

SUMMARY
Obesity is a top public health concern and a molecule that safely treats obesity is urgently needed. Disulfiram (known commercially as Antabuse), an FDA-approved treatment for chronic alcohol addiction, exhibits anti-inflammatory properties and helps protect against certain types of cancer. Here, we show that in mice disulfiram treatment prevented body weight gain and abrogated the adverse impact of an obesogenic diet on insulin responsiveness, while mitigating liver steatosis and pancreatic islet hypertrophy. Additionally, disulfiram treatment reversed established diet-induced obesity and metabolic dysfunctions in middle-aged mice. Reductions in feeding efficiency and increases in energy expenditure were associated with body weight regulation in response to long-term disulfiram treatment. Loss of fat tissue and an increase in liver fenestrations were also observed in rats on disulfiram. Given the potent anti-obesogenic effects in rodents, repurposing disulfiram in the clinic may represent a new strategy to treat obesity and its metabolic comorbidities.
Graphical Abstract
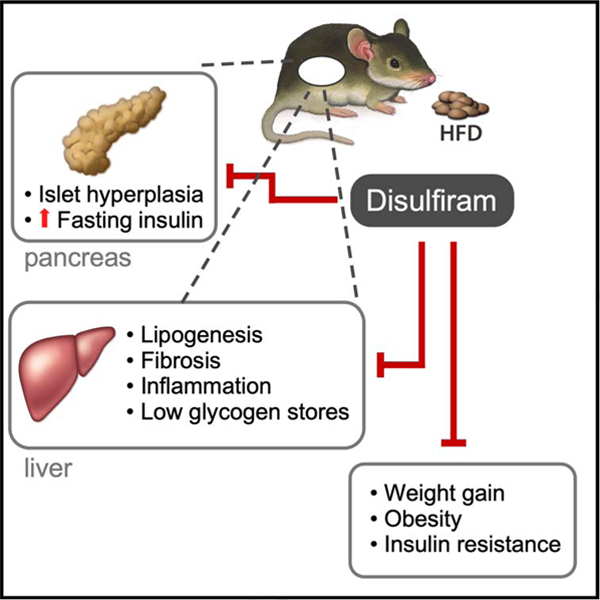
eTOC:
In this study, Bernier et al. report on the off-label use of disulfiram to combat diet-induced obesity in mice through normalization of body weight and improvement of various physiological outcomes related to body composition and insulin responsiveness.
INTRODUCTION
Obesity has reached epidemic proportions in developed countries and represents a major public health challenge. The consumption of high-fat and high-carbohydrate diets induces oxidative stress and inflicts mitochondrial damage that ultimately results in chronic low-grade inflammation and the initiation of changes in the structure and function of multiple peripheral organs, including the liver, pancreas, endothelium, kidney and cardiovascular system (Altunkaynak et al., 2008; Cogger et al., 2016; Putti et al., 2015; Sachdeva et al., 2009; Terauchi et al., 2007; Panchal et al., 2011). Metabolic syndrome represents a cluster of risk factors for premature mortality that include central adiposity, elevated blood pressure, impaired glucose tolerance, insulin resistance and dyslipidemia (Dandona 2005; Lonardo et al., 2015; Marseglia et al., 2014). For many individuals with obesity, behavioral and lifestyle modifications are insufficient for long-term weight-loss maintenance and, therefore, depend on surgical procedures and/or pharmacological interventions that alter either appetite or absorption of calories. The current US FDA-approved weight loss medications for obesity management include orlistat, lorcaserin, phentermine/topiramate-ER, buproprion, metformin, naltrexone, and liraglutide (Yanovski and Yanovski, 2014). However, battling obesity requires long-term use of safe and effective drugs, and ideally ones that can alleviate its co-morbidities, such as improving systemic glucose and lipid homeostasis.
For the last five decades, disulfiram (DSF, also known chemically as tetraethyl thiuramdisulfide and commercially as Antabuse) has been widely used for aversive treatment of alcoholism. It is an FDA-approved drug that is well-tolerated with few side effects. DSF and related members of the dithiocarbamate family, such as pyrrolidinedithiocarbamate (PDTC), were initially discovered as inhibitors of the transcription factor NF-κB in intact cells (Schreck, 1992; Muñoz et al., 1996) and subsequently demonstrated to lower the levels of free glutathione (Wang et al., 2009) and interleukin-18 in acute pancreatitis (Kast 2008). PDTC promotes an increase in hepatic glycogen synthesis and lowers fasting blood glucose in rats with experimental type 2 diabetes (Zhu et al., 2012); besides, it confers protection against oxidative damage and apoptosis in pancreatic β-cells and improves insulin production in a rat model of type 2 diabetes (Ding et al., 2014). Although DSF is effective in combatting inflammation in vivo, it is unclear whether the drug would also be useful in preventing the deregulation of homeostatic mechanisms caused by consumption of a high-fat diet.
In this study, we evaluated the long-term use of DSF as a drug repurposed for the prevention and treatment of diet-induced obesity in mice. In addition, we analyzed the treated mice for changes in body weight, morphometric measures, metabolic performance and the structure of the pancreas and liver by assessing a number of physiological and histological parameters. Some of these outcomes were also investigated in rats. Overall, our results indicate that DSF normalizes body weight both prophylactically and therapeutically while reversing metabolic abnormalities commonly associated with diet-induced obesity.
RESULTS AND DISCUSSION
We assessed the effect of DSF on phenotypic and metabolic parameters in 14-week-old male C57BL/6J mice fed either a standard diet (SD; 4.85 kCal/g food) or a high-fat diet (HFD; 7.05 kCal/g food) supplemented with a low (HFDL,100 mg/kg food/day) or a high dose (HFDH, 200 mg/kg food/day) of DSF for periods up to 64 weeks. The maximum dosage of 500 mg per day PO for a 70 kg adult corresponds to 7.14 mg DSF/kg BW (http://www.pdr.net/drug-summary/Antabuse-disulfiram-681). Following the allometric scaling of Reagan-Shaw et al. (Reagan-Shaw et al., 2008), this amounts to 2.64 mg DSF/30 g mouse PO. Based on the fact that DSF is added in the food and each mouse eats ~ 3 g food per day, we estimate that each animal ingests about 0.3 mg (low dose) or 0.6 mg (high dose) of DSF daily, which corresponds to 57–113 mg DSF per day PO in humans and viewed as sub- to low-maintenance doses.
The body weight of the mice receiving DSF in their food was consistently lower than their respective SD and HFD controls (Figures S1A and 1A). To determine the physiological responses and adaptations to DSF treatment, we applied a well-established timetable routinely used in our laboratory (Figure 1B). A non-significant difference in daily food intake between DSF and control groups was observed throughout the duration of the study (Figures S1B and 1C), which was combined with substantial reduction in feeding efficiency, as defined as change in BW over number of calories ingested, in DSF-treated animals fed either SD or a HFD diet (Figures S1C and 1D). To exclude food palatability as a confounding factor, fasted animals in their home cages when given a predetermined amount of food during the dark cycle and the amount of food consumed in an hour was recorded and found to be greater in mice on DSF compared to the control groups (data not shown). Diet supplementation with DSF contributed to a significant increase in the lean-to-fat ratio regardless of the diet (Figures S1D and 1E), coincident with lower body fat mass (Figures S1E and S1F, Figures S1P and S1Q).
Figure 1.
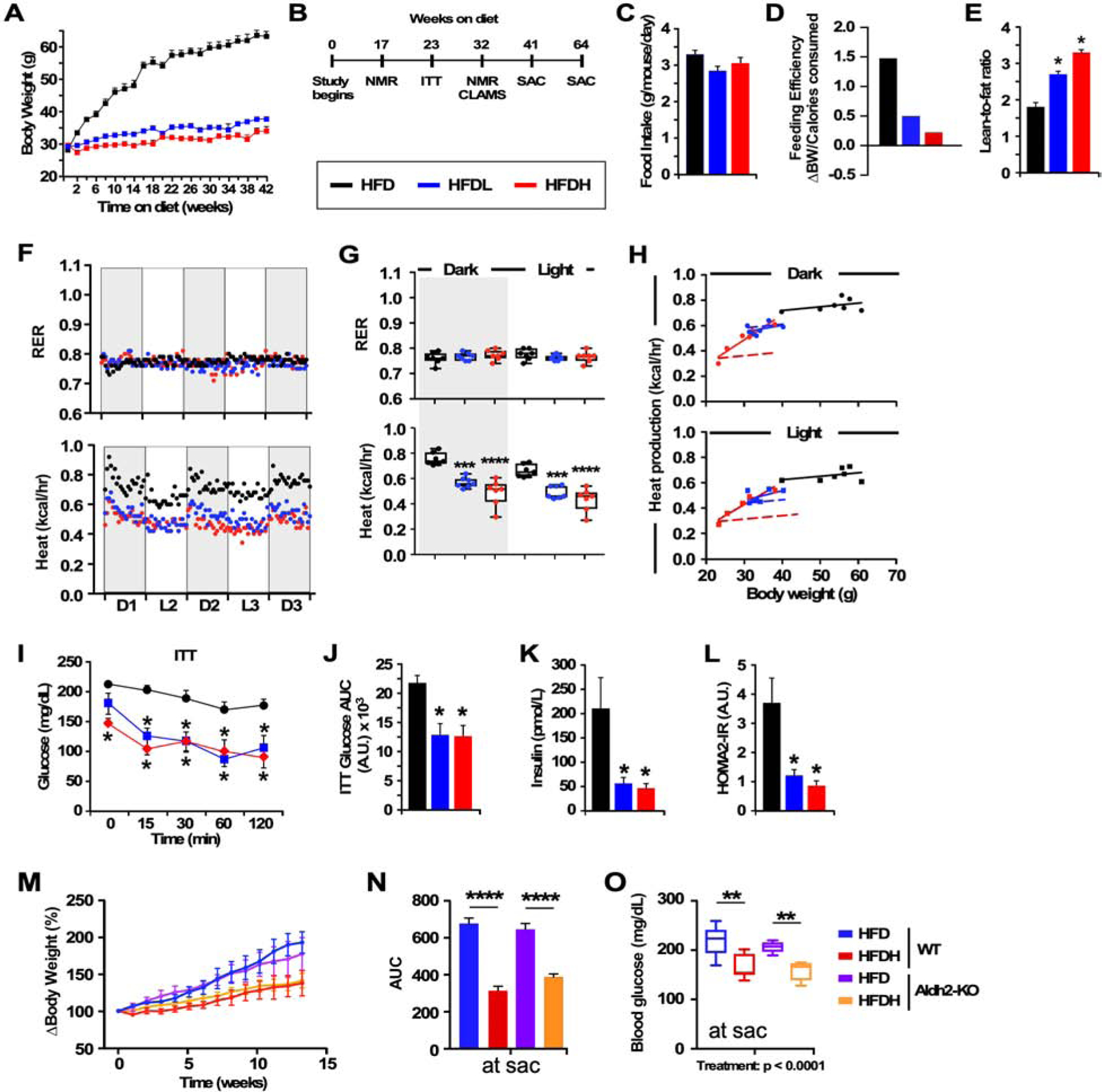
DSF improves health and insulin responsiveness in mice fed a high-fat diet (HFD).
(A) Average body weight trajectory in HFD-fed mice supplemented without or with low (HFDL) and high (HFDH) doses of DSF, n =14–22 per group.
(B) Timetable for phenotypic, metabolic and biochemical analyses.
(C) Average daily food consumption across 42 weeks.
(D) Feeding efficiency expressed as change in BW over average number of calories consumed per day.
(E) Lean-to-fat ratio measured by nuclear magnetic resonance spectroscopy.
(F) Metabolic cage analyses for the determination of the Respiratory Exchange Ratio (RER) and average heat production across 60 h. D, dark cycle; L, light cycle.
(G) Average signals associated with D1-D3 (dark) and L1-L2 (light) represented as scatter plots. *** and ****, P < 0.001 and 0.0001 vs. HFD using one-way ANOVA coupled with Dunnett’s post-hoc test.
(H) Relationship between heat production and body weight in HFD, HFDL, HFDH mice. Dotted line represents the transposition of the HFD slope to that of HFDH; 48 weeks of age, 32 weeks on diet, n = 7 per group.
(I-J) Insulin tolerance test (I) with area under the curve (AUC) (J); 39 weeks of age, 23 weeks on diet, n = 6 per group.
(K) Insulin levels after a 16-h fast.
(L) The homeostatic model assessment calculation of insulin resistance (HOMA-IR); 45 weeks of age, 23 weeks on diet, n =4–6 per group. *, P < 0.05 vs. HFD.
(M-O) Response to DSF supplementation in Aldh2-KO mice fed HFD. (M) Body weight gain trajectories over the course of 13-weeks expressed as % change over baseline with (N) AUC; mean ± SEM. (O) Fasting blood glucose collected at the time of sacrifice. WT mice fed HFD +/− DSF, n = 7/group; Aldh2-KO mice on HFD +/− DSF, n = 4–5/group. Statistics for the overall effects of treatment, genotype, and the interaction (treatment x genotype) represent the P value from a two-way ANOVA for each measurement; ** and ****, P < 0.01 and 0.0001, Tukey post-hoc test.
We next assessed the impact of DSF on in vivo measurement of energy metabolism by placing mice in metabolic chambers for a period of 60 h. Substrate utilization was examined by measuring the respiratory exchange ratio (RER). An RER approaching 0.7 is indicative of lipid utilization while an RER of 1 indicates that carbohydrates are used as energy sources (Hasek et al., 2010). Mice on the HFD had an RER of ~0.78 for the duration of the experiment, whether DSF was present or not (Figure 1F), with distinctive diurnal patterns of O2 consumption, CO2 production (Figure S1T) and heat generation (Figure 1F). The parameters measured in the 3 dark cycles and the 2 light cycles were averaged and depicted in Figure 1G and Figures S1H and S1S. HFDL and HFDH mice had significantly lower O2 utilization and CO2 production (Figures S1R and S1S) that translated into less energy expenditure (EE) compared to HFD-fed controls (Figures 1F–1G). SD-fed mice had RER levels of ~0.9–0.95 in the presence or absence of DSF (Figure S1G). A significant but weaker impact of DSF on the diurnal patterns of O2 consumption, CO2 production, and heat generation was also observed in SDL and SDH mice compared to SD controls (Figures S1G–S1H). Ambulatory activity was not impacted by DSF treatment in mice fed SD or HFD (data not shown). To assess the influence of body size on energy balance, we applied the analytical procedure proposed by Mina et al. (Mina et al., 2018) and examined whether EE of DSF-treated animals and controls was identically affected by mass (Figures 1H and S1I). The difference in the slopes suggested that each group of mice have different associations between EE and mass, thus preventing the use of conventional ANCOVA for the analysis (Mina et al., 2018; Fernández-Verdejo et al., 2019). We observed a robust difference in the association between these parameters especially in HFDH mice which had a more pronounced energy dissipation per unit of body mass compared to HDFL and HFD controls in both the dark and light cycles (Figure 1H). Transposing the slope of HFD to that of HFDH revealed a strong interaction effect of DSF well above the ‘predicted’ association between the measured parameters seen with HFD-fed controls (Figure 1H, dotted vs. solid red lines). However, DSF had a substantially weaker impact on the effect of mass on EE in SD mice (Figure S1I). There was no alteration in locomotor activity between DSF-treated mice and controls regardless of the diet (data not shown). These results indicate that long-term treatment with DSF leads to body weight regulation through lower feeding efficiency and improved energy expenditure without impacting fuel selection or voluntary locomotor activity.
We determined the effect of DSF on glucose homeostasis by performing insulin tolerance tests (ITT) in 39-week-old mice maintained on the various diets for 23 weeks. DSF treatment did not improve either the baseline glucose levels or the rate of glucose clearance and its associated glucose AUC in SD-fed mice relative to controls (Figures S1M–S1O), while HFD-fed mice on DSF showed a dose-dependent reduction in glucose levels at baseline (Figure S1X) coupled with significant increase in the rate of glucose disposal (glucose values expressed either as mg/dL, Figure 1I, or percent change of glucose levels from baseline, Figure S1Y) together with a significant reduction in AUC (Figure 1J). After a 6-h fast, glucose and plasma insulin levels were significantly lower in HFD-fed mice on DSF than in control mice on HFD (Figures S1Z and 1K), but not in SD-fed mice with and without DSF (Figures S1P and S1Q). Consistent with these results, the HOMA2-IR values showed that DSF helped restored insulin sensitivity in the HFD cohorts to the same level as SD-fed mice (Figure 1L versus S1R).
The associations between plasma adipokines (e.g., leptin and adiponectin), weight gain, and insulin resistance led us to assess the impact of DSF on circulating adiponectin and leptin levels (Table S1). Irrespective of the diet type, DSF significantly reduced the levels of both adipokines at 29 weeks of treatment, although consumption of a HFD mitigated the lowering effects of DSF on adiponectin levels. Little alteration, if any, in the levels of IL-6 and TNFα was observed with DSF added in either diet (Table S1). When similar determinations were carried out after 63 weeks of treatment, DSF failed to induce significant differences compared to controls (Table S1). Thus, the observed improvement of insulin sensitivity evoked by DSF treatment in HFD-fed mice did not occur as a result of reduced circulating pro-inflammatory cytokine levels.
Aldehyde dehydrogenase 2 (ALDH2) is a mitochondrial enzyme whose dysregulation has been associated with a number of human pathologies including metabolic disorders and cancer (Chen et al., 2014). ALDH2 also plays a key role in detoxifying acetaldehyde in the liver. Because of the ability of metabolic products of DSF to inhibit mitochondrial ALDH activity in vivo (Deitrich and Erwin, 1971), we investigated whether inhibition of ALDH2 could account for the health benefits of DSF in HFD-fed mice. Wild-type (WT) and Aldh2 knockout (Aldh2-KO) male mice (8–10 weeks of age) were fed an HFD or an HFD supplemented with DSF (HFDH; 200 mg/kg BW) for 13 weeks, after which body weight gain, food consumption, and glucose metabolism were investigated. Preliminary experiments confirmed the full knockout of Aldh2 by RT-PCR (Figure S1W). At baseline, Aldh2-KO mice weighted less than their WT counterparts (22.9 ± 0.4 g vs 25.0 ± 0.4 g, n = 9–14 per group, p < 0.003); however, a significantly slower rate of weight gain was observed in mice of both genotypes when fed HFD+DSF vs. HFD alone (Figures 1M and 1N). When measured over a 5-week period, all groups of mice consumed a daily average of 3.45 ± 0.24 g food. At sacrifice, mice on DSF weighted significantly less than the controls (Figure S1X, top panel) although their liver weight, expressed as a percentage of total body mass, did not differ from HFD controls regardless of genotype (Figure S1X, bottom panel). A significant reduction in fasting blood glucose accompanied DSF supplementation (Figure 1O). These observations suggest that the loss of ALDH2 did not interfere with DSF’s ability to confer metabolic benefits in mice fed an obesogenic diet.
The consumption of an HFD is associated with liver damage due to lipid accumulation and local inflammation. Histochemical analyses were carried out on frozen fixed liver tissues of mice to determine the effects of DSF on diet-induced increases in hepatic steatosis and fibrosis. Mice on an HFD for 41 weeks displayed considerable levels of fat and collagen infiltration, and lower glycogen deposition (Figure 2A). A similar signature was observed in the liver of SD-fed mice, although to a lesser degree (Figure S2A). In both diet groups, DSF completely prevented this pro-inflammatory profile and the loss in glycogen (Figures 2A and S2A) and scanning electron microscopy confirmed the sharp reduction in hepatic fat content upon DSF treatment (Figures 2B and S2B, arrows). HFD-feeding caused a considerable increase in the number of hepatic stellate cells (Figure 2B), which are considered to play a role in storing dietary fat and promoting fibrosis (Weiskirchen and Tacke, 2016). Supplementation with DSF decreased stellate cell number in both diet groups (Figures 2B and S2B), with little to no change in the porosity of the hepatic sinusoidal endothelium (fenestration frequency or the number of fenestrations per unit area) (data not shown). DSF blunted the large increase in hepatic triglyceride levels evoked by HFD (Figure 2C). Together, these findings indicate that long-term use of DSF maintains hepatic homeostasis by preventing diet-induced liver dysfunction and injury.
Figure 2.
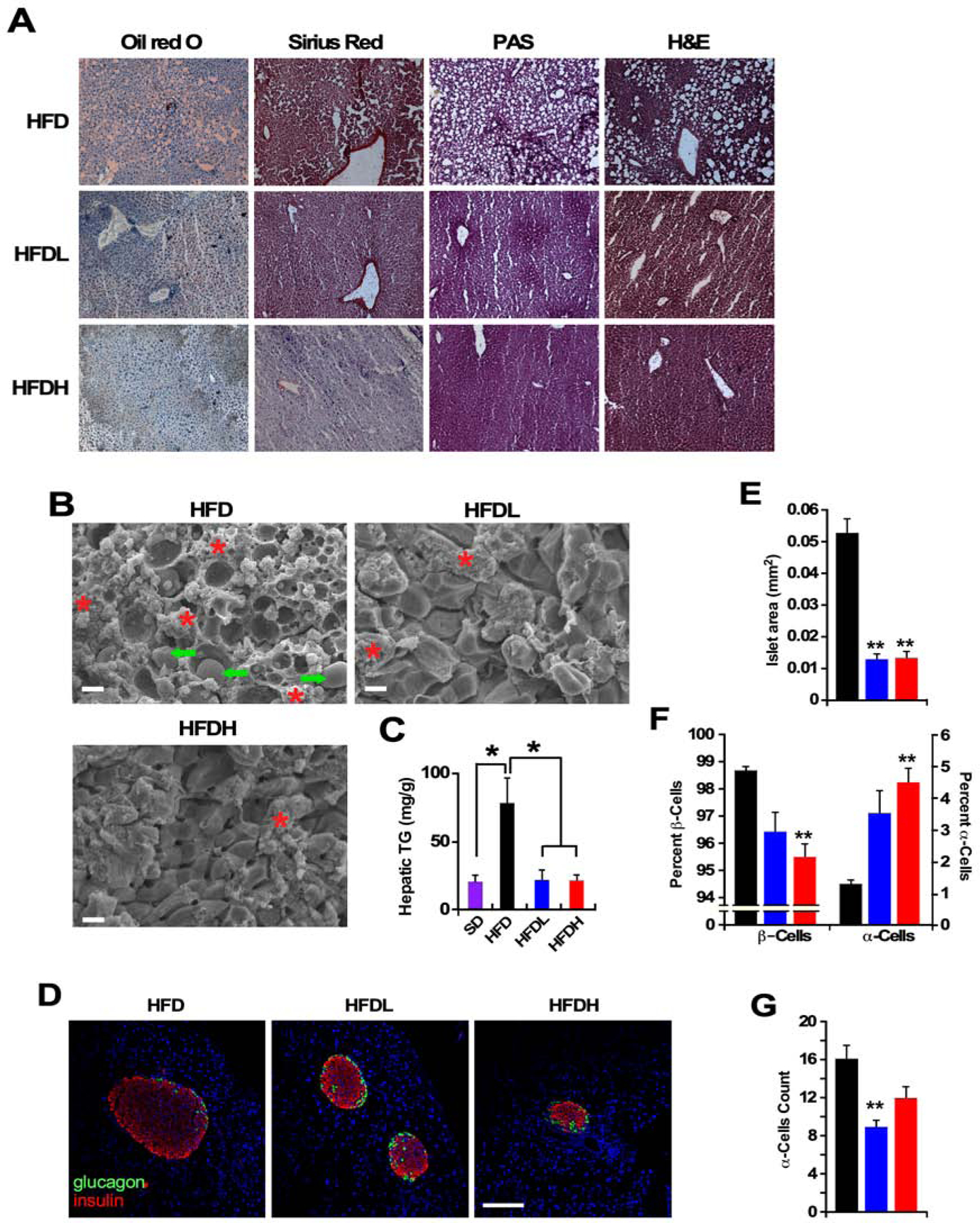
Disulfiram confers protection of liver and pancreas in HFD-fed mice.
(A) Representative photomicrographs of fixed liver sections after staining with Oil red O, Sirius Red, periodic acid-Schiff (PAS), and H&E (x400). (n =5 per group. This was performed on at least 10–15 fields per slide, 3 slides/liver).
(B) Representative SEM images of fixed liver sections (x1000). Scale bar = 10 μm. Arrows denote lipid droplets and * represent hepatic stellate cells. (n = 5 per group, multiple fields per micrograph/animal).
(C) Hepatic triglyceride (TG) levels after 40 weeks on diet, n = 3–4 per group. Data are shown as mean ± SEM (n = 5 per group). * P ≤0.05 compared to diet without DSF; #, P ≤0.05 compared to low DSF.
(D) Representative images of immunostaining for insulin (red), glucagon (green), and TOPRO-3 (blue) in mouse islets after 40 weeks on the indicated dietary intervention. Scale bar = 100 μm.
(n =5 per group. This was performed on at least 10 to 15 sections separated by at least 200 μm from each other section, were assessed for signal intensity per animal).
(E) Quantification of total islet size.
(F) Percentage of β-cells (insulin positive) and α-cells (glucagon positive).
(G) Number of glucagon-producing α-cells per islet.
See also Figure S2.
Evidence suggests that pancreatic islet cell hyperplasia precedes the development of insulin resistance. Therefore, the effect of DSF on the pancreatic β-cell function was evaluated using a morphometric and immunohistochemical approach. HFD-fed mice on DSF exhibited a significant reduction in total islet size and the number of hyperplastic islets (Figures 2D and 2E), together with a large accumulation of α-cells (glucagon positive) --at the expense of β-cells (insulin positive) -- in HFDH mice compared to HFD controls (Figure 2F). The number of α-cells per islet in HFDH mice was akin to HFD controls despite their smaller total islet size (Figure 2G versus 2E). In SD-fed mice, DSF supplementation also diminished islet size (Figures S2C and S2D) without altering the percentage of α-cells and β-cells (Figure S2E). The total number of α-cells per islet was consistent with total islet size in all groups of animals (Figure S2F). It would appear that the observed improvement in insulin sensitivity evoked by DSF stemmed from the lowering in β-cell hyperplasia and correction of diet-induced dysregulation in insulin secretion.
To determine whether DSF could be used as a therapeutic treatment for metabolic disorders, rather than just prophylactically, we induced obesity by way of feeding 9-month-old male and female C57BL/6J mice with an HFD for 12 weeks and then randomly dividing them into four experimental groups (n=12 animals per group/sex). Each group of mice was then exposed to a study diet for an additional 12 weeks. The four study diets were as followed: HFD supplemented with either 0, 100 or 200 mg/kg DSF (HFD, HFDL, HFDH), or standard diet alone (SD) (see Figure 3A for the study protocol). At the termination of the study, mice were killed and serum and tissues were collected for further analysis. At the time of onset of the DSF treatment, both HFDL and HFDH mice showed a substantial reduction in feeding efficiency compared to weight-matched HFD controls (Figure 3B) even though they were consuming the same amount of food as the HFD groups (average 3.0 ± 0.1 and 2.4 ± 0.1 g/mouse/day for males and females, respectively). As expected, mice on chronic HFD for the initial 12 weeks gained a large amount of weight (Figures 3C and 3D, second hatched bars for each sex) and adiposity (Figures 3E–3J) and exhibited features of insulin resistance such as elevated blood glucose after a 6-h fast (Figure 3K) and delayed blood glucose clearance during an ITT (Figure S3A). Mice that remained on HFD for an additional 12 weeks showed an exacerbation in these phenotypes, whereas replacement by SD led to the normalization of body weight, adiposity, and fasting blood glucose levels (Figures 3C–3K, open bars; Table S2). More importantly, the body weight gain and various morphometric changes caused by HFD were reversed in mice fed DSF (Figures 3C–3K, and Figures S3B and S3C, blue and red bars vs. black bars). Similarly, both doses of DSF resulted in reduced adiposity and increased lean/fat ratio when measured by either NMR (Figures 3E–3G) or MRI (Figures 3H–3J), as well as lower circulating blood glucose levels (Figure 3K). The rapid drop in body weight in HFDL and HFDH mice, coupled with a gain in metabolic benefits, mirrored that of mice returning to SD feeding.
Figure 3.
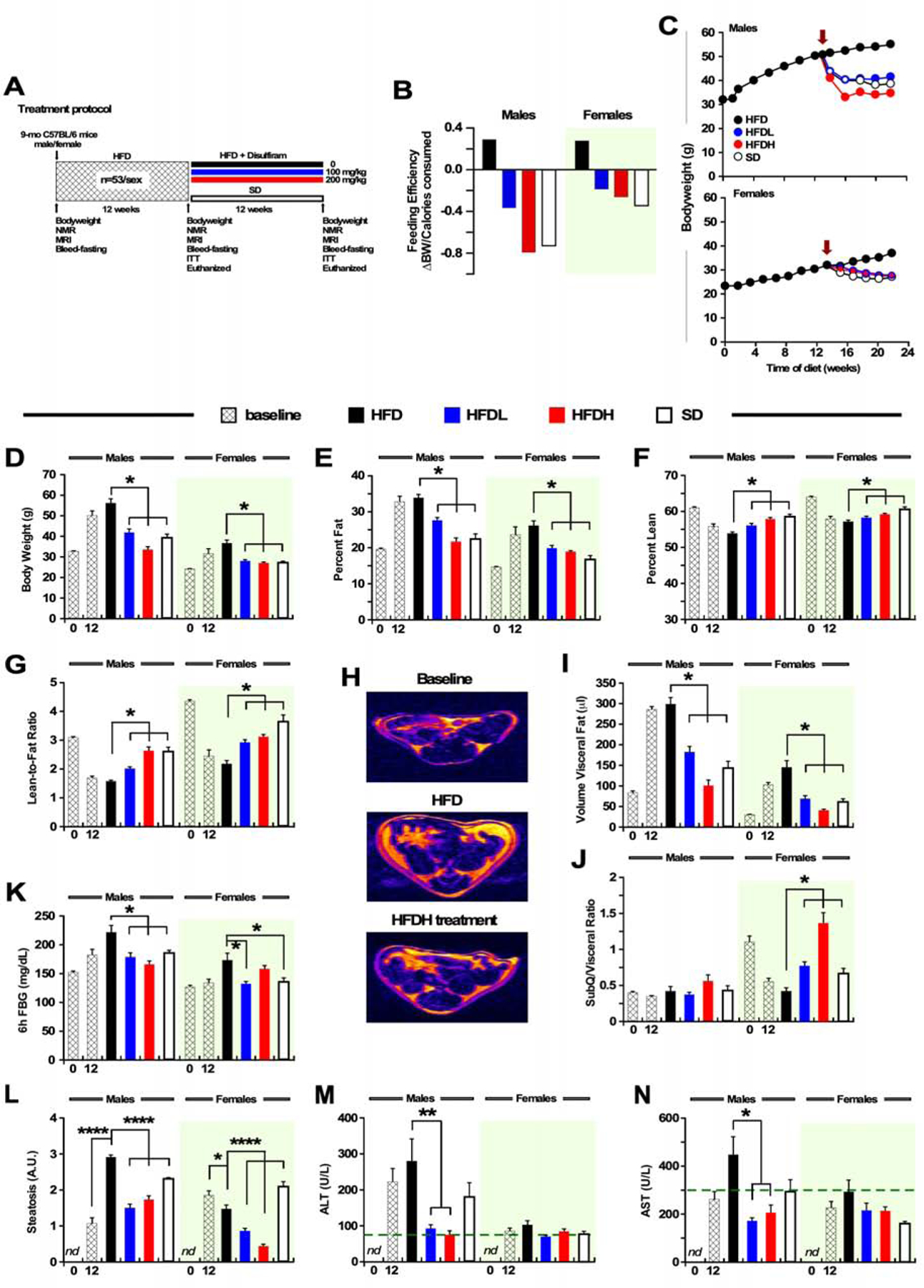
DSF Treatment Suppresses Body Weight Gain and Improves Adiposity and Glucose Intolerance in Mice at Mid-life Fed a High-fat Diet for the Previous 3 Months.
(A) Treatment protocol.
(B) Feeding efficiency expressed as change in BW over average number of calories consumed per day for 12 weeks (week 12–24). The green shading indicates the results obtained in females, and it applies to all panels throughout the figure.
(C) Body weight trajectories in male (top panel) and female (bottom panel) mice. Red arrow indicates the start of the DSF supplementation or SD feeding.
(D) Body weight at baseline and after HFD for 12 weeks (hatched bars), and at the study completion after an additional 12 weeks of treatment.
(E-F) Percent fat mass (E) and lean body mass (F) measured by NMR.
(G) Lean-to-fat ratio calculated from panels E and F.
(H) Representative MRI images from a male mouse. (n =53 per sex, 12 weeks on HFD) and following the completion of the intervention period (n =12 per sex and diet).
(I-J) Quantitative measures of visceral fat volume (I) and calculated ratio of subcutaneous/visceral fat volumes (J) from abdominal MRI scans. Panels B-I: n = 8–12 for males and n = 9–12 for females in each experimental group unless indicated otherwise.
(K) Blood glucose after a 6-h fast, n = 6–8 for males and n = 5–7 for females.
(L) Relative degree of steatosis in fixed liver sections from male (n =3–6) and female (n =4–6) mice. The total number of images per diet group ranged from 60–120. Representative stained liver sections can be found in Figure S3H.
(M-N) Serum liver enzymes (ALT and AST) at the conclusion of the 12-week treatment in both male (n =5–11) and female (n =5–11) mice. Dotted line indicates the upper normal limit in mice. All data are means ± SEM, * P ≤ 0.05. The treatment groups included: HFD, HFD-fed without DSF supplementation; HFDL and HFDH, diet supplementation with low and high doses of DSF, respectively; SD, standard diet. See also Figure S3 and Table S2.
The circulating levels of LDL, HDL, cholesterol, and triglycerides were unchanged between HFD mice on DSF and HFD controls (Figures S3D–S3G); however, the return of HFD-fed female mice to an SD led to significant reductions in serum cholesterol and triglyceride content (Figures S3F and S3G). The HFD-mediated increase in hepatosteatosis (Figures 3K and S3H) in male mice was reversed when mice were switched to HFDL and HFDH diets, similar to animals switched to an SD. These changes correlated with high circulating levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), circulating liver enzymes associated with hepatic diseases and liver cell damage, a process blocked by DSF supplementation (Figures 3L and 3M). This was also true, albeit to a lesser extent, in female mice. Taken together, these results indicate the ability of DSF to not only prevent, but actually reverse the metabolic and physiological disturbances that occur in middle-aged mice on HFD, suggesting that this could provide an effective therapeutic approach for human patients with obesity and metabolic dysfunction.
Low-energy diets rapidly reduce body weight and improve/normalize glycemic control. We investigated the long-term (12 weeks) effects of diet switching on physiological parameters and food consumption in 1-year-old male mice. Obese mice maintained on an HFD at baseline for an additional 12 weeks gained 6.5 ± 1.4% body weight, which they rapidly lost upon a second diet switching to HFD supplemented with DSF (200 mg/kg BW) (Figure 4A, upper panel, orange arrow). At the onset of the second switch, energy intake was sharply reduced within the first 2 weeks (g food/week/mouse, 23.4 ± 0.9 vs. 11.1 ± 2.5, p = 0.027) before returning to ~90% food consumption of baseline (21.0 ± 0.3 g/week/mouse) (Figure 4A, lower panel). Nonetheless, a stable 20% reduction of body weight was observed at the conclusion of the 12-week protocol (Figure 4A, upper panel), in agreement with the prophylactic use of DSF in HFD-fed mice (Figure 1A). Moreover, diet switching at baseline (t=0) from an HFD to either an SD or HFD+DSF led to a progressive loss of body weight, reaching 70.4 ± 2.0% and 67.8 ± 3.0% of baseline, respectively, after 12 weeks of treatment (Figure 4B, lower panel). Body weight sharply increased when HFD+DSF mice were refed HFD, while diet switching of SD mice to HFD+DSF further reduced their body weight to 66.6 ± 1.5% of baseline. Upon the second diet switch, reversal animals consumed 17.2 ± 0.1 (SD-> HFD+DSF) and 21.2 ± 0.1 (HFD+DSF-> HFD) g food/week/mouse, respectively, indicating that persistent reduction in food intake may account at least in part of the effects of DSF on weight loss. Feeding efficiency, defined as the ratio of BW change divided by calories consumed (Scheibye-Knudsen et al., 2014), decreased over time when HFD+DSF mice were put on HFD alone (Figure 4C). We surmise that such increase in metabolism stemmed from active esterification of available fatty acids in white adipose tissues (WAT) during the 12-week refeeding with HFD (Figure 4D).
Figure 4.
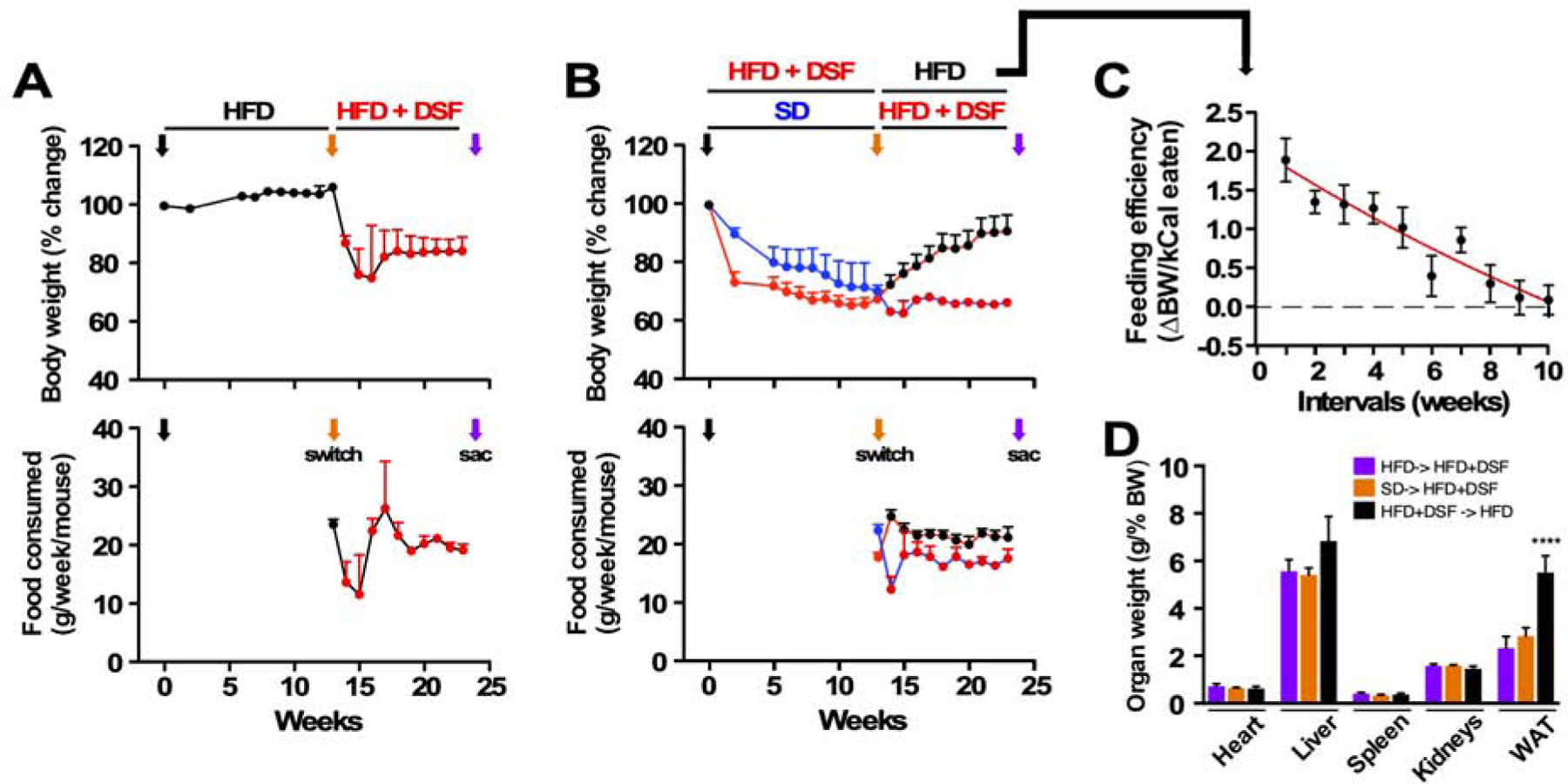
Diet switching reveals reversible actions of DSF against diet-induced metabolic dysfunction.
(A) Trajectories of body weight as percent change from baseline (upper panel) and average weekly food consumption per mouse (lower panel) fed HFD.
(B) Trajectories of body weight as percent change from baseline (upper panel) and average weekly food consumption per mouse (lower panel) fed either SD or HFD+DSF. Black arrow, diet switch from HFD to the indicated diet at the start of the experiment (t= 0); orange arrow, second diet switch; purple arrow, sac.
(C) Feeding efficiency shown by the ratio of body weight change per the number of calories ingested (n = 8, mean ± SEM).
(D) Organ weights at sac as percent of body weight (HFD-> HFD+DSF: n = 13; SD-> HFD+DSF: n = 4; HFD+DSF-> HFD: n = 8, mean ± SEM). ***, P < 0.001.
We next performed studies in an additional preclinical model of obesity to determine the translatability of these effects of DSF. To achieve this goal, 3-month-old male Sprague Dawley rats, identified as prone to the development of obesity, were maintained for 12 weeks on either standard chow alone or chow supplemented with low and high doses of DSF (100 and 200 mg/kg BW). The three groups of animals ate the same amount of food (Figure 5A, upper panel and Figure S4A) and gained weight at the same rate except for a 2-week delay from onset of the DSF intervention (Figure 5A, lower panel). Body composition was then evaluated at 2, 4 and 8 weeks of treatment by EchoMRI, and the data normalized by BW. A significant reduction in fat mass (Figure 5B) and increase in lean tissue mass translated into higher lean-to-fat ratio in DSF-treated rats (Figures S4B and S4C). At the conclusion of the study, fasting blood glucose and insulin levels were found not to be substantially impacted by DSF (Figures S4D and S4E) while liver triglycerides trended lower in DSF-treated rats (Figure S4F). Total body weight and quadriceps muscle weight were not affected by DSF supplementation (Figure 5C), whereas high dose of DSF promoted both liver weight gain and reduction in three fat depots compared to controls (Figure 5C), consistent with DSF-mediated fat mass loss (Figure 5B).
Figure 5.
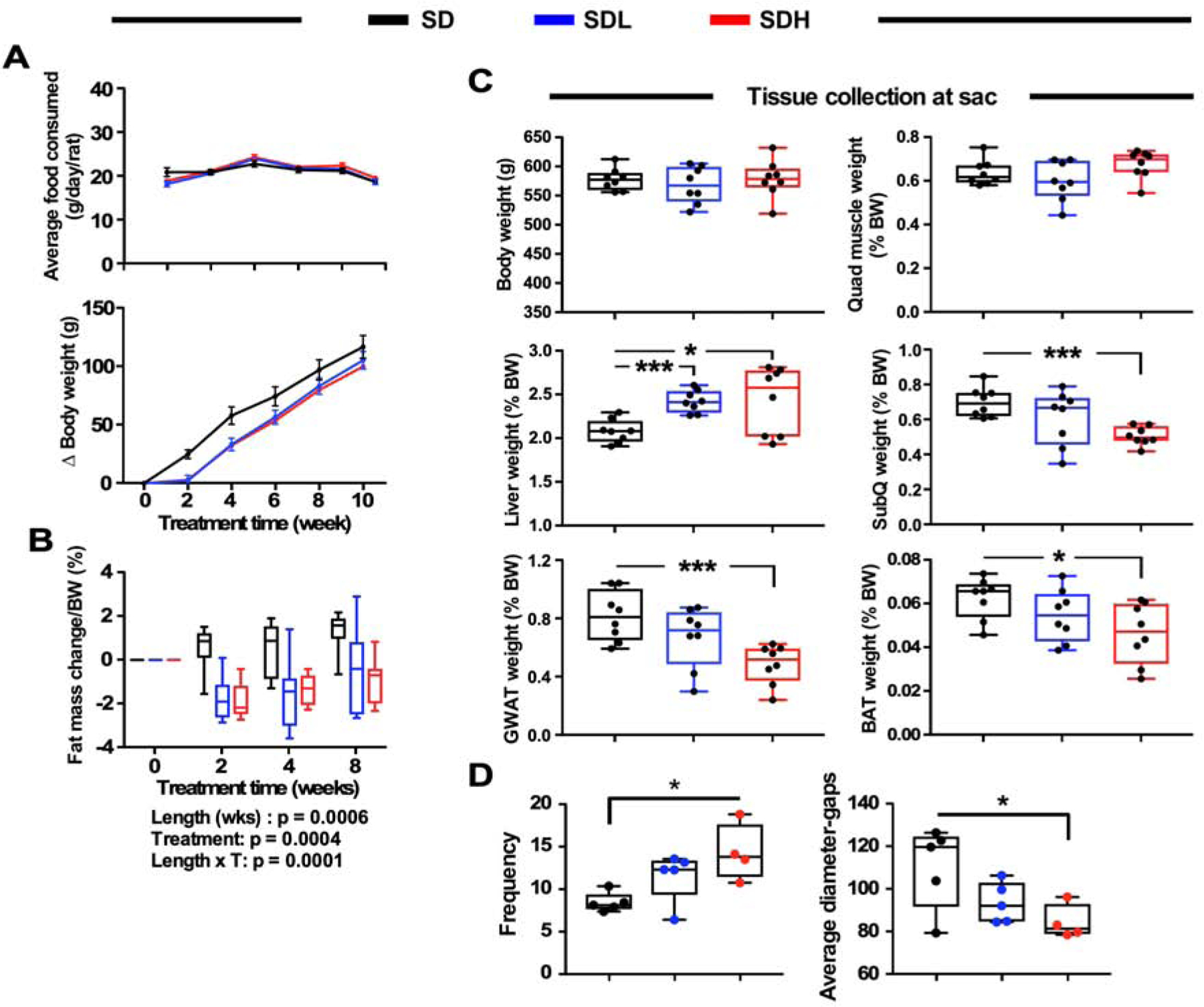
Effects of a 12-week treatment with DSF in Sprague-Dawley rats.
(A) Upper panel, trajectories of daily food consumption by rats on the indicated diets; lower panel, trajectories of change in body weight gain from baseline of the same rats.
(B) Changes in fat mass after normalization for BW at the indicated time points. Baseline values were set at ‘0’. Data are represented in box and whisker plot format (n =8 per group). Statistics for the overall effects of treatment length, DSF treatment (T), and the interaction represent the P value from a two-way ANOVA with Tukey’s post-hoc tests.
(C) Organ weights at the time of killing (sac) as percent of total body weight.
(D) Frequency of fenestrations in liver (left) and average diameter-gaps (right). Most of the data are represented in box and whisker plot format (n =8 per group). *, *** = P < 0.05 and 0.001 vs. control chow diet.
See also Figure S4.
Fenestrations present in the liver sinusoidal endothelial cells mediate the clearance of dietary macronutrients (e.g., fatty acids, gut-derived molecules) from the circulation and facilitate the transfer of substrates between blood and hepatocytes (Cogger et al., 2016). We found that DSF significantly increased frequency in fenestrations while decreasing their average diameter (Figure 5D). Together, these observations support the idea that DSF treatment improves organismal response through preservation of hepatic function.
Overall, our results demonstrate that DSF treatment initiated at early adulthood triggers a range of health benefits culminating in improvements in whole body physiology and metabolism in mice and rats; besides, the drug provides benefits by reversing diet-induced metabolic dysfunction in middle-aged male and female obese mice partly via the normalization of body weight, loss in adiposity, and reduction in fasting blood glucose. An increase in EE may partly explain the beneficial effects of DSF on energy metabolism.
One of the strengths of the study is the demonstration that long-term DSF supplementation mitigates diet-induced obesity and related co-morbidities including hepatic fibrosis and steatosis and pancreatic β-cell hyperplasia in mice. Long-term exposure to DSF has led to small, non-significant differences in daily food consumption, while markedly reducing feeding efficiency (∆ body weight / number of calories consumed). This energy imbalance did not change metabolic fuel selection, was independent of diet composition, and dramatically improved overall body composition by decreasing fat mass. Indirect calorimetry studies showed that the dissipation of excess energy appears more efficient in HFDH > HFDL than HFD animals, resulting in negative energy balance and associated loss in body weight. Thermogenesis could account for the larger EE in response to chronic DSF treatment via activation of brown adipose tissue and beige adipocyte recruitment especially under obesogenic conditions. We are currently testing this hypothesis.
A trend toward normalization of ALT and AST levels in the blood upon DSF treatment is consistent with an effective protection from diet-induced liver damage (e.g., hepatosteatosis). Strikingly, histological staining and SEM images of fixed liver sections of mice demonstrate the maintenance of normal liver architecture and morphology, e.g, fenestration, and prevention of hepatosteatosis and associated complications, including the derangement of liver glycogen storage, collagen deposition, fibrosis, and infiltration of inflammatory cells. Furthermore, the number of fenestrations in the liver sinusoidal endothelial cells of rats was improved in response to DSF. Defenestration in the liver sinusoidal endothelium occurs as part of the normal aging process and in response to HFD, which leads to dyslipidemia and hence, atherosclerosis (Fraser et al., 2012; Le Couteur et al., 2002).
A liver-pancreas endocrine axis has been previously described whereby hepatic insulin resistance induces β-cell hyperplasia (Escribano et al., 2009). Owing to the fact that DSF improves insulin sensitivity (HOMA-IR index) and clearly reduces both plasma insulin levels and islet size in HFD-fed mice, we suspect that DSF may also prevent islet hypertrophy that occurs with normal aging (Helman et al., 2016; Weir, 2016). Future studies are underway to elucidate the potential role of DSF in combatting aging by mitigating age-associated metabolic disturbances and systemic low-grade inflammation.
Chronic consumption of a high-fat diet elicits pathological alterations stemming from increased adiposity and low-grade inflammation rather than body weight gain per se. The capacity of DSF at reducing fat deposition through lower feeding efficiency and greater EE per unit of body mass is likely responsible for weight loss and protection from the adverse metabolic effects of adiposity. The coincident increase in insulin sensitivity upon DSF treatment may have thwarted lipase-mediated triglyceride hydrolysis in adipocytes as evidenced by the lack of non-esterified fatty acids and glycerol surge in serum in the intervention study.
DSF is rapidly metabolized in vivo to diethyldithiocarbamate and subsequently converted by several human and rat CYP450 enzymes to active metabolites responsible for ALDH2 inhibition (reviewed by Koppaka et al., 2012). The in vivo potencies of these DSF metabolites depend on reduced glutathione levels and their oxidative desulfuration, which reverses ALDH2 inhibition (Madan et al., 1994). Thus, it appears that a DSF metabolite rather than the parent molecule may be responsible for combatting obesity and restoring systemic metabolic balance. The metabolic adaption to DSF was preserved in Aldh2-KO mice, which may be attributed to new players in DSF signaling.
To date, there are 44 clinical studies in which DSF has been used for various conditions ranging from alcohol and cocaine use disorders (n=20), cancer (n=17), and HIV (n=5) (www.ClinicalTrials.gov), but none of the trials monitor weight loss as an outcome. The reported effects of DSF on CNS dopamine regulation and/or hepatic effects which are then transmitted to the CNS are indeed possible, if not likely. There is a single report describing the use of DSF in individuals with obesity and binge eating disorder through downmodulation of the CNS dopaminergic reward system (Farci et al., 2015). In that study, twelve patients were treated with DSF (250 mg daily) for 16 weeks resulting in lower frequency of binge eating episodes. Although calorie intake was not controlled for in this open trial, there was a significant 2.6% reduction in body weight as a secondary outcome measure (Farci et al., 2015). The scarcity of information about individuals on DSF losing weight may stem from the fact that patients suffering from addiction and craving are likely to have episodes of anxiety disorders, metabolic dysregulation, and malnutrition that likely mask the weight-loss properties of DSF. It is unclear how efficacious DSF might be for individuals with morbid obesity in the clinic and, therefore, a controlled clinical study for DSF with a primary endpoint of weight loss is warranted, but the selection of individuals for such a study would have to be carefully considered, including perhaps a range of BMIs.
This work identifies important physiological and metabolic effects of DSF which may provide therapeutic benefits for non-alcoholic patients. However, like with any other drugs and interventions, the genetic diversity, age, sex, diet, and environmental factors all contribute to people being free from or harmed by adverse events to any given therapy. So, these factors need to also be considered when considering DSF for future therapy as an anti-obesity agent. Indeed, molecular profiling (e.g., CYP450 polymorphism, serum biomarkers) will be required to determine the appropriateness of DSF as a weight reduction therapy in humans. In this context, it should be noted that it does not appear that the beneficial impact of DSF on metabolic plasticity is mechanistically linked to ALDH2 inhibition, which is considered its mechanism of action as a treatment for chronic alcoholism. This is an important consideration if one worries that DSF or its active derivative may cause an adverse reaction to alcohol consumption by an individual who is taking the drug as a treatment for obesity.
In summary, DSF treatment normalizes body weight and reverses metabolic dysfunctions in obese middle-aged mice of both sexes. This new knowledge is of key importance given current proposals to repurpose drugs for improving metabolic health and other biomarkers of healthspan in humans.
Limitations of the Study
In the current study, no attempts were made to assess whether DSF promoted brown fat activation or elicited an effect on the mitochondrial uncoupling systems, which could explain the increase in energy expenditure upon DSF treatment. Along those lines, it would have been interesting to have considered changes in nutrient absorption by measuring feces energy content by bomb calorimetry. Further, the inclusion of pair-feeding groups would have controlled for the small, non-significant change in energy intake, as small differences in food intake over a long period of time can gradually produce significant effects on body weight (Guo and Hall, 2011). Moreover, glucose and insulin tolerance tests and endpoint analysis in collected tissues were not performed in the intervention study to establish the contribution of body weight loss vs. the direct pharmacodynamic action of DSF. And the use of DSF in mouse dietary models of nonalcoholic steatohepatitis were not tested to see if this common co-morbidity of obesity would also be ameliorated. Finally, while we ruled out a role of Aldh2 inhibition as the mechanism of action of DSF on the normalization of body weight under obesogenic conditions, the detailed molecular mechanism(s) for the potent anti-obesity effects in our study were not identified.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact —
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Rafael de Cabo (decabora@mail.nih.gov).
Materials Availability—
This study did not generate new unique reagents.
Data and Code Availability —
This study did not generate datasets or code.
EXPERIMENTAL MODEL DETAILS
Husbandry, Diets, and Dietary Intervention in Mice
Male C57BL/6J mice were obtained from the NIA Aging colony (Charles Rivers, Bethesda, MD) at 8 weeks of age. Mice were grouped into cages of four and were fed house chow (2018 Teklad Global 18% Protein Rodent Diet 2018S, Harlan Teklad) until they reached 16 weeks of age. Mice were then randomized to one of six study diets with n=14–20 animals per diet. The diets were as followed: standard diet (SD) of AIN-93G (carbohydrate:protein:fat ratio of 64:19:17 percent of kcal), a SD supplemented with low dose of disulfiram (DSF) (SDL; 100 mg/kg/day), a SD supplemented with high dose of DSF (SDH; 200 mg/kg/day), a high fat diet consisting of AIN-93G modified to provide 60% of calories from fat (HFD; carbohydrate:protein:fat ratio of 16:23:61), a HFD supplemented with low dose of DSF (HFDL; 100 mg/kg/day), or a HFD supplemented with high dose of DSF (HFDH; 200 mg/kg/day). DSF was included in diets at a concentration of 1 g/kg (SDL) and 2 g/kg (SDH), as well as 2.33 g/kg (HFDL) and 2.67 g/kg (HFDH), respectively, which was formulated to provide mice with approximate daily doses of 100 and 200 mg/kg body weight. Study diets were purchased from Dyets, Inc., and DSF was obtained from Sigma-Aldrich. Study diets were produced and pelleted as followed: The base diet, made as a powder, was irradiated. The premix was split into equal portions and the proper amounts of cornstarch, sucrose, and disulfiram were added to each. Each diet was then mixed as a powder until homogenous, at which point 10% water was added in to facilitate pelleting. Once pelleted, the diets were dried for 12–15 h at 10 °C above ambient temperature.
All groups had ad libitum access to their prescribed diet and water throughout the study. Body weight and food intake were monitored bi-weekly. Animal rooms were maintained at 20–22°C with 30–70% relative humidity and a 12-hour light/dark cycle. Animals were inspected twice daily for health issues and veterinary care was provided as needed. The criteria for euthanasia were based on an independent assessment by a veterinarian according to AAALAC guidelines. A subset of mice was killed at 58 weeks of age (40 weeks on the diet) and a second group at 80 weeks of age (64 weeks on the diet), and tissues were collected for further analysis. For euthanasia and subsequent tissue collection, mice were on their normal feeding cycle (study diet ad libitum) and not fasted. All animal protocols were approved by the Animal Care and Use Committee (444-TGB-2016) of the National Institute on Aging.
For the fasting/1-h refeeding protocol, mice were fasted for 36 h in their normal housing room. Then, 2 h after lights out (dark cycle), pre-weighed pellet of experimental diet was placed on the floor of the cage. After 60 min, the pellet was removed and weighed to determine how much they ate during the 1-h refeeding period. The aim of this assay was to test palatability of the diet supplemented with DSF.
For the intervention protocol, seven-month-old retired breeder male and female C57BL/6J mice were obtained from the Jackson’s Laboratory and single housed under temperature-controlled conditions with 12 h light/12 h dark cycle with ad libitum access to food (2018S, see above) and water. Beginning at 9 months of age (defined as baseline), mice were maintained on high-fat diet (see above for composition) for the next 12 weeks (defined as HFD period) with bodyweight and food consumption measured bi-weekly. After the HFD period, 6 mice of both sexes were sacrificed and the remaining animals were randomly divided into four groups (n =12/group per sex) for the intervention leg of the study, which consisted of: 1) HFD diet alone; 2) HFD supplemented with 100 mg/kg/day DSF (HFDL); 3) HFD supplemented with 200 mg/kg/day DSF (HFDH) and; 4) Standard diet (SD, see above for composition). All animals were sacrificed after an additional 12 weeks of treatment and tissues were collected for further analysis. All animal protocols were approved by the Animal Care and Use Committee (444-TGB-2016) of the National Institute on Aging.
Homozygous Aldh2-KO mice on a C57BL/6N background were generated at the NIAAA facility as described previously (Kwon et al., 2014). C57BL/6N mice were purchased from the NCI (Frederick, MD) and bred at the NIAAA facility to generate WT controls. Eight‐ to 10‐week‐old male WT and Aldh2-KO mice were used in this study. Mice were provided with high fat AIN-93G purified rodent diet (cat. # 101920, Dyets) supplemented or not with 2.67 g/kg food of DSF (200 mg/kg BW). All animal studies were approved by the NIAAA Animal Care and Use Committee (protocol number #NIAAA-LLD-BG-1).
Mouse genomic DNA was isolated from collected ear tissue following digestion at 55°C in DirectPCR-Lysis Reagent (Viagen Biotech, Inc.) containing Proteinase K (0.2 mg/ml), followed by heat inactivation for 45 min. The presence of mutated Aldh2 alleles was confirmed by PCR amplification as previously described (Kitagawa et al., 2000) using the following primer sequences: Forward 5’-CCGTACTGACTGTCCCATGCAGTGCT-3’; Reverse (Aldh2-WT) 5’-TCCGCCAATCGGTACAACAGCCG-3’; and Reverse (Aldh2-mut) 5’-GGTGGATGTGGAATGTGTGCGAGGC-3’. These primer pairs produced 208 bp (WT) and 280 bp (Aldh2-mut) products as predicted. PCR conditions were as follows: 30 cycles at 95°C for 45 s, 56°C for 45 s, and 72°C for 60 s.
Husbandry, Diets, and Dietary Intervention in Rats
Twenty-four adult male Sprague Dawley rats, aged approximately 3 months, were obtained from The Animal Resources Centre (WA, Australia) and housed in the Charles Perkins Centre at the University of Sydney. Rats were housed individually (in split cages) on a 12-hour light/dark cycle at 22–24° C with free access to water. After a one-week acclimatization period, rats were randomly split into three dietary groups (n = 8/group) and provided with either a standard chow diet (AIN-93G) alone or supplemented with low dose (100 mg/kg BW) or high dose (200 mg/kg BW) of disulfiram. Animals were weighed every four days and food consumption was measured every other day. Whole body fat and lean mass were measured at 2, 4, and 8 weeks by magnetic resonance imaging (EchoMRI 900 – EchoMRI LLC, Houston, Texas, USA). The rats were awake during the MRI procedure and positioned snuggly into a plastic tube before being placed into the machine. Health assessments were routinely performed to ensure that university veterinary care requirements were met. All protocols for this part of the study were approved by the University of Sydney Animal Welfare Office, ethics number: 2018/1365.
Rats were fasted for 24 h before tissue collection. Rats were deeply anaesthetized with Lethabard (60 mg/Kg) and fasting blood glucose levels were measured (Accu-Check Performa, Roche, Australia). Tissues were harvested, weighed, and snap-frozen in liquid nitrogen. Fasted serum insulin levels were measured by ELISA, following manufacturer’s instructions (Crystal Chem, IL, USA).
METHOD DETAILS
Liver Histology
Fresh pieces of liver were embedded in Optimal Cutting Temperature (OCT) compound and frozen in liquid nitrogen. Liver sections were cut at 10 µm using a cryostat (Leica Biosystems) and kept at −80°C until further analysis. Fresh frozen liver sections were fixed in 10% neutral buffered formalin (Thermo Fisher Scientific) and then stained with Oil red O using a protocol adapted from Mehlem et al. (Mehlem et al., 2013). Staining with Periodic Acid Schiff (Polysciences 24200–1) and Picrosirius Red (Polysciences 24901–500) was carried out according to the manufacturer’s instructions, n =5 per group; age = 57 weeks; diet = 41 weeks. For the intervention study, livers were fixed in 4% paraformaldehyde and embedded in paraffin by a commercial service (Histoserv). Hematoxylin and eosin (H&E) staining was performed per standardized protocols. Quantification of steatosis in H&E sections was performed by two independent observers blinded to the treatment groups. This was performed on at least 10 fields per animal with the data representing the average of two scores. Steatosis was determined by assigning an equally weighted arbitrary score of 0 (no visible steatosis), 1 (< 33% of the section showing steatosis), 2 (>33% but < 66% of the section showing steatosis) to 3 (>66% of the section showing steatosis). n = 5 per group; age = 58 weeks; diet = 40 weeks.
Immunohistochemistry of Pancreatic Islets
The pancreatic tissue was processed and embedded in paraffin. Pancreatic sections were cut at 5-μm thickness using a microtome, and the sections were adhered to poly-L-lysine coated microscope slides (Fisher Thermo Scientific). In order to obtain a systematic appreciation without bias of the entire pancreas, whole pancreas was sectioned and every 20th section was saved onto a slide.
Pancreatic sections were immunostained according to a previously described protocol (Cai et al., 2014). Briefly, following antigen retrieval with 1× citrate buffer (Biogenex) at 98°C for 20 min, sections were blocked in 5% bovine serum albumin (Sigma-Aldrich) and 0.1% Tween-20 in 1× Tris-buffered saline (pH 7.4) for 1 h at room temperature. Then, the tissue was incubated with the primary insulin antibody (guinea pig anti- insulin; 1:800; Sigma-Aldrich) and glucagon antibody (1:1000; mouse anti-glucagon; Sigma-Aldrich) overnight in the cold room. After washing, sections were incubated for 1 h with fluorescent secondary antibodies (1:1,000 dilution; Invitrogen) along with 4’,6-diamidino-2-phenylindole (1:5,000 dilution; Invitrogen) for nuclear staining. No fluorescent staining was observed in any sections when the primary antibodies were omitted. Sections were imaged with an LSM-710 confocal microscope (Carl Zeiss MicroImaging) in single planes. Bar = 100μm. n = 5 per group; age = 57 weeks; diet = 41 weeks.
Quantification of immunohistochemistry images was performed in Matlab (Mathworks) using novel software in conjunction with Matlab’s image processing toolbox, as described previously (Martin et al., 2009; Kim et al., 2011). Intensity readings of each image ranged from 0 to 256, with 256 being the greatest pixel density and hence the highest staining intensity. The region of interest (ROI) was drawn around each islet after background subtraction. The pixels within the ROI boundaries and above the set threshold of 8 were selected, from which actual islet area was calculated. The normalized variance of the ROI was used to calculate an artificial ellipse from which the major and minor axes were determined. The major axis is the longest diameter that can be drawn in the ellipse, and the minor axis is the shortest diameter, both giving an accurate approximation for the range of the actual islet diameter. Islet sizing, alpha cell numbers, alpha cell percentage and beta cell percentage were performed in an unbiased, random fashion. All the quantification was verified using our newly developed pancreatic islet analytical software program, Pancreas++ (Chen et al., 2013).
Body Composition
Measurements of lean body mass, fat and fluid mass in live mice were acquired by nuclear magnetic resonance (NMR) using the Minispec LF90 (Bruker Optics).
Magnetic Resonance Imaging
MRI was performed on the mice to quantify fat content throughout the body. Mice were anesthetized by inhalation of 1.5% isoflurane in 100% oxygen, delivered via a facemask. Respiration and core temperature were monitored using a MR-compatible monitoring and gating system (Model 1025, SA Instruments) and core temperature was maintained at 37 °C by a stream of warm air. Images were acquired using a 7 Tesla pre-clinical MRI scanner (Bruker Biospec) with an axial 3D RARE sequence. Parameters included echo time TE = 11.61 ms, RARE factor = 8, effective TE = 46.44 ms, repetition time TR = 125 ms, number of averages NEX = 2, field-of-view (FOV) = 7×4×10 cm (L/R × A/P × H/F) and matrix size 256×128×128, resulting in a voxel size of 234×313×781 microns. The duration of each scan was 6 min. MRI was performed in all mice after the completion of the HFD period (n =53 per sex, 12 weeks on HFD) and following the completion of the intervention period (n =12 per sex and diet). To analyze the fat content, a user scrolled through each package of axial slices, and determined the mid-section of the kidneys. This single slice is then analyzed. Using the “threshold” function within ImageJ, the pixel intensity was measured, representative of the fat content. To distinguish visceral and subcutaneous fat, the user created a region-of-interest around the peritoneum and measured the visceral content. The subcutaneous content was calculated by subtracting visceral fat pixel area from total fat pixel area. The pixel areas are then converted into μL units.
Determination of Serum Markers
To quantify the insulin levels, whole blood was obtained after a 16-h fast and spun at 14,000 rpm for 10 min to pellet blood cells. Serum was aliquoted and stored at −80°C until further analysis. Glucose was measured in whole blood using the Ascensia Elite glucose meter (Bayer) while insulin was measured by enzyme-linked immunosorbent assay (ELISA, Crystal Chem, Inc.,) according to the kit manufacturers’ instructions. Insulin resistance was estimated using the HOMA2 calculator software available from the Oxford Centre for Diabetes, Endocrinology and Metabolism Diabetes Trials Unit website (www.dtu.ox.ac.uk).
From the Intervention protocol, non-fasted animals were killed either after a 12-week period on a HFD or 12 weeks later on a study diet. Blood samples were collected and allowed to clot at room temperature for 30 min and then centrifuged at 14,000 rpm, 4 °C for 15 min to obtain serum. Serum was frozen at −80 °C until further analysis. On the day of the experiment, serum was defrosted on ice and serum activity of alanine aminotransferase (ALT) and aspartate aminotransferase (AST), and concentrations of cholesterol, triglycerides, LDL, and HDL were determined by using an automated analyzer (Roche Diagnostics) as per the manufacturer’s instructions. The averaged range limits for ALT and AST are 17–77 U/L and 54–298 U/L, respectively, and account for variation in age, sex, breed or strain (https://www.ahc.umn.edu/rar/refvalues.html). n = 4–8 per group.
Insulin Tolerance Test (ITT)
Mice were fasted for 3 h and then injected i.p. with 1.5 IU.kg−1 of insulin (Novo Nordisk), and blood glucose levels were measured at 0, 15, 30, 60, and 120 min. n = 6 per group; age = 39 weeks; diet = 23 weeks.
Metabolic Assessment
Mouse metabolic rate was assessed by indirect calorimetry in open-circuit oxymax chambers using the Comprehensive Lab Animal Monitoring System (Columbus Instruments) as previously described (Minor et al., 2011) (n = 6 per group; age = 48 weeks; diet = 32 weeks). In brief, mice were housed singly with ad libitum water and food and were maintained at 20–22 °C under a 12:12 h light-dark cycle (light period 06:30–18:30). Oxygen consumption was determined by measuring oxygen concentration in air entering the chamber compared with air leaving the chamber. Constant airflow (0.6 L/min) was drawn through the chamber and monitored by a mass-sensitive flow meter. The concentrations of oxygen and carbon dioxide were monitored at the inlet and outlet of the sealed chambers to calculate oxygen consumption. Measurement in each chamber was recorded for 30 s at 30 min intervals for a total of 60 h. The second dark:light cycle is represented in the plots. Movement (both horizontal and vertical) was also monitored. n = 5–7 per group; age = 48 weeks; diet = 32 weeks.
Liver triglyceride assay
Triglycerides were extracted based on previously established methods (Folch et al., 1957) with some minor modifications. Approximately 20 – 30 mg of finely powdered liver tissue was added to an ice-cold 10 mL plastic tube and placed on dry ice. Four mL of chloroform/methanol (2:1) was added to the tube and lipids were extracted for 2 h while spinning on a roller. After incubation, 2 mL of 0.6 M NaCl was added and the tubes were briefly vortexed and centrifuged at 2,000 RPM for 10 min at RT. The bottom layer was carefully transferred to a fresh 7 mL glass vial and dried overnight. The samples were resuspended in 500 μL of RNA grade ethanol and vortexed for approximately 5 s. Liver triglyceride concentrations were determined by ELISA using serial dilutions of Precimat Glycerol (Roche; 2.29 nmol/L) as standards. Five ul of standards and samples were added to a 96-well plate and the plate heated for 20 min at 37 °C followed by addition of 300 μL of warm Triglyceride reagent (Pointe Scientific, Inc; Cat #1730711) to each well. The plate was incubated for 30 min at 37 °C and absorbance was measured at 490 nm using a plate reader. Data are expressed as nmol/mg of tissue.
QUANTIFICATION AND STATISTICAL ANALYSIS
No statistical method was used to predetermine sample size. All statistical analyses were done with two-tailed unpaired Student’s t-test or one-way ANOVA, followed by Tukey post hoc comparison, unless otherwise stated. These analyses reflect fold change from SD- or HFD-fed control animals and were performed using either Excel for Mac (Microsoft Corp.), IBM SPSS Statistics, SigmaStat 3.0 (Aspire Software International), or Prism v.6 (GraphPad Software). Data are expressed as means ± standard error of the mean (SEM). Values of P ≤ 0.05 were considered statistically significant. Collection, analysis and quantification of images from liver histology, immunohistochemistry of pancreatic islets and magnetic resonance imaging were performed by investigators blind to the different experimental conditions.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-IGFBP2 | Novus Biologicals | Cat# NBP1-57914; RRID: AB_ |
| Rabbit anti-phospho-NFκB p65 | Cell Signaling Technology | Cat# 3039; RRID: AB_ |
| Rabbit anti-NFκB p65 subunit | Epitomics | Cat# 1546-1; RRID: AB_ |
| Rabbit anti-IL-1β | Cell Signaling Technology | Cat# 12507; RRID: AB_ |
| Rabbit anti-acetylated SOD2 (K68) | Abcam | Cat# ab137037; RRID: AB_ |
| Rabbit anti-SOD2 | Abcam | Cat# ab13533; RRID: AB_ |
| Mouse anti-SIRT1 | Sigma-Aldrich | Cat# S5196; RRID: AB_ |
| Rabbit anti-SIRT3 | Cell Signaling Technology | Cat# 5490; RRID: AB_ |
| Mouse anti-GAPDH | Santa Cruz Biotechnology | Cat# sc-32233; RRID: AB_ |
| Mouse anti-acetyl-lysine | EMD Millipore | Cat# 05-515; RRID: AB_ |
| Rabbit anti-HNE fluorophore | EMD Millipore | Cat# 393206; RRID: AB_ |
| Mouse anti-ALDH2 | Abcam | Cat# ab166697; RRID: AB_ |
| Rabbit anti-ALDH2 | Abcam | Cat# ab108306; RRID: AB_ |
| Mouse anti-rabbit HRP | Santa Cruz Biotechnology | Cat# sc2357; RRID: AB_ |
| Bovine anti-mouse HRP | Santa Cruz Biotechnology | Cat# sc2371; RRID: AB_ |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Mouse liver samples | This paper | N/A |
| Mouse serum samples | This paper | N/A |
| Rat liver samples | This paper | N/A |
| Rat serum samples | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| AIN-93G diet | Dyets, Inc. | Cat# 110700 |
| HFD AIN-93G diet | Dyets, Inc. | Cat# 101920 |
| Disulfiram | Sigma-Aldrich | Cat# 86720 |
| Insulin solution | Novo Nordisk | Cat# EMEA/H/C/000424 |
| RIPA Buffer with EDTA and EGTA (500ml) | Boston BioProducts | Cat#BP-115DG |
| Phosphatase Inhibitor Cocktail 1 | Sigma-Adrich | Cat#P2580-5ML |
| Phosphatase Inhibitor Cocktail 2 | Sigma-Adrich | Cat#P5726-5ML |
| Phosphatase Inhibitor Cocktail 3 | Sigma-Adrich | Cat#P0044-5ML |
| Phenylmethanesulfonyl fluoride solution | Sigma-Adrich | Cat#93482-50ML-F |
| 2x Laemmli Sample Buffer | Bio-Rad | Cat#161-0737 |
| 10x Tris/Glycine/SDS | Bio-Rad | Cat#1610772 |
| 4-15% Criterion™ TGX™ Precast Midi Protein Gel | Bio-Rad | Cat#5671085 |
| Precision Plus Protein™ Kaleidoscope™ Standards | Bio-Rad | Cat#1610375 |
| Trans-Blot Turbo RTA Midi Nitrocellulose Transfer Kit | Bio-Rad | Cat#1704271 |
| PBS (10X) | Boston BioProducts | Cat#BM-221 |
| Tween-20 | Boston BioProducts | Cat#P934 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| High-Capacity cDNA reverse transcription kit | Thermo Fisher Scientific | Cat#4368814 |
| TaqMan™ Gene Expression Master Mix | Thermo Fisher Scientific | Cat#4369016 |
| Immobilon Western Chemiluminescent HRP Substrate Kit | MilliporeSigma | Cat#WBKLS0500 |
| Critical Commercial Assays | ||
| Mitochondrial ALDH2 activity | MitoSciences | Cat# ab115348 |
| Mouse insulin ELISA kit | Crystal Chem, Inc. | Cat# 90080 |
| Rat insulin ELISA kit | Crystal Chem, Inc. | Cat# 90010 |
| Pierce BCA Protein Assay Kit | Thermo Scientific | Cat# 23225 |
| Deposited Data | ||
| Mass Spectrometry proteomics data | ProteomeXchange Consortium | PXD016793 |
| Liver microarray analysis | Gene Expression Omnibus (GEO) repository | GSE110200 |
| Experimental Models: Cell Lines | ||
| Mouse NIH3T3 fibroblasts | ATCC | CRL-1658 |
| Experimental Models: Organisms/Strains | ||
| Male C57BL/6J mice | NIA Aging colony | N/A |
| Male and female C57BL/6J mice | The Jackson Laboratory | JAX 000664 |
| C57BL/6N mice | National Cancer Institute Mouse repository | |
| Aldh2-KO mice on C57BL/6N background | NIAAA facility | N/A |
| Male Sprague Dawley rats | Animal Resources Center WA, Australia | ArcCrl:CD(SD)IGS |
| Oligonucleotides | ||
| m_Aldh2-WT_FW | IDT | CCGTACTGACTGTCCCATGCAGTGCT |
| m_Aldh2-WT_Rev | IDT | TCCGCCAATCGGTACAACAGCCG |
| m_Aldh2-mut_Rev | IDT | GGTGGATGTGGAATGTGTGCGAGGC |
| m_Igfbp2_FW | IDT | CAGACGCTACGCTGCTATCC |
| m_Igfbp2_Rev | IDT | CCCTCAGAGTGGTCGTCATCA |
| m_Avpr1a_FW | IDT | GCTGGCGGTGATTTTCGTG |
| m_Avpr1a_Rev | IDT | GCAAACACCTGCAAGTGCT |
| m_Cyp2c29_FW | IDT | CATCGACCTCCTCCCCACTAGC |
| m_Cyp2c29_Rev | IDT | GGTTGGGAAACTCCTTGCTGTCA |
| m_Socs2_FW | IDT | AGTTCGCATTCAGACTACCTACT |
| m_Socs2_Rev | IDT | TGGTACTCAATCCGCAGGTTAG |
| m_Raet1b_ FW | IDT | TGGACACTCACAAGACCAATG |
| m_Raet1b_Rev | IDT | CCCAGGTGGCACTAGGAG |
| m_Acot11_ FW | IDT | TGGGGAGCTGAGCATTGGA |
| m_Acot11_ Rev | IDT | GGCCGACACTAATGGTATGGT |
| m_Cidea_ FW | IDT | ATCACAACTGGCCTGGTTACG |
| m_Cidea_ Rev | IDT | TACTACCCGGTGTCCATTTCT |
| m_Hprt_FW | IDT | TGGGAGGCCATCACATTGT |
| m_Hprt_Rev | IDT | GCTTTTCCAGTTTCACTAATGACA |
| m_18s_FW | IDT | GTAACCCGTTGAACCCCATT |
| m_18s_Rev | IDT | CCATCCAATCGGTAGTAGCG |
| Recombinant DNA | ||
| Software and Algorithms | ||
| Prism 7.0 | GraphPad | http://www.graphpad.com/scientific-software/prism; RRID:SCR_015807 |
| Microsoft Excel (version 16.19) | Microsoft Corp. | https://www.microsoft.com/en-gb/; RRID:SCR_016137 |
| MetaboAnalyst (versions 3.0, 4.0) | Web-based resource (McGill University, Canada) | https://www.metaboanalyst.ca/MetaboAnalyst/faces/home.xhtml |
| ImageJ | National Institutes of Health (NIH) | https://imagej.nih.gov/ij/; RRID:SCR_003070 |
| Canvas Draw 6 for macOS | Canvas GFX | RRID:SCR_014288 |
| Other | ||
| Teklad Rodent Diet | ENVIGO | Cat#8604 |
| Accu-Chek Aviva Plus Meter | Roche | N/A |
| stainless steel beads, 5mm (200) | Qiagen | Cat#69989 |
| Non-Fat powdered Milk | Boston BioProducts | Cat#P-1400 |
| Minispec LF90 | Bruker Optics | https://www.bruker.com/products/mr/td-nmr/minispec-lf-series.html |
| Oxymax Open Circuit Indirect Calorimeters | Columbus Instruments | http://www.colinst.com/docs/OxymaxBrochure.pdf |
| Breeze2 Glucometer | Bayer | http://personalcare.manualsonline.com/ Bayer HealthCare Blood Glucose Meter |
| Amersham Imager 600 | GE Healthcare Life Sciences | Cat# 29083461 |
Highlights:
Prophylactic and therapeutic effects of disulfiram (DSF) on diet-induced obesity in mice and rats
DSF reduces feeding efficiency and restores insulin responsiveness
DSF alleviates hepatosteatosis and pancreatic islet hyperplasia in obese mice
The metabolic benefits of DSF are preserved in Aldh2 KO mice fed an obesogenic diet
Context and Significance:
The paper by Bernier et al. focuses on the effects of disulfiram on diet-induced obesity and the downstream metabolic sequelae. Because obesity has reached epidemic proportions in developed countries and represents a major public health challenge, pharmacologic approaches to treat obesity-associated metabolic disorders is of general interest. Disulfiram treatment prevented body weight gain in mice fed a high fat diet. Histological analysis indicate that the liver and pancreatic islets are the primary sites of action the metabolic protective effects of disulfiram. The results also show beneficial effects of disulfiram as a treatment to lower body weight in diet-induced obese mice. The main implications of the findings are that disulfiram can potentially be repurposed in patients to treat obesity and related comorbidities.
ACKNOWLEDGMENTS
This work was supported by funding from the Intramural Research Program of the National Institute on Aging/NIH, and NIH Award R35HL135820 (C.F.-H.). NIH/NIA grants AG031782 and AG038072 (A.M.C.). S.J.M. was supported by a National Medical Health and Research Council of Australia CJ Martin Early Career Fellowship (RGMS ID 2010-01671). M.L. is a Cancer Institute New South Wales Future Research Leader Fellow (15/FRL/1-06A). This work was supported by grants from the NHMRC (APP1120475). E.Y.K. was supported by a grant from the KRIBB Research Initiative Program (Korean Biomedical Scientist Fellowship Program), Korea Research Institute of Bioscience and Biotechnology, Republic of Korea. Special thanks to the members of the Translational Gerontology Branch, Laboratory of Clinical Investigation, and the Comparative Medicine Section of the National Institute on Aging. We are particularly grateful to Dawn Nines, Dawn Boyer, Olga Carlson, and Alice E. Kane for their excellent work, and Marc Raley at NIA Visual Media Service.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
ADDITIONAL RESOURCES
Not applicable
REFERENCES
- Altunkaynak ME, Özbek E, Altunkaynak BZ, Can İ, Unal D, and Unal B (2008). The effects of high-fat diet on the renal structure and morphometric parametric of kidneys in rats. J. Anat 212, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Daimon CM, Cong WN, Wang R, Chirdon P, de Cabo R, Sevigny J, Maudsley S, and Martin B (2014). Longitudinal analysis of calorie restriction on rat taste bud morphology and expression of sweet taste modulators. J. Gerontol. A Biol. Sci. Med. Sci 69, 532–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Martin B, Cai H, Fiori JL, Egan JM, Siddiqui S, and Maudsley S (2013). Pancreas++ : Automated Quantification of Pancreatic Islet Cells in Microscopy Images. Front. Physiol 3, 482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogger VC, Mohamad M, Solon-Biet SM, Senior AM, Warren A, O’Reilly JN, Tung BT, Svistounov D, McMahon AC, Fraser R, et al. (2016). Dietary macronutrients and the aging liver sinusoidal endothelial cell. Am. J. Physiol. Heart Circ. Physiol 310, H1064–H1070. [DOI] [PubMed] [Google Scholar]
- Dandona P, Aljada A, Chaudhuri A, Mohanty P, and Garg R (2005). Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 111, 1448–1454. [DOI] [PubMed] [Google Scholar]
- Deitrich RA, and Erwin VG (1971). Mechanism of the inhibition of aldehyde dehydrogenase in vivo by disulfiram and diethyldithiocarbamate. Mol. Pharmacol 7, 301–307. [PubMed] [Google Scholar]
- Ding H, Zhao R, Yin X, Liu J, Zhao Q, Bernier M, and Zhu T (2014). Pyrrolidine dithiocarbamate protects pancreatic β cells from oxidative damage through regulation of FoxO1 activity in type 2 diabetic rats. Acta Biochim. Biophys. Sin 46, 582–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery MG, Jubert C, Thummel KE, and Kharasch ED (1999). Duration of cytochrome P-450 2E1 (CYP2E1) inhibition and estimation of functional CYP2E1enzyme half-life after single-dose disulfiram administration in humans. J. Pharmacol. Exp. Ther 291, 213–219. [PubMed] [Google Scholar]
- Escribano O, Guillén C, Nevado C, Gómez-Hernández A, Kahn CR, and Benito M (2009). Beta-Cell hyperplasia induced by hepatic insulin resistance: role of a liver-pancreas endocrine axis through insulin receptor A isoform. Diabetes 58, 820–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farci AM, Piras S, Murgia M, Chessa A, Restivo A, Gessa GL, and Agabio R (2015). Disulfiram for binge eating disorder: an open trail. Eat. Behav 16, 84–87. [DOI] [PubMed] [Google Scholar]
- Fernández-Verdejo R, Ravussin E, Speakman JR, and Galgani JE (2019). Progress and challenges in analyzing rodent energy expenditure. Nat. Methods 16, 797–799. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, and Sloane-Stanley GH (1957). A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem 226, 497–509. [PubMed] [Google Scholar]
- Fraser R, Cogger VC, Dobbs B, Jamieson H, Warren A, Hilmer SN, and Le Couteur DG (2012). The liver sieve and atherosclerosis. Pathology 44, 181–186. [DOI] [PubMed] [Google Scholar]
- Frye RF, and Branch RA (2002). Effect of chronic disulfiram administration on the activities of CYP1A2, CYP2C19, CYP2D6, CYP2E1, and N-acetyltransferase in healthy human subjects. Br. J. Clin. Pharmacol 53, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giménez-Gómez P, Pérez-Hernández M, O’Shea E, Caso JR, Martín-Hernandez D, Cervera LA, Centelles MLG, Gutiérrez-Lopez MD, and Colado MI (2019). Changes in brain kynurenine levels via gut microbiota and gut-barrier disruption induced by chronic ethanol exposure in mice. FASEB J 33, 12900–12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González Esquivel D, Ramírez-Ortega D, Pineda B, Castro N, Ríos C, and Pérez de la Cruz V (2017). Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacology 112, 331–345. [DOI] [PubMed] [Google Scholar]
- Guo JM, Liu AJ, Zang P, Dong WZ, Ying L, Wang W, Xu P, Song XR, Cai J, Zhang SQ, et al. (2013). ALDH2 protects against stroke by clearing 4-HNE. Cell Res 23, 915–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasek BE, Stewart LK, Henagan TM, Boudreau A, Lenard NR, Black C, Shin J, Huypens P, Malloy VL, Plaisance EP, et al. (2010). Dietary methionine restriction enhances metabolic flexibility and increases uncoupled respiration in both fed and fasted states. Am. J. Physiol. Regul. Integr. Comp. Physiol 299, R728–R739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helman A, Avrahami D, Klochendler A, Glaser B, Kaestner KH, Ben-Porath I, and Dor Y (2016). Effects of ageing and senescence on pancreatic β-cell function. Diabetes Obes. Metab 18 Suppl 1, 58–62. [DOI] [PubMed] [Google Scholar]
- Kast RE (2008). Ritonavir and disulfiram may be synergistic in lowering active interleukin-18 levels in acute pancreatitis, and thereby hasten recovery. JOP 9, 350–353. [PubMed] [Google Scholar]
- Kim W, Doyle ME, Liu Z, Lao Q, Shin YK, Carlson OD, Kim HS, Thomas S, Napora JK, Lee EK, et al. (2011). Cannabinoids inhibit insulin receptor signaling in pancreatic beta-cells. Diabetes 60, 1198–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD, and Vasiliou V (2012). Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev 64, 520–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HJ, Won YS, Park O, Chang B, Duryee MJ, Thiele GE, Matsumoto A, Singh S, Abdelmegeed MA, Song BJ, et al. (2014). Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology 60, 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Couteur DG, Fraser R, Cogger VC, and McLean AJ (2002). Hepatic pseudocapillarisation and atherosclerosis in ageing. Lancet 359, 1612–1615. [DOI] [PubMed] [Google Scholar]
- Lonardo A, Ballestri S, Marchesini G, Angulo P, and Loria P (2015). Nonalcoholic fatty liver disease: a precursor of the metabolic syndrome. Dig. Liver Dis 47, 181–190. [DOI] [PubMed] [Google Scholar]
- Madan A, Williams TD, and Faiman MD (1994). Glutathione- and glutathione-S-transferase-dependent oxidative desulfuration of the thione xenobiotic diethyldithiocarbamate methyl ester. Mol. Pharmacol 46, 1217–1225. [PubMed] [Google Scholar]
- Marseglia L, Manti S, D’Angelo G, Nicotera A, Parisi E, Di Rosa G, Gitto E, and Arrigo T (2014). Oxidative stress in obesity: a critical component in human diseases. Int. J. Mol. Sci 16, 378–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B, Golden E, Carlson OD, Pistell P, Zhou J, Kim W, Frank BP, Thomas S, Chadwick WA, Greig NH, et al. (2009). Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington’s disease. Diabetes 58, 318–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlem A, Hagberg CE, Muhl L, Eriksson U, and Falkevall A (2013). Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc 8, 1149–1154. [DOI] [PubMed] [Google Scholar]
- Mina AI,, LeClair RA, LeClair KB, Cohen DE, Lantier L, and Banks AS (2018). CalR: A Web-Based Analysis Tool for Indirect Calorimetry Experiments. Cell Metab 28, 656–666.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor RK, Baur JA, Gomes AP, Ward TM, Csiszar A, Mercken EM, Abdelmohsen K, Shin YK, Canto C, Scheibye-Knudsen M, et al. (2011). SRT1720 improves survival and healthspan of obese mice. Sci. Rep 1, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz C, Pascual-Salcedo D, Castellanos MC, Alfranca A, Aragonés J, Vara A, Redondo JM, and de Landázuri MO (1996). Pyrrolidine dithiocarbamate inhibits the production of interleukin-6, interleukin-8, and granulocyte-macrophage colony-stimulating factor by human endothelial cells in response to inflammatory mediators: modulation of NF-kappa B and AP-1 transcription factors activity. Blood 88, 3482–3490. [PubMed] [Google Scholar]
- Panchal SK, Poudyal H, Iyer A, Nazer R, Alam MA, Diwan V, Kauter K, Sernia C, Campbell F, Ward L, et al. (2011). High-carbohydrate, high-fat diet-induced metabolic syndrome and cardiovascular remodeling in rats. J. Cardiovasc. Pharmacol 57, 611–624. [DOI] [PubMed] [Google Scholar]
- Poulsen HE, Jorgensen L, and Thomsen P (1987). Prevention of acetaminophen hepatotoxicity by disulfiram. Pharmacol. Ther 33, 83. [DOI] [PubMed] [Google Scholar]
- Putti R, Sica R, Migliaccio V, and Lionetti L (2015). Diet impact on mitochondrial bioenergetics and dynamics. Front. Physiol 6, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, and Ahmad N (2008). Dose translation from animal to human studies revisited. FASEB J 22, 659–661. [DOI] [PubMed] [Google Scholar]
- Sachdeva MM, and Stoffers DA (2009). Meeting the demand for insulin: molecular mechanisms of adaptive postnatal beta-cell mass expansion. Mol. Endocrinol 23, 747–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, Dunn CA, Singh N, Veith S, Hasan-Olive MM, et al. (2014). A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab 20, 840–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreck R, Meier B, Männel DN, Dröge W, and Baeuerle PA (1992). Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J. Exp. Med 175, 1181–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi Y, Takamoto I, Kubota N, Matsui J, Suzuki R, Komeda K, Hara A, Toyoda Y, Miwa I, Aizawa S, et al. (2007). Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J. Clin. Invest 117, 246–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Xie Y, Bernier M, and Wainer IW (2009). Determination of free and protein-bound glutathione in HepG2 cells using capillary electrophoresis with laser-induced fluorescence detection. J. Chromatogr. A 1216, 3533–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir GC (2016). Islet-cell biology in 2015: Understanding secretion, ageing and death in β cells. Nat. Rev. Endocrinol 12, 72–74. [DOI] [PubMed] [Google Scholar]
- Weiskirchen R, and Tacke F (2016). Interleukin-33 in the pathogenesis of liver fibrosis: alarming ILC2 and hepatic stellate cells. Cell. Mol. Immunol 14, 143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiskirchen R, and Tacke F (2014). Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr 3, 344–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanovski SZ, and Yanovski JA (2014). Long-term Drug Treatment for Obesity: A Systematic and Clinical Review. JAMA 311, 74–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T, Zhao R, Zhang L, Bernier M, and Liu J (2012). Pyrrolidine dithiocarbamate enhances hepatic glycogen synthesis and reduces FoxO1-mediated gene transcription in type 2 diabetic rats. Am. J. Physiol. Endocrinol. Metab 302, E409–E416. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets or code.