

SUMMARY
Tumor and stromal interactions consist of reciprocal signaling through cytokines, growth factors, direct cell-cell interactions, and extracellular vesicles (EVs). Small EVs (≤200 nm) have been considered critical messengers of cellular communication during tumor development. Here, we demonstrate that gain-of-function (GOF) p53 protein can be packaged into small EVs and transferred to fibroblasts. GOF p53 protein is selectively bound by heat shock protein 90 (HSP90), a chaperone protein, and packaged into small EVs. Inhibition of HSP90 activity blocks packaging of GOF, but not wild-type, p53 in small EVs. GOF p53-containing small EVs result in their conversion to cancer-associated fibroblasts. In vivo studies reveal that GOF p53-containing small EVs can enhance tumor growth and promote fibroblast transformation into a cancer-associated phenotype. These findings provide a better understanding of the complex interactions between cancer and stromal cells and may have therapeutic implications.
Graphical abstract
In Brief
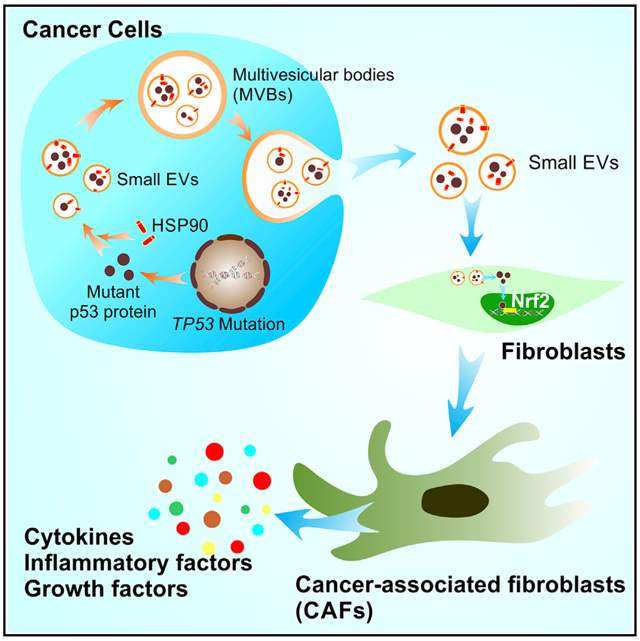
Ma et al. report that gain-of-function (GOF) p53 protein can be packaged into small EVs and transferred to stromal fibroblasts. The packaging of GOF p53 into small EVs is regulated by HSP90. Small EVs with GOF p53 activate Nrf2-mediated pathways in fibroblasts and induce their conversion to a cancer-associated phenotype.
INTRODUCTION
Small extracellular vesicles (EVs) have emerged as important modes of extracellular communication (Tkach and Théry, 2016). These small (≤200 nm) secreted vesicles are characterized by high levels of CD63 and heat shock protein (HSP) expression (Kowal et al., 2016; Pols and Klumperman, 2009; Théry et al., 2002) while canonically lacking intracellular content such as the endoplasmic reticular protein GRP94. Small EVs can transport a wide range of molecules such as microRNAs, translatable mRNAs, DNA, and fully functional proteins (Gyuris et al., 2019; Jeppesen et al., 2019; Thakur et al., 2014; van Niel et al., 2018; Zhang et al., 2015). Considering their multi-functional role in cellular processes, small EVs are increasingly recognized as major players in cancer pathogenesis, including promoting a pro-tumor microenvironment (Azmi et al., 2013; Bell and Taylor, 2016; Hong et al., 2017; Wendler et al., 2017). Isolating and targeting the specific population of EVs that induce a tumor-permissive microenvironment through the exchange of materials with tumor-associated stroma is an important step in decreasing tumor progression and metastasis.
TP53 is the most commonly mutated gene in cancer, with more than 50% of all tumors containing TP53 mutations, resulting in loss or inactivation of its protein product (Olivier et al., 2010). Researchers have identified various p53 mutations that result in dominant-negative inhibition of wild-type (WT) p53 expression (Milner and Medcalf, 1991). Whereas many of these mutations result in functional inactivation of the p53 protein, certain mutations produce a gain-of-function (GOF) phenotype, resulting in pathogenic effects beyond loss of p53 function (Oren and Rotter, 2010). Considering these effects of p53 signaling, packaging of mutant p53 protein into EVs or spread of p53 protein between cells via an alternate mechanism can impact a variety of stromal cell processes, producing alterations in the tumor microenvironment. TP53R273H mutation has been reported in exosomal DNA from patients with pancreatic ductal adenocarcinoma or intraductal papillary mucinous neoplasm, suggesting that small EVs are potential biomarkers of cancer risk (Yang et al., 2017).
Several studies have shown that GOF p53 protein can be detected in cancer-associated fibroblasts (CAFs) (Addadi et al., 2010; Du and Che, 2017; Hosein et al., 2010). Importantly, expression of GOF p53 in fibroblasts promotes their conversion to the cancer-associated phenotype, and tumor cells co-injected with mutant p53-expressing fibroblasts exhibit increased growth and metastasis (Addadi et al., 2010). However, the mechanisms by which fibroblasts acquire mutant p53 are not well understood. Some investigators have suggested that the presence of mutant p53 protein in fibroblasts is a possible result of genomic instability (Liu et al., 1996). One study showed that stromal fibroblasts can harbor TP53 mutation in sporadic breast cancer and accelerate the disease process (Patocs et al., 2007). Although this finding was challenged by a following study (Campbell et al., 2008), the expression of mutant p53 protein in fibroblasts strongly suggests that exogenous p53 sources could contribute to this observation.
As described herein, we found that the presence of p53 in cancer-derived small EVs along with GOF p53 protein was substantially greater than that of WT p53. We also demonstrated the capacity for GOF p53 protein to be transferred from tumor cells to normal fibroblasts. By investigating the change in gene expression in fibroblasts after treatment with small EVs with or without GOF p53 protein, we observed that expression of multiple CAF-associated genes was increased after treatment with GOF p53-containing small EVs. Furthermore, the interaction of HSP90 with GOF p53, but not WT p53, was important for the packaging of p53 into small EVs. Additionally, in tumor-bearing mice, GOF p53-containing EVs resulted in increased tumor size as well as increased CAF-associated gene expression. These results demonstrate the relevance of small EVs containing GOF p53 in promoting a pro-tumor microenvironment through the conversion of fibroblasts to CAFs, as well as a molecular mechanism by which selective packaging of GOF p53 in small EVs occurs.
RESULTS
GOF p53 protein is present in fibroblasts and is packaged in small EVs
To investigate whether fibroblasts in the tumor microenvironment contain mutant p53 protein, we examined a tissue microarray of high-grade serous ovarian cancer (HGSOC) samples based on immunohistochemistry (IHC) because HGSOC is characterized most frequently with TP53 mutation (96%) (The Cancer Genome Atlas Research Network, 2011). In the patients with a missense mutation of TP53 (indicated by strong and diffuse immune-expression of p53) (Yemelyanova et al., 2011), we found p53-positive staining in adjacent regions of fibroblasts (Figure 1A). To further illustrate whether hotspot mutant p53 protein can be found in fibroblasts, we obtained patient tumor tissues and performed whole-genome sequencing on those samples (Lee et al., 2020) (Table S1). By carrying out IHC staining using a p53 (DO-1) antibody, we demonstrated positive p53 staining in fibroblasts in tumor samples harboring hotspot mutations (TP53Y234C [n = 1], TP53G245D [n = 1], TP53I195T [n = 1], TP53R273H [n = 1], TP53R282W [n = 1], and TP53Y163C [n = 1]) (Figure 1B) (Baugh et al., 2018; Tuna et al., 2020). In addition, there were p53-positive fibroblasts in tumors with less common mutations (TP53V216M and TP53K132R, n = 1, respectively) (Figure S1A). However, we did not observe any p53-positive fibroblasts in patients with TP53C176F (n = 2) and TP53C135W (n = 1) mutations (Figure S1A). Tumors with WT TP53 (n = 2), nonsense mutation (n = 1), or a splice site mutation (X187, n = 1) did not show positive p53 staining in fibroblasts (Figure S1B).
Figure 1. Presence of p53 protein in fibroblasts and small EVs.

(A) Immunohistochemical staining of tumor (left panel) and stromal (right panel) cells for p53 in patient-derived high-grade serous ovarian cancer (HGSOC). Red arrows show p53-positive fibroblasts. Scale bars, 500 μm (left) and 100 μm (right).
(B) Immunohistochemical staining for p53 protein in HGSOC samples with hotspot TP53 mutations. Red arrows show p53-positive fibroblasts. Scale bars, 100 μm (left) and 50 μm (right).
(C) Size of the small EVs used as determined via nanoparticle tracking analysis (NTA). Transmission electron microscopy (TEM) images of small EVs and western blotting for the small EV-positive (CD63, TSG101, and Alix) and negative (GRP94) markers. The small EVs were isolated from HT29 cells. Scale bar, 100 nm
(D) Expression of p53 protein in a panel of 10 cell lines and small EVs derived from these cell lines. H1299 is a p53 null cell line that was used as a negative control.
(E) Expression of p53 protein in serum small EV extract (sEVE) from ovarian cancer patients. HT29 whole-cell extract (WCE) was used as a positive control for GRP94.
To determine whether the p53 protein can be secreted within small EVs, we selected 10 cell lines from six common tumor types (breast, prostate, ovarian, uterine, lung, and colorectal) with WT TP53 (A2780, MCF7, RKO, A549, and lymph node carcinoma of the prostate [LNCaP]) or mutant GOF TP53 (KLE [TP53R175H mutation], T47D [TP53L194F mutation], HT29 [TP53R273H mutation], H1975 [TP53R273H mutation], and DU145 [TP53P223L/V274F mutation]) for analysis. We used a TP53 null cell line (H1299) as a negative control. First, we characterized the small EVs isolated from the cell culture supernatant using nanoparticle tracking analysis (NTA), transmission electron microscopy (TEM), and western blotting. The size mode of isolated small EVs was 132 nm according to NTA, and we observed a cup-shaped structure of small EVs via TEM (Figure 1C). We selected CD63, Alix, and TSG101 as positive markers for small EVs and GRP94 as a negative marker for them (Figures 1C-1E and S1C). We used a p53 (DO-1) antibody that can recognize both WT and mutant human p53 protein. The western blotting results revealed that the p53 protein expression level was higher in mutant GOF TP53 cell lines than WT TP53 cell lines (Figure 1D). Small EVs containing p53 protein were detectable in four of the five GOF TP53 cell lines, whereas the WT cell lines had either undetectable or extremely low levels of p53 (Figure 1D). Considering that GOF p53 protein is much more stable than WT p53 protein (Blagosklonny, 2000), these findings suggest that this increased stability is an important factor in the observed increased amounts of GOF p53 in small EVs. Next, we isolated small EVs from serum samples obtained from 13 patients with HGSOC and examined them for the presence of p53 protein using western blotting. Characterization of these small EVs isolated from serum samples is shown in Figure S1D. Overall, we detected p53 protein in 10 of 13 serum samples (Figures 1E and S1E).
GOF p53 protein can be transferred to fibroblasts via small EVs
Next, we subjected the samples to a 30% sucrose/deuterium oxide (D2O) cushion ultracentrifugation (SUC) to purify the small EVs from potential protein contamination. Compared to the p53 level in small EVs isolated by UC, p53 expression was not reduced after SUC purification (Figure 2A); this suggests that free p53 protein is not a contaminant for the p53 detected in small EVs. To clarify whether p53 protein is bound to the surface of small EVs or resides inside them, we treated the purified small EVs with proteinase K (PK; 50 μg/mL) to digest the proteins loosely attached to the EV membrane (Diaz et al., 2018). To digest the proteins located inside vesicles, additional 1% Triton X-100 was added into the samples to permeabilize the EV membrane (Figure 2B). Western blotting results revealed that PK only did not reduce p53 protein levels in the EVs, suggesting that the lipid bilayers of small EVs protect p53. The addition of Triton X-100 resulted in complete digestion of p53 protein as well as EV markers (Figure 2B). We further conducted immune-gold electron microscopy to show that p53 was not a membrane-bound protein in small EVs (Figures 2C and S2A). Immuno-gold staining for CD63, a tetraspanin, and an exosomal marker was used as a positive control (Figure 2C), and negative controls are shown in Figure S2A.
Figure 2. Transfer of p53 protein to fibroblasts from cancer cells via small EVs.

(A) Schema of 30% sucrose/D2O ultracentrifugation assay and western blotting for p53 expression after purification.
(B) Schema of proteinase K digestion assay and western blotting for p53 expression in small EVs. PK, proteinase K; X-100, Triton X-100.
(C) Immuno-gold staining for CD63 and p53 in small EVs isolated from HT29 cells. Small EVs were from the same vial and aliquoted equally for CD63 and p53 staining. Gold size of anti-rabbit IgG for CD63 protein, 25 nm; gold size of anti-mouse IgG for p53 protein, 10 nm. Scale bar, 100 nm.
(D) mCherry-tagged p53 expression in NoF 151 fibroblasts after co-culture with tumor cells. HT29 WCEs were used as a negative control for mCherry-tagged p53 expression.
(E) Expression of PDX1, Cre recombinase, and p53 protein in KPC cells.
(F) Characterization of small EVs isolated from KPC cells according to NTA, TEM, and western blotting. Scale bar, 100 nm.
(G) Schema of in vivo study related to Trp53−/− mice. I.P., intraperitoneal.
(H) The bioluminescence image from the IVIS imager showing the tumor take rate 10 days after cell injection.
(I) The location, tumor weight (g), and number of nodules of KPC xenograft tumors. Data are represented as mean ± SD.
(J) Immunohistochemical staining for p53 protein using a CM5p antibody and fibroblast markers (αSMA and fibronectin) on adjacent slides of KPC xenograft tumor tissues. Scale bars, 100 μm and 50 μm.
Next, we asked whether GOF p53 can be transferred from cancer cells to stromal cells via small EVs. We established HT29 colorectal cancer cells expressing mCherry-tagged p53 or a control vector using lentiviral transduction. First, we confirmed that small EVs derived from HT29-mCherry-p53 cells contain mCherry-tagged p53 protein using western blotting (Figure 2D). We then co-cultured these cells with normal ovarian fibroblast 151 (NoF 151) cells (Yang et al., 2006). As shown in Figure 2D, mCherry-tagged p53 was detectable via western blotting within the fibroblasts co-cultured with mCherry-tagged p53-expressing HT29 cells. In this experiment, we used HT29 cell lysate as a negative control for mCherry-tagged p53 expression. To study the transferring of GOF p53 protein in vivo, we inoculated Trp53−/− mice with KPC (lox-stop-lox [LSL]-KrasG12D/+; LSL-Trp53R172H/+; Pdx1-Cre) pancreatic ductal adenocarcinoma (PDAC) luciferase cells. KPC cells express mutant GOF p53 protein in the presence of pancreatic and duodenal homeobox 1 (PDX1) (Morton et al., 2010) (Figure 2E). PDX1 leads to Cre recombinase expression, which excises LSL to enable the transcription of the mutant Trp53 gene (Lee et al., 2016). We isolated small EVs from KPC cells and demonstrated the presence of mutant p53 protein in small EVs derived from KPC cells (Figure 2F). As shown in Figure 2G, we injected the cells intraperitoneally and sacrificed the mice after 4 weeks to obtain tumor tissues. Ten days after inoculation, we examined the tumor take rate using in vivo imaging system (IVIS) bioluminescence (Figure 2H). The tumors (1.77 ± 0.99 g) were distributed in the pancreas, peritoneum, ovaries, uterus, and small intestine and the total number of nodules was 17.5 ± 10.2 (Figure 2I). We performed IHC staining for p53 protein using CM5p antibody and fibroblast markers (alpha smooth muscle actin [αSMA] and fibronectin) on adjacent paraffin-embedded slides. The results showed that R172H mutant p53 protein was present in fibroblasts of KPC cell xenograft mice (Figures 2J and S2B). There were no p53-positive fibroblasts observed in C57BL/6 mice (n = 3) xenografted with WT Trp53 ID8 cells in which we did not detect p53 expression in their small EVs (Figures S2C). These results demonstrated that p53 protein can be transferred to fibroblasts from mutant GOF TP53 cancer cells.
GOF p53-containing small EVs induce a cancer-associated phenotype
To further determine the impact of GOF p53-containing small EVs on fibroblasts, we generated HT29 cells stably transduced with nontargeting or TP53 short hairpin RNA (shRNA) plasmids. NTA revealed that the size of small EVs isolated from HT29-non-targeting control sequence (ntsh) cells (ntsh-sEVs) and HT29-TP53 shRNA cells (shP53-sEVs) were 113 nm and 117 nm, respectively (Figure 3A). We further characterized the small EVs using a TEM assay and observed a cup-shaped structure in small EVs (Figure 3A). The p53 protein amount was significantly reduced both in whole-cell extract (WCE) and within the small EVs (Figure 3B). A NTA assay revealed no significant differences in small EV secretion after knockdown of TP53 (Figure S3A). Compared with mutant TP53 HT29 cells, WT TP53 RKO colorectal cancer cells secreted fewer EVs per cell (Figure S3A). In addition, the total protein amount carried by small EVs was not impacted by the knockdown of TP53, as revealed in an analysis using the Qubit protein assay kit (Figure S3B). Surprisingly, the protein amount per 106 small EVs in RKO cells was markedly higher than that in mutant TP53 HT29 cells (Figure S3B). We then performed RNA sequencing further to explore the gene signature differences between ntsh-sEVs and shP53-sEVs. The gene expression fold-change (FC) data showed that the most frequently upregulated and downregulated genes were long noncoding RNAs (lncRNAs; e.g., lnc-MYO1F-2 and lnc-XBP-1) in shP53-sEVs relative to ntsh-sEVs (Figures 3C and S3C). Additionally, annotation of RNA-seq data from ntsh-sEVs and shP53-sEVs revealed similar constitution of RNA types in two groups, including lncRNAs (the most enriched ones), precursor RNAs, small nucleolar RNAs, small nuclear RNAs, γRNAs, and others (ribosomal RNA, miscellaneous RNAs, and vault RNAs) (Figure 3D).
Figure 3. The induction of a CAF phenotype by GOF p53 from cancer cells.

(A) Characterizations of small EVs from HT29-ntsh and HT29-shP53 cells using NTA and TEM assays. Scale bar, 100 nm.
(B) The p53 knockdown efficiency in HT29 cells and the p53 expression in relatively small EVs. Pixel densities for p53 bands in comparison with GAPDH and CD63 bands normalized to 1.0 are shown. CD63, Alix, and TSG101 were positive markers for small EVs, whereas GRP94 was a negative marker for them. Data are represented as mean ± SD. *p < 0.05; **p < 0.01.
(C) Volcano plot of differentially enriched variables in small EVs representing the logarithm of fold change (log2) on the x axis and −log10 of the p value on the y axis.
(D) Distribution of different types of RNAs in ntsh-sEVs and shP53-sEVs.
(E) The frequently enriched upregulated pathways, interaction pathways, and the upstream regulators in NoF 151 fibroblasts following treatment with GOF p53-containing small EVs.
(F) FAP, IL1B, TGFB, and CXCL12 mRNA expression levels in NoF 151 fibroblasts after treatment with ntsh-sEVs and shP53-sEVs in the presence or absence of Nrf2 inhibitor (Nrf2i) ML385. Data are represented as mean ± SD. *p < 0.05; **p < 0.01; n.s., not significant.
(G) Results of a human cytokine array performed using supernatants of NoF 151 fibroblasts after treatment with ntsh-sEVs or shP53-sEVs.
Next, we harvested ntsh-sEVs and shP53-sEVs and added them to NoF 151 fibroblasts. After 48 h of treatment with 10 μg/mL small EVs twice daily, the fibroblasts were harvested and subjected to an mRNA array. The mRNA array data revealed that multiple markers of the cancer-associated phenotype were upregulated in the fibroblasts treated with ntsh-sEVs relative to shP53-sEVs (Table S2). These markers include ACTA2 (actin alpha 2, smooth muscle), TGFB (transforming growth factor β [TGF-β]), and PDGFB. The pathways most upregulated in fibroblasts treated with ntsh-sEVs were related to direct p53 effectors, platelet-derived growth factor (PDGF) signaling, and TGF-β signaling (Figure 3E). Furthermore, the upstream regulator of ntsh-sEVs treated fibroblasts whose downstream targets were altered includes TP53, demonstrating that p53-containing small EVs impacts p53 signaling and transcriptional regulation in fibroblasts (Figure 3E). To validate the results of the mRNA array, we treated NoF 151 fibroblasts with ntsh-sEVs or shP53-sEVs for 48 h and performed quantitative real-time reverse-transcriptase polymerase chain reaction (quantitative real-time RT-PCR). The data revealed an upregulation of FAP (fibroblast activation protein alpha) as well as significantly increased expression of CXCL12 (C-X-C motif chemokine ligand 12) and IL1B (interleukin-1β [IL-1β]) after treatment with ntsh-sEVs relative to untreated fibroblasts. Also, we observed no induction of these genes in fibroblasts treated with shP53-sEVs (Figure 3F). Additionally, the small EVs isolated from WT TP53 RKO cells did not exhibit an enhanced effect on these markers in fibroblasts (Figure S3D). A previous report has demonstrated that Nrf2 protein is involved in the regulation of inflammatory cytokine secretion in fibroblasts, and Nrf2 is an important partner for GOF p53 in cancer development (Mantovani et al., 2019; Martin et al., 2011). We then analyzed the Kelch-like erythroid cell-derived protein with cap ‘n’ collar homology (ECH)-associated protein 1 (KEAP-1)-NRF2 interactions with the up- and downregulated networks and annotated functional interactions that are associated with upregulated genes by GOF p53-containing small EVs (Figure 3E). To further characterize the roles of Nrf2 in the induction of CAF phenotype change, we exposed NoF 151 fibroblasts to an Nrf2 inhibitor, ML385 (2 μM for 48 h). The quantitative real-time RT-PCR results suggested that ML385 can significantly abrogate the effects of GOF p53-containing small EVs on the induction of CAF-related gene expression (Figure 3F). We further evaluated cytokine secretion in the cell culture supernatant of NoF 151 fibroblasts after treatment with ntsh-sEVs or shP53-sEVs. Among 36 cytokines examined, expression of eight of them (C–C motif chemokine ligand 2 [CCL2], CXCL1, granulocyte colony-stimulating factor [G-CSF], granulocyte-macrophage colony-stimulating factor [GM-CSF], IL-1β, IL-6, IL-8, and macrophage migration inhibitory factor [MIF]) was higher in cells treated with small EVs than in those not treated with EVs (Figure 3G). Also, CCL2, CXCL1, G-CSF, GM-CSF, IL-1β, and IL-6 were expressed at higher levels in cells treated with ntsh-sEVs than in those treated with shP53-sEVs (Figure 3G).
The packaging of GOF p53, but not WT p53, into small EVs is regulated by HSP90 binding
Because we observed a greater abundance of GOF p53 protein than WT p53 in small EVs, we investigated two possible explanations for this difference. Due to the misfolded nature of mutant p53 protein, it has a broader variety of binding partners than does WT p53 (Rangel et al., 2014). Therefore, we first examined the effect of a p53-binding partner on the EV-packaging process. In particular, HSP70 and HSP90 exhibit much greater binding affinity with mutant p53 than other partners (Wiech et al., 2012). This binding induces the stabilization of the misfolded protein, similar to how these HSPs stabilize protein that has been denatured by excessive heat (Vabulas et al., 2010). Furthermore, these proteins are present in high amounts within small EVs and serve as markers of EV enrichment (Théry et al., 2018). Therefore, we sought to determine the impact of inhibition of HSP70 and HSP90 activity on EV p53 quantity. We treated mutant TP53 HT29 cells and KLE endometrial carcinoma cells with a HSP70 inhibitor, VER-155008 (5 μM for 48 h), and evaluated the presence of EV p53 using western blotting. We found that the expression of EV p53 did not change after HSP70 inhibition (Figures 4A and S4A). Next, we subjected these cell lines to treatment with 17-AAG (200 nM for 48 h), an inhibitor of HSP90, which resulted in a decrease in EV p53 protein expression, suggesting that HSP90 plays an important role in the packaging of p53 into small EVs (Figures 4B and S4B).
Figure 4. Inhibition of HSP90, but not HSP70, regulates the packaging of GOF p53 into small EVs.
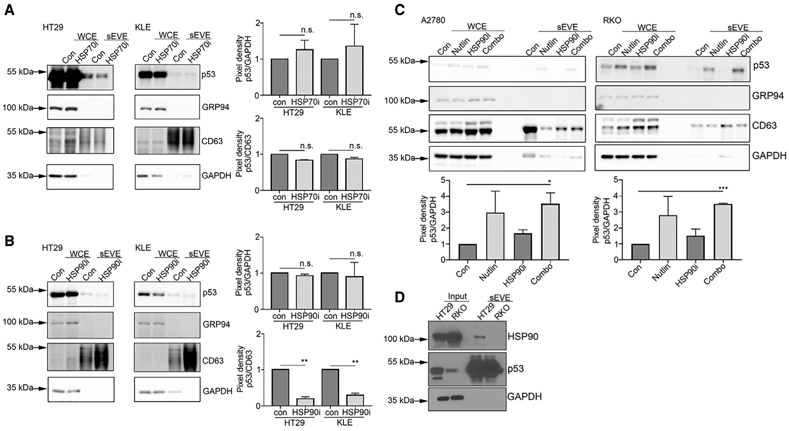
(A) WCE and sEVE were used to determine the level of p53 protein after treatment with the HSP70 inhibitor (HSP70i) VER-155008. Data are represented as mean ± SD. n.s., not significant.
(B) Expression of GOF p53 protein in small EVs in HT29 and KLE cell-derived sEVE after treatment with the HSP90 inhibitor (HSP90i) 17-AAG. Data are represented as mean ± SD. **p < 0.01; n.s., not significant.
(C) WT p53 protein expression in A2780 and RKO cells after treatment with nutlin-3A, an HSP90i, or a combination of the two. Pixel densities for all p53 bands in comparison with GAPDH and CD63 bands normalized to 1.0 are shown. Data are represented as mean ± SD. *p < 0.05; ***p < 0.001.
(D) Results of a co-immunoprecipitation assay performed using HT29 and RKO cells and their relatively small EVs.
We next sought to determine whether stabilization of p53 protein influences the amount of WT p53 in small EVs. GOF p53 is stabilized through multiple mechanisms, such as escape from negative regulation by mouse double minute 2 homolog (MDM2), and therefore accumulates in high quantities in cells (Blagosklonny, 2000). To test this, we treated the WT TP53 ovarian cancer cell line A2780, which exhibited a very low level of EV p53 protein expression, with nutlin-3A, which can induce increased stability of p53 protein (Crane et al., 2015). This resulted in much greater accumulation of p53 in the WCEs, as well as within the EV compartment, compared with no nutlin-3A treatment (Figure 4C). This demonstrated that the quantity of EV p53 is dependent, at least partially, on p53 stability.
To determine whether the quantity of WT p53 in small EVs decreases due to inhibition of HSP90, we selected WT TP53 A2780 and RKO cells for the experiments. These two cell lines exhibited much greater amount of p53 protein in small EVs after treatment with nutlin-3A than untreated groups, consistent with previous observations. However, when we treated these cells with the combination of nutlin-3A and an HSP90 inhibitor, we observed no reduction in p53 protein expression in small EVs (Figures 4C and S4C). To determine whether these results were due to differences in HSP90 and p53 binding in the EV compartment, we performed co-immunoprecipitation in small EVs of mutant TP53 HT29 cells and WT TP53 RKO cells. The data showed that HSP90 was bound to p53 in HT29-, but not RKO-derived, small EVs, demonstrating that HSP90 and GOF p53, but not WT p53, interact within the EV compartment (Figure 4D).
GOF p53-containing small EVs promote tumor growth and CAF conversion in mice
To determine the in vivo impact of GOF p53-containing small EVs on tumor growth and CAF phenotype change, we established a mouse subcutaneous tumor model using the HT29 cell line. One week after implantation, we directly injected the resulting subcutaneous tumors with ntsh-sEVs or shP53-sEVs. The tumors injected with ntsh-sEVs had significantly higher tumor weights than did those injected with shP53-sEVs (Figure 5A). Additionally, they had larger volumes, although the difference did not reach statistical significance (Figure 5A). The tumor tissues were then harvested and embedded in paraffin and then sectioned and stained for p53 (DO-1), αSMA, and fibronectin. IHC results showed that mutant p53 protein expression level in the two groups was similar (Figure 5B). Also, the results revealed that injection of GOF p53-containing small EVs resulted in increased expression of αSMA, pointing to the capacity of these EVs to induce a cancer-associated phenotype (Figure 5B).
Figure 5. Small GOF p53-containing EVs promote tumor growth and CAF transformation in mouse models.

(A) Tumor weights and volumes in mice inoculated with HT29 cells subcutaneously. Data are represented as mean ± SD. *p < 0.05; n.s., not significant.
(B) Comparison of p53, αSMA, and fibronectin expression in mice tumors injected with ntsh-sEVs and shP53-sEVs according to IHC. The IHC score was analyzed based on the expression area and calculated using the IHC toolbox in the ImageJ software program. Data are represented as mean ± SD. *p < 0.05; n.s., not significant. Scale bar, 100 μm.
(C) Tumor weights obtained from mice inoculated with HT29-ntsh and -shP53 cells orthotopically. Data are represented as mean ± SD. n.s., not significant.
(D) Comparison of p53, αSMA, and fibronectin expression in the HT29-ntsh and HT29-shP53 groups based on an IHC assay. HPF, high-power field. The IHC score was analyzed based on the expression area and calculated using the IHC toolbox in the ImageJ software program. Data are represented as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001. Scale bar, 100 μm.
(E) The populations of fibroblasts positive for the surface maker PDGFR-α as revealed by FACS flow cytometry. SSC, side scatter; FSC-A, forward scatter area.
(F) The quantitative real-time RT-PCR results for Trp53, FAP, IL1B, CXCL12, MMP2, and MMP9 in fibroblasts isolated from tumors using FACS. Data are represented as mean ± SD.
See also Figure S5.
To further determine the endogenous effects of GOF p53 on cancer-associated phenotype conversion, we established an orthotopic colorectal cancer model by injecting HT29-ntsh or HT29-shP53 cells into the cecal wall in nude mice (Tseng et al., 2007). The tumor weights did not differ significantly between the two groups (Figure 5C), but the p53 expression level was considerably lower in the HT29-shP53 group than in the HT29-ntsh group (Figure 5D). The negative staining for p53 protein in cecum tissues is shown in Figure S5A. Importantly, the expression of αSMA and fibronectin was substantially lower in the HT29-shP53 group than in the HT29-ntsh group (Figure 5D).
We further isolated fresh fibroblasts from tumors in HT29-ntsh and HT29-shP53 orthotopic mouse models via fluorescence-activated cell sorting (FACS) (Sharon et al., 2013). We used a fluorescence-labeled antibody against the surface marker platelet-derived growth factor receptor alpha (PDGFR-α) to mark the fibroblasts while antibodies against surface markers in immune, epithelial, and endothelial cells to exclude any positive populations of these cells (Figure 5E). We used two mice per group for the fibroblast isolation and immediately extracted the total mRNA samples from fibroblasts using TRIzol reagent. Quantitative real-time RT-PCR results showed that in the HT29-ntsh mouse model, Trp53 gene expression was increased, which was consistent with our mRNA array data. We also observed increased expression of FAP, the cytokines IL1B and CXCL12, and the profibrogenic factors matrix metallopeptidase 2 (MMP2) and MMP9 (Figure 5F).
We have shown that HSP90 was an important chaperone for the packaging of GOF p53 into small EVs. Therefore, we next investigated the effects of HSP90 inhibition on CAF conversion in an orthotopic HT29 colorectal cancer model. In this model, mice given 17-AAG had smaller tumor burdens than did the control group, but the difference did not reach statistical significance (Figure S5B). Immunohistochemical staining of harvested tumors demonstrated that the protein score for p53, αSMA, and fibronectin did not differ significantly between the control and treatment groups (Figure S5C). However, we observed positive staining for GOF p53 in stromal cells in the control group, but not in the treatment group (Figure S5C). These findings suggest that inhibition of HSP90 activity could influence the transfer of GOF p53 from tumor cells to stromal cells in this model.
DISCUSSION
The role of the tumor microenvironment in tumorigenesis (Allinen et al., 2004; Liao et al., 2018; Wang et al., 2017), tumor growth (Lyssiotis and Kimmelman, 2017; Whiteside, 2008), drug resistance (Hazlehurst et al., 2003; Sun, 2016; Trédan et al., 2007), immunosuppression (Arina et al., 2016; Cohen et al., 2017; Wang and DuBois, 2015), and metastasis (Mlecnik et al., 2016; Quail and Joyce, 2013; Yang and Lin, 2017) is becoming increasingly recognized as an important target for therapeutic intervention. Fully elucidating the molecular components that modulate the microenvironment is critical for determining how to eliminate these sanctuaries for cancer cells. Herein, we demonstrate the capacity of cancer cells to spread GOF p53 to the stromal compartment through small EVs and create a permissive environment for tumor growth. In particular, fibroblasts that take up these GOF p53-containing small EVs are re-educated to adopt a cancer-associated phenotype, marked by increased expression of αSMA, fibronectin, and FAP. Notably, EV p53 protein upregulates the PDGF and TGF-β signaling pathways in fibroblasts. The relationship between p53 and Nrf2-mediated pathway was described previously, with WT p53 counteracting transcription of Nrf2 target genes (Faraonio et al., 2006). Conversely, mutant p53 cooperates with Nrf2, influencing the levels of oxidative stress within the tumor cells and microenvironment (Hamada et al., 2017; Kalo et al., 2012). Nrf2 is implicated to have a role in fibroblast cell division (Jódar et al., 2011) and regulates the expression of inflammatory cytokines such as IL-6 and IL-8 (Martin et al., 2011). Furthermore, our data showed that Nrf2 activation was associated with the conversion of fibroblasts to a cancer-associated phenotype, suggesting that the small EV p53/Nrf2 axis is an important contributor to the re-education of fibroblasts. Enrichment of lncRNAs in small EVs could be another important factor in reshaping the tumor microenvironment. The role of lncRNAs in broadly affecting the immune response, inflammatory response regulation, and T cell differentiation is recognized to occur through diverse mechanisms (Heward and Lindsay, 2014). lncRNAs can also play an important role in the IL-1β-induced inflammatory response in lung fibroblasts (Hadjicharalambous et al., 2018).
Previous studies demonstrated that p53 could facilitate the secretion of EVs by inducing expression of STEAP3 metalloreductase (TSAP6) and maspin in H460 human non-small cell lung cancer cell lines with a WT TP53 allele (Yu et al., 2006, 2009). However, in contrast with these reports, we observed that the knockdown of GOF p53 protein resulted in a trend of greater small EV secretion than did the HT29-ntsh cells. However, compared with WT TP53 RKO cell line, HT29 cells exhibited a substantial increase in small EV secretion per cell. Given the large genomic differences among different cell lines, other regulators should also be considered for the small EV secretion. Determining the impact of TP53 on EV secretion will require additional work in the future.
CAFs are major contributors to remodeling of the tumor microenvironment (Gascard and Tlsty, 2016; Shiga et al., 2015; Xing et al., 2010). CAFs are characterized by expression of markers such as αSMA (Orimo and Weinberg, 2007), PDGFR-α and PDGFR-β (Togo et al., 2013), FAP (Park et al., 1999), and fibronectin (Tomasek et al., 2002). This is achieved through multiple mechanisms, including secretion of growth factors and cytokines such as CXCL12 (Orimo et al., 2005), TGF-β (Kalluri and Zeisberg, 2006), and tumor necrosis factor α (TNF-α) (Silzle et al., 2004). CAFs can be found in nearly every tumor type (Shiga et al., 2015). Therefore, eliminating CAFs, or converting them back to a normal fibroblast phenotype, holds great promise in the treatment of cancer. Our results demonstrated that tumor cells can convert fibroblasts to a cancer-associated phenotype via small EVs. Given these findings, blocking the transfer of GOF p53 protein in small EVs from cancer cells to fibroblasts could convert these cells from the cancer-associated phenotype. In showing that the mechanism of GOF p53 packaging in small EVs is dependent on binding with HSP90, we revealed a selective approach to targeting small EVs from cancer cells while leaving WT p53 in normal cells unperturbed. HSP90 inhibitors are being evaluated in clinical trials (Evans et al., 2010; Sidera and Patsavoudi, 2014), and the results presented herein offer key insights into which patients may benefit the most from treatment with these inhibitors, as well as which drugs may pair well with HSP90 inhibition. Numerous studies have established a firm link between CAFs and immune evasion (Lakins et al., 2018; Tao et al., 2017; Ziani et al., 2018). Therefore, HSP90 inhibitors and immune therapy may be a rational combination therapy regimen for cancer patients with certain p53 mutations. However, the efficacy of this and other combinations remains to be explored.
Whether GOF p53 in small EVs affects stromal cells other than fibroblasts in the microenvironment, such as endothelial and immune cells, requires further investigation. However, given the dominant-negative capacity of GOF p53, as well as the ability of these mutated proteins to activate aberrant transcriptional programs, the impact of GOF dissemination to the stroma is likely to be widespread. Furthermore, the effects of p53 in small EVs that exit the localized environment and enter the bloodstream may affect physiology at a systemic level. Gaining an understanding of where GOF p53-containing small EVs go once in the bloodstream and determining whether certain subpopulations of small EVs are enriched in GOF p53 are important next steps. Considering that half of cancers have some form of alteration of TP53, these findings and future discoveries hold great potential in broadening our understanding of tumor biology.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for all original resources and reagents presented in this manuscript should be directed to the Lead Contact, Anil K. Sood (asood@mdanderson.org)
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
The mRNA array and RNA Sequencing data have been deposited to the GEO repository with the accession number GSE164248 and GSE148698, respectively.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Four to eight-week female homozygous Trp53tm1Tyj knockout (Trp53−/−) mice (B6.129S2-Trp53tm1Tyj) were purchased from The Jackson Laboratory. NCRNU-Female nude (NCr) mice of 6 to 8-week were obtained from Taconic. All mice were housed at MDACC animal facility under specific pathogen-free conditions. All animal-related experiments were approved by the Institutional Animal Care and Use Committee of MDACC.
KPC PDAC cells xenograft mouse model
KPC PDAC cells were kindly provided by Professor Anirban Maitra (Department of Pathology, Division of Pathology and Laboratory Medicine, MDACC) and labeled with firefly luciferase (Genecopoeia Cat. # LPP-FLUC-Lv100c) as a genetic reporter. A total of 2 × 106 KPC PDAC cells were injected into the peritoneal cavity of Trp53−/− mice. The tumor take rate was monitored using the IVIS imaging system. Four weeks after cell injection, the mice were sacrificed and dissected for harvesting tumor tissues. The tumor tissues from the pancreas and outside pancreas (e.g., peritoneal, intestines, uterine, and ovarian) were collected for OCT frozen tissues and paraffin-fixed tissues.
Subcutaneous mouse model
HT29 colorectal cells (3 × 106) were injected into the right flanks of NCRNU-Female nude mice (two groups of 13) and allowed to grow for 10 days. Afterward, mice were injected intratumorally with small EVs harvested from HT29-ntsh and HT29-shP53 cells. Small EVs were harvested via ultracentrifugation as outlined above. The first injection consisted of small EVs from 3 × 106 HT29 cells/mouse grown in McCoy’s 5a medium modified + 2% Exo-free FBS + 0.1% gentamycin. The next rounds of injection consisted of small EVs from 6 × 106 HT29 cells grown in the same condition. Forty-eight hours later, cultured media was harvested, and small EVs were isolated. Tumors were injected with 5 mg of small EVs re-suspended in PBS. Once the tumors grew to a size in which tumor was no longer able to be injected due to density, the mice were sacrificed, and the tumor volume and weight were measured.
Orthotopic mouse model
For orthotopic implantation of tumors, HT29-ntsh and HT29-shP53 cells were harvested from culture flasks and prepared at a concentration of 1 × 106 in 50 μL of Hank’s balanced salt solution. The implantation was performed as previously reported (Tseng et al., 2007). Briefly, after induction of anesthesia with ketamine and isoflurane and sterilization of the abdominal wall with alcohol swabs, a small abdominal incision was made, and the cecum was exposed. Warm saline was used to keep the isolated cecum moist. The cancer cells (1 × 106) in 50 μL of Hank’s balanced salt solution were inoculated into the cecal wall of NCRNU-Female nude mice using a 27-gauge needle. The cecum was then returned to the peritoneal cavity and the abdominal wall was closed using three interrupted stitches of 4-0 absorbable suture. After 6 weeks, the mice were killed by cervical dislocation and the tumors were harvested. For in vivo inhibition experiments, HT29 luciferase cells (1 × 106) were inoculated into NCRNU-Female nude mouse cecum as described above. The HSP90 inhibitor (17-AAG, 25 mg/kg, daily, 5 days/week, i.p. injection) was administered 10 days after cell injection. After 4-week treatment, the mice were sacrificed, and the tumors were harvested.
Cell culture
The cell lines LNCaP, A2780, H1975, and H1299 were maintained in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 0.1% gentamycin. The breast cancer cell line MCF7 was cultured in Eagle’s Minimum Essential Medium with 0.01 mg/mL human recombinant insulin, 10% FBS, and 0.1% gentamycin; RKO cells were cultured in Eagle’s Minimum Essential Medium, 10% FBS and 0.1% gentamycin; A549 lung cancer cells were maintained in F-12K medium supplemented with 10% FBS and 0.1% gentamycin; DU145 cells were cultured in Eagle’s Minimum Essential Medium, 10% FBS and 0.1% gentamycin; KLE cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium, 10% FBS and 0.1% gentamycin; HT29 cells were maintained in McCoy’s 5a medium modified with 10% FBS and 0.1% gentamycin; T47D cells were maintained in RPMI-1640 medium with 10% FBS, 0.1% gentamycin, and 0.2 U/mL bovine insulin. NoF 151 fibroblasts were cultured in a 1:1 mixture of Medium 199 (Sigma-Aldrich, Cat. # M5017-10X1L) and medium MCDB 105 (Sigma-Aldrich, Cat. # M6395-10X1L) supplemented with 10% FBS, 1% penicillin-streptomycin, and 5 μg epidermal growth factor. KPC PDAC and HEK293T cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM) medium, 10% FBS, and 0.1% gentamycin. A universal mycoplasma detection kit (ATCC, Cat. # 30-1012K) was used to confirm that all the cells were mycoplasma-free. Cell line validation was performed using short tandem repeat (STR) fingerprinting by Cytogenetics and Cell Authentication Core of MDACC. All the cells were cultured in an incubator at 37°C with 5% CO2.
Patient samples
Tissue microarray slides of ovarian cancer patients (Female, median age 61 years, range 24 to 87 years) were obtained from the Department of Pathology at MDACC. The p53 staining was performed by the Department of Pathology at MDACC and evaluated by gynecologic pathologists. A total of 13 serum samples of ovarian cancer patients (Female, median age 57 years, range 36 to 72 years) were provided by the Multidisciplinary Gynecologic Cancer Translational Research Tumor Bank at MDACC. The formalin-fixed paraffin-embedded (FFPE) tissue slides of ovarian cancer patients with whole-genome sequencing were retrieved from the Multidisciplinary Gynecologic Cancer Translational Research Tumor Bank at MDACC. All the specimens were approved for processing and analyzing by the MD Anderson IRB protocol.
METHOD DETAILS
Isolation of small EVs
Cell lines were grown in suitable medium, 2% Exo-free FBS (System Biosciences, Cat. # EXO-FBS-250A-1), and 0.1% gentamycin for 48 hr. The medium was then spun down at 300 × g for 10 min and 2,000 × g for 20 min and filtered through a 0.22-μm filter to remove any remaining cell debris or large vesicles. For a cell panel, small EVs isolation reagent (Thermo Fisher Scientific, Cat. # 4478359) was added at a 1:2 ratio to the medium, then incubated at 4°C overnight. The solution was then spun at 20,000 × g for 30 min to pellet small EVs. Harvested small EVs were then lysed using small EVs resuspension buffer (Life Technologies, Carlsbad, CA), and the protein quantity in small EVs was measured using a Qubit protein assay. The ultracentrifugation method was used in all other experiments. After filtration through a 0.22-μm filter, the medium was spun down at 100,000 × g for 2 hr at 4°C in an ultracentrifuge. The resulting pellet was resuspended in phosphate-buffered saline (PBS) and then spun again at 100,000 × g for 2 hr at 4°C. The resulting pellet was then suspended in 20 μL of PBS buffer, and the protein quantity was assessed using a Qubit protein assay. EV protein samples were stored at −80°C until used. For western blotting assay, CD63 (System Biosciences, Cat. # EXOAB-CD63A-1, 1:1000), Alix (Santa Cruz, Cat. # sc-53538, 1:1000), and TSG101 (Abcam, Cat. # ab30871, 1:1000) were used as positive markers for small EVs. GRP94 (Santa Cruz, Cat. # sc-32249, 1:1000) was used as a negative marker for small EVs.
To isolate small EVs from serum, 500 μL to 1 mL of each patient’s serum sample was collected and stored at −80°C. The serum was diluted with 50 mL PBS and then centrifuged for 30 min at 2,000 × g, 4°C. The supernatant was transferred to clean tubes without stirring the pellet and centrifuged 40 min at 10,000 × g, 4°C. The supernatant was then transferred to fresh tubes and diluted with PBS in a large volume (60 mL). After filtration with a 0.22-μm filter, the supernatant was transferred to ultracentrifuge tubes and centrifuged for 2 hr at 100,000 × g, and 4°C. Next, we discarded the supernatant and suspended the pellet in 3 mL of PBS followed by spinning for 2 hr, 100,000 × g, 4°C (repeated one more time). The final pellet was suspended with 20 μL of PBS and stored at −80°C.
30% sucrose/deuterium oxide (D2O) cushion
To further purify small EVs, the small EVs isolated by ultracentrifugation were passed through a 30% sucrose cushion (Tris/sucrose/D20:30 g protease-free sucrose, 2.4 g Tris base in 100 mL D2O, pH 7.4, filtered through 0.22-μm filtration). Briefly, we loaded 5 mL of 30% sucrose cushion in an SW32 tube and added the diluted small EV samples (in 25 mL PBS total) gently above the sucrose cushion without disturbing the interface. The samples were then centrifuged using the SW32 rotor for 2 hr at 100,000 × g, 4°C. A total of 4.5 mL of the Tris/sucrose/D2O cushion was collected using a 5-mL syringe with an 18-gauge needle slowly. The sucrose cushion containing small EVs were transferred to a fresh ultracentrifuge tube, and the cushion was diluted to 60 mL total with PBS. The samples were centrifuged using a Ti45 rotor for 2 hr at 100,000 × g, 4°C. Next, we carefully discarded the supernatant without disturbing the pellet and suspended the pellet in 3 mL of PBS. The suspension was centrifuged using a Ti45 rotor for 2 hr 100,000 × g, 4°C. The final pellet (purified small EVs) was suspended in 20 μL of PBS and store at −80°C.
Proteinase K digestion assay
Purified small EV pellets were suspended in PBS and split into three aliquots. Each aliquot was processed as follows: 1) control EVs in PBS only, incubated at 37°C for 30 min; 2) Proteinase K treated EVs, incubated with 50 μg/mL proteinase K at 37°C for 30 min; 3) Proteinase K plus Triton X-100 treated EVs, incubated with 50 μg/mL proteinase K and 1% Triton X-100 at 37°C for 30 min. After incubation, Halt Protease Inhibitor Cocktail (100X) was added to the samples to terminate the digestion. Samples were then incubated on ice for 10 min. The digested samples were then re-suspended in 3 mL of PBS and subsequently spun down for 2 hr at 100,000 × g, 4°C to remove the digested peptides. The final pellets were re-suspended with 20 μL of PBS and lysed using RIPA buffer and proceeded to western blotting analysis.
Immuno-gold labeling and transmission electron microscopy (TEM)
The immuno-gold labeling and TEM were performed by the High Resolution Electron Microscopy Facility of MDACC. For immuno-gold labeling, freshly isolated small EVs were fixed with 2% glutaraldehyde in 0.1 M PBS, pH 7.3. Then, the formvar-carbon-coated mesh nickel grids were treated with a poly-L-lysine solution for 5 min, and excess was blotted with filter paper to allow the grids to dry. The fixed samples were then added onto formvar-carbon-coated mesh nickel grids to allow small EVs to absorb for 1 hr. Grids were rinsed with several drops of PBS 5 times, 3 min each. Then, the grids were blocked with 2% BSA and 0.1% saponin for 30 min. Primary antibodies (CD63, Abcam, Cat. # ab217345, 1:5 dilution, or p53, Santa Cruz, Cat. # sc-126, 1:5 dilution) were added onto the grids to incubate overnight at 4°C. The next day, the grids were rinsed 5 times with PBS, 4 min each, and incubated with secondary gold antibodies (Goat-anti-Rabbit IgG (H&L), 25 nm, Electron Microscopy Sciences, Cat. # 25116, or goat-anti-Mouse IgG (H&L), 10 nm, Electron Microscopy Sciences, Cat. # 25129) (1:20 dilution) for 2 hr at room temperature. The grids were then washed 8 times, 2 min each, and incubated with 2% glutaraldehyde in PBS for 15 min. Grids were then rinsed with distilled water 8 times, 2 min each. Grids were incubated on Millipore-filtered 1% uranyl acetate in ddH2O for 60 s. Excess uranyl acetate was blotted from grids with filter paper and allowed it to dry before the TEM examination. Samples were examined under a JEM 1010 transmission electron microscope (JEOL USA, Inc.) at an accelerating voltage of 80 kV.
Cloning of mCherry-p53 plasmid
The pLenti6/V5-p53_R273H plasmids were purchased from Addgene (Plasmid # 22934) and the interested insert was amplified using the following primers: Forward 5′-ATTACTCGAGCGATGGAGGAGCCGCAGTCAGA-3′; Reverse 5′-ATTAGAATTCTCAGTCTGAGTCAGGCCCTT-3′. After gel purification, the ampllified products was digested with Xho I and EcoR I enzymes. The destination vector (pLVX-mCherry-C1) was purchased from Clontech (Cat. # 632561) and digested with Xho I and EcoR I enzymes. DNA ligations were performed using a Rapid DNA Ligation Kit (ThermoFisher Scientific, Cat. # K1423) according to the manufacturer’s instructions. The resulting mCherry-p53 constructs were then transformed into Stellar competent cells and growing in ampicillin-containing (100 μg/mL) Luria Broth Base for 12 hr at 37°C. The mCherry-p53 plasmids were then purified using a QIAGEN Plasmid Plus Midi Kit (Cat. # 12945) and confirmed through sequencing.
Lentiviral transduction
Stable knockdown of TP53 in HT29 cells was achieved via the transduction of the lentiviral vector pLKO-p53-shRNA-427 (Addgene, Plasmid # 25636). The control vector pLKO.1 was purchased from Addgene (Plasmid # 1864). Overexpression of mCherry-tagged p53 protein in HT29 cells was achieved via the transduction of mCherry-p53 plasmids. To produce viral particles, 2 × 106 293T cells were plated on 10-cm plates and grown in DMEM, 15% FBS, and 0.1% gentamycin. The medium was then replaced with 5 mL of serum-free DMEM per plate. Five μg of lentiviral plasmid, 2.5 μg pMD2.G plasmid (Addgene, Plasmid # 12259), and 2.5 μg psPAX2 plasmid (Addgene, Plasmid # 12260) were incubated in 250 μL of DMEM for 5 min. Simultaneously, 30 μL of FuGENE transfection reagent (Promega, Cat. # E2311) was incubated in 250 μL of DMEM for 5 min. Then, the plasmid mixture was dropwise added into the transfection reagent and incubated at room temperature for 15 min. This mixture was pipetted into the 293T cells. Four hours later, the medium was changed to 10 mL of complete medium, and the cells were left to generate a virus for 3 days. Next, the medium was harvested, spun down at 500 × g for 5 min, and passed through a 0.45-μm filter. Two milliliters of the virus-containing medium were then added to a six-well plate containing the cell type of interest at a confluency of approximately 60%. The cells were left in a viral medium for 3 days, and the selection antibiotic was added, according to the cell line’s sensitivity. For HT29, 3 μg of puromycin/mL was added, and the cells were left to grow for 3 days in puromycin selection medium.
RNA sequencing (RNA-seq)
RNA samples from small EVs were extracted using an exoRNeasy Midi and Maxi Kit (QIAGEN, Cat. # 77144) following the manufacturer’s instructions. Biological replicates were prepared for differential expression analysis. An equal amount of RNA (15 ng) was prepared for sequencing. The quantity and quality of the RNA from small EVs were determined by Qubit RNA HS Assay Kit (Thermo Fisher Scientific, Cat. # Q32852). The samples were stored at −80°C until analyzed. The library construction for RNAs and the sequencing were performed by Novogene Corporation Inc. (Sacramento, CA) on Illumina NovaSeq 6000 platform. Total RNA sample was processed through a round of sequencing adaptor ligation, reverse transcription, PCR enrichment, and purification before sequencing. The quality control was performed on each step to guarantee the reliability of the data. The read alignment and annotation were performed by The MD Anderson Bioinformatics & Computational Biology department. Post-alignment normalization of the RNA-Seq data and differential expression analysis was performed in R version 4.0.3 using limma package and its function Voom from Bioconductor (limma: Linear Models for Microarray and RNA-Seq Data User’s Guide). The R code follows the steps from https://ucdavis-bioinformatics-training.github.io/2018-June-RNA-Seq-Workshop/thursday/DE.html. Statistical significance was defined as a p value < 0.05. A multi-dimensional scaling (MDS) with R was used to plot the RNA-Seq data in two dimensions, thus visualizing the relative distance of the data. For further visual data exploration, a heatmap displaying most differenced genes were generated in R using the heatmap.2 function of gplots library. The volcano plot was generated in R displaying the distribution of fold changes and p value for the comparison performed.
Quantitative Real-Time Reverse Transcription PCR
Cells were trypsinized until detachment, collected in complete medium, spun down at 1200 rpm for 5 min at 4°C, and 350 μL of TRIzol (Thermo Fisher Scientific, Cat. # 15596018) was added to cell pellets. Total RNA was isolated using the Direct-zol RNA isolation kit (Zymo Research, Cat. # R2062), and the concentration was determined using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific); 100 to 1,000 nanograms of RNA was used as a template for cDNA. The cDNA was made using a Verso cDNA synthesis kit (Thermo Fisher Scientific, Cat. # AB1453B). Upon synthesis, 2 μL of cDNA was combined with 4 μL of 10 μM forward and reverse primers and 10 μL of 2 x SYBR Green PCR Master Mix (Thermo Fisher Scientific, Cat. # A25778) per well. This mixture was then run in a real-time PCR machine (Applied Biosystems, Foster City, CA) using the following program: 50°C for 2 min, 95°C for 15 min, 95°C for 15 s, and 60°C for 1 min × 40 cycles. The Ct values determined using the PCR machine were compared, which yielded the ΔΔCt. This was employed to determine the relative change in mRNA expression across samples. 18 s ribosomal RNA was used as a reference gene for normalizing quantitative real-time RT-PCR data. The sequence of primers was shown in Table S3. Each experiment was performed with n = 3.
Western blotting
Cells used in the present study were scraped and collected by centrifuging for 5 min at 1200 rpm, 4°C, then the spin was repeated after washing the cells with ice-cold PBS. Cell pellets were then suspended in RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, Cat. # 89900) and quantified using a Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Cat. # 23225). Equal amounts of protein (20 μg for WCE and 5 μg for sEVE) were run on a sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gel (8%–12%), then transferred to a nitrocellulose membrane, incubated in 5% non-fat milk (in TBS-T) for 1 hr at room temperature, then incubated with primary antibody overnight at 4°C. Membrane blots were then washed with TBS-T for 3 × 10 min and incubated with a secondary antibody (1:2000 in blocking buffer) for 1 hr at room temperature. ECL was then added to the blots for 1 min, then developed using an X-ray film or Azure Biosystems (Dublin, CA, USA) to quantify protein. For detecting multiple protein markers, we either restore the membrane using Restore Plus Western Blot Stripping Buffer (Thermo Fisher Scientific, Cat. # 46430) or run the same protein samples on separate gels. Each experiment was repeated at least twice otherwise indicated, and the pixel density of bands of interest was analyzed by ImageJ software.
Fluorescence-activated cell sorting (FACS)
Fibroblast isolation from tumors was performed following a protocol for FACS described previously (Sharon et al., 2013). Mice from HT29-ntsh and HT29-shP53 orthotopic models were killed using cervical dislocation, and their tumors were collected using sterilized surgical tools. When dissecting the tumor tissue, the necrotic and adjacent normal regions were carefully removed. After mincing the tissues thoroughly, an Accumax solution (StemCell Technologies, Inc., Cat. # 7921) was added to the tissues and then incubated in a 37°C water bath for 15 min. To lyse the red blood cells in cell suspension, 5 mL of ACK lysis buffer (Thermo Fisher Scientific, Cat. # A1049201) was used to resuspend the cell pellet and incubated them for 5 min at room temperature. Then, 50 mL of FACS buffer (Calcium/Magnesium-free PBS plus 0.5% bovine serum albumin) was then added to the mixture to neutralize the lysis solution, followed by centrifugation at 450 × g, 5 min to pellet the cells. The cells were then re-suspended in FACS buffer, and endogenous Fc in tissue samples was blocked by adding FcBlock (anti-mouse CD16/CD32, 1:50). At this point, the cells in solution were ready for staining. The following antibodies directly conjugated to fluorophore were used in the staining: PE CD140a (PDGFRA) monoclonal antibody (APA5) (1:100) for labeling fibroblasts, APC anti-mouse CD45 Antibody for labeling immune cells (1:100), eFluor 450 CD326 (EpCAM) Monoclonal Antibody (G8.8, 1:100) for labeling epithelial cells, and PE/Cy7 Anti-CD31 antibody [390] (1:100) for labeling endothelial cells. Ghost Dye Red 710 (1:100) was used for cell viability. Un-stained cells and single-color control beads were used for controls accordingly. The stained cells were analyzed using a BD FACSAria cell sorter (BD Biosciences, San Jose, CA).
Co-immunoprecipitation
Co-immunoprecipitation was carried out using a Universal Magnetic Co-IP kit (Active Motif, Cat. # 54002). Small EVs from GOF TP53 HT29 and wt TP53 RKO cells were isolated as described above. Small EVs were lysed using kit lysis buffer, and 5 μg of p53 antibody (Santa Cruz, Cat. # sc-126) per sample was used for the pull-down of p53 and HSP90. The resulting proteins immunoprecipitated with p53 were then run on a denaturing gel, as described above. The presence of immunoprecipitated HSP90 in the same samples was determined by probing the blot with an anti-HSP90 antibody (Cell Signaling Technology, Cat. # 4877, 1:1000).
mRNA array
A mRNA array was carried out on the Clariom D platform (Affymetrix, Santa Clara, CA). Normal Fibroblasts isolated from a patient with ovarian cancer were plated on a six-well plate and treated with 10 μg/mL small EVs harvested from HT29-ntsh or -shP53 cells for 48 hr. The samples were in biological triplicate for both groups. The fibroblasts were then harvested and total RNA was isolated using a Direct-zol kit (Zymo Research, Cat. # R2062). RNA quality control was performed on the Total RNA Analysis platform (Agilent, Santa Clara, CA). A total of 500 ng RNA was used for the mRNA array (50 ng/μL) suspended in water. The mRNA array was performed using the Clariom D assay. The CEL files generated from Affymetrix RNA microarray image analysis software were processed through Transcriptome Analysis Console 4.0 which normalizes (and applies the log2 function to) array signals using a robust multiarray averaging algorithm. One-way analysis of variance (ANOVA) was used to identify the differential expressed mRNAs in a comparative analysis with a p value less than 0.05 and an absolute fold change greater than 1.1. Fold change in gene expression between the fibroblasts treated with ntsh-sEVs and in those treated with shP53-sEVs was used for NetWalker analysis (Komurov et al., 2012) (https://www.netwalkersuite.org/). Networks of genes that were highest scoring for upregulation and downregulation based on the gene expression data were identified and functional pathways scored and annotated. The upstream regulators of the downregulated genes in fibroblasts treated with shP53-EVs were analyzed using Ingenuity Pathway Analysis software (QIAGEN, CA).
Tumor immunohistochemistry
For all animal experiments, paraffin-embedded specimens were sliced into 5 μm sections, placed on glass slides, and deparaffinized in xylene. For antigen retrieval, the deparaffinized samples on slides were placed in a Coplin jar with 1 × Diva decloaker buffer (Biocare Medical, Cat. # SKU: DV2004). The Coplin jar containing the slides was then transferred to a cooker and heated to 100°C for 45 min. Afterward, the slides were cooled to room temperature for 20 min. The cooled slides were rinsed with PBS. After that, the endogenous peroxidase activity in tissues was quenched by incubating the sections for 12 min with 3% H2O2 in absolute methanol. The slides were subsequently washed in PBS and incubated with blocking serum (4% Fish Gelatin, Aurion, Cat. # 900.033) for 30 min at room temperature. For staining mouse tissue, the slides were then blocked with AffiniPure Fab Fragment Goat Anti-Mouse IgG (H+L) (Jackson ImmunoResearch, 1:10 in 4% Fish Gelatin) for 1 hr at room temperature before applying a primary antibody derived from mouse. The pretreated sections were then exposed to an anti-p53 antibody (for tumor protein p53 [Homo sapiens], we used DO-1 antibodies [Santa Cruz, Cat. # sc-126; 1:150]; for transformation-related protein 53 [Mus musculus], we used CM5p antibodies [Leica Biosystems, Cat. # NCL-L-p53-CM5p; 1:200]), anti-αSMA antibody (Abcam, Cat. # ab5694, 1:100), or anti-fibronectin antibody (Abcam, Cat. # ab2413, 1:200) overnight at 4°C. After incubation with the antibodies, the sections were washed with PBS 3 times and incubated with a peroxidase-conjugated secondary antibody for 1 hr at room temperature. Peroxidase was visualized by incubating the sections with DAB for 1 to 5 min. Coverslips were placed on the slides and affixed with Permount mounting medium (Thermo Fisher Scientific, Cat. # SP15-100) before evaluating the samples using light microscopy. The IHC images were acquired using a Leica camera (Wetzlar, Germany) and five mid-power (20 ×) microscopic fields per slide were examined using the IHC toolbox in the ImageJ software program.
Co-culture experiments
NoF 151 fibroblasts isolated from patient samples were plated on a companion six-well plate (Falcon, BD Biosciences) at a concentration of 1 × 105 cells/well. Once cells attached, a 0.22-μm insert (Falcon, BD Biosciences) was placed on top of each well and allowed to equilibrate for 6 hr. Next, HT29 cells transduced with mCherry-tagged p53 in a pLVX-mCherry-C1 Lentiviral vector, or transduced with a control vector, were plated onto the insert at a concentration of 1 × 106 cells/well. The co-cultured cells were allowed to grow for 48 hr; then protein from each group of cells was then isolated and probed via western blotting using an anti-p53 antibody (Santa Cruz, Cat. # sc-126).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism 8 (San Diego, CA). Differences were assessed using the Mann-Whitney U test or Student t test dependent on data distribution and variance homogeneity between two groups. For mRNA array and RNA-Seq analysis, the logarithmic fold change and adjusted p value were used to evaluate the differential expression of genes. All p values were two-tailed otherwise indicated, and p < 0.05 was considered significant. All data were presented with mean ± SD.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-p53 (DO-1) | Santa Cruz | Cat. # sc-126; RRID: AB_628082 |
| Anti-p53 (CM5p) | Leica Biosystems | Cat. # NCL-L-p53-CM5p; RRID: AB_2744683 |
| Anti-GRP94 | Santa Cruz | Cat. # sc-32249; RRID: AB_627676 |
| Anti-CD63 (for western blotting) | System Biosciences | Cat. # EXOAB-CD63A-1; RRID: AB_2561274 |
| Anti-CD63 (for immune-gold staining) | Abcam | Cat. # ab217345; RRID: AB_2754982 |
| Anti-Alix | Santa Cruz | Cat. # sc-53538; RRID: AB_673821 |
| Anti-TSG101 | Abcam | Cat. # ab30871; RRID: AB_2208084 |
| Anti-PDX1 | Cell Signaling Technology | Cat. # 5679; RRID: AB_10706174 |
| Anti-Cre | Cell Signaling Technology | Cat. # 15036; RRID: AB_2798694 |
| Anti-αSMA | Abcam | Cat. # ab5694; RRID: AB_2223021 |
| Anti-Fibronectin | Abcam | Cat. # ab2413; RRID: AB_2262874 |
| Anti-HSP90 | Cell Signaling Technology | Cat. # 4877; RRID: AB_2233307 |
| Anti-GAPDH | Sigma-Aldrich | Cat. # G8795; RRID: AB_1078991 |
| Goat-anti-Rabbit IgG (H&L) | Electron Microscopy Sciences | Cat. # 25116; RRID: N/A |
| Goat-anti-Mouse IgG (H&L) | Electron Microscopy Sciences | Cat. # 25129; RRID: N/A |
| AffiniPure Fab Fragment Goat Anti-Mouse IgG (H+L) |
Jackson ImmunoResearch | Cat. # 115-007-003; RRID: AB_2338476 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG + IgM (H+L) |
Jackson ImmunoResearch | Cat. # 115-035-068; RRID: AB_2338505 |
| Peroxidase AffiniPure F(ab’)2 Fragment Goat Anti-Rabbit IgG, F(ab’)2 fragment specific |
Jackson ImmunoResearch | Cat. # 111-036-047; RRID: AB_2337945 |
| HRP Goat Anti-Rat Ig | BD Biosciences | Cat. # 554017; RRID: AB_395211 |
| ECL Anti-Rabbit IgG, Horseradish Peroxidase | GE Healthcare | Cat. # GENA934; RRID: AB_2722659 |
| ECL Anti-Mouse IgG, Horseradish Peroxidase | GE Healthcare | Cat. # NA931; RRID: AB_772210 |
| CD140a (PDGFRA) Monoclonal Antibody (APA5), PE, eBioscience | Thermo Fisher Scientific | Cat. # 12-1401-81; RRID: AB_657615 |
| APC anti-mouse CD45 Antibody | BioLegend | Cat. # 103111; RRID: AB_312976 |
| CD326 (EpCAM) Monoclonal Antibody (G8.8), eFluor 450, eBioscience | Thermo Fisher Scientific | Cat. # 48-5791-82; RRID: AB_10717090 |
| Anti-CD31 antibody [390] (PE/Cy7) | Abcam | Cat. # ab46733; RRID: AB_868905 |
| Bacterial and virus strains | ||
| Stellar Competent Cells | Clontech | Cat. # 636763 |
| Firefly Luciferase Lentifect Purified Lentiviral Particles | Genecopoeia | Cat. # LPP-FLUC-Lv100c |
| Biological samples | ||
| Patients with HGSOC (serum and tissues) | MDACC | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Nutlin-3A | Sigma-Aldrich | Cat. # SML0580 |
| 17-AAG | SelleckChem | Cat. # S1141-25MG |
| ML385 | Millipore Sigma | Cat. # SML1833-5MG |
| VER-155008 | Cayman Chemical | Cat. # 1134156-31-2 |
| Tris base | Thermo Fisher Scientific | Cat. # BP152-5 |
| NaCl | Thermo Fisher Scientific | Cat. # AC424290050 |
| Glycine | Thermo Fisher Scientific | Cat. # BP381-5 |
| Sucrose | Sigma-Aldrich | Cat. # 84097-1KG |
| Deuterium oxide | Sigma-Aldrich | Cat. # 151882 |
| Glutaraldehyde solution | Sigma-Aldrich | Cat. # G7651-10ML |
| Proteinase K | Sigma-Aldrich | Cat. # P6556-100MG |
| Ampicillin | Sigma-Aldrich | Cat. # 10835269001 |
| Triton X-100 | Thermo Fisher Scientific | Cat. # BP151-500 |
| Xho I enzyme | NEB | Cat. # R0146S |
| EcoR I enzyme | NEB | Cat. # R0101S |
| Fish Gelatin | Aurion | Cat. # 900.033 |
| 10% Formalin | Thermo Fisher Scientific | Cat. # 23-305510 |
| Diva Decloaker, 10X | Biocare Medical | Cat. # SKU: DV2004 |
| Halt Protease Inhibitor Cocktail (100X) | Thermo Fisher Scientific | Cat. # 78438 |
| Luria Broth Base | Thermo Fisher Scientific | Cat. # 12795027 |
| Fisher Chemical Permount Mounting Medium | Thermo Fisher Scientific | Cat. # SP15-100 |
| TRIzol Reagent | Thermo Fisher Scientific | Cat. # 15596018 |
| SYBR Green | Thermo Fisher Scientific | Cat. # A25778 |
| FuGENE HD Transfection Reagent | Promega | Cat. # E2311 |
| Accumax | StemCell Technologies, Inc. | Cat. # 7921 |
| ACK lysing buffer | Thermo Fisher Scientific | Cat. # A1049201 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher Scientific | Cat. # 89900 |
| Ghost Dye Red 710 | Tonbo Biosciences | Cat. # 13-0871-T100 |
| Epidermal Growth Factor from murine submaxillary gland | Sigma-Aldrich | Cat. # E4127-5X.1MG |
| Critical commercial assays | ||
| Universal Magnetic Co-IP Kit | Active Motif | Cat. # 54002 |
| Human Cytokine Array Kit | R&D Systems | Cat. # ARY005B |
| exoRNeasy Midi and Maxi Kit | QIAGEN | Cat. # 77144 |
| Total Exosome Isolation Reagent | Thermo Fisher Scientific | Cat. # 4478359 |
| Restore Plus Western Blot Stripping Buffer | Thermo Fisher Scientific | Cat. # 46430 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat. # 23225 |
| Direct-zol RNA Kits | Zymo Research | Cat. # R2062 |
| QIAGEN Plasmid Plus Midi kit | QIAGEN | Cat. # 12945 |
| Rapid DNA Ligation Kit | Thermo Fisher Scientific | Cat. # K1423 |
| Qubit Protein Assay Kit | Thermo Fisher Scientific | Cat. # Q33212 |
| Qubit RNA HS Assay Kit | Thermo Fisher Scientific | Cat. # Q32852 |
| Verso cDNA Synthesis Kit | Thermo Fisher Scientific | Cat. # AB1453B |
| Universal Mycoplasma Detection Kit | ATCC | Cat. # 30-1012K |
| Deposited data | ||
| mRNA Array | This paper | GEO: GSE164248 |
| Uncropped western blotting | This paper | Document S1 |
| RNA Sequencing Data | This paper | GEO: GSE148698 |
| Experimental models: cell lines | ||
| H1975 | Kindly provided by Heymach lab (MDACC) | N/A |
| H1299 | Kindly provided by Heymach lab (MDACC) | N/A |
| RKO | Kindly provided by Lee Ellis lab (MDACC) | N/A |
| A549 | Kindly provided by Heymach lab (MDACC) | N/A |
| HT29 | Kindly provided by Lee Ellis lab (MDACC) | N/A |
| A2780 | MDACC cell line bank | N/A |
| LNCaP | MDACC cell line bank | N/A |
| DU145 | ATCC cell line bank | N/A |
| KLE | MDACC cell line bank | N/A |
| T47D | MDACC cell line bank | N/A |
| HEK293T | MDACC cell line bank | N/A |
| MCF7 | Kindly provided by Gabriel Lopez lab (MDACC) | N/A |
| NoF 151 fibroblasts | Kindly provided by Jinsong Liu lab (MDACC) | N/A |
| KPC PDAC cells | Kindly provided by Anirban Maitra lab (MDACC) | N/A |
| Experimental models: organisms/strains | ||
| Mouse: B6.129S2-Trp53tm1Tyj | The Jackson Laboratory | Stock No: 002101 |
| Mouse: NCRNU-F nude (NCr) | Taconic | Model #: NCRNU-F |
| Oligonucleotides | ||
| Primers for quantitative real-time RT-PCR, see Table S3 |
This paper | N/A |
| Primers for mCherry-p53 cloning plasmids, Forward: ATTACTCGAGCGATGGAGGAGCCGCAGTCAGA | This paper | N/A |
| Recombinant DNA | ||
| pLKO-p53-shRNA-427 | Todd Waldman Lab | Addgene, Plasmid # 25636 |
| pLKO.1 puro | David Sabatini Lab | Addgene, Plasmid # 1864 |
| pLenti6/V5-p53_R273H | Bernard Futscher Lab | Addgene, Plasmid # 22934 |
| pLVX-mCherry-C1 Lenti-viral vector | Clontech | Cat. # 632561 |
| Plasmid: mCherry-p53 | This paper | N/A |
| Software and algorithms | ||
| Prism Version 8.00 | GraphPad Software | https://www.graphpad.com/ |
| Ingenuity Pathway Analysis | QIAGEN Bioinformatics | https://digitalinsights.qiagen.com/ |
| NetWalker | Kakajan Komurov, Dursun S, Erdin S, Ram P.T. | https://www.netwalkersuite.org/ |
| ImageJ | NIH Image | https://imagej.nih.gov/ij/index.html |
| R version 4.0.3 limma package | Bioconductor | https://www.bioconductor.org/packages/devel/bioc/vignettes/limma/inst/doc/usersguide.pdf |
| Transcriptome Analysis Console (TAC) Software | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/life-science/microarray-analysis/microarray-analysis-instruments-software-services/microarray-analysis-software/affymetrix-transcriptome-analysis-console-software.html |
| CorelDraw Graphic 2018 | CorelDraw | http://www.coreldraw.com/e/ |
| Adobe Photoshop CC 2018 | Adobe | https://www.adobe.com/ |
Highlights.
Cancer cells with mutant TP53 can package mutant p53 protein in small EVs
Small EVs with GOF p53 can convert fibroblasts into a cancer-associated phenotype
Packaging of GOF p53 into small EVs is regulated by HSP90
Small EVs with GOF p53 promote tumor growth in vivo
ACKNOWLEDGMENTS
This publication is part of the NIH Extracellular RNA Communication Consortium paper package and was supported by the NIH Common Fund’s ex-RNA Communication Program. This work was supported in part by NIH grants R35CA209904, UH3TR000943, P50CA217685, P50CA098258, CA177909, U01CA213759, CA227622, and P30CA016672; the Ovarian Cancer Research Alliance; the Blanton-Davis Ovarian Cancer Research Program; the American Cancer Society Research Professor Award; the Frank McGraw Memorial Chair in Cancer Research; and the MD Anderson Ovarian Cancer Moon Shot Program. S.M. was supported by the Foundation for Women’s Cancer research grant (sponsor award number FP00009883). S.Y.W. was supported by the Cancer Prevention & Research Institute of Texas Research Training Program (grants RP101502, RP140106, and RP170067). E.S. was supported by Ovarian Cancer Research Alliance (OCRA number FP00006137). We acknowledge Scientific Publications, Research Medical Library at The University of Texas MD Anderson Cancer Center for reviewing and editing this manuscript. We acknowledge High Resolution Electron Microscopy Facility (NIH grant P30CA016672) at The University of Texas MD Anderson Cancer Center for performing the TEM studies. We acknowledge The Flow Cytometry and Cellular Imaging Core Facility (FCCICF) (NCI grant P30CA16672) at The University of Texas MD Anderson Cancer Center for performing the cell sorting.
Footnotes
DECLARATION OF INTERESTS
A.K.S. a consultant for Kiyatec, Astra Zeneca, and Merck, has received research funding from M-Trap, and is a Bio-Path Holdings stockholder. The remaining authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2021.108726.
REFERENCES
- Addadi Y, Moskovits N, Granot D, Lozano G, Carmi Y, Apte RN, Neeman M, and Oren M (2010). p53 status in stromal fibroblasts modulates tumor growth in an SDF1-dependent manner. Cancer Res. 70, 9650–9658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, et al. (2004). Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 6, 17–32. [DOI] [PubMed] [Google Scholar]
- Arina A, Corrales L, and Bronte V (2016). Enhancing T cell therapy by overcoming the immunosuppressive tumor microenvironment. Semin. Immunol 28, 54–63. [DOI] [PubMed] [Google Scholar]
- Azmi AS, Bao B, and Sarkar FH (2013). Exosomes in cancer development, metastasis, and drug resistance: a comprehensive review. Cancer Metastasis Rev. 32, 623–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh EH, Ke H, Levine AJ, Bonneau RA, and Chan CS (2018). Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 25, 154–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell E, and Taylor MA (2016). Functional roles for exosomal microRNAs in the tumour microenvironment. Comput. Struct. Biotechnol. J 15, 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV (2000). p53 from complexity to simplicity: mutant p53 stabilization, gain-of-function, and dominant-negative effect. FASEB J. 14, 1901–1907. [DOI] [PubMed] [Google Scholar]
- Campbell IG, Qiu W, Polyak K, and Haviv I (2008). Breast-cancer stromal cells with TP53 mutations. N. Engl. J. Med 358, 1634–1635, author reply 1636. [DOI] [PubMed] [Google Scholar]
- Cohen N, Shani O, Raz Y, Sharon Y, Hoffman D, Abramovitz L, and Erez N (2017). Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene 36, 4457–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane EK, Kwan SY, Izaguirre DI, Tsang YT, Mullany LK, Zu Z, Richards JS, Gershenson DM, and Wong KK (2015). Nutlin-3a: a potential therapeutic opportunity for TP53 wild-type ovarian carcinomas. PLoS ONE 10, e0135101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz G, Bridges C, Lucas M, Cheng Y, Schorey JS, Dobos KM, and Kruh-Garcia NA (2018). Protein digestion, ultrafiltration, and size exclusion chromatography to optimize the isolation of exosomes from human blood plasma and serum. J. Vis. Exp (134), 57467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, and Che G (2017). Genetic alterations and epigenetic alterations of cancer-associated fibroblasts. Oncol. Lett 13, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CG, Chang L, and Gestwicki JE (2010). Heat shock protein 70 (hsp70) as an emerging drug target. J. Med. Chem 53, 4585–4602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraonio R, Vergara P, Di Marzo D, Pierantoni MG, Napolitano M, Russo T, and Cimino F (2006). p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem 281, 39776–39784. [DOI] [PubMed] [Google Scholar]
- Gascard P, and Tlsty TD (2016). Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 30, 1002–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyuris A, Navarrete-Perea J, Jo A, Cristea S, Zhou S, Fraser K, Wei Z, Krichevsky AM, Weissleder R, Lee H, et al. (2019). Physical and molecular landscapes of mouse glioma extracellular vesicles define heterogeneity. Cell Rep. 27, 3972–3987.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadjicharalambous MR, Roux BT, Feghali-Bostwick CA, Murray LA, Clarke DL, and Lindsay MA (2018). Long non-coding RNAs are central regulators of the IL-1β-induced inflammatory response in normal and idiopathic pulmonary lung fibroblasts. Front. Immunol 9, 2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada S, Taguchi K, Masamune A, Yamamoto M, and Shimosegawa T (2017). Nrf2 promotes mutant K-ras/p53-driven pancreatic carcinogenesis. Carcinogenesis 38, 661–670. [DOI] [PubMed] [Google Scholar]
- Hazlehurst LA, Landowski TH, and Dalton WS (2003). Role of the tumor microenvironment in mediating de novo resistance to drugs and physiological mediators of cell death. Oncogene 22, 7396–7402. [DOI] [PubMed] [Google Scholar]
- Heward JA, and Lindsay MA (2014). Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 35, 408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong CS, Sharma P, Yerneni SS, Simms P, Jackson EK, Whiteside TL, and Boyiadzis M (2017). Circulating exosomes carrying an immunosuppressive cargo interfere with cellular immunotherapy in acute myeloid leukemia. Sci. Rep 7, 14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosein AN, Wu M, Arcand SL, Lavallée S, Hébert J, Tonin PN, and Basik M (2010). Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res. 70, 5770–5777. [DOI] [PubMed] [Google Scholar]
- Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, Liebler DC, Ping J, Liu Q, Evans R, et al. (2019). Reassessment of exosome composition. Cell 177, 428–445.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jódar L, Mercken EM, Ariza J, Younts C, González-Reyes JA, Alcaín FJ, Burón I, de Cabo R, and Villalba JM (2011). Genetic deletion of Nrf2 promotes immortalization and decreases life span of murine embryonic fibroblasts. J. Gerontol. A Biol. Sci. Med. Sci 66, 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, and Zeisberg M (2006). Fibroblasts in cancer. Nat. Rev. Cancer 6, 392–401. [DOI] [PubMed] [Google Scholar]
- Kalo E, Kogan-Sakin I, Solomon H, Bar-Nathan E, Shay M, Shetzer Y, Dekel E, Goldfinger N, Buganim Y, Stambolsky P, et al. (2012). Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J. Cell Sci 125, 5578–5586. [DOI] [PubMed] [Google Scholar]
- Komurov K, Dursun S, Erdin S, and Ram PT (2012). NetWalker: a contextual network analysis tool for functional genomics. BMC Genomics 13, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J, Arras G, Colombo M, Jouve M, Morath JP, Primdal-Bengtson B, Dingli F, Loew D, Tkach M, and Théry C (2016). Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 113, E968–E977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakins MA, Ghorani E, Munir H, Martins CP, and Shields JD (2018). Cancer-associated fibroblasts induce antigen-specific deletion of CD8 + T Cells to protect tumour cells. Nat. Commun 9, 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Komar CA, Bengsch F, Graham K, and Beatty GL (2016). Genetically engineered mouse models of pancreatic cancer: the KPC model (LSL-Kras(G12D/+) ;LSL-Trp53(R172H/+) ;Pdx-1-Cre), its variants, and their application in immuno-oncology drug discovery. Curr. Protoc. Pharmacol 73, 14.39.11–14.39.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Zhao L, Rojas C, Bateman NW, Yao H, Lara OD, Celestino J, Morgan MB, Nguyen TV, Conrads KA, et al. (2020). Molecular analysis of clinically defined subsets of high-grade serous ovarian cancer. Cell Rep. 31, 107502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao CP, Booker RC, Brosseau JP, Chen Z, Mo J, Tchegnon E, Wang Y, Clapp DW, and Le LQ (2018). Contributions of inflammation and tumor microenvironment to neurofibroma tumorigenesis. J. Clin. Invest 128, 2848–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PK, Kraus E, Wu TA, Strong LC, and Tainsky MA (1996). Analysis of genomic instability in Li-Fraumeni fibroblasts with germline p53 mutations. Oncogene 12, 2267–2278. [PMC free article] [PubMed] [Google Scholar]
- Lyssiotis CA, and Kimmelman AC (2017). Metabolic interactions in the tumor microenvironment. Trends Cell Biol. 27, 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani F, Collavin L, and Del Sal G (2019). Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 26, 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SF, Esser PR, Weber FC, Jakob T, Freudenberg MA, Schmidt M, and Goebeler M (2011). Mechanisms of chemical-induced innate immunity in allergic contact dermatitis. Allergy 66, 1152–1163. [DOI] [PubMed] [Google Scholar]
- Milner J, and Medcalf EA (1991). Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell 65, 765–774. [DOI] [PubMed] [Google Scholar]
- Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M, Church SE, Maby P, Vasaturo A, Angelova M, et al. (2016). The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci. Transl. Med 8, 327ra26. [DOI] [PubMed] [Google Scholar]
- Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, Jamieson NB, Oien KA, Lowy AM, Brunton VG, et al. (2010). Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 107, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, and Hainaut P (2010). TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol 2, a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren M, and Rotter V (2010). Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol 2, a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo A, and Weinberg RA (2007). Heterogeneity of stromal fibroblasts in tumors. Cancer Biol. Ther 6, 618–619. [DOI] [PubMed] [Google Scholar]
- Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, and Weinberg RA (2005). Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348. [DOI] [PubMed] [Google Scholar]
- Park JE, Lenter MC, Zimmermann RN, Garin-Chesa P, Old LJ, and Rettig WJ (1999). Fibroblast activation protein, a dual specificity serine protease expressed in reactive human tumor stromal fibroblasts. J. Biol. Chem 274, 36505–36512. [DOI] [PubMed] [Google Scholar]
- Patocs A, Zhang L, Xu Y, Weber F, Caldes T, Mutter GL, Platzer P, and Eng C (2007). Breast-cancer stromal cells with TP53 mutations and nodal metastases. N. Engl. J. Med 357, 2543–2551. [DOI] [PubMed] [Google Scholar]
- Pols MS, and Klumperman J (2009). Trafficking and function of the tetraspanin CD63. Exp. Cell Res 315, 1584–1592. [DOI] [PubMed] [Google Scholar]
- Quail DF, and Joyce JA (2013). Microenvironmental regulation of tumor progression and metastasis. Nat. Med 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangel LP, Costa DC, Vieira TC, and Silva JL (2014). The aggregation of mutant p53 produces prion-like properties in cancer. Prion 8, 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon Y, Alon L, Glanz S, Servais C, and Erez N (2013). Isolation of normal and cancer-associated fibroblasts from fresh tissues by fluorescence activated cell sorting (FACS). J. Vis. Exp (10), e4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, and Takeyama H (2015). Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers (Basel) 7, 2443–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidera K, and Patsavoudi E (2014). HSP90 inhibitors: current development and potential in cancer therapy. Recent Patents Anticancer Drug Discov. 9, 1–20. [PubMed] [Google Scholar]
- Silzle T, Randolph GJ, Kreutz M, and Kunz-Schughart LA (2004). The fibroblast: sentinel cell and local immune modulator in tumor tissue. Int. J. Cancer 108, 173–180. [DOI] [PubMed] [Google Scholar]
- Sun Y (2016). Tumor microenvironment and cancer therapy resistance. Cancer Lett. 380, 205–215. [DOI] [PubMed] [Google Scholar]
- Tao L, Huang G, Song H, Chen Y, and Chen L (2017). Cancer associated fibroblasts: an essential role in the tumor microenvironment. Oncol. Lett 14, 2611–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, Zheng Y, Hoshino A, Brazier H, Xiang J, et al. (2014). Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 24, 766–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théry C, Zitvogel L, and Amigorena S (2002). Exosomes: composition, biogenesis and function. Nat. Rev. Immunol 2, 569–579. [DOI] [PubMed] [Google Scholar]
- Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin-Smith GK, et al. (2018).Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 7, 1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkach M, and Théry C (2016). Communication by extracellular vesicles: where we are and where we need to go. Cell 164, 1226–1232. [DOI] [PubMed] [Google Scholar]
- Togo S, Polanska UM, Horimoto Y, and Orimo A (2013). Carcinoma-associated fibroblasts are a promising therapeutic target. Cancers (Basel) 5, 149–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, and Brown RA (2002). Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol 3, 349–363. [DOI] [PubMed] [Google Scholar]
- Trédan O, Galmarini CM, Patel K, and Tannock IF (2007). Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst 99, 1441–1454. [DOI] [PubMed] [Google Scholar]
- Tseng W, Leong X, and Engleman E (2007). Orthotopic mouse model of colorectal cancer. J. Vis. Exp (10), 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuna M, Ju Z, Yoshihara K, Amos CI, Tanyi JL, and Mills GB (2020). Clinical relevance of TP53 hotspot mutations in high-grade serous ovarian cancers. Br. J. Cancer 122, 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabulas RM, Raychaudhuri S, Hayer-Hartl M, and Hartl FU (2010). Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb. Perspect. Biol 2, a004390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel G, D’Angelo G, and Raposo G (2018). Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol 19, 213–228. [DOI] [PubMed] [Google Scholar]
- Wang D, and DuBois RN (2015). Immunosuppression associated with chronic inflammation in the tumor microenvironment. Carcinogenesis 36, 1085–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, et al. (2017). Role of tumor microenvironment in tumorigenesis. J. Cancer 8, 761–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendler F, Favicchio R, Simon T, Alifrangis C, Stebbing J, and Giamas G (2017). Extracellular vesicles swarm the cancer microenvironment: from tumor-stroma communication to drug intervention. Oncogene 36, 877–884. [DOI] [PubMed] [Google Scholar]
- Whiteside TL (2008). The tumor microenvironment and its role in promoting tumor growth. Oncogene 27, 5904–5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiech M, Olszewski MB, Tracz-Gaszewska Z, Wawrzynow B, Zylicz M, and Zylicz A (2012). Molecular mechanism of mutant p53 stabilization: the role of HSP70 and MDM2. PLoS ONE 7, e51426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing F, Saidou J, and Watabe K (2010). Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front. Biosci 15, 166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, and Lin PC (2017). Mechanisms that drive inflammatory tumor microenvironment, tumor heterogeneity, and metastatic progression. Semin. Cancer Biol 47, 185–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Rosen DG, Zhang Z, Bast RC Jr., Mills GB, Colacino JA, Mercado-Uribe I, and Liu J (2006). The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc. Natl. Acad. Sci. USA 103, 16472–16477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Che SP, Kurywchak P, Tavormina JL, Gansmo LB, Correa de Sampaio P, Tachezy M, Bockhorn M, Gebauer F, Haltom AR, et al. (2017). Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Cancer Biol. Ther 18, 158–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yemelyanova A, Vang R, Kshirsagar M, Lu D, Marks MA, Shih IeM., and Kurman RJ (2011). Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod. Pathol 24, 1248–1253. [DOI] [PubMed] [Google Scholar]
- Yu X, Harris SL, and Levine AJ (2006). The regulation of exosome secretion: a novel function of the p53 protein. Cancer Res. 66, 4795–4801. [DOI] [PubMed] [Google Scholar]
- Yu X, Riley T, and Levine AJ (2009). The regulation of the endosomal compartment by p53 the tumor suppressor gene. FEBS J. 276, 2201–2212. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li S, Li L, Li M, Guo C, Yao J, and Mi S (2015). Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinformatics 13, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziani L, Chouaib S, and Thiery J (2018). Alteration of the antitumor immune response by cancer-associated fibroblasts. Front. Immunol 9, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mRNA array and RNA Sequencing data have been deposited to the GEO repository with the accession number GSE164248 and GSE148698, respectively.