed] [Google Scholar]
- Salditt‐Georgieff M, Jelinek W, Darnell JE, Furuichi Y, Morgan M, Shatkin A (1976) Methyl labeling of HeLa cell hnRNA: a comparison with mRNA. Cell 7: 227–237 [DOI] [PubMed] [Google Scholar]
- Schaefer M, Pollex T, Hanna K, Tuorto F, Meusburger M, Helm M, Lyko F (2010) RNA methylation by Dnmt2 protects transfer RNAs against stress‐induced cleavage. Genes Dev 24: 1590–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schosserer M, Minois N, Angerer TB, Amring M, Dellago H, Harreither E, Calle‐Perez A, Pircher A, Gerstl MP, Pfeifenberger S et al (2015) Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat Commun 6: 6158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanmugam R, Fierer J, Kaiser S, Helm M, Jurkowski TP, Jeltsch A (2015) Cytosine methylation of tRNA‐Asp by DNMT2 has a role in translation of proteins containing poly‐Asp sequences. Cell Discov 1: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Yang J, Watzinger P, Kötter P, Entian KD (2013) Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res 41: 9062–9076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Lafontaine DLJ (2015) ‘View From A Bridge’: a new perspective on eukaryotic rRNA base modification. Trends Biochem Sci 40: 560–575 [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, et al (2011) Fast, scalable generation of high‐quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spåhr H, Habermann B, Gustafsson CM, Larsson N‐G, Hallberg BM (2012) Structure of the human MTERF4‐NSUN4 protein complex that regulates mitochondrial ribosome biogenesis. Proc Natl Acad Sci USA 109: 15253–15258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM, Preiss T (2012) Widespread occurrence of 5‐methylcytosine in human coding and non‐coding RNA. Nucleic Acids Res 40: 5023–5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler M, Fire A (2011) Wobble base‐pairing slows in vivo translation elongation in metazoans. RNA 17: 2063–2073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemoto C, Spremulli LL, Benkowski LA, Ueda T, Yokogawa T, Watanabe K (2009) Unconventional decoding of the AUA codon as methionine by mitochondrial tRNA Met with the anticodon f 5 CAU as revealed with a mitochondrial in vitro translation system. Nucleic Acids Res 37: 1616–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Fan X, Xing J, Liu Z, Jiang B, Dou Y, Gorospe M, Wengong W (2015) NSun2 delays replicative senescence by repressing p27 (KIP1) translation and elevating CDK1 translation. Aging 7: 1143–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuorto F, Liebers R, Musch T, Schaefer M, Hofmann S, Kellner S, Frye M, Helm M, Stoecklin G, Lyko F (2012) RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat Struct Mol Biol 19: 900–905 [DOI] [PubMed] [Google Scholar]
- Tuorto F, Herbst F, Alerasool N, Bender S, Popp O, Federico G, Reitter S, Liebers R, Stoecklin G, Gröne H‐J et al (2015) The tRNA methyltransferase Dnmt2 is required for accurate polypeptide synthesis during haematopoiesis. EMBO J 34: 2350–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Haute L, Dietmann S, Kremer L, Hussain S, Pearce SF, Powell CA, Rorbach J, Lantaff R, Blanco S, Sauer S et al (2016) Deficient methylation and formylation of mt‐tRNAMet wobble cytosine in a patient carrying mutations in NSUN3. Nat Commun 7: 12039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A, Hofmeister O, Rolland SG, Maiser A, Aasumets K, Schmitt S, Schorpp K, Feuchtinger A, Hadian K, Schneider S et al (2019) Mitochondrial Alkbh1 localizes to mtRNA granules and its knockdown induces the mitochondrial UPR in humans and C. elegans . J Cell Sci 132: jcs223891 [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Tsuruis H, Ueda T, Furushimag R, Takamiyas S, Kitan K, Nishikawall K, Watanabe K (1994) Primary and Higher Order Structures of Nematode (Ascaris suum) Mitochondrial tRNAs Lacking Either the T or D Stem*. J Biol Chem 269: 22902–22906 [PubMed] [Google Scholar]
- Xi Y, Li W (2009) BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10: 232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao T, Wang Y, Luo H, Liu L, Wei G, Chen X, Sun Y, Chen X, Skogerbø G, Chen R (2012) A differential sequencing‐based analysis of the C. elegans noncoding transcriptome. RNA 18: 626–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakubovskaya E, Guja KE, Mejia E, Castano S, Hambardjieva E, Choi WS, Garcia‐Diaz M (2012) Structure of the essential MTERF4:NSUN4 protein complex reveals how an MTERF protein collaborates to facilitate rRNA modification. Structure 20: 1940–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Yang Y, Sun B‐F, Chen Y‐S, Xu J‐W, Lai W‐Y, Li A, Wang X, Bhattarai DP, Xiao W et al (2017) 5‐methylcytosine promotes mRNA export – NSUN2 as the methyltransferase and ALYREF as an m5C reader. Cell Res 27: 606–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Liu Z, Yi J, Tang H, Xing J, Yu M, Tong T, Shang Y, Gorospe M, Wang W (2012) The tRNA methyltransferase NSun2 stabilizes p16INK4 mRNA by methylating the 3′‐untranslated region of p16. Nat Commun 3: 712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorbas C, Nicolas E, Wacheul L, Huvelle E, Heurgue‐Hamard V, Lafontaine DL (2015) The human 18S rRNA base methyltransferases DIMT1L and WBSCR22‐TRMT112 but not rRNA modification are required for ribosome biogenesis. Mol Biol Cell 26: 2080–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
BS‐seq: Gene Expression Omnibus (GEO) GSE144822 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE144822).
RNA‐seq: Gene Expression Omnibus (GEO) GSE146256 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE146256)
Ribo‐seq: Gene Expression Omnibus (GEO) GSE146256 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE146256)