

Abstract
The acute and chronic effects of alcohol on the brain and behavior are linked to alterations in inhibitory synaptic transmission. Alcohol’s most consistent effect at the synaptic level is probably a facilitation of γ-aminobutyric acid (GABA) release, as seen from several rodent studies. The impact of alcohol on GABAergic neurotransmission in human neurons is unknown, due to a lack of a suitable experimental model. Human neurons can also be used to model effects of genetic variants linked with alcohol use disorders (AUDs). The A118G single nucleotide polymorphism (SNP rs1799971) of the OPRM1 gene encoding the N40D (D40 minor allele) mu-opioid receptor (MOR) variant has been linked with individuals who have an AUD. However, while N40D is clearly associated with other drugs of abuse, involvement with AUDs is controversial. In this study, we employed Ascl1-and Dlx2-induced inhibitory neuronal cells (AD-iNs) generated from human iPS cell lines carrying N40D variants, and investigated the impact of ethanol acutely and chronically on GABAergic synaptic transmission. We found that N40 AD-iNs display a stronger facilitation (versus D40) of spontaneous and miniature inhibitory postsynaptic current frequency in response to acute ethanol application. Quantitative immunocytochemistry of Synapsin 1+ synaptic puncta revealed a similar synapse number between N40 and D40 iNs, suggesting an ethanol modulation of presynaptic GABA release without affecting synapse density. Interestingly, D40 iNs exposed to chronic intermittent ethanol application caused a significant increase in mIPSC frequency, with only a modest enhancement observed in N40 iNs. These data suggest that the MOR genotype may confer differential sensitivity to synaptic output, which depends on ethanol exposure time and concentration for AD-iNs and may help explain alcohol dependence in individuals who carry the MOR D40 SNPs. Furthermore, this study supports the use of human neuronal cells carrying risk-associated genetic variants linked to disease, as in vitro models to assay the synaptic actions of alcohol on human neuronal cells.
Keywords: alcohol use disorder (AUD), human disease modeling, induced neuronal cell (iN), induced pluripotent stem (iPS) cell, mu-opioid receptor (MOR), synaptic transmission
Introduction
Alcohol use disorders (AUDs), which include alcohol abuse and dependence, are among the most common types of life-threatening neuropsychiatric disorders, impacting ~15% of the population (Sridhar, 2012; Grant et al., 2017; World Health Organization, 2019). Despite the high prevalence of AUDs, the precise molecular mechanism(s) that lead to the development of dependence and tolerance remain poorly understood (Lewohl et al., 2000; Hanchar et al., 2006). One limitation to our current mechanistic understanding of AUDs is the lack of an appropriate human model system that permits functional studies.
Despite the lack of understanding of alcohol’s impact on human neurons, rodent studies have provided us with invaluable information. Ethanol, also known as ethyl alcohol, produces its clearest presynaptic effects at GABAergic synapses in several brain regions in rodents (Lovinger, 2018) Specifically, early electrophysiological studies established that ethanol consistently and reproducibly potentiated GABA-mediated inhibitory transmission, and this was described as one of the clearest synaptic effects of the drug (Wan, Berton, Madamba, Francesconi, & Siggins, 1996; Weiner, Gu, & Dunwiddie, 1997). This effect was observed at GABAergic synapses in many brain regions, including the hippocampus (Ariwodola & Weiner, 2004; Sanna et al., 2004), basolateral amygdala (Butler, Chapell, & Weiner, 2014; Karkhanis, Alexander, McCool, Weiner, & Jones, 2015; Talani & Lovinger, 2015), central amygdala (Bajo, Cruz, Siggins, Messing, & Roberto, 2008), cerebellum (Kelm, Criswell, & Breese, 2008), dorsal striatum (Wilcox et al., 2014), nucleus (Hyytiä & Koob, 1995), spinal cord (Ziskind-Conhaim, Gao, & Hinckley, 2003), and ventral tegmental area (VTA) (Theile, Morikawa, Gonzales, & Morrisett, 2008). It was hypothesized that one mechanism of the rewarding properties of ethanol could be a result of a disinhibitory mechanism involving opioid receptors that are expressed on VTA interneurons (Xiao, Zhang, Krnjević, & Ye, 2007).
AUDs are heterogeneous in etiology, pathophysiology, and response to treatment. In addition to environmental factors, AUDs are genetically complex, without an obvious pattern of Mendelian inheritance, and are moderately heritable as observed from family-based twin studies (~49%) (Kendler, Heath, Neale, Kessler, & Eaves, 1992; McGue, Pickens, & Svikis, 1992; Reed, Page, Viken, & Christian, 1996; Heath et al., 1997; Prescott & Kendler, 1999; True et al., 1999; Verhulst, Neale, & Kendler, 2015). It is likely that a combination of genetic variants and environment, each responsible for a small effect, are involved. Genome-wide association studies (GWAS) have identified several risk loci for AUDs (Dick et al., 2006; Edenberg, 2007; Hart & Kranzler, 2015). One variant associated with AUDs is a non-synonymous single nucleotide polymorphism (SNP) in OPRM1, which encodes the μ-opioid receptor (MOR), A118G (rs1799971; Asn40Asp or N40D), although this remains controversial (LaForge, Yuferov, & Kreek, 2000; Mague & Blendy, 2010; Sloan et al., 2018). The A118 OPRM1 major allele that codes for the MOR (N40) is found at the highest frequency in individuals of all ethnic backgrounds, and the minor allele G118 OPRM1 (D40 MOR) is found at a lower frequency overall. However, the minor allele SNP occurs in different frequencies in different ethnic groups, from ~3% in African, ~16% in European, and 39–42% in Asian ancestry (Zerbino et al., 2018). There have been several studies aimed at understanding the functional consequences of the MOR D40 variant on receptor activation in mouse models, primates, and humans (Bond et al., 1998; Befort et al., 2001; Beyer, Koch, Schröder, Schulz, & Höllt, 2004; Miller et al., 2004; Zhang, Wang, Johnson, Papp, & Sadée, 2005; Kroslak et al., 2007; Mague et al., 2009; Mague & Blendy, 2010; Mahmoud et al., 2011; Margas, Zubkoff, Schuler, Janicki, & Ruiz-Velasco, 2007). Both animal and human studies reveal that the N40D substitution destroys an N′-terminal glycosylation site and reduces the surface expression for MORs (Ray et al., 2011; Wang, Huang, Ung, Blendy, & Liu-Chen, 2012; Wang, Huang, Blendy, & Liu-Chen, 2014; Halikere et al., 2019). However, another study using a humanized mouse model of MOR N40D is not in complete agreement with this finding (Bilbao et al., 2015). Thus, to gain insight into mechanisms underlying drug abuse, it is imperative to understand how the D40 variant impacts MOR signaling and synaptic function in human neuronal context.
Extensive work has explored the relationship between OPRM1 A118G and various substance abuse disorders, specifically alcohol abuse disorders (Bergen et al., 1997; Gelernter, Kranzler, & Cubells, 1999; Town et al., 1999; Schinka et al., 2002; Crowley et al., 2003; Luo, Kranzler, Zhao, & Gelernter, 2003; Kim et al., 2004; Loh, Fann, Chang, Chang, & Cheng, 2004; Nishizawa et al., 2006; Szeto, Tang, Lee, & Stadlin, 2001; Türkan et al., 2019). This has profound treatment implications, since naltrexone, an opioid receptor antagonist, decreases relapse in alcoholic individuals (Jonas et al., 2014) and is one of the few adjunctive therapies approved for the treatment of subjects with AUDs. Analysis of G-allele carriers diagnosed with an AUD responded more strongly to naltrexone, with lower rates of relapse and a longer time to return to heavy drinking (Oslin et al., 2003). However, a more recent study failed to observe the same effect (Oslin et al., 2015; Sloan et al., 2018), but reported that G-allele carriers consume more drinks per day, on average. This study’s findings may differ from previous meta-analyses because the authors omitted studies that were not randomized clinical trials, which could explain the discrepancies between studies. This could be explained by multiple genetic components, for example, the selection of specific ethnic backgrounds included in the study, since the minor allele frequency of rs1799971 is variable among different ethnic groups, and potentially confounded by the presence or absence of other linked genetic variants. To control for this possibility, we have modeled contrasting rs1799971 genotypes in a CRISPR-edited, isogenic set of human-induced pluripotent (iPS) cells (Halikere et al., 2019).
Several studies have employed the use of patient-specific iPS cell technologies to model and elucidate the mechanisms underlying AUDs (reviewed by Prytkova, Goate, Hart, & Slesinger, 2018; Scarnati, Halikere, & Pang, 2019). Electrophysiological recordings conducted in several brain regions, from animal models, report a potentiation of GABAA receptor response following exposure to alcohol (Aguayo, 1990; Jia, Chandra, Homanics, & Harrison, 2008; Nie et al., 2004; Nie, Madamba, & Siggins, 2000; Yeh & Kolb, 1997). However, recordings performed in iPS cell-derived human neuronal cells contradict these findings (Lieberman, Kranzler, Levine, & Covault, 2017). However, these recordings were performed in mixed excitatory/inhibitory co-cultures, and a puffing procedure was used to specifically measure the effect of ethanol on postsynaptic GABAA receptors. There was no apparent strengthening of the GABAA response following acute exposure to alcohol. This suggests species-specific mechanisms governing GABAA receptor function, which supports the importance of human neuronal models in understanding the synaptic role of alcohol. Finally, the mechanisms of how GABAergic release and GABAA receptor function are impacted by alcohol application in human neurons carrying functionally important and commonly found SNPs that have been linked to alcohol abuse are critical for understanding the initiation and progression of AUDs. This includes OPRM1 A118G, which has been linked to AUDs through GWAS (Hoehe et al., 2000; Berrettini, 2013).
The aim of this study is to elucidate ethanol’s cellular and synaptic impact, both acutely and chronically, on human neurons of the GABAergic variety, carrying MOR N40D gene variants. Specifically, we were focused on the effect of ethanol on GABAergic transmission in iNs expressing MOR N40D, independent of opioidergic signaling. We found that acute application of ethanol caused an increase in sIPSC and mIPSC frequency for N40-harboring iNs, while only a modest increase was observed in D40 human iNs. In agreement with these data, we also observed a significant decrease in spontaneous action potential firing frequency for N40-containing iNs, due to an increased GABA release. Interestingly, we observed a significant increase in inhibitory synaptic release exclusively in iNs harboring D40 MOR allelic variants, following a 10-day chronic intermittent ethanol (CIE) exposure paradigm, intended to mimic a diurnal drinking pattern common in AUD subjects. Our findings suggest that ethanol treatment alone results in a differential sensitivity to acute and chronic treatment between genotypes. These results may provide a better mechanistic understanding of the interaction between alcohol and opioid signaling in humans.
Materials and methods
Stem cell culture and rapid neuronal induction
The original selection criteria and generation of human iPS cells from lymphocytes of subjects carrying MOR N40D were described previously (Halikere et al., 2019). This study utilized isogenic human stem cell lines that were both generated and validated previously (Halikere et al., 2019). Briefly, CRISPR/Cas9 genome editing was employed to convert rs1799971 in the 03SF subject iPS cell line from homozygous minor allele (GG) to major allele (AA). The two subclones used in this study were C12 (GG118, unedited control) and A10 (AA118, edited), both of which had been processed through identical CRISPR/Cas9 procedures. A detailed description describing the generation and characterization of the isogenic iPS cell lines are provided in Halikere et al., 2019.
iPS cells were cultured on Matrigel™ Matrix (Corning Life Sciences; catalog# 354234)-coated plates in mTeSR™ medium (Stem Cell Technologies; catalog# 85851). Matrigel was used at a 1:100 dilution in Minimum Essential Medium (MEM; Gibco, catalog# 51200038). The medium was refreshed daily and the cultures were passaged as single cells every 4–6 days. Briefly, for passaging and differentiation, iPS cells were washed with MEM and dissociated using Accutase™ (Stem Cell Technologies; catalog# AT-104). Cells were resuspended in mTeSR™ with 5 μM ROCK inhibitor (Stemolecule™ Y27632, Stemgent). iPS cells were re-plated dropwise to a Matrigel-coated dish containing mTeSR™ and 5 μM ROCK inhibitor at a density of 20,000 cells/cm2 for maintenance cultures and 50,000 cells/cm2 for differentiation.
The protocol for generating GABAergic human AD-iN cells was described elsewhere (Yang et al., 2017; Halikere et al., 2019). Briefly, iPS cells were plated as dissociated cells on 1:100 Matrigel™ Matrix-coated plates in mTeSR™ medium with 5 μM ROCK inhibitor. Infection was performed in suspension. When iPS cells reached ~70% confluency, cells were passaged and re-plated, at ~33.3% ratio of total iPS cells collected following passaging, in mTeSR™ media containing the doxycycline-inducible lentiviruses expressing transcription factors (Ascl1 and Dlx2) and the reverse tetracycline-controlled transactivator (rtTA) at 37 °C, 5% CO2. Infection proceeded for approximately 6–7 h, upon which culture medium was replaced with Neurobasal™ medium (GIBCO by Life Technologies, catalog# 21103–049) containing B27 and l-glutamine supplemented with 2 μg/mL of doxycycline (MP Biomedicals, catalog# 198955) and 5 μM ROCK inhibitor to induce TetO expression. The protocol for generating lentiviruses was previously described (Yang et al., 2017). Puromycin (1 μg/mL; Sigma Aldrich, catalog# P8833) and hygromycin (50 ng/μL; Sigma Aldrich, catalog# H9773) selection was conducted for the following 2 days, and on day 5, the iN cells (~200,000 iNs/coverslip) were dissociated with Accutase™ and plated on glass coverslips with a monolayer of passage three primary mouse astrocytes (~50,000 cells/coverslip) isolated from postnatal day 1–3 pups, as described (Maximov, Pang, Tervo, & Südhof, 2007; Oni et al., 2016; Yang et al., 2017). On day 6, fresh Neurobasal™ media containing B27, l-glutamine, Ara-C (4 mM; Sigma Aldrich, catalog# C6645) 10 ng/mL of BDNF, NT3, and GDNF (Peprotech™). Following day 6, 50% of the culture medium was replaced every 5 days with fresh Neurobasal™ medium containing B27, l-glutamine, BDNF, NT3, and GDNF. Molecular, morphological, and functional analyses were conducted ~5 weeks (days in vitro [DIV] 40) following initial induction with doxycycline.
Chronic intermittent ethanol (CIE) exposure
CIE exposure was performed for a period of 10 days and initiated at DIV40. Briefly, primary glial culture (passage 2) medium was exchanged for Neurobasal™ medium containing B27 and l-glutamine, and this medium was exchanged daily. Conditioned glial medium was used for the CIE exposure paradigm and supplemented with 10 ng/mL of BDNF, NT3, GDNF, and 75 mM ethanol (control plates were fed with identical medium but without ethanol). Ethanol (200 Proof, Decon Laboratories, catalog# 2716) was used. The iNs were fed with the above medium every 24 h for the duration of 10 days. Following the 10-day CIE exposure, iNs were subjected to functional analysis. Evaporation of ethanol following 24-h medium changes in unsealed culture dishes is expected to produce a gradual reduction in ethanol concentration each day. Thus, the daily replenishment of ethanol followed by a gradual loss due to evaporation in unsealed culture dishes mimics a pattern of ethanol exposure more similar to human drinkers (Lieberman, Levine, Kranzler, Abreu, & Covault, 2012). Evaporation of ethanol was examined in 6-well culture dishes with culture media by measurement of ethanol concentrations (0, 40, and 75 mM) at 0, 12, and 24 h using an AM1 Alcohol Analyzer (Analox Instruments, Ltd.). Five independent 6-well culture dishes were used for analysis. The calculation of half-life was performed as previously described (Chen, Ezzeddine, & Shyu, 2008). Briefly, results were plotted as the log of the fraction remaining, normalizing to the initial (T = 0) concentration. The linear regression fit is plotted as a line, and the half-life was calculated from the slope of the fit, as −log(2)/slope.
Immunocytochemistry and imaging analysis
Human iN cells were fixed in ice-cold 4% paraformaldehyde in phosphate-buffered saline (PBS) and permeabilized using 0.2% Triton X-100 in PBS at room temperature. Cells were then washed three times in PBS and blocked in 4% bovine serum albumin (BSA) with 1% normal goat serum (blocking buffer) in PBS at room temperature. Primary antibodies were diluted in blocking buffer and incubated overnight at 4 °C. The next day, samples were washed five times with PBS at room temperature and incubated with secondary antibody in blocking buffer at room temperature. Following incubation, coverslips were washed three times in PBS, with a final wash in deionized water. Coverslips were then mounted onto glass slides with Mounting Media containing DAPI (Fluoroshield, catalog# F6057). Primary antibodies used include chicken anti-MAP2 (Sigma Aldrich AB5543, 1:1000), rabbit anti-Synapsin (E028, 1:3000), mouse anti-Oct 4 (Millipore Sigma MAB4401, 1:2000), mouse anti-Tra-1–60 (Millipore Sigma MAB4360, 1:1000), and mouse anti-Gad-67 (Abcam ab26116, 1:500). Secondary antibodies used included goat anti-chicken IgY-Alexa Fluor 546 (Invitrogen A-11040, 1:500), goat anti-rabbit IgG-Alexa Fluor 488 (Invitrogen A-31556, 1:500) and goat anti-mouse IgG-Alexa Fluor 546 (Invitrogen A-11030, 1:500). Images were taken on a Zeiss LSM700 confocal microscope. All images presented are maximum intensity projections of 5 plane z-stacks obtained at a step size of 1 μm.
For synaptic quantification, 63X images were taken for iNs stained for MAP2 and Synapsin. The number of Synapsin+ puncta, as well as puncta size and fluorescence intensity of Synapsin staining, were identified using Intellicount (Fantuzzo, Miorabella, et al., 2017, a high-throughput, fully automated synapse quantification program.
Electrophysiology
Functional analyses of iN cells were conducted using whole-cell patch-clamp electrophysiology as described elsewhere (Oni et al., 2016; Yang et al., 2017; Halikere et al., 2019). For current-clamp recordings, a K-gluconate internal solution was used, which consisted of (in mM): 126 K-gluconate, 4 KCl, 10 HEPES, 0.05 EGTA, 4 ATP-magnesium, 0.3 GTP-sodium, and 10 phosphocreatine. For voltage-clamp recordings, a Cs2+-based solution was used, which consisted of (in mM): 40 CsCl, 3.5 KCl, 10 HEPES, 0.05 EGTA, 90 K-gluconate, 1.8 NaCl, 1.7 MgCl2, 2 ATP-magnesium, 0.4 GTP-sodium, and 10 phosphocreatine. The pH was adjusted to 7.2 with KOH for the solution for the current-clamp recordings and with CsOH for the solution for the voltage-clamp recordings, and osmolarity was adjusted to 270–290 mOsm. The bath solution consisted of (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, and 10 glucose. The pH was adjusted to 7.4 with NaOH. Spontaneous inhibitory postsynaptic currents (sIPSCs) were recorded at a holding potential of −70 mV in the presence of 20 μM CNQX (Tocris, catalog# 0190) added to the perfusion solution. Miniature inhibitory postsynaptic currents (mIPSCs) were recorded at a holding potential of −70 mV in the presence of 20 μM CNQX and 1 μM tetrodotoxin (TTX; VWR, catalog#89160–628). Inhibitory identity was confirmed via perfusion of 50 μM picrotoxin (PTX; Tocris, catalog# 11–281). The following MOR agonist was used for the sIPSC recordings: DAMGO ([D-Ala2, N-MePhe4, Gly-ol]-enkephalin) (Hellobio, cat# HB2409) 6 μM. All recordings were conducted at room temperature because recordings made at elevated temperatures resulted in poor recording quality and shorter recording durations. Cells were excluded from analysis if series resistance (Rs) changed by more than 20% along the duration of the recording. In addition, any recordings where the access resistance was greater than 25 MΩ was also excluded from the analysis. Electrophysiological data were analyzed using Clampfit 10.5 (Molecular Devices).
For examining the acute impact of ethanol on synaptic transmission in human neurons, following baseline recordings, freshly made ethanol solution (40 mM) was perfused into the recording chamber for a duration of 5 min. For recordings under CIE conditions, coverslips with N40 or D40 neurons were moved into the chamber perfused with 40 mM ethanol during the entire electrophysiological recording period. For the control condition of CIE, ethanol was absent from sister N40 or D40 iN culture wells during the exposure paradigm and subsequent recordings.
Statistical analysis
All data are presented as the mean ± standard error of the mean (SEM), unless otherwise noted. All experiments were repeated on at least three biological replicates, with each replicate consisting of an independent infection/differentiation. The effects of acute and chronic ethanol application were normalized to basal (baseline) activity in the absence of drug, i.e., drug effects are defined as the relative changes to baseline values (in the absence of drug). The effects of ethanol before and after the application in the same group of cells were compared using paired t tests (within-genotype differences). On normalized post-treatment data, one-way ANOVA tests were used to evaluate the ethanol effects between genotypes. To indicate statistical significance, the following symbols were used (*p < 0.05, **p < 0.01, ***p < 0.001) (NS = no statistical significance found). In the event that genotypic and within-genotype differences were found (#) are used to indicate significance for between-genotype effects and (*) for within-genotype effects.
Results
Generation of GABAergic induced neuronal (iN) cells from human isogenic iPS cell lines for MOR genetic variants N40 and D40
To investigate the acute and chronic effects of alcohol application on stem-cell derived human neurons carrying MOR N40D variants, induced neuronal (iN) cell technology (Pang et al., 2011; Yang et al., 2017; Halikere et al., 2019) was used to prepare GABAergic iN cultures from isogenic iPS cell lines homozygous for either MOR N40 or MOR D40 (Halikere et al., 2019) (Fig. 1A). The pluripotency of the source iPS cell lines was confirmed by co-localized immunocytochemistry (ICC) for OCT4 and Tra-1–60 (Fig. 1B). Expression of lentivirus-encoded, GABAergic lineage-specific transcription factors Ascl1 and Dlx2 (Yang et al., 2017) was induced with doxycycline. Lentiviral constructs carrying Ascl1 and Dlx2 contain the selectable antibiotic resistance cassettes puromycin and hygromycin, respectively (Yang et al., 2017). Thus, infected cells were selected for reprogrammed neurons from uninfected iPS. This protocol generated nearly 100% GABAergic neurons with a high degree of neuronal morphology, expression of neuron-specific markers (MAP2 and TuJ 1, not shown), and evidence of synapse formation (Synapsin 1, Fig. 1C). To confirm the identity of the AD-iNs, we performed immunolabeling with antibodies specific for GAD67 (a marker for inhibitory GABAergic neurons), and found that both N40 and D40 AD-iNs stained positive for GAD67 (Fig. 1D). To further prove the neuronal identity of the AD-iNs used in this study, we employed whole-cell voltage clamp recordings to monitor miniature synaptic release (mIPSC) in the presence and absence of the GABAA antagonist picrotoxin (PTX) (Supplemental Fig. S1). Perfusion of PTX (50 μM) into the recording chamber completely abolished synaptic release, further confirming the inhibitory nature of the AD-iNs used in this study. Taken together, mature human-specific AD-iNs were successfully generated from isogenic human stem lines carrying MOR allelic variants.
Fig. 1. Generation of GABAergic-induced neuronal (iN) cells from human isogenic iPS cell lines for MOR genetic variants N40 and D40.
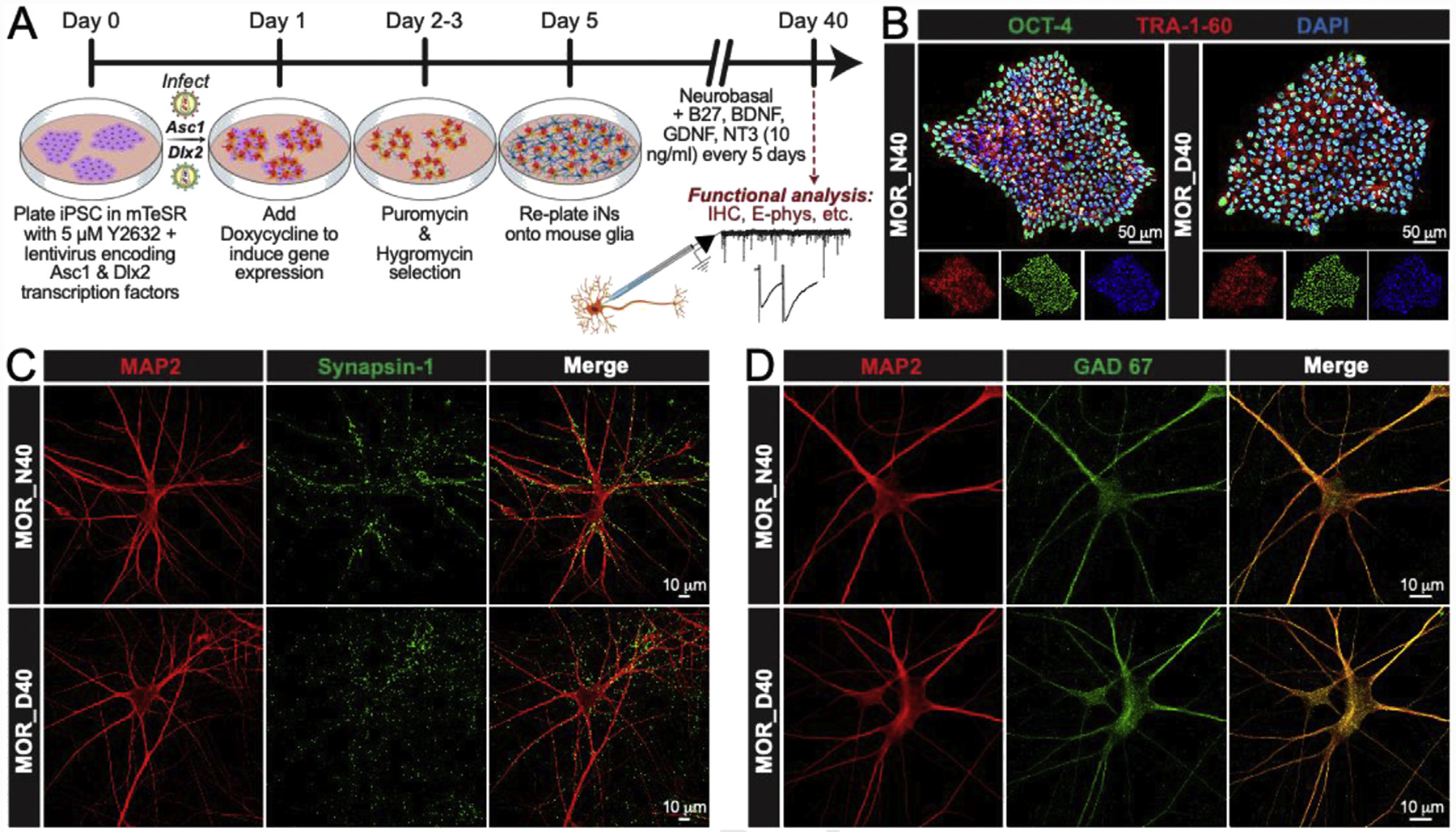
(A) Experimental design. Schedule of experiments required for direct conversion of iPS cells to GABAergic-induced neurons (AD-iNs) (B) Oct 4 (green), Tra-1–60 (red), and DAPI (blue) immunocytochemistry (ICC) for N40 and D40 isogenic iPS cells depicting pluripotency (scale = 50 μm) (C) MAP2 (red) and Synapsin (green) ICC of iN cells generated from N40 and D40 isogenic iPS cell lines (scale = 10 μm) (D) Immunofluorescence of MAP2 (red) and GAD 67 (green) of iN cells produced from N40 and D40 isogenic iPS cell lines (scale = 10 μm).
The MOR genetic variant N40D does not affect synapse formation in human AD-iNs
In order to assess any differences in synaptic sensitivity to ethanol application, it is important to first understand whether the development of synapses (synaptic number) is comparable between N40 and D40 iNs. Therefore, we immunolabeled N40 and D40 AD-iNs at DIV40 to measure the number of synapses in each neuron by counting Synapsin 1+ puncta in close proximity to MAP2+ mature neuronal cell bodies and dendrites (Fig. 2A and B). We observed no significant (p = 0.9) difference in the number of MAP2-correlated Synapsin-positive puncta, and no differences in the mean Synapsin puncta intensity, mean Synapsin puncta area, or MAP2 fluorescence intensity (Fig. 2C). However, the average MAP2+ cellular area of D40 AD-iNs was significantly larger when compared to N40 (Fig. 2C, ***p ≤ 0.001). Ultimately, our analysis failed to uncover any evidence of significant differences in synaptic numbers between N40 and D40 iNs. Thus, even though MOR D40 influences average MAP2 area, it does not impact the development of synapses in human AD-iNs.
Fig. 2. The MOR genetic variant N40D does not affect synapse formation in human AD-iNs.
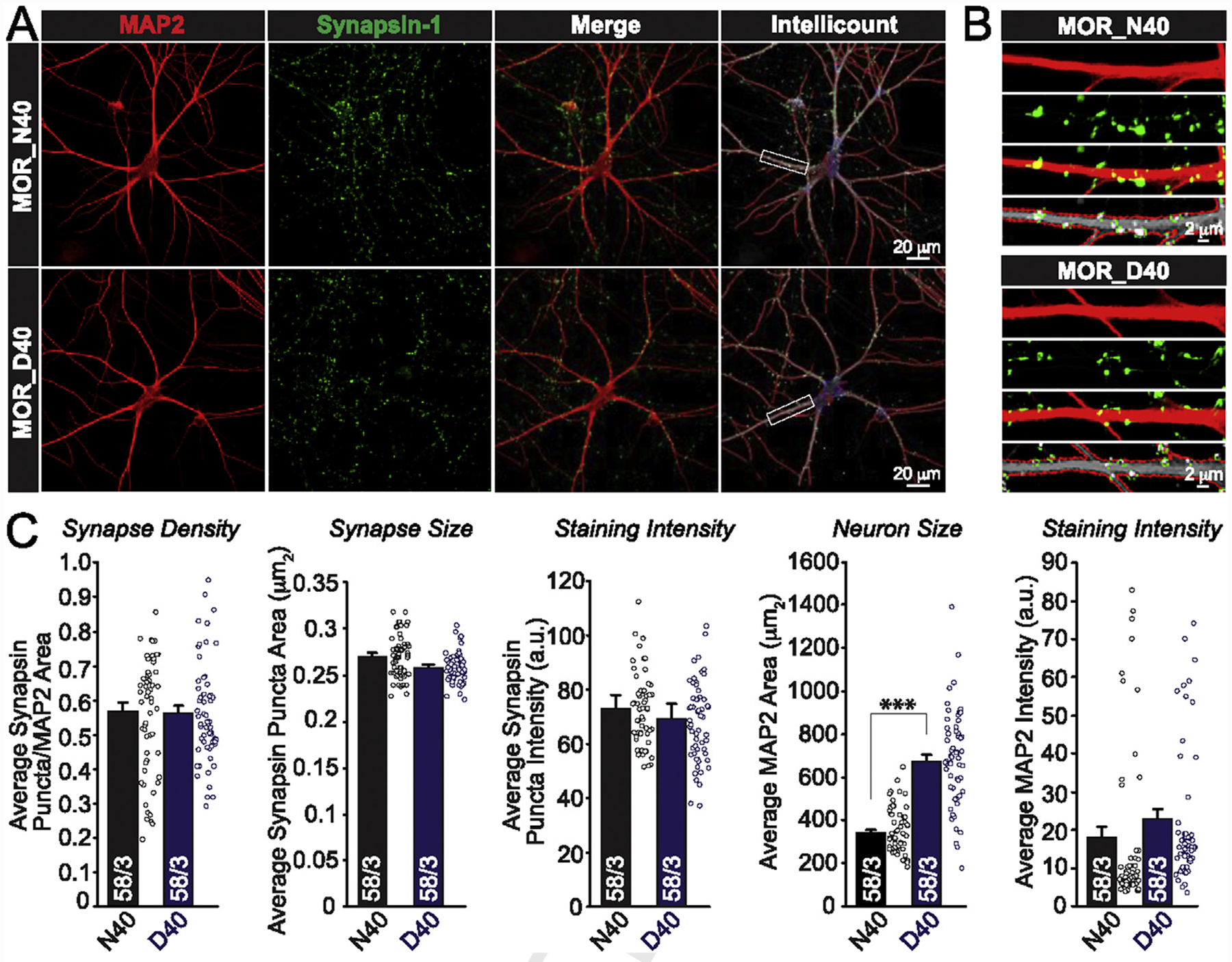
(A) Top: Representative confocal images of N40 AD-iNs that were employed for Intellicount analysis. MAP2 (red), Synapsin (green), and Merged images (scale = 20 μm). Bottom: Representative confocal images of D40 AD-iNs that were employed for Intellicount analysis. MAP2 (red), Synapsin (green), and Merged images (scale = 20 μm) (B) Zoomed-in images of the boxed areas in (A) (scale = 2 μm) (C) Quantifications of MAP2-correlated synapse density, synapse size, and staining intensity (Synapsin and MAP2) are not different between genotypes. Average MAP2 area (μm2) is significantly different between iNs harboring N40D MOR allelic variants (***p < 0.001). Paired t test was used to evaluate within-genotype statistical differences (*p < 0.05, **p < 0.01, ***p < 0.001) (NS = no statistical significance found).
Acute ethanol application causes an increase in the overall inhibitory tone in MOR N40 AD-iNs
There have been several studies (Barr et al., 2007; Vallender, Rüedi-Bettschen, Miller, & Platt, 2010; Ramchandani et al., 2011; Bilbao et al., 2015; Henderson-Redmond, Lowe, Tian, & Morgan, 2018) examining the functional consequence of ethanol application on neurons carrying MOR N40D allelic variants. This has mainly been done in primates, rodent overexpression models, or MOR humanized mice harboring MOR N40D (Bilbao et al., 2015). However, no functional or electrophysiological analyses on ethanol-treated human cultured GABAergic neurons carrying MOR genetic variants have been reported. To determine whether N40 and D40 AD-iNs respond differently to acute application, we perfused cultures with HEPES-based recording solution supplemented with 40 mM ethanol. Recordings were performed in voltage-clamp, and the AMPA/kainate receptor antagonist CNQX (20 μM) was perfused into the bath for all recordings to isolate the true inhibitory postsynaptic current (IPSC). Furthermore, this allowed us to clamp the AD-iNs at −70 mV rather than 0 mV. Spontaneous inhibitory postsynaptic currents (sIPSCs) were monitored for 5 min prior to ethanol application to measure the baseline responses in both N40 and D40 AD-iNs (Control, Fig. 3A). Following the addition of 40 mM ethanol, we observed a robust increase in the frequency of sIPSCs in MOR N40 AD-iNs, relative to controls. Interestingly, only a modest non-significant increase was observed in D40, relative to N40 AD-iNs (Fig. 3B and C). There were no changes observed for sIPSC amplitude in both genotypes tested (Fig. 3D). There were no significant differences observed in baseline sIPSC frequency between N40 and D40 AD-iNs (Fig. 3E). We next measured the sIPSC kinetics and observed no differences in the average sIPSC rise time (Fig. 3F) between genotypes before and after acute ethanol application. Interestingly, we observed a significant increase in the average sIPSC decay time prior to ethanol application (Fig. 3G, *p ≤ 0.05) between MOR N40 and D40 AD-iNs, and this difference was not detected following ethanol application. These data suggest that acute ethanol application results in an enhancement in network and/or synaptic activity in a relatively homogenous population of inhibitory N40 AD-iNs, suggesting an increase in the overall inhibitory tone.
Fig. 3. Acute ethanol application causes an increase in the overall inhibitory tone in human iNs carrying MOR N40.
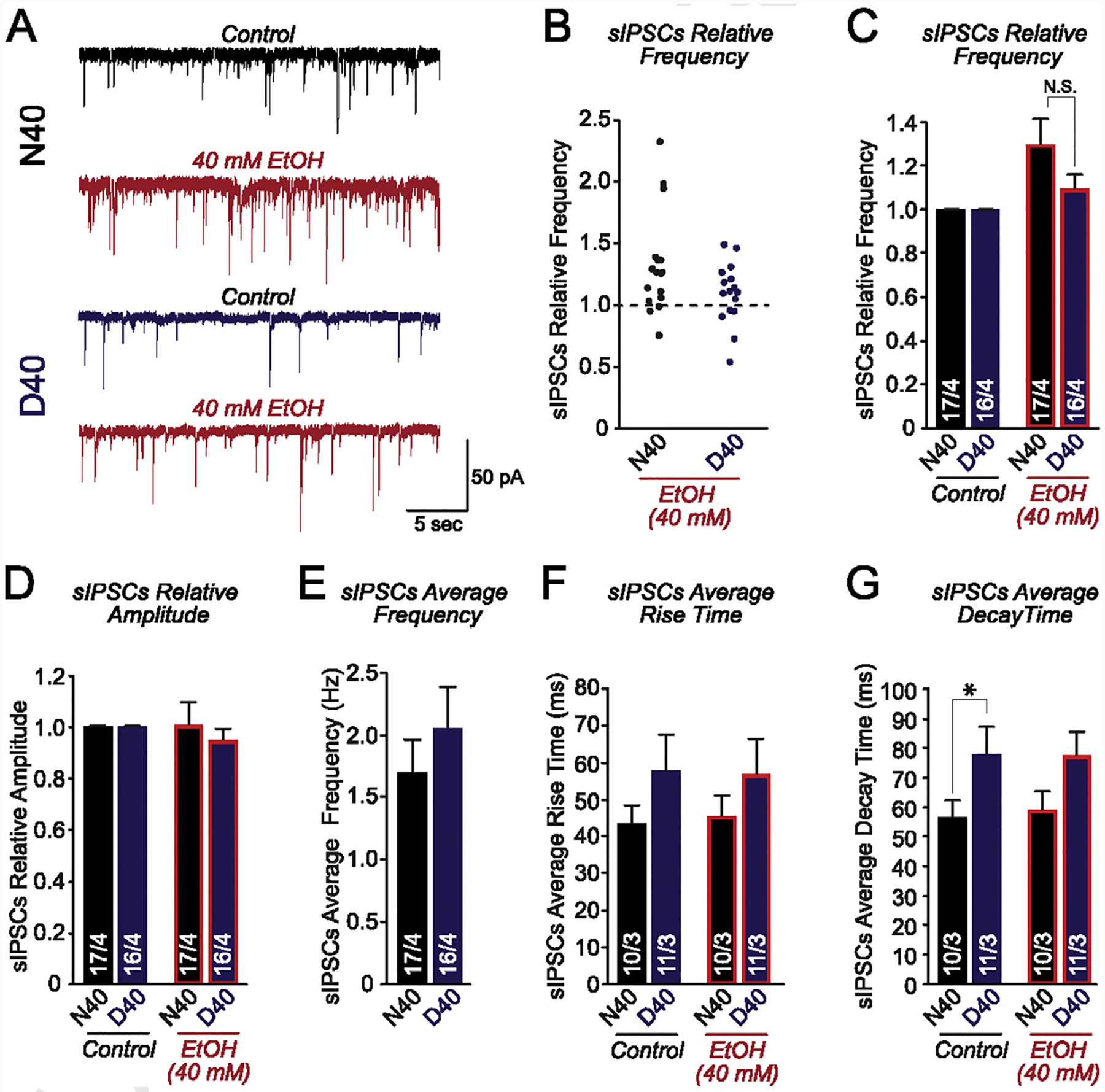
(A) Representative traces of sIPSCs recorded before [N40 (black) and D40 (blue)] and after acute 40 mM ethanol application (red) (B) Jittered-X graph illustrating the changes in sIPSC frequency in response to acute ethanol application for N40 and D40 AD-iNs. These data are relative to control sIPSC responses (dotted line). Each data point is a recording from an individual iN (C) Quantification of sIPSC frequency changes in response to acute 40 mM ethanol application for iPS-derived N40 and D40 AD-iNs; data are normalized to control (N40EtOH vs. control: p = 0.5; D40EtOH vs. control: p = 0.8) (N40EtOH vs. D40EtOH: p = 0.1) (D) Quantification of sIPSC amplitude changes in response to acute 40 mM ethanol application for iPS-derived N40 and D40 AD-iNs; data are normalized to control (N40EtOH vs. control: p = 0.9; D40EtOH vs. control: p = 0.7) (N40EtOH vs. D40EtOH: p = 0.9) (E) Average sIPSC control frequency comparing N40 to D40 AD-iNs (N40EtOH vs. D40EtOH: p = 0.3) (F) Quantification of average sIPSC rise time changes in response to acute 40 mM ethanol application for iPS cell-derived N40 and D40 AD-iNs (N40EtOH vs. control: p = 0.7; D40EtOH vs. control: p = 0.9; N40 vs. D40 controls: p = 0.2; N40EtOH vs. D40EtOH: p = 0.2) (G) Quantification of average sIPSC decay time changes in response to acute 40 mM ethanol application for iPS cell-derived N40 and D40 AD-iNs (N40EtOH vs. control: p = 0.7; D40EtOH vs. control: p = 0.9; N40 vs. D40 controls: *p ≤ 0.5; N40EtOH vs. D40EtOH: p = 0.09). Data are depicted as means ± SEM. Numbers of cells/number of independently generated cultures analyzed are depicted within the bars. Paired t test was used to evaluate within-genotype statistical differences, and one-way ANOVA was used to evaluate between-genotype statistical differences (*p < 0.05, **p < 0.01, ***p < 0.001) (NS = no statistical significance found).
We next aimed to further test this by assaying spontaneous action potential (sAP) firing in response to acute 40 mM ethanol application (Supplemental Fig. S2). Action potentials were monitored for 5 min prior to ethanol application to determine the baseline sAP frequency (Supplemental Fig. S2A, B) and amplitude (Supplemental Fig. S2A, C). There were no differences observed between baseline action potential characteristics across genotypes. Acute ethanol was then perfused into the recording chamber and spikes were measured for sAP frequency (Supplemental Fig. S2A, D) and amplitude (Supplemental Fig. S2A & E). Ethanol was equilibrated into the bath for 5 min, and responses were separated into either early (0–2.5 min) or late (2.5–5 min) bins. Following perfusion of 40 mM ethanol, we observed a decrease in the frequency of sAPs for early and late responses, exclusively in N40 AD-iNs, with only a modest decrease observed for D40 AD-iNs (Supplemental Fig. S2D). The late effect of acute ethanol application displayed significantly different genotypic differences between N40 and D40 for sAP frequency (Supplemental Fig. S2D, *p ≤ 0.05). Furthermore, there was no difference observed in the amplitude of sAPs for both genotypes (Supplemental Fig. S2E). Only action potentials that displayed an overshoot were included for analysis. These data suggest that in response to ethanol application, AD-iNs containing MOR N40 variants display an enhanced release of GABA (Fig. 3), which increases the overall inhibitory tone. Since the AD-iNs generated are of the GABAergic variety, an increase of GABA release following ethanol application (Fig. 3) will activate GABAA receptors throughout the AD-iN population, thus ultimately shutting down neuronal excitability. These data further demonstrate that ethanol application causes a robust increase of GABA release, more prominently in iNs harboring the MOR major allele variant, N40, thus illustrating genotype-dependent sensitivity to acute ethanol application.
Next, we aimed to gauge whether N40 and D40 AD-iNs respond differently to MOR activation following 5 min of acute (40 mM) ethanol application (Supplemental Fig. S3). To execute this, we employed a MOR-specific agonist DAMGO ([D-Ala 2, N-MePhe4, Gly-ol]-enkephalin) in combination with 40 mM ethanol, to study its role in modulating inhibitory synaptic output following ethanol exposure alone. DAMGO suppressed the ethanol-mediated increase in sIPSC frequency in both N40 and D40 AD-iNs (Supplemental Fig. S3A, B). However, the suppression of sIPSC frequency was more robust in D40 AD-iNs when compared to N40 iNs cells, following ethanol application (Supplemental Fig. S3B, **p ≤ 0.01). There was no difference observed in sIPSC amplitude following DAMGO application in either genotypes (Supplemental Fig. S3C). These data suggest a genotype-dependent regulation of MOR signaling following acute ethanol application.
Acute ethanol application increases miniature GABA release in MOR N40 AD-iNs
In response to acute 40 mM ethanol application, we observed a significant increase in sIPSCs, which was accompanied by a decrease in sAP frequency, more prominently in MOR N40 AD-iNs. Next, we aimed to investigate the synaptic output of MOR N40D AD-iNs by monitoring miniature inhibitory postsynaptic currents (mIPSCs) in response to acute 40 mM ethanol application (Fig. 4). We found that acute ethanol application more robustly increased mIPSC frequency (Fig. 4A–C) in N40 versus D40 AD-iN cells, with no change in mIPSC amplitude (Fig. 4A, D). The frequency increase observed following ethanol application in N40 AD-iNs was significantly different when compared to D40 AD-iNs (Fig. 4C, *p ≤ 0.05). Furthermore, there was no significant difference in baseline mIPSC frequency between N40 and D40 iNs (Fig. 4E). We also measured the mIPSC kinetics and observed no differences in the average mIPSC rise time (Fig. 4F) and decay time (Fig. 4G) between MOR N40 and D40 AD-iNs before or after acute ethanol application. These data suggest an ethanol-mediated increase in synaptic release in N40 AD-iNs, ultimately indicating a presynaptic effect. Taken together, acute (40 mM) ethanol application facilitates inhibitory synaptic transmission exclusively in N40 iNs, illustrating a genotypic difference in acute ethanol sensitivity for MOR variants.
Fig. 4. Acute ethanol application increases miniature GABA release in AD-iNs harboring MOR N40.
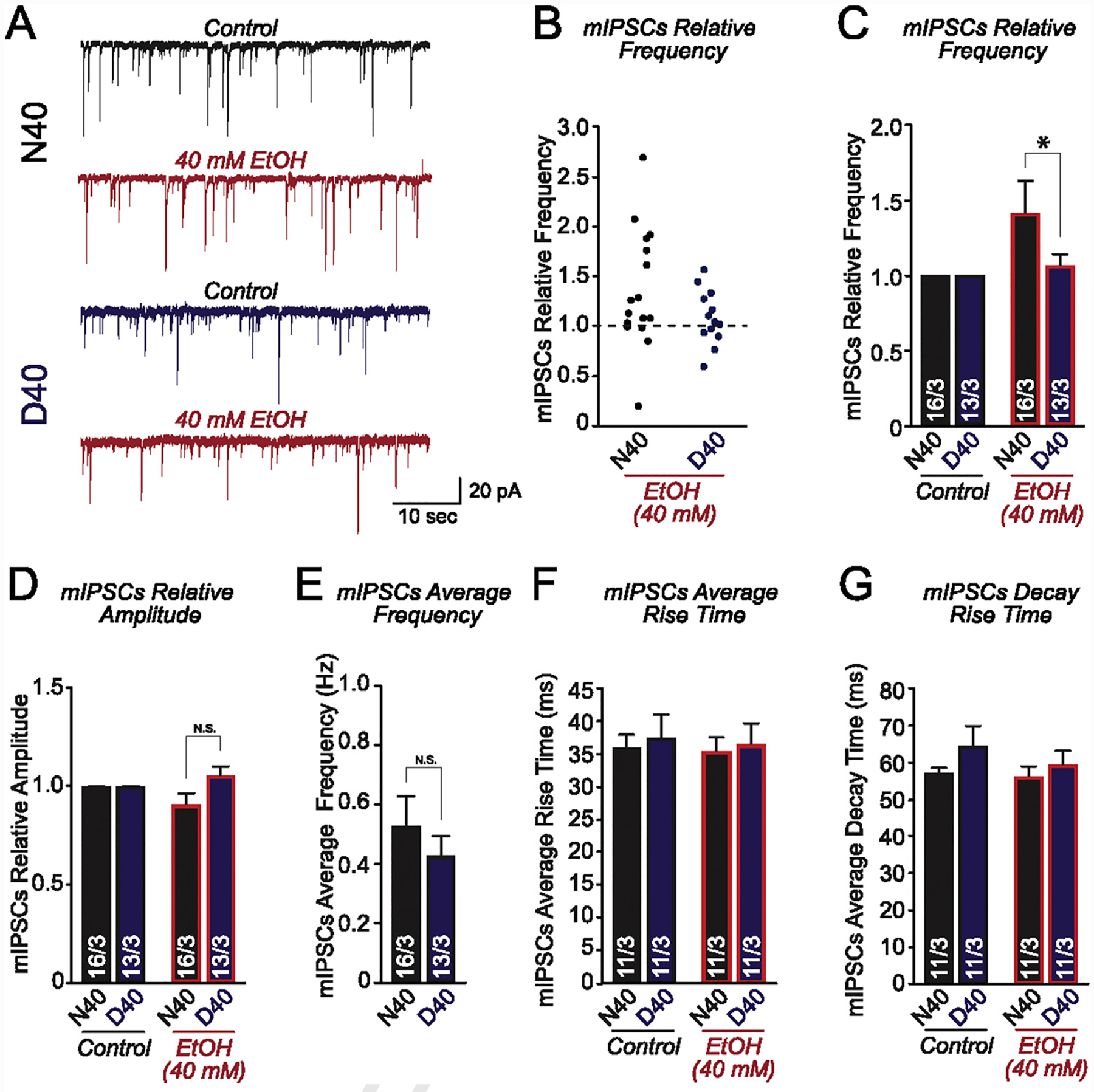
(A) Representative traces of mIPSCs recorded before [N40 (black) and D40 (blue)] and after acute 40 mM ethanol application (red) (B) Jittered-X graph illustrating the changes in mIPSC frequency in response to acute ethanol application for N40 and D40 AD-iNs. These data are relative to control sIPSC responses (dotted line). Each data point is a recording from an individual iN (C) Quantification of mIPSC frequency changes in response to acute 40 mM ethanol application for iPS-derived N40 and D40 AD-iNs; data are normalized to control (N40EtOH vs. control: p = 0.5, D40EtOH vs. control: p = 0.8) (N40EtOH vs. D40EtOH: *p ≤ 0.05) (D) Quantification of mIPSC amplitude changes in response to acute 40 mM ethanol application for iPS-derived N40 and D40 AD-iNs; data are normalized to control (N40EtOH vs. control: p = 0.2; D40EtOH vs. control: p = 0.7) (N40EtOH vs. D40EtOH: p = 0.1) (E) Average mIPSC control frequency comparing N40 to D40 AD-iNs (N40EtOH vs. D40EtOH: p = 0.4) (F) Quantification of average mIPSC rise time changes in response to acute 40 mM ethanol application for iPS cell-derived N40 and D40 AD-iNs (N40EtOH vs. control: p = 0.8; D40EtOH vs. control: p = 0.8; N40 vs. D40 controls: p = 0.7; N40EtOH vs. D40EtOH: p = 0.7) (G) Quantification of average mIPSC decay time changes in response to acute 40 mM ethanol application for iPS cell-derived N40 and D40 AD-iNs (N40EtOH vs. control: p = 0.8; D40EtOH vs. control: p = 0.4; N40 vs. D40 controls: p = 0.2; N40EtOH vs. D40EtOH: p = 0.5). Data are depicted as means ± SEM. Numbers of cells/number of independently generated cultures analyzed are depicted within the bars. Paired t test was used to evaluate within-genotype statistical differences, and one-way ANOVA was used to evaluate between-genotype statistical differences (*p < 0.05, **p < 0.01, ***p < 0.001) (NS = no statistical significance found).
10-day chronic intermittent ethanol (CIE) application selectively increases GABAergic transmission in MOR D40 AD-iNs
Our data suggest that acute ethanol enhances GABAergic neurotransmission in MOR N40 AD-iNs. Furthermore, it has been established (Lieberman et al., 2012) that chronic ethanol exposure in human iPS-derived neural cells caused significant increases in mRNA expression of NMDA receptor subunit genes. This was only observed after 7 days of chronic ethanol exposure in neurons generated from alcoholic individuals, and not from controls (Lieberman et al., 2012). We next aimed to address whether a CIE paradigm (a model that mimics binge-like levels of brain ethanol exposure) directly alters GABA release in AD-iNs, and whether the effect depends on MOR N40D variants (Fig. 5). The CIE paradigm is illustrated in Fig. 5A, as adapted from a previous study (Guo, Chen, Carreon, & Qiang, 2012). CIE in vitro results in the evaporation of ethanol from culture media containing ethanol over the period of 24 h prior to replenishment. We measured the ethanol concentration at 0, 12, and 24 h after media replenishment (Fig. 5B) and found the half-life of 75 mM ethanol in solution is ~7 h (Fig. 5C). Interestingly, the sister well without ethanol application would be “contaminated” with a low concentration of ethanol, peaking at 12 h (~10 mM) (Fig. 5B). To determine the impact of CIE exposure on synaptic transmission, we recorded mIPSCs from CIE and sister-cultured AD-iNs in the same culture plate (Fig. 5B) (Note: we normally culture control and treatment groups in the same plate to reduce batch effects in in vitro experiments). In order to bypass any homeostatic scaling changes in synaptic transmission (Lovinger & Abrahao, 2018), N40 or D40 AD-iNs subjected to 10-day CIE were incubated in 40 mM ethanol for the entire electrophysiological recording periods, while recordings in control neurons cultured in sister wells were done without ethanol (Fig. 5A). We observed no difference for control AD-iNs in terms of average mIPSC frequency (Fig. 5C) and amplitude (Fig. 5D). Interestingly, CIE AD-iNs showed an opposite phenotype to that observed with acute ethanol application (Fig. 3). The frequency of mIPSCs was significantly increased in D40 AD-iNs compared to controls (Fig. 5C, ***p ≤ 0.001), with only a modest increase observed in N40 (Fig. 5C). In addition, there was a significant genotypic difference between N40 and D40 AD-iNs following CIE (Fig. 5C, # # #p ≤ 0.001). There were no changes observed in the amplitude of mIPSCs for both N40 and D40 AD-iNs (Fig. 5D), indicating a presynaptic effect for CIE application exclusively in D40 AD-iNs. These data suggest a differential sensitivity to ethanol exposure between MOR N40 and D40 AD-iNs, which is both concentration- and duration-dependent.
Fig. 5. 10-day chronic intermittent ethanol (CIE) application selectively increases GABAergic transmission in MOR D40 iNs.
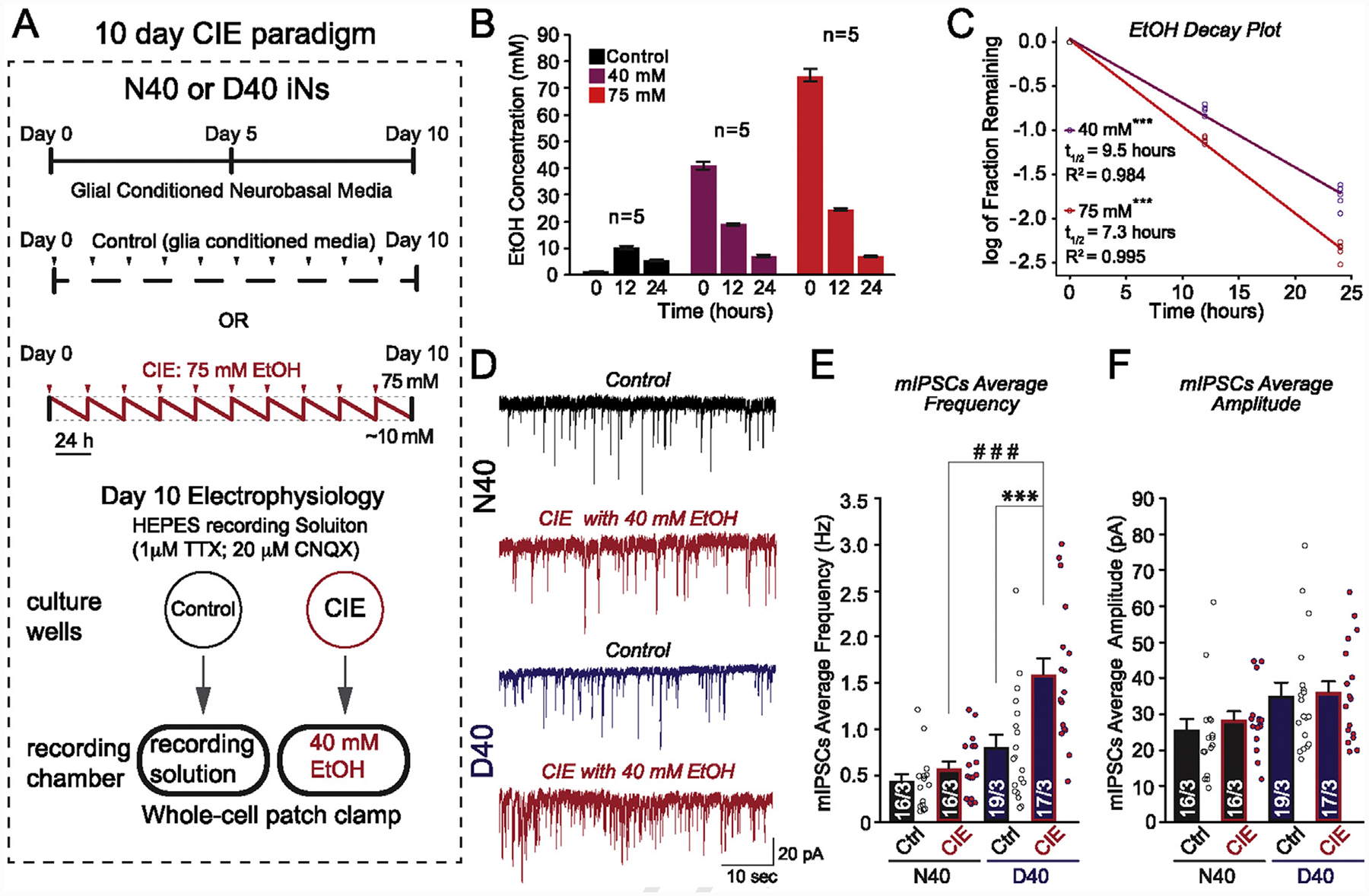
(A) Experimental design. Ctrl, control; iNs were treated with glial-conditioned medium with the indicated components daily for a period of 10 days. CIE, chronic intermittent ethanol; iNs were treated with the same medium as control, supplemented with 75 mM ethanol, daily for a period of 10 days (saw tooth red line represents decreasing ethanol concentration as a result of evaporation; red arrows indicate addition of fresh 75 mM ethanol). Following day 10, mIPSCs were recorded: Ctrl iNs were recorded in HEPES recording medium with the indicated components. CIE iNs were recorded in HEPES recording medium supplemented with 40 mM ethanol (acute application) (B) Evaporation of ethanol was examined in culture dishes not containing neural cells by measurement of medium ethanol concentrations (Control, 40 mM, and 75 mM) at 0, 12, and 24 h post-ethanol addition using an AM1 Alcohol Analyzer (C) Half-life of ethanol was determined by plotting the log of the fraction remaining, normalized to initial ethanol concentration. The linear regression fit is plotted as a line (***p value of the fit40mM < 0.001; ***p value of the fit75mM < 0.001). The half-life was calculated from the slope of the fit, as −log (2)/slope (t1/2 for 40 mM = 9.51 h with a 95% confidence interval of 8.85–10.27 h; r2 = 0.984) (t1/2 for 75 mM = 7.05 h with a 95% confidence interval of 6.77–7.34 h; r2 = 0.995) (D) Representative traces of mIPSCs recorded before [N40 (black) and D40 (blue)] and after CIE exposure + 40 mM ethanol acute re-application (red) (E) Quantification of average mIPSC frequency changes in response to CIE and acute 40 mM ethanol re-application for iPS-derived N40 and D40 AD-iNs. Each dot (data point) is a recording from an individual iN (N40EtOH vs. control: p = 0.2; D40EtOH vs. control: ***p ≤ 0.001); (N40EtOH vs. D40EtOH: # # #p ≤ 0.001) (F) Quantification of average mIPSC amplitude changes in response to CIE and acute 40 mM ethanol re-application for iPS-derived N40 and D40 AD-iNs. Each dot (data point) is a recording from an individual iN (N40EtOH vs. control: p = 0.4; D40EtOH vs. control: p = 0.8); (N40EtOH vs. D40EtOH: p = 0.08). Numbers of cells/number of independently generated cultures analyzed are depicted within the bars. Paired t test was used to evaluate within-genotype statistical differences, and one-way ANOVA was used to evaluate between-genotype statistical differences (*p < 0.05, **p < 0.01, ***p < 0.001) (NS = no statistical significance found).
Discussion
Our study provides experimental evidence describing the functional consequences of the MOR N40D SNP on ethanol sensitivity in human neuronal context, and this preparation permits the study of synaptic modulations that may be involved in the pathogeneses of AUDs. We hypothesized that GABAergic human neurons expressing MOR N40D genetic variants would display differential sensitivities to ethanol that would depend on time and concentration of ethanol exposure. First, we generated AD-iN cells from isogenic stem cell lines harboring either MOR N40 or D40 genetic variants. Second, we demonstrated that isogenic AD-iN cells express the appropriate inhibitory neuronal marker (Fig. 1) and develop a similar number of synapses between MOR N40 and D40 iNs (Figs. 1 and 2). Third, we demonstrated that acute application of ethanol caused an increase in excitability and synaptic release, which was observed exclusively in N40 iNs (Figs. 3 and 4). The increase in the frequency of mIPSCs suggests that acute ethanol application impacts presynaptic GABAergic terminals in iNs expressing the MOR N40 genetic variant, with no effect on amplitude (Fig. 4). Finally, a CIE exposure paradigm for a duration of 10 days caused a significant increase in mIPSC frequency, exclusively in D40 AD-iN cells (Fig. 5). Our findings suggest that the exposure time and concentration for ethanol on AD-iNs (acute versus chronic; also 40 mM versus a maximum of 75 mM) confers differential sensitivity to synaptic output, which is dependent on the MOR genotype. These data will help elucidate the synaptic consequences of the N40D SNP, in response to ethanol, in human neurons.
The approach of using human iN cells to investigate the synaptic impact of acute and chronic ethanol application on human neurons with a major functional OPRM1 SNP is novel for two reasons: 1) Human studies aimed at understanding the correlation of MOR N40D and an AUD suggest that the G allele confers genetic vulnerability to alcohol dependence (Anton et al., 2008; Bart et al., 2005; Chamorro et al., 2012; Hendershot, Claus, & Ramchandani, 2016; Oslin et al., 2003; Ray & Hutchison, 2004, 2007). However, some clinical studies have not discovered an association between the A118G SNP and alcohol dependence (Arias, Feinn, & Kranzler, 2006; Bergen et al., 1997; Gelernter et al., 2007; Ooteman et al., 2009; Rouvinen-Lagerström et al., 2013; Tidey et al., 2008). Comparisons made between individuals who carry the MOR N40D SNP, which comes from different ethnic backgrounds, can potentially influence the phenotype. Our study employs isogenic stem cell lines, which allows us to specifically study the impact of MOR N40D, without known contributions from any differences in the genetic background or those that can be attributed to ethnicity (Halikere et al., 2019). Thus, the identification of differential sensitivities to acute and chronic ethanol for AD-iNs is interpreted as a direct consequence of only the MOR N40D variant. 2) The disease of addiction is a progressive process. Interactions with various neurotransmitter and neuromodulator systems promote the pharmacological effects of alcohol. Neuromodulators, such as endogenous opioids, play a pivotal role in the reinforcing and rewarding effects of alcohol, leading to addiction. Our study utilizes a homogenous population of GABAergic neurons, which allows us to probe the functional consequence of acute and chronic ethanol exposure on a relatively homogenous population of AD-iNs carrying MOR N40D genetic variants. This suggests that the MOR plays a role in the ethanol response independent of opioid binding and MOR activation. However, exogenous MOR-specific agonists can be supplied to the cultures, and MOR functionality can be directly probed in response to ethanol-mediated facilitation of GABAergic synaptic release. In this study, we observed a more robust inhibition of sIPSC frequency in D40 AD-iNs following acute ethanol application in response to DAMGO (Supplemental Fig. S3). Ultimately, the use of human stem cell-derived iNs yields novel information about human-specific mechanisms of a highly prevalent OPRM1 SNP, A118G, in response to ethanol.
Ethanol exerts its effects through a plethora of molecular targets to produce intoxication. However, the clearest presynaptic effect for ethanol is at GABAergic synapses in several brain regions (Lovinger, 2018). The direct action of ethanol at GABAergic presynaptic terminals was shown in isolated “neuron-bouton” preparations (Kelm, Criswell, & Breese, 2007; Zhu & Lovinger, 2006), where ethanol produced a rapid and robust increase in the frequency of sIPSC and mIPSCs (Zhu & Lovinger, 2006). These data suggest that ethanol exerts a direct effect on GABAergic presynaptic terminals, which seems to be independent of modulatory or retrograde signals from postsynaptic neurons. Our study builds on these findings in a human-specific context and represents a significant advance in elucidating the neurobiological mechanisms underlying the ethanol sensitivity for human neurons expressing MOR N40D.
Clinical and rodent studies aimed at understanding the impact of OPRM1 A118G on AUDs have yielded mixed conclusions. Work using humanized 118GG mice shows an increased rate of ethanol intake when compared to 118AA mice (Henderson-Redmond et al., 2018). This work supports both clinical (Anton et al., 2008; Bart et al., 2005; Chamorro et al., 2012; Hendershot et al., 2016; Oslin et al., 2003; Ray & Hutchison, 2004, 2007) and rodent studies that suggest that G minor allele carriers may bestow a genetic susceptibility toward AUDs. However, these studies hypothesize that ethanol reinforcement observed for G allele carriers (animals and humans) is mediated by the endogenous opioid system (reviewed by Herz, 1997), ultimately increasing dopamine release along mesolimbic dopamine neurocircuitry (reviewed by Koob, 1992). Our recent functional study, utilizing patient-derived iPS cells harboring OPRM1 A118G, found that in response to an opioid agonist, MOR N40D allele iNs consistently exhibited stronger suppression, which suggests that this SNP causes a gain of MOR function. These data show altered opioid responsivity and/or dependence between human MOR genetic variants (Halikere et al., 2019). The current study found an altered ethanol response on GABAergic neurotransmission between MOR genetic variants that depends on the time and concentration of exposure (Figs. 4 and 5). Interestingly, our data indicate that MOR signaling is likely not involved in the ethanol sensitivity observed in AD-iNs. It is possible that ligand-independent MOR activity might contribute to the differences seen between MOR N40- and D40-harboring AD-iNs. However, a recent study looked at the impact of the opioid antagonist naltrexone on inhibitory synaptic transmission in AD-iNs and found no effect (Halikere et al., 2019). This suggests that, in the absence of ligand, MOR is not influencing synaptic output. Another possibility is the presence of iNs in our cultures that are capable of releasing endogenous opioids. We have briefly examined this but failed to detect any endogenous opioids in our cultures. These data may ultimately help predict altered ethanol sensitivity and/or dependence for individuals who carry MOR N40D allelic variants. Finally, opioid receptors have recently been shown to be diffusely distributed and laterally mobile on the axon surface (Jullié et al., 2020), and the D40 variant could impact the lateral mobility of the receptor. Thus, an interaction of MOR with GABAA receptors (or possibly GABAB) could impact the gating and conductance of the channel. This is entirely speculative and needs to be tested experimentally.
One technical complication of the CIE experiment is worth noting: for better control of batch effects in vitro cultures, we routinely culture treatment (e.g., CIE) and control neurons in the same plate and in the same incubator. We measured the half-life of ethanol (40 and 75 mM) along a period of 24 h in the cell culture incubator (Fig. 5C). However, we need to be cognizant of the ability of ethanol to evaporate and “contaminate” the untreated control, which we observed (Fig. 5B). In this study, our focus was on the genotype (AA vs. GG OPRM1), so differences in human iNs in responding to CIE, including the evaporation of ethanol into the control condition, were the same between N40 and D40 iN conditions. Therefore, we conclude that it had minimal effect on interpretation in this study.
This study highlights the benefits of using iPS and iN cell technology for in-depth analysis of gene variants that have been associated with a particular human disease. To date, we have not yet tested the synaptic effect of acute or chronic ethanol application on other cell types such as excitatory or dopaminergic neurons. Therefore, it remains unknown whether the differential sensitivities we observe in response to ethanol, between MOR N40 and D40, is characteristic to inhibitory iNs or observed across other neuronal cell types. This can only be determined experimentally. In addition, a combination of neuronal cell types can be combined with the use of microfluidic devices Fantuzzo, De Filippis et al., 2017) to mimic reward neurocircuitry. However, our focus in this study was to understand how ethanol sensitivity impacts synaptic function in inhibitory iNs that mimic those in reward circuitry. Therefore, characterization of MOR N40D in other neuronal cell types will be addressed in future studies. This work identifies neurophysiological differences in the human neuronal response to alcohol that is influenced by common variation in human OPRM1. Our findings may help explain differential alcohol sensitivities and/or dependence in humans. This model provides a platform to understand alcohol’s physiological impact on human neurons and allows researchers to dissect drugs of abuse in relation to addiction-associated SNPs. Ultimately, the use of human stem cells to understand the interplay of alcohol and MOR gene variants may provide the necessary information required to develop patient-specific, therapeutic interventions for subjects with AUDs.
Supplementary Material
Acknowledgments
We thank RUCDR Infinite Biologics for generating the iPS cells from human subjects and assisting with CRISPR/Cas9 gene targeting on 03SF iPS cell line. We thank Dr. Jay Tischfield for critical suggestions on experiments and manuscript. Research is supported by grants from NIH-NIAAA R01 AA023797 as well as Collaborative Studies on the Genetics of Alcoholism/COGA 5U10AA008-26. MSS is supported by NIH-NIAAA T32 AA028254. AJB is supported by The National Institute of General Medicine Sciences (NIGMS, NIH T32 GM008339. Pang laboratory at The Child Health Institute of New Jersey (CHINJ) is funded by the Robert Wood Johnson (RWJ) Foundation Grant #74260.
Footnotes
Conflict of Interest
All authors have no conflict of interest to declare.
Appendix A. Supplementary data
Supplementary data related to this article can be found at https://doi.org/10.1016/j.alcohol.2020.05.004.
References
- Aguayo LG (1990). Ethanol potentiates the GABAA-activated Cl− current in mouse hippocampal and cortical neurons. European Journal of Pharmacology, 187(1), 127–130. 10.1016/0014-2999(90)90349-b. [DOI] [PubMed] [Google Scholar]
- Anton RF, Oroszi G, O’Malley S, Couper D, Swift R, Pettinati H, et al. (2008). An evaluation of mu-opioid receptor (OPRM1) as a predictor of naltrexone response in the treatment of alcohol dependence: Results from the combined pharmacotherapies and behavioral interventions for alcohol dependence (COMBINE) study. Archives of General Psychiatry, 65(2), 135–144. 10.1001/archpsyc.65.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias A, Feinn R, & Kranzler HR (2006). Association of an Asn40Asp (A118G) polymorphism in the mu-opioid receptor gene with substance dependence: A meta-analysis. Drug and Alcohol Dependence, 83(3), 262–268. 10.1016/j.drugalcdep.2005.11.024. [DOI] [PubMed] [Google Scholar]
- Ariwodola OJ, & Weiner JL (2004). Ethanol potentiation of GABAergic synaptic transmission may be self-limiting: Role of presynaptic GABA(B) receptors. Journal of Neuroscience, 24(47), 10679–10686. 10.1523/JNEUROSCI.1768-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajo M, Cruz MT, Siggins GR, Messing R, & Roberto M (2008). Protein kinase C epsilon mediation of CRF- and ethanol-induced GABA release in central amygdala. Proceedings of the National Academy of Sciences of the United States of America, 105(24), 8410–8415. 10.1073/pnas.0802302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr CS, Schwandt M, Lindell SG, Chen SA, Goldman D, Suomi SJ, et al. (2007). Association of a functional polymorphism in the mu-opioid receptor gene with alcohol response and consumption in male rhesus macaques. Archives of General Psychiatry, 64(3), 369–376. 10.1001/archpsyc.64.3.369. [DOI] [PubMed] [Google Scholar]
- Bart G, Kreek MJ, Ott J, LaForge KS, Proudnikov D, Pollak L, et al. (2005). Increased attributable risk related to a functional mu-opioid receptor gene polymorphism in association with alcohol dependence in central Sweden. Neuropsychopharmacology, 30(2), 417–422. 10.1038/sj.npp.1300598. [DOI] [PubMed] [Google Scholar]
- Befort K, Filliol D, Decaillot FM, Gaveriaux-Ruff C, Hoehe MR, & Kieffer BL (2001). A single nucleotide polymorphic mutation in the human mu-opioid receptor severely impairs receptor signaling. Journal of Biological Chemistry, 276(5), 3130–3137. 10.1074/jbc.M006352200. [DOI] [PubMed] [Google Scholar]
- Bergen AW, Kokoszka J, Peterson R, Long JC, Virkkunen M, Linnoila M, et al. (1997). Mu opioid receptor gene variants: Lack of association with alcohol dependence. Molecular Psychiatry, 2(6), 490–494. 10.1038/sj.mp.4000331. [DOI] [PubMed] [Google Scholar]
- Berrettini W (2013). Opioid pharmacogenetics of alcohol addiction. Cold Spring Harbor Perspectives In Medicine, 3(7). 10.1101/cshperspect.a012203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer A, Koch T, Schröder H, Schulz S, & Höllt V (2004). Effect of the A118G polymorphism on binding affinity, potency and agonist-mediated endocytosis, desensitization, and resensitization of the human mu-opioid receptor. Journal of Neurochemistry, 89(3), 553–560. 10.1111/j.1471-4159.2004.02340.x. [DOI] [PubMed] [Google Scholar]
- Bilbao A, Robinson JE, Heilig M, Malanga CJ, Spanagel R, Sommer WH, et al. (2015). A pharmacogenetic determinant of mu-opioid receptor antagonist effects on alcohol reward and consumption: Evidence from humanized mice. Biological Psychiatry, 77(10), 850–858. 10.1016/j.biopsych.2014.08.021. [DOI] [PubMed] [Google Scholar]
- Bond C, LaForge KS, Tian M, Melia D, Zhang S, Borg L, et al. (1998). Single-nucleotide polymorphism in the human mu opioid receptor gene alters beta-endorphin binding and activity: Possible implications for opiate addiction. Proceedings of the National Academy of Sciences of the United States of America, 95(16), 9608–9613. 10.1073/pnas.95.16.9608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler TR, Chappell AM, & Weiner JL (2014). Effect of β3 adrenoceptor activation in the basolateral amygdala on ethanol seeking behaviors. Psychopharmacology, 231(1), 293–303. 10.1007/s00213-013-3238-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro A-J, Marcos M, Mirón-Canelo J-A, Pastor I, González-Sarmiento R, & Laso F-J (2012). Association of μ-opioid receptor (OPRM1) gene polymorphism with response to naltrexone in alcohol dependence: A systematic review and meta-analysis. Addiction Biology, 17(3), 505–512. 10.1111/j.1369-1600.2012.00442.x. [DOI] [PubMed] [Google Scholar]
- Chen C-YA, Ezzeddine N, & Shyu A-B (2008). Messenger RNA half-life measurements in mammalian cells. Methods in Enzymology, 448, 335–357. 10.1016/S0076-6879(08)02617-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JJ, Oslin DW, Patkar AA, Gottheil E, DeMaria PA, O’Brien CP, et al. (2003). A genetic association study of the mu opioid receptor and severe opioid dependence. Psychiatric Genetics, 13(3), 169–173. 10.1097/00041444-200309000-00006. [DOI] [PubMed] [Google Scholar]
- Dick DM, Jones K, Saccone N, Hinrichs A, Wang JC, Goate A, et al. (2006). Endophenotypes successfully lead to gene identification: results from the collaborative study on the genetics of alcoholism. Behavior Genetics, 36(1), 112–126. 10.1007/s10519-005-9001-3. [DOI] [PubMed] [Google Scholar]
- Edenberg HJ (2007). The genetics of alcohol metabolism: Role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Research & Health, 30(1), 5–13. [PMC free article] [PubMed] [Google Scholar]
- Fantuzzo JA, De Filippis L, McGowan H, Yang N, Ng Y-H, Halikere A, et al. (2017). μNeurocircuitry: Establishing in vitro models of neurocircuits with human neurons. Technology, 5(2), 87–97. 10.1142/S2339547817500054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantuzzo JA, Mirabella VR, Hamod AH, Hart RP, Zahn JD, & Pang ZP (2017). Intellicount: high-throughput quantification of fluorescent synaptic protein puncta by machine learning. ENeuro, 4(6). 10.1523/ENEURO.0219-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Gueorguieva R, Kranzler HR, Zhang H, Cramer J, Rosenheck R, et al. (2007). Opioid receptor gene (OPRM1, OPRK1, and OPRD1) variants and response to naltrexone treatment for alcohol dependence: Results from the VA cooperative study. Alcoholism: Clinical and Experimental Research, 31(4), 555–563. 10.1111/j.1530-0277.2007.00339.x. [DOI] [PubMed] [Google Scholar]
- Gelernter J, Kranzler H, & Cubells J (1999). Genetics of two mu opioid receptor gene (OPRM1) exon I polymorphisms: Population studies, and allele frequencies in alcohol- and drug-dependent subjects. Molecular Psychiatry, 4(5), 476–483. 10.1038/sj.mp.4000556. [DOI] [PubMed] [Google Scholar]
- Grant BF, Chou SP, Saha TD, Pickering RP, Kerridge BT, Ruan WJ, et al. (2017). Prevalence of 12-month alcohol use, high-risk drinking, and DSM-IV alcohol use disorder in the United States, 2001–2002 to 2012–2013: Results from the national epidemiologic survey on alcohol and related conditions. JAMA Psychiatry, 74(9), 911–923. 10.1001/jamapsychiatry.2017.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Chen Y, Carreon S, & Qiang M (2012). Chronic intermittent ethanol exposure and its removal induce a different miRNA expression pattern in primary cortical neuronal cultures. Alcoholism: Clinical and Experimental Research, 36(6), 1058–1066. 10.1111/j.1530-0277.2011.01689.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halikere A, Popova D, Scarnati MS, Hamod A, Swerdel MR, Moore JC, et al. (2019). Addiction associated N40D mu-opioid receptor variant modulates synaptic function in human neurons. Molecular Psychiatry, 25(7), 1406–1419. 10.1038/s41380-019-0507-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanchar HJ, Chutsrinopkun P, Meera P, Supavilai P, Sieghart W, Wallner M, et al. (2006). Ethanol potently and competitively inhibits binding of the alcohol antagonist Ro15–4513 to alpha 4/6beta3delta GABAA receptors. Proceedings of the National Academy of Sciences of the United States of America, 103(22), 8546–8551. 10.1073/pnas.0509903103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart AB, & Kranzler HR (2015). Alcohol dependence genetics: Lessons learned from genome-wide association studies (GWAS) and post-GWAS analyses. Alcoholism: Clinical and Experimental Research, 39(8), 1312–1327. 10.1111/acer.12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath AC, Bucholz KK, Madden PA, Dinwiddie SH, Slutske WS, Bierut LJ, et al. (1997). Genetic and environmental contributions to alcohol dependence risk in a national twin sample: Consistency of findings in women and men. Psychological Medicine, 27(6), 1381–1396. 10.1017/s0033291797005643. [DOI] [PubMed] [Google Scholar]
- Hendershot CS, Claus ED, & Ramchandani VA (2016). Associations of OPRM1 A118G and alcohol sensitivity with intravenous alcohol self-administration in young adults. Addiction Biology, 21(1), 125–135. 10.1111/adb.12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson-Redmond AN, Lowe TE, Tian XB, & Morgan DJ (2018). Increased ethanol drinking in “humanized” mice expressing the mu opioid receptor A118G polymorphism are mediated through sex-specific mechanisms. Brain Research Bulletin, 138, 12–19. 10.1016/j.brainresbull.2017.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz A (1997). Endogenous opioid systems and alcohol addiction. Psychopharmacology, 129(2), 99–111. 10.1007/s002130050169. [DOI] [PubMed] [Google Scholar]
- Hoehe MR, Köpke K, Wendel B, Rohde K, Flachmeier C, Kidd KK, et al. (2000). Sequence variability and candidate gene analysis in complex disease: Association of mu opioid receptor gene variation with substance dependence. Human Molecular Genetics, 9(19), 2895–2908. 10.1093/hmg/9.19.2895. [DOI] [PubMed] [Google Scholar]
- Hyytiä P, & Koob GF (1995). GABAA receptor antagonism in the extended amygdala decreases ethanol self-administration in rats. European Journal of Pharmacology, 283(1–3), 151–159. 10.1016/0014-2999(95)00314-b. [DOI] [PubMed] [Google Scholar]
- Jia F, Chandra D, Homanics GE, & Harrison NL (2008). Ethanol modulates synaptic and extrasynaptic GABAA receptors in the thalamus. Journal of Pharmacology and Experimental Therapeutics, 326(2), 475–482. 10.1124/jpet.108.139303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas DE, Amick HR, Feltner C, Bobashev G, Thomas K, Wines R, et al. (2014). Pharmacotherapy for adults with alcohol use disorders in outpatient settings: A systematic review and meta-analysis. Journal of the American Medical Association, 311(18), 1889–1900. 10.1001/jama.2014.3628. [DOI] [PubMed] [Google Scholar]
- Jullié D, Stoeber M, Sibarita J-B, Zieger HL, Bartol TM, Arttamangkul S, et al. (2020). A discrete presynaptic vesicle cycle for neuromodulator receptors. Neuron, 105(4), 663–677. 10.1016/j.neuron.2019.11.016.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karkhanis AN, Alexander NJ, McCool BA, Weiner JL, & Jones SR (2015). Chronic social isolation during adolescence augments catecholamine response to acute ethanol in the basolateral amygdala. Synapse, 69(8), 385–395. 10.1002/syn.21826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm MK, Criswell HE, & Breese GR (2007). Calcium release from presynaptic internal stores is required for ethanol to increase spontaneous gamma-aminobutyric acid release onto cerebellum Purkinje neurons. Journal of Pharmacology and Experimental Therapeutics, 323(1), 356–364. 10.1124/jpet.107.126144. [DOI] [PubMed] [Google Scholar]
- Kelm MK, Criswell HE, & Breese GR (2008). The role of protein kinase A in the ethanol-induced increase in spontaneous GABA release onto cerebellar Purkinje neurons. Journal of Neurophysiology, 100(6), 3417–3428. 10.1152/jn.90970.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendler KS, Heath AC, Neale MC, Kessler RC, & Eaves LJ (1992). A population-based twin study of alcoholism in women. Journal of the American Medical Association, 268(14), 1877–1882. [PubMed] [Google Scholar]
- Kim S-G, Kim C-M, Kang D-H, Kim Y-J, Byun W-T, Kim S-Y, et al. (2004). Association of functional opioid receptor genotypes with alcohol dependence in Koreans. Alcoholism: Clinical and Experimental Research, 28(7), 986–990. 10.1097/01.alc.0000130803.62768.ab. [DOI] [PubMed] [Google Scholar]
- Koob GF (1992). Drugs of abuse: Anatomy, pharmacology and function of reward pathways. Trends in Pharmacological Sciences, 13(5), 177–184. 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- Kroslak T, Laforge KS, Gianotti RJ, Ho A, Nielsen DA, & Kreek MJ (2007). The single nucleotide polymorphism A118G alters functional properties of the human mu opioid receptor. Journal of Neurochemistry, 103(1), 77–87. 10.1111/j.1471-4159.2007.04738.x. [DOI] [PubMed] [Google Scholar]
- LaForge KS, Yuferov V, & Kreek MJ (2000). Opioid receptor and peptide gene polymorphisms: Potential implications for addictions. European Journal of Pharmacology, 410(2–3), 249–268. 10.1016/s0014-2999(00)00819-0. [DOI] [PubMed] [Google Scholar]
- Lewohl JM, Wang L, Miles MF, Zhang L, Dodd PR, & Harris RA (2000). Gene expression in human alcoholism: Microarray analysis of frontal cortex. Alcoholism: Clinical and Experimental Research, 24(12), 1873–1882. [PubMed] [Google Scholar]
- Lieberman R, Kranzler HR, Levine ES, & Covault J (2017). Examining the effects of alcohol on GABAA receptor mRNA expression and function in neural cultures generated from control and alcohol dependent donor induced pluripotent stem cells. Alcohol, 66, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman R, Levine ES, Kranzler HR, Abreu C, & Covault J (2012). Pilot study of iPS-derived neural cells to examine biologic effects of alcohol on human neurons in vitro. Alcoholism: Clinical and Experimental Research, 36(10), 1678–1687. 10.1111/j.1530-0277.2012.01792.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh EW, Fann CSJ, Chang YT, Chang CJ, & Cheng ATA (2004). Endogenous opioid receptor genes and alcohol dependence among Taiwanese Han. Alcoholism: Clinical and Experimental Research, 28(1), 15–19. 10.1097/01.ALC.0000106303.41755.B8. [DOI] [PubMed] [Google Scholar]
- Lovinger DM (2018). Presynaptic ethanol actions: Potential roles in ethanol seeking. Handbook of Experimental Pharmacology, 248, 29–54. 10.1007/164_2017_76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, & Abrahao KP (2018). Synaptic plasticity mechanisms common to learning and alcohol use disorder. Learning & Memory, 25(9), 425–434. 10.1101/lm.046722.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Kranzler HR, Zhao H, & Gelernter J (2003). Haplotypes at the OPRM1 locus are associated with susceptibility to substance dependence in European-Americans. American Journal of Medical Genetics Part B, Neuropsychiatric Genetics, 120B(1), 97–108. 10.1002/ajmg.b.20034. [DOI] [PubMed] [Google Scholar]
- Mague SD, & Blendy JA (2010). OPRM1 SNP (A118G): Involvement in disease development, treatment response, and animal models. Drug and Alcohol Dependence, 108(3), 172–182. 10.1016/j.drugalcdep.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mague SD, Isiegas C, Huang P, Liu-Chen L-Y, Lerman C, & Blendy JA (2009). Mouse model of OPRM1 (A118G) polymorphism has sex-specific effects on drug-mediated behavior. Proceedings of the National Academy of Sciences of the United States of America, 106(26), 10847–10852. 10.1073/pnas.0901800106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud S, Thorsell A, Sommer WH, Heilig M, Holgate JK, Bartlett SE, et al. (2011). Pharmacological consequence of the A118G μ opioid receptor polymorphism on morphine- and fentanyl-mediated modulation of Ca2+ channels in humanized mouse sensory neurons. Anesthesiology, 115(5), 1054–1062. 10.1097/ALN.0b013e318231fc11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margas W, Zubkoff I, Schuler HG, Janicki PK, & Ruiz-Velasco V (2007). Modulation of Ca2+ channels by heterologously expressed wild-type and mutant human micro-opioid receptors (hMORs) containing the A118G single-nucleotide polymorphism. Journal of Neurophysiology, 97(2), 1058–1067. 10.1152/jn.01007.2006. [DOI] [PubMed] [Google Scholar]
- Maximov A, Pang ZP, Tervo DGR, & Südhof TC (2007). Monitoring synaptic transmission in primary neuronal cultures using local extracellular stimulation. Journal of Neuroscience Methods, 161(1), 75–87. 10.1016/j.jneumeth.2006.10.009. [DOI] [PubMed] [Google Scholar]
- McGue M, Pickens RW, & Svikis DS (1992). Sex and age effects on the inheritance of alcohol problems: A twin study. Journal of Abnormal Psychology, 101(1), 3–17. 10.1037//0021-843x.101.1.3. [DOI] [PubMed] [Google Scholar]
- Miller GM, Bendor J, Tiefenbacher S, Yang H, Novak MA, & Madras BK (2004). A mu-opioid receptor single nucleotide polymorphism in rhesus monkey: Association with stress response and aggression. Molecular Psychiatry, 9(1), 99–108. 10.1038/sj.mp.4001378. [DOI] [PubMed] [Google Scholar]
- Nie Z, Madamba SG, & Siggins GR (2000). Ethanol enhances γ-aminobutyric acid responses in a subpopulation of nucleus accumbens neurons: Role of metabotropic glutamate receptors. Journal of Pharmacology and Experimental Therapeutics, 293(2), 654–661. [PubMed] [Google Scholar]
- Nie Z, Schweitzer P, Roberts AJ, Madamba SG, Moore SD, & Siggins GR (2004). Ethanol augments GABAergic transmission in the central amygdala via CRF1 receptors. Science, 303(5663), 1512–1514. 10.1126/science.1092550. [DOI] [PubMed] [Google Scholar]
- Nishizawa D, Han W, Hasegawa J, Ishida T, Numata Y, Sato T, et al. (2006). Association of mu-opioid receptor gene polymorphism A118G with alcohol dependence in a Japanese population. Neuropsychobiology, 53(3), 137–141. 10.1159/000093099. [DOI] [PubMed] [Google Scholar]
- Oni EN, Halikere A, Li G, Toro-Ramos AJ, Swerdel MR, Verpeut JL, et al. (2016). Increased nicotine response in iPSC-derived human neurons carrying the CHRNA5 N398 allele. Scientific Reports, 6, 34341. 10.1038/srep34341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooteman W, Naassila M, Koeter MWJ, Verheul R, Schippers GM, Houchi H, et al. (2009). Predicting the effect of naltrexone and acamprosate in alcohol-dependent patients using genetic indicators. Addiction Biology, 14(3), 328–337. 10.1111/j.1369-1600.2009.00159.x. [DOI] [PubMed] [Google Scholar]
- Oslin DW, Berrettini W, Kranzler HR, Pettinati H, Gelernter J, Volpicelli JR, et al. (2003). A functional polymorphism of the mu-opioid receptor gene is associated with naltrexone response in alcohol-dependent patients. Neuropsychopharmacology, 28(8), 1546–1552. 10.1038/sj.npp.1300219. [DOI] [PubMed] [Google Scholar]
- Oslin DW, Leong SH, Lynch KG, Berrettini W, O’Brien CP, Gordon AJ, et al. (2015). Naltrexone vs placebo for the treatment of alcohol dependence: A randomized clinical trial. JAMA Psychiatry, 72(5), 430–437. 10.1001/jamapsychiatry.2014.3053. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Yang N, Vierbuchen T, Ostermeier A, Fuentes DR, Yang TQ, et al. (2011). Induction of human neuronal cells by defined transcription factors. Nature, 476(7359), 220–223. 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott CA, & Kendler KS (1999). Genetic and environmental contributions to alcohol abuse and dependence in a population-based sample of male twins. American Journal of Psychiatry, 156(1), 34–40. 10.1176/ajp.156.1.34. [DOI] [PubMed] [Google Scholar]
- Prytkova I, Goate A, Hart RP, & Slesinger PA (2018). Genetics of alcohol use disorder: A role for induced pluripotent stem cells? Alcoholism: Clinical and Experimental Research, 42(9), 1572–1590. 10.1111/acer.13811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramchandani VA, Umhau J, Pavon FJ, Ruiz-Velasco V, Margas W, Sun H, et al. (2011). A genetic determinant of the striatal dopamine response to alcohol in men. Molecular Psychiatry, 16(8), 809–817. 10.1038/mp.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray LA, & Hutchison KE (2004). A polymorphism of the mu-opioid receptor gene (OPRM1) and sensitivity to the effects of alcohol in humans. Alcoholism: Clinical and Experimental Research, 28(12), 1789–1795. 10.1097/01.alc.0000148114.34000.b9. [DOI] [PubMed] [Google Scholar]
- Ray LA, & Hutchison KE (2007). Effects of naltrexone on alcohol sensitivity and genetic moderators of medication response: A double-blind placebo-controlled study. Archives of General Psychiatry, 64(9), 1069–1077. 10.1001/archpsyc.64.9.1069. [DOI] [PubMed] [Google Scholar]
- Ray R, Ruparel K, Newberg A, Wileyto EP, Loughead JW, Divgi C, et al. (2011). Human Mu Opioid Receptor (OPRM1 A118G) polymorphism is associated with brain mu-opioid receptor binding potential in smokers. Proceedings of the National Academy of Sciences of the United States of America, 108(22), 9268–9273. 10.1073/pnas.1018699108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed T, Page WF, Viken RJ, & Christian JC (1996). Genetic predisposition to organ-specific endpoints of alcoholism. Alcoholism: Clinical and Experimental Research, 20(9), 1528–1533. 10.1111/j.1530-0277.1996.tb01695.x. [DOI] [PubMed] [Google Scholar]
- Rouvinen-Lagerström N, Lahti J, Alho H, Kovanen L, Aalto M, Partonen T, et al. (2013). μ-Opioid receptor gene (OPRM1) polymorphism A118G: Lack of association in Finnish populations with alcohol dependence or alcohol consumption. Alcohol and Alcoholism, 48(5), 519–525. 10.1093/alcalc/agt050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna E, Talani G, Busonero F, Pisu MG, Purdy RH, Serra M, et al. (2004). Brain steroidogenesis mediates ethanol modulation of GABAA receptor activity in rat hippocampus. Journal of Neuroscience, 24(29), 6521–6530. 10.1523/JNEUROSCI.0075-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarnati MS, Halikere A, & Pang ZP (2019). Using human stem cells as a model system to understand the neural mechanisms of alcohol use disorders: Current status and outlook. Alcohol, 74, 83–93. 10.1016/j.alcohol.2018.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinka JA, Town T, Abdullah L, Crawford FC, Ordorica PI, Francis E, et al. (2002). A functional polymorphism within the mu-opioid receptor gene and risk for abuse of alcohol and other substances. Molecular Psychiatry, 7(2), 224–228. 10.1038/sj.mp.4000951. [DOI] [PubMed] [Google Scholar]
- Sloan ME, Klepp TD, Gowin JL, Swan JE, Sun H, Stangl BL, et al. (2018). The OPRM1 A118G polymorphism: Converging evidence against associations with alcohol sensitivity and consumption. Neuropsychopharmacology, 43(7), 1530–1538. 10.1038/s41386-017-0002-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridhar D (2012). Health policy: Regulate alcohol for global health. Nature, 482(7385), 302. 10.1038/482302a. [DOI] [PubMed] [Google Scholar]
- Szeto CY, Tang NL, Lee DT, & Stadlin A (2001). Association between mu opioid receptor gene polymorphisms and Chinese heroin addicts. NeuroReport, 12(6), 1103–1106. 10.1097/00001756-200105080-00011. [DOI] [PubMed] [Google Scholar]
- Talani G, & Lovinger DM (2015). Interactions between ethanol and the endocannabinoid system at GABAergic synapses on basolateral amygdala principal neurons. Alcohol, 49(8), 781–794. 10.1016/j.alcohol.2015.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theile JW, Morikawa H, Gonzales RA, & Morrisett RA (2008). Ethanol enhances GABAergic transmission onto dopamine neurons in the ventral tegmental area of the rat. Alcoholism: Clinical and Experimental Research, 32(6), 1040–1048. 10.1111/j.1530-0277.2008.00665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidey JW, Monti PM, Rohsenow DJ, Gwaltney CJ, Miranda R, McGeary JE, et al. (2008). Moderators of naltrexone’s effects on drinking, urge, and alcohol effects in non-treatment-seeking heavy drinkers in the natural environment. Alcoholism: Clinical and Experimental Research, 32(1), 58–66. 10.1111/j.1530-0277.2007.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Town T, Abdullah L, Crawford F, Schinka J, Ordorica PI, Francis E, et al. (1999). Association of a functional mu-opioid receptor allele (+118A) with alcohol dependency. American Journal of Medical Genetics, 88(5), 458–461. [PubMed] [Google Scholar]
- True WR, Xian H, Scherrer JF, Madden PA, Bucholz KK, Heath AC, et al. (1999). Common genetic vulnerability for nicotine and alcohol dependence in men. Archives of General Psychiatry, 56(7), 655–661. 10.1001/archpsyc.56.7.655. [DOI] [PubMed] [Google Scholar]
- Türkan H, Karahalil B, Kadıoğlu E, Eren K, Gürol DT, & Karakaya AE (2019). The association between the OPRM1 A118G polymorphism and addiction in a Turkish population. Arhiv za Higijenu Rada i Toksikologiju, 70(2), 97–103. 10.2478/aiht-2019-70-3153. [DOI] [PubMed] [Google Scholar]
- Vallender EJ, Rüedi-Bettschen D, Miller GM, & Platt DM (2010). A pharmacogenetic model of naltrexone-induced attenuation of alcohol consumption in rhesus monkeys. Drug and Alcohol Dependence, 109(1–3), 252–256. 10.1016/j.drugalcdep.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhulst B, Neale MC, & Kendler KS (2015). The heritability of alcohol use disorders: A meta-analysis of twin and adoption studies. Psychological Medicine, 45(5), 1061–1072. 10.1017/S0033291714002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan FJ, Berton F, Madamba SG, Francesconi W, & Siggins GR (1996). Low ethanol concentrations enhance GABAergic inhibitory postsynaptic potentials in hippocampal pyramidal neurons only after block of GABAB receptors. Proceedings of the National Academy of Sciences of the United States of America, 93(10), 5049–5054. 10.1073/pnas.93.10.5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-J, Huang P, Blendy JA, & Liu-Chen L-Y (2014). Brain region- and sex-specific alterations in DAMGO-stimulated [(35) S]GTPγS binding in mice with Oprm1 A112G. Addiction Biology, 19(3), 354–361. 10.1111/j.1369-1600.2012.00484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-J, Huang P, Ung A, Blendy JA, & Liu-Chen L-Y (2012). Reduced expression of the μ opioid receptor in some, but not all, brain regions in mice with OPRM1 A112G. Neuroscience, 205, 178–184. 10.1016/j.neuroscience.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Gu C, & Dunwiddie TV (1997). Differential ethanol sensitivity of subpopulations of GABAA synapses onto rat hippocampal CA1 pyramidal neurons. Journal of Neurophysiology, 77(3), 1306–1312. 10.1152/jn.1997.77.3.1306. [DOI] [PubMed] [Google Scholar]
- Wilcox MV, Carlson VC, Sherazee N, Sprow GM, Bock R, et al. (2014). Repeated binge-like ethanol drinking alters ethanol drinking patterns and depresses striatal GABAergic transmission. Neuropsychopharmacology, 39(3), 579–594. 10.1038/npp.2013.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization. (2019). Global status report on alcohol and health 2018. Retrieved from https://www.who.int/substance_abuse/publications/global_alcohol_report/en/.
- Xiao C, Zhang J, Krnjević K, & Ye JH (2007). Effects of ethanol on midbrain neurons: Role of opioid receptors. Alcoholism: Clinical and Experimental Research, 31(7), 1106–1113. 10.1111/j.1530-0277.2007.00405.x. [DOI] [PubMed] [Google Scholar]
- Yang N, Chanda S, Marro S, Ng Y-H, Janas JA, Haag D, et al. (2017). Generation of pure GABAergic neurons by transcription factor programming. Nature Methods, 14(6), 621–628. 10.1038/nmeth.4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh HH, & Kolb JE (1997). Ethanol modulation of GABA-activated current responses in acutely dissociated retinal bipolar cells and ganglion cells. Alcoholism: Clinical and Experimental Research, 21(4), 647–655. [PubMed] [Google Scholar]
- Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, et al. (2018). Ensembl 2018. Nucleic Acids Research, 46(D1), D754–D761. 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Wang D, Johnson AD, Papp AC, & Sadée W (2005). Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. Journal of Biological Chemistry, 280(38), 32618–32624. 10.1074/jbc.M504942200. [DOI] [PubMed] [Google Scholar]
- Zhu PJ, & Lovinger DM (2006). Ethanol potentiates GABAergic synaptic transmission in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. Journal of Neurophysiology, 96(1), 433–441. 10.1152/jn.01380.2005. [DOI] [PubMed] [Google Scholar]
- Ziskind-Conhaim L, Gao B-X, & Hinckley C (2003). Ethanol dual modulatory actions on spontaneous postsynaptic currents in spinal motoneurons. Journal of Neurophysiology, 89(2), 806–813. 10.1152/jn.00614.2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.