

Abstract

Small-molecule antimicrobial peptidomimetic amphiphiles represent a promising class of novel antimicrobials with the potential for widespread therapeutic application. To investigate the role of spatial positioning for key hydrophobic and hydrophilic groups on the antimicrobial efficacy and selectivity, positional isomers of the lead biphenyl antimicrobial peptidomimetic compound 1 were synthesized and subjected to microbial growth inhibition and mammalian toxicity assays. Positional isomer 4 exhibited 4–8× increased efficacy against the pathogenic Gram-negative bacteria Pseudomonas aeruginosa and Escherichia coli (MIC = 2 μg/mL), while isomers 2, 3, and 7 exhibited a 4× increase in activity against Acinetobacter baumannii (MIC = 4 μg/mL). Changes in molecular shape had a significant impact on Gram-negative antibacterial efficacy and the resultant spectrum of activity, whereas all structural isomers exhibited significant efficacy (MIC = 0.25–8 μg/mL) against Gram-positive bacterial pathogens (e.g., methicillin-resistant Staphylococcus aureus, Streptococcus pneumoniae, and Enterococcus faecalis).
Keywords: antimicrobial, peptidomimetic, amphiphile, membrane disruption, structural isomers
The increasing prevalence of bacterial resistance to antimicrobial pharmaceuticals presents an ever-growing crisis because of the reliance of modern medicine on antimicrobial treatments.1−4 The issue has been identified as a major concern by multiple organizations and governments, whom have implemented “global action plans” and various initiatives to combat and ameliorate the rise of antimicrobial resistance.5−8 There exists an urgent need for the development of novel, inexpensive antimicrobials for the treatment of drug-resistant bacterial infections.9,10
Small-molecule antimicrobial peptidomimetics (SMAPs) have garnered significant interest in recent years because of their ability to effectively disrupt microbial membranes and biofilms in addition to their enhanced synthetic accessibility and metabolic stability relative to traditional antimicrobial peptides.11,12 A basic, albeit limited, understanding of the membrane disruption mechanism behind amphiphilic antimicrobial peptides13−17 has led to the development of small-molecule mimics that contain hydrophilic and hydrophobic residues (Figure 1). The spatial arrangement of these residues creates an overall amphiphilic structure, which is essential for inducing cellular membrane disruption. Furthermore, the balance and arrangement of these hydrophobic and hydrophilic residues is highly important for modulating the antimicrobial efficacy and the bacterial/mammalian membrane selectivity (i.e., cytotoxicity).18−20
Figure 1.
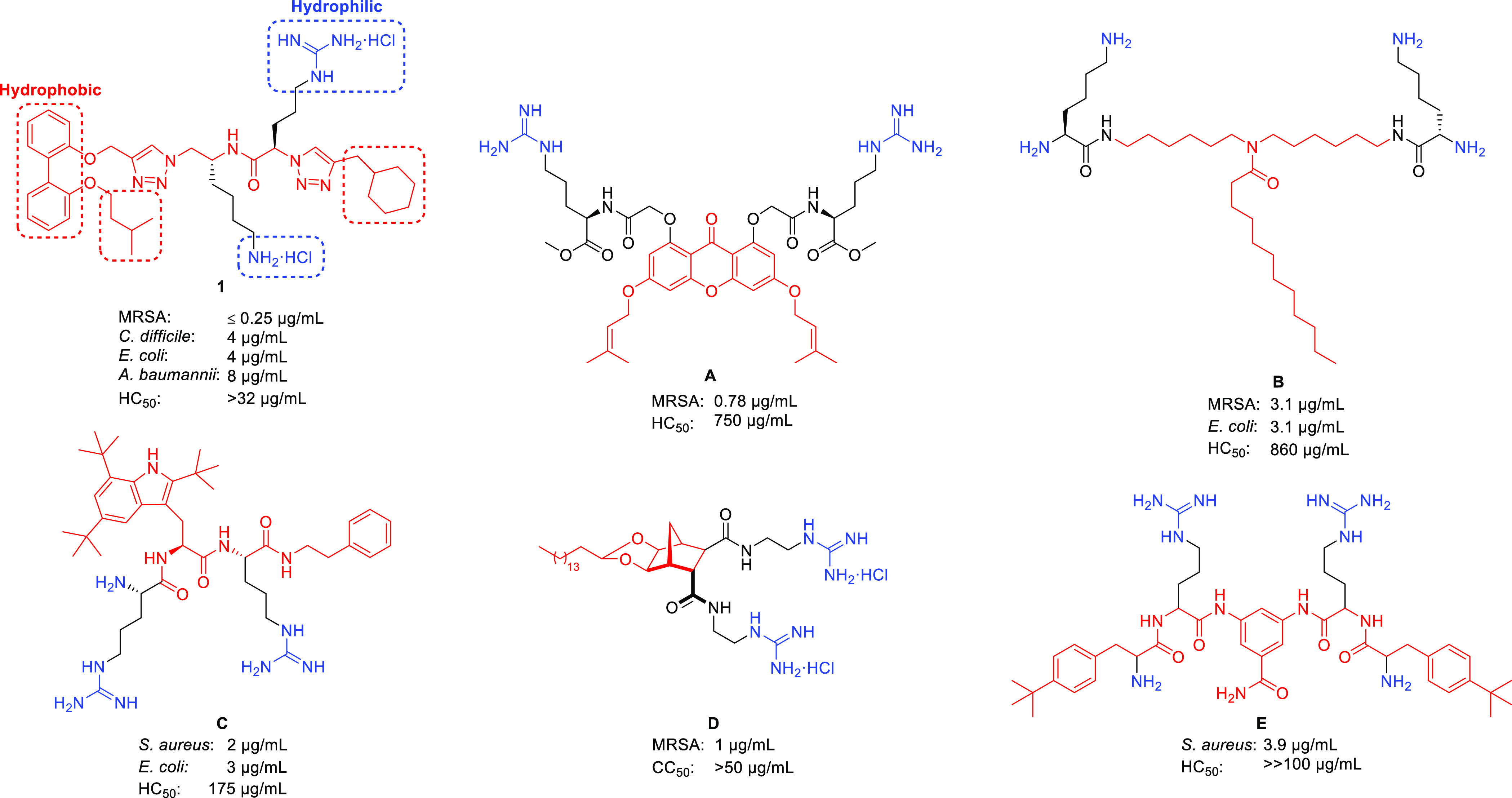
Lead biphenyl antimicrobial amphiphile 1(25) and other small-molecule antimicrobial peptidomimetics A,18B,19C,29D,23,30 and E(21) – corresponding antibacterial activities (MIC) and hemolysis values (HC50) are reported in μg/mL.
The oligomerization and self-aggregation of these small-molecule amphiphiles plays a crucial role in their antimicrobial activity and membrane-disrupting ability.21−24 As such, varying the spatial positioning of the relevant hydrophobic and hydrophilic substituents could have a marked impact on the concerted membrane disruption effected by the self-aggregation and membrane interactions of these small-molecule amphiphiles.
Recent work in our laboratory has focused on SMAPs (e.g., compound 1, Figure 1) that comprise a hydrophobic biphenyl aromatic core along with pendant hydrophobic functionalities and a 1,2,3-triazole containing peptidomimetic chain that incorporates hydrophilic, cationic lysine and arginine residues.25−28 These biphenyl peptidomimetic amphiphiles have displayed potent minimum inhibitory concentration (MIC) values against both Gram-positive and Gram-negative bacteria (Figure 1). Their positive results in membrane-disruption assays and their extremely fast time-kill kinetics were indicative of membrane-active antimicrobial compounds.25
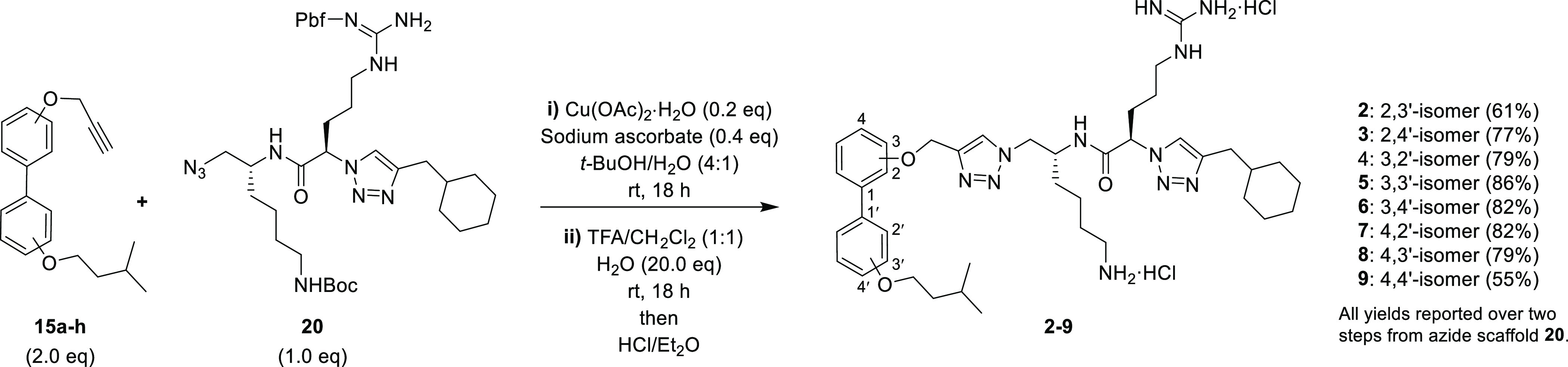
These compounds exhibit a 2,2′-disubstitution pattern on the biphenyl aromatic core (compound 1, Figure 2), wherein the peptidomimetic chain (R) constitutes one substituent and the hydrophobic isopentyl chain comprises the other substituent. In order to investigate the role of the biphenyl substituent positioning on the antimicrobial activity and membrane selectivity of lead compound 1, a systematic investigation was conducted wherein all biphenyl positional isomers of compound 1 (i.e., compounds 2–9, Figure 2) were synthesized and subjected to in vitro bacterial and fungal growth inhibition assays, in conjunction with mammalian cytotoxicity and hemolysis assays.
Figure 2.

Positional isomers of the lead biphenyl amphiphile 1 synthesized for this study.
To access the nine isomers in an efficient manner, a divergent pathway was utilized which allowed for construction of the requisite peptidomimetic chain followed by late-stage attachment of the various disubstituted biphenyl cores by Cu-catalyzed azide–alkyne cycloaddition (CuAAC). The required biphenyl cores were accessed via Suzuki cross-coupling of the corresponding iodophenols and phenol boronic acid pinacol esters—with the isopentyl substituent attached to one of the two aryl fragments prior to coupling to allow for selective placement. Alkylation of the phenol boronic esters m-10 and p-10 with 1-bromo-3-methylbutane proceeded smoothly with K2CO3 and catalytic (n-Bu)4NI in CH3CN at 80 °C, furnishing the aromatic precursor fragments m-11 and p-11 in excellent yields (Scheme 1).
Scheme 1. Synthesis of Aromatic Precursors m-11, p-11, and 13.

Alkylation of o-10 would not proceed under these conditions (due to suspected intramolecular hydrogen-bonding)—while alkylation of o-10 in DMF with Cs2CO3 (or NaH) provided the desired product, the purification was complicated by hydrolysis of the boronic ester during silica gel chromatography. To circumvent this issue, alkylation of o-12 was performed to give aryl iodide 13 (Scheme 1), which functioned as the 2-isopentyloxy aryl fragment for subsequent couplings.
With the requisite aryl fragments in hand, palladium-on-carbon (10% w/w – Pd/C) catalyzed Suzuki coupling31 was performed to construct monoalkylated biphenols 14a–h (Scheme 2). Boronic esters m-11 and p-11 were successfully coupled with o-, m- or p-iodophenol (i.e., o-,m- or p-12) to give the monoalkylated biphenols 14a, 14b, 14d, 14e, 14g, and 14h in good yields. In a reverse fashion, aryl iodide 13 was coupled with m- or p-phenol boronic acid pinacol ester (i.e., m- or p-10), yielding the monoalkylated biphenols 14c and 14f.
Scheme 2. Synthesis of Monoalkylated Biphenols 14a–h by Pd/C-Catalyzed Suzuki Cross-Coupling.

O-Propargylation of monoalkylated biphenols 14a–h with propargyl bromide (Scheme 3) was implemented to generate biaryl alkynes 15a–h. While some of the monoalkylated biphenol substrates reacted quite readily (e.g., 18 h reflux for compound 15h), most analogues required multiple days at reflux with increased equivalents of propargyl bromide to ensure reaction completion.
Scheme 3. Synthesis of Biphenyl Alkynes 15a–h via O-Propargylation of Monoalkylated Biphenols 14a–h.

The known β-azido-amine 17(26) was synthesized from the N-protected lysinol derivative 16(26) utilizing a modified two-step procedure (Scheme 4). The required 1,2,3-triazole acid compound 19 was made by a CuAAC reaction between 3-cyclohexyl-1-propyne and the known α-azido acid 18.25 Construction of the peptidomimetic chain fragment 20 (containing a terminal azide handle for late-stage divergence) was accomplished by EDCI/HOBt promoted peptide coupling of acid 19 and β-azido-amine 17 (Scheme 4).
Scheme 4. Synthesis of Key Peptide Fragment 20.

The final positional isomers 2–9 were then constructed from the corresponding biaryl alkynes 15a–h and the peptidomimetic chain fragment 20 via a CuAAC reaction; subsequent trifluoroacetic-acid-mediated N-deprotection and treatment with ethereal HCl gave the final isomers 2–9 as their dihydrochloride salts (Scheme 5). The purity of all final derivatives was >90% as determined by RP-HPLC analysis (Figures S61–S68, Supporting Information).
Scheme 5. Synthesis of Positional Isomers 2–9.
Positional isomers 1–9 were then subjected to two rounds of microbial growth inhibition screening. Preliminary testing was performed in-house against pathogenic Gram-positive bacteria and E. coli (Table 1), while secondary screening was performed by the Community for Open Antimicrobial Drug Discovery32 against pathogenic Gram-negative bacteria, fungi, and MRSA (Table 2). Preliminary screening revealed potent antibacterial efficacy against S. aureus, E. faecalis, and S. pneumoniae; isomers 1–9 generally exhibited MIC values between 2–4 μg/mL for these species, including methicillin-resistant S. aureus (MRSA) and vancomycin intermediate susceptibility S. aureus (VISA) – Table 1. Moderate antibacterial efficacy was observed for all isomers against vancomycin-resistant E. faecalis (VRE) and C. difficile, with most MIC values ranging from 8–16 μg/mL. Furthermore, secondary screening revealed extremely potent MIC values (≤0.25 μg/mL) for all isomers against a different strain of MRSA (ATCC 43300) – Table 2. Compound 2 (2,3′-isomer) consistently exhibited the lowest MIC values against Gram-positive bacteria (where discernible), whereas compounds 8 and 9 displayed the highest MIC values against S. pneumoniae/C. difficile (compound 8 – 3,4′-isomer) and E. faecalis (compound 9 – 4,4′-isomer). These results support the notion of an optimal biaryl positioning for the key hydrophilic (cationic peptide chain) and hydrophobic (isopentyl moiety) biaryl substituents because compounds with an increased distance between these two substituents (e.g., compounds 8 and 9) exhibited slightly diminished antibacterial efficacy against the Gram-positive bacterial pathogens.
Table 1. Preliminary Antibacterial Screeninga.
|
S. aureus |
E.
faecalis |
S. pneumoniae | E. coli |
C. difficile |
|||||
|---|---|---|---|---|---|---|---|---|---|
| compound | ATCC 29213 | NCTC 10442b | Mu50c | ATCC 29212 | ATCC 51299d | ATCC 49619 | ATCC 10418 | ATCC 700057 | NSW 132 (RT027)e |
| 1 | 2 | 2 | 4 | 4 | 8 | 1 | 4 | 16 | 8 |
| 2 | 2 | 2 | 2 | 2 | 8 | 1 | 2 | 16 | 16 |
| 3 | 4 | 2 | 4 | 4 | 8 | 1 | 4 | 16 | 16 |
| 4 | 4 | 4 | 2 | 4 | 8 | 2 | 4 | 16 | 8 |
| 5 | 4 | 4 | 4 | 4 | 8 | 2 | 4 | 16 | 16 |
| 6 | 4 | 8 | 2 | 4 | 8 | 2 | 32 | 16 | 16 |
| 7 | 2 | 4 | 4 | 4 | 16 | 1 | 8 | 16 | 8 |
| 8 | 4 | 4 | 4 | 4 | 16 | 4 | 16 | 32 | 16 |
| 9 | 4 | 4 | 4 | 8 | 16 | 2 | 32 | 16 | 8 |
| vancomycin | 1 | 1 | 8 | 2 | >8 | 0.5 | >8 | 1 | 0.5 |
| colistin | - | - | - | - | - | - | 1 | - | - |
Values are reported as MIC values in μg/mL.
Methicillin-resistant S. aureus (MRSA).
Vancomycin intermediate susceptibility S. aureus (VISA).
Vancomycin-resistant E. faecalis (VRE).
C. difficile PCR Ribotype 027 (RT027).
Table 2. Secondary Antimicrobial Screeninga (Bacteria and Fungi).
| S. aureus | P. aeruginosa | K. pneumoniae | A. baumannii | E. coli | C. albicans | C. neoformans var. grubii | |
|---|---|---|---|---|---|---|---|
| compound | ATCC 43300b | ATCC 27853 | ATCC 700603c | ATCC 19606 | ATCC 25922 | ATCC 90028 | ATCC 208821 |
| 1 | ≤0.25 | 16 | 32 | 16 | 8 | 32 | >32 |
| 2 | ≤0.25 | 8 | >32 | 4 | 8 | 32 | >32 |
| 3 | ≤0.25 | 8 | 32 | 4 | 4 | >32 | >32 |
| 4 | ≤0.25 | 2 | >32 | 16 | 2 | 16 | >32 |
| 5 | ≤0.25 | 32 | >32 | 32 | 32 | 8 | >32 |
| 6 | ≤0.25 | 16 | 32 | 32 | 8 | 8 | >32 |
| 7 | ≤0.25 | 8 | 32 | 4 | 8 | 16 | >32 |
| 8 | ≤0.25 | 32 | >32 | 32 | 32 | 16 | >32 |
| 9 | ≤0.25 | 8 | 32 | 32 | 8 | 1 | >32 |
| vancomycin | 1 | - | - | - | - | - | - |
| colistin | - | 0.25 | 0.25 | 0.25 | 0.125 | - | - |
| fluconazole | - | - | - | - | - | 0.125 | 8 |
| amphotericin B | - | - | - | - | - | 1.56 | 1.56 |
Testing performed by the Community for Open Antimicrobial Drug Discovery (CO-ADD) – values are reported as MIC values in μg/mL.
Methicillin-resistant S. aureus (MRSA).
Multidrug-resistant K. pneumoniae.
Screening against the more resilient Gram-negative bacteria produced increased variation in the observed MIC values—a substantial increase in the Gram-negative antibacterial efficacy was observed for compounds 3 and 4 (relative to lead compound 1, see Table 2). Compound 4 (3,2′-isomer) exhibited the strongest efficacy against P. aeruginosa and E. coli (MIC = 2 μg/mL) among the tested isomers, representing a 4- to 8-fold increase in antibacterial activity. Furthermore, a 4-fold increase in antibacterial efficacy against A. baumannii was observed for compounds 2, 3, and 7, all exhibiting potent MIC values of 4 μg/mL. The multidrug-resistant K. pneumoniae was the most resilient Gram-negative pathogen, requiring concentrations of 32 μg/mL (or more) for each isomer to inhibit bacterial growth.
While significant increases in antibacterial efficacy were observed against most of the tested Gram-negative species, no one isomer exhibited increased potency for all strains. The growth inhibition results show that the structural arrangement of these biphenyl amphiphiles has a substantial effect on the antimicrobial potency. Additionally, these results highlight the strain-specific efficacy observed among amphiphilic, membrane-disrupting antimicrobials in general, particularly against Gram-negative bacteria due to their protective outer-membrane.13
Antimicrobial efficacy was observed for the yeast C. albicans; interestingly, compound 9 displayed the strongest growth inhibition (MIC = 1 μg/mL) by far, with isomers 1, 2, and 3 displaying substantially weaker efficacy (MIC ≥ 32 μg/mL) – Table 2. Despite the observed potent antifungal activity for compound 9 (4,4′-isomer) against C. albicans, all isomers were inactive at the tested concentrations against the pathogenic yeast C. neoformans var. grubii – compound 9 was therefore found to act as a potential selective antifungal for C. albicans. The findings further illustrate how the structural arrangement of an amphiphilic compound can have a significant impact on its antimicrobial efficacy and spectrum of activity.
Positional isomers 1–9 were analyzed for mammalian toxicity by conducting a cytotoxicity assay (human embryonic kidney cells) and hemolysis assays (both human and sheep red blood cells) – see Table 3. Strong hemolytic activity against human erythrocytes was observed for isomer 9 (average HC50 = 10.5 μg/mL), while isomers 2 and 3 exhibited reduced, albeit still significant, hemolytic activity (HC50 > 32 μg/mL with maximum hemolysis below 65% at the tested concentrations). The remaining isomers all exhibited significant hemolytic activity against human erythrocytes (Table 3) – while HC50 values could not be determined for most isomers (i.e., failed curve-fitting due to single data point activity), the maximal percentage of hemolysis observed (i.e., the DMax value) indicates substantial hemolytic activity. Sheep erythrocyte hemolysis assays were conducted at compound testing concentrations of 5 and 50 μg/mL (Table 3); however, minimal hemolysis was observed at the lower testing concentration (5 μg/mL), and all compounds (except isomer 4) displayed >50% hemolysis when tested at 50 μg/mL. Compound 4 exhibited 38% hemolysis of sheep erythrocytes at 50 μg/mL, revealing reduced but significant hemolysis. Interestingly, the data between the human and sheep hemolysis assays did not correlate exactly; for example, compound 9 was clearly the most potent hemolytic agent for human erythrocytes, whereas compound 5 was the most potent hemolytic agent against sheep erythrocytes (Table 3). Previous studies25 on compound 1 and related analogues have shown that the balance between hydrophobic and hydrophilic structural residues is highly crucial for inducing bacterial/mammalian membrane selectivity and thus reducing hemolytic activity.
Table 3. Cytotoxicity and Hemolysis Assay Data.
| cytotoxicitya (HEK-293)b |
hemolysisa (human erythrocytes) |
hemolysis (sheep erythrocytes) |
||||||
|---|---|---|---|---|---|---|---|---|
| CC50c (DMax)d |
HC50c (DMax)d |
5 μg/mLe |
50 μg/mLe |
|||||
| compound | i | ii | i | ii | mean | SD | mean | SD |
| 1 | 24.4 (80) | >32 (14) | >32 (63) | >32 (56) | 2.1% | 0.1% | 66.6% | 10.6% |
| 2 | >32 (31) | >32 (28) | >32 (59) | >32 (38) | 2.6% | 1.0% | 72.1% | 11.2% |
| 3 | >32 (18) | >32 (13) | >32 (63) | >32 (41) | 2.7% | 0.4% | 63.0% | 8.3% |
| 4 | >32 (38) | >32 (36) | >32 (101) | >32 (81) | 1.4% | 0.6% | 38.2% | 7.6% |
| 5 | 28.1 (59) | 29.4 (55) | >32 (109) | >32 (94) | 3.7% | 0.4% | 76.7% | 9.2% |
| 6 | >32 (30) | >32 (18) | >32 (107) | >32 (102) | 2.3% | 0.3% | 66.5% | 11.3% |
| 7 | >32 (42) | >32 (40) | >32 (103) | 21.1 (82) | 2.0% | 0.6% | 66.4% | 9.0% |
| 8 | >32 (43) | >32 (43) | >32 (108) | >32 (88) | 2.1% | 0.4% | 40.8% | 7.0% |
| 9 | >32 (41) | >32 (25) | 9.8 (94) | 11.1 (111) | 1.4% | 0.1% | 57.2% | 11.6% |
| tamoxifen | 9.0 (SD = 2.2) | - | - | - | - | - | - | |
| melittin | - | - | 8.5 (SD = 2.5) | - | - | - | - | |
Testing performed by the Community for Open Antimicrobial Drug Discovery (CO-ADD).
Human embryonic kidney cells – (ATCC CRL-1573).
Values represent two assay replicates (i and (ii) and are reported in μg/mL. Any values with >32 μg/mL represent an inactive compound or partially active compound (lower DMax value) or an active compound where curve-fitting failed due to activity at only the highest tested concentration (higher DMax value).
DMax = maximum percentage inhibition response observed for all tested concentrations (i.e., ≤ 32 μg/mL).
Compound testing concentration for sheep erythrocyte hemolysis assays (performed in triplicate) – assay values represent the mean percentage hemolysis and standard deviation (SD) for each testing concentration.
Cytotoxicity assays revealed substantially reduced mammalian cytotoxicity for isomers 2–4 and 6–9; these compounds exhibited CC50 values >32 μg/mL with maximum percentage inhibition values below 50% (Table 3). For example, isomer 3 exhibited a maximum cytotoxic growth inhibition of 13% and 18% in two separate replicate assays, indicating a CC50 value much higher than the 32 μg/mL testing limit. While the hemolytic activity of isomers 1–9 precludes these compounds from being used as systemic antimicrobials, they demonstrate promise as potential therapeutics for topical skin infections and other nonsystemic applications because of their reduced cytotoxicity and potent antimicrobial efficacy against both Gram-positive and Gram-negative bacteria. Furthermore, optimization of the hemolytic/cytotoxic activity has not yet been conducted for this class of SMAPs; as such, these compounds serve as a potential lead for the development of unique, membrane-active antimicrobial chemotherapeutics with increased membrane selectivity (i.e., therapeutic safety).
Importantly, substantial differences in antimicrobial efficacy and mammalian cytotoxicity were observed for the various positional isomers of lead compound 1. Compound 4 displayed significant increases (4–8×) in activity against the pathogenic P. aeruginosa and E. coli species, while similar potency increases (4×) were seen for positional isomers 2, 3, and 7 against A. baumannii. These findings highlight the importance of structure and substituent arrangement for antimicrobial amphiphiles, that is, small changes in substituent orientation and positioning can lead to large changes in a compound’s antimicrobial efficacy and spectrum of activity, and this was particularly evident against the more resilient Gram-negative bacteria. Changes in the structural arrangement of these biphenyl amphiphiles had a significant effect on the compounds’ ability to inhibit the growth of Gram-negative pathogenic bacteria.
Acknowledgments
The authors are grateful for secondary antimicrobial screening and human cytotoxicity/hemolysis assays that were performed by CO-ADD (The Community for Antimicrobial Drug Discovery), funded by the Wellcome Trust (U.K.) and The University of Queensland (Australia).
Glossary
ABBREVIATIONS
- CC50
50% cytotoxic concentration
- CuAAC
Cu-catalyzed azide alkyne cycloaddition
- EDCI
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- HC50
50% hemolytic concentration
- HOBt
1-hydroxybenzotriazole
- MIC
minimum inhibitory concentration
- MRSA
methicillin-resistant Staphylococcus aureus
- Pd/C
palladium-on-carbon (10% w/w)
- SMAP
small-molecule antimicrobial peptidomimetic
- RP-HPLC
reverse-phase high-performance liquid chromatography
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00611.
General synthetic information and procedures (S2 and S3); synthesis and characterization of all intermediates (S5) and final compounds 2–9 (S17); experimental procedures for microbial growth inhibition, cytotoxicity and hemolysis assays (S25); 1H and 13C NMR spectra of all intermediates (S28) and final compounds 2–9 (S49); HPLC chromatograms for final compounds 2–9 (S58) (PDF)
Author Contributions
A.J.T., P.A.K., and S.G.P conceived the study design. A.J.T. managed the project, synthesized and characterized all compounds, interpreted the biological data and wrote the manuscript. P.P. performed all in-house biological assays (growth inhibition and hemolysis). P.A.K. and S.G.P oversaw the project and participated in helpful discussions regarding the project implementation and manuscript revisions. T.V.R. provided useful discussions and insight regarding the microbiological testing. All authors have given approval to the final version of the manuscript.
The authors thank the Australian National Health and Medical Research Council (NHMRC) for financial support (Grant No. APP1124032).
The authors declare no competing financial interest.
Supplementary Material
References
- World Health Organization . Antibiotic Resistance Fact Sheet. 2020. http://www.who.int/mediacentre/factsheets/antibiotic-resistance/en/ (accessed Oct. 2, 2020).
- Centers for Disease Control and Prevention . Antibiotic Resistance Threats in the United States 2019. 2019. https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed Oct. 2, 2020).
- Aslam B.; Wang W.; Arshad M. I.; Khurshid M.; Muzammil S.; Rasool M. H.; Nisar M. A.; Alvi R. F.; Aslam M. A.; Qamar M. U.; et al. Antibiotic resistance: a rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. 10.2147/IDR.S173867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens E.; Demain A. L. The antibiotic resistance crisis, with a focus on the United States. J. Antibiot. 2017, 70 (5), 520–526. 10.1038/ja.2017.30. [DOI] [PubMed] [Google Scholar]
- World Health Organization . Global Action Plan on Antimicrobial Resistance. 2015. https://www.who.int/publications/i/item/global-action-plan-on-antimicrobial-resistance (accessed Oct. 2, 2020). [DOI] [PubMed]
- Centers for Disease Control and Prevention . Antibiotic Resistance (AR) Solutions Initiative. 2020. https://www.cdc.gov/drugresistance/solutions-initiative/index.html (accessed Oct. 2, 2020).
- Organization for Economic Cooperation and Development (OECD) . Stemming the Superbug Tide - Policy Brief. 2018. https://www.oecd.org/health/health-systems/Stemming-the-Superbug-Tide-Policy-Brief-2018.pdf (accessed Sept. 25, 2020).
- Australian Government . Australia’s National Antimicrobial Resistance Strategy - 2020 and Beyond. 2020. https://www.amr.gov.au/resources/australias-national-antimicrobial-resistance-strategy-2020-and-beyond (accessed Oct. 2, 2020).
- World Health Organization . Lack of new antibiotics threatens global efforts to contain drug-resistant infections. 2020. https://www.who.int/news-room/detail/17-01-2020-lack-of-new-antibiotics-threatens-global-efforts-to-contain-drug-resistant-infections (accessed Oct. 2, 2020).
- World Health Organization . 2019 - Antibacterial Agents in Clinical Development. 2019. https://apps.who.int/iris/bitstream/handle/10665/330420/9789240000193-eng.pdf (accessed Oct. 2, 2020).
- Ghosh C.; Haldar J. Membrane-Active Small Molecules: Designs Inspired by Antimicrobial Peptides. ChemMedChem 2015, 10 (10), 1606–1624. 10.1002/cmdc.201500299. [DOI] [PubMed] [Google Scholar]
- Kuppusamy R.; Willcox M.; Black D. S.; Kumar N. Short Cationic Peptidomimetic Antimicrobials. Antibiotics (Basel, Switz.) 2019, 8 (2), 44. 10.3390/antibiotics8020044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Koh J.-J.; Liu S.; Lakshminarayanan R.; Verma C. S.; Beuerman R. W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73. 10.3389/fnins.2017.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Kizhakkedathu J. N.; Straus S. K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8 (1), 4. 10.3390/biom8010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojsoska B.; Jenssen H. Peptides and Peptidomimetics for Antimicrobial Drug Design. Pharmaceuticals 2015, 8 (3), 366–415. 10.3390/ph8030366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira V.; Feio M. J.; Bastos M. Role of lipids in the interaction of antimicrobial peptides with membranes. Prog. Lipid Res. 2012, 51 (2), 149–177. 10.1016/j.plipres.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Wimley W. C. Describing the Mechanism of Antimicrobial Peptide Action with the Interfacial Activity Model. ACS Chem. Biol. 2010, 5 (10), 905–917. 10.1021/cb1001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Koh J.-J.; Aung T. T.; Lim F.; Li J.; Zou H.; Wang L.; Lakshminarayanan R.; Verma C.; Wang Y.; et al. Symmetrically Substituted Xanthone Amphiphiles Combat Gram-Positive Bacterial Resistance with Enhanced Membrane Selectivity. J. Med. Chem. 2017, 60 (4), 1362–1378. 10.1021/acs.jmedchem.6b01403. [DOI] [PubMed] [Google Scholar]
- Konai M. M.; Samaddar S.; Bocchinfuso G.; Santucci V.; Stella L.; Haldar J. Selectively targeting bacteria by tuning the molecular design of membrane-active peptidomimetic amphiphiles. Chem. Commun. 2018, 54 (39), 4943–4946. 10.1039/C8CC01926F. [DOI] [PubMed] [Google Scholar]
- Hoque J.; Konai M. M.; Sequeira S. S.; Samaddar S.; Haldar J. Antibacterial and Antibiofilm Activity of Cationic Small Molecules with Spatial Positioning of Hydrophobicity: An in Vitro and in Vivo Evaluation. J. Med. Chem. 2016, 59 (23), 10750–10762. 10.1021/acs.jmedchem.6b01435. [DOI] [PubMed] [Google Scholar]
- Thota C. K.; Berger A. A.; Harms B.; Seidel M.; Böttcher C.; von Berlepsch H.; Xie C.; Süssmuth R.; Roth C.; Koksch B. Short self-assembling cationic antimicrobial peptide mimetics based on a 3,5-diaminobenzoic acid scaffold. Pept. Sci. 2020, 112 (1), e24130 10.1002/pep2.24130. [DOI] [Google Scholar]
- Koh J.-J.; Lin S.; Bai Y.; Sin W. W. L.; Aung T. T.; Li J.; Chandra V.; Pervushin K.; Beuerman R. W.; Liu S. Antimicrobial activity profiles of Amphiphilic Xanthone derivatives are a function of their molecular Oligomerization. Biochim. Biophys. Acta, Biomembr. 2018, 1860 (11), 2281–2298. 10.1016/j.bbamem.2018.05.006. [DOI] [PubMed] [Google Scholar]
- Hickey S. M.; Ashton T. D.; Boer G.; Bader C. A.; Thomas M.; Elliott A. G.; Schmuck C.; Yu H. Y.; Li J.; Nation R. L.; et al. Norbornane-based cationic antimicrobial peptidomimetics targeting the bacterial membrane. Eur. J. Med. Chem. 2018, 160, 9–22. 10.1016/j.ejmech.2018.09.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquette A.; Mason A. J.; Bechinger B. Aggregation and membrane permeabilizing properties of designed histidine-containing cationic linear peptide antibiotics. J. Pept. Sci. 2008, 14 (4), 488–495. 10.1002/psc.966. [DOI] [PubMed] [Google Scholar]
- Tague A. J.; Putsathit P.; Hammer K. A.; Wales S. M.; Knight D. R.; Riley T. V.; Keller P. A.; Pyne S. G. Cationic biaryl 1,2,3-triazolyl peptidomimetic amphiphiles: synthesis, antibacterial evaluation and preliminary mechanism of action studies. Eur. J. Med. Chem. 2019, 168, 386–404. 10.1016/j.ejmech.2019.02.013. [DOI] [PubMed] [Google Scholar]
- Tague A. J.; Putsathit P.; Hutton M. L.; Hammer K. A.; Wales S. M.; Knight D. R.; Riley T. V.; Lyras D.; Keller P. A.; Pyne S. G. Cationic biaryl 1,2,3-triazolyl peptidomimetic amphiphiles targeting Clostridioides (Clostridium) difficile: Synthesis, antibacterial evaluation and an in vivo C. difficile infection model. Eur. J. Med. Chem. 2019, 170, 203–224. 10.1016/j.ejmech.2019.02.068. [DOI] [PubMed] [Google Scholar]
- Wales S. M.; Hammer K. A.; King A. M.; Tague A. J.; Lyras D.; Riley T. V.; Keller P. A.; Pyne S. G. Binaphthyl-1,2,3-triazole peptidomimetics with activity against Clostridium difficile and other pathogenic bacteria. Org. Biomol. Chem. 2015, 13 (20), 5743–5756. 10.1039/C5OB00576K. [DOI] [PubMed] [Google Scholar]
- Wales S. M.; Hammer K. A.; Somphol K.; Kemker I.; Schroder D. C.; Tague A. J.; Brkic Z.; King A. M.; Lyras D.; Riley T. V.; et al. Synthesis and antimicrobial activity of binaphthyl-based, functionalized oxazole and thiazole peptidomimetics. Org. Biomol. Chem. 2015, 13 (44), 10813–10824. 10.1039/C5OB01638J. [DOI] [PubMed] [Google Scholar]
- Isaksson J.; Brandsdal B. O.; Engqvist M.; Flaten G. E.; Svendsen J. S. M.; Stensen W. A Synthetic Antimicrobial Peptidomimetic (LTX 109): Stereochemical Impact on Membrane Disruption. J. Med. Chem. 2011, 54 (16), 5786–5795. 10.1021/jm200450h. [DOI] [PubMed] [Google Scholar]
- Hickey S. M.; Ashton T. D.; Khosa S. K.; Robson R. N.; White J. M.; Li J.; Nation R. L.; Yu H. Y.; Elliott A. G.; Butler M. S.; et al. Synthesis and evaluation of cationic norbornanes as peptidomimetic antibacterial agents. Org. Biomol. Chem. 2015, 13 (22), 6225–6241. 10.1039/C5OB00621J. [DOI] [PubMed] [Google Scholar]
- Schmidt B.; Riemer M. Suzuki–Miyaura Coupling of Halophenols and Phenol Boronic Acids: Systematic Investigation of Positional Isomer Effects and Conclusions for the Synthesis of Phytoalexins from Pyrinae. J. Org. Chem. 2014, 79 (9), 4104–4118. 10.1021/jo500675a. [DOI] [PubMed] [Google Scholar]
- Blaskovich M. A. T.; Zuegg J.; Elliott A. G.; Cooper M. A. Helping Chemists Discover New Antibiotics. ACS Infect. Dis. 2015, 1 (7), 285–287. 10.1021/acsinfecdis.5b00044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.