

Juvenile dermatomyositis (JDM) is a rare systemic autoimmune vasculopathy characterised by weakness in proximal muscles and pathognomonic skin rashes.1 Clinically, some patients are refractory to any available treatments or became steroids dependent.2 The adverse reactions of long-term use of steroids are severe; therefore, more effective and safer medications are urgently needed. JAK inhibitors (JAKi) can reduce interferon (IFN)-induced STAT1 phosphorylation and block the JAK-STAT pathway, demonstrating a therapeutic potential of inflammation control in JDM.3 The successful uses of JAKi were reported in adult dermatomyositis (DM) and two patients with JDM.3–5 Here, we want to share the JAKi using experiences of 25 refractory JDM cases who were diagnosed and classified according to Bohan and Peter’s criteria and treated between November 2017 and May 2019. Written informed consents were obtained from the guardians of all patients before starting the treatment.
Among 25 cases, 44% (11/25) patients were female, the mean age of onset was 4.6±3.3 years and the mean age to start add-on JAKi treatment was 7.2±4.0 years. The mean disease course of the 25 JDM patients before JAKi treatment is 21.0 months (range: 14.0–36.5). All cases are refractory JDMs, including 32% (8/25) ineffective patients and 68% (17/25) glucocorticoid-dependent cases. After routine treatment fails, they received JAKi for 3–18 months as an off-label use. In subsequent JAKi treatment, 28% (7/25) used tofacitinib, and 72% (18/25) used ruxolitinib. In patients of <25 kg (n=11), the initial dosage was 2.5 mg twice daily, and in patients of ≥25 kg (n=14), the initial dosage was 5 mg twice daily, and one patient required the maximum dosage of 7.5 mg twice daily.
The 25 patients were followed for a median of 7.0 months (range: 3–21 months). Ninety-six per cent (24/25) had rash when JAKi was added, and all showed improved rashes, including 66.7% (16/24) cases of complete resolution. In patients with rash, rashes started to improve after 1.0 (0.6–2.0) weeks of JAKi and showed obvious improvement after 2.5 (2.0–4.0) weeks of JAKi. No clinically observable rash could be seen after 12.0 (8.0–24.0) weeks of JAKi. The Cutaneous Assessment Tool Binary Method score decreased dramatically from 7.0 (3.0–10.0) to 0.0 (0.0–1.0) (p<0.001). figure 1 shows a girl with a long-term refractory rash that disappeared after gradually increasing the JAKi dose to 7.5 mg twice daily. Up to the last follow-up in August 2019, 28% (7/25) patients discontinued glucocorticoids including this girl. There is currently only one case relapsed, the rash disappeared after 8 weeks of JAKi but recurred 4 weeks later and JAKi was stopped at 12 weeks.
Figure 1.
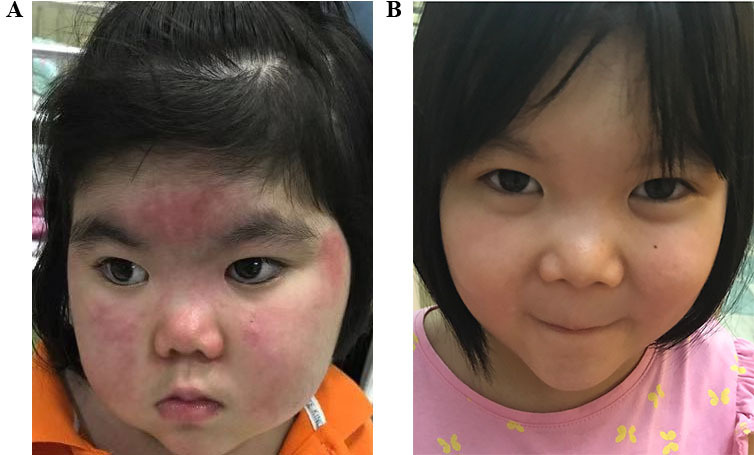
A typical case of a girl >25 kg who received an initial dose of 5 mg twice daily of Janus kinase inhibitor (JAKi) but required the maximum dose of 7.5 mg twice daily. She received this dose for 6 months, and the dose was gradually tapered. In the meantime, glucocorticoids were also tapered, and the patient showed an increased growth rate. (A) The typical skin lesions before treatment with JAKi. (B) Those lesions had mostly disappeared after treatment.
Additionally, 10/25 (40%) patients had decreased muscle strength, and 4% (1/25) had continuous high levels of muscle enzymes. After treatment, seven cases improved in childhood myositis assessment scale (CMAS) score (from 18.6±15.0 to 35.7±6.3, p=0.018). Two patients did not change in CMAS score (pretreatment/post-treatment score=47) but reported improvement in fatigue and activity tolerance. One patient was unevaluable for CMAS score before JAKi treatment due to joint contracture. As for biochemical indicators, CK and/or LDH were abnormal in 12 patients when JAKi was added. Median CK levels were normal before and after treatment. LDH decreased from 361.5 (306.3–463.3) U/L to 291.0 (275.8–394.8) U/L (p=0.034) in 12 patients, but two patients showed LDH increase, from 340 to 395 U/L and from 307 to 420 U/L, respectively. More details of patients’ clinical characters and index changes are in the online supplemental material. During our observation period, no increase in the infection rates with Epstein-Barr virus, cytomegalovirus, varicella-zoster virus and tuberculosis occurred as reported by another study.6 No thromboembolic event was observed as well.
annrheumdis-2020-218582supp001.pdf (57.5KB, pdf)
This is the first case series study summarising the JAKi treatment on patients with refractory JDM. In our observation, JAKi improved refractory rash and muscle involvement, helped to reduce or stop glucocorticoid and no obvious side effects were found. Therefore, our study suggested that JAKi might be an idea choice in children with refractory JDM.
Footnotes
Handling editor: Josef S Smolen
Contributors: YD and BH contribute the same in this study. ZZ and JL are the cocorrespondence authors. ZZ and JL designed the research study; YD and BH wrote the manuscript; YD, YW, JH and YC collected the clinical data. JL and ZZ revised the manuscript. All authors read and approved the final manuscript.
Funding: This study was funded by the Beijing Municipal Administration of Hospitals Clinical Medicine Development of special funding support (#XMLX201813) and the Capital’s Funds for Health Improve-ment and Research (#2020-2-2102).
Competing interests: None declared.
Patient consent for publication: Parental/guardian consent obtained.
Ethics approval: According to the off-lable principle, written informed consents of receiving JAKi treatment were obtained from the guardians of all patients before starting the treatment. The study was approved by the ethics committee of the Capital Institute of Pediatrics (SHERLL2019065).
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
References
- 1. Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts). N Engl J Med 1975;292:344–7. 10.1056/NEJM197502132920706 [DOI] [PubMed] [Google Scholar]
- 2. Bellutti Enders F, Bader-Meunier B, Baildam E, et al. Consensus-Based recommendations for the management of juvenile dermatomyositis. Ann Rheum Dis 2017;76:329–40. 10.1136/annrheumdis-2016-209247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oddis CV, Reed AM, Aggarwal R, et al. Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum 2013;65:314–24. 10.1002/art.37754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Papadopoulou C, Hong Y, Omoyinmi E, et al. Janus kinase 1/2 inhibition with baricitinib in the treatment of juvenile dermatomyositis. Brain 2019;142:e8 10.1093/brain/awz005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aeschlimann FA, Frémond M-L, Duffy D, et al. A child with severe juvenile dermatomyositis treated with ruxolitinib. Brain 2018;141:e80. 10.1093/brain/awy255 [DOI] [PubMed] [Google Scholar]
- 6. You H, Xu D, Zhao J, et al. Jak inhibitors: prospects in connective tissue diseases. Clin Rev Allergy Immunol 2020;28. 10.1007/s12016-020-08786-6 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
annrheumdis-2020-218582supp001.pdf (57.5KB, pdf)