

Abstract
Respiratory syncytial virus (RSV) causes severe pathology of the lower respiratory tract in infants, immunocompromised people, and elderly. Despite decades of research, there is no licensed vaccine against RSV, and many therapeutic drugs are still under development. Detailed understanding of molecular and cellular mechanisms of the RSV infection pathology can accelerate the development of efficacious treatment. Current studies on the RSV pathogenesis are based on the analysis of biopsies from the infected patients; however deeper understanding of molecular and cellular mechanisms of the RSV pathology could be achieved using animal models. Mice are the most often used model for RSV infection because they exhibit manifestations similar to those observed in humans (bronchial obstruction, mucous hypersecretion, and pulmonary inflammation mediated by lymphocytes, macrophages, and neutrophils). Additionally, the use of mice is economically feasible, and many molecular tools are available for studying RSV infection pathogenesis at the molecular and cellular levels. This review summarizes new data on the pathogenesis of RSV infection obtained in mouse models, which demonstrated the role of T cells in both the antiviral defense and the development of lung immunopathology. T cells not only eliminate the infected cells, but also produce significant amounts of the proinflammatory cytokines TNFα and IFNγ. Recently, a new subset of tissue-resident memory T cells (TRM) was identified that provide a strong antiviral defense without induction of lung immunopathology. These cells accumulate in the lungs after local rather than systemic administration of RSV antigens, which suggests new approaches to vaccination. The studies in mouse models have revealed a minor role of interferons in the anti-RSV protection, as RSV possesses mechanisms to escape the antiviral action of type I and III interferons, which may explain the low efficacy of interferon-containing drugs. Using knockout mice, a significant breakthrough has been achieved in understanding the role of many pro-inflammatory cytokines in lung immunopathology. It was found that in addition to TNFα and IFNγ, the cytokines IL-4, IL-5, IL-13, IL-17A, IL-33, and TSLP mediate the major manifestations of the RSV pathogenesis, such as bronchial obstruction, mucus hyperproduction, and lung infiltration by pro-inflammatory cells, while IL-6, IL-10, and IL-27 exhibit the anti-inflammatory effect. Despite significant differences between the mouse and human immune systems, mouse models have made a significant contribution to the understanding of molecular and cellular mechanisms of the pathology of human RSV infection.
Keywords: molecular mechanisms, respiratory syncytial virus, pathogenesis, experimental models
INTRODUCTION
Respiratory syncytial virus (RSV; order Mononegavirales, family Pneumoviridae, genus Orthopneumovirus) is one of the most common pathogens [1] that cause severe upper and lower respiratory tract infection in children [2], elderly [3], and patients with immunodeficiencies [4]. Individuals with bronchial asthma and chronic obstructive pulmonary disease also often suffer from this infection [5]. As many as 3 million RSV-infected children are hospitalized annually, resulting in 66,000-199,000 deaths [6]. The economic impact caused by the RSV infection in the USA alone was estimated to be 1.15 billion US dollars [7].
No efficacious RSV vaccine has been developed so far [8]. Although preparations based on specific monoclonal antibodies (mAbs) are used for immunoprevention, but their wider application is limited by the high cost [9]. New vaccines are being developed; at present, 16 candidate vaccines undergo various stages of clinical trials, six of which have reached phase II [10]. It may be expected that a preventive RSV vaccine will be registered for market in the next few years.
Anti-RSV therapeutics are also being developed, such as virus inhibitors based on small interfering RNAs (siRNAs) [11], nanoparticles [12], peptide compounds [13], and other small molecules [14]. One of the promising approaches is the use of siRNAs which specifically suppress important genes of the virus, thereby blocking viral replication. Among them is ALN-RSV01, which contains siRNA molecules specifically recognizing the viral nucleocapsid-encoding n gene. In clinical studies, the intranasal administration of ALN-RSV01 for 5 days significantly (by 38%) decreased the number of patients with the verified RSV infection [15]. Despite these positive results, Alnylam Pharmaceuticals discontinued the trial for ALN-RSV01, making it difficult to predict when this preparation will be registered.
Understanding molecular and cellular mechanisms of the RSV infection pathogenesis is essential necessary for creating efficacious and safe therapeutics and preventive agents, the development of which is impossible without the use of experimental animal models. Many models of RSV infection have been proposed by now are developed in animals such as mice, rats, ferrets, calves, sheep, chimpanzees, etc. [16, 17]. Although chimpanzees are the only species naturally susceptible to human RSV, mice are commonly used to model this infection [16, 17] due to the ease-of-use and low cost of veterinary care, as well as the availability of diverse scientific tools (mAbs, probes, specialized reagents, and equipment) for revealing mechanisms of the pathogenesis. Here, we summarized new data on the RSV infection pathogenesis obtained in mouse models.
RSV STRUCTURE AND LIFE CYCLE
RSV genome is a single-stranded non-segmented negative-sense RNA molecule that contains 10 genes encoding 11 proteins: NS1, NS2, N, P, M, SH, G, F, M2-1, M2-2, and L (the m gene encodes two proteins – M2-1 and M2-2). Genomic RNA is encapsulated in the nucleocapsid consisting of the N protein, RNA polymerase (L protein), its cofactor (P protein), and M2-1 protein. The M protein surrounds the nucleocapsid and interacts with the lipid bilayer of the virion envelope and the cytoplasmic domain of the F protein. Several RSV glycoproteins are embedded into the envelope, such as the fusion protein F, protein G, and small hydrophobic protein SH. The M2-2 protein and two nonstructural proteins (NS1 and NS2) are lacking in the virion structure [18].
The RSV life cycle begins after virion attachment to the target cell followed by the fusion between the viral and the host cell membranes. The crucial role in this event plays glycoproteins F and G. The G protein exists in two forms. The membrane-bound form enables virion attachment to the cells via binding to the cognate receptor or cell surface attachment factors. Of note, in the recent studies, cell surface proteins that bind the G protein are designated as attachment factors, whereas cell proteins initiating the fusion process between the virus and the cell are designated as cell receptors [13, 18]. The soluble form of the G protein (sG) functions as an antigenic trap for binding the anti-G protein antibodies, which is necessary for evading the host immune system [19].
Some of the most studied attachment factors are heparan sulfates (HSGAGs) belonging to glycosaminoglycans (GAGs). They are disaccharide polymers bound to the transmembrane proteins on the surface of many cell types. The binding between the G protein and HSGAGs occurs via electrostatic interactions of positively charged heparin-binding domain of the G protein to the negatively charged HSGAGs [20]. Other attachment factors have also been identified, e.g., the chemokine CX3CR1 receptor that binds to the CX3C motif in the G protein [21]. The F protein can also be involved in the virion attachment by binding to the factors such as HSGAG [22], ICAM1 [23], EGFR [24], and nucleolin [25] (the latter functions as both the attachment factor and the receptor) [25].
Although the F protein participates in the attachment, its major function is to ensure the fusion between the viral and cell membranes. This glycoprotein is synthesized by the infected host cells as a precursor molecule (F0) to be further proteolytically cleaved into the F1 and F2 subunits covalently bound into a heterodimer. The functional F protein on the virion surface is a trimer composed of F1/F2 heterodimers in the prefusion conformation. The fusion peptide (FP) with the adjacent α-helical HR-N motif is located at the N-terminus of the F1 subunit, whereas the C-terminus contains a transmembrane domain (TM) with the α-helical HR-C motif that ensures protein anchoring in the viral envelope. During the fusion, FP interacts with the membrane of the target cell, resulting in the F protein rearrangement from the prefusion to the postfusion conformation. During such rearrangement, the HR-C and HR-N motifs approach each other and form a hairpin structure that brings the membranes of the host cell and the virion into close contact followed by their final fusion. Recent studies have shown that the virion-cell fusion is a two-step process: macropinocytosis occurs first followed by the immediate membrane fusion inside the endosome [13, 18]. The SH glycoprotein does not play any role in the attachment and fusion events [13, 18].
After the fusion, the viral genome enters the cytosol, where it is transcribed into mRNAs that are translated into the viral proteins. There are three viral proteins involved in the viral genome replication: N, P, and L. The P and L proteins act as subunits of the RNA-dependent RNA polymerase, whereas the N protein binds to the genomic RNA and protects it from cell nucleases. Viral mRNAs are transcribed by the same enzyme involved in the replication of viral genomic RNA [26].
The virion assembly involves cell actin cytoskeleton, which transports viral glycoproteins (F, G, and SH) to the apical surface of the cell plasma membrane. The N protein binds to the de novo synthesized viral genomic RNA together with the RNA-dependent RNA polymerase, thus forming the ribonucleoprotein complex that additionally associates with the M protein, which is also transported to the apical surface of the cell plasma membrane (i.e., to the location of viral glycoproteins). These events result in the formation of viral particles that bud off from the infected cells as mature infectious virions [13, 18].
PATHOLOGICAL CHANGES OF THE RESPIRATORY TRACT INDUCED BY THE RSV INFECTION IN HUMANS
In humans, RSV targets the upper and lower respiratory tract. The infection occurs via direct contact between the viral particles and the respiratory epithelium. After the incubation period (4-5 days), the viral replication proceeds in the nasopharyngeal epithelium, followed by the virus spread into the lower respiratory tract. The severity of the disease varies from minor symptoms of common cold to the airway obstruction, hypoxia, and pneumonia [27].
RSV is mainly replicated in the respiratory ciliated epithelium, as well as in the type II and I alveolar pneumocytes [28]. Histological data on the changes in the respiratory tract have been obtained by the analysis of post-mortem lung samples from patients who died from severe RSV infection. Such changes include perivascular and peribronchial infiltration by mononuclear and T cells, pneumonia symptoms, necrosis of bronchiole epithelium, obstruction of bronchial lumen by cell conglomerates (desquamated epithelial cells, macrophages, and neutrophils), and mucus hypersecretion [28]. On the contrary, CD4+ and CD8+ T cells have been rarely found in the respiratory tract [29]. Analysis of bronchoalveolar lavage (BAL) samples from children with the RSV-induced bronchiolitis revealed a significantly elevated content of neutrophils [30]. In addition, the BAL samples were also enriched in the pro-inflammatory cytokines, such as TNFα, IL-6, IL-1a, IL-8, MIP-1a, MCP-1, RANTES, IFNγ, IL-4, IL-5, IL-10, IL-9, and IL-17 [31, 32], which impact the pathogenesis of the RSV infection.
CURRENT CONCEPTS ON THE PATHOGENESIS OF RSV INFECTION
Both clinical data from patients and results of animal studies (mice, rats, ferrets, calves, sheep, etc.) [16, 17] are used to reveal the pathogenesis of RSV infection. The biggest limitation of RSV studies in animal models is that all used species are only partially sensitive to RSV. The virus poorly replicates in the animal respiratory tract, resulting in mild symptoms. Chimpanzees are the only animal species naturally susceptible to human RSV [16, 17]. However, the use of chimpanzees for modeling the RSV infection is limited due to the high cost of animal care; hence, most of such experiments are conducted in rodents, particularly, mice. A great body of evidence on the molecular and cellular mechanisms of the RSV infection pathogenesis has been obtained in the mouse models. BALB/c mice are commonly used, in which RSV replicates (although in a limited manner) in the respiratory tract (table). It was also found that the viral load is higher and the pathological symptoms are more pronounced in the aged mice [33]. In most studies, mice are infected intranasally at a dose of 104-107 pfu (plaque-forming units) per animal. Generally, the dose of 106 pfu is sufficient to induce visible symptoms (mucus hypersecretion, lung infiltration with pro-inflammatory cells, etc.). The three laboratory RSV strains commonly used for modeling the infection are A2, line 19, and long; however, some studies used clinical isolates of RSV received from patients (table) [34].
Table.
Models used in studying RSV infection pathogenesis
| References | Mouse strain; RSV strain | RSV dose, pfu | Results |
|---|---|---|---|
| Pathological changes in the respiratory tract induced by the RSV infection | |||
| [35] | 20 mouse strains; RSV long | 103-104 | RSV was for the first time shown to reproduce in the mouse respiratory tract: CBA/CaHN and DBA/2N mouse strains showed the highest resistance and the highest susceptibility, respectively, to the viral replication |
| [36, 37] | BALB/c; RSV A2 | 104-106 | RSV-induced pathological changes in the respiratory tract were described: viral reproduction peaked on days 3-4 in the upper respiratory tract and on days 4-6 post-infection in the lower respiratory tract; replication took place in the alveolar rather than bronchial epithelium; the loss of the host activity and body weight (up to 20%) was found to be a crucial manifestation of the virus-induced pathology; lung infiltration with lymphocytes and macrophages occurred along with the thickening of bronchial wall and respiratory epithelium desquamation; multinuclear giant cells were observed in the bronchial epithelium; anti-RSV antibodies developed 2 weeks post-infection |
| [39] | BALB/c; RSV A2 | 106-107 | RSV infection induces bronchial hyperreactivity: viral replication in the respiratory tract peaked on days 3-5 post-infection, but was almost absent on day 7; the maximum bronchial hyperreactivity and mucus hypersecretion were observed on day 5 post-infection and were sustained for 42 days; bronchial hyperreactivity (BHR) significantly correlated with the mucus hypersecretion and intensity of lung infiltration with pro-inflammatory cells, but not with the viral load; pathological changes in the lung tissue were most prominent on days 4-5 post-infection and were sustained for 154 days; RSV infection was associated with the upregulated expression of pro-inflammatory cytokines (TNFα, IL-6, IFNγ, IL-4, IL-10, KC, MIG, RANTES, MIP-1a, and eotaxin) |
| [40] | BALB/c; RSV long | 107 | RSV caused pulmonary inflammation and BHR, which were manifested on days 2-10 days post-infection; RSV induced leukotriene production that mediated mucus hypersecretion and BHR |
| [42] | BALB/cJ; RSV rA2-line19F | 105 | chimeric RSV A2 line 19 strain encoding F protein was created that replicated more actively in the respiratory tract compared to the long and A2 strains; the virus induced the IL-13-dependent mucus hypersecretion and BHR, as well as IFNα production (to a lesser extent) |
| [34] | BALB/cJ; laboratory strains: RSV A2, RSV line 19, RSV long; Clinical RSV isolates: A2001/2-20, A2001/3-12, A1997/12-35, A1998/3-2, A1998/12-21, A2000/3-4 | 105 | different RSV strains caused varying severity of pulmonary pathology: strain A2 was mainly observed in the alveolar regions, whereas virus clinical isolates were found in the bronchial epithelium; clinical isolates A2001/2-20 and A2001/3-12 elicited IL-13-mediated BHR and mucus hypersecretion, as well as more pronounced lung pathology compared to the laboratory strains; the knockout of the IL-13 gene markedly lowered mucus hypersecretion in the mouse bronchial epithelium infected with the viral isolates; RSV induced IL-13 expression in alternatively activated macrophages (and not in Th2 cells), as the IL-13 knockout caused no increase in the IFNγ level in the lungs |
| [38] | BALB/c; rHRSV-Cherry rHRSV-Luc | 5 × 104 | RSV reproduction was visualized in the respiratory tract of live mice: viral reproduction peaked on day 3 and days 4-5 post-infection in the upper and lower respiratory tract, respectively; both lungs were equally affected; RSV at the used dose caused no prominent pathological manifestations |
| [43] | BALB/c; RSV A2 | 5 × 106 TCD50* | RSV-induced BHR developed via the IL-13-independent pathway: RSV infection caused body weight loss, BHR, lung infiltration with macrophages and lymphocytes (but not with eosinophils and neutrophils), and hyperplasia of mucus-producing goblet cells; RSV infection upregulated expression of IFNγ and TNFα, but not of IL-13, which suggested development of BHR via the IL-13-independent pathway |
| Role of T cells in the antiviral defense and lung immunopathology | |||
| [44] | nu/nu BALB/c; RSV A2 | 5 × 104 | for the first time were shown that T cells provide RSV clearance in the lungs |
| [50] | BALB/c; RSV A2 | 2 × 105 | for the first time were shown that T cells play a role in of RSV-induced lung immunopathology: introduction of T cells into infected mice resulted in lower viral clearance, but exacerbated lung pathology |
| [51] | BALB/c; RSV A2 | 107 | depletion of CD4 and CD8 T cells resulted in the elevated viral reproduction accompanied by the decrease in the pathology manifestation |
| [62] | BALB/c; RSV A2 | 105 | the role of tissue-resident memory CD8 T cells (TRM) in the antiviral defense in lungs was shown for the first time: local (intranasal) immunization with the viral antigen resulted in a higher number of mouse lung TRM cells that caused no prominent lung immunopathology compared to other subsets of memory T cells; intranasal administration of CD8 TRM cells induced a marked antiviral effect, whereas administration of CD4 TRM cells suppressed expression of pro-inflammatory TNFα and ameliorated pathological changes, but did not affect viral replication |
| [52] | BALB/c; RSV A2 | 106 | IFNγ and TNFα produced by the memory CD8 T cells did not affect viral replication, but caused pulmonary immunopathology: in the absence of CD4 T cells and antibodies, memory CD8 T cells exhibited protective antiviral response, but induced lung immunopathology after RSV infection; lung immunopathology developed due to IFNγ and TNFα produced by CD8 T cells, as their neutralization with specific mAbs abrogated lung immunopathology, but did not affect the viral load; TRM cells exhibited the antiviral activity without inducing lung immunopathology |
| Role of humoral immunity | |||
| [72] | BALB/c; RSV A2 | 106 | anti-IgG mAbs lowered the viral load and the severity of pulmonary inflammation |
| [70] | BALB/c; RSV A2 | 108 | Higher virus-neutralizing potential was identified for mAb targeting the antigenic site Ø in the F protein vs. mAb against site II (experimental analog of palivizumab) |
| Role of interferons | |||
| [81] | BALB/c; RSV A2 | 105-107 | the role of STAT1 (transcription factor required for type I and type II IFN signaling) in the anti-RSV defense was demonstrated: the STAT1 gene knockout resulted in the elevated viral load in the lungs and promoted BHR, mucus hypersecretion, and infiltration of pro-inflammatory cells into the lungs; production of Muc5ac (major component of bronchial epithelial secretion), as well as of cytokines IL-5, IL-13, IFNγ, and IL-17A was increased in the infected STAT1-deficient mice |
| [63] | C57BL/6 deficient by TLR1, TLR2, TLR4, TLR6, and MyD88; RSV A2 | 2,4 × 106 | RSV was shown to interact with the TLR receptors and to activate innate immunity: the knockout of the TLR2 and TLR6 genes resulted in lower neutrophil infiltration into the lungs and increase in the viral load; RSV interacted with TLR2 and TLR6 and activated NF-κB-dependent production of cytokines and chemokines, but not of IFN-I; RSV induced IFN-I production via the macrophage TLR3 signaling cascade |
| [84] | neonatal BALB/c; RSV A2 | 2 × 105 TCD50 per gram body weight | aberrant interferon response to RSV infection was observed in the neonatal mice, that promoted Th2-mediated lung immunopathology: expression of pulmonary IFNα and IFNβ was 2-4 times higher in adult vs. neonatal mice; the number of IFN-producing plasmacytoid dendritic cells (DCs) was 10 times higher in the lungs of adult vs. neonatal mice; IFNα administration to the neonatal mice prior to the infection resulted in the lower BHR and less pronounced lung immunopathology, as well as caused a decline in the number of Th2 cells and in the amount of cytokines released by these cells (IL-4 and IL-13), while no effect on the level of Th1 cytokines (IFNγ and IL-12) was observed |
| [85] | neonatal BALB/c; RSV A2 | 2 × 105 TCD50 per gram body weight | a dual role for IFNα as an antiviral agent and an activator of adaptive immunity was demonstrated: IFNα administered intranasally to the neonatal mice resulted in a lower viral load after primary RSV infection and in the reduced inflammation in the lungs after reinfection; compared to adult mice, neonatal animals produced no mucosal IgA antibodies in the respiratory tract after the RSV infection, while administration of exogenous IFNα activated production of protective IgA |
| Role of cytokines | |||
| [91] | BALB/cJ; RSV line 19 | 1,5 × 106 | intranasally administrated IFNγ lowered the viral load in the lungs, but promoted production of pro-inflammatory cytokine IL-6 and chemokine CXCL1 |
| [92] | C57BL/6 and BALB/c; RSV A2 | 106 | IFNγ exerted a protective effect during RSV infection: the IFNγ gene knockout resulted in the increased RSV replication after both primary and secondary infections |
| [61] | BALB/c; RSV A2 | 106 | the beneficial role of IL-10 in the RSV-induced lung inflammation was demonstrated: RSV infection elicited production of IL-10 by the airway CD4+ and CD8+ T cells; the IL-10 gene knockout augmented lung immunopathology, but did not affect viral replication |
| [41] | BALB/cJ; RSV A2, RSV line 19 | 105-107 | IL-13 participate in the RSV-induced BHR: both RSV strains (line 19 and A2) induced inflammation in the lungs; RSV line 19, but not A2, induced IL-13 expression, BHR, and mucus hypersecretion; IL-13 gene knockout profoundly reduced BHR and mucus hypersecretion |
| [97] | BALB/c; RSV strains 01/2-20, 12/11-19 and 12/12-6 | 3 × 106 | the role for thymic stromal lymphopoietin (TSLP) in RSV-induced BHR and mucus production was uncovered: RSV infection resulted in the elevated IL-13 expression and increased number of type 2 innate lymphoid (ILC2) cells producing this cytokine; mice deficient by the TSLP receptor (TSLPR) demonstrated upregulated IL-13 production, as well as augmented BHR and mucus hyperproduction; the TSLPR gene knockout did not affect the RSV replication |
| [98] | BALB/c; RSV line 19 | 105 | IL-17A was shown to be involved in the pathogenesis of RSV-induced inflammation: RSV upregulated expression of the IL-17A gene, but not of the IL-17F gene, in CD4+ T cells; neutralization of the IL-17A protein with mAbs or the IL-17A gene knockout did not affect BHR, but lowered hypersecretion and intensity of viral replication in the airways; the IL-17A gene knockout led to the decreased neutrophil infiltration into the lungs |
| [99] | BALB/c; RSV A2 | 8 × 105 | IL-6 and IL-27 were found to suppress RSV-induced inflammation: expression of IL-6 and IL-27 in the respiratory tract was elevated during RSV infection; inactivation of IL-6 and IL-27 by mAbs resulted in the increased count of CD8 T cells producing the pro-inflammatory cytokines TNFα and IFNγ that aggravated pathological events in the lungs; administration of IL-27 promoted Treg maturation and ameliorated inflammatory process |
| [104] | neonatal BALB/c; RSV A2 | 2 × 105 TCD50 per gram body weight | the role of IL-33 in the RSV infection was demonstrated: expression of IL-13 and IL-33 in the lungs of neonatal mice was substantially higher than in adult animals; the content of pulmonary ILC2 cells in the neonatal mice was elevated 3-4-fold compared to adult animals; neutralization of IL-33 with specific mAbs in the neonatal mice resulted in the decreased number of ILC2 cells and lower expression of IL-13 and ameliorated lung pathology, but did not affect the viral load; administration of recombinant IL-33 during RSV infection aggravated pathological events in the respiratory tract; the knockout of ST2 gene (receptor for IL-33) fully abrogated the Th2-mediated lung immunopathology during RSV infection |
| Role of reactive oxygen species (ROS) | |||
| [107] | BALB/c; RSV A2 | 5 × 106 | the gene encoding IRG1 factor and ROS were shown to contribute to the RSV-induced lung inflammation and tissue damage: RSV infection activated IRG1 gene controlling ROS formation in the lungs; IRG1 knockdown with siRNA lowered ROS production in the lungs and abrogates inflammation |
Note: * TCD50, 50% tissue cytopathic dose.
The first studies on the mouse models of RSV infection were published in the late 1970s - early 1980s [35-37]. The very first study by Prince et al. [35] investigated the ability of RSV to replicate in the mouse respiratory tract. The authors infected 20 mouse strains with RSV (strain long) at a dose of ~104 pfu. CBA/CaHN mice was found to be most resistant and DBA/2N turned out to be most susceptible, whereas BALB/c mice displayed intermediate susceptibility to this RSV strain. However, after the ability of RSV to replicate in the mouse respiratory tract had been documented, no studies on the pathological changes occurred during the infection (table) [35].
Several years later, two research groups [36, 37] independently confirmed that RSV (strain A2) was able to infect the upper and lower respiratory tract in BALB/c mice, with the viral replication peaking on days 4-6 post-infection. It was also shown that the viral replication occurred mainly in the alveolar (but not bronchial) epithelium and resulted in the respiratory tract pathological changes, such as lung infiltration with macrophages and neutrophils, thickening of bronchial wall, and desquamation of the respiratory epithelium (table) [36, 37]. Later [38], RSV replication was intravitally visualized in the respiratory tract of live mouse using recombinant RSV rHRSV-Cherry and rHRSV-Luc strains carrying reporter genes. It was confirmed that RSV reproduction in the lower respiratory tract peaked on days 4-5 post-infection, and the virus targeted both lungs equally (table) [38].
The impact of RSV infection on the bronchial hyperreactivity (BHR) has been speculated for a long time. Jafri et al. [39] demonstrated that mice, infected with high dose RSV (~107 pfu) developed BHR that peaked on day 5 post-infection which lasted for more than 40 days. It was assumed that BHR was induced due to the mucus hypersecretion by the bronchial epithelium, as well as to the lung infiltration with pro-inflammatory cells (lymphocytes, macrophages, and neutrophils) detected for over 150 days post-infection. However, no correlation between the BHR and viral load in the lungs was found (table) [39]. One year later, Bitko et al. confirmed that RSV at a dose of 107 pfu triggered BHR in mice, which was believed to be due to the elevated production of leukotrienes interacting with the cognate receptors on the bronchial smooth muscle cells and eliciting bronchoconstriction. Moreover, leukotrienes provoked mucus hypersecretion in the respiratory tract epithelium, which also contributed to the BHR development (table) [40].
However, not all RSV strains trigger BHR and mucus hypersecretion. It was demonstrated that the RSV line 19 strain was more mucogenic and induced prominent BHR even at a dose of 104 pfu, as well as upregulated IL-13 production responsible for the BHR development and mucus hypersecretion (this effect was abrogated in the IL-13 knockout animals) (table) [41]. These data were confirmed in another study that used chimeric RSV rA2-line19F strain (modified strain A2 with the F protein derived from the line 19 strain). Compared to the strain A2, the chimeric virus was able to activate IL-13 production, thus inducing mucus hypersecretion and BHR (table) [42]. Hence, surface glycoprotein F might account for the pathogenicity of RSV strains. This hypothesis was further confirmed by Stokes et al. [34], who investigated the possibility to induce the respiratory tract pathology not only by the laboratory RSV strains (A2, line 19, and long), but also by clinical isolates. Some clinical isolates induced no pathological changes in mice; however, 2 out of 6 examined isolates triggered pathological changes manifested as lung infiltration by the pro-inflammatory cells, as well as IL-13-mediated BHR and mucus production (table) [34]. These data demonstrated different severity of pathological changes observed in clinical practice during epidemiological seasons. Taking into consideration that IL-13 and IFNγ are mainly produced by T cells and act as mutual antagonists, it was proposed that RSV induced IL-13 expression in alternatively activated macrophages rather than in Th2 cells, because the IL-13 gene knockout did not result in the increased pulmonary IFNγ levels after RSV infection in mice [34].
Later, it was shown that the RSV strain A2 triggered BHR and mucus hypersecretion in the bronchial epithelium, but only when applied at a high dose (5 × 106 TCID50/mouse). No elevated IL-13 expression was observed during the infection, so that the aforementioned pathological manifestations have probably developed via the TNFα-mediated pathway (table) [43]. These data indicated that the development of the same pathological changes might occur via different molecular mechanisms.
The impact of T cells. The use of mouse models allowed to demonstrate the role of T cells in the antiviral defense against the RSV infection. Mice lacking the thymus and, therefore, T cells (nu/nu BABLB/c) or exposed to gamma-irradiation were more susceptible to the RSV infection and developed more severe pathology compared to the animals with functionally active T cells [44]. The adoptive transfer of the virus-primed T cells to these mice accelerated RSV clearance from the respiratory tract. Primed T cells exhibited the antiviral activity due to their cytotoxic effects independently of the humoral immunity, because the virus clearance in the lungs occurred in the absence of virus-specific antibodies [44]. However, the presence of virus-specific antibodies accelerated RSV clearance from the lungs, indicating an important role not only of the cell-mediated immunity, but also of the humoral component of the antiviral defense [44]. Similar observations were made in clinical practice. It was found that an increased number of CD8 T cells in BAL samples correlated with lower viral load in human respiratory tract during RSV-infection [45].
Later, the role of T cells in the RSV infection pathogenesis has been repeatedly confirmed (see reviews elsewhere [46-48]). According to the obtained data, during the primary RSV infection, dendritic cells (DCs) engulf and present virus-derived antigens to activate virus-specific CD8 T cells in the local draining lymph node. CD8 T cells then migrate to the respiratory tract and provide protection. Moreover, the number of activated T cells in the bronchial aspirate from the RSV infection patients peaked on day 10 after the onset of disease symptoms; the virus-specific CD8 T cells accumulated mainly in the lung parenchyma rather than circulated in the bloodstream. During the infection, activated T cells produce pro-inflammatory cytokines IFNγ and TNFα. After resolution of infection, CD8 T cells are maintained for several months as memory cells and provide defense against subsequent infections. Upon the reinfection, T cells accumulate in the lungs at a much faster rate primarily due to the migration rather than cell proliferation (Fig. 1a) and exert cytotoxic effects against the virus-infected cells by triggering apoptosis of the latter or by directly destroying them with granzymes and perforins (Fig. 1b). Moreover, production of IFNγ and TNFα initiates the death of the infected cells [46, 47].
Fig. 1.
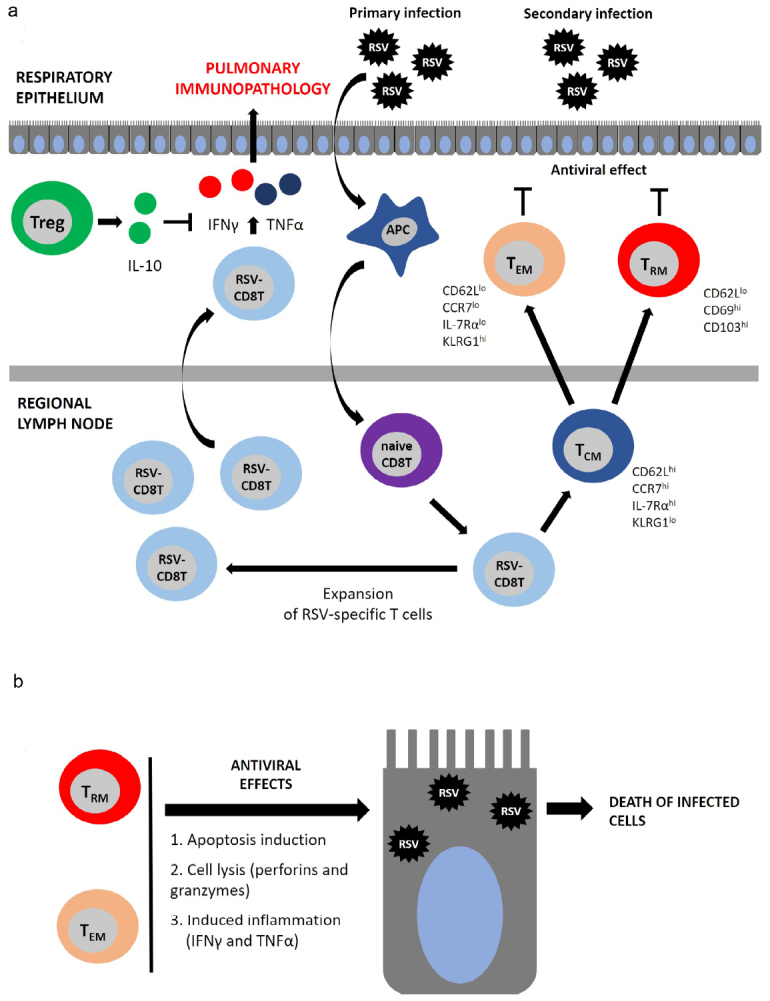
The role of T cells in the antiviral defense and pulmonary immunopathology. a) CD8 T cell-mediated response develops after primary RSV infection. Viral antigens are presented by the antigen-presenting cells (APCs), most commonly, by DCs. Naïve CD8 T cells (naive CD8 T) become activated and differentiate into RSV-specific CD8 T cells (RSV-CD8 T), which then proliferate peaking on days 8-10 post-infection. RSV-CD8 T cells produce large amounts of pro-inflammatory cytokines IFNγ and TNFα, which mediate pulmonary immunopathology. After reaching the plateau, the number of RSV-CD8 T cells declines and they give rise to the memory T cells represented by several phenotypic subsets: 1) TCM – central memory T cells (CD62Lhi/CCR7hi/IL-7Rαhi/KLRG1l°) circulate in the bloodstream and accumulate in the secondary lymphoid tissues; 2) TEM – effector memory T cells (CD62Ll°/CCR7l°/IL-7Rαl°/KLRG1hi) are mainly located in the lungs, but can circulate in the bloodstream; 3) TRM – tissue-resident memory T cells (CD62Ll°/CD69hi/CD103hi) are found solely in the lungs and are unable to exit into the circulation. b) TEM and TRM exert protective antiviral response by triggering apoptosis and direct lysis of infected cells.
Several memory T cell subsets have been identified, including TCM – central memory T cells located in the secondary lymphoid organs, and TEM – effector memory T cells migrating to the respiratory tract and realize effector functions. Both TCM and TEM subsets are simultaneously found in the circulation and the respiratory tract. In addition, another T cell type was found solely in the lungs – TRM (tissue-resident memory CD8 T cells) serving as the first-line defense against repeated infections [45-47, 49] (Fig. 1a).
The role of T cells in the RSV-induced pulmonary pathology. Despite their antiviral activity, T cells can elicit pulmonary immunopathology [50]. It was shown that depletion of CD8 T cells resulted in the elevated viral replication in the lungs, accompanied by the amelioration of pathological changes in the respiratory tract [51]. On the contrary, an increased number of CD8 T cells aggravated pathological events in the lungs, but lowered the viral load [52]. According to the recent data, this type of immunopathology involves cytokines TNFα and IFNγ produced by the virus-specific T cells. TNFα neutralization with specific mAbs prior to the infection resulted in the alleviation of pathological changes in the lungs [52]. Similar mAb-mediated neutralization of IFNγ or the knockout of the IFNγ gene decreased the severity of lung immunopathology after RSV infection (table; Fig. 1a) [52, 53].
It is remarkable that such T cell-mediated immunopathology is typical for the RSV infection, but not for all respiratory viruses. For instance, memory CD8 T cells provide protection against influenza and coronavirus (strain MA15), causing no severe lung immunopathology [54, 55].
The data on the role of T cells in the immunopathology obtained in mouse models correlate with the clinical observations. It was found that the elevated number CD8 T cells in BAL led to a more severe pathology of the respiratory tract of patients with the RSV infection [56]. The increase in the count of CD8 T cells after the bone marrow transplantation correlated with a lower RSV titer in the nasal washings, while the appearance of CD8 T cells negatively affected the patients’ respiratory function, being a marker of pathological changes in the lungs [57].
Regulatory T cells (Treg) play a role in negating the pathology mediated by CD8 T cells. In particular, they alleviate the severity of lung pathology. Treg depletion during RSV infection resulted in the increased number of CD8 T cells, elevated production of TNFα and IFNγ, and more pronounced lung pathology [58]. On the contrary, increasing Treg number led to the opposite effect [59] due to the upregulated production of the anti-inflammatory cytokine IL-10. Neutralization of IL-10 with mAbs or the knockout of the corresponding gene restore lung pathology (table; Fig. 1a) [60, 61].
Recent studies have shown that TRM cells are able to provide the antiviral defense without inducing lung immunopathology. Kinnear et al. [62] intranasally administered TRM cells isolated from infected mouse lungs to naïve mice prior to the RSV infection, which substantially lowered the viral load in the lungs and alleviated lung pathology. In this case, CD8 TRM cells exhibited the antiviral defense, whereas CD4 TRM cells did not affect RSV reproduction, but exhibited the anti-inflammatory effects by downregulating expression of TNFα (table) [62]. It should be noted that formation of TRM cells in the lungs occurred after local (rather than systemic) immunization with viral antigens [49]. Most likely, local (e.g., aerosol) vaccination route will hold promise for preventing RSV infection.
It should be mentioned that the pro-inflammatory cytokine TNFα is produced not only by CD8 T cells as a result of viral infection, but by macrophages as well. Macrophages isolated from TLR2-deficient mice produced much less TNFα after the virus-dependent stimulation compared to the macrophages isolated from the wild-type mice [63]. Respiratory epithelial cells also release TNFα after virus stimulation, as it was shown in the in vitro study using anti-TNFα mAbs [64]. Thus, like CD8 T cells, other cell types (macrophages and epithelial cells) are involved in the RSV-induced lung immunopathology.
The impact of humoral immunity. The role of adaptive humoral immunity in the RSV infection pathogenesis has not been examined in animal models due to the profound differences in the structure and functions of human and mouse antibodies. Numerous data have been obtained in the human RSV infection model (volunteers infected with the purified virus). It was found that class IgA antibodies isolated from the nasal washings and serum IgG antibodies demonstrated the anti-viral protection [65], IgA antibodies exhibiting more prominent and long-lasting anti-RSV effect than IgG antibodies [66].
The neutralizing antibodies are mainly produced against the F protein, which, according to the immunological mapping, has seven major antigenic sites: Ø, I, II, III, IV, V, and VIII. The sites Ø, V, and VIII exist solely in the prefusion conformation of the F protein [67, 68]. It was demonstrated that the majority of virus-neutralizing antibodies (>60%) isolated from the B cells of patients recovered from the RSV infection were directed against the Ø, V [69] and VIII sites [68]. At the same time, antibodies specific to the sites II, III, and IV possess much lower (100 to 1000-fold) virus-neutralizing potential.
It may be due to the fact that the antigenic sites Ø, V and VIII are found in the prefusion rather than postfusion conformation of the F protein. Antibody binding to such sites stabilizes F protein, hindering its rearrangement into the postfusion conformation and preventing the virion-cell fusion [67, 68].
Palivizumab contains antibodies against the antigenic site II found in the postfusion F protein isoform. mAbs against the Ø, V, and VIII sites had a much higher (100-1000-fold) virus-neutralizing potential compared with palivizumab (table) [68, 70]. New data on the antigenic structure of F protein will allow not only to create more efficient palivizumab analogues, but also to design efficacious RSV vaccines.
The neutralizing antibodies have been also developed against another surface viral glycoprotein – G protein, that target its central conserved domain [71]; however, the soluble form of this protein may negate the antiviral effects of these antibodies (table) [72].
IgE antibodies are also induced during the RSV-infection, but they provide no antiviral effect, and, conversely, play an unfavorable role in the infection pathogenesis. RSV-specific IgE antibodies initiate histamine and leukotriene release from the mast cells, thereby eliciting inflammatory reactions [73].
The role of interferons. Three types of interferon have been identified in humans: IFN-I, IFN-II, and IFN-III, each of them exerting biological activity via binding to the cognate receptor. IFN-I and IFN-III are directly involved in the host antiviral defense [74, 75]. Interferon production is induced after interaction of RSV glycoproteins with the TLR molecules (TLR2, TLR6, TLR3, TLR4, and TLR7) found on the surface of leukocytes, plasmacytoid DCs, and alveolar macrophages [76]. The F protein is recognized by TLR4 that elicits IFN-I production [77]. The knockout of the TLR4 gene lowered mouse potential to eliminate RSV [76]. TLR2 and TLR6 receptors are also involved in the recognition of RSV ligands. The knockout of the corresponding genes resulted in the elevated viral load (table; Fig. 2) [63]. Endosomal TLR3 and TLR7 receptors, as well as the cytosolic NLR family member Nod2, recognize viral RNA and trigger IFN-I production [78-80].
Fig. 2.

Immunopathogenesis of RSV infection. RSV infection activates proliferation of CD8 T cells producing IFNγ and TNFα involved in the emergence of major pathology manifestations. IFNγ not only participates in the pathology development, but also accounts for the host antiviral defense. RSV induces necrosis in the respiratory epithelium, resulting in the release of cytokines IL-33 and thymic stromal lymphopoietin (TSLP) that activate ILC2 cells producing their own cytokines (IL-5 and IL-13). IL-5 and IL-13 are involved in the development of pathological manifestations by causing pulmonary eosinophilic inflammation, mucus hypersecretion, and BHR. Polarization of Th2 and Th17 cells occurs during RSV infection due to specific cytokine microenvironment. Th2 cells produce cytokines IL-4, IL-5, and IL-13, which trigger BHR, mucus hypersecretion, and pulmonary eosinophilic inflammation, whereas Th17 cells secrete IL-17A that induces pulmonary neutrophilic inflammation and mucus hypersecretion. Immunoregulatory functions are executed by the cytokine IL-10 released by regulatory T cells (Treg). IL-10 suppresses the pro-inflammatory activity of CD8 T cells. Similar activity was described for IL-6 and IL-27, which are able to directly suppress CD8 T cells and activate Treg. RSV is recognized by surface TLR proteins on the macrophages (MPhs) and plasmacytoid DCs (pDCs) producing limited amounts of type I interferons (IFN-I) with the antiviral activity.
The role of interferons in the RSV infection pathogenesis has been demonstrated in mouse models. Inactivation of the gene for the transcription factor STAT1 (involved in IFN-I/II-mediated signaling) resulted in the elevated viral load in the lungs, more severe BHR and mucus hypersecretion in the bronchial epithelium, and increased lung infiltration by the pro-inflammatory cells [81] (table; Fig. 2). Analysis of patients’ biological samples showed that IFN-I and IFN-III become upregulated within the two days post-infection [82]. It should be mention that type III interferons (IFN-III) include four members: IFN-λ1, IFN-λ2, IFN-λ3 (also known as IL-29, IL-28A, and IL-28B), and IFN-λ4. Selvaggi et al. [82] demonstrated that children hospitalized with the verified RSV infection had a markedly upregulated expression of the IFN-λ1–IFN-λ3 genes in the nasal washings, while expression levels of IFN-λ1 directly correlated with the disease severity and impaired lung function. At the same time, the impact of the RSV infection on the IFN-λ4 expression in the bronchial epithelium has not been studied yet.
Compared to other respiratory viruses, RSV induces mild interferon response, especially in children [83]. The data obtained in the RSV infection model in the neonatal mice confirmed it: infection of mice at the age of 5 days vs. adult mice (6-8 weeks) induced no IFNα and IFNβ expression in the lungs, while administration of exogenous IFNα abrogated pathological changes (table) [84]. Another study showed that IFNα administered intranasally in the neonatal mice before the infection resulted in a markedly decreased viral load in the lungs via elevated production of protective IgA antibodies in the respiratory tract mucosa (table) [85].
On the one hand, aberrant IFN response in early age may be accounted for by a low number of plasmacytoid DCs and macrophages, which are the major source of IFN-I in the respiratory tract [84, 86]. The data obtained in mouse models corroborate the results of the large-scale INFANT clinical study showing that in children, the RSV infection does not induce any prominent IFN response in the respiratory tract mucosa [87]. The aforementioned studies demonstrate the similarity between the human and mouse immune responses to RSV, thereby justifying the use of mouse models to study the properties of innate immunity in humans.
Despite a large body of accumulated data on the antiviral activity of IFN-I and IFN-III, their efficacy for the anti-RSV therapy remains controversial due to the ability of RSV to evade the IFN response [48]. In particular, two viral nonstructural proteins (NS1 and NS2) bind to the cytosolic RIG-I receptor inside the infected cells and suppress the downstream signaling pathways. Moreover, it was found that the viral G protein is also involved in the inhibition of the IFN-related antiviral effects via suppressor proteins SOCS1 and SOCS3, which are activated during RSV infection through the TLR-dependent pathway and block IFN production [88]. Therefore, RSV deploys several mechanisms not only to block the antiviral signaling from the IFN receptors, but also to suppress production of endogenous IFNs, thereby limiting their medical application for treating the RSV infection. In some cases, IFN may even promote more severe course of RSV infection [82].
The impact of cytokines. RSV infection leads to the upregulated production of some pro-inflammatory cytokines (IFNγ, TNFα, IL-4, IL-6, IL-8, IL-17A, etc.) both during the natural infection in humans [89] and in mouse models [39]. The majority of pro-inflammatory cytokines are secreted by the virus-specific CD8 T cells, as well as by macrophages, epithelial cells and ILC2 cells [39, 45, 52].
Patients with the RSV infection have elevated IFNγ levels in nasal washings and blood serum [89]. Moreover, severe infection course is associated with low IFNγ levels [90]. Similar data were obtained in mice. Thus, the intranasal delivery of IFNγ in mice promoted virus clearance in the lungs [91], and conversely, ablation of the IFNγ gene resulted in more pronounced RSV replication (table; Fig. 2) [92]. Taken together, these data suggest the protective antiviral effect of IFNγ. However, this cytokine may also play an unfavorable pathogenetic role, as its administration during RSV infection aggravated inflammation and caused bronchial obstruction in mice (table; Fig. 2) [91], whereas IFNγ neutralization with specific mAbs resulted in the reduced lung immunopathology [52]. Hence, IFNγ exerts protective effects during RSV infection, but its role in the development of lung immunopathology remains controversial.
TNFα also affects lung immunopathology during the RSV infection. In particular, its neutralization with mAbs does not affect RSV replication, but reduces lung inflammation (Fig. 2) [52].
The production of both pro-inflammatory and regulatory (e.g., IL-10) cytokines is increased during the RSV infection. Thus, upregulated IL-10 expression was observed in the mouse models [61] and clinical samples [89]. IL-10 is mainly secreted by Treg cells [61] and exhibit the anti-inflammatory effects that may partially ameliorate the virus-induced inflammation in the respiratory tract. Mice deficient by the IL10 gene experience more severe pulmonary inflammation during the RSV infection, because IL-10 suppresses production of the pro-inflammatory cytokines IFNγ and TNFα by CD8 T cells (table; Fig. 2) [61]. Similar observations were obtained in clinical practice, indicating that downregulation of the IL10 gene and genes involved in the IL-10 signaling pathway correlates with the disease severity [93].
It was also found in mouse models that the IL-4 production was associated with the RSV infection severity [94]. Similar data were obtained by assessing nasal washings from patients with the RSV infection [95]. In contrast, neutralizing IL-4 with mAbs abrogated infection-related inflammation (Fig. 2) [94], because this cytokine suppresses the antiviral cytotolytic activity of CD8 T cells [96]. Another Th2 cytokine – IL-13 – is involved in the development of mucus hypersecretion and BHR; knocking out the IL-13 gene abolished these infection symptoms (table; Fig. 2) [41]. Apparently, IL-13 production by ILC2 cells is regulated by TSLP, because its neutralization downregulates IL-13 expression and reduces mucus hypersecretion (table; Fig. 2) [97].
The cytokine IL-17A, which is produced mainly by Th17 cells, induces pulmonary neutrophil inflammation and mucus hypersecretion in mice infected with RSV (table; Fig. 2) [98]. Secretion by the epithelial cells and macrophages of another pro-inflammatory cytokine, IL-6 (necessary for Th17 skewing), is also elevated during the RSV infection. Despite the pro-inflammatory properties of IL-6, its neutralization by mAbs prior to the infection resulted in the aggravated pathological process. This might be due to the fact that IL-6 neutralization is associated with the increase in the number of virus-specific CD8+ T cells in the lungs and, consequently, with the increased production of the pro-inflammatory cytokines TNFα and IFNγ, which is accompanied by the decreased secretion of regulatory cytokines IL-10 and IL-27, as well as significant reduction in the IL-17A concentration (table; Fig. 2) [99]. Based on these results, no clinical trials assessing the efficacy of anti-IL-6 and anti-IL-17 therapy in RSV infection have been conducted. Likewise, the IL-27 level is upregulated during murine RSV-infection, but its inactivation with specific mAbs results in the aggravated lung pathology due to the increased number of CD8+ T cells producing IFNγ and TNFα. Hence, IL-6 and IL-27 play a similar role in the RSV infection immunopathogenesis by decreasing the activity of CD8+ T cells and activating Tregs (table; Fig. 2) [99].
Despite the lack of studies on the clinical efficacy of anti-IL-6 therapy in patients with the RSV infection, a number of papers have been published recently on the use of anti-IL-6 mAbs (Tocilizumab, Sarilumab, and Siltuximab) for treating COVID-19 patients; however, none of the 10 clinical trials demonstrated prominent favorable effects of such therapy [100, 101]. The use of anti-IL-17 mAbs in the COVID-19 therapy has been also discussed [102], although no related publications are available so far. Anti-IL-17 mAbs were applied to treat pulmonary neutrophil inflammation in moderate to severe bronchial asthma, but no clinical efficacy was demonstrated for Brodalumab (mAbs targeting the IL-17RA subunit of the receptor for IL-17A, IL-17F, and IL-25). Examination of another anti-IL-17 preparation (anti-IL-17A mAb CJM112) is currently underway [103].
Recently, the role of epithelium-produced cytokines (IL-33 and TSLP) in the RSV infection pathogenesis has been revealed. It was found that neonatal mice demonstrated an elevated expression of the pro-inflammatory cytokine IL-33 in the lung tissue [104]. IL-33 activates ILC2 cells that produce large amounts of IL-13, which contributes to the development of type 2 immune response against the RSV infection. IL-33 neutralization or knockout of the gene for its cognate receptor resulted in the alleviated lung pathology, but did not impact the viral replication (table; Fig. 2) [104]. Similar data were obtained in our studies: IL-33 silencing by siRNA resulted in the reduced lung pathology, without affecting the viral load [105]. TSLP as another bronchial epithelial cytokine is also involved in the RSV-induced pulmonary pathology, and its inactivation lowers the number of ILC2 cells in the respiratory tract [97].
Reactive oxygen species. A convincing body of evidence shows that ROS serve as crucial cues in lung inflammation and tissue damage (mainly by affecting bronchial epithelium) in the RSV infection. In 2004, it was demonstrated that RSV triggers production of ROS that induce the inflammatory cascade via activating STAT transcription factors in the epithelial cells [106]. The IRG1 gene is involved in the ROS regulation. RSV infection of the bronchial cells in vitro resulted in the IRG1 activation and elevated ROS production. These data were corroborated in the in vivo experiments in RSV-infected mice. Thus, the IRG1 gene knockdown with specific siRNA lowered ROS production, which reduced lung inflammation (table) [107].
LIMITATIONS OF MOUSE MODELS OF RSV INFECTION
Despite obvious similarities in the features of RSV infection in humans and mice, there are substantial differences. Mouse models are often criticized for the fact that RSV is not a natural mouse pathogen, which accounts for its poor replication in the mouse respiratory tract. Hence, pathological changes are induced only after infecting mice with a high dose of the virus. Moreover, human RSV infection is often accompanied by a marked neutrophilic inflammation of the respiratory tract, so that the percentage of neutrophils in the BAL samples reaches up to 80%, whereas in mice, these leukocytes do not dominate and comprise no more than 20%. Natural human RSV infection induces both Th1- and Th2-dependent immune responses, which are accompanied by the eosinophilic infiltration of the respiratory tract, whereas in mice, it triggers solely Th1 immune response, so that the lungs virtually lack eosinophils [48].
Despite the aforementioned limitations, modeling the RSV infection in mice allows to reproduce main clinically relevant pathological symptoms, such as BHR, mucus hypersecretion, and infiltration of pro-inflammatory cells (mainly lymphocytes and macrophages) into the lungs. The studies conducted using such in vivo models contribute to the understanding molecular and cellular mechanisms of human RSV pathology.
Abbreviations
- BAL
bronchoalveolar lavage
- BHR
bronchial hyperreactivity
- DC
dendritic cell
- HSGAG
heparan sulfate
- mAb
monoclonal antibody
- pfu
plaque-forming units
- RSV
respiratory syncytial virus
- ROS
reactive oxygen species
- siRNA
small interfering RNA
- Treg
regulatory T cells
- TRM
tissue-resident memory T cells
Funding
The study was supported by the Russian Science Foundation (project no. 18-74-10002).
Ethics declarations
The authors declare no conflict of interests. This article does not contain description of studies with the involvement of humans or animal subjects.
References
- 1.Troeger C., Blacker B., Khalil I. A., Rao P. C., Cao J., et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018;18:1191–1210. doi: 10.1016/S1473-3099(18)30310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fauroux B., Simões E. A. F., Checchia P. A., Paes B., Figueras-Aloy J., et al. The burden and long-term respiratory morbidity associated with respiratory syncytial virus infection in early childhood. Infect. Dis. Ther. 2017;6:173–197. doi: 10.1007/s40121-017-0151-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shi T., Denouel A., Tietjen A. K., Campbell I., Moran E., et al. Global disease burden estimates of respiratory syncytial virus – associated acute respiratory infection in older adults in 2015: a systematic review and meta-analysis. J. Infect. Dis. 2019;222:S577–S583. doi: 10.1093/infdis/jiz059. [DOI] [PubMed] [Google Scholar]
- 4.Ebbert J. O., Limper A. H. Respiratory syncytial virus pneumonitis in immunocompromised adults: clinical features and outcome. Respiration. 2005;72:263–269. doi: 10.1159/000085367. [DOI] [PubMed] [Google Scholar]
- 5.Edwards M. R., Walton R. P., Jackson D. J., Feleszko W., Skevaki C., et al. The potential of anti-infectives and immunomodulators as therapies for asthma and asthma exacerbations. Allergy. 2017;73:50–63. doi: 10.1111/all.13257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi T., McAllister D. A., O’Brien K. L., Simoes E. A. F., Madhi S. A., et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: a systematic review and modelling study. Lancet. 2017;390:946–958. doi: 10.1016/S0140-6736(17)30938-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Halasa N. B., Williams J. V., Wilson G. J., Walsh W. F., Schaffner W., Wright P. F. Medical and economic impact of a respiratory syncytial virus outbreak in a neonatal intensive care unit. Pediatr. Infect. Dis. J. 2005;24:1040–1044. doi: 10.1097/01.inf.0000190027.59795.ac. [DOI] [PubMed] [Google Scholar]
- 8.Muralidharan A., Li C., Wang L., Li X. Immunopathogenesis associated with formaldehyde-inactivated RSV vaccine in preclinical and clinical studies. Expert Rev. Vaccines. 2017;16:351–360. doi: 10.1080/14760584.2017.1260452. [DOI] [PubMed] [Google Scholar]
- 9.Wang D., Cummins C., Bayliss S., Sandercock J., Burls A. Immunoprophylaxis against respiratory syncytial virus (RSV) with palivizumab in children: a systematic review and economic evaluation. Health Technol. Assess. 2008;12:1–86. doi: 10.3310/hta12360. [DOI] [PubMed] [Google Scholar]
- 10.Mazur N. I., Higgins D., Nunes M. C., Melero J. A., Langedijk A. C., et al. The respiratory syncytial virus vaccine landscape: lessons from the graveyard and promising candidates. Lancet Infect. Dis. 2018;18:e295–e311. doi: 10.1016/S1473-3099(18)30292-5. [DOI] [PubMed] [Google Scholar]
- 11.Khaitov M. R., Litvin L. S., Shilovskiy I. P., Bashkatova Yu. N., Fayzuloev E. B., Zverev V. V. RNA interference. New approaches to develop antivirals [in Russian] Immunologiya. 2010;31:69–76. [Google Scholar]
- 12.Osminkina L. A., Timoshenko V. Y., Shilovsky I. P., Kornilaeva G. V., Shevchenko S. N., et al. Porous silicon nanoparticles as scavengers of hazardous viruses. J. Nanoparticle Res. 2014;16:1–10. doi: 10.1007/s11051-014-2430-2. [DOI] [Google Scholar]
- 13.Shilovskiy I. P., Andreev S. M., Kozhikhova K. V., Nikolskii A. A., Khaitov M. R. Prospects for the use of peptides against respiratory syncytial virus. Mol. Biol. 2019;53:484–500. doi: 10.1134/S0026893319040125. [DOI] [PubMed] [Google Scholar]
- 14.Heylen E., Neyts J., Jochmans D. Drug candidates and model systems in respiratory syncytial virus antiviral drug discovery. Biochem. Pharmacol. 2017;127:1–12. doi: 10.1016/j.bcp.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 15.DeVincenzo J., Lambkin-Williams R., Wilkinson T., Cehelsky J., Nochur S., et al. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA. 2010;107:8800–8805. doi: 10.1073/pnas.0912186107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Taylor G. Animal models of respiratory syncytial virus infection. Vaccine. 2017;35:469–480. doi: 10.1016/j.vaccine.2016.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altamirano-Lagos M. J., Díaz F. E., Mansilla M. A., Rivera-Pérez D., Soto D., et al. Current animal models for understanding the pathology caused by the respiratory syncytial virus. Front. Microbiol. 2019;10:873. doi: 10.3389/fmicb.2019.00873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Battles M. B., McLellan J. S. Respiratory syncytial virus entry and how to block it. Nat. Rev. Microbiol. 2019;17:233–245. doi: 10.1038/s41579-019-0149-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bukreyev A., Yang L., Collins P. L. The secreted g protein of human respiratory syncytial virus antagonizes antibody-mediated restriction of replication involving macrophages and complement. J. Virol. 2012;86:10880–10884. doi: 10.1128/JVI.01162-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feldman S. ., Hendry R. M., Beeler J. A. Identification of a linear heparin binding domain for human respiratory syncytial virus attachment glycoprotein G. J. Virol. 1999;73:6610–6617. doi: 10.1128/JVI.73.8.6610-6617.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tripp R. A., Jones L. P., Haynes L. M., Zheng H. Q., Murphy P. M., Anderson L. J. CX3C chemokine mimicry by respiratory syncytial virus G glycoprotein. Nat. Immunol. 2001;2:732–738. doi: 10.1038/90675. [DOI] [PubMed] [Google Scholar]
- 22.Feldman S. A., Audet S., Beeler J. A. The fusion glycoprotein of human respiratory syncytial virus facilitates virus attachment and infectivity via an interaction with cellular heparan sulfate. J. Virol. 2000;74:6442–6447. doi: 10.1128/jvi.74.14.6442-6447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Behera A. K., Matsuse H., Kumar M., Kong X., Lockey R. F., Mohapatra S. S. Blocking intercellular adhesion molecule-1 on human epithelial cells decreases respiratory syncytial virus infection. Biochem. Biophys. Res. Commun. 2001;280:188–195. doi: 10.1006/bbrc.2000.4093. [DOI] [PubMed] [Google Scholar]
- 24.Currier M. G., Lee S., Stobart C. C., Hotard A. L., Villenave R., et al. EGFR interacts with the fusion protein of respiratory syncytial virus strain 2-20 and mediates infection and mucin expression. PLoS Pathog. 2016;12:1–22. doi: 10.1371/journal.ppat.1005622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tayyari F., Marchant D., Moraes T. J., Duan W., Mastrangelo P., Hegele R. G. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat. Med. 2011;17:1132–1135. doi: 10.1038/nm.2444. [DOI] [PubMed] [Google Scholar]
- 26.Collins, P. L., Fearns, R., and Graham, B. S. (2013) Respiratory syncytial Virus: virology, reverse genetics, and pathogenesis of disease, in Challenges and Opportunities for Respiratory Syncytial Virus Vaccines. Current Topics in Microbiology and Immunology (Anderson, L., and Graham, B., eds) Vol. 372, Springer, Berlin, Heidelberg, 10.1007/978-3-642-38919-1_1. [DOI] [PMC free article] [PubMed]
- 27.Ralston S., Hill V. Incidence of apnea in infants hospitalized with respiratory syncytial virus bronchiolitis. J. Pediatr. 2009;155:728–733. doi: 10.1016/j.jpeds.2009.04.063. [DOI] [PubMed] [Google Scholar]
- 28.Johnson J. E., Gonzales R. A., Olson S. J., Wright P. F., Graham B. S. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod. Pathol. 2007;20:108–119. doi: 10.1038/modpathol.3800725. [DOI] [PubMed] [Google Scholar]
- 29.Welliver T. P., Garofalo R. P., Hosakote Y., Hintz K. H., Avendano L., et al. Severe human lower respiratory tract illness caused by respiratory syncytial virus and influenza virus is characterized by the absence of pulmonary cytotoxic lymphocyte responses. J. Infect. Dis. 2007;195:1126–1136. doi: 10.1086/512615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith P. K., Wang S. Z., Dowling K. D., Forsyth K. D. Leucocyte populations in respiratory syncytial virus-induced bronchiolitis. J. Paediatr. Child Health. 2001;37:146–151. doi: 10.1046/j.1440-1754.2001.00618.x. [DOI] [PubMed] [Google Scholar]
- 31.Rosenberg H. F., Domachowske J. B. Inflammatory responses to respiratory syncytial virus (RSV) infection and the development of immunomodulatory pharmacotherapeutics. Curr. Med. Chem. 2012;19:1424–1431. doi: 10.2174/092986712799828346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoppelenburg A. J., De Roock S., Hennus M. P., Bont L., Boes M. Elevated Th17 response in infants undergoing respiratory viral infection. Am. J. Pathol. 2014;184:1274–1279. doi: 10.1016/j.ajpath.2014.01.033. [DOI] [PubMed] [Google Scholar]
- 33.Mosquera R. A., Stark J. M., Atkins C. L., Colasurdo G. N., Chevalier J., et al. Functional and immune response to respiratory syncytial virus infection in aged BALB/c mice: a search for genes determining disease severity. Exp. Lung Res. 2014;40:40–49. doi: 10.3109/01902148.2013.859334. [DOI] [PubMed] [Google Scholar]
- 34.Stokes K. L., Chi M. H., Sakamoto K., Newcomb D. C., Currier M. G., et al. Differential pathogenesis of respiratory syncytial virus clinical isolates in BALB/c mice. J. Virol. 2011;85:5782–5793. doi: 10.1128/JVI.01693-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prince G. A., Horswood R. L., Berndt J., Suffin S. C., Chanock R. M. Respiratory syncytial virus infection in inbred mice. Infect. Immun. 1979;26:764–766. doi: 10.1128/IAI.26.2.764-766.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor G., Stott E. J., Hughes M., Collins A. P. Respiratory syncytial virus infection in mice. Infect. Immun. 1984;43:649–655. doi: 10.1128/IAI.43.2.649-655.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graham B. S., Perkins M. D., Wright P. F., Karzon D. T. Primary respiratory syncytial virus infection in mice. J. Med. Virol. 1988;26:153–162. doi: 10.1002/jmv.1890260207. [DOI] [PubMed] [Google Scholar]
- 38.Rameix-Welti M. A., Le Goffic R., Hervé P. L., Sourimant J., Rémot A., et al. Visualizing the replication of respiratory syncytial virus in cells and in living mice. Nat. Commun. 2014;5:5104. doi: 10.1038/ncomms6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jafri H. S., Chávez-Bueno S., Mejías A., Gómez A. M., Ríos A., et al. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J. Infect. Dis. 2004;189:1856–1865. doi: 10.1086/386372. [DOI] [PubMed] [Google Scholar]
- 40.Bitko V., Musiyenko A., Shulyayeva O., Barik S. Inhibition of respiratory viruses by nasally administered siRNA. Nat. Med. 2005;11:50–55. doi: 10.1038/nm1164. [DOI] [PubMed] [Google Scholar]
- 41.Lukacs N. W., Moore M. L., Rudd B. D., Berlin A. A., Collins R. D., et al. Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus. Am. J. Pathol. 2006;169:977–986. doi: 10.2353/ajpath.2006.051055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore M. L., Chi M. H., Luongo C., Lukacs N. W., Polosukhin V. V., et al. A chimeric A2 strain of respiratory syncytial virus (RSV) with the fusion protein of RSV strain line 19 exhibits enhanced viral load, mucus, and airway dysfunction. J. Virol. 2009;83:4185–4194. doi: 10.1128/JVI.01853-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shilovskiy I. P., Nikolskii A. A., Nikonova A. A., Gaisina A. R., Vishniakova L. I., et al. Respiratory syncytial virus infection in mice inducing airway disfunction associated with lung tissue infl ammation as a model of human pathology. Immunologiya. 2019;40:72–83. doi: 10.24411/0206-4952-2019-15008. [DOI] [Google Scholar]
- 44.Cannon M. J., Stott E. J., Taylor G., Askonas B. A. Clearance of persistent respiratory syncytial virus infections in immunodeficient mice following transfer of primed T cells. Immunology. 1987;62:133–138. [PMC free article] [PubMed] [Google Scholar]
- 45.Jozwik A., Habibi M. S., Paras A., Zhu J., Guvenel A., et al. RSV-specific airway resident memory CD8+ T cells and differential disease severity after experimental human infection. Nat. Commun. 2015;6:10224. doi: 10.1038/ncomms10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schmidt M. E., Varga S. M. The CD8 T cell response to respiratory virus infections. Front. Immunol. 2018;9:678. doi: 10.3389/fimmu.2018.00678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schmidt M. E., Varga S. M. Cytokines and CD8 T cell immunity during respiratory syncytial virus infection. Cytokine. 2018;133:15448. doi: 10.1016/j.cyto.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Russell C. D., Unger S. A., Walton M., Schwarze J. The human immune response to respiratory syncytial virus infection. Clin. Microbiol. Rev. 2017;30:481–502. doi: 10.1128/CMR.00090-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morabito K. M., Ruckwardt T. R., Redwood A. J., Moin S. M., Price D. A., Graham B. S. Intranasal administration of RSV antigen-expressing MCMV elicits robust tissue-resident effector and effector memory CD8+ T cells in the lung. Mucosal Immunol. 2017;10:545–554. doi: 10.1038/mi.2016.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cannon M. J., Openshaw P. J. M., Askonas B. A. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J. Exp. Med. 1988;168:1163–1168. doi: 10.1084/jem.168.3.1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Graham B. S., Bunton L. A., Wright P. F., Karzon D. T. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J. Clin. Investig. 1991;88:1026–1033. doi: 10.1172/JCI115362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmidt M. E., Knudson C. J., Hartwig S. M., Pewe L. L., Meyerholz D. K., et al. Memory CD8 T cells mediate severe immunopathology following respiratory syncytial virus infection. PLoS Pathog. 2018 doi: 10.1371/journal.ppat.1006810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ostler T., Davidson W., Ehl S. Virus clearance and immunopathology by CD8+ T cells during infection with respiratory syncytial virus are mediated by IFN-γ. Eur. J. Immunol. 2002;32:2117–2123. doi: 10.1002/1521-4141(200208)32:8<2117::AID-IMMU2117>3,0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 54.Slütter B., Pewe L. L., Kaech S. M., Harty J. T. Lung airway-surveilling CXCR3hi memory CD8+ T cells are critical for protection against influenza A virus. Immunity. 2013;39:939–948. doi: 10.1016/j.immuni.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Channappanavar R., Fett C., Zhao J., Meyerholz D. K., Perlman S. Virus-specific memory CD8 T cells provide substantial protection from lethal severe acute respiratory syndrome coronavirus infection. J. Med. Virol. 2014;88:11034–11044. doi: 10.1128/JVI.01505-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Connors T. J., Ravindranath T. M., Bickham K. L., Gordon C. L., Zhang F., et al. Airway CD8+ T cells are associated with lung injury during infant viral respiratory tract infection. Am. J. Respir. Cell Mol. Biol. 2016;54:822–830. doi: 10.1165/rcmb.2015-0297OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.El Saleeby C. M., Suzich J., Conley M. E., DeVincenzo J. P. Quantitative effects of palivizumab and donor-derived T cells on chronic respiratory syncytial virus infection, lung disease, and fusion glycoprotein amino acid sequences in a patient before and after bone marrow transplantation. Clin. Infect. Dis. 2004;39:17–20. doi: 10.1086/421779. [DOI] [PubMed] [Google Scholar]
- 58.Fulton R. B., Meyerholz D. K., Varga S. M. Foxp3 + CD4 regulatory T cells limit pulmonary immunopathology by modulating the CD8 T cell response during respiratory syncytial virus infection. Eur. J. Immunol. 2010;185:2382–2392. doi: 10.4049/jimmunol.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu J., Ruckwardt T. J., Chen M., Nicewonger J. D., Johnson T. R., Graham B. S. Epitope-specific regulatory CD4 T cells reduce virus-induced illness while preserving CD8 T-cell effector function at the site of infection. J. Virol. 2010;84:10501–10509. doi: 10.1128/JVI.00963-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun J., Madan R., Karp C. L., Braciale T. J. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nat. Med. 2009;15:277–284. doi: 10.1038/nm.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loebbermann J., Schnoeller C., Thornton H., Durant L., Sweeney N. P., et al. IL-10 regulates viral lung immunopathology during acute respiratory syncytial virus infection in mice. PLoS One. 2012 doi: 10.1371/journal.pone.0032371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kinnear E., Lambert L., McDonald J. U., Cheeseman H. M., Caproni L. J., Tregoning J. S. Airway T cells protect against RSV infection in the absence of antibody. Mucosal Immunol. 2018;11:249–256. doi: 10.1038/mi.2017.79. [DOI] [PubMed] [Google Scholar]
- 63.Murawski M. R., Bowen G. N., Cerny A. M., Anderson L. J., Haynes L. M., et al. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J. Virol. 2009;83:1492–1500. doi: 10.1128/JVI.00671-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Morrison P. T., Thomas L. H., Sharland M., Friedland J. S. RSV-infected airway epithelial cells cause biphasic up-regulation of CCR1 expression on human monocytes. J. Leukoc. Biol. 2007;81:87–95. doi: 10.1189/jlb.1006611. [DOI] [PubMed] [Google Scholar]
- 65.Bagga B., Cehelsky J. E., Vaishnaw A., Tomwilkinson T., Meyers R., et al. Effect of preexisting serum and mucosal antibody on experimental respiratory syncytial virus (RSV) challenge and infection of adults. J. Infect. Dis. 2015;212:1719–1725. doi: 10.1093/infdis/jiv281. [DOI] [PubMed] [Google Scholar]
- 66.Habibi M. S., Jozwik A., Makris S., Dunning J., Paras A., et al. Impaired antibody-mediated protection and defective IgA b-cell memory in experimental infection of adults with respiratory syncytial virus. Am. J. Respir. Crit. Care Med. 2015;191:1040–1049. doi: 10.1164/rccm.201412-2256OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Graham B. S. Immunological goals for respiratory syncytial virus vaccine development. Immunity. 2019;51:429–442. doi: 10.1016/j.immuni.2019.08.007. [DOI] [PubMed] [Google Scholar]
- 68.Mousa J. J., Kose N., Matta P., Gilchuk P., Crowe J. E. A novel pre-fusion conformation-specific neutralizing epitope on the respiratory syncytial virus fusion protein. Nat. Microbiol. 2017;2:16271. doi: 10.1038/nmicrobiol.2016.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gilman M. S. A., Castellanos C. A., Chen M., Ngwuta J. O., Goodwin E., et al. Rapid profiling of RSV antibody repertoires from the memory B cells of naturally infected adult donors. Sci. Immunol. 2016 doi: 10.1126/sciimmunol.aaj1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao M., Zheng Z. Z., Chen M., Modjarrad K., Zhang W., et al. Discovery of a prefusion respiratory syncytial virus F-specific monoclonal antibody that provides greater in vivo protection than the murine precursor of palivizumab. J. Virol. 2017;91:e00176–17. doi: 10.1128/JVI.00176-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jones H. G., Ritschel T., Pascual G., Brakenhoff J. P. J., Keogh E., et al. Structural basis for recognition of the central conserved region of RSV G by neutralizing human antibodies. PLoS Pathog. 2018 doi: 10.1371/journal.ppat.1006935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Caidi H., Miao C., Thornburg N. J., Tripp R. A., Anderson L. J., Haynes L. M. Anti-respiratory syncytial virus (RSV) G monoclonal antibodies reduce lung inflammation and viral lung titers when delivered therapeutically in a BALB/c mouse model. Antiviral Res. 2018;154:149–157. doi: 10.1016/j.antiviral.2018.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee Chung H., Jang Y. Y. High serum ige level in the children with acute respiratory syncytial virus infection is associated with severe disease. Clin. Microbiol. Rev. 2016;30:481–502. doi: 10.1128/CMR.00090-16. [DOI] [Google Scholar]
- 74.McNab F., Mayer-Barber K., Sher A., Wack A., O’Garra A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015;15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khaitov M. R., Shilovskiy I. P., Khaitov R. M. Type III interferons [in Russian] Usp. Sovr. Biol. 2010;130:147–153. [Google Scholar]
- 76.Kurt-Jones E. A., Popova L., Kwinn L., Haynes L. M., Jones L. P., et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 77.Zeng R., Cui Y., Hai Y., Liu Y. Pattern recognition receptors for respiratory syncytial virus infection and design of vaccines. Virus Res. 2012;167:138–145. doi: 10.1016/j.virusres.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 78.Rudd B. D., Burstein E., Duckett C. S., Li X., Lukacs N. W. Differential role for TLR3 in respiratory syncytial virus-induced chemokine expression. J. Med. Virol. 2005;79:3350–3357. doi: 10.1128/JVI.79.6.3350-3357.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lukacs N. W., Smit J. J., Mukherjee S., Morris S. B., Nunez G., Lindell D. M. Respiratory virus-induced TLR7 activation controls IL-17–associated increased mucus via IL-23 regulation. Eur. J. Immunol. 2010;185:2231–2239. doi: 10.4049/jimmunol.1000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bhoj V. G., Sun Q., Bhoj E. J., Somers C., Chen X., et al. MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA. 2008;105:14046–14051. doi: 10.1073/pnas.0804717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hashimoto K., Durbin J. E., Zhou W., Collins R. D., Ho S. B., et al. Respiratory syncytial virus infection in the absence of STAT1 results in airway dysfunction, airway mucus, and augmented IL-17 levels. J. Allergy Clin. Immunol. 2005;116:550–557. doi: 10.1016/j.jaci.2005.03.051. [DOI] [PubMed] [Google Scholar]
- 82.Selvaggi C., Pierangeli A., Fabiani M., Spano L., Nicolai A., et al. Interferon lambda 1-3 expression in infants hospitalized for RSV or HRV associated bronchiolitis. J. Infect. 2014;68:467–477. doi: 10.1016/j.jinf.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun Y., Jain D., Koziol-White C. J., Genoyer E., Gilbert M., et al. Immunostimulatory defective viral genomes from respiratory syncytial virus promote a strong innate antiviral response during infection in mice and humans. PLoS Pathog. 2015 doi: 10.1371/journal.ppat.1005122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cormier S. A., Shrestha B., Saravia J., Lee G. I., Shen L., et al. Limited type I interferons and plasmacytoid dendritic cells during neonatal respiratory syncytial virus infection permit immunopathogenesis upon reinfection. J. Med. Virol. 2014;88:9350–9360. doi: 10.1128/JVI.00818-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hijano D. R., Siefker D. T., Shrestha B., Jaligama S., Vu L. D., et al. Type I interferon potentiates IgA immunity to respiratory syncytial virus infection during infancy. Sci. Rep. 2018;8:11034. doi: 10.1038/s41598-018-29456-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Goritzka M., Makris S., Kausar F., Durant L. R., Pereira C., et al. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J. Exp. Med. 2015;212:699–714. doi: 10.1084/jem.20140825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scotta M. C., Machado D. G., Oliveira S. G., de Moura A., Estorgato G. R., et al. Evaluation of nasal levels of interferon and clinical severity of influenza in children. J. Clin. Virol. 2019;114:37–42. doi: 10.1016/j.jcv.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 88.Yu C. F., Peng W.-M., Schlee M., Barchet W., Eis-Hübinger A. M., et al. SOCS1 and SOCS3 target IRF7 degradation to suppress TLR7-mediated type I IFN production of human plasmacytoid dendritic cells. Eur. J. Immunol. 2018;200:4024–4035. doi: 10.4049/jimmunol.1700510. [DOI] [PubMed] [Google Scholar]
- 89.Christiaansen A. F., Syed M. A., Ten Eyck P. P., Hartwig S. M., Durairaj L., et al. Altered Treg and cytokine responses in RSV-infected infants. Pediatr. Res. 2016;80:702–709. doi: 10.1038/pr.2016.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thwaites R. S., Coates M., Ito K., Ghazaly M., Feather C., et al. Reduced nasal viral load and IFN responses in infants with respiratory syncytial virus bronchiolitis and respiratory failure. Am. J. Respir. Crit. Care Med. 2018;198:1074–1084. doi: 10.1164/rccm.201712-2567OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eichinger K. M., Empey K. M. Data describing IFNγ-mediated viral clearance in an adult mouse model of respiratory syncytial virus (RSV) Data Brief. 2017;14:272–277. doi: 10.1016/j.dib.2017.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee Y. M., Miyahara N., Takeda K., Prpich J., Oh A., et al. IFN-γ production during initial infection determines the outcome of reinfection with respiratory syncytial virus. Am. J. Respir. Crit. Care Med. 2008;177:208–218. doi: 10.1164/rccm.200612-1890OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mejias A., Dimo B., Suarez N. M., Garcia C., Suarez-Arrabal M. C., et al. Whole blood gene expression profiles to assess pathogenesis and disease severity in infants with respiratory syncytial virus infection. PLoS Med. 2013 doi: 10.1371/journal.pmed.1001549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tang Y. W., Graham B. S. Anti-IL-4 treatment at immunization modulates cytokine expression, reduces illness, and increases cytotoxic T lymphocyte activity in mice challenged with respiratory syncytial virus. J. Clin. Invest. 1994;94:1953–1958. doi: 10.1172/JCI117546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Vu L. D., Siefker D., Jones T. L., You D., Taylor R., et al. Elevated levels of type 2 respiratory innate lymphoid cells in human infants with severe respiratory syncytial virus bronchiolitis. Am. J. Respir. Crit. Care Med. 2019;200:1414–1423. doi: 10.1164/rccm.201812-2366OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bukreyev A., Belyakov I. M., Prince G. A., Yim K. C., Harris K. K., et al. Expression of interleukin-4 by recombinant respiratory syncytial virus is associated with accelerated inflammation and a nonfunctional cytotoxic T-lymphocyte response following primary infection but not following challenge with wild-type virus. J. Virol. 2005;79:9515–9526. doi: 10.1128/JVI.79.15.9515-9526.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stier M. T., Bloodworth M. H., Toki S., Newcomb D. C., Goleniewska K., et al. Respiratory syncytial virus infection activates IL-13-producing group 2 innate lymphoid cells through thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2016;138:814–824. doi: 10.1016/j.jaci.2016.01.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mukherjee S., Lindell D. M., Berlin A. A., Morris S. B., Shanley T. P., et al. IL-17Induced pulmonary pathogenesis during respiratory viral infection and exacerbation of allergic disease. Am. J. Pathol. 2011;179:248–258. doi: 10.1016/j.ajpath.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pyle C. J., Uwadiae F. I., Swieboda D. P., Harker J. A. Early IL-6 signalling promotes IL-27 dependent maturation of regulatory T cells in the lungs and resolution of viral immunopathology. PLoS Pathog. 2017 doi: 10.1371/journal.ppat.1006640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Antwi-Amoabeng D., Kanji Z., Ford B., Beutler B. D., Riddle M. S., Siddiqui F. Clinical outcomes in COVID-19 patients treated with tocilizumab: an individual patient data systematic review. J. Med. Virol. 2020 doi: 10.1002/jmv.26038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lan S. H., Lai C. C., Huang H. T., Chang S. P., Lu L. C., Hsueh P. R. Tocilizumab for severe COVID-19: a systematic review and meta-analysis. Int. J. Antimicrob. Agents. 2020;56:106103. doi: 10.1016/j.ijantimicag.2020.106103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pacha O., Sallman M. A., Evans S. E. COVID-19: a case for inhibiting IL-17? Nat. Rev. Immunol. 2020;20:345–346. doi: 10.1038/s41577-020-0328-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shilovskiy I., Nikolskii A., Kurbacheva O., Khaitov M. Modern view of neutrophilic asthma molecular mechanisms and therapy. Biochemistry (Moscow) 2020;85:854–868. doi: 10.1134/S0006297920080027. [DOI] [PubMed] [Google Scholar]
- 104.Saravia J., You D., Shrestha B., Jaligama S., Siefker D., et al. Respiratory syncytial virus disease is mediated by age-variable IL-33. PLoS Pathog. 2015 doi: 10.1371/journal.ppat.1005217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Krasnikh L. M., Gaisina A. R., Shilovskiy I. P., Nikonova A. A., Mitin A. N., et al. The study of pharmacological efficiency of sirna tarfeted to il-33 on the mouse model of virus-induced exacerbations of bronchial asthma. Russ. J. Biopharm. 2018;10:49–55. [Google Scholar]
- 106.Liu T., Castro S., Brasier A. R., Jamaluddin M., Garofalo R. P., Casola A. Reactive oxygen species mediate virus-induced STAT activation: role of tyrosine phosphatases. J. Biol. Chem. 2004;279:2461–2469. doi: 10.1074/jbc.M307251200. [DOI] [PubMed] [Google Scholar]
- 107.Ren K., Lv Y., Zhuo Y., Chen C., Shi H., et al. Suppression of IRG-1 reduces inflammatory cell infiltration and lung injury in respiratory syncytial virus infection by reducing production of reactive oxygen species. J. Virol. 2016;90:7313–7322. doi: 10.1128/JVI.00563-16. [DOI] [PMC free article] [PubMed] [Google Scholar]