

Abstract
The myocardium is metabolically flexible, however, impaired flexibility is associated with cardiac dysfunction in conditions including diabetes and heart failure. The mitochondrial pyruvate carrier (MPC) complex, composed of MPC1 and MPC2, is required for pyruvate import into the mitochondria. Here we show that MPC1 and MPC2 expression is downregulated in failing human and mouse hearts. Mice with cardiac-specific deletion of MPC2 (CS-MPC2−/−) exhibited normal cardiac size and function at 6-weeks old, but progressively developed cardiac dilation and contractile dysfunction, which was completely reversed by a high fat, low carbohydrate “ketogenic diet.” Diets with higher fat content, but enough carbohydrate to limit ketosis, also improved heart failure, while direct ketone body provisioning provided only minor improvements in cardiac remodeling in CS-MPC2−/− mice. An acute fast also improved cardiac remodeling. Together, our study reveals a critical role for mitochondrial pyruvate utilization in cardiac function, and highlights the potential of dietary interventions to enhance cardiac fat metabolism to prevent or reverse cardiac dysfunction and remodeling in the setting of MPC-deficiency.
The myocardium requires vast amounts of chemical energy stored in nutrients to fuel cardiac contraction. To maintain this high metabolic capacity, the heart is extremely flexible and can adapt to altered metabolic fuel supplies during diverse developmental, nutritional, or physiologic conditions. Cardiac mitochondria are capable of oxidizing fatty acids, pyruvate (derived from either glucose or lactate), ketone bodies, or amino acids when needed. Whereas fatty acids are considered a predominant fuel source for normal adult hearts1,2, several physiological conditions can increase the importance of other substrates for cardiac metabolism. For example, the mammalian fetal heart relies mostly on anaerobic glycolysis until oxygen is abundant and the oxidative capacity of the heart matures ~15–20 days postnatally in rodents3–5. In the adult heart, myocardial lactate6 and ketone body7–9 extraction and oxidation can be greatly enhanced depending upon the physiological condition.
A hallmark of heart failure in mice and humans is a metabolic switch away from mitochondrial oxidative metabolism10–13. Fatty acid oxidation (FAO) is reduced in the failing heart as a result of deactivating the expression of a wide transcriptional program for FAO enzymes and transporters10,14–16 and other mitochondrial metabolic enzymes10,12,13. The deactivation of mitochondrial metabolism in pathological heart remodeling leads to an increased reliance on glycolysis17, but decreased glucose/pyruvate oxidation18 results in a mismatch that may cause energetic defects, altered redox status, or accumulation of metabolic intermediates with signaling and physiological effects.
Many aspects of cardiac pyruvate/lactate metabolism in heart remain to be fully understood. For pyruvate to enter the mitochondrial matrix and be oxidized, it must be transported across the inner mitochondrial membrane by the mitochondrial pyruvate carrier (MPC); a hetero-oligomer composed of MPC1 and MPC2 proteins19,20. Pyruvate oxidation occurs in the mitochondrial pyruvate dehydrogenase (PDH) complex and previous studies have shown that impaired cardiac PDH activity in mouse heart limits metabolic flexibility21–24. However, PDH deactivation does not cause overt cardiac remodeling or dysfunction in the absence of further cardiac stress21–24. Another metabolic fate for pyruvate is carboxylation which is an anaplerotic reaction capable of replenishing TCA cycle intermediates. In cardiac myocytes, pyruvate carboxylation can occur in the cytosol via malic enzyme 1, or in the mitochondrial matrix via malic enzymes 2 or 3, or pyruvate carboxylase. Because MPC deletion could affect both pyruvate carboxylation and oxidation, we hypothesized that impaired MPC activity would have a greater impact on pyruvate metabolism and regulation of cardiac metabolic flexibility compared to modulating PDH activity alone.
In the present study, we demonstrate that MPC expression is decreased in failing human and mouse hearts, and that genetic deletion of the MPC in mice leads to cardiac remodeling and dysfunction. Interestingly, this heart failure can be prevented or even reversed by providing a high-fat, low carbohydrate “ketogenic” diet. A 24 h fast in mice also provided significant improvement in heart remodeling. Diets with higher fat content, but enough carbohydrates to limit ketosis also significantly improved heart failure in mice lacking cardiac MPC expression. Gene expression, metabolomic analyses, and cardiac respiration analyses all suggest improved myocardial fat metabolism, rather than increased ketone body metabolism, as the mechanism driving these improvements in heart failure. These results demonstrate that mitochondrial pyruvate carrier deficiency induces cardiac dysfunction in mice, and that dietary manipulations can prevent or reverse cardiomyopathy in this model.
RESULTS
Mitochondrial Pyruvate Carrier Is Downregulated in Human and Mouse Heart Failure
We first examined the expression of MPC proteins in heart samples of human patients obtained at the time of left-ventricular assist device (LVAD) implantation or cardiac transplantation. We compared these samples to cardiac donor tissue from hearts that were non-failing but deemed unsuitable for transplant. As expected, qRT-PCR analyses indicated that failing human hearts exhibited increased expression of natriuretic peptides and fibrotic collagens compared to non-failing controls (Extended Data Fig. 1a) as well as decreased expression of PPARGC1A, PPARA and mitochondrial FAO enzymes (Extended Data Fig. 1b). Failing hearts also expressed lower levels of MPC1 and MPC2 compared to non-failing controls at both the mRNA and protein level (Fig. 1a–c). Other mitochondrial proteins such as VDAC1 and the complex I subunit NDUFB8 and complex II subunit SDHB were also reduced in the failing hearts (Fig. 1c), further supporting the notion that heart failure is associated with a general loss of mitochondrial abundance and oxidative function10–16. However, the complex III subunit UQCRC2 and complex 5 subunit ATP5A were not downregulated in the failing hearts (Fig. 1c), thus the MPC proteins trended to be, or were significantly reduced depending on how expression is normalized to other mitochondrial or cytosolic proteins (Extended Data Fig. 1c–d). Interestingly, failing hearts showed improvements in metabolic gene expression after LVAD placement, but natriuretic peptides and collagens were not significantly improved (Fig. 1a–b and Extended Data Fig. 1a–b). Mpc1 and Mpc2 were also reduced in WT mouse hearts subjected to the heart failure model of transverse aortic constriction and myocardial infarction (TAC+MI) compared to sham (Extended Data Fig. 1e). Thus, consistent with recent data25, human heart failure is associated with decreased cardiac MPC expression. While this reduced expression could be due to the general downregulation of mitochondrial oxidative pathways, we suspect that MPC loss might contribute to the reduced glucose/pyruvate oxidation that occurs in heart failure despite elevated rates of glycolysis17,18.
Fig. 1: MPCs downregulated in human heart failure, deletion of cardiac MPC2 results in TCA cycle dysfunction.
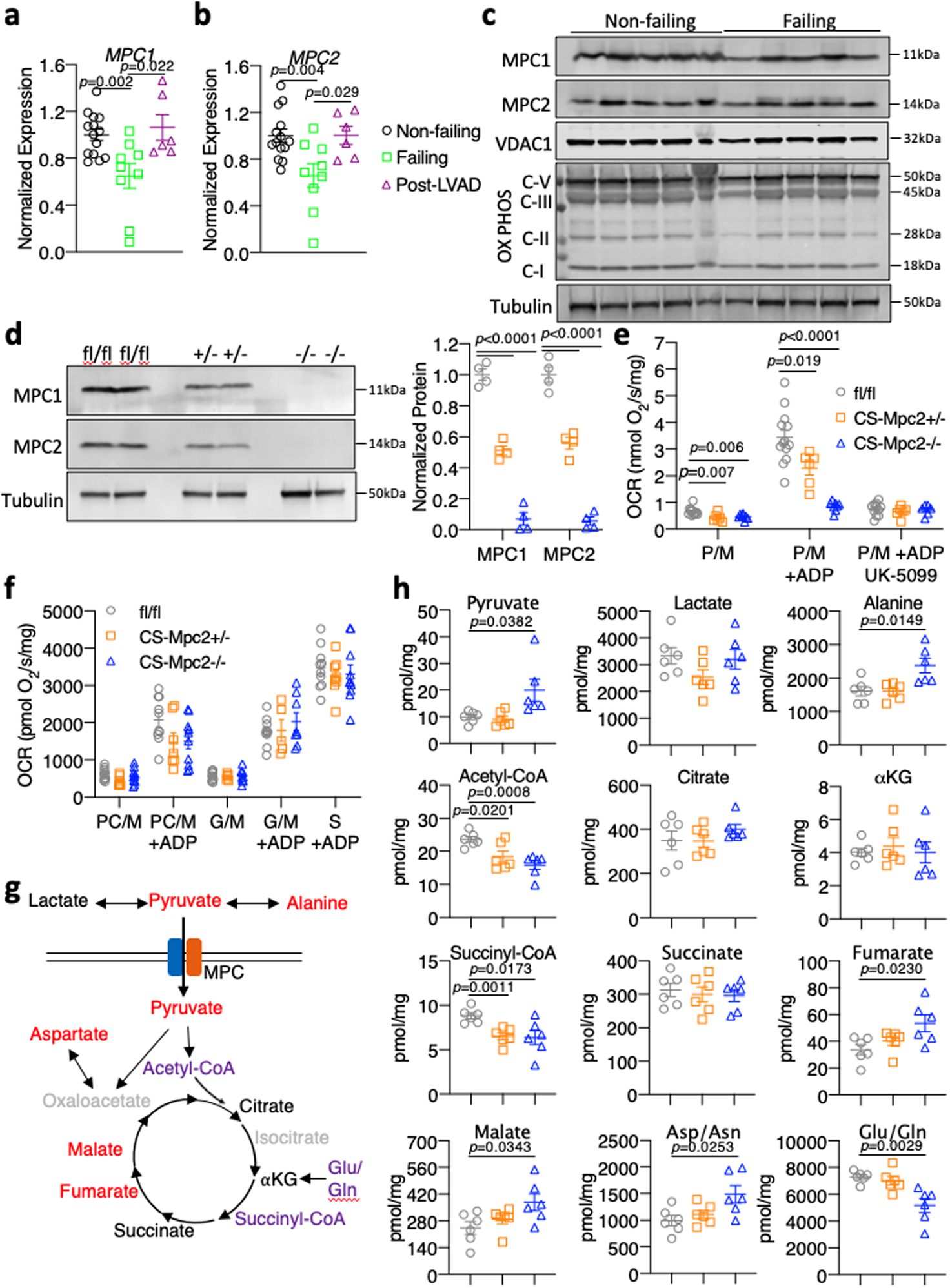
a-b, Gene expression measured by qRT-PCR for MPC1 and MPC2 normalized to RPLP0 from human hearts of non-failing, failing, and failing hearts Post-left ventricular assist device (LVAD) implant (n=14, 9, and 6 for Non-failing, Failing, and Post-LVAD, respectively). c, Western blot images for MPC1, MPC2, VDAC1, OX PHOS subunits, and αTubulin in non-failing and failing human heart tissue (n=5). d, Representative western blots of MPC1, MPC2, and αTubulin of mouse heart tissue and densitometry quantification (n=4). e, Oxygen consumption rates (OCR) stimulated by pyruvate/malate (P/M) of isolated cardiac mitochondria before and after addition of ADP and 5μM of the MPC-inhibitor UK-5099 (n=13, 6, and 8 for fl/fl, CS-Mpc2+/−, and CS-Mpc2−/−, respectively). f, Oxygen consumption rates stimulated by palmitoyl carnitine/malate (PC/M), glutamate/malate (G/M) or succinate (S) before or after the addition of ADP measured from isolated cardiac mitochondria (n=10, 8, and 10 for fl/fl, CS-Mpc2+/−, and CS-Mpc2−/−, respectively). g, Schematic of TCA cycle alterations measured by metabolomic analyses of heart tissue. red=increased, purple=decreased, black=unchanged (comparing fl/fl to CS-Mpc2−/−), and grey=unmeasured. h, TCA cycle intermediates (Pyruvate, Lactate, Alanine, Acetyl-CoA, Citrate, α-ketoglutarate, Succinyl-CoA, Succinate, Fumarate, Malate, Aspartate/Asparagine, and Glutamate/Glutamine) measured by mass-spectrometry from 6-week old heart tissue (n=6). Mean ± s.e.m. shown within dot plot. Each symbol represents an individual sample. Two-tailed unpaired Student’s t test.
CS-MPC2−/− Mice Display Altered Pyruvate Metabolism and TCA Cycle Defects
To determine whether this decrease in cardiac MPC expression was an adaptive process in heart failure or contributes to the cardiac remodeling and dysfunction, we generated cardiac-specific Mpc2 knockouts (CS-MPC2−/−) using our established Mpc2 floxed mouse26–28 and mice expressing Cre under the endogenous myosin light chain 2v promoter29. CS-MPC2−/− mice had complete loss of cardiac Mpc2 gene expression (Extended Data Fig. 1f). Loss of MPC2 led to destabilization of MPC1 protein as well, and neither MPC2 nor MPC1 protein was detected in CS-MPC2−/− mouse hearts (Fig. 1d). CS-MPC2−/− heart mitochondria displayed drastically reduced pyruvate stimulated oxygen consumption rates (OCR), and were resistant to inhibitory effects of the MPC inhibitor UK-5099 on respiration (Fig. 1e). Mpc2 flox heterozygotes expressing Cre (CS-MPC2+/− mice) displayed a ~50% decrease in MPC expression and pyruvate-stimulated respiration (Fig. 1d–e and Extended Data Fig. 1f). Isolated mitochondria from CS-MPC2+/− and CS-MPC2−/− hearts displayed normal OCR on palmitoylcarnitine/malate, glutamate/malate, and succinate (Fig. 1f), suggesting a specific defect in mitochondrial pyruvate metabolism. CS-MPC2−/− mice also displayed slight, but significantly elevated blood lactate levels (Extended Data Fig. 1g), consistent with the heart as an appreciable lactate-consuming organ or suggesting that the CS-MPC2−/− hearts were producing lactate.
To more thoroughly investigate how loss of MPC expression altered mitochondrial metabolism, targeted metabolomics analyses were conducted on hearts from 6-week old female mice (Fig. 1g–h and Supplemental Table 1). CS-MPC2−/− hearts contained decreased acetyl-CoA levels, and an accumulation of TCA cycle intermediates upstream of acetyl-CoA (fumarate, malate, and oxaloacetate [aspartate measured as surrogate])(Fig. 1g–h and Supplemental Table 1). Interestingly, pyruvate accumulated, but myocardial lactate concentrations were unaltered, leading to a decreased lactate/pyruvate ratio in the CS-MPC2−/− hearts (Fig 1g–h and Supplemental Table 1). The elevated blood lactate (Extended Data Fig. 1g) and unchanged myocardial lactate concentrations potentially suggests elevated lactate release from the CS-MPC2−/− hearts, or that these hearts were not efficiently using lactate from the blood. These results are similar to lactate and pyruvate concentrations of perfused rat hearts treated with the MPC inhibitor α-cyanocinnimate30. Transamination to alanine is another potential fate of pyruvate, and CS-MPC2−/− hearts displayed significantly elevated alanine concentrations (Fig. 1g–h and Supplemental Table 1). The decreased lactate/pyruvate ratio suggests a decreased cytosolic NADH/NAD+ redox ratio. However, analysis of other mitochondrial redox-linked reactions does not provide a consistent conclusion on redox status as the αketoglutarate/succinyl-CoA ratio was increased, and the succinate/fumarate ratio was decreased in CS-MPC2−/− hearts (Supplemental Table 1). Altogether, these findings suggest that loss of cardiac MPC results in defective mitochondrial pyruvate metabolism, alterations in TCA cycle flux, and potentially dysregulated redox status.
CS-MPC2−/− Mice Develop Dilated Cardiomyopathy
Hearts from 6-week old CS-MPC2−/− mice appeared normal by echocardiography, but heart weight and hypertrophic gene expression were slightly elevated in these young mice (Fig. 2a–c, Extended Data Fig. 1h–j, and Extended Data Fig. 2a–h). Cardiac enlargement and decreased contractile function was well-evident at 10-weeks and further worsened at 16-weeks of age (Fig. 2a–d and Extended Data Fig. 2a–h). Increased ventricular mass was confirmed at sacrifice (Fig. 2e–f), as was increased lung weight indicative of lung edema (Fig. 2g). CS-MPC2−/− hearts also showed dramatically altered gene expression markers of heart failure and fibrosis (Fig. 2h–i). Importantly, CS-MPC2+/− heterozygotes displayed normal cardiac size, function, and hypertrophic gene expression (Fig. 2a–i and Extended Data Fig. 2a–h), suggesting that MPC haploinsufficiency or Cre expression alone was not provoking this heart failure phenotype. WT C57BL6/J mice treated with the MPC inhibitor MSDC-0602, currently in development to treat diabetes and nonalcoholic steatohepatitis28, also did not show cardiac enlargement or cardiac hypertrophic gene expression (Extended Data Fig. 2i–j), although beneficial pharmacologic effect on fatty liver disease was observed in these mice28. Together, these results indicate that complete loss of MPC expression, but not partial loss or pharmacologic MPC inhibition, results in cardiac remodeling and dysfunction.
Fig. 2: CS-MPC2−/− mice develop dilated cardiomyopathy.
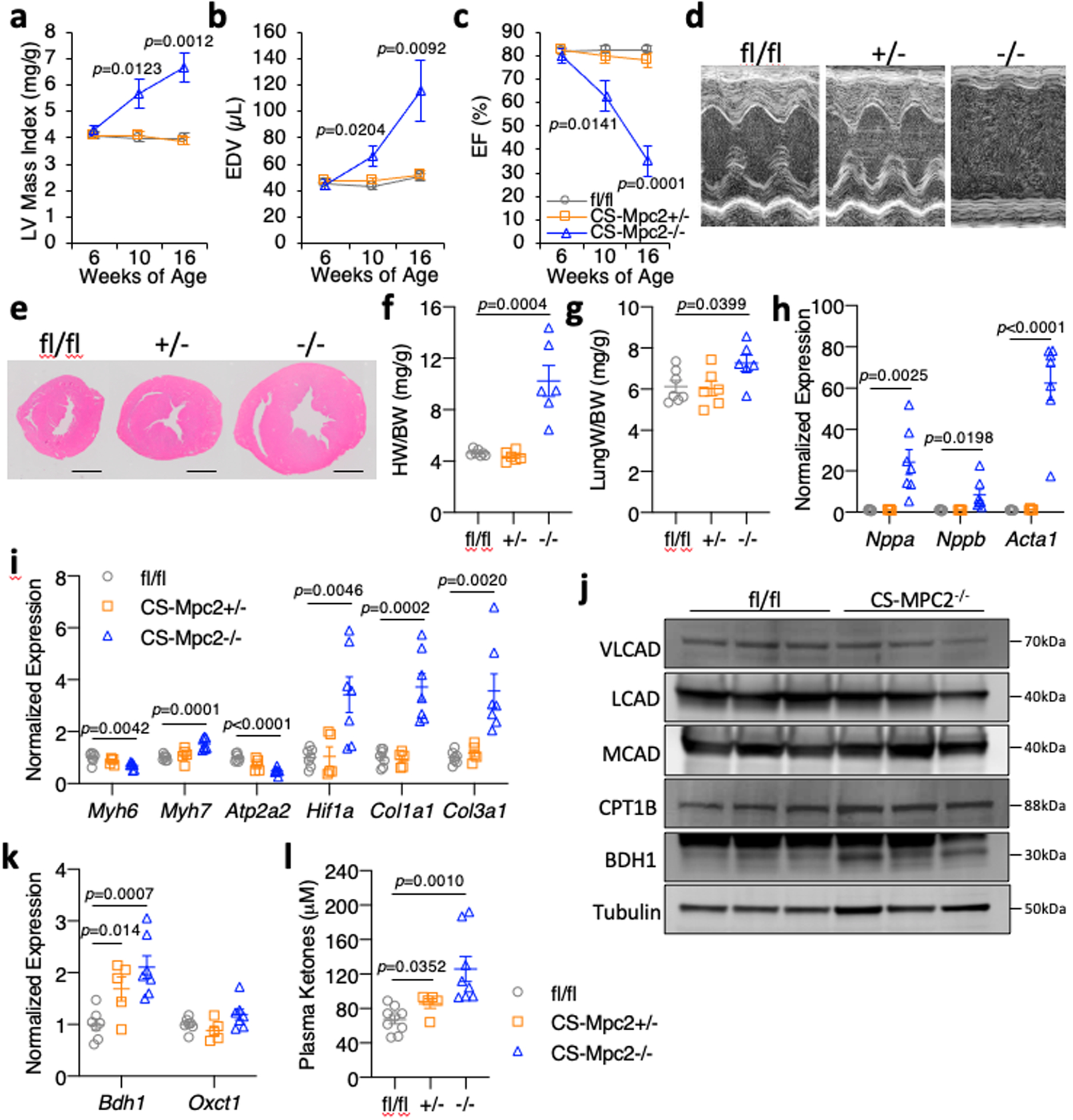
a-c, Echocardiography measures of left ventricular (LV) mass index, end-diastolic volume (EDV), and ejection fraction (EF) of mice at 6, 10, and 16-weeks of age (n=7, 10, and 9 for fl/fl, CS-Mpc2+/−, and CS-Mpc2−/−, respectively). d, Representative M-mode electrocardiogram images of 16-week old mice. e, Representative short-axis heart images stained by H&E (scale bar = 1mm). For d and e, experiments were repeated four times with small independent groups of littermate mice, with similar results obtained. f-g, Heart weight and lung weight normalized to body weight from 16-week old mice (n=7, 6, and 6 for fl/fl, CS-Mpc2+/−, and CS-Mpc2−/−, respectively). h-i, Gene expression markers of cardiac hypertrophy/failure from 16-week old mouse hearts (n=7, 5, and 7 for fl/fl, CS-Mpc2+/−, and CS-Mpc2−/−, respectively). j, Western blot images of VLCAD, LCAD, MCAD, CPT1B, BDH1, and αTubulin from whole cardiac lysates (n=3). k, Gene expression for Bdh1 and Oxct1 from 16-week old mouse hearts (n=7, 5, and 7 for fl/fl, CS-Mpc2+/−, and CS-Mpc2−/−, respectively). l, Plasma total ketone body levels from 16-week old mice (n=9, 5, and 8 for fl/fl, CS-Mpc2+/−, and CS-Mpc2−/−, respectively). Mean ± s.e.m. shown within dot plot. Each symbol represents an individual sample. Two-tailed unpaired Student’s t test.
Interestingly, other than Cpt1b, this cohort of CS-MPC2−/− hearts did not show major downregulation of FAO enzymes and transporters associated with FAO as is typical for failing hearts (Fig. 2j and Extended Data Fig. 2k). However, many other PPARα target genes were downregulated in these failing hearts (Extended Data Fig. 2l). CS-MPC2−/− hearts exhibited increased expression of BDH1 at the gene and protein level (Fig. 2j–k) and significantly elevated plasma ketone bodies were found in CS-MPC2−/− mice (Fig. 2l). Together, this elevated ketosis and increased BDH1 expression suggests increased ketone body metabolism in the failing CS-MPC2−/− hearts31,32, which was recently shown to be an adaptive and protective process in heart failure33.
High Fat, Low Carbohydrate “Ketogenic Diet” Prevents Heart Failure in CS-MPC2−/− Mice
We hypothesized that the cardiac remodeling and dysfunction in CS-MPC2−/− mice could be improved by providing nutrients in the diet that could be better used by CS-MPC2−/− hearts. To test this, fl/fl and CS-MPC2−/− mice were fed a low-carbohydrate (1.8% kcal), high-fat (93.9% kcal) “ketogenic diet” (KD) or a low-fat (LF) control diet from 6 weeks until 17 weeks of age. KD resulted in the expected increase in ketosis (Fig. 3a, Supplemental Table 2), as well as limited weight gain, decreased blood glucose, and decreased plasma insulin concentrations in both fl/fl and CS-MPC2−/− mice (Extended Data Fig. 3a–c, Supplemental Table 2). LF-fed CS-MPC2−/− mice displayed extreme cardiac enlargement and dysfunction upon echocardiography (Fig. 3b–d and Extended Data Fig. 3d–n), which was even worse when compared to chow-fed CS-MPC2−/− mice (see Fig. 2), potentially due to the increased content of refined sucrose in the LF diet. Strikingly, KD-fed CS-MPC2−/− mice displayed virtually normal cardiac size and function during echocardiography studies at 10- and 16-weeks of age (Fig. 3b–d, Extended Data Fig. 3d–n, and Supplemental Video 1). The severe cardiac dysfunction in LF-fed CS-MPC2−/− mice was associated with loss of body weight by 17 weeks of age (Extended Data Fig. 3a), which was driven by loss of adipose tissue fat mass (Extended Data Fig. 3o–s). Nearly 35% of LF-fed CS-MPC2−/− mice died prior to 17 weeks of age, but all CS-MPC2−/− mice fed KD survived (Fig. 3e). Extreme cardiac enlargement, increased lung edema, and increased cardiomyocyte cross-sectional area were observed in LF-fed CS-MPC2−/− mice, which were all completely prevented by feeding KD (Fig. 3f–i). Gene expression markers for heart failure and fibrosis, as well as trichrome fibrosis staining, were all elevated in LF-fed, and completely corrected in KD-fed, CS-MPC2−/− hearts (Fig. 3f and Fig. 3j–n). LF-fed CS-MPC2−/− hearts also displayed altered hypertrophic growth signaling by increased ERK phosphorylation, decreased AMPKα phosphorylation, increased mTOR phosphorylation, and increased S6 ribosomal protein phosphorylation (Fig. 3o), consistent with increased protein synthesis required to drive pathologic cardiac hypertrophy34. Feeding CS-MPC2−/− mice KD completely prevented this aberrant hypertrophic growth signaling (Fig. 3o). Altogether, these results show that ketogenic diet is able to completely prevent cardiac remodeling and dysfunction of CS-MPC2−/− mice.
Fig. 3: Ketogenic diet can prevent heart failure in CS-MPC2−/− mice.
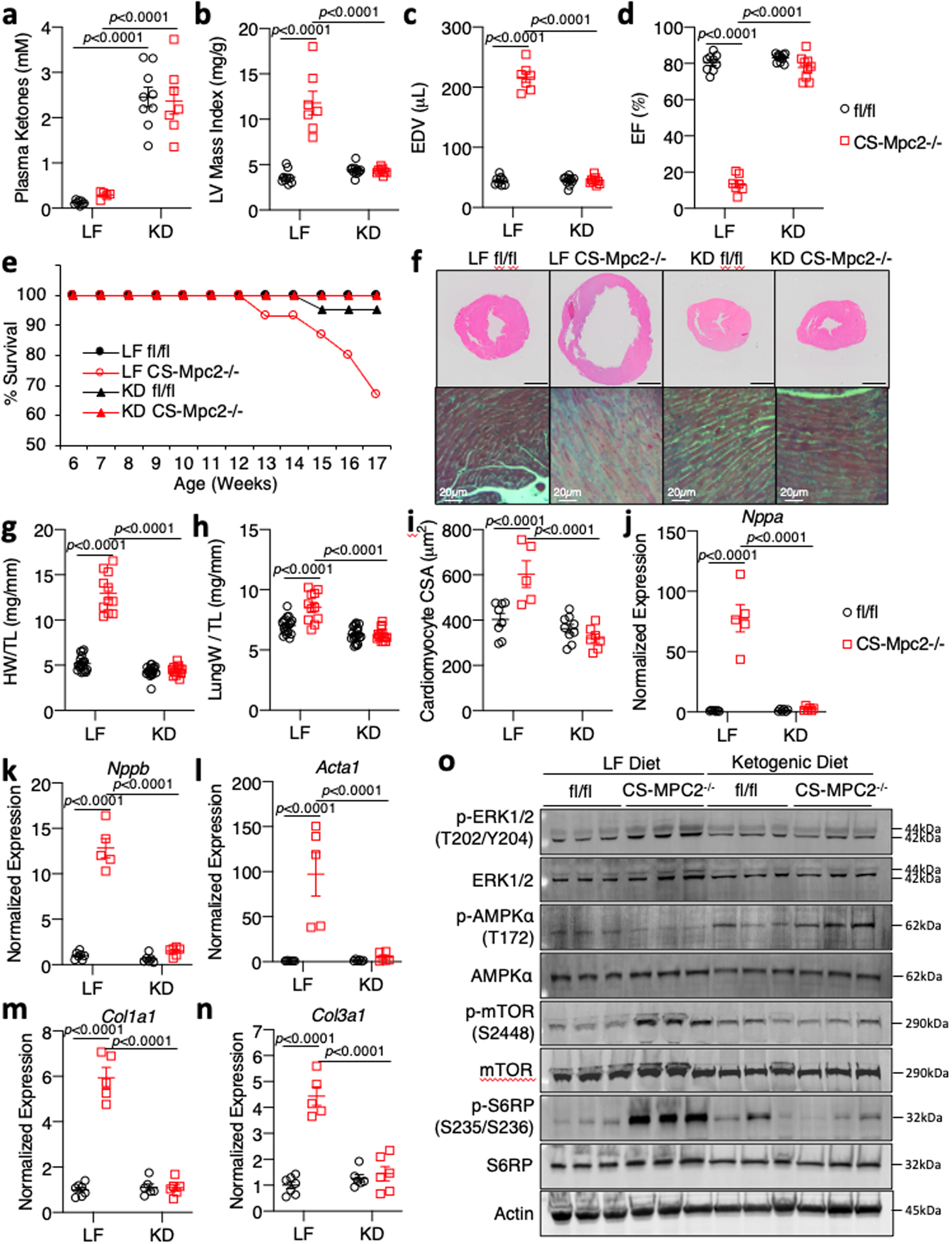
a, Plasma total ketone bodies from low fat (LF)- or ketogenic diet (KD)-fed mice (n=8, 5, 9, and 7 for fl/fl LF, CS-Mpc2−/− LF, fl/fl KD, and CS-Mpc2−/− KD, respectively). b-d, Echocardiography measures of left ventricular (LV) mass index, end-diastolic volume (EDV), and ejection fraction (EF) of LF- or KD-fed mice at 16-weeks of age (n=9, 7, 11, and 9 for fl/fl LF, CS-Mpc2−/− LF, fl/fl KD, and CS-Mpc2−/− KD, respectively). e, Survival curve of LF- or KD-fed mice (initial n=19, 15, 21, and 14 for fl/fl LF, CS-Mpc2−/− LF, fl/fl KD, and CS-Mpc2−/− KD, respectively). f, Representative short-axis H&E images and magnified trichrome stains of hearts from LF- or KD-fed 17-week old mice (black scale bar = 1mm; similar data reproduced with seven independent groups of littermate mice). g-h, Heart weight and lung weight normalized to tibia length of LF- or KD-fed 17-week old mice (n=19, 11, 20, and 14 for fl/fl LF, CS-Mpc2−/− LF, fl/fl KD, and CS-Mpc2−/− KD, respectively). i, Cardiac myocyte cross-sectional area (CSA) measured from H&E images (n=8, 5, 9, and 7 for fl/fl LF, CS-Mpc2−/− LF, fl/fl KD, and CS-Mpc2−/− KD, respectively). j-n, Gene expression markers of cardiac hypertrophy/failure and fibrosis from mouse hearts (n=7, 5, 6, and 6 for fl/fl LF, CS-Mpc2−/− LF, fl/fl KD, and CS-Mpc2−/− KD, respectively). o, Western blot images for signaling pathways associated with cardiac hypertrophic growth (PhosphoERK, Total ERK, PhosphoAMPKα, Total AMPKα, Phospho-mTOR, Total mTOR, Phospho-S6-Ribosomal Protein, Total S6-Ribosomal Protein, and β-Actin) from hearts of LF- or KD-fed mice (n=3). Mean ± s.e.m. shown within dot plot. Each symbol represents an individual sample. Two-way ANOVA with Tukey’s multiple-comparisons test.
Ketogenic Diet Downregulates Cardiac Ketone Body Oxidation and Enhances Fat Metabolism
The chow-fed CS-MPC2−/− mice showed elevated ketone bodies and increased BDH1 expression (Fig. 2j–l), consistent with recent work suggesting an increase in ketone body oxidation in failing hearts31,32. LF-fed CS-MPC2−/− mice also displayed an increase in plasma ketone bodies (Fig. 3a), and the failing hearts from these mice showed an upregulation of the ketolytic enzymes Bdh1 and Oxct1, as well as increases in C4-OH-carnitine and 3-hydroxybutyrate-CoA (Fig. 4a–f). Interestingly, hearts from both fl/fl and CS-MPC2−/− mice show decreased BDH1 and Oxct1 expression after KD-feeding (Fig. 4b–d), in agreement with a previous report demonstrating that the myocardium downregulates ketone body oxidation during ketogenic diet35. Along these lines, the levels of succinyl-CoA, succinate, and succinate/succinyl-CoA ratio all suggest increased ketolytic flux in failing LF-fed CS-MPC2−/− hearts that is reduced by KD-feeding (Fig. 4g–i and Supplemental Table 3). KD-feeding also normalized the levels of free CoA-SH, malonyl-CoA, as well as the expression of malonyl-CoA-generating enzymes Acaca and Acacb in CS-MPC2 hearts (Fig. 4j–n and Supplemental Table 3). Altogether, these results suggest that the improvements in cardiac remodeling and function from KD-feeding were not related to enhanced ketone metabolism.
Fig. 4: Ketogenic diet downregulates cardiac ketone body catabolism.
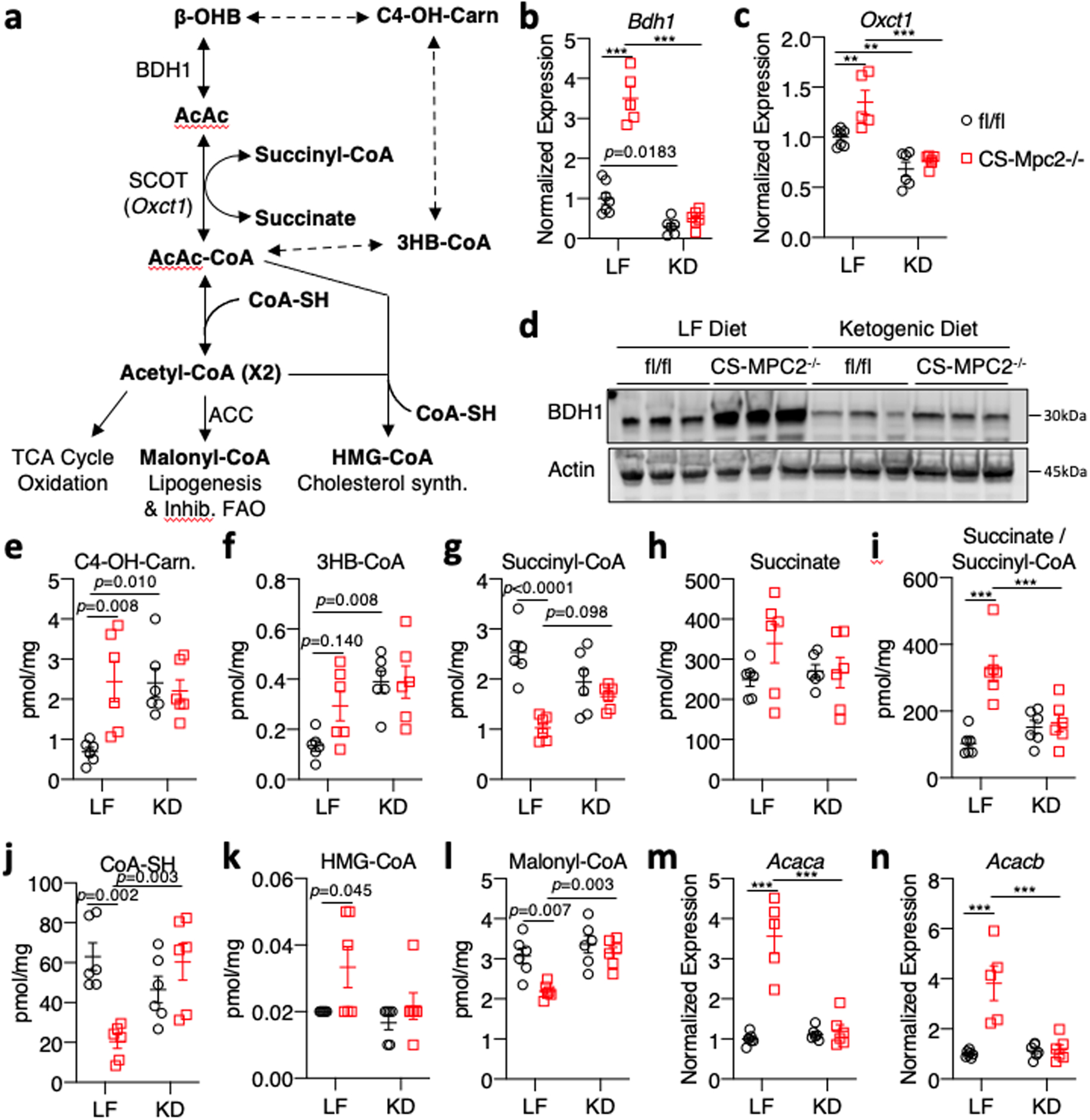
a, Schematic of oxidative and non-oxidative ketone body catabolism. b-c, Gene expression for the ketolytic enzymes Bdh1 and Oxct1 from hearts of low fat (LF)- or ketogenic diet (KD)-fed fl/fl or CS-Mpc2−/− mice (n=7, 5, 6, and 6 for fl/fl LF, CS-Mpc2−/− LF, fl/fl KD, and CS-Mpc2−/− KD, respectively)(Oxct1: p=0.0058 for LF fl/fl vs CS-Mpc2−/−, p=0.0083 for fl/fl LF vs KD). d, Western blot images of BDH1 and Actin from heart tissue of LF- or KD-fed mice (n=3). e-l, Cardiac concentrations of metabolites associated with ketone body catabolism measured in hearts from LF- or KD-fed mice (n=6). m-n, Gene expression for Acaca and Acacb normalized to Rplp0 from hearts of LF- and KD-fed mice (n=7, 5, 6, and 6 for fl/fl LF, CS-Mpc2−/− LF, fl/fl KD, and CS-Mpc2−/− KD, respectively). Mean ± s.e.m. shown within dot plot. Each symbol represents an individual sample. Two-way ANOVA with Tukey’s multiple-comparisons test. *P < 0.05, **P < 0.01, ***P < 0.001.
The failing LF-fed CS-MPC2−/− hearts displayed an accumulation of acylcarnitines and depletion of free carnitine, which was normalized by KD-feeding (Fig. 5a–d, and Supplemental Table 3). Accumulation of acylcarnitines suggests a decrease in their transport into the mitochondrial matrix and oxidation, as has been shown previously in ischemia and heart failure36,37. Indeed, failing LF-fed CS-MPC2−/− hearts displayed decreased expression of Ppargc1a, Ppara, and many of their target genes for fatty acid transport and metabolism (Fig. 5e–l). KD-feeding rescued or strongly elevated the expression of Ppargc1a, Ppara, and its gene targets related to FAO in both fl/fl and CS-MPC2−/− hearts (Fig. 5e–l). Interestingly, the Ppara target gene, Hmgcs2, which generates ketone bodies and is normally expressed almost exclusively in the liver, was strongly induced in KD-fed fl/fl and CS-MPC2−/− hearts (Fig. 5l). Cumulatively, these results suggest that KD-feeding does not enhance cardiac ketone body metabolism, but rather stimulates FAO, which may be responsible for the improved cardiac remodeling and performance.
Fig. 5: Ketogenic diet enhances cardiac fatty acid metabolism.
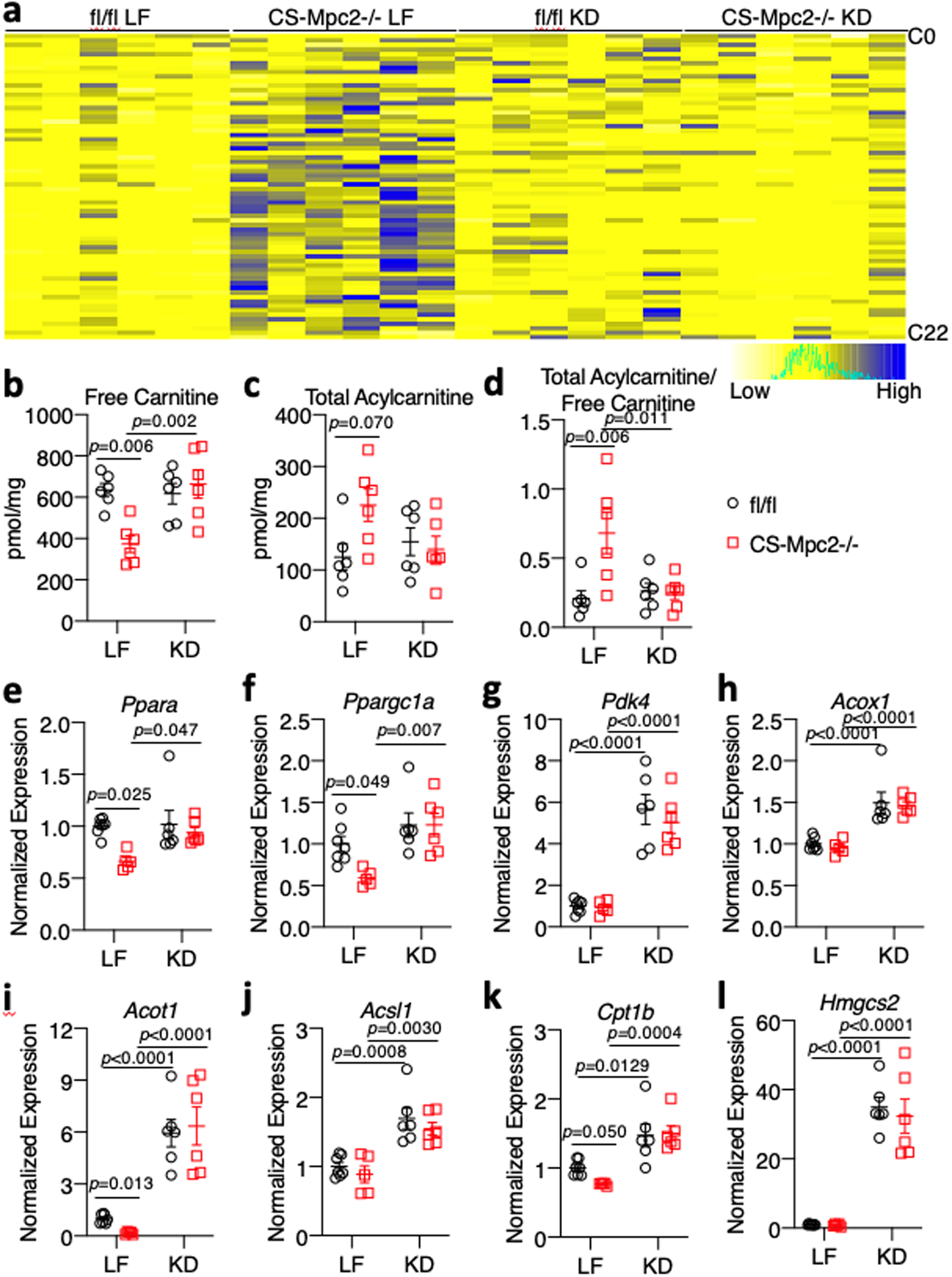
a, Heatmap of acylcarnitine species measured in hearts of low fat (LF)- or ketogenic diet (KD)-fed fl/fl or CS-Mpc2−/− mice (n=6). b-d, Concentrations of free carnitine, total acylcarnitines, and the acylcarnitine/free carnitine ratio measured by mass-spectrometry of heart tissue (n=6). e-l, Gene expression markers of PPARα and fatty acid oxidation (Ppara, Ppargc1a, Pdk4, Acox1, Acot1, Acsl1, Cpt1b, and Hmgcs2) from heart tissue of LF- or KD-fed mice (n=7, 5, 6, and 6 for fl/fl LF, CS-Mpc2−/− LF, fl/fl KD, and CS-Mpc2−/− KD, respectively). Mean ± s.e.m. shown within dot plot. Each symbol represents an individual sample. Two-way ANOVA with Tukey’s multiple-comparisons test.
Exogenous Ketone Bodies Moderately Attenuate Cardiac Remodeling in CS-MPC2−/− Mice
We also wanted to assess whether increased ketosis without altering dietary fat intake was able to improve cardiac function. To test this, CS-MPC2−/− mice were maintained on chow diet and injected i.p. with saline vehicle or 10 mmol/kg β-hydroxybutyrate (βHB) daily for two weeks (Extended Data Fig. 4a). Over this timeframe, vehicle treated CS-MPC2−/− mice displayed worsened LV dilation and contractile function, which were either limited or slightly improved by daily βHB administration (Extended Data Fig. 4b–h). At sacrifice, 4 hours after the last βHB injection, plasma ketone concentrations were significantly elevated (Extended Data Fig. 4i), but not nearly to the same degree as when fed a ketogenic diet (see Fig. 3a). Heart weight (Extended Data Fig. 4j) and hypertrophic/fibrotic gene expression (Extended Data Fig. 4k) were only modestly improved by administering ketone bodies daily on top of carbohydrate-rich chow diet.
In a second attempt to raise ketosis without altering dietary fat, we fed mice a diet supplemented with 16.5%kcal D-β-hydroxybutyrate-(R)-1,3 butanediol monoester “ketone ester” (KE). For this experiment, mice were fed control or KE diet from 9–15 weeks of age. KE diet slightly raised plasma ketone bodies (Extended Data Fig. 5a), but did not improve cardiac size or contractile function measured by echocardiography (Extended Data Fig. 5b–e), or heart weight at sacrifice (Extended Data Fig. 5f). Lastly, cardiac gene expression of markers of heart failure were only modestly improved by KE diet (Extended Data Fig. 5g–i). Thus, two different ways to enhance ketosis without altering dietary fat intake did not drastically improve cardiac size or function in CS-MPC2−/− mice. These results suggest that provision of ketones per se is not sufficient to improve heart failure in KD-fed CS-MPC2−/− mice.
High-Fat Diets Significantly Improve Heart Failure in CS-MPC2−/− Mice
To dissect the importance of dietary fat and myocardial FAO, we also fed fl/fl and CS-MPC2−/− mice two diets that were higher in fat, but with moderate levels of carbohydrate and protein, which only modestly increased plasma ketone body concentrations compared to LF-fed fl/fl mice (Fig. 6a–b). Feeding CS-MPC2−/− mice a ~42% medium chain triglyceride (MCT) or a 60% high-fat (HF) diet was able to significantly improve cardiac enlargement and contractile function as measured by echocardiography (Fig. 6c–d, Extended Data Fig. 6a–i, and Supplemental Video 2). Heart weight, lung edema, and hypertrophic/fibrotic gene expression were also significantly improved by MCT and HF diets (Fig. 6e–m). Interestingly, in CS-MPC2−/− hearts the HF diet was also capable of enhancing expression of Ppara and its target genes (Extended Data Fig. 6j–l), as well as lowering Bdh1 expression (Fig. 6n) compared to LF diet. Thus, diets enriched with higher levels of fat but enough carbohydrate and protein to limit ketosis were also able to significantly improve or even prevent cardiac remodeling and dysfunction in CS-MPC2−/− mice.
Fig. 6: High fat diets also prevent cardiac remodeling and dysfunction in CS-MPC2−/− mice.
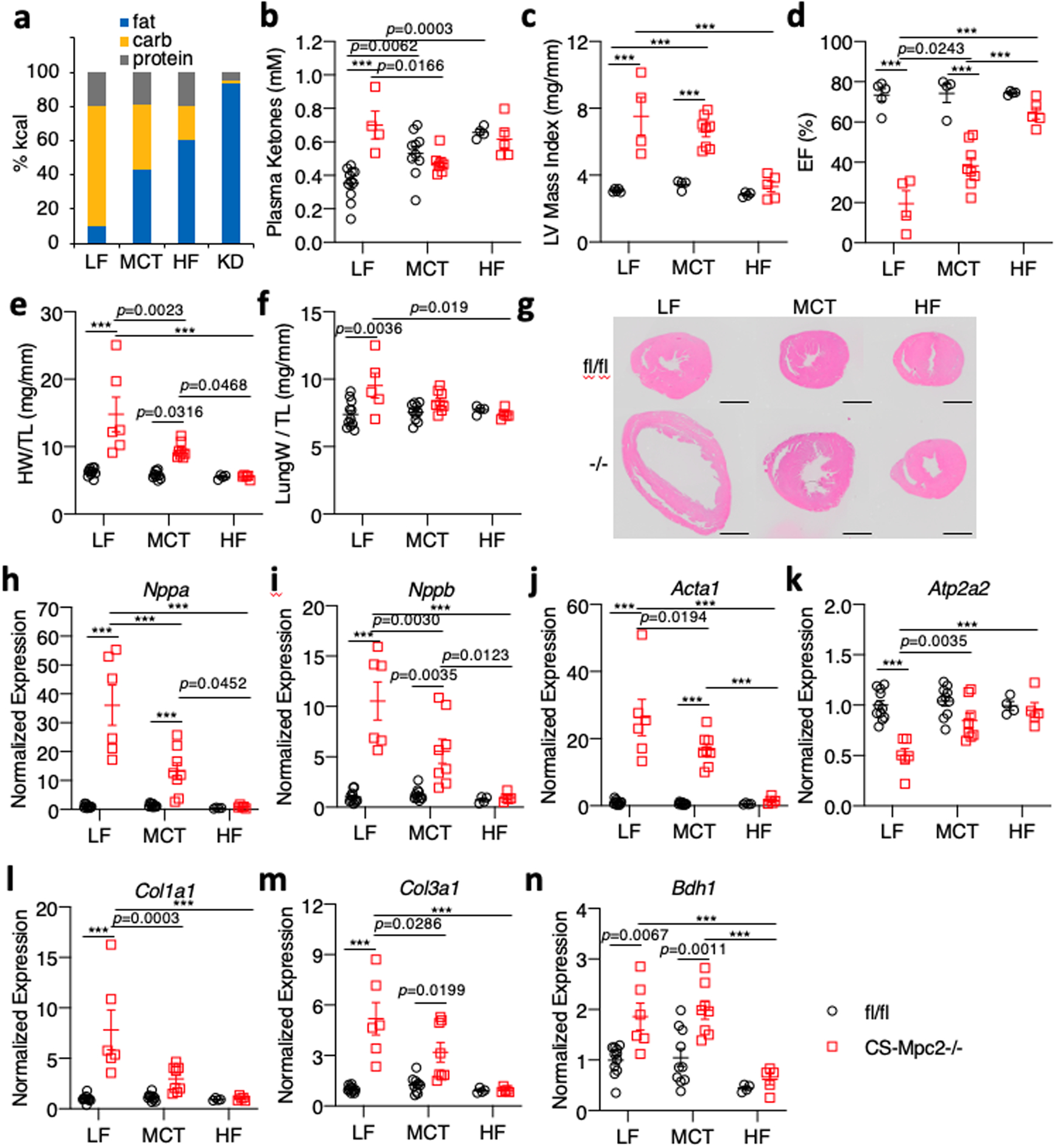
a, Comparison of diet macronutrient composition for low fat (LF), medium chain triglyceride (MCT), high fat (HF), and ketogenic diet (KD). b, Plasma total ketone body concentrations measured from mice after LF, MCT, or HF diet feeding (n=11, 4, 10, 8, 4, and 5 for fl/fl LF, CS-Mpc2−/− LF, fl/fl MCT, CS-Mpc2−/− MCT, fl/fl HF, and CS-Mpc2−/− HF, respectively). c-d, Echocardiography measures of left ventricular (LV) mass index and ejection fraction (EF) of mice fed LF, MCT, or HF diets (n=5, 4, 4, 8, 4, and 5 for fl/fl LF, CS-Mpc2−/− LF, fl/fl MCT, CS-Mpc2−/− MCT, fl/fl HF, and CS-Mpc2−/− HF, respectively). e-f, Heart weight and lung weight normalized to tibia length (n=11, 6, 10, 8, 4, and 5 for fl/fl LF, CS-Mpc2−/− LF, fl/fl MCT, CS-Mpc2−/− MCT, fl/fl HF, and CS-Mpc2−/− HF, respectively). g, Representative short-axis heart images stained with H&E (scale bar = 1mm; similar results obtained during four independent experiments of littermate mice). h-n, Gene expression markers of hypertrophy, heart failure, fibrosis, and the ketolytic enzyme Bdh1 from mouse hearts (n=11, 6, 10, 8, 4, and 5 for fl/fl LF, CS-Mpc2−/− LF, fl/fl MCT, CS-Mpc2−/− MCT, fl/fl HF, and CS-Mpc2−/− HF, respectively). Mean ± s.e.m. shown within dot plot. Each symbol represents an individual sample. Two-way ANOVA with Tukey’s multiple-comparisons test. Exact p values given unless ***P < 0.0001.
A 24-Hour Fast Improves Cardiac Remodeling Concordant with Enhanced Fat Oxidation
Like ketogenic diet, prolonged fasting increases the cardiac reliance on fatty acid oxidation and reduces ketolytic flux despite increased cardiac ketone body delivery35. We aged fl/fl and CS-MPC2−/− mice to 16-weeks, and then either fasted them for 24 h or allowed them to continue consuming chow ad libitum. As expected, the 24 h fast reduced blood glucose levels, and strongly enhanced plasma concentrations of non-esterified fatty acids and ketone bodies in both fl/fl and CS-MPC2−/− mice (Fig. 7a–c). Interestingly, fasting completely corrected the elevated blood lactate concentrations (Extended Data Fig. 7a), but had no effect on the elevated cardiac glycogen concentrations observed in fed CS-MPC2−/− mice (Extended Data Fig. 7b). Fasting also did not markedly alter plasma TAG concentrations (Extended Data Fig. 7c). Strikingly, the 24 h fast significantly improved CS-MPC2−/− heart weights (Fig. 7d) and several gene expression markers of hypertrophy, failure, and fibrosis (Fig 7e–g, and Extended Data Fig. 7d–e). Similar to ketogenic diet feeding, despite mM concentrations of circulating ketone bodies, 24 h fasting resulted in reduced expression of the ketolytic enzymes Bdh1 and Oxct1 in the heart compared to the fed condition (Fig. 7h–i). While many PPARα target genes and genes important for fatty acid oxidation, including Cpt1b, Acadl, and Acadm, were reduced in the fed CS-MPC2−/− hearts, these were all normalized or even enhanced by the 24 h fast (Fig. 7j–l and Extended Data Fig. 7f–h). Acaca, which encodes acetyl-CoA carboxylase and generates malonyl-CoA which inhibits mitochondrial fat oxidation, was elevated in the fed CS-MPC2−/− hearts, and reduced by fasting (Extended Data Fig. 7i).
Fig. 7: Improved cardiac remodeling during fasting is associated with enhanced fat oxidation.
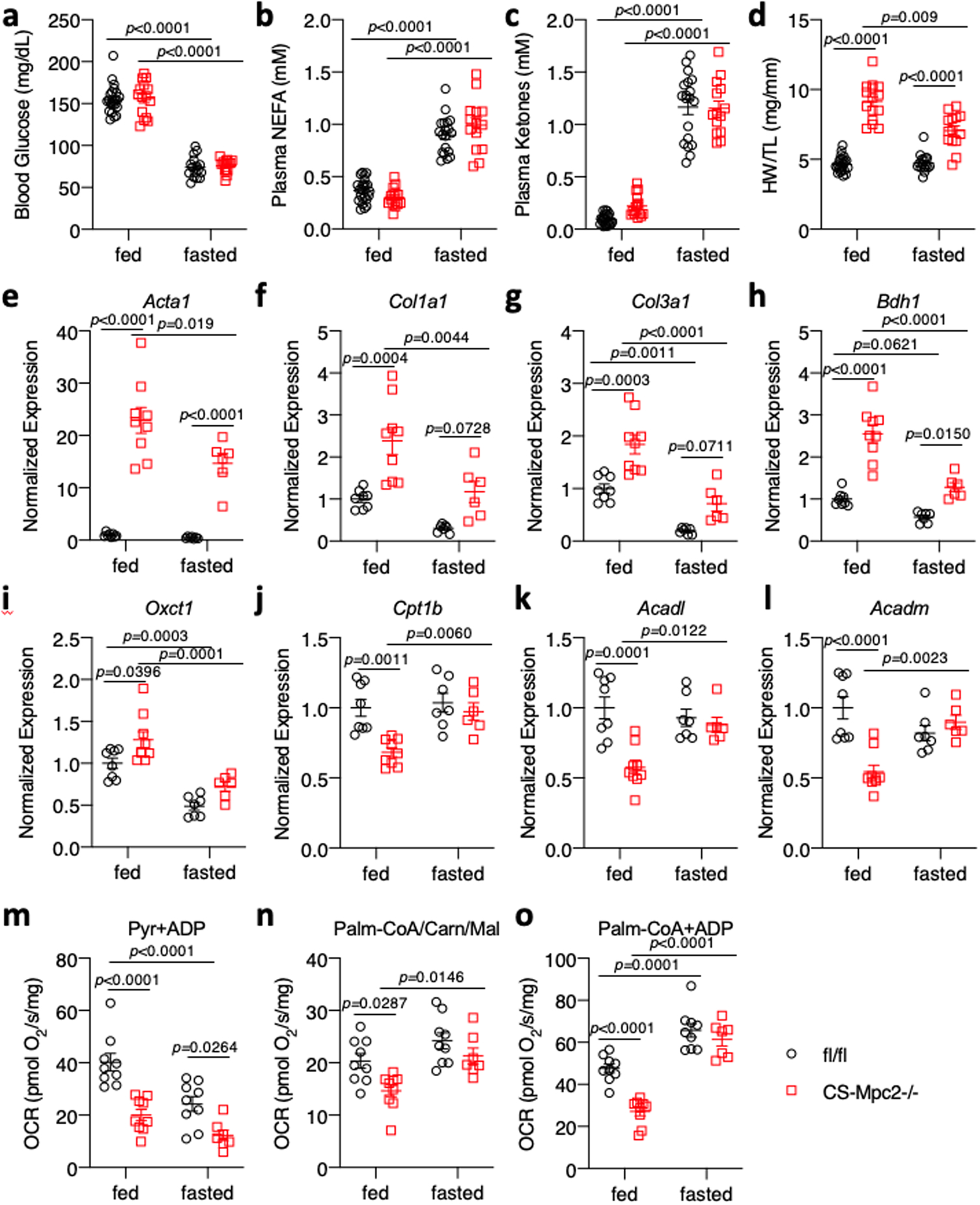
a-c, Blood glucose, plasma non-esterified fatty acid (NEFA), and plasma total ketone body concentrations from fed or 24 h-fasted mice (n=22, 15, 16, and 14 for fl/fl fed, CS-Mpc2−/− fed, fl/fl fasted, and CS-Mpc2−/− fasted, respectively). d, Heart weight normalized to tibia length after feeding or fasting (n=22, 15, 16, and 14 for fl/fl fed, CS-Mpc2−/− fed, fl/fl fasted, and CS-Mpc2−/− fasted, respectively). e-l, Cardiac gene expression markers of heart failure, fibrosis, or ketone and fatty acid metabolizing enzymes (n=8, 9, 7, and 6 for fl/fl fed, CS-Mpc2−/− fed, fl/fl fasted, and CS-Mpc2−/− fasted, respectively). m-o, Oxygen consumption rates (OCR) measured from permeabilized cardiac muscle fibers using pyruvate (Pyr) or palmitoyl-CoA with carnitine and malate (Palm-CoA/Carn/Mal) as substrates (n=9, 9, 9, and 7 for fl/fl fed, CS-Mpc2−/− fed, fl/fl fasted, and CS-Mpc2−/− fasted, respectively). Data are presented as mean ± s.e.m. within dot plot. Each symbol in dot plot represents an individual sample. Two-way ANOVA with Tukey’s multiple-comparisons test.
A subset of hearts were used to generate permeabilized cardiac muscle fibers to assess their ability to oxidize pyruvate and palmitoyl-CoA. As expected, the CS-MPC2−/− heart fibers displayed lower OCR from pyruvate in both the fed and fasted conditions (Fig. 7m). Fasting reduced pyruvate-stimulated respiration in the fl/fl hearts and surprisingly also tended to reduce OCR from pyruvate in the CS-MPC2−/− hearts. In the fed condition, failing CS-MPC2−/− hearts displayed reduced OCR with palmitoyl-CoA as substrate during both state 2 and ADP-stimulated state 3 conditions (Fig. 7n–o). However, palmitoyl-CoA respiration was strongly enhanced in the CS-MPC2−/− hearts by the 24 h fast (Fig. 7n–o). In summary, a 24 h fast significantly reduced the size of CS-MPC2−/− hearts and suppressed the expression of hypertrophic and fibrotic genes, which was associated with a downregulation of ketolytic enzymes and an enhancement of cardiac fat oxidation.
Ketogenic Diet Can Reverse Heart Failure in CS-MPC2−/− Mice
We also assessed if 3 weeks feeding the high fat, low carbohydrate KD could reverse existing heart failure. We allowed CS-MPC2−/− mice to consume chow until 16 weeks of age, then assigned them to either LF- or KD-feeding for 3 weeks (Fig. 8a). All CS-MPC2−/− mice displayed cardiac dilation and poor contractile function during the 16-week echocardiograms, which remained or was worsened by 3 weeks of LF diet feeding (Fig. 8b–d and Extended Data Fig. 8a–h). However, 3-weeks of KD-feeding greatly improved the LV dilation and contractile function of the previously failing CS-MPC2−/− hearts (Fig. 8b–d, Extended Data Fig. 8a–h, and Supplemental Video 3). The 3 weeks of KD-feeding strongly elevated ketosis (Fig. 8e) and heart weight, lung edema, and cardiac gene expression markers of pathological remodeling and fibrosis were all drastically reversed by 3-weeks of KD-feeding (Fig. 8f–h). Lastly, the 3-weeks of KD-feeding reduced cardiac gene expression of ketolytic enzymes and induced PPARα target genes encoding fat oxidation enzymes (Fig. 8i–j). Thus, ketogenic diet consumption for only 3-weeks and the concordant increase in fat metabolism was associated with reverse remodeling of the failing CS-MPC2−/− hearts to essentially normal size.
Fig. 8: Ketogenic diet can reverse heart failure in CS-Mpc2−/− mice.
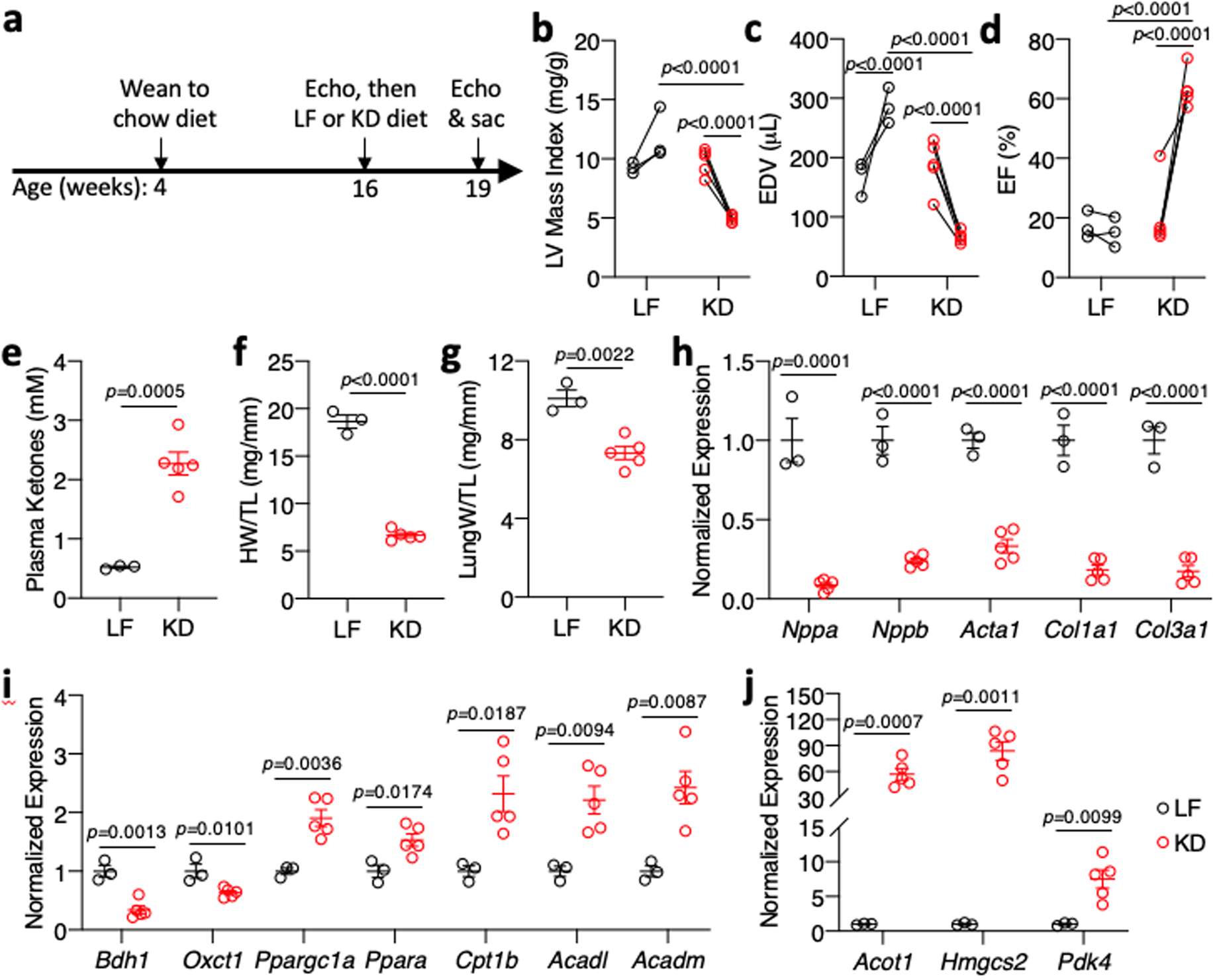
a, Timeline for heart failure reversal experiment, in which CS-MPC2−/− mice were switched to low fat (LF) or ketogenic diet (KD) at 16 weeks of age for 3 weeks. b-d, Echocardiography measures of left ventricular (LV) mass index, end-diastolic volume (EDV), and ejection fraction (EF) of CS-MPC2−/− mice PRE and POST LF or KD feeding (n=3 LF, 5 KD; data presented as PRE-POST with first data point at 16-weeks old and second data point at 19-weeks old after 3 weeks of LF or KD). e, Plasma total ketone values from CS-MPC2−/− mice fed LF or KD (n=3 LF, 5 KD). f-g, Heart weight and lung weight normalized to tibia length (n=3 LF, 5 KD). h-j, Cardiac gene expression of hypertrophy, heart failure, fibrosis, and ketone body and fatty acid metabolizing genes (n=3 LF, 5 KD). Data presented either as PRE-POST, or mean ± s.e.m. shown within dot plot. Each symbol represents an individual sample. Two-tailed paired Student’s t test to compare PRE vs. POST, two-tailed unpaired Student’s t test to compare LF vs. KD.
DISCUSSION
Myocardial fuel metabolism is altered in hypertrophy and heart failure, characterized as a generalized decrease in the ability to oxidize fatty acids and pyruvate in the mitochondrion17,18,38. The import of pyruvate into the mitochondria occurs via the mitochondrial pyruvate carrier, which was identified in 2012 as a hetero-oligomeric complex of MPC1 and MPC2 proteins19,20. An early study conducted prior to the cloning of MPC proteins and using a chemical inhibitor estimated that cardiac MPC expression was quite high, and MPC activity would be rate-limiting for pyruvate oxidation in heart mitochondria39. Subsequent studies agreed that inhibitor-binding of cardiac mitochondria was very high (indicating high cardiac MPC expression), but did not suggest pyruvate transport to be the limiting factor for pyruvate oxidation40,41. Studies regarding the importance of MPC activity in cardiac function or development of heart failure have been quite limited. Expression of MPC1 and MPC2 was shown to be an important marker of surviving myocardium near the border of infarct zones in a pig model, and this study also identified increased MPC expression in human hearts with ischemic heart failure42. While this current work was in preparation, another report showed that failing human hearts exhibited decreased expression of the MPC proteins25, which we have confirmed in this current study. Thus, myocardial MPC expression in heart failure may depend on ischemic vs non-ischemic etiology, as well as location in relation to infarct zone. Together with two companion papers43,44, we show that complete deletion of the MPC in myocardium leads to a severe, progressive cardiac remodeling and dilated heart failure. However, pharmacologic MPC inhibition or loss of one MPC2 allele and approximately 50% of the MPC protein did not affect cardiac function. These findings suggest that partial inhibition of MPC activity in the heart can be overcome metabolically and is not sufficient to cause pathologic remodeling as long as other cardiac stressors are not present. However, the work of Fernandez-Caggiano and colleagues demonstrates that MPC1 overexpression in a TAC model improves hypertrophy43, suggesting that MPC deactivation in the context of pressure overload plays a role in pathological remodeling.
Previous work has shown that modulating the expression or activity of PDH limits cardiac metabolic flexibility by decreasing glucose oxidation and increasing FAO21–24. Interestingly, these models of decreased PDH activity did not result in overt cardiac dysfunction. One possible explanation for why MPC-deletion is more severe is that blocking pyruvate entry could also impact pyruvate carboxylation (anaplerosis) and the replenishing of TCA cycle intermediates. Although the effects of deleting pyruvate carboxylase in the myocardium are unknown, this pathway is known to be active in the heart45. However, the majority of pyruvate carboxylation in the heart likely occurs by NADP+-dependent malic enzyme46 generating malate in the cytosol. Additionally, the abundance of most TCA cycle metabolites was normal or even elevated in the CS-MPC2−/− hearts (Fig. 1g–h, Supplemental Tables 1 & 3), suggesting no defect in anaplerosis. Another possibility is that a small amount of pyruvate is able to enter the mitochondrial matrix in the absence of the MPC, potentially through pyruvate-alanine cycling as we have described in the liver26.
The current studies cannot definitively explain why CS-MPC2−/− mice develop heart failure. The simplest explanation would be an inability to oxidize pyruvate results in an energetic deficit. Interestingly, the failing CS-MPC2−/− hearts display decreased AMPK phosphorylation (Fig. 3o), suggesting that their metabolic stress does not involve dysregulated AMP/ATP levels. Another possibility is that a decrease in mitochondrial pyruvate metabolism results in an accumulation of metabolic intermediate(s) that enhance hypertrophic signaling. One example of this would be the oncometabolite 2-hydroxyglutarate (2-HG), which has been implicated in driving cardiac hypertrophy and impairing contractility47,48. We found that failing LF-fed CS-MPC2−/− hearts contained almost 2-fold higher concentrations of total 2-HG (Supplemental Table 3). However, hearts from KD-fed mice also had higher total 2-HG compared to LF-fed fl/fl mice (Supplemental Table 3). Unfortunately, our mass-spectrometry analyses did not distinguish between D- and L-2-HG, as only D-2-HG appears to be responsible for inducing cardiomyopathy47,48. Two recent studies have suggested that cardiac hypertrophy is associated with enhanced glucose flux into the pentose phosphate pathway, generating reducing equivalents as NADPH and potentially other metabolites that signal to mTOR to stimulate protein synthesis49,50. While we have not identified specific signals, we can confirm that the failing CS-MPC2−/− hearts display enhanced mTOR activation and downstream signaling to support hypertrophic growth (Fig. 3o). The decreased AMPK phosphorylation in CS-MPC2−/− hearts likely does not indicate “energetic stress”, but is consistent with elevated mTOR activation, as AMPK is a repressor of mTOR activity. However, the relationship between AMPK and cardiac hypertrophy is not completely clear as genetic mouse models of AMPK depletion do not lead to hypertrophy51,52 and can even protect against isoproterenol-induced hypertrophy53. Additionally, while acute pharmacologic AMPK activation inhibits mTOR, chronic AMPK activation can induce cardiac hypertrophy54. Lastly, a recent study also showed that enhancing fat oxidation via acetyl-CoA carboxylase 2 deletion was able to reduce altered glucose metabolism and prevent cardiac hypertrophy50. Therefore, as our current study suggests, altered glucose and pyruvate metabolism seems to drive pathologic remodeling, while enhanced fat oxidation appears to correct this cardiac remodeling. Further study is required to dissect what metabolites are altered by decreased MPC activity that ultimately increase hypertrophic growth.
Recent studies have described improvements in cardiac function with ketone body infusion in both a dog model and human patients with heart failure33,55. Additionally, genetic mouse models of BDH1 or OXCT1 suggest that increased ketone metabolism is a protective adaptation in heart failure33,56,57. Interestingly, a ketogenic diet was unable to improve cardiac hypertrophy in a mouse model of defective FAO caused by carnitine palmitoyltransferase 2 deletion58 suggesting that enhancing ketolysis per se cannot rescue heart failure in that model. Several lines of evidence suggest that the prevention or reversal of heart failure in CS-MPC2−/− mice were driven by enhanced fatty acid metabolism rather than ketone body utilization. Injecting CS-MPC2−/− mice daily with β-hydroxybutyrate did slightly ameliorate cardiac remodeling, but feeding a ketone-ester-supplemented chow did not improve cardiac size or function. Diets that were enriched with fat, but not overtly ketogenic, were also able to significantly prevent heart failure in CS-MPC2−/− mice. While hearts can extract and metabolize ketone bodies in proportion to their delivery, ketones and fatty acids are in competition for oxidation7–9 and in agreement with a previous report in normal mouse hearts35, we show that fasting or ketogenic diet decreased the expression of the ketolytic enzymes BDH1 and Oxct1 and likely reduced ketolytic flux. KD-feeding and fasting were also associated with upregulation of PPARα-target genes related to FAO and corrected the cardiac accumulation of acylcarnitines. Fasted CS-MPC2−/− hearts also displayed increased oxidation of palmitoyl-CoA consistent with enhanced fat oxidation.
It should also be noted that the MPC has been suggested to also be a mitochondrial importer/exporter of ketone bodies59, which may further suggest that the ameliorative effects of ketogenic diet on MPC hearts are not due to enhanced cardiac ketolysis. However, there is genetic evidence that the MPC is not the sole mitochondrial ketone transporter. Cardiac β–hydroxybutyrate flux into the TCA cycle was actually increased in MPC1−/− hearts44, indicating that the MPC is not required for cardiac mitochondrial ketone body import. Ketone bodies are produced and released almost exclusively in the liver, and hepatic MPC1/2 knockout mice display normal or even enhanced plasma ketone body concentrations26,60, suggesting no defect in mitochondrial ketone export. Whether genetic loss of the MPC impacts mitochondrial ketone import/export will require future study.
Lastly, it is interesting that the degree of heart failure improvement appears to also track with a reduction in dietary carbohydrate. Hearts from CS-MPC2−/− mice showed even worse failure after refined LF diet feeding compared to chow (Fig. 3 and Extended Data Fig. 3 compared to Fig. 2 and Extended Data Fig. 2), potentially due to the large amount of sucrose in the LF diet compared to complex carbohydrates in chow. Fasting also lowered blood glucose concentrations and is known to reduce cardiac glucose uptake and oxidation35. Collectively, we believe the present data using a variety of model systems suggest that enhanced FAO and limiting the provision of carbohydrate to be the predominant mechanism for preventing or reversing cardiac dysfunction in CS-MPC2−/− mice.
Conclusions and Limitations of Study
These studies describe that the MPC is deactivated in failing human and mouse hearts and that cardiac deletion of MPC2 in mice results in progressive cardiac hypertrophy and dilated heart failure. Interestingly, heart failure in CS-MPC2−/− mice could be prevented or even reversed by feeding a ketogenic diet and that an acute fast was also able to initiate reverse remodeling. These improvements appear to be predominantly mediated by increasing cardiac fat oxidation and limiting provision of carbohydrate, rather than enhancing ketone metabolism. Some mechanistic aspects of the cause of heart failure observed in mice lacking MPC in the heart remain to be teased apart. A limitation of the models we used is that the circulating ketone concentrations generated by ketone injection or feeding ketone ester diet are not as high as when feeding a ketogenic diet or fasting. Thus, it is difficult to say whether a more pronounced level of ketosis would also improve the CS-MPC2−/− hearts.
METHODS
Human Heart Tissue Collection.
Human heart tissue was collected with written informed consent received from participants as part of an Institutional Review Board (IRB)-approved protocol (#201101858) at the Washington University School of Medicine. Failing human left ventricular heart tissue was obtained from the Washington University Translational Cardiovascular Tissue Core at the time of left ventricular assist device (LVAD) placement, or post-LVAD placement at the time of cardiac transplantation. Non-failing human heart tissue was obtained from Mid-America Transplant (St. Louis, MO) from hearts deemed unsuitable for transplantation due to donor age, non-occlusive coronary artery disease, or high-risk behavioral profile. The collected piece of cardiac tissue had fat removed, was rinsed in saline, and then was snap-frozen in liquid nitrogen and stored at −80°C until analyzed.
Animals.
All animal procedures were performed in accordance with National Institutes of Health guidelines and approved by the Institutional Animal Care and Use Committees at the Washington University School of Medicine and Saint Louis University School of Medicine (Protocol 2845). The use of mice conformed to guidelines set forth in the NIH’s Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011). Generation of mice with the Mpc2 gene flanked by loxP sites has been described previously26. To create cardiac myocyte specific deletion, these mice were crossed with a knock-in mouse in which one allele of the myosin light chain 2v (Mlc2v) gene was replaced with Cre recombinase61, which was obtained from the Jackson Laboratory (Bar Harbor, ME). All mice were from a C57BL6/J background. Unless specifically noted, all experiments were performed with a mixture of male and female littermate mice. Most studies were performed with mice beginning at 6-weeks-old and ending at 16–17 weeks of age (chow fed, ketogenic diet study, and MCT/HFD study). The fed/fasted study was performed on mice that were chow-fed for 16 weeks before acute 24 h fast. For the ketogenic diet reversal study, 16-week-old chow-fed mice were switched to either LF or KD for 3 weeks and euthanized at 19 weeks of age. For the TAC+MI studies, 4–5 week-old wildtype C57BL6/J females were purchased from the Jackson Laboratory (Bar Harbor, ME) , thus were not necessarily littermates.
Animal Care.
Mice were housed in a climate-controlled barrier facility maintained at 22–24°C and 40–60% humidity in ventilated cages with a 12-hour light/dark cycle with light period from 0600 to 1800 local time. Ad-libitum access to drinking water was provided by individual bottles in each cage. Mice were housed in cages with corn-cob bedding or switched to aspen chip bedding during special diet studies or fasting prior to sacrifice. All mice were group-housed, up to 5 mice per cage, with cloth nestlets to use for enrichment. Mice on special diets were also provided with a Nylabone (Central Garden & Pet Co., Neptune City, NJ) for both enrichment and to maintain teeth when fed soft, higher fat diets. With all diets, mice were provided ad-libitum access to food, except for a brief 4-hour fast prior to euthanasia. Unless specifically noted, all special diets were initiated at 6-weeks of age. All diets were provided on a wire rack above the cage bedding, with the exception of the ketogenic diet paste which was spread into a glass petri dish, placed on the bottom of the cage, and replaced every 2–3 days. Mice fed standard chow received PicoLab® Rodent Diet 20 (#5053, LabDiet, St. Louis, MO) which comprised of 62.1%kcal carbohydrate, 13.2%kcal fat, and 24.7%kcal protein. The refined low-fat (LF) diet was composed of 70%kcal carbohydrate, 10%kcal fat, and 20%kcal protein (D12450B, Research Diets, New Brunswick, NJ). The high fat, low carbohydrate, low protein “ketogenic diet” (KD) was composed of 1.8%kcal carbohydrate, 93.4%kcal fat, and 4.7%kcal protein (F3666, Bio-Serv, Flemington, NJ). The medium-chain triglyceride (MCT) diet was composed of 37.9%kcal carbohydrate, 43%kcal fat (depleted of long-chain fatty acids), and 19.1%kcal protein (TD.00308, Envigo, Madison, WI). High-fat (HF) diet was composed of 20%kcal carbohydrate, 60%kcal fat, and 20%kcal protein (D12492, Research Diets, New Brunswick, NJ). Hearts were also analyzed from a cohort of WT mice fed a high trans-fat, fructose, cholesterol (HTF-C) diet (D09100301, Research Diets, New Brunswick, NJ) with or without insulin-sensitizing MPC-inhibitor MSDC-0602K treatment, which were previously published with respect to nonalcoholic steatohepatitis28. For the ketone ester (KE) diet experiment, control diet consisted of 63%kcal carbohydrate, 10%kcal fat, and 24%kcal protein (104403, Dyets, Bethlehem, PA), and the KE diet was composed of the same diet except 16.5%kcal of the carbohydrates were replaced with D-β-hydroxybutyrate-(R)-1,3 butanediol monoester “ketone ester” (16.5%kcal ketone-ester, 46.5%kcal carbohydrate, 10%kcal fat, and 24%kcal protein)(104404, Dyets, Bethlehem, PA). Control or KE diet were fed from 9–15 weeks of age.
Unless specifically noted, mice were euthanized after a 4 hour fast by CO2 asphyxiation and blood was collected via cannulation of the inferior vena cava into EDTA-treated tubes. Tissues were then excised, rinsed in PBS, weighed, and snap frozen in liquid nitrogen. Plasma was collected by spinning blood tubes at 8,000 × g for 8 minutes and then freezing the plasma supernatant in liquid nitrogen. For the fed vs 24 h fasted experiment, 16-week-old fl/fl and CS-MPC2−/− mice were either fasted ~8:00 AM, or allowed to continue feeding on normal chow ad libitum. The following day at ~8:00 AM, mice were sacrificed by CO2 asphyxiation, and blood and tissue collected as above, except for a small piece of LV tissue which was processed for tissue respiration studies as explained below.
Gene Expression Analysis.
Levels of gene expression were determined by qPCR. Total RNA was extracted from snap frozen tissues using RNA-Bee (Tel-Test, Friendswood, TX). ~50 mg of tissue was homogenized in RNA-Bee for 3–5 minutes using a 3 mm stainless steel bead at 30 Hz using a TissueLyser II (Qiagen, Hilden, Germany). RNA abundance and quality were assessed by Nanodrop (ThermoFisher Scientific, Waltham, MA). 1 μg of sample was then reverse transcribed into cDNA by Superscript VILO (ThermoFisher Scientific, Waltham, MA) using an Eppendorf Mastercycler® X50 thermocycler (Hauppauge, NY). Relative quantification of target gene expression was measured in duplicate using Power SYBR Green (ThermoFisher Scientific, Waltham, MA), using an ABI 7500 Real-Time PCR System (ThermoFisher Scientific, Waltham, MA). Target gene Ct values were normalized to reference gene (Rplp0) Ct values by the 2−ΔΔCt method. Oligonucleotide primer sequences used for qPCR are listed in Supplemental Table 4.
Western Blotting and Protein Expression Analysis.
Protein extracts were prepared by homogenizing ~50 mg of frozen tissue in an NP-40-based lysis buffer (15 mM NaCl, 25 mM Tris Base, 1 mM EDTA, 0.2% NP-40, 10% glycerol) supplemented with 1X cOmplete™ protease inhibitor cocktail (Roche, Basel, Switzerland) and a phosphatase inhibitor cocktail (1 mM Na3VO4, 1 mM NaF, and 1mM PMSF). Tissue was homogenized in this buffer for 3–5 minutes using a pre-chilled 3 mm stainless steel bead at 30 Hz using a TissueLyser II (Qiagen, Hilden, Germany). Protein concentrations were measured using a Pierce MicroBCA kit (ThermoFisher Scientific, Waltham, MA), and detected with a BioTek Synergy plate reader and Gen5 software (BioTek Instruments, Winooski, VT). 50 μg of protein lysate was electrophoresed on precast Criterion 4–15% polyacrylamide gels (Biorad, Hercules, CA), and transferred onto 0.45 μm Immobilon PVDF membranes (MilliporeSigma, St. Louis, MO). Membranes were blocked with 5% Bovine Serum Albumin (Sigma, St. Louis, MO) in TBS-T for at least 1 hour.
Primary antibodies were then used at 1:1000 (or 1:5000 for VLCAD and LCAD) in 5%BSA-TBS-T overnight while rocking at 4°C. Antibodies for human MPC1 and MPC2 were from Cell Signaling (Danvers, MA), while anti-mouse MPC1 and MPC2 antibodies were a gift from Dr. Michael Wolfgang26,62,63. Antibodies for VLCAD64, LCAD65, and MCAD66 were gifts from Drs. Daniel Kelly or Arnold Strauss. Anti-CPT1B antibody was from Alpha Diagnostic International (San Antonio, TX). Anti-BDH1 and Anti-OX PHOS cocktail antibodies were from ThermoFisher Scientific (Waltham, MA). Phospho-ERK1/2 (Thr202/Tyr204), Total ERK1/2, Phospho-AMPKα (Thr172), Total AMPKα, Phospho-mTOR (Ser2448), Total mTOR, Phospho-S6 Ribosomal Protein (Ser235/236), Total S6 Ribosomal Protein were from Cell Signaling (Danvers, MA). VDAC1 antibody was from Abcam (Cambridge, United Kingdom). Anti-α-Tubulin and β-Actin antibodies were from Sigma (St. Louis, MO). After primary antibody incubation, membranes were washed 3–5X for 5 min in TBS-T, and probed with IRDye secondary antibodies at 1:10,000 (Li-Cor Biosciences, Lincoln, NE) in 5%BSA-TBS-T for 1 hour. Membranes were imaged on an Odyssey® imaging system and analyzed with Image Studio™ Lite software (Li-Cor Biosciences, Lincoln, NE).
Mitochondrial Isolation and High Resolution Respirometry.
Mitochondria were isolated by differential centrifugation from whole mouse hearts by homogenization with 10 passes of a glass-on-glass Dounce on ice with 4 mL of buffer containing 250 mM sucrose, 10 mM Tris Base, and 1 mM EDTA (pH 7.4). Homogenates were then spun at 1,000 × g for 5 min at 4°C to pellet nuclei and undisrupted cell debris. The supernatant was then spun at 10,000 × g for 10 min to pellet the mitochondrial fraction. The mitochondrial pellet was washed twice in homogenization buffer minus the EDTA with 10,000 × g 10 min spins. After the final wash, mitochondrial pellets were solubilized in ~150 μL Mir05 respiration buffer (0.5 mM EGTA, 3 mM MgCl2, 60 mM Lactobionic acid, 20 mM Taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, and 1 g/L fatty acid free Bovine Serum Albumin; pH 7.1). Mitochondrial protein concentration was then measured using a Pierce MicroBCA kit (ThermoFisher Scientific, Waltham, MA), and detected with a Synergy plate reader and Gen5 software (BioTek Instruments, Winooski, VT).
To measure oxygen consumption rates, 50 μg of mitochondrial protein was added to each 2 mL chamber of an Oxygraph O2k equipped with DatLab software (Oroboros Instruments, Innsbruck, Austria). Substrates used to assess pyruvate-stimulated respiration were 5 mM sodium pyruvate, 2 mM malate, 2.5 mM ADP+Mg2+, and then 5 μM UK-5099. To assess respiration on other substrates, 50 μM palmitoyl-DL-carnitine and 2 mM malate ± 2.5 mM ADP+Mg2+, or 10 mM glutamate and 2 mM malate ± 2.5 mM ADP+Mg2+, or 5 mM succinate + 2.5 mM ADP+Mg2+ were used. Oxygen consumption rates were measured as pmol O2/s/mg mitochondrial protein.
For the fed vs 24 h fasted experiment, immediately after euthanasia hearts were briefly stored in BIOPS solution (10 mM Ca/K2-EGTA, 5.77 mM Na2ATP, 6.56 mM MgCl2, 20 mM Taurine, 15 mM Na2Phosphocreatine, 20 mM Imidazole, 0.5 mM DTT, and 50 mM MES hydrate) on ice. 1–4 mg of LV muscle was teased apart into fibers and permeabilized with 50 μg/mL saponin in 2 mL BIOPS on ice for 20 minutes. After permeabilization, muscle fibers were washed in 2 mL Mir05 buffer on ice for 15 minutes. Muscle fibers were patted dry, weighed, and added to the 2 mL chamber of an Oxygraph O2k containing Mir05 buffer supplemented with 20 mM creatine and 25 μM blebbistatin. Before chambers were sealed, O2 was injected to hyperoxygenate the buffer (to ~375 μM) to prevent lack of O2 diffusion into tissue from limiting O2 consumption rates. After chambers were sealed and baseline respiration rates equilibrated to zero, substrates were added in the following order: 2 mM carnitine, 2.5 mM malate, 50 μM palmitoyl-CoA, 7 mM ADP+Mg2+, 5 mM sodium pyruvate, 5 mM succinate. Oxygen consumption rates were measured as pmol/s/mg tissue weight.
Blood and Plasma Metabolite and Hormone Measurements.
Immediately prior to euthanasia, a snip of the tail was made with a razor blade and a drop of mixed venous blood was used to measure blood glucose using a Contour Next EZ (Bayer Ascensia Diabetes Care, Parsippany, NJ) glucometer. A second drop of blood was then used to measure blood lactate concentrations using a Lactate Plus meter (Nova Biomedical, Waltham, MA). Plasma insulin concentrations were measured from 10 μL of plasma by Singulex Erenna® assay (Sigma, St. Louis, MO) performed by the Washington University Core Lab for Clinical Studies. Total ketone bodies were measured from 4 μL plasma using the Total Ketone AutoKit (FujiFilm Wako, Mountain View, CA) according to kit directions. Optic density (OD) at 405 and 600 nm were measured every minute for 5 minutes, and absorbance changes were normalized to a 300 μM standard. Non-esterified “free” fatty acids were measured from 2 μL plasma using a non-esterified fatty acid kit according to manufacturer’s directions (FujiFilm Wako, Mountain View, CA). OD at 560 nm was measured and normalized to a standard curve. Triglycerides were measured from 5 μL plasma using Infinity assay kit according to manufacturer’s directions (ThermoFisher Scientific, Waltham, MA). OD and 540 was measured and related to the OD of a standard curve. OD for all assays was measured in clear 96-well plates using a Synergy plate reader and Gen5 software (BioTek Instruments, Winooski, VT).
Targeted Metabolomics for Amino Acids, Acylcarnitines, Organic acids, and Short Chain Acyl-CoAs.
Mice used for targeted metabolomic analyses were fasted for 3 hours, anesthetized with 100 μg/g sodium pentobarbital injected i.p., and euthanized by excision of the beating heart. Hearts were snap frozen in liquid nitrogen and stored at −80°C until they were collectively processed and analyzed. Flash frozen hearts were pulverized to a fine powder in a liquid nitrogen chilled percussion mortar and pestle and weighed into pre-chilled 2ml tubes. A chilled 5mm homogenizing bead was added to samples and tissue was diluted to 50 mg/ml with 50% acetonitrile containing 0.3% formate (for acylcarnitines, amino acids, and organic acids) or isopropanol/phosphate buffer (for CoAs), homogenized for 2 min at 30 Hz using a TissueLyser II (Qiagen, Hilden, Germany) and aliquoted for metabolite assays. For all metabolite analyses, tissues and homogenates were kept on ice, centrifuged at 4°C, and when ready to measure, were placed in an autosampler kept at 4°C.
Amino acids and acylcarnitines were analyzed by flow injection electrospray ionization tandem mass spectrometry and quantified by isotope or pseudo-isotope dilution similar to previous67–69, which are based on methods developed for fast ion bombardment tandem mass spectrometry70. Extracted heart samples were spiked with a cocktail of heavy-isotope internal standards (Cambridge Isotope Laboratories, Tewksbury, MA; or CDN Isotopes, Pointe-Claire, Canada) and deproteinated with methanol. The methanol supernatants were dried and esterified with either acidified methanol or butanol for acylcarnitine or amino acid analysis, respectively. Mass spectra for acylcarnitine and amino acid esters were obtained using precursor ion and neutral loss scanning methods, respectively. The spectra were acquired in a multi-channel analyzer (MCA) mode to improve signal-to-noise. The data were generated using a Waters TQ (triple quadrupole) detector equipped with Acquity™ UPLC system and a data system controlled by MassLynx 4.1 operating system (Waters, Milford, MA). For the amino acids analysis, the mass spectrometer settings were as follows: ionization mode - positive electrospray, capillary voltage - 3.6 V, cone voltage - 14 V, extractor voltage - 2 V, RF lens voltage - 0.1 V, collision energy - 14–25 V, source temperature - 130°C, desolvation temperature - 200°C, desolvation gas flow - 550 L/hr, and cone gas flow - 50 L/hr. For the acylcarnitine analysis, the mass spectrometer settings were as follows: ionization mode - positive electrospray, capillary voltage - 3.5 V, cone voltage - 25 V, extractor voltage - 2 V, RF lens voltage - 0.1 V, collision energy - 30 V, source temperature - 130°C, desolvation temperature - 200°C, desolvation gas flow - 550 L/hr, and cone gas flow - 50 L/hr. Ion ratios of analyte to respective internal standard computed from centroided spectra are converted to concentrations using calibrators constructed from authentic aliphatic acylcarnitines and amino acids (Sigma, St. Louis, MO; Larodan, Solna, Sweden) and Dialyzed Fetal Bovine Serum (Sigma, St. Louis, MO).
Organic acids were analyzed by capillary gas chromatography/mass spectrometry (GC/MS) using isotope dilution techniques employing Trace Ultra GC coupled to ISQ MS operating under Xcalibur 2.2 (ThermoFisher Scientific, Austin, TX)71. The mass spectrometer settings were as follows: ionization mode - electron ionization, ion source temperature - 250°C, and the transfer line temperature - 275°C. The supernatants of tissue homogenates were spiked with a mixture of heavy isotope labeled internal standards and the keto acids were stabilized by ethoximation. The organic acids were acidified and extracted into ethyl acetate. The extracts were dried and derivatized with N,O-bis(Trimethylsilyl) trifluoroacetamide. The organic acids were quantified using ion ratios determined from single ion recordings of fragment ions which are specific for a given analyte and its internal standard. These ratios were converted to concentrations using calibrators constructed from authentic organic acids (Sigma, St. Louis, MO). The heatmap for acylcarnitines was generated by shinyheatmap72.
Short chain acyl CoA were analyzed by LC-MS/MS using a method based on a previously published report73. The extracts were spiked with 13C2-Acetyl-CoA, centrifuged and filtered through the Millipore Ultrafree-MC 0.1 μm centrifugal filters before being injected onto the Chromolith FastGradient RP-18e HPLC column, 50 × 2 mm (MilliporeSigma, St. Louis, MO) and analyzed on a Waters Xevo TQ-S triple quadrupole mass spectrometer coupled to an Acquity UPLC system (Waters, Milford, MA). The mass spectrometer settings were as follows: ionization mode - positive electrospray, capillary voltage - 3.7 V, cone voltage - 50 V, source offset voltage - 50 V, collision energy - 28 V, dwell time - 0.06 seconds, desolvation temperature – 500°C, desolvation gas flow - 600 L/hr, cone gas flow - 150 L/hr, and nebulizer pressure - 7 bar. The following MRM transitions were monitored: Acetyl- CoA - 810.2->303.1, 13C2-Acetyl-CoA – 812.2->305.1, Succinyl-CoA - 868.2 ->361.1, and Malonyl-CoA – 854.2->347.1.
Mouse Echocardiography.
In vivo cardiac size and function were measured by echocardiography performed with a Vevo 2100 Ultrasound System equipped with a 30-MHz linear-array transducer (VisualSonics Inc, Toronto, Ontario, Canada)74. Mice were lightly anesthetized by i.p. injection of 0.005 ml/g of 2% Avertin (2,2,2-tribromoethanol; Sigma, St. Louis, MO). If required, one-fifth of the initial dose was given as a maintenance dose at regular intervals. Hair was removed from the left anterior chest by shaving, and mice were then placed onto a warming pad in a left lateral decubitus position. Normothermia (37°C) was maintained and monitored by a rectal thermometer. Ultrasound gel was applied to the chest and care was taken to maintain adequate transducer contact while avoiding excessive pressure on the chest. Two-dimensional and M-mode images were obtained in the long- and short-axis views. Images were retrieved off-line and analyzed using the Vevo LAB software package (VisualSonics Inc, Toronto, Ontario, Canada). Measurements were averaged from three separate images for each mouse. LV volumes were calculated from M-mode measurements using standard techniques75,76. Immediately after completion of imaging, mice were allowed to recover from anesthesia on a warming pad and returned to their home cage. For echocardiography during the ketone ester diet experiment, procedures were the same as above except mice were anesthetized by 1–2% inhaled isoflurane, and imaging was performed with a Vevo 770 Ultrasound System equipped with a 30-MHz linear-array transducer (VisualSonics Inc, Toronto, Ontario, Canada).
Histology.
Short-axis slices of the LV were fixed in 10% neutral buffered formalin overnight and processed by the Anatomic and Molecular Pathology core laboratory of the Washington University School of Medicine or the Research Microscopy & Histology Core of the Saint Louis University School of Medicine. The short-axis heart slices were embedded in paraffin blocks and sectioned onto glass slides. Slides were then stained for either Hematoxylin and Eosin (H&E) or Mason’s trichrome stains. Cardiomyocyte cross-sectional area (CSA) was measured from H&E images of the left ventricle taken at 10X magnification. At least 3 images of each heart were taken, and 10 myocytes per image measured. CSA was measured with ImageJ software. The individual taking images and measuring cardiomyocytes size was blinded to genotype and diet.
Body Composition Analysis.
Mouse body composition analysis was performed using an EchoMRI 3-in-1 system (EchoMRI™, Houston, TX). Briefly, after machine calibration with an olive oil standard, conscious mice were restrained in a plastic tube and placed into the instrument bore. Fat, lean, free water, and total water mass was then determined. Imaging required <5 min per mouse, and following imaging, mice were immediately placed back into their home cage.
TAC+MI Surgically-Induced Heart Failure Model.
7-week old female WT C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME) were subjected to TAC+MI surgery as performed previously74,77. Mice were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine injected i.p. and were then restrained supine, intubated, and ventilated with a respirator (Harvard Apparatus, Holliston, MA). After shaving of the left anterior chest, the intercostal muscles were dissected, and aorta identified and freed by blunt dissection. 7–0 silk suture was placed around the transverse aorta and tied around a blunt 26-gauge needle. The needle was then removed after placement of the constrictor. Immediately following the first procedure, the LV and left main coronary artery system were exposed and the apical portion of the LAD was ligated with 9–0 silk suture. The surgical incision was closed, and the mice were recovered on a warmer until arousal from anesthesia whence they were returned to their home cage. All surgeries were performed in under 20 minutes. Mice were sacrificed 4 weeks after sham or TAC+MI surgery by CO2 asphyxiation after a 4 hour fast for collection of plasma and tissues.
Ketone Body Injection.
12-week old CS-MPC2−/− mice underwent echocardiography as detailed above and were then randomized into two groups to receive daily i.p. injections of either saline vehicle or 10 mmol/kg R-3-hydroxybutyric acid sodium salt (βHB) (Sigma, St. Louis, MO), which was pH’d to ~7.0. After 2 weeks of daily i.p. injection, echocardiography was repeated following the same procedures as detailed above. The following day, mice received a final saline or βHB injection, were fasted for 4 hours, and were euthanized by CO2 asphyxiation for collection of plasma and tissues.
Cardiac Glycogen Assay.
Glycogen concentrations were measured in a similar fashion as performed previously78. 15–60 mg of heart tissue was placed into 2 mL Eppendorf tubes and boiled in 300 μL of 30% KOH at 100°C for 30 minutes, with vortex mixing every 10 minutes. Tubes were cooled on ice and then 100 μL of 2% Na2SO4 and 800 μL of 100% EtOH was added and tubes vortexed. Tubes were boiled again for 5 minutes to aid in the precipitation of glycogen and then tubes centrifuged at 16,000 × g for 5 min and supernatant aspirated. The pellet was dissolved in 1 mL of 80% EtOH, vortexed and recentrifuged 16,000 × g for 5 min, and this wash was repeated 1 more time (3 total pelleting steps). The final pellets were resuspended in 200 μL of 0.3 mg/mL amyloglucosidase (Sigma, St. Louis, MO) in 0.2M sodium acetate. Serial dilutions of 10 mg/mL oyster glycogen (Sigma, St. Louis, MO) were prepared as standards. Samples and standards were then incubated in a 40°C water bath for 3 h. Samples and standards were then diluted 1:1 with H2O, and 5 μL of each added to a 96-well plate. 200 μL of glucose assay buffer (0.3 M Triethanolamine, pH~7.5, 4 mM MgCl2, 2 mM ATP, 0.9 mM NADP+, and 5 μg/mL Glucose-6-phosphate dehydrogenase [all from Sigma, St. Louis, MO]) was then added to each well, and the absorbance at 340 nm measured. 1 μg of Hexokinase (Sigma, St. Louis, MO) was then added to each well, and the plate incubated at room temperature in the dark for 30 min, then the absorbance again read at 340nm. The glycogen concentration was calculated from the difference in absorbance readings and plotted in relation to the oyster glycogen standards.
Statistical Analysis.
All data are presented as dot plots with mean +/− s.e.m., or as PRE-POST data points. All data was first entered into Microsoft Excel v16.4 and then imported into GraphPad Prism 8.4.2 for graphing and statistical analysis. Multiple comparisons were analyzed using a 2-way ANOVA with Tukey’s multiple-comparisons test. An unpaired, 2-tailed Student’s t test was used for comparison of 2 groups. A P value of less than 0.05 was considered statistically significant.
Reporting Summary.
Further information on research design and technical information is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1: Human heart failure gene expression and characterization of 6-week old CS-MPC2−/− mice.
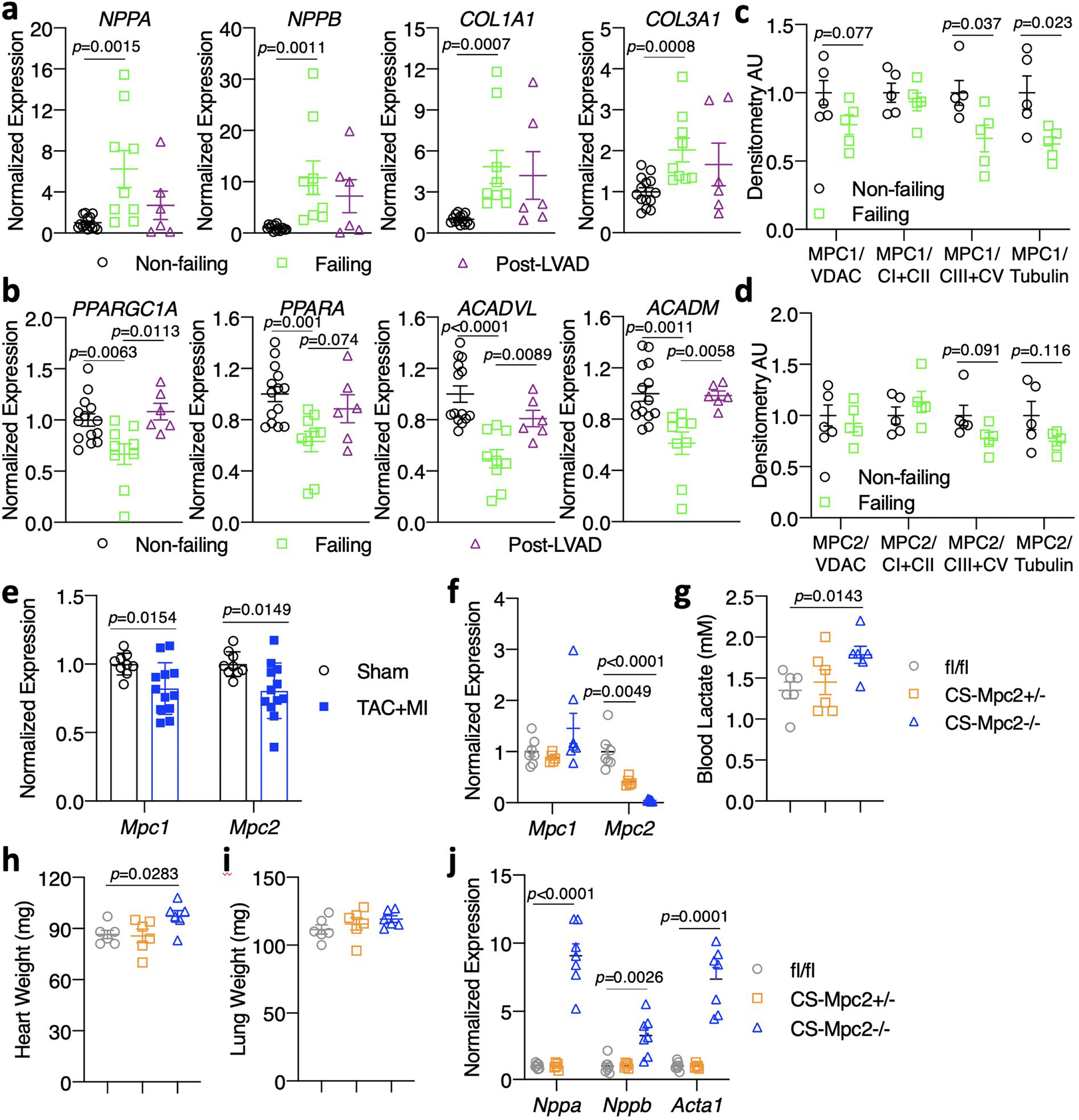
Gene expression from human non-failing, failing, and failing hearts after left ventricular assist device (LVAD) placement (n=14, 9, and 6 for Non-failing, Failing, and Post-LVAD, respectively). c-d, MPC1 and MPC2 protein expression quantification from non-failing and failing human hearts normalized to either VDAC, complex I and II, complexes III and IV, or Tubulin (n=5). e, Gene expression for Mpc1 and Mpc2 from wildtype C57BL6/J mouse hearts after sham or transverse aortic constriction plus myocardial infarction (TAC+MI) surgery (n=9 sham, 12 TAC-MI). f, Mouse heart gene expression for Mpc1 and Mpc2 (n=7, 5, 7 for fl/fl, +/−, −/− respectively). g, Blood lactate measured after a 4 h fast prior to sacrifice in 6-week old mice (n=6). h-i, Heart weight and lung weight of 6-week old mice (n=6). j, Mouse heart gene expression of heart failure, and hypertrophy genes from 6-week old mice (n= 7, 5, 7 for fl/fl, +/−, −/− respectively). Data are presented as mean ± s.e.m. within dot plot. Each data point represents one individual mouse or sample. Two-tailed unpaired Student’s t test.
Extended Data Fig. 2: Heart failure develops in CS-MPC2−/− mice, but not CS-MPC2+/− or mice treated with the MPC inhibitor MSDC-0602K.
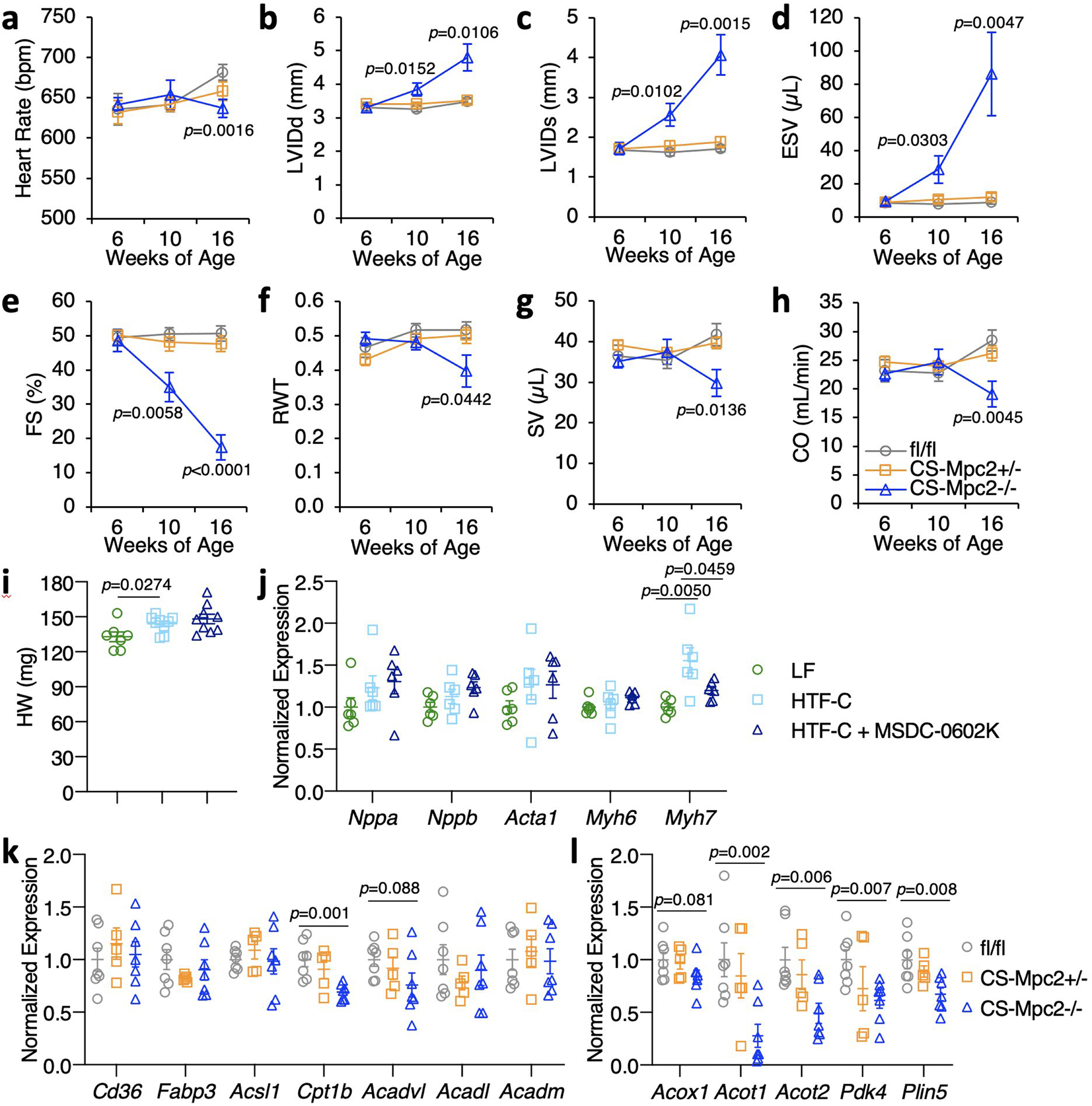
a-h, Serial echocardiography data of chow-fed mice at 6, 10, and 16 weeks of age. Left ventricular internal diameter at end diastole (LVIDd) and end systole (LVIDs), end systolic volume (ESV), fractional shortening (FS), relative wall thickness (RWT), stroke volume (SV), and cardiac output (CO) (n=7, 10, and 9 for fl/fl, +/−, and −/−, respectively). i, Heart weights from WT mice fed low fat (LF) diet or a high trans-fat, fructose, cholesterol (HTF-C) diet +/− 330 ppm MSDC-0602, an insulin-sensitizing MPC inhibitor (n=7, 9, and 9 for LF, HTF-C, and HTF-C+MSDC-0602K, respectively). j, Heart gene expression of hypertrophy gene markers from WT mice fed LF, HTF-C, or HTF-C + MSDC-0602 diets (n=6 for all groups). k-l, Heart gene expression for fatty acid transport and oxidation genes and PPAR⍺ target genes from chow-fed 16-week old mice after a 4 h fast (n= 7, 5, and 7 for fl/fl, +/−, and −/−, respectively). Data are presented as mean ± s.e.m., or mean ± s.e.m. within dot plot. Each data point represents one individual mouse or sample. Two-tailed unpaired Student’s t test.
Extended Data Fig. 3: Ketogenic diet prevents heart failure in CS-MPC2−/− mice.
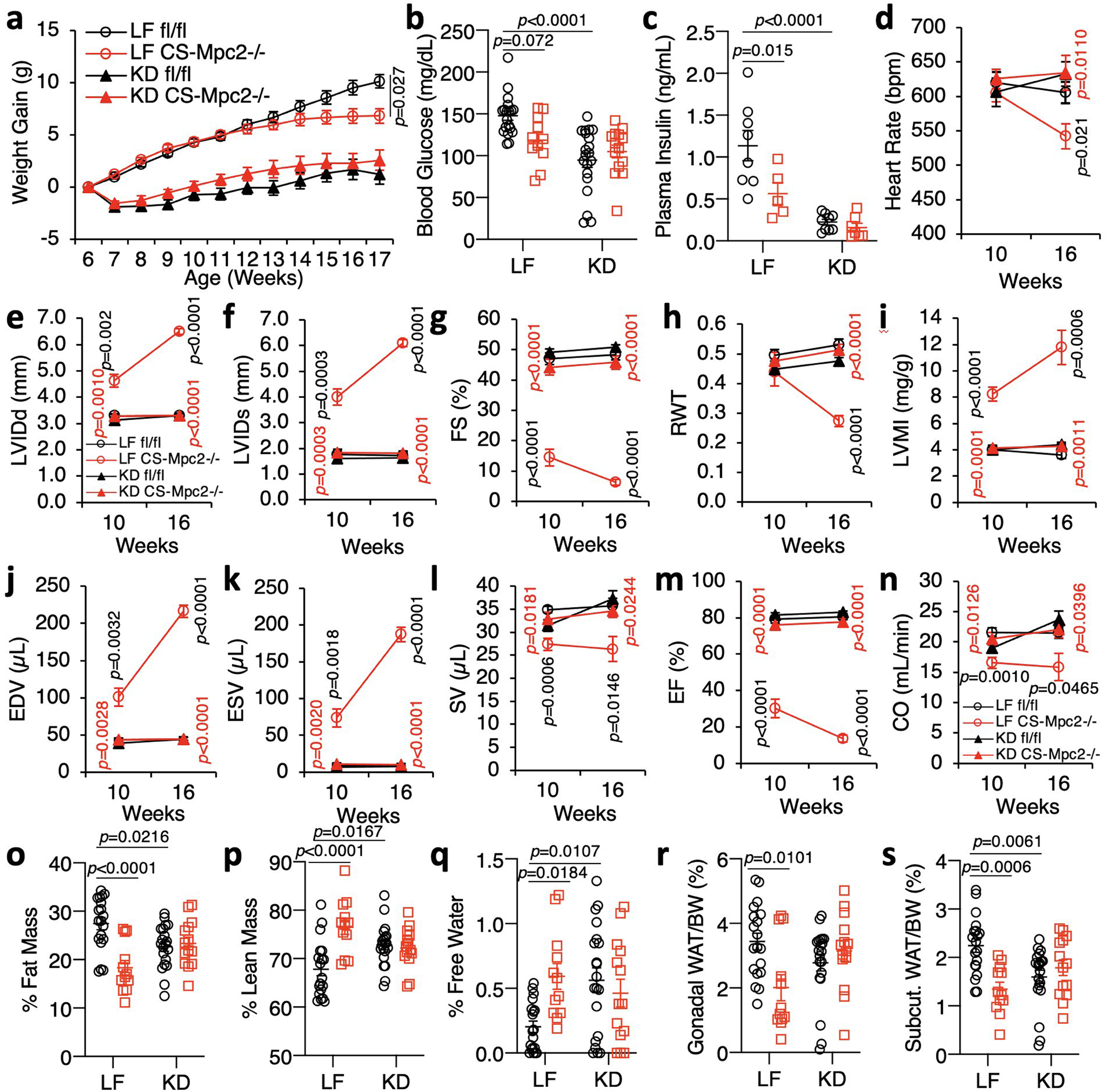
a, Body weights of mice fed low fat (LF) or ketogenic diet (KD) from 6–17 weeks of age (initial n=19, 15, 21, and 14 for fl/fl LF, CS-Mpc2−/− LF, fl/fl KD, and CS-Mpc2−/− KD, respectively)(fl/fl LF vs KD p<0.0001; CS-Mpc2−/− LF vs KD p<0.0001). b-c, Blood glucose and plasma insulin measured after a 4 h fast (n=19, 11, 20, and 14, respectively for glucose and 8, 5, 9, and 7, respectively for insulin). d-n, Echocardiography data at 10 and 16 weeks of age. Left ventricular internal diameter at end diastole (LVIDd) and end systole (LVIDs), fractional shortening (FS), relative wall thickness (RWT), end diastolic volume (EDV), end systolic volume (ESV), stroke volume (SV), ejection fraction (EF), and cardiac output (CO) (n=9, 7, 12, and 9 for fl/fl LF, CS-Mpc2−/− LF, fl/fl KD, and CS-Mpc2−/− KD, respectively). o-q, % Fat mass, % lean mass, and % free water body composition measured by echoMRI (n= 19, 12, 20, and 14, respectively). r-s, Gonadal and inguinal white adipose tissue (WAT) weights normalized to body weight (n=19, 12, 20, and 14, respectively). Data are presented as mean ± s.e.m. or mean ± s.e.m. within dot plot. Each data point in dot plot represents one individual mouse sample. Two-way ANOVA with Tukey’s multiple comparisons test. For d-n, black p values indicate LF-fed fl/fl vs. CS-Mpc2−/−, red p values indicate LF vs. KD for CS-Mpc2−/− for each echocardiography date.
Extended Data Fig. 4: Ketone body injection modestly reduces cardiac remodeling in CS-MPC2−/− mice.
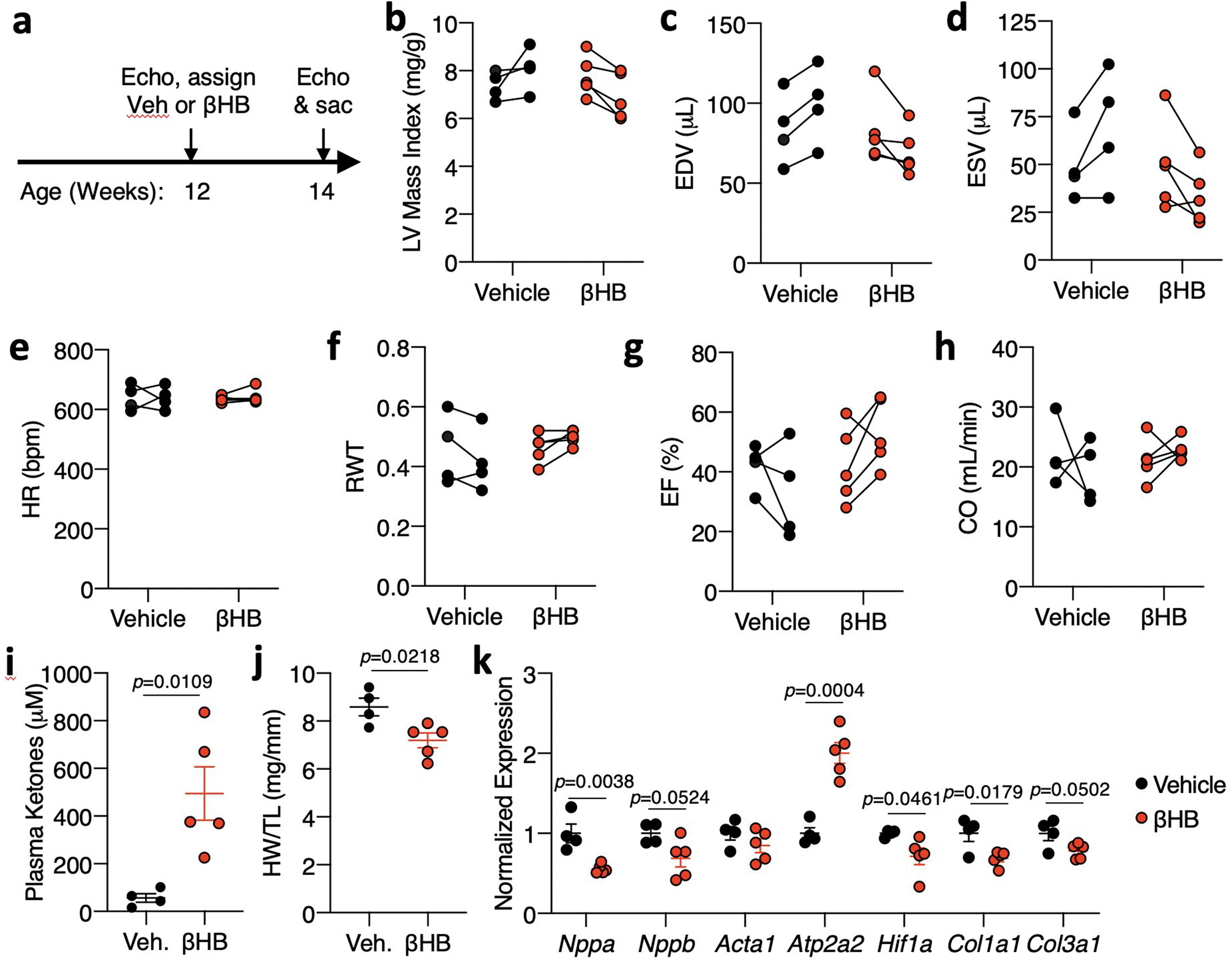
a, Timeline for β-hydroxybutyrate (βHB) injection experiment in which CS-MPC2−/− mice were injected i.p. with saline vehicle or 10 mmol/kg βHB daily from 12 to 14 weeks of age. b-h, Echocardiography measurements before and after 2 weeks of daily i.p. injection of saline vehicle (Veh) or 10mmol/kg β-hydroxybutyrate. Left ventricular (LV) mass index, end-diastolic volume (EDV), end-systolic volume (ESV), heart rate (HR), relative wall thickness (RWT), ejection fraction (EF), and cardiac output (CO) (n=4 Veh, 5 βHB). i, Plasma total ketone body concentrations (n= 4 Veh, 5 βHB). j, Heart weight normalized to tibia length (n= 4 Veh, 5 βHB). k, Gene expression markers of hypertrophy, heart failure, and fibrosis from hearts after 2 weeks of daily vehicle or βHB treatment (n= 4 Veh, 5 βHB). Data presented either as PRE-POST, or mean ± s.e.m. shown within dot plot. Each symbol represents an individual sample. Two-tailed unpaired Student’s t test.
Extended Data Fig. 5: Ketone ester diet does not improve cardiac remodeling or function in CS-MPC2−/− mice.
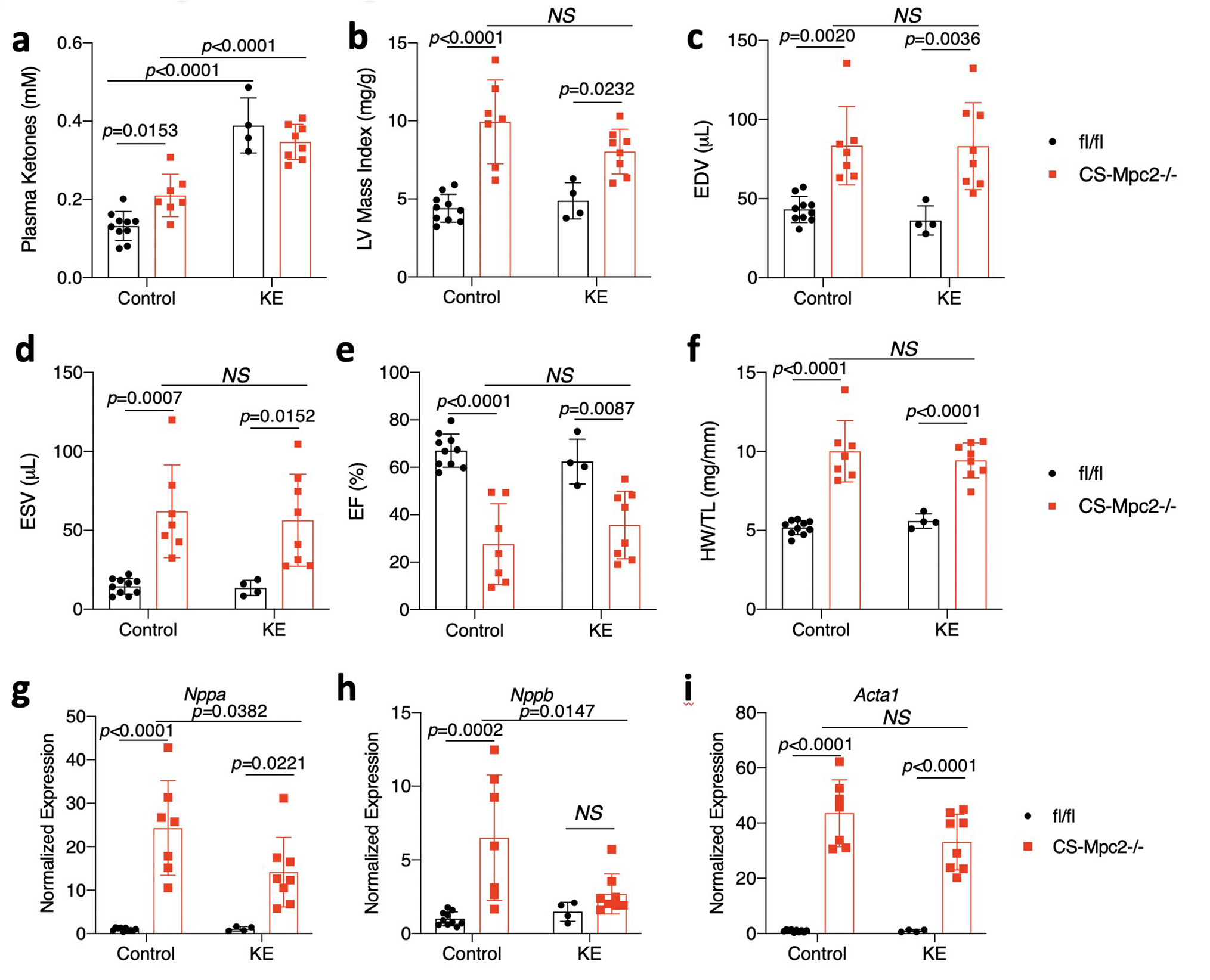
a, Plasma ketone bodies measured from mice fed either control or ketone ester (KE)-supplemented diet (n=10, 7, 4, and 8, respectively). b-e, Echocardiography measurements after 6 weeks of KE diet feeding. Left ventricular (LV) mass index, end-diastolic volume (EDV), end-systolic volume (ESV), and ejection fraction (EF) (n= 10, 7, 4, and 8, respectively). f, Heart weight normalized to tibia length (n= 10, 7, 4, and 8, respectively). g-i, Cardiac gene expression markers of hypertrophy and heart failure (Nppa, Nppb, Acta1) (n= 10, 7, 4, and 8, respectively). Data presented as mean ± s.e.m. shown within dot plot. Each symbol represents an individual sample. Two-way ANOVA with Tukey’s multiple comparisons test.
Extended Data Fig. 6: High fat diets also greatly improve cardiac remodeling and function of CS-MPC2−/− mice.
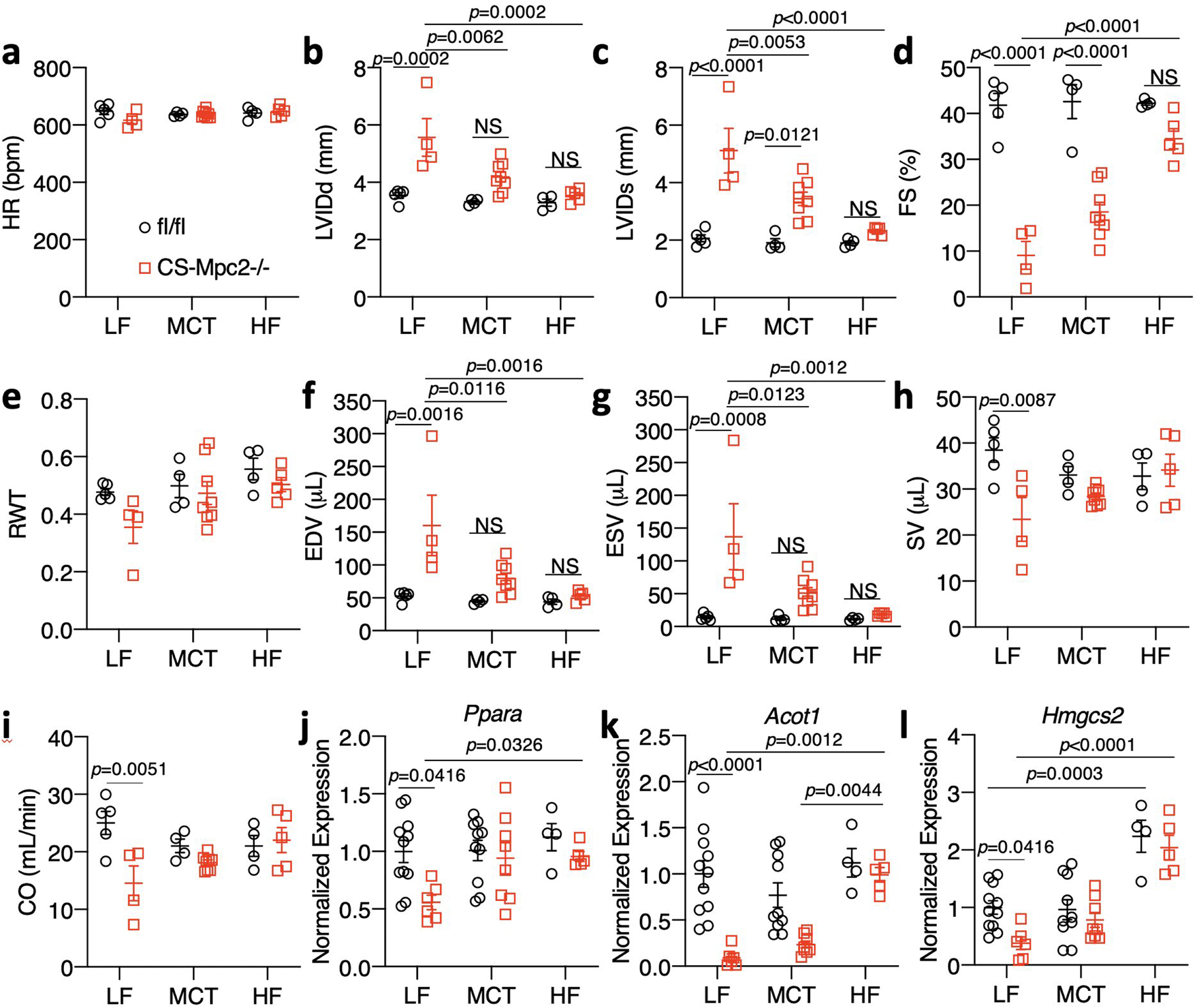
a-I, Echocardiography measurements taken at 16 weeks of age after 10 weeks of low fat (LF), medium chain triglyceride (MCT), or high-fat (HF) feeding. Left ventricular internal diameter at end diastole (LVIDd) and end systole (LVIDs), fractional shortening (FS), relative wall thickness (RWT), end diastolic volume (EDV), end systolic volume (ESV), stroke volume (SV), and cardiac output (CO) (n=5, 4, 4, 8, 4, and 5, respectively). j-l, Cardiac gene expression for Ppara and it’s targets Acot1 and Hmgcs2 (n=11, 6, 10, 8, 4, and 5, respectively). Data are presented as mean ± s.e.m. within dot plot. Each data point represents an individual mouse. Two-way ANOVA with Tukey’s multiple comparisons test.
Extended Data Fig. 7: A 24 hour fast improves cardiac remodeling by enhancing fat oxidation.
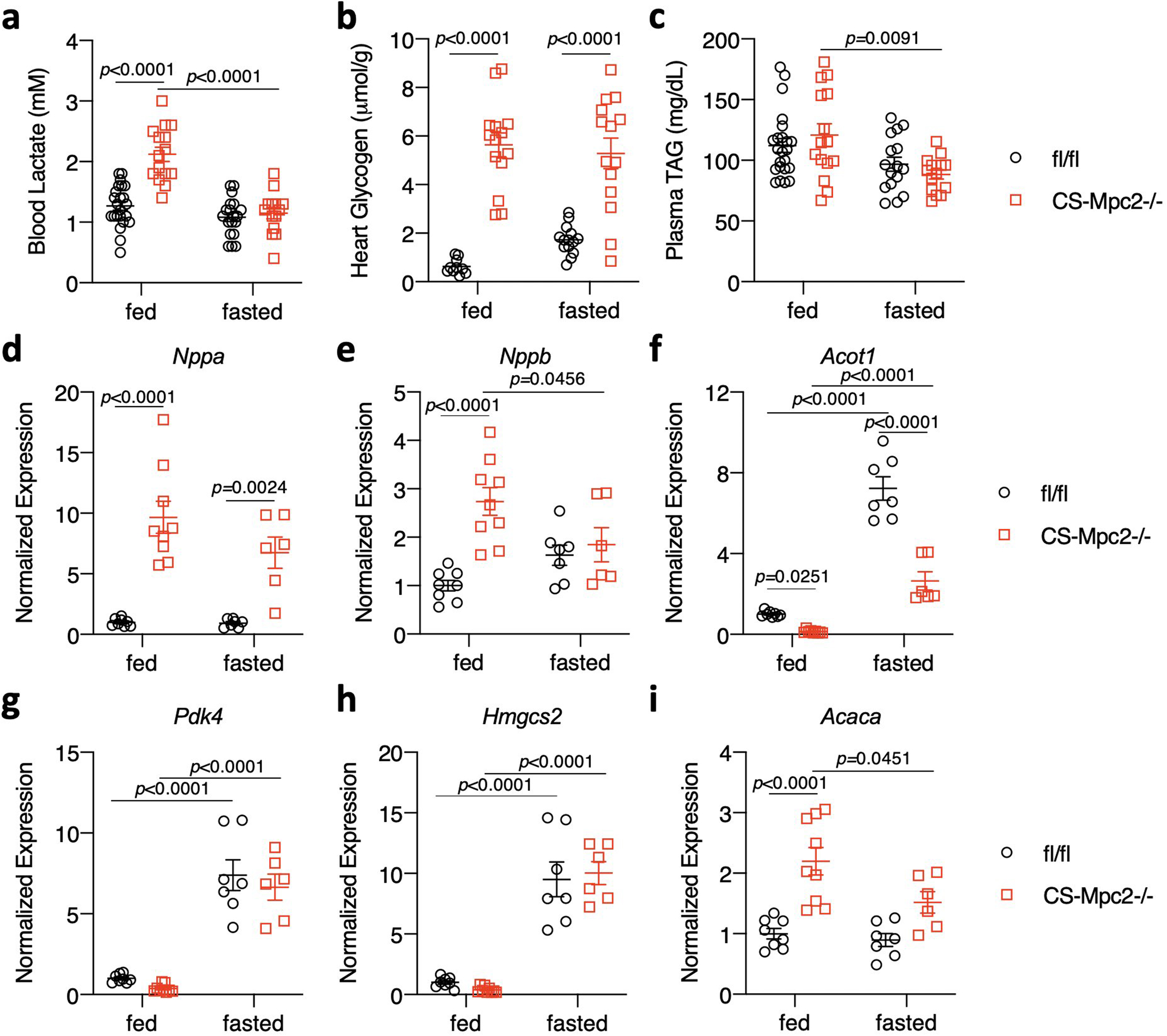
a, Blood lactate of fed or fasted mice just prior to euthanasia (n=22, 15, 16, and 14, respectively). b, Cardiac glycogen concentrations in hearts of fed and fasted mice (n=10, 14, 15, and 14, respectively). c, Plasma TAG from fed or fasted mice (n= 22, 15, 16, and 14, respectively). d-i, Cardiac gene expression for natriuretic peptides and PPAR⍺-target and fatty acid metabolism genes (n=8, 9, 7, and 6, respectively). Data are presented as mean ± s.e.m. within dot plot. Each symbol on dot plot represents an individual sample. Two-way ANOVA with Tukey’s multiple comparisons test.
Extended Data Fig. 8: Ketogenic diet reverses heart failure in CS-MPC2−/− mice.
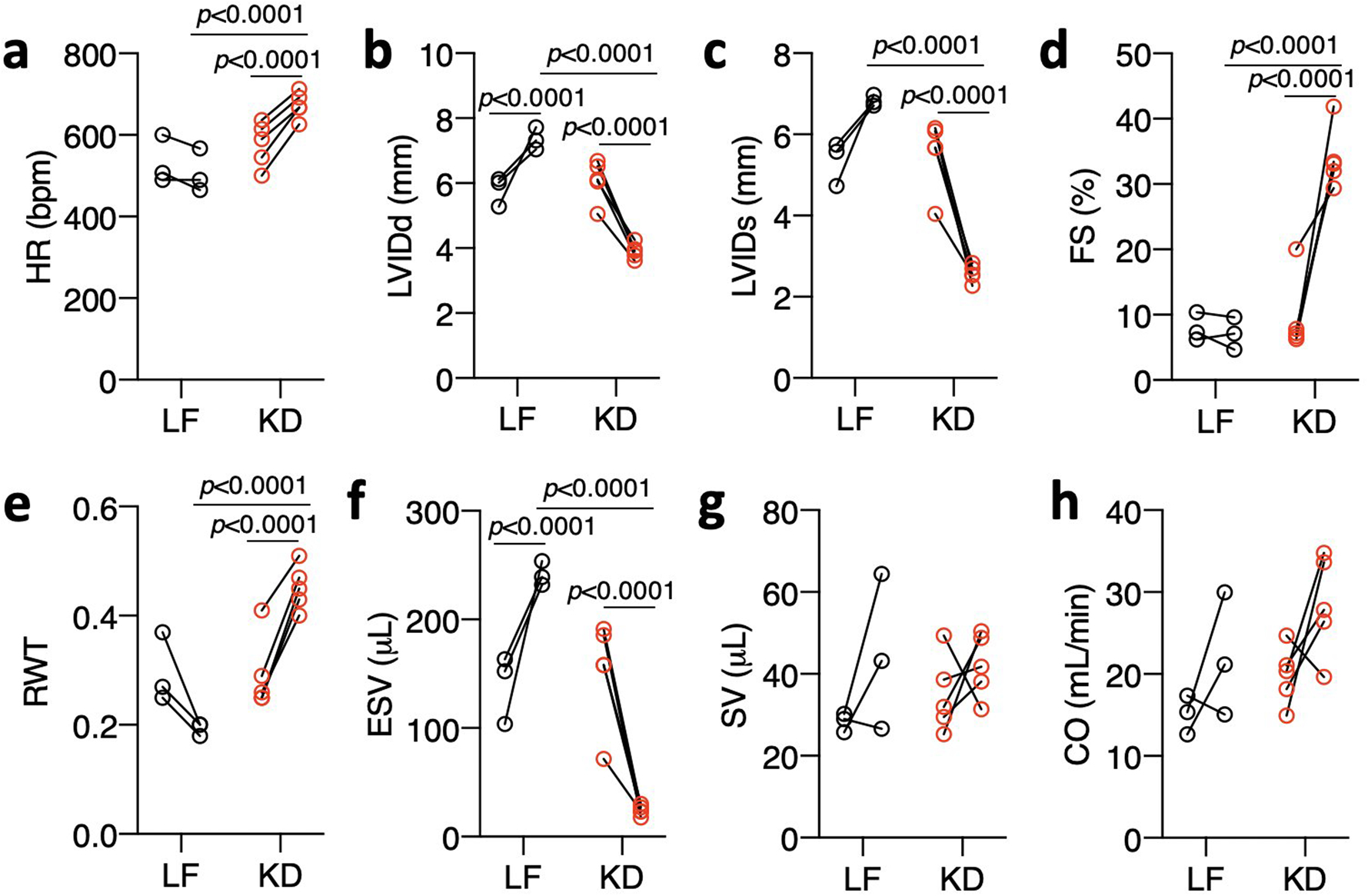
a-h, Echocardiography measurements before and after 3 weeks of LF or KD-feeding in 16-week-old CS-MPC2−/− mice with established heart failure (n=3 LF, 5 KD). Data are presented as PRE-POST. Each data point represents an individual mouse. Paired two-tailed student’s t-test for PRE vs. POST. Unpaired two-tailed student’s t-test for LF vs. KD.
Supplementary Material
ACKNOWLEDGEMENTS
Sadly, Dr. Richard (Bud) Veech passed away at the age of 84 during the preparation of this manuscript. We thank him for providing the ketone ester diet and his enthusiasm towards this project. This work was supported by core resources of the Nutrition Obesity Research Center (NORC) (P30 DK56341), Diabetes Research Center (DRC) (P30 DK020579), and Institute for Clinical and Translational Sciences (ICTS) (UL1 TR002345) at the Washington University School of Medicine. NIH grants K99/R00 HL136658 (to KSM), R01 HL133178 (to RWG), and R01 HL119225 and R01 DK104735 (to BNF) supported these studies.
Footnotes
DATA AVAILABILITY STATEMENT
All data from these studies are contained within this manuscript, the figures, and extended/supplemental figures and tables. Data are also available from the corresponding author upon reasonable request.
COMPETING INTERESTS
KSM previously received research support from Cirius Therapeutics, and BNF is a stockholder and scientific advisory board member of Cirius Therapeutics. RLV held patents on the synthesis and uses of ketone esters, and MTK is a co-inventor in the synthesis of ketone esters. All other authors have declared that no conflict of interest exists.
REFERENCES
- 1.Bing RJ, Siegel A, Ungar I & Gilbert M Metabolism of the human heart. II. Studies on fat, ketone and amino acid metabolism. Am J Med 16, 504–515, doi: 10.1016/0002-9343(54)90365-4 (1954). [DOI] [PubMed] [Google Scholar]
- 2.Wisneski JA, Gertz EW, Neese RA & Mayr M Myocardial metabolism of free fatty acids. Studies with 14C-labeled substrates in humans. J Clin Invest 79, 359–366, doi: 10.1172/JCI112820 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopaschuk GD & Spafford MA Energy substrate utilization by isolated working hearts from newborn rabbits. Am J Physiol 258, H1274–1280, doi: 10.1152/ajpheart.1990.258.5.H1274 (1990). [DOI] [PubMed] [Google Scholar]
- 4.Glatz JF & Veerkamp JH Postnatal development of palmitate oxidation and mitochondrial enzyme activities in rat cardiac and skeletal muscle. Biochim Biophys Acta 711, 327–335, doi: 10.1016/0005-2760(82)90042-x (1982). [DOI] [PubMed] [Google Scholar]
- 5.Lopaschuk GD, Spafford MA & Marsh DR Glycolysis is predominant source of myocardial ATP production immediately after birth. Am J Physiol 261, H1698–1705, doi: 10.1152/ajpheart.1991.261.6.H1698 (1991). [DOI] [PubMed] [Google Scholar]
- 6.Kaijser L & Berglund B Myocardial lactate extraction and release at rest and during heavy exercise in healthy men. Acta Physiol Scand 144, 39–45, doi: 10.1111/j.1748-1716.1992.tb09265.x (1992). [DOI] [PubMed] [Google Scholar]
- 7.Vanoverschelde JL et al. Competition between palmitate and ketone bodies as fuels for the heart: study with positron emission tomography. Am J Physiol 264, H701–707, doi: 10.1152/ajpheart.1993.264.3.H701 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Jeffrey FM, Diczku V, Sherry AD & Malloy CR Substrate selection in the isolated working rat heart: effects of reperfusion, afterload, and concentration. Basic Res Cardiol 90, 388–396 (1995). [DOI] [PubMed] [Google Scholar]
- 9.Stanley WC, Meadows SR, Kivilo KM, Roth BA & Lopaschuk GD beta-Hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-CoA content. Am J Physiol Heart Circ Physiol 285, H1626–1631, doi: 10.1152/ajpheart.00332.2003 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Garnier A et al. Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J Physiol 551, 491–501, doi: 10.1113/jphysiol.2003.045104 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heinke MY et al. Changes in myocardial protein expression in pacing-induced canine heart failure. Electrophoresis 20, 2086–2093, doi: 10.1002/(SICI)1522-2683(19990701)20:10<2086::AID-ELPS2086>3.0.CO;2-4 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Ide T et al. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 88, 529–535, doi: 10.1161/01.res.88.5.529 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Marin-Garcia J, Goldenthal MJ & Moe GW Abnormal cardiac and skeletal muscle mitochondrial function in pacing-induced cardiac failure. Cardiovasc Res 52, 103–110, doi: 10.1016/s0008-6363(01)00368-6 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Sack MN et al. Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 94, 2837–2842, doi: 10.1161/01.cir.94.11.2837 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Warren JS, Oka SI, Zablocki D & Sadoshima J Metabolic reprogramming via PPARalpha signaling in cardiac hypertrophy and failure: From metabolomics to epigenetics. Am J Physiol Heart Circ Physiol 313, H584–H596, doi: 10.1152/ajpheart.00103.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barger PM, Brandt JM, Leone TC, Weinheimer CJ & Kelly DP Deactivation of peroxisome proliferator-activated receptor-alpha during cardiac hypertrophic growth. J Clin Invest 105, 1723–1730, doi: 10.1172/JCI9056 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taegtmeyer H & Overturf ML Effects of moderate hypertension on cardiac function and metabolism in the rabbit. Hypertension 11, 416–426 (1988). [DOI] [PubMed] [Google Scholar]
- 18.Zhabyeyev P et al. Pressure-overload-induced heart failure induces a selective reduction in glucose oxidation at physiological afterload. Cardiovasc Res 97, 676–685, doi: 10.1093/cvr/cvs424 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Herzig S et al. Identification and functional expression of the mitochondrial pyruvate carrier. Science 337, 93–96, doi: 10.1126/science.1218530 (2012). [DOI] [PubMed] [Google Scholar]
- 20.Bricker DK et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100, doi: 10.1126/science.1218099 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chambers KT et al. Chronic inhibition of pyruvate dehydrogenase in heart triggers an adaptive metabolic response. J Biol Chem 286, 11155–11162, doi: 10.1074/jbc.M110.217349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao G et al. Overexpression of pyruvate dehydrogenase kinase 4 in heart perturbs metabolism and exacerbates calcineurin-induced cardiomyopathy. Am J Physiol Heart Circ Physiol 294, H936–943, doi: 10.1152/ajpheart.00870.2007 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Gopal K et al. Cardiac-Specific Deletion of Pyruvate Dehydrogenase Impairs Glucose Oxidation Rates and Induces Diastolic Dysfunction. Front Cardiovasc Med 5, 17, doi: 10.3389/fcvm.2018.00017 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun W et al. Cardiac-Specific Deletion of the Pdha1 Gene Sensitizes Heart to Toxicological Actions of Ischemic Stress. Toxicol Sci 151, 193–203, doi: 10.1093/toxsci/kfw035 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sheeran FL, Angerosa J, Liaw NY, Cheung MM & Pepe S Adaptations in Protein Expression and Regulated Activity of Pyruvate Dehydrogenase Multienzyme Complex in Human Systolic Heart Failure. Oxid Med Cell Longev 2019, 4532592, doi: 10.1155/2019/4532592 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCommis KS et al. Loss of mitochondrial pyruvate carrier 2 in the liver leads to defects in gluconeogenesis and compensation via pyruvate-alanine cycling. Cell Metab 22, 682–694, doi: 10.1016/j.cmet.2015.07.028 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCommis KS et al. An ancestral role for the mitochondrial pyruvate carrier in glucose-stimulated insulin secretion. Mol Metab 5, 602–614, doi: 10.1016/j.molmet.2016.06.016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCommis KS et al. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology 65, 1543–1556, doi: 10.1002/hep.29025 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen J, Kubalak SW & Chien KR Ventricular muscle-restricted targeting of the RXRalpha gene reveals a non-cell-autonomous requirement in cardiac chamber morphogenesis. Development 125, 1943–1949 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Halestrap AP & Denton RM The specificity and metabolic implications of the inhibition of pyruvate transport in isolated mitochondria and intact tissue preparations by alpha-Cyano-4-hydroxycinnamate and related compounds. Biochem J 148, 97–106 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aubert G et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 133, 698–705, doi: 10.1161/CIRCULATIONAHA.115.017355 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bedi KC Jr. et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 133, 706–716, doi: 10.1161/CIRCULATIONAHA.115.017545 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horton JL et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 4, e124079, doi: 10.1172/jci.insight.124079 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sciarretta S, Forte M, Frati G & Sadoshima J New Insights Into the Role of mTOR Signaling in the Cardiovascular System. Circ Res 122, 489–505, doi: 10.1161/CIRCRESAHA.117.311147 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wentz AE et al. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J Biol Chem 285, 24447–24456, doi: 10.1074/jbc.M110.100651 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ford DA, Han X, Horner CC & Gross RW Accumulation of unsaturated acylcarnitine molecular species during acute myocardial ischemia: metabolic compartmentalization of products of fatty acyl chain elongation in the acylcarnitine pool. Biochemistry 35, 7903–7909, doi: 10.1021/bi960552n (1996). [DOI] [PubMed] [Google Scholar]
- 37.Martin MA et al. Myocardial carnitine and carnitine palmitoyltransferase deficiencies in patients with severe heart failure. Biochim Biophys Acta 1502, 330–336, doi: 10.1016/s0925-4439(00)00061-2 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Stanley WC, Recchia FA & Lopaschuk GD Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85, 1093–1129, doi: 10.1152/physrev.00006.2004 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Shearman MS & Halestrap AP The concentration of the mitochondrial pyruvate carrier in rat liver and heart mitochondria determined with alpha-cyano-beta-(1-phenylindol-3-yl)acrylate. Biochem J 223, 673–676 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hildyard JC, Ammala C, Dukes ID, Thomson SA & Halestrap AP Identification and characterisation of a new class of highly specific and potent inhibitors of the mitochondrial pyruvate carrier. Biochim Biophys Acta 1707, 221–230, doi: 10.1016/j.bbabio.2004.12.005 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Bunger R & Mallet RT Mitochondrial pyruvate transport in working guinea-pig heart. Work-related vs. carrier-mediated control of pyruvate oxidation. Biochim Biophys Acta 1151, 223–236 (1993). [DOI] [PubMed] [Google Scholar]
- 42.Fernandez-Caggiano M et al. Analysis of Mitochondrial Proteins in the Surviving Myocardium after Ischemia Identifies Mitochondrial Pyruvate Carrier Expression as Possible Mediator of Tissue Viability. Mol Cell Proteomics 15, 246–255, doi: 10.1074/mcp.M115.051862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandez-Caggiano M K. A; Francois AA; Prysyazhna O; Eykyn T; Krasemann S; Crespo-Leiro MG; Garcia Vieites M; Domenech N; Eaton P Mitochondrial pyruvate carrier abundance mediates pathological cardiac hypertrophy. Nature Metabolism (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y T. PV; Cochran JD; Robillard Frayne I; Maximilian Marx J; Soto J; Pewa AD; Tayyari F; Teesch LM; Rauckhorst AJ; Gray LR; Puchalska P; Funari TR; McGlauflin R; Zimmerman K; Kutschke WJ; Cassier T; Hitchcock S; Lin K; Kato KM; Stueve JL; Haff L; Weiss RM; Rutter J; Taylor EB; Crawford PA; Lewandowski ED; Des Rosiers C; Abel ED Mitochondrial pyruvate carriers are required for myocardial stress adaptation. Nature Metabolism (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malloy CR, Sherry AD & Jeffrey FM Evaluation of carbon flux and substrate selection through alternate pathways involving the citric acid cycle of the heart by 13C NMR spectroscopy. J Biol Chem 263, 6964–6971 (1988). [PubMed] [Google Scholar]
- 46.Sundqvist KE, Hiltunen JK & Hassinen IE Pyruvate carboxylation in the rat heart. Role of biotin-dependent enzymes. Biochem J 257, 913–916, doi: 10.1042/bj2570913 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Karlstaedt A et al. Oncometabolite d-2-hydroxyglutarate impairs alpha-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc Natl Acad Sci U S A 113, 10436–10441, doi: 10.1073/pnas.1601650113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Akbay EA et al. D-2-hydroxyglutarate produced by mutant IDH2 causes cardiomyopathy and neurodegeneration in mice. Genes Dev 28, 479–490, doi: 10.1101/gad.231233.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karlstaedt A, Khanna R, Thangam M & Taegtmeyer H Glucose 6-Phosphate Accumulates via Phosphoglucose Isomerase Inhibition in Heart Muscle. Circ Res 126, 60–74, doi: 10.1161/CIRCRESAHA.119.315180 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ritterhoff J et al. Metabolic Remodeling Promotes Cardiac Hypertrophy by Directing Glucose to Aspartate Biosynthesis. Circ Res 126, 182–196, doi: 10.1161/CIRCRESAHA.119.315483 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sung MM et al. AMPK deficiency in cardiac muscle results in dilated cardiomyopathy in the absence of changes in energy metabolism. Cardiovasc Res 107, 235–245, doi: 10.1093/cvr/cvv166 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xing Y et al. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J Biol Chem 278, 28372–28377, doi: 10.1074/jbc.M303521200 (2003). [DOI] [PubMed] [Google Scholar]
- 53.Zarrinpashneh E et al. AMPKalpha2 counteracts the development of cardiac hypertrophy induced by isoproterenol. Biochem Biophys Res Commun 376, 677–681, doi: 10.1016/j.bbrc.2008.09.057 (2008). [DOI] [PubMed] [Google Scholar]
- 54.Kim M et al. Mutation in the gamma2-subunit of AMP-activated protein kinase stimulates cardiomyocyte proliferation and hypertrophy independent of glycogen storage. Circ Res 114, 966–975, doi: 10.1161/CIRCRESAHA.114.302364 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nielsen R et al. Cardiovascular Effects of Treatment With the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 139, 2129–2141, doi: 10.1161/CIRCULATIONAHA.118.036459 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Uchihashi M et al. Cardiac-Specific Bdh1 Overexpression Ameliorates Oxidative Stress and Cardiac Remodeling in Pressure Overload-Induced Heart Failure. Circ Heart Fail 10, doi: 10.1161/CIRCHEARTFAILURE.117.004417 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Schugar RC et al. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol Metab 3, 754–769, doi: 10.1016/j.molmet.2014.07.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pereyra AS et al. Loss of cardiac carnitine palmitoyltransferase 2 results in rapamycin-resistant, acetylation-independent hypertrophy. J Biol Chem 292, 18443–18456, doi: 10.1074/jbc.M117.800839 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Halestrap AP Pyruvate and ketone-body transport across the mitochondrial membrane. Exchange properties, pH-dependence and mechanism of the carrier. Biochem J 172, 377–387 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gray LR et al. Hepatic Mitochondrial Pyruvate Carrier 1 Is Required for Efficient Regulation of Gluconeogenesis and Whole-Body Glucose Homeostasis. Cell Metab 22, 669–681, doi: 10.1016/j.cmet.2015.07.027 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen J et al. Selective requirement of myosin light chain 2v in embryonic heart function. J Biol Chem 273, 1252–1256 (1998). [DOI] [PubMed] [Google Scholar]
- 62.Vigueira PA et al. Mitochondrial pyruvate carrier 2 hypomorphism in mice leads to defects in glucose-stimulated insulin secretion. Cell Rep 7, 2042–2053, doi: 10.1016/j.celrep.2014.05.017 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bowman CE, Zhao L, Hartung T & Wolfgang MJ Requirement for the mitochondrial pyruvate carrier in mammalian development revealed by a hypomorphic allelic series. Mol Cell Biol 36, 2089–2104, doi: 10.1128/MCB.00166-16 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Exil VJ et al. Very-long-chain acyl-coenzyme a dehydrogenase deficiency in mice. Circ Res 93, 448–455, doi: 10.1161/01.RES.0000088786.19197.E4 (2003). [DOI] [PubMed] [Google Scholar]
- 65.Hainline BE, Kahlenbeck DJ, Grant J & Strauss AW Tissue specific and developmental expression of rat long-and medium-chain acyl-CoA dehydrogenases. Biochim Biophys Acta 1216, 460–468, doi: 10.1016/0167-4781(93)90015-6 (1993). [DOI] [PubMed] [Google Scholar]
- 66.Kelly DP et al. Nucleotide sequence of medium-chain acyl-CoA dehydrogenase mRNA and its expression in enzyme-deficient human tissue. Proc Natl Acad Sci U S A 84, 4068–4072, doi: 10.1073/pnas.84.12.4068 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.An J et al. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat Med 10, 268–274, doi: 10.1038/nm995 (2004). [DOI] [PubMed] [Google Scholar]
- 68.Ferrara CT et al. Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling. PLoS Genet 4, e1000034, doi: 10.1371/journal.pgen.1000034 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Millington DS & Stevens RD Acylcarnitines: analysis in plasma and whole blood using tandem mass spectrometry. Methods Mol Biol 708, 55–72, doi: 10.1007/978-1-61737-985-7_3 (2011). [DOI] [PubMed] [Google Scholar]
- 70.Chace DH et al. Rapid diagnosis of phenylketonuria by quantitative analysis for phenylalanine and tyrosine in neonatal blood spots by tandem mass spectrometry. Clin Chem 39, 66–71 (1993). [PubMed] [Google Scholar]
- 71.Jensen MV et al. Compensatory responses to pyruvate carboxylase suppression in islet beta-cells. Preservation of glucose-stimulated insulin secretion. J Biol Chem 281, 22342–22351, doi: 10.1074/jbc.M604350200 (2006). [DOI] [PubMed] [Google Scholar]
- 72.Khomtchouk BB, Hennessy JR & Wahlestedt C shinyheatmap: Ultra fast low memory heatmap web interface for big data genomics. PLoS One 12, e0176334, doi: 10.1371/journal.pone.0176334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gao L et al. Simultaneous quantification of malonyl-CoA and several other short-chain acyl-CoAs in animal tissues by ion-pairing reversed-phase HPLC/MS. J Chromatogr B Analyt Technol Biomed Life Sci 853, 303–313, doi: 10.1016/j.jchromb.2007.03.029 (2007). [DOI] [PubMed] [Google Scholar]
- 74.Weinheimer CJ, Lai L, Kelly DP & Kovacs A Novel mouse model of left ventricular pressure overload and infarction causing predictable ventricular remodelling and progression to heart failure. Clin Exp Pharmacol Physiol 42, 33–40, doi: 10.1111/1440-1681.12318 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Teichholz LE, Kreulen T, Herman MV & Gorlin R Problems in echocardiographic volume determinations: echocardiographic-angiographic correlations in the presence of absence of asynergy. Am J Cardiol 37, 7–11, doi: 10.1016/0002-9149(76)90491-4 (1976). [DOI] [PubMed] [Google Scholar]
- 76.Kronik G, Slany J & Mosslacher H Comparative value of eight M-mode echocardiographic formulas for determining left ventricular stroke volume. A correlative study with thermodilution and left ventricular single-plane cineangiography. Circulation 60, 1308–1316, doi: 10.1161/01.cir.60.6.1308 (1979). [DOI] [PubMed] [Google Scholar]
- 77.Weinheimer CJ et al. Load-Dependent Changes in Left Ventricular Structure and Function in a Pathophysiologically Relevant Murine Model of Reversible Heart Failure. Circ Heart Fail 11, e004351, doi: 10.1161/CIRCHEARTFAILURE.117.004351 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Suzuki Y et al. Insulin control of glycogen metabolism in knockout mice lacking the muscle-specific protein phosphatase PP1G/RGL. Mol Cell Biol 21, 2683–2694, doi: 10.1128/MCB.21.8.2683-2694.2001 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.