

Abstract
Background & Aims:
Currently, only a few genetic variants explain the heritability of fatty liver disease. Quantitative trait loci (QTL) analysis of mouse strains has identified the susceptibility locus Ltg/NZO (liver triglycerides from New Zealand obese [NZO] alleles) on chromosome 18 as associating with increased hepatic triglycerides. Herein, we aimed to identify genomic variants responsible for this association.
Methods:
Recombinant congenic mice carrying 5.3 Mbp of Ltg/NZO were fed a high-fat diet and characterized for liver fat. Bioinformatic analysis, mRNA profiles and electrophoretic mobility shift assays were performed to identify genes responsible for the Ltg/NZO phenotype. Candidate genes were manipulated in vivo by injecting specific microRNAs into C57BL/6 mice. Pulldown coupled with mass spectrometry-based proteomics and immunoprecipitation were performed to identify interaction partners of IFGGA2.
Results:
Through positional cloning, we identified 2 immunity-related GTPases (Ifgga2, Ifgga4) that prevent hepatic lipid storage. Expression of both murine genes and the human orthologue IRGM was significantly lower in fatty livers. Accordingly, liver-specific suppression of either Ifgga2 or Ifgga4 led to a 3–4-fold greater increase in hepatic fat content. In the liver of low-fat diet-fed mice, IFGGA2 localized to endosomes/lysosomes, while on a high-fat diet it associated with lipid droplets. Pulldown experiments and proteomics identified the lipase ATGL as a binding partner of IFGGA2 which was confirmed by co-immunoprecipitation. Both proteins partially co-localized with the autophagic marker LC3B. Ifgga2 suppression in hepatocytes reduced the amount of LC3B-II, whereas overexpression of Ifgga2 increased the association of LC3B with lipid droplets and decreased triglyceride storage.
Conclusion:
IFGGA2 interacts with ATGL and protects against hepatic steatosis, most likely by enhancing the binding of LC3B to lipid droplets.
Keywords: Fatty liver, Positional cloning, Immunity-related GTPases, miRNA, NAFLD
Graphical abstract
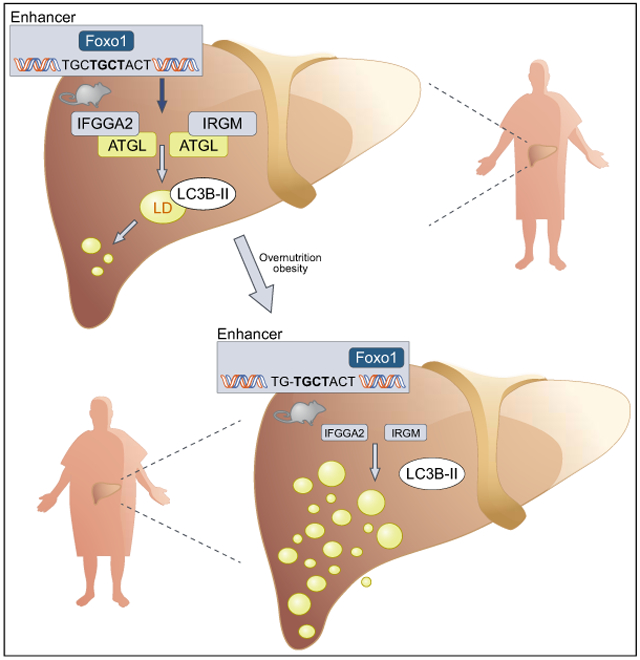
Lay summary:
The genetic basis of non-alcoholic fatty liver disease remains incompletely defined. Herein, we identified members of the immunity-related GTPase family in mice and humans that act as regulators of hepatic fat accumulation, with links to autophagy. Overexpression of the gene Ifgga2 was shown to reduce hepatic lipid storage and could be a therapeutic target for the treatment of fatty liver disease.
Introduction
The prevalence of obesity is increasing globally. Excessive hepatic deposition of triglycerides is termed hepatic steatosis, which ranges from simple steatosis through fibrosis, non-alcoholic steatohepatitis to cirrhosis.1 Non-alcoholic fatty liver disease (NAFLD) is strongly associated with metabolic abnormalities such as insulin resistance, fasting hyperglycemia and altered hepatokines.2
The cause of NAFLD is multifactorial including genetic and environmental factors.3 Several human studies have emphasized the highly heritable nature of the disease.4,5 Polymorphisms in PNPLA35-7 and TM6SF28 are considered to be major genetic determinants of NAFLD susceptibility. However, the identified genetic variants explain only a small proportion of the risk of NAFLD. Thus, various factors such as epigenetic mechanisms and gene-gene interactions must be considered when looking at heritability in the general population.9
A genome-wide scan of mouse outcross populations that differ in their phenotype enables the identification of quantitative trait loci (QTL): genomic segments conferring susceptibility to several metabolic traits. Additionally, positional cloning enables introgression of an identified QTL into a specific mouse background to finally verify pathogenic genes such as Tsc210 and Ifi202b.,11 To identify common gene variants responsible for metabolic traits, e.g. hepatic fat accumulation, the Collective Diabetes Cross of the German Center for Diabetes Research (DZD) was performed. This is a comprehensive linkage study of an intercross of the New Zealand obese (NZO) mouse and lean strains (C57BL/6 [B6], DBA, C3H, 129P2), which differ in their diabetes susceptibility.12 The NZO mouse, characterized by adiposity, insulin resistance and hyperglycemia, is an ideal model to provide further insights into the genetic nature of the human metabolic syndrome.11,13
The aim of the current study was to identify genomic variants responsible for the susceptibility locus Ltg/NZO (liver triglycerides from NZO alleles) on chromosome 18, which associates with elevated hepatic lipids. We show that Ifgga2 und Ifgga4 are the most likely candidates mediating the Ltg/NZO phenotype. The hepatic expression of both genes and the human orthologue IRGM (immunity-related GTPase M) were markedly reduced in livers of NZO-allele carriers and patients with NAFLD, respectively. IFGGA2 interacts with the lipase ATGL which could be responsible for preventing ectopic fat accumulation in the liver.
Materials and methods
Animals
The generation of the F1 and N2 mice and housing was described.12 Recombinant congenic mice (RCS) were repeatedly backcrossed to the NZO strain. RCS (N/N vs. N/B) selected for Ltg/NZO (5.3 Mbp) were bred by repeated backcrossing of mice (N/B) with NZO females and characterized in the F4.N10 generation. Siblings were intercrossed to generate homozygous mice (N/N vs. B/B, F8.N9). The Ltg/NZO locus was also transferred to a B6 background and characterized in F2.N8. At 3 weeks of age, animals received high-fat diet (HFD) (D12451, Research Diets Inc.).
Animal experiments were performed referring to the ARRIVE guidelines and approved by the ethics committee of the State Agency of Environment, Health, and Consumer Protection (State of Brandenburg, Germany, 2347-21-2012, 2347-10-2014 and 2347-33-2017) and Ethics Committee of the State Ministry of Agriculture, Nutrition and Forestry (State of North Rhine-Westphalia, Germany, 84-02.04.2013.A118).
Genotyping
Genomic DNA-isolation from mouse tails and genotyping was performed as described12 with self-designed KASP assays (LGC) according to manufacturer's recommendations (Table S2).
Haplogroup analysis
Haplogroup analysis was performed as described.14,15 In brief, mouse single nucleotide polymorphisms (SNPs) were used (https://www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1303). The chromosomal region was dissected into intervals (250 kb) to determine the frequency of polymorphic SNPs between mouse strains.
Gene expression analysis
Total RNA was isolated using miRNeasy Micro Kit (Qiagen) or RNeasy Mini Kits (Qiagen), cDNA prepared by M-MLV Reverse Transcriptase-Kit (Promega). Hydrolysis probes were applied for quantitative reverse transcription PCR (qRT-PCR). Actb, Ppia or HPRT were used as reference genes.
Gene expression analysis of potential miR-Ifgga2 target genes was performed using the universal probe library (UPL, Roche; Table S3). Relative gene expression was analyzed using the ΔCt method.16
Microarray analysis
Genome-wide expression analysis was performed by OakLabs GmbH (Germany) applying SurePrint G3 Mouse GE 8x60k Microarray gene chips (Agilent Technologies). Results of analysis are available on GEO: GSE146721 and GSE146724.
Recombinant viruses
Specific micro RNAs (miRNAs or miR) targeting Ifgga2 or Ifgga4 were designed using BLOCK-iT™ RNAi Designer tool (Thermo Fisher Scientific). Each engineered pre-miRNA double-stranded DNA oligonucleotide was subcloned into the pcDNA6.2-GW/EmGFP-miR vector (BLOCK-iTTM Pol II miR RNAi Expression Vector Kit, (Invitrogen)). The miRNA expression cassette was cloned into the pdsAAV-LP1-EGFPmut for liver-specific overexpression under the control of the LP1 promoter.17 Within hepatocytes, the pre-miRNA forms an intramolecular loop structure similar to endogenous pre-miRNAs and is processed by Dicer into the mature miRNA. Recombinant viruses (Vigene Bioscience; 5 × 1011 viral genomes) were injected into the tail vein of B6 mice (8 weeks).
Plasma and supernatant analysis
Triglyceride, glycerol (TR0100, Sigma-Aldrich) and NEFA (NEFA-HR(2), Wako Chemicals GmbH) were measured in plasma and supernatant of liver explants and insulin concentration in plasma by ELISA (80-Insmsu-E01, ALPCO Diagnostics) following manufacturer's protocol.18
Non-invasive quantification of liver fat by CT
Liver fat content was measured by CT (Hitachi-Aloka LCT-200) as described.19
Detection of triglycerides in liver and hepatocytes
Triglycerides were quantified enzymatically using RandoxTR-210 (Randox).12 In brief, livers and McArdle hepatocytes or primary hepatocytes were homogenized in 10 mmol/l sodium dihydrogen phosphate, 1 mmol/l EDTA and 1% (v/v) polyoxyethylene-10-tridecyl ether and incubated for 5 min at 70°C. After centrifugation (4°C) supernatant was diluted 1:10, incubated at 70°C for 5 min and then placed on ice for 5 min before being centrifuged again. Clear supernatant was used for the enzymatic assay.
Quantification of hepatic lipids by liquid chromatography-mass spectrometry
Liver tissue was homogenized and equivalents of 1 or 0.5 mg were subjected to lipid extraction using methanol/chloroform (2:1, v:v) for quantification of ceramides and sphingomyelins or diacylglycerols (DAGs), respectively. Ceramides and sphingomyelins were measured in tandem mass spectrometry mode as described.20 DAGs were analyzed in mass spectrometry mode monitoring the following [M+Na]+ ions: C15:0/C15:0 DAG (m/z 563.465), C32:0 DAG (m/z 591.496), C34:2 DAG (m/z 615.496), C16:0/C18:1 DAG (m/z 617.512), C17:0/C17:0-d5 DAG (m/z 624.559), C36:4 DAG (m/z 639.496), C36:3 DAG (m/z 641.512) and C36:2 DAG (m/z 643.527).
Identification of putative regulatory SNPs/Indels
To determine promoter or enhancer regions (2 kb or 50 kb down-/upstream) we utilized histone modification datasets (GSM100140, GSM769014 and GSM769015). Within these putative regulatory regions, genetic variants (SNPs/Indels) between parental strains (Wellcome Trust Sanger Institute, REL-1505) were combined with position weight matrices (PWM) from the JASPER database.21 Thereby, genetic variants in potential transcription factor binding sites (TFBS) were identified. TFBS were predicted by R version 3.4 and the package TFBS Tools.22
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was carried out using 32P-end-labeled double-stranded DNA oligos and nuclear extracts from HEK293T cells overexpressing HA-mFoxo1 or from wild-type cells. Oligos for enhancer variants were annealed and labeled at 5′end. For binding reactions, 5 μg of nuclear extract were pre-incubated in EMSA binding buffer (10 mM Tris-HCl pH 8.0, 50 mM KCl, 10% glycerol, 2 mM DTT, 2 mM EDTA, 1 mM MgCl2,0.1 mg/ml BSA, 0.1 μg/μl poly dI·dC) for 20 min on ice. Complex formation was initiated by adding 1 μl labeled DNA probe. After 30 min incubation at 25°C samples were directly loaded on a 5% native PAA-gel. Dried gels were visualized by autoradiography.
Transfection of cells and staining
McArdle or HeLa cells were seeded on poly-L-lysine coated cover slips and cultured in DMEM (4.5 g/l glucose, Life Technologies) containing 10% FCS until reaching a confluency of 60–70%. McArdle hepatocytes were transfected with plasmids by using Viromer Red (Lipocalyx) according to manufacturer's instructions. For induction of lipid droplet synthesis, 24 h after transfection, cells were incubated with 50 μM of lipid complex (oleic acid:taurocholic acid; 1.95:0.5 mM, Sigma) with or without 2 μg BODIPY in DMEM (4.5 g/L glucose) with 10% FCS. After 8 h, cells were fixed and stained as described.18
Plasmid transfection into HeLa cells was performed with Lipofectamine (Invitrogen) according to manufacturer's protocol, after 24 h fixed and stained as described.18
Primary and secondary antibodies and dilutions used for immunostaining are listed in Table S4.
Isolation and transfection of primary hepatocytes
Primary hepatocytes were isolated as described.23 Cells were cultured in Williams E Medium with 4% FCS (Biochrome), 100 nM Dexamethason, 0.5 nM insulin for 4 h. Afterwards, medium was changed to Williams E Medium without supplements and small interfering RNA (siRNA) transfection was performed (10 nmol, Dharmacon) using INTERFERin (VWR). After overnight incubation, transfection medium was replaced by Oleat-BSA complex or BSA alone in culture medium to induce lipid droplet synthesis for additional 24 h before harvesting.
Ex vivo autophagic flux
Autophagic flux in liver explants was assessed as described.24 In brief, explants were incubated in DMEM ± 10% FBS containing lysosomal inhibitors leupeptin (Sigma; 200 μM) and NH4Cl (Merck; 20 mM) incubated for 2 h at 37°C and lysed.
Subcellular fractionation of livers and protein localization profile
Isolation of hepatic cellular fractions of mice and peptide analysis was published.25 Mass spectrometry-based proteomics data are available online (http://nafld-organellemap.org).
Isolation of lipid droplets of McArdle hepatocytes
After transfection cells were incubated with lipid complex and the droplet fraction was isolated as described.26
Proteomic analysis
Proteins were eluted from Chromotek GFP-Trap beads by boiling in SDS lysis buffer containing 2% (w/v) SDS, 100 mM Tris-HCl (pH 8.5) for 5 min at 95°C. Proteome preparation was performed by the StageTip (iST) method.27 Raw mass spectrometry data were processed with MaxQuant version 1.6.1.13 using default settings (false discovery rate 0.01, oxidized methionine and acetylation [protein N-term] as variable modifications, and carbamidomethyl as fixed modification). Label-free quantitation (LFQ) and “Match between runs” were enabled.
Determination of protein concentration
Tissue lysates were prepared in RIPA buffer and cells lysates in 20 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% TritonX-100, 2.5 mM NaPP, 1 mM β-glycerolphosphat, 1 mM Na3VO4, 1 mM NaF and protein concentrations detected with Pierce™ BCA™ Protein Assay Kit (Thermo Scientific) as described.18
Western blot
Western blotting was performed as described.18 Dilutions of primary and secondary antibodies are listed in Table S5.
Co-immunoprecipitation
Immunoprecipitation of IFGGA2-GFP or IRGM-Myc was performed by GFP- or Myc-Trap beads (Chromotek) (Table S5).
Histology and H&E staining
Paraffin sections were stained with antibodies (Table S4). H&E staining of sections was performed as described.28
Human liver samples
The human study (registered clinical trial, NCT01477957) was conducted in accordance with the Declaration of Helsinki as reported.29
Statistical analysis
Animal number was calculated by a priori power analysis. Statistical analysis of 2 groups was performed by unpaired Student's t test or unpaired t test with Welch's correction. One-way analysis of variance (ANOVA) with Kruskal-Wallis testing was used for comparisons of more than 2 groups. p <0.05 was regarded as statistically significant, and calculated with Prism 8 (GraphPad Software).
For further details regarding the materials and methods used, please refer to the CTAT.
Results
Identification of Ltg/NZO, a fatty liver locus
In the Collective Diabetes Cross project, 4 lean mouse strains were crossed with the obese NZO strain, followed by genome-wide linkage analysis.12,15 Male NZO mice developed increased liver weight and higher triglyceride levels on HFD in comparison to the other strains (Fig. S1A). Genome-wide linkage analysis identified an exclusive QTL for liver weight (Fig. 1A) and liver triglycerides (Fig. S1B) on chromosome 18 (LOD score of 8.0 and 6.8, respectively; Fig.1A). N2 mice homozygous at marker rs4231907 (17 cM/64.9 Mbp) had elevated liver weight and triglyceride content compared to heterozygous mice (Fig. S1C). Thus, the locus was designated Ltg/NZO (liver triglycerides from NZO alleles).
Fig. 1. Identification of a susceptibility locus for fatty liver on chromosome 18.

(A) Genome-wide linkage analysis of (NZOxB6)N2 population for liver weight revealed a QTL on chromosome 18. LOD score curves for liver weight and triglycerides. Solid horizontal (p <0.01) and dashed lines (p <0.05) indicate threshold of significance calculated with 1,000 permutations. (B) Identification of the critical region of Ltg/NZO (red box) and liver triglycerides in (NZOxB6)N2 mice. (C,E) Liver weight and triglyceride concentrations of RCS Ltg/NZO.5.3N/N (n = 9) and Ltg/NZO.5.3N/B (n = 18) (F4.N10) as well as Ltg/NZO.5.3N/N (n = 12) and Ltg/NZO.5.3B/B (n =12) (F8.N9). Data analyzed by unpaired t test with Welch's correction and presented as mean ± SD. **p <0.01; ***p <0.001. (D,F) Histology of liver sections from 12-week-old mice stained for PLIN2 (green) and TO-PRO (blue, nuclei) as well as H&E staining. B6, C57BL/6; Ltg/NZO, liver triglycerides from NZO alleles; NZO, New Zealand obese; QTL, quantitative trait loci.
Fine-mapping and characterization of Ltg/NZO
Positional cloning was performed to identify the critical segment of Ltg/NZO (Fig. 1B). The highest hepatic triglyceride concentration was detected in RCS homozygous (N/N) between 58-63.3 Mbp. Mice carrying the critical region of 5.3 Mbp in a homo- (Ltg/NZO.5.3N/N, Ltg/NZO.5.3B/B) or heterozygous (Ltg/NZO.5.3N/B) manner were generated. Ltg/NZO.5.3N/N showed increased liver weight and significantly elevated liver triglyceride levels (Fig. 1C) and larger lipid droplets indicated by PLIN2 and H&E staining (Fig. 1D) in comparison to Ltg/NZO.5.3N/B mice. The same characteristics were observed after an overnight fast without any impact on plasma parameters (Table S1). Overall DAG concentrations and specific DAG species were induced in Ltg/NZO.5.3N/N compared to Ltg/NZO.5.3N/B mice; concentrations of ceramides and sphingomyelins were not altered (Fig. S1D,E). Comparing mice harboring homozygous alleles at the critical Ltg/NZO locus revealed a stronger difference in hepatic triglyceride accumulation (Fig. 1E,F). On the genetic B6-background, Ltg/NZON/N resulted in significantly higher hepatic fat content in comparison to Ltg/NZOB/B, indicating that Ltg/NZO acts independently of adiposity (Fig. S2A-C).
Identification of immunity-related GTPases as responsible gene variants
The region spanning 58–63.3 Mbp of Ltg/NZO harbors 106 annotated genes and gene models (NCBI). To identify the responsible gene(s), we included sequence and expression data from other backcross populations, genome-wide expression data, and genomic sequence information ((NZOxDBA)N2, (NZOxC3H)N2 and (NZOx129P2)N2).12 In the (NZOxC3H)N2 progeny, liver weight and triglycerides (LOD score 13.6) were also increased by NZO alleles at 63 Mbp on chromosome 18. In the (NZOxDBA)N2 progeny, liver weight showed significant associations after adjusting for blood glucose as a covariate (LOD 6.3). The (NZOx129P2)N2 progeny did not show a QTL on chromosome 18 (Fig. S3A), suggesting that genetic variance in Ltg/NZO is caused by alleles identical between B6, C3H and DBA mice, but different to 129P2 and NZO mice. Based on this assumption, a haplotype analysis of the critical region was generated by using SNP information from the Wellcome Trust Sanger Institute.30,31 The QTL was dissected into regions that are identical by descent between NZO and 129P2 vs. B6-, C3H- and DBA-polymorphic regions. The number of polymorphic SNPs were determined for each 250 kb.14 Regions exceeding a threshold of 100 SNPs/window were considered as polymorphic according to B6, C3H, DBA ≠ NZO, 129P2 (Fig. 2A). Only genes fulfilling these criteria were considered as candidates, reducing the number to 45 genes.
Fig. 2. Fine-mapping of the Ltg/NZO locus in different NZO crosses.

(A) Haplotype map of SNPs within the critical region of Ltg/NZO (58-63.3 Mbp). Black line: total SNPs (all SNPs annotated for B6 reference genome with calls for C3H, DBA, 129P2 and NZO); red line: SNPs according to B6, C3H, DBA ≠ NZO, 129P2. QTL was dissected into windows of 250 kb. Red boxes indicate 100 SNPs per window polymorphic according to B6, C3H, DBA ≠ NZO, 129P2. (B) Differentially expressed genes of parental strains and RCS that are higher (blue line) or lower (red line) in NZO. (C) Hepatic expression of Ifgga2 and Ifgga4 in parental strains (normalized to Eef2, Kruskal-Wallis test; n = 3–5) and (D) RCS (normalized to Ppia, unpaired t test with Welch's correction; n = 12). Data presented as mean ± SD. (E) Alteration of Foxo1 binding motif in an active enhancer marked by histone modifications. (F) EMSA in HEK293 cells. Data not marked with the same letter are significantly different. ***p <0.001. B6, C57BL/6; Ltg/NZO, liver triglycerides from NZO alleles; NZO, New Zealand obese; SNPs, single nucleotide polymorphisms.
Next, we implemented transcriptome data from livers of all 5 parental strains. Three genes were differentially expressed and only 2 of them (Ifgga2, Ifgga4) were located within the critical haplotype block. The transcriptome of Ltg/NZO.5.3N/N and Ltg/NZO.5.3N/B livers revealed 6 differentially expressed genes (Fig. 2B). The final overlap resulted in candidate genes Ifgga2 (Gm4951) and Ifgga4 (RIKEN cDNA F830016B08Rik) (Fig. S3B), members of the family of immunity-related GTPases (IRG).
Expression of Ifgga2 and Ifgga4 was higher in livers of B6, DBA and C3H compared to NZO and 129P2 mice (Fig. 2C). Similarly, Ltg/NZO.5.3N/N showed a significantly lower hepatic expression of Ifgga2 and Ifgga4 in comparison to Ltg/NZO.5.3N/B by 5- and 12-fold and to Ltg/NZO.5.3B/B mice by 13- and 41-fold, respectively (Fig. S3C, Fig. 2D).
A Foxo1 binding site appears to be responsible for strain-specific expression of the Irg gene cluster
Next, we investigated the molecular cause for the reduced expression of Ifgga2 and Ifgga4 in NZO compared to B6 livers. We assumed a common factor within a cis-regulatory region upstream of Ifgga2 as the genetic cause for the strain-specific gene regulation of the cluster (Fig. S3D). Transcription factors often bind to nucleosome-depleted regions which are flanked by nucleosomes carrying H3K4me1 and H3K27ac marks.32 By combining data obtained by chromatin immunoprecipitation sequencing (ChIP-seq) from ENCODE33 and PWM, we identified an activated enhancer harboring a Foxo1 binding motif containing a single nucleotide deletion at position 60,211,556 bp (Build 38) in NZO and 129P2 but not in the other strains (Fig. 2E). Although the deletion was not located in the core sequence, the capacity of Foxo1 to bind to its motif is reduced as shown by EMSA in nuclear extracts of HEK293 cells overexpressing Foxo1 (Fig. 2F) and could thereby be causative for the observed hepatic expression pattern for Ifgga2 and Ifgga4.
Ifgga2 suppression in mice increases hepatic fat storage
To test whether the lower abundance of IRGs is causative for the increased hepatic lipid accumulation induced by Ltg/NZO, Ifgga2 and Ifgga4 were downregulated specifically in livers of B6 mice by a virus expressing a designed miRNA which is processed within cells like an endogenous miRNA and targets Ifgga2 (Fig. 3A) or Ifgga4 (Fig. S4A). AAV-miR-Ifgga2 or AAV-miR-Ifgga4 significantly suppressed their expression compared to AAV-control mice 8 weeks after virus injection (Fig. 3B; Fig. S4B). This suppression resulted in a 4-fold or 3-fold greater increase in hepatic fat content compared to control, respectively, according to CT analysis (Fig. 3C; Fig. S4C), as well as the detection of increased hepatic triglyceride levels (Fig. 3D; Fig. S4D). The increased hepatic fat content was accompanied by larger lipid droplets as indicated by PLIN2 and H&E staining (Fig. 3E; Fig. S4E). Supporting these findings, primary hepatocytes accumulated significantly more triglycerides than control cells upon Ifgga2 knockdown and lipid loading (Fig. 3F). In order to test if the designed miRNA of Ifgga2 targets other mRNAs which could theoretically be responsible for the observed phenotype, we performed an in silico analysis to predict mRNA targets. We identified 11 potential targets which were not altered between the livers of AAV-control and AAV-miR-Ifgga2 mice (Fig. S5).
Fig. 3. In vivo downregulation of Ifgga2 in the liver elevates fat storage.

(A) Sequence of miRNA and its Ifgga2 targeted sequence. (B) Hepatic expression of Ifgga2 in AAV-control or AAV-miR-Ifgga2 mice (n = 8; normalized to Ppia). (C) Changes of liver fat detected by CT (n = 16) and (D) hepatic triglycerides (n = 8) 8 weeks after virus injection. Data analyzed by unpaired t test with Welch's correction and presented as mean ± SD. (E) Liver sections of virus-treated mice stained for PLIN2 (green, TO-PRO (blue)) and with H&E. (F) Triglycerides in primary hepatocytes transfected with indicated siRNA and treated ± BSA-Oleate complex. Data analyzed by Kruskal-Wallis test and presented as mean ± SD. **p <0.001; ***p <0.001. AAV, adeno-associated virus; miRNA, microRNA; siRNA, small interfering RNA.
Subcellular localization of Ifgga2
The cellular localization of IFGGA2 was investigated in sub-fractions derived from livers of B6 mice fed a low-fat diet (LFD) or HFD. We used the data generated by a mass-spectrometric workflow developed to monitor abundance and cellular distributions of ~6,000 hepatic proteins.25 IFGGA4 was not identified in the dataset. IFGGA2 displayed a lysosomal/endosomal profile on LFD and was mainly present in the lipid droplet fraction on HFD (Fig. 4A). This observation was confirmed by immunocytochemistry of untreated and lipid-loaded McArdle hepatocytes expressing IFGGA2. In the absence of oleic acid:- taurocholic acid complex (hereinafter referred to as oleate) in the culture medium, IFGGA2-Myc was detected in intracellular membranes and partially co-localized with endosomal markers (Rab18, Fig. 4B; Rab11 and transferrin receptor, Fig. S6A). Also, in HeLa cells, IFGGA2-Myc showed a partial overlap with LAMP1 and the transferrin receptor (Fig. S6B). In oleate treated cells (8 h), IFGGA2-GFP localized on the lipid droplet surface as indicated by the co-staining of PLIN2 (Fig. 4C).
Fig. 4. Hepatic localization of IFGGA2 upon lipid challenge.

(A) Diet-dependent changes of IFGGA2 localization in liver fractions. Mice on LFD (white), mice on HFD (black). (B) Staining of IFGGA2-Myc transfected McArdle hepatocytes under basal conditions for IFGGA2-Myc (green) and Rab18 (red). (C) Staining of PLIN2 (red) in IFGGA2-GFP (green) transfected McArdle hepatocytes after lipid load. HFD, high-fat diet; LFD, low-fat diet.
Interaction of IFGGA2 and ATGL
To gain insights into cellular functions and associated proteins for IFGGA2, we performed a pulldown experiment followed by mass spectrometry-based proteomics of McArdle hepatocytes overexpressing IFGGA2-GFP ± oleate. Several putative binding partners of IFGGA2 were identified. The lipase ATGL showed a significant enrichment in the pulldown from oleate-treated cells (Fig. 5A, Fig. S7), which was confirmed by co-immunoprecipitation experiments of hepatocytes overexpressing IFGGA2-GFP and ATGL-His. Obviously, the interaction of IFGGA2 and ATGL was more pronounced after oleate treatment (Fig. 5B). Similarly, immunocytochemical staining showed a co-localization of IFGGA2 and ATGL on the surface of lipid droplets after lipid load (Fig. 5C).
Fig. 5. Interaction of IFGGA2 with ATGL and localization of IFGGA2 and LC3B upon lipid load.

(A) Volcano plot of mass spectrometry-based proteomics of GFP-pulldown of McArdle hepatocytes transfected with IFGGA2-GFP ± oleate. (B) Co-immunoprecipitation experiment of co-transfected McArdle hepatocytes. (C) Staining of IFGGA2-GFP (green) and ATGL-His (red) in lipid-loaded McArdle hepatocytes. (D) Staining of IFGGA2-Myc (green) and LC3B (red) in McArdle hepatocytes after lipid load. Bodipy visualizes lipid droplets (white). (E) Quantification of LC3B-positive lipid droplets dependent on the amount of IFGGA2 in McArdle hepatocytes. Data analyzed by unpaired t test with Welch's correction and presented as mean ± SD. (F) Triglyceride levels in transfected McArdle hepatocytes overexpressing either pEGFP, IFGGA2-GFP or ATGL-His after lipid load and fasting (n = 8). Data analyzed by Kruskal-Wallis test and presented as mean ± SD. *p <0.05; ***p <0.001.
IFGGA2 expression elevates the amount of the autophagy marker LC3B on lipid droplets
As earlier studies described that members of the IRG family34 as well as ATGL35 are involved in processes of autophagy, we tested if IFGGA2 induces the degradation of lipids by lipophagy. IFGGA2 is located at the surface of lipid droplets upon lipid load and partially co-localized with LC3B (Fig. 5D). In control cells, 32.5% of lipid droplets displayed a positive LC3B signal, whereas IFGGA2-Myc-expressing cells exhibited 71.1% lipid droplets with an LC3B association (Fig. 5E), indicating that IFGGA2 enhanced LC3B-binding to lipid droplets. In response to treatment with oleate followed by a 24 h incubation in serum-free medium, McArdle hepatocytes overexpressing IFGGA2-GFP or ATGL-His stored significantly lower levels of triglycerides than control cells (Fig. 5F), suggesting that appropriate IFGGA2 expression might protect against hepatic steatosis. Furthermore, in lipid droplet fractions of IFGGA2-GFP expressing McArdle hepatocytes, LC3B-II, ATGL, CGI-58 were more abundant in comparison to cells lacking IFGGA2 (Fig. S8A). This difference was more pronounced after serum starvation, thus under conditions when more IFGGA2-GFP was present in the lipid droplet fraction (Fig. S8B). Fig. S9 shows the comparison of the subcellular distribution of IFGGA2 and LC3B in the liver fractions of mice kept on LFD or HFD for 3 and 12 weeks. LC3B was mainly detected in lysosomal/endosomal fractions and both proteins show a higher abundance in the lipid droplet fraction in response to 3 weeks HFD feeding than on LFD. In contrast, after 12 weeks on HFD, IFGGA2 and LC3B differ in their localization; only IFGGA2 disappears from the intracellular membranes and is more or less exclusively detectable on lipid droplets (Fig. S9).
Tendency towards decreased lipophagy after suppression of IFGGA2
In order to test if IFGGA2 induces lipophagy, its expression was downregulated in primary hepatocytes via a specific siRNA (Fig. S10A) and cells were treated without serum ± the autophagy inhibitor BafilomycinA for 2 h. BafilomycinA increased the LC3B-II signal intensity in control cells, whereas this effect was weaker in si-Ifgga2 transfected cells. The evaluation of autophagic flux by calculating the ratio of LC3B-II of serum-free plus BafilomycinA and serum-free conditions provided evidence for a lower degree of autophagy after downregulation of Ifgga2, however this effect did not reach significance. No clear results were detected for other autophagy markers (ATG5, BECLIN1, p62) (Fig. 6A). Only after 24 h treatment with serum-free medium plus BafilomycinA was a tendency towards an elevated p62 content visible in si-Ifgga2 cells (Fig. 6B). In addition, liver explants of AAV-miR-Ifgga2 mice were either incubated with serum-containing or with serum-free medium ± Leupeptin/NH4Cl, inhibitors of autophagy. The LC3B-II concentrations of control samples were increased overall, but were not significantly higher in AAV-control than in AAV-miR-Ifgga2 explants (Fig. 6C, Fig. S10B,C). In livers of Ltg/NZO.5.3N/N, the amount of LC3B-II was significantly lower compared to Ltg/NZO.5.3N/B mice (Fig. 6D, Fig. S11). However, p62 and ATG5 did not show differences. Thus, we cannot conclusively claim that IFGGA2 influences the autophagy rate.
Fig. 6. Suppression of IFGGA2 reduces autophagy.

(A) Western blotting and quantification of autophagy proteins in siRNA-transfected primary hepatocytes treated ± BafilomycinA for 2 h (n = 3). (B) Analysis and quantification of p62 in siRNA-transfected hepatocytes treated ± BafilomycinA for 24 h (n = 2–3). (C) Ex vivo flux assay from liver explants of AAV-Control (n = 4) and AAV-miR-Ifgga2 (n = 3) mice. Quantification also includes data of Fig. S11. Data are expressed as fold of control. (D) Analysis of autophagy proteins in livers of Ltg/NZO.5.3N/N (n = 10) and Ltg/NZO.5.3N/B (n = 11) mice after 16 h fasting. Quantification also includes data of Fig. S12. Data are expressed as fold of control (N/N), analyzed by unpaired t test with Welch's correction and presented as mean ± SD. ***p <0.001. AAV, adeno-associated virus; Ltg/NZO, liver triglycerides from New Zealand obese alleles; miR, microRNA; siRNA, small interfering RNA.
Obese patients with NAFLD exhibit low expression of the human orthologue IRGM
Humans express 2 IRG genes, IRGC (immunity-related GTPase cinema) and IRGM on chromosomes 19 and 5, respectively. The human genomic segment on chromosome 5 is syntenic to the Irg gene cluster on murine chromosome 18 where IRGM maps only 80 kb away from the closest syntenic marker Dctn4 (Fig. 7A)36. Therefore, IRGM appears to be the most plausible human orthologue of Ifgga2 and Ifgga4. Additionally, expression analysis in liver biopsies of obese patients with NAFLD and healthy lean controls29 showed no difference in IRGC expression between the groups, but the expression of IRGM was lower in patients with NAFLD compared to healthy controls (Fig. 7B). Thus, IRGM expression is also negatively correlated with hepatic fat content in humans (Pearson correlation: r = −0.327; p = 0.092).
Fig. 7. Expression of human IRGC and IRGM genes.

(A) Cluster of immunity-related GTPases on mouse chromosome 18 and human chromosome 5. (B) Expression of IRGC and IRGM in liver biopsies of lean controls (Control, n = 7) and humans with non-alcoholic fatty liver disease (NAFLD, n = 11). Data were normalized to HPRT, analyzed by unpaired t test and presented as mean ± SD. *p <0.05. (C) Co-immunoprecipitation of co-transfected McArdle hepatocytes. (D) Scheme of the hypothesis how the interaction of the proteins IFGGA2, LC3B and ATGL promotes lipophagy. NAFLD, non-alcoholic fatty liver disease.
Interestingly, the human IRGM protein interacts with ATGL in a similar way to the murine IFGGA2, as demonstrated by the co-immunoprecipitation shown in Fig. 7C, which supports the assumption that the IRGM gene is the human orthologue of Ifgga2.
Discussion
A novel fatty liver locus (Ltg/NZO) was identified wherein the NZO allele is linked to increased hepatic fat content. The IRGs Ifgga2 and Ifgga4 are the most likely candidates mediating the phenotype. They were markedly suppressed in livers of NZO allele carriers presumably due to a single nucleotide deletion in a Foxo1-binding motif within an enhancer upstream of Ifgga2. Downregulation of either Ifgga2 or Ifgga4 in B6 mice increased hepatic fat accumulation and overexpression of Ifgga2 in McArdle hepatocytes decreased fat storage. These findings provide direct functional evidence for the important role of Ifgga2 and Ifgga4 in regulating ectopic fat storage. The potential mechanism might be mediated via the interaction of IFGGA2 with the lipase ATGL and an increased association of LC3B with lipid droplets upon its overexpression.
The identification of disease-associated QTL and the subsequent positional cloning is a powerful non-hypothesis driven approach to identify candidate genes for metabolic traits.12 Here, a genome-wide linkage analysis revealed a single QTL for liver weight and liver triglycerides in mice of (NZOxB6)N2 progeny. As causative genes of Ltg/NZO, Ifgga2 and Ifgga4 (human orthologue IRGM)37 were identified. Direct functional evidence that sufficient Ifgga2 expression prevents fatty liver development was provided by its overexpression in McArdle hepatocytes which resulted in reduced triglyceride storage. Correspondingly, downregulation of Ifgga2 or Ifgga4 in the liver increased hepatic fat content, thereby phenocopying NZO and Ltg/NZO.53NN mice.
The cause of the reduced expression of Ifgga2 and Ifgga4 in NZO mice appears to be a single nucleotide deletion in a Foxo1-binding motif in the NZO genome within an active enhancer element upstream of Ifgga2. In line with this, we identified a putative active enhancer element (ENCODE) 50 kb upstream of IRGM showing multiple Foxo1-binding motifs in the human genome (Fig. S12).
We hypothesize that IFGGA2 associates with membranes containing LC3B and that a lipid load of cells or the feeding of HFD enhances the interaction of IFGGA2 and ATGL. The data indicate that IFGGA2 brings ATGL near to LC3B and induces lipophagy through this interaction (Fig. 7D). Our hypothesis is based on the following facts: (i) Overexpression of Ifgga2 in hepatocytes increased association of LC3B with lipid droplets, whereas suppression of Ifgga2 resulted in a lower abundance of LC3B-II. (ii) In subcellular fractions, IFGGA2 and LC3B associated within the same endosomal/lysosomal fractions; upon lipid load and HFD, IFGGA2 and to a lesser extent LC3B translocated to lipid droplets. (iii) Irgm1 has been described as playing a crucial role in the stimulation of early stages of autophagy in macrophages.34 (iv) IFGGA2 interacts with ATGL, a known inducer of lipophagy in the liver.38 ATGL contains a LC3B-interacting region and its binding to autophagosomes/LC3B-II supports the translocation of ATGL to the surface of lipid droplets and enhances its activity.38
Autophagy is an intracellular pathway that targets – besides other intracellular components – lipids (lipophagy) to the lysosomes for degradation. Autophagy is increased in lean compared to obese mice, as indicated by the enhanced conversion of LC3B-I to LC3B-II, preventing the development of hepatic steatosis and insulin resistance.39 In fact, suppression of Ifgga2 expression in primary hepatocytes slightly reduced the formation of LC3B-II (Fig. 6A), liver explants of mice in which Ifgga2 expression was downregulated exhibited a lower concentration of LC3B-II (Fig. 6C), and livers of Ltg/NZO.5.3 NN mice with low expression of Ifgga2 had a lower concentration of LC3B-II (Fig. 6D; Fig. S11). Moreover, the human orthologue IRGM was reported to induce autophagy by facilitating the recruitment of Syntaxin17 to autophagosomes by interacting with LC3B.40 Therefore, it might be possible that the crosstalk of IFGGA2, LC3B and ATGL promotes lipophagy. However, it is also likely that IFGGA2 binding to ATGL modifies its activity by other mechanisms. Firstly, the association of IFGGA2 with ATGL could interrupt the binding of the lipase to the inhibitors G0S2 or HILPDA, which have been shown to directly interact with ATGL.41 Secondly, IFGGA2 could affect sorting of proteins to lipid droplets, thereby limiting their growth. Thirdly, IFGGA2 could interfere with the action of enzymes of the glycerol-3-phosphate pathway which not only act at the endoplasmic reticulum but also associate directly with lipid droplets (GPAT4, DGAT1, DGAT2, AGPAT3 and lipin) and mediate triglyceride biosynthesis.42 Further experiments and biochemical analyses are required to clarify how IFGGA2 acts at the lipid droplet and decreases ectopic fat storage in the liver.
Expression of IRGM is significantly reduced in the livers of obese individuals with NAFLD compared to control individuals. Two human cohorts showed that IRGM variants are associated with an increased risk of NAFLD. The studies indicated that the frequency of the risk allele is highly depended on ethnic groups; 45.6% carry the risk allele in a cohort of Han-Chinese43 but only 2.5% in an European cohort.44 The less functional TT-variant of IRGM (rs10065172) was linked to an increased risk of steatosis in obese Italian children.44 Furthermore, the downregulation of IRGM in HepG2 cells decreased autophagic flux and was accompanied by increased lipid accumulation.43 Thus, compounds that can increase the expression of IRGM could have therapeutic potential for fatty liver diseases.
In conclusion, our data show that Ifgga2 and Ifgga4 and their human orthologue IRGM are key players that protect against hepatic steatosis.
Supplementary Material
Highlights.
Positional cloning identified Ifgga2 and Ifgga4 as inhibitor genes of fatty liver.
The expression of the human orthologue IRGM is significantly lower in individuals with NAFLD.
IFGGA2 interacts with ATGL and co-localizes with this lipase at lipid droplets.
IFGGA2 increases the amount of LC3B on lipid droplets.
Acknowledgements
We gratefully thank J. Würfel, C. Gumz, F. Gabler, A. Helms and A. Teichmann of the German Institute of Human Nutrition Potsdam-Rehbruecke (Germany) for their skillful technical assistance. We thank R. Zechner for providing the Atgl-His plasmid, A. M. Davidoff for kindly providing the LP1 promoter (St. Jude Children's Research Hospital, Memphis, TN) and A. Attie (University of Wisconsin) for discussions on this project.
This work was supported by grants from the German Ministry of Education and Research, the State of Brandenburg, and the State of North-Rhine-Westfalia (82DZD00302, AS; 82DZD00202, HA) and by the German Research Foundation (DFG; SFB 958, FH, AS). N.K. is funded by Emmy-Noether DFG (KR5166/1-1).
Abbreviations
- AAV
adeno-associated virus
- B6
C57BL/6
- ChIP
chromatin immunoprecipitation
- DAGs
diacylglycerols
- EMSA
electrophoretic mobility shift assay
- HFD
high-fat diet
- IRG
immunity-related GTPases
- IRGC
immunity-related GTPase cinema
- IRGM
immunity-related GTPase M
- LFD
low fat diet
- Ltg/NZO
liver triglycerides from NZO alleles
- NAFLD
non-alcoholic fatty liver disease
- NZO
New Zealand obese
- miRNA
microRNA
- PWM
position weight matrices
- QTL
quantitative trait loci
- RCS
recombinant congenic mice
- siRNA
small interfering RNA
- SNPs
single nucleotide polymorphisms
- TFBS
transcription factor binding site
Footnotes
Conflict of interest
The authors declare no conflicts of interest that pertain to this work.
Please refer to the accompanying ICMJE disclosure forms for further details.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhep.2020.04.031.
References
- [1].Choudhury J, Sanyal AJ. Clinical aspects of fatty liver disease. Semin Liver Dis 2004;24:349–362. [DOI] [PubMed] [Google Scholar]
- [2].Tilg H, Moschen AR, Roden M. NAFLD and diabetes mellitus. Nat Rev Gastroenterol Hepatol 2017;14:32–42. [DOI] [PubMed] [Google Scholar]
- [3].Macaluso FS, Maida M, Petta S. Genetic background in nonalcoholic fatty liver disease: a comprehensive review. World J Gastroenterol 2015;21:11088–11111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Palmer ND, Musani SK, Yerges-Armstrong LM, Feitosa MF, Bielak LF, Hernaez R, et al. Characterization of European ancestry nonalcoholic fatty liver disease-associated variants in individuals of African and Hispanic descent. Hepatology 2013;58:966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bruschi FV, Tardelli M, Claudel T, Trauner M. PNPLA3 expression and its impact on the liver: current perspectives. Hepat Med 2017;9:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sookoian S, Castaño GO, Burgueño AL, Gianotti TF, Rosselli MS, Pirola CJ. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J Lipid Res 2009;50:2111–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kozlitina J,Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature 2009;461:747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang C-Y, Stapleton DS, Schueler KL, Rabaglia ME, Oler AT, Keller MP, et al. Tsc2, a positional candidate gene underlying a quantitative trait locus for hepatic steatosis. J Lipid Res 2012;53:1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Vogel H, Scherneck S, Kanzleiter T, Benz V, Kluge R, Stadion M, et al. Loss of function of Ifi202b by a microdeletion on chromosome 1 of C57BL/6J mice suppresses 11β-hydroxysteroid dehydrogenase type 1 expression and development of obesity. Hum Mol Genet 2012;21:3845–3857. [DOI] [PubMed] [Google Scholar]
- [12].Vogel H, Kamitz A, Hallahan N, Lebek S, Schallschmidt T, Jonas W, et al. A collective diabetes cross in combination with a computational framework to dissect the genetics of human obesity and type 2 diabetes. Hum Mol Genet 2018;27:3099–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kleinert M, Clemmensen C, Hofmann SM, Moore MC, Renner S, Woods SC, et al. Animal models of obesity and diabetes mellitus. Nat Rev Endocrinol 2018;14:140–162. [DOI] [PubMed] [Google Scholar]
- [14].Schmidt C, Gonzaludo NP, Strunk S, Dahm S, Schuchhardt J, Kleinjung F, et al. A meta-analysis of QTL for diabetes-related traits in rodents. Physiol Genomics 2008;34:42–53. [DOI] [PubMed] [Google Scholar]
- [15].Schallschmidt T, Lebek S, Altenhofen D, Damen M, Schulte Y, Knebel B, et al. Two novel candidate genes for insulin secretion identified by comparative genomics of multiple backcross mouse populations. Genetics 2018;210:1527–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- [17].Nathwani AC, Tuddenham EGD, Rangarajan S, Rosales C, McIntosh J, Linch DC, et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med 2011;365:2357–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Rödiger M, Werno MW, Wilhelmi I, Baumeier C, Hesse D, Wettschureck N, et al. Adiponectin release and insulin receptor targeting share trans-Golgi-dependent endosomal trafficking routes. Mol Metab 2018;8:167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lubura M, Hesse D, Neumann N, Scherneck S, Wiedmer P, Schürmann A. Non-invasive quantification of white and brown adipose tissues and liver fat content by computed tomography in mice. PLoS One 2012;7:e37026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kachler K, Bailer M, Heim L, Schumacher F, Reichel M, Holzinger CD, et al. Enhanced acid sphingomyelinase activity drives immune evasion and tumor growth in non–small cell lung carcinoma. Cancer Res 2017;77:5963–5976. [DOI] [PubMed] [Google Scholar]
- [21].Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, et al. Jaspar 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res 2014;42:D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tan G, Lenhard B. TFBSTools: an R/bioconductor package for transcription factor binding site analysis. Bioinformatics 2016;32:1555–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Saussenthaler S, Ouni M, Baumeier C, Schwerbel K, Gottmann P, Christmann S, et al. Epigenetic regulation of hepatic Dpp4 expression in response to dietary protein. J Nutr Biochem 2019;63:109–116. [DOI] [PubMed] [Google Scholar]
- [24].Henagan TM, Laeger T, Navard AM, Albarado D, Noland RC, Stadler K, et al. Hepatic autophagy contributes to the metabolic response to dietary protein restriction. Metabolism 2016;65:805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Krahmer N, Najafi B, Schueder F, Quagliarini F, Steger M, Seitz S, et al. Organellar proteomics and phospho-proteomics reveal subcellular reorganization in diet-induced hepatic steatosis. Dev Cell 2018;47: 205–221.e7. [DOI] [PubMed] [Google Scholar]
- [26].Brasaemle DL, Wolins NE. Isolation of lipid droplets from cells by density gradient centrifugation. Curr Protoc Cell Biol 2016;2016:3.15.1–3.15.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat Methods 2014;11:319–324. [DOI] [PubMed] [Google Scholar]
- [28].Baumeier C, Schlüter L, Saussenthaler S, Laeger T, Rödiger M, Alaze SA, et al. Elevated hepatic DPP4 activity promotes insulin resistance and non-alcoholic fatty liver disease. Mol Metab 2017;6:1254–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab 2015;21:739–746. [DOI] [PubMed] [Google Scholar]
- [30].Keane TM, Goodstadt L, Danecek P, White MA, Wong K, Yalcin B, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 2011;477:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yalcin B, Wong K, Agam A, Goodson M, Keane TM, Gan X, et al. Sequence-based characterization of structural variation in the mouse genome. Nature 2011;477:326–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pundhir S, Poirazi P, Gorodkin J. Emerging applications of read profiles towards the functional annotation of the genome. Front Genet 2015;6:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].ENCODE Project Consortium, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature 2012;489:57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006;313:1438–1441. [DOI] [PubMed] [Google Scholar]
- [35].Sathyanarayan A, Mashek MT, Mashek DG. ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Rep 2017;19:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bekpen C, Hunn JP, Rohde C, Parvanova I, Guethlein L, Dunn DM, et al. The interferon-inducible p47 (IRG) GTPases in vertebrates: loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol 2005;6:R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].MacMicking JD. IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol 2004;25:601–609. [DOI] [PubMed] [Google Scholar]
- [38].Martinez-Lopez N, Garcia-Macia M, Sahu S, Athonvarangkul D, Liebling E, Merlo P, et al. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab 2016;23:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab 2010;11:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kumar S, Jain A, Farzam F, Jia J, Gu Y, Choi SW, et al. Mechanism of Stx17 recruitment to autophagosomes via IRGM and mammalian Atg8 proteins. J Cell Biol 2018;217:997–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Padmanabha Das KM, Wechselberger L, Liziczai M, De La Rosa Rodriguez M, Grabner GF, Heier C, et al. Hypoxia-inducible lipid droplet-associated protein inhibits adipose triglyceride lipase. J Lipid Res 2018;59:531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang H, Airola MV, Reue K. How lipid droplets “TAG” along: glycerolipid synthetic enzymes and lipid storage. Biochim Biophys Acta Mol Cell Biol Lipids 2017;1862:1131–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lin Y-C, Chang P-F, Lin H-F, Liu K, Chang M-H, Ni Y-H. Variants in the autophagy-related gene IRGM confer susceptibility to non-alcoholic fatty liver disease by modulating lipophagy. J Hepatol 2016;65:1209–1216. [DOI] [PubMed] [Google Scholar]
- [44].Bellini G, Miraglia del Giudice E, Nobili V, Rossi F. The IRGM rs10065172 variant increases the risk for steatosis but not for liver damage progression in Italian obese children. J Hepatol 2017;67:653–655. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.