

Abstract
A new series with the tetrahydroisoquinoline-fused benzodiazepine (TBD) ring system combined with the surrogates of (1-methyl-1H-pyrrol-3-yl)benzene (“MPB”) payloads were designed and executed for conjugation with a monoclonal antibody for anticancer therapeutics. DNA models helped in rationally identifying modifications of the “MPB” binding component and guided structure–activity relationship generation. This hybrid series of payloads exhibited excellent in vitro activity when tested against a panel of various cancer cell lines. One of the payloads was appended with a lysosome-cleavable peptide linker and conjugated with an anti-mesothelin antibody via a site-specific conjugation method mediated by the enzyme bacterial transglutaminase (BTGase). Antibody–drug conjugate (ADC) 50 demonstrated good plasma stability and lysosomal cleavage. A single intravenous dose of ADC 50 (5 or 10 nmol/kg) showed robust efficacy in an N87 gastric cancer xenograft model.
Keywords: PBD, DNA minor groove binders, DNA binding models, ADC, cytotoxicity
Antibody–drug conjugates (ADCs) have ascended as powerful biologics that combine the high affinity and specificity of monoclonal antibodies (mAbs) with the promising antitumor efficacy of the payloads.1,2 Over the last two decades, ADCs have continued to evolve as an attractive modality for the treatment of hematological malignancies and solid tumors.3 Currently, there are nine approved and marketed ADCs.4 A cytotoxic agent, often called a payload, that is generally too toxic for systemic administration is combined with mAbs with the help of linkers to specifically target tumor cells. On the basis of the mechanism of action, payloads fall into three categories: antimitotic, DNA-interacting, and transcription-inhibiting. The antimitotic class of payloads used in ADCs act by interacting with tubulin and include maytansinoids, auristatins, tubulysins, and many others.5 The DNA-interacting class includes calicheamicin, duocarmycin, etc.6 The transcription inhibitors include amatoxins that bind to RNA polymerase II.7 Antimitotic and DNA-interacting payloads are most broadly used in ADCs. DNA-interacting agents act by binding to the minor groove and lead to intercalation, scission, alkylation, or cross-linking of strands. Hence, they are also called DNA minor groove binders (MGBs). Another important class of payloads used in ADCs is camptothecins.8 Camptothecin and its derivatives bind to the topoisomerase I/DNA complex as their mechanism of action.9
One of the MGBs that is highly pursued in the ADC architecture is the pyrrolo[2,1-c][1,4]benzodiazepine (PBD) class of compounds (Figure 1, example payload 1).10−17 PBD dimers bind in the minor groove of DNA, where they form interstrand cross-links with guanines. Rahman et al.18 reported that some non-covalent heteroaryl pharmacophores have a strong preference for GC-rich DNA sequences either alone or when combined with PBDs. Compounds 2a and 2b are the two most advanced candidates from Rahman et al.18 Notably, 2b demonstrated significant antitumor efficacy in breast (MDA-MB-231) and pancreatic (MIA PaCa-2) xenograft models.18
Figure 1.

Example structures of PBD dimer,17 “PBD-py-MPB”,18 and TBD dimer.19−21
As part of our ADC discovery efforts19−21 and inspired by the report by Rahman et al.,18 we sought to pursue PBD–4-(1-methyl-1H-pyrrol-3-yl)benzeneamine (“MPB” as termed by Rahman et al.18) hybrid payloads for the ADC modality (Figure 1, example payloads 2a and 2b). The inherent toxicity of these payloads to normal cells motivated us to prosecute these agents for a targeted therapeutic approach like ADC. Our prior experience with PBD molecules led us to hypothesize that some examples in this class might be too potent to achieve an acceptable therapeutic index. An appealing aspect of these hybrid molecules was the potential to tune the properties by preparing a diverse set of analogues through variation of the MPB portion of the molecules. We pursued the reported “PBD–MPB” hybrid payloads in the context of our fused benzodiazepine with a tetrahydroisoquinoline ring system, herein called TBD (Figure 1, example payload 3).
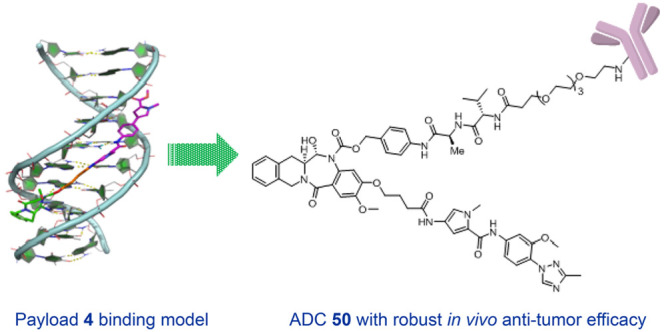
For benchmarking purposes, we made 4 and 5, which are the TBD versions of 2a and 2b, respectively, from the original publication by Rahman et al.18 Initial characterization of the payloads was based on an in vitro evaluation against a suite of cellular proliferation assays for lung (H226), gastric (N87), ovarian (OVCAR3), and colon (HCT116) cancer cell lines as described in our recent publication.22 Compounds 4 and 5 showed quite potent (single- to double-digit and subpicomolar) activities in these cell lines (Table 1). A double-stranded B-DNA binding model for 4 was generated using the sequence 5′-AAGAAGGCAA-3′, as reported by Rahman et al.18 We hypothesized that 4, which is a TBD variant of 2a, would also bind to the 5′-AAGAAGGCAA-3′ sequence that was demonstrated to be the preferred binding sequence for 2b.18Figure 2A,B illustrates the binding mode of 4 in the DNA minor groove. The C11–N10 imine of TBD makes a covalent bond with the exocyclic amine of G3 of the DNA, while the “py-MPB” portion of the payload makes non-covalent interactions and nicely follows the curvature of the DNA minor groove.
Table 1. Compounds and Cytotoxicity Data (IC50 in nM).


The IC50 value was the result of multiple determinations (n ≥ 2).
The IC50 value was obtained after single determination (n = 1).
Figure 2.

(A) Binding model of 4 in double-helical B-DNA. The inset shows the sequence used in the DNA model development. (B) Surface representation of the DNA model showing the snug fit of 4 in the minor groove. Hydrogen bonds are represented by yellow dotted lines. The covalently bound TBD portion of the payload, the linker, and the non-covalently bound “py-MPB” portion are shown in green, orange, and magenta, respectively. The figure was prepared using PyMOL (The PyMOL molecular graphics system, Version 2.3.0, Schrödinger, LLC).
The DNA-binding model of 4 revealed opportunities to modify the “MPB” portion of the payloads. A set of internally available anilines was evaluated to rationally replace the “MPB” moiety. Among these, we envisioned a (methyltriazolo)benzene replacement (compound 6) for the “MPB” moiety. We hypothesized that the flat (methyltriazolo)benzene group would nicely complement the minor groove, with one of the ring nitrogens in the triazole ring being in close proximity to the exocyclic amine of G7 in the DNA model to engage in a hydrogen-bonding interaction dynamically. While the H226 cell line activity was retained or improved in 6 compared with its progenitor 4, the other three cell lines showed diminished activity, with the worst being the OVCAR3 cell line. Compound 7 was made to explore the impact of removing the methyl group in the triazole ring, and it turned out to be equipotent to its methyl counterpart in the H226, N87, and HCT116 cell lines and ∼3-fold more potent in the OVCAR3 cell line. As an N-linked imidazole derivative, 8 lacks the hydrogen-bond acceptor close to the exocyclic amine of G7 and showed a considerable loss of activity across all four cell lines compared with 6. C-linked imidazole 9 had further-diminished activities across all four cell lines. The substantial drop in the activity is perhaps partly due to the possibility of tautomerization where hydrogen bonding with the exocyclic amine of G7 is absent in one set of tautomers. The perturbed coplanarity between the two terminal rings may also play a role in reducing the activity. For oxazole 10, which has the ring nitrogen to complement the exocyclic amine of G7, the activity was restored in all of the cell lines except H226 compared to its triazole counterpart 6.
Next, we kept the methyltriazolo ring constant and surveyed the structure–activity relationship (SAR) around the N-linked phenyl group. o-Fluoro analogue 11 showed comparable activity in the H226 cell line and improved activities in the other three cell lines relative to 6. Slightly bulky ortho substituents negatively impacted the activities in all four cell lines (compounds 12–16). o-Hydroxy analogue 17 showed activities comparable to those of 6 in the H226 and HCT116 cell lines. Pyridyl derivative 18 showed an enhancement in activity in the N87 cell line relative to its phenyl counterpart 7. Compound 19 with a difluoromethyl group instead of the methyl group in 13 showed an improvement in the activities in the N87 and OVCAR3 cell lines. Desmethyl analogue 20 showed a modest drop in activity across all of the tested cell lines except N87 compared with its parent analogue 13. Dimethyl derivative 21 exhibited a dramatic loss in the activities across all four cell lines, likely hinting at the need for coplanarity of the distal biaryl ring system. We revisited N-linked imidazole (cf. compound 8) and introduced substitutions in the linking phenyl and terminal imidazole rings. Compounds 22–26 are the outcomes of this effort, and the combination of chloroimidazole with either a fluorophenyl or pyridine linking ring yielded acceptable activity profiles in all four cell lines. N-Phenylpyrazole 27 showed double-digit picomolar activities in three of the four cell lines.
Tricyclic analogues 28 and 29 showed activity profiles comparable to that of 6. Tricyclic derivative 30 is not as flat as 28 and 29 and showed a marked drop in activities across all four cell lines. Tricyclic analogue 31 exhibited further-diminished activities in all four cell lines. While all three rings in the tricyclic ring system in 30 fit well in the minor groove of the DNA model, the morpholinotriazole portion of the tricyclic ring system in 31 projected outside the minor groove. Aza-N-methylbenzimidazole 32 had activities in the single-digit nanomolar range in all four cell lines, which once again may partly be due to the putative suboptimal fit of the bicyclic ring in the minor groove of the DNA.
The synthetic route for payloads 4 to 32 is shown in Scheme 1 (see the Supporting Information for details). An effort was started to produce a fit-for-purpose synthetic route capable of delivering tens to hundreds of grams of monomer precursors. This was undertaken to supply ample quantities of material for SAR studies while also providing a blueprint toward a large-scale process. The route begins with aldehyde 33, obtained from vanillin in three steps, all conducted in continuous flow mode.23 The phenol was protected as a tosyl ester, which imparted crystallinity to many of the early-stage intermediates and greatly reduced the number of purifications by chromatography. Aldehyde 34 was oxidized to the acid through a Pinnick oxidation (92%), and through the intermediacy of the acid chloride, the acid was coupled with tetrahydroisoquinoline 36 to give amide 37 (82%) under Schotten–Baumann conditions. The nitro group of 37 was reduced with hydrogen gas catalyzed by palladium on carbon doped with 1% iron (99%). The aniline was protected as an allyloxycarbamate (Alloc), again under Schotten–Baumann conditions (98%). Freshly distilled Alloc-Cl was found to eliminate the formation of urea-like byproducts that formed from old lots of Alloc-Cl. The primary alcohol of 38 was oxidized with TEMPO/NCS to afford the aldehyde, which cyclized in situ to form tetracycle 39 as a single aminal diastereomer (86%). We found this to be a strategic place to store bulk material, as it is the last crystalline intermediate in the synthesis. The aminal oxygen in 39 was protected as a TBS ether (89%), and then the tosyl group was removed under the action of potassium carbonate in methanol (82%) to afford phenol 40, which was then alkylated with methyl bromobutyrate and hydrolyzed to afford key intermediate acid 41 in a robust, scalable 10-step process. This acid was then diversified by amide bond formation followed by deprotection of the Alloc group, which led to spontaneous elimination of the protected hemiaminal, affording the imine products 4–32. It was felt that unmasking the imine functionality as late as possible was important to minimize any potential stability concerns with the potentially reactive imine and also, for safety reasons, to minimize the number of steps that required handling of these potently cytotoxic compounds.
Scheme 1. Synthetic Route for 4–32.

On the basis of our in vitro cytotoxicity SAR and the desired physicochemical properties, 13 was selected as an exemplar for installing the linker for conjugation with an antibody since this starting point might lead us to tune potency up or down if we found the corresponding ADC to be either not potent enough or too poorly tolerated. This synthesis commenced in a manner similar to that of 4–32, albeit on a differentially protected scaffold (Scheme 2). Amide coupling between chiral amine 42 and acid 43 afforded nitro–amide 44, which was next reduced to the corresponding aniline, and the cathepsin-cleavable valine–alanine–benzyl carbamate linker was installed to give 45. Cleavage of the TBS ether, oxidation to the hemiaminal, and a subsequent protecting-group manipulation led to phenol 46, from which the MGB motif was added, similarly to the nonconjugated analogues, to afford 48. Removal of the Alloc protecting group on the linker followed by installation of a poly(ethylene glycol) 4 (PEG4) spacer gave linker–payload 49. Conjugation to an antibody that targets the mesothelin24 antigen using site-specific conjugation mediated by BTGase25 as described in the Supporting Information provided ADC 50 (Figure 3). The final purified ADC 50 was >98% monomer with a drug to antibody ratio (DAR) of 2.0.
Scheme 2. Synthetic Route for Payload–Linker 49.

Figure 3.

Structure of ADC 50.
ADC 50 was incubated with cathepsin B at 37 °C for 4 h to check whether this ADC could be cleaved enzymatically by a lysosomal protease. Cleavage of 98% of the payload occurred within 4 h. ADC 50 was found to be quite stable upon incubation with mouse serum at 37 °C, with minimal loss of payload (<0.1%) over 96 h. The in vitro potency of ADC 50 is comparable to that of the corresponding payload by itself.
The efficacy of anti-mesothelin ADC 50 was evaluated in a xenograft model of mesothelin-positive N87 human gastric tumors while anti-FucGM126 ADC 50 was administered as a nontargeted isotype-ADC control. Administration of a single iv dose of anti-mesothelin ADC 50 at 5 or 10 nmol/kg (0.4 or 0.8 mg/kg) is highly efficacious, whereas the nontargeted anti-FucGM1 ADC 50 demonstrated only minor tumor growth inhibition at 10 nmol/kg iv (0.8 mg/kg) (Figure 4A). Anti-mesothelin ADC 50 was well-tolerated up to 10 nmol/kg with only a transient body weight loss observed relative to vehicle-treated mice (Figure 4B).
Figure 4.

Antitumor efficacy was measured in an established N87 human gastric cancer xenograft model in mice. (A) Time course of tumor volume after administration of a single iv dose of anti-mesothelin-ADC 50. (B) Time course of median percent body weight change from the same experiment. A dose of 5 or 10 nmol/kg is equivalent to 0.4 or 0.8 mg/kg, respectively.
In summary, inspired by a literature report of the conventional payload efficiency of a hybrid PBD, we pursued modified hybrid payloads in the context of ADC modality. PBD-based payloads are extremely potent and quite toxic to normal cells, and hence, a targeted therapeutic approach like the ADC modality is warranted to increase their safety when used in anticancer therapeutics. With the help of molecular modeling studies, we executed some TBD version of PBD payloads combined with the new (methyltriazolo)benzene MGBs. This new series of payloads showed great promise in terms of in vitro activities in a select panel of cancer cell lines. One of the new hybrid payloads was appended with a linker and conjugated with an antibody. This ADC demonstrated robust in vivo antitumor efficacy and acceptable tolerability in a mouse xenograft model.
Acknowledgments
The authors thank Ayesha Nazeer, Colin Chong, Remie Mandawe, and Joseph Naglich for their help with in vivo and in vitro studies and the Compound Management & Cellular Technologies Groups for their great support.
Glossary
Abbreviations
- TBD
tetrahydroisoquinoline-fused benzodiazepine
- “MPB”
(1-methyl-1H-pyrrol-3-yl)benzene
- ADC
antibody–drug conjugate
- BTGase
bacterial transglutaminase
- mAb
monoclonal antibody
- MGB
DNA minor groove binder
- PBD
pyrrolo[2,1-c][1,4]benzodiazepine
- IC50
half-maximal inhibitory concentration
- Alloc
allyloxycarbamate
- TEMPO
2,2,6,6-tetramethylpiperidin-1-oxyl
- TBS
tert-butyldimethylsilyl
- NCS
N-chlorosuccinimide
- DAR
drug to antibody ratio
- PEG4
poly(ethylene glycol) 4.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00578.
Synthetic procedures, NMR spectra, molecular modeling procedures, serum and cathepsin B stability assay procedures, in vivo experimental details, and antibody conjugation experimental details (PDF)
Author Present Address
¶ P.S.: Computational Sciences, GlaxoSmithKline, 1250 South Collegeville Road, Collegeville, PA 19426, USA.
Author Present Address
■ I.M.: Discovery Chemistry, Bristol Myers Squibb Research and Early Development, 100 Binney Street, Cambridge, MA 02142, USA.
Author Present Address
△ C.I. and K.M.P.: ViiV Healthcare, 36 East Industrial Road, Branford, CT 06405, USA.
Author Present Address
● D.R.L.: Arvinas Inc., 5 Science Park, 395 Winchester Avenue, New Haven, CT 06511, USA.
Author Present Address
★ M.A.S., B.Z., Y.T., and M.D.E.: Chemical Process Development, Bristol Myers Squibb, 1 Squibb Drive, New Brunswick, NJ 08903, USA.
Author Present Address
⊡ PMC: GPCR Therapeutics, Seoul 08790, South Korea.
Author Present Address
⬢ C.X.: Lead Discovery and Optimization, Bristol Myers Squibb Research and Early Development, P.O. Box 4000, Princeton, NJ 08543, USA.
Author Present Address
◇ P.H.: Department of Protein Chemistry, Genentech Research and Early Development, 1 DNA Way, 41-4106, South San Francisco, CA 94080, USA.
Author Present Address
▲ G.V.: Discovery Chemistry, Bristol Myers Squibb Research and Early Development, P.O. Box 4000, Princeton, NJ 08543, USA.
The authors declare no competing financial interest.
Supplementary Material
References
- Chau C. H.; Steeg P. S.; Figg W. D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. 10.1016/S0140-6736(19)31774-X. [DOI] [PubMed] [Google Scholar]
- Vezina H. E.; Cotreau M.; Han T. H.; Gupta M. Antibody–Drug Conjugates as Cancer Therapeutics: Past, Present, and Future. J. Clin. Pharmacol. 2017, 57, S11–S25. 10.1002/jcph.981. [DOI] [PubMed] [Google Scholar]
- Goli N.; Bolla P. K.; Talla V. Antibody–drug conjugates (ADCs): Potent biopharmaceuticals to target solid and hematological cancers—an overview. J. Drug Delivery Sci. Technol. 2018, 48, 106–117. 10.1016/j.jddst.2018.08.022. [DOI] [Google Scholar]
- Theocharopoulos C.; Lialios P.-P.; Gogas H.; Ziogas D. C. An overview of antibody–drug conjugates in oncological practice. Ther. Adv. Med. Oncol. 2020, 12, 1–20. 10.1177/1758835920962997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Lin Z.; Arnst K. E.; Miller D. D.; Li W. Tubulin Inhibitor-Based Antibody–Drug Conjugates for Cancer Therapy. Molecules 2017, 22, 1281. 10.3390/molecules22081281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y.; Ho M. DNA damaging agent-based antibody–drug conjugates for cancer therapy. Antibody Ther. 2018, 1, 43–53. 10.1093/abt/tby007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl A.; Lutz C.; Hechler T.. Amatoxins as RNA Polymerase II Inhibiting Antibody–Drug Conjugate (ADC) Payloads. In Cytotoxic Payloads for Antibody–Drug Conjugates; Thurston D. E., Jackson P. J. M., Eds.; Royal Society of Chemistry: Cambridge, U.K., 2019; pp 398–426. [Google Scholar]
- Sahota S.; Vahdat L. T. Sacituzumab govitecan: an antibody−drug conjugate. Expert Opin. Biol. Ther. 2017, 17, 1027–1031. 10.1080/14712598.2017.1331214. [DOI] [PubMed] [Google Scholar]
- Thomas A.; Pommier Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. 10.1158/1078-0432.CCR-19-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley J. A. The development of pyrrolobenzodiazepines as antitumor agents. Expert Opin. Invest. Drugs 2011, 20, 733–744. 10.1517/13543784.2011.573477. [DOI] [PubMed] [Google Scholar]
- Bose D. S.; Thompson A. S.; Ching J.; Hartley J. A.; Berardini M. D.; Jenkins T. C.; Neidle S.; Hurley L. H.; Thurston D. E. Rational design of a highly efficient irreversible DNA interstrand cross-linking agent based on the pyrrolobenzodiazepine ring system. J. Am. Chem. Soc. 1992, 114, 4939–4941. 10.1021/ja00038a089. [DOI] [Google Scholar]
- Jeffrey S. C.; Burke P. J.; Lyon R. P.; Meyer D. W.; Sussman D.; Anderson M.; Hunter J. H.; Leiske C. I.; Miyamoto J. B.; Nicholas N. D.; Okeley N. M.; Sanderson R. J.; Stone I. J.; Zeng W.; Gregson S. J.; Masterson L.; Tiberghien A. C.; Howard P. W.; Thurston D. E.; Law C. L.; Senter P. D. A potent anti-CD70 antibody–drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconjugate Chem. 2013, 24, 1256–1263. 10.1021/bc400217g. [DOI] [PubMed] [Google Scholar]
- Miller M. L.; Fishkin N. E.; Li W.; Whiteman K. R.; Kovtun Y.; Reid E. E.; Archer K. E.; Maloney E. K.; Audette C. A.; Mayo M. F.; Wilhelm A.; Modafferi H. A.; Singh R.; Pinkas J.; Goldmacher V.; Lambert J. M.; Chari R. V. J. A new class of antibody–drug conjugates with potent DNA alkylating activity. Mol. Cancer Ther. 2016, 15, 1870–1878. 10.1158/1535-7163.MCT-16-0184. [DOI] [PubMed] [Google Scholar]
- Antonow D.; Thurston D. E. Synthesis of DNA-Interactive Pyrrolo[2,1-c][1,4]benzodiazepines (PBDs). Chem. Rev. 2011, 111, 2815–2864. 10.1021/cr100120f. [DOI] [PubMed] [Google Scholar]
- Cipolla L.; Araújo A. C.; Airoldi C.; Bini D. Pyrrolo[2,1-c][1,4]benzodiazepine as a scaffold for the design and synthesis of anti-tumour drugs. Anti-Cancer Agents Med. Chem. 2009, 9, 1–31. 10.2174/187152009787047743. [DOI] [PubMed] [Google Scholar]
- Gerratana B. Biosynthesis, synthesis, and biological activities of pyrrolobenzodiazepines. Med. Res. Rev. 2012, 32, 254–293. 10.1002/med.20212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantaj J.; Jackson P. J. M.; Rahman K. M.; Thurston D. E. From anthramycin to pyrrolobenzodiazepine (PBD)-containing antibody–drug conjugates (ADCs). Angew. Chem., Int. Ed. 2017, 56, 462–488. 10.1002/anie.201510610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman K. M.; Jackson P. J. M.; James C. H.; Basu B. P.; Hartley J. A.; de la Fuente M.; Schatzlein A.; Robson M.; Pedley B.; Pepper C.; Fox K. R.; Howard P. W.; Thurston D. E. GC-Targeted C8-Linked Pyrrolobenzodiazepine-Biaryl Conjugates with Femtomolar in Vitro Cytotoxicity and in Vivo Antitumor Activity in Mouse Models. J. Med. Chem. 2013, 56, 2911–2935. 10.1021/jm301882a. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; McDonald I. M.; Chowdari N. S.; Tram H.; Borzilleri R. M.; Gangwar S.. Benzodiazepine dimers, conjugates thereof, and methods of making and using. US 9527871, 2016.
- McDonald I. M.; Chowdari N. S.; Johnson W. J.; Zhang Y.; Borzilleri R. M.; Gangwar S.. Heteroarylene-bridged benzodiazepine dimers, conjugates thereof, and methods of making and using. US 9526801, 2016.
- McDonald I. M.; Sivaprakasam P.; Iwuagwu C. I.; Peese K. M.; Cheng H.; Chowdari N. S.; Gangwar S.. Antiproliferative Compounds and Conjugates made therefrom. US 0110873, 2018.
- Chowdari N. S.; Zhang Y.; McDonald I. M.; Johnson W. J.; Langley D. R.; Sivaprakasam P.; Mate R.; Huynh T.; Kotapati S.; Deshpande M.; Pan C.; Menezes D.; Wang Y.; Rao C.; Sarma G.; Warrack B. M.; Rangan V. S.; Mei-Chen S.; Cardarelli P.; Deshpande S.; Passmore D.; Rampulla R.; Mathur A.; Borzilleri R.; Rajpal A.; Vite G.; Gangwar S. Design, Synthesis and Structure Activity Relationships of Novel Tetrahydroisoquinolino Benzodiazepine Dimer Antitumor Agents and Their Application in Antibody–Drug Conjugates. J. Med. Chem. 2020, 63, 13913–13950. 10.1021/acs.jmedchem.0c01385. [DOI] [PubMed] [Google Scholar]
- Rakshit S.; Lakshminarasimhan T.; Guturi S.; Kanagavel K.; Kanusu U. R.; Niyogi A. G.; Sidar S.; Luzung M. R.; Schmidt M. A.; Zheng B.; Eastgate M. D.; Vaidyanathan R. Nitration using fuming HNO3 in sulfolane: synthesis of 6-nitrovanillin in flow mode. Org. Process Res. Dev. 2018, 22, 391–398. 10.1021/acs.oprd.8b00020. [DOI] [Google Scholar]
- Terret J. A.; Pogue S. L.; Toy K.; Yang L.; Rao C. R.; Chen B.. Human antibodies that bind mesothelin, and uses thereof. US 8,425,904, 2013.
- Strop P.; Delaria K.; Foletti D.; Witt J. M.; Hasa-Moreno A.; Poulsen K.; Casas M. G.; Dorywalska M.; Farias S.; Pios A.; Lui V.; Dushin R.; Zhou D.; Navaratnam T.; Tran T.-T.; Sutton J.; Lindquist K. C.; Han B.; Liu S.-H.; Shelton D. L.; Pons J.; Rajpal A. Site-specific conjugation improves therapeutic index of antibody drug conjugates with high drug loading. Nat. Biotechnol. 2015, 33, 694–696. 10.1038/nbt.3274. [DOI] [PubMed] [Google Scholar]
- Ponath P.; Menezes D.; Pan C.; Chen B.; Oyasu M.; Strachan D.; LeBlanc H.; Sun H.; Wang X.-T.; Rangan V. S.; Deshpande S.; Cristea S.; Park K.-S.; Sage J.; Cardarelli P. M. A novel, fully human anti-fucosyl-GM1 antibody demonstrates potent in vitro and in vivo antitumor activity in preclinical models of small cell lung cancer. Clin. Cancer Res. 2018, 24, 5178–5189. 10.1158/1078-0432.CCR-18-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.