

Abstract
Protein N-terminal methyltransferases (NTMTs) catalyze the methylation of the α-N-terminal amines of proteins starting with an X–P–K/R motif. NTMT1 has been implicated in various cancers and in aging, implying its role as a potential therapeutic target. Through structural modifications of a lead NTMT1 inhibitor, BM30, we designed and synthesized a diverse set of inhibitors to probe the NTMT1 active site. The incorporation of a naphthyl group at the N-terminal region and an ortho-aminobenzoic amide at the C-terminal region of BM30 generates the top cell-potent inhibitor DC541, demonstrating increased activity on both purified NTMT1 (IC50 of 0.34 ± 0.02 μM) and the cellular α-N-terminal methylation level of regulator of chromosome condensation 1 (RCC1, IC50 value of 30 μM) in human colorectal cancer HT29 cells. Furthermore, DC541 exhibits over 300-fold selectivity to several methyltransferases. This study points out the direction for the development of more cell-potent inhibitors for NTMT1.
Keywords: Protein N-terminal methyltransferase, Peptidomimetic inhibitor, Structure-based discovery, Cell-potent inhibitor, Methyltransferases
Protein α-N-terminal methylation, a conserved modification on ribosomal proteins and muscle light chains across almost all species, has recently been uncovered on a diverse set of proteins involved in cell division, DNA damage repair, and chromatin remodeling.1−6 Specifically, α-N-terminal methylation modulates the dynamic association between the regulator of chromosome condensation 1 (RCC1) and chromatin during mitosis.1,7 Likewise, a similar function of α-N-terminal methylation has been observed on centromere proteins A and B.5,7 Unlike the protein methylation on the side chain of either lysine or arginine residues, methylation of the α-N-terminus alters both the hydrophobicity and the charge state under physiological conditions, which may be the underlying mechanism of its function in regulating protein–protein and protein–DNA interactions.3 Protein N-terminal methyltransferases (NTMTs) are the enzymes that catalyze the transfer of a methyl group from the cofactor, S-adenosyl-l-methionine (SAM), onto the α-N-terminal amines of substrate proteins with a specific N-terminal motif X–P–K/R, where X can be any amino acid other than D/E.8−11 With the important biological function of α-N-terminal methylation, the dysregulation of NTMT1 has been implicated in many diseases. including malignant melanoma and colorectal and brain cancer.3,8 Knockdown or knockout of NTMT1 results in hypersensitivity of breast cancer cell lines to DNA damage and premature aging,12,13 implying the role of NTMT1 in DNA damage repair.
Despite recent progress on protein α-N-terminal methylation, its physiological and pharmacological roles are still in their infancy compared with protein lysine methylation and arginine methylation. Therefore, it is crucial to have specific and cell-permeable inhibitors for NTMT1 to elucidate its biological functions and therapeutic potentials NTMT1 bisubstrate inhibitors including the inhibitor NAH–C4–GPKRIA displayed high potency at 130 pM, but their poor cell permeability and protease stability prevent their application in cellular contexts (Figure 1).14−17 Recently, we reported the first potent peptidomimetic inhibitor BM30 (IC50 = 0.89 ± 0.10 μM) by targeting the unique substrate-binding pocket of NTMT1 (Figure 1).18 Even after the introduction of cell-permeable peptides at the C-terminus of BM30 to generate DC432, the cellular inhibitory effect on the N-terminal methylation was modest until 1 mM was reached.18 Herein we reported the structure-based discovery of NTMT1 inhibitors with enhanced cellular potency.
Figure 1.
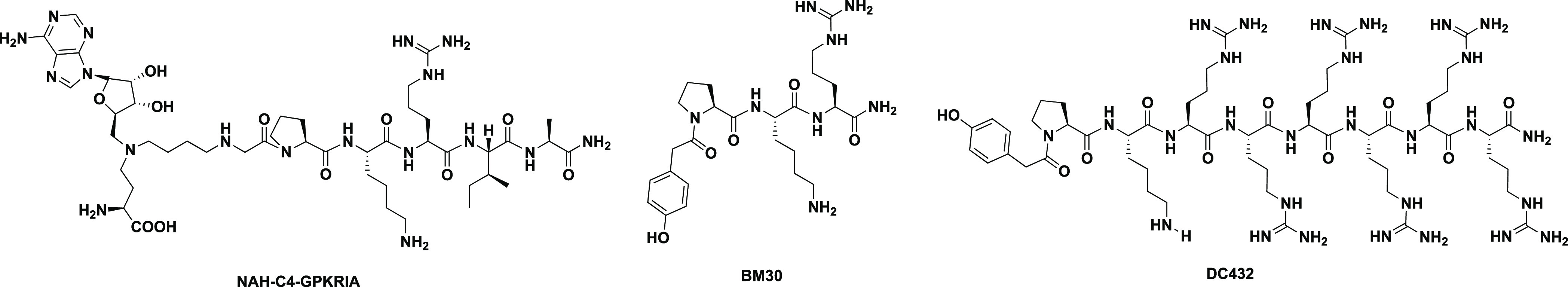
Representative structures of reported NTMT1 inhibitors.
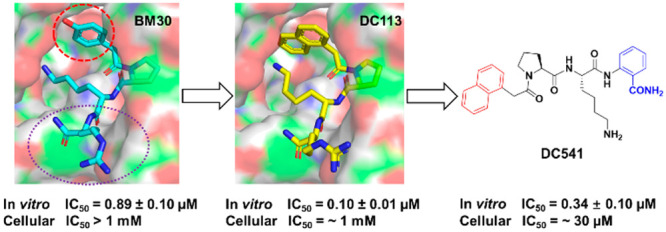
To improve the cellular potency of the peptidomimetic inhibitor BM30, we focused on the modifications on the N-terminal and C-terminal regions of the lead compound BM30, as Pro2 and Lys3 in the middle region of BM30 are less tolerable. Our rationale stemmed from a close examination of the NTMT1–BM30–SAH ternary complex (PDB ID: 6WH8).18 Additional space around the phenyl group of BM30 was identified through an inspection of the binding pocket of the 4-hydroxyphenyl group at the first position (R1) (Figure 2). Thus we hypothesized that the introduction of a bicyclic group in the first position would simultaneously increase both the potency and the hydrophobicity to facilitate cell permeability. For the C-terminal region, we first replaced the Arg4 with other amino acids to investigate their effects on inhibition. Then, non-amino-acid moieties were introduced in an attempt to enhance the stability and cell penetration.
Figure 2.

Structure-based design of new inhibitors.
In general, peptidomimetics, except compounds 15, 18, and 19, were prepared through the standard coupling reaction of various carboxylic acids with the tripeptide Pro–Lys–X (X = Arg, Lys, Gly, Ala, Glu, His, Phe, Trp, Tyr, Me–Arg, Me–Lys, Me–Ala, 3-aminobenzoic acid, or 2-aminobenzoic acid) or the dipeptide Pro–Lys on Rink resin.18 A subsequent acidic treatment with a cleavage cocktail consisting of trifluoroacetic acid (TFA)/2,2′-(ethylenedioxy)diethanethiol/water (H2O)/triisopropylsilane (94:2.5:2.5:1) followed by purification through reverse-phase high-performance liquid chromatography (RP-HPLC) provided the products. Compounds 15, 18, and 19 were synthesized in the solution phase, as shown in Scheme 1.
Scheme 1. Synthetic Route for Compounds 15, 18, and 19.

Reagents and conditions: (a) HCl·Lys(Boc)–COMe, HBTU, HOBt, DIPEA, DMF. (b) i. LiOH·H2O, MeOH/H2O, 0 °C to rt; ii. R-NH2, HBTU, HOBt, DIPEA, DMF; iii. 4 N HCl in dioxane, 0 °C to rt, 50–68% in three steps.
All synthesized peptidomimetics were evaluated in an S-5′-adenosyl-l-homocysteine (SAH) hydrolase (SAHH)-coupled fluorescence assay under the conditions of saturated SAM (100 μM) and the Michaelis constant (Km) value of the peptide substrate (GPKRIA).19 The inhibitory activity of BM30 was redetermined as a positive control, showing a comparable half-maximal inhibitory concentration (IC50) value to that previously reported (Table 1).18
Table 1. Modifications at N-Terminal Regiona.
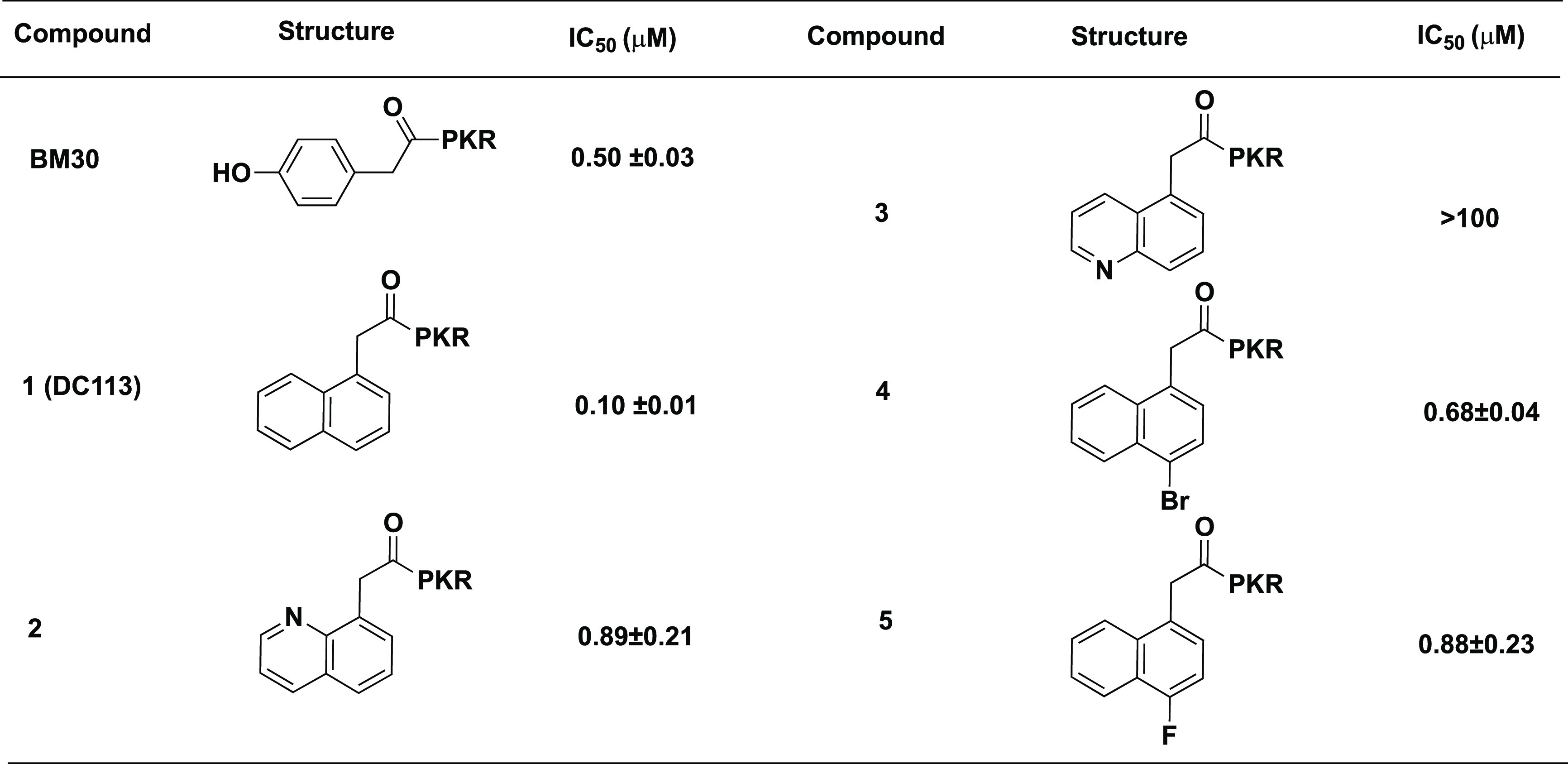
IC50 values were determined in triplicate (n = 3) and are presented as the mean ± standard deviation (SD).
N-Terminal Region. To increase the hydrophobicity of the inhibitor, the naphthyl group was incorporated in the first position to replace the 4-hydroxyphenyl group of BM30 to generate compound 1 (DC113) (Table 1). It showed about five-fold increased activity compared with BM30. Next, an 8-quinolinyl group was introduced to replace the naphthyl group to produce 2, yielding an eight-fold loss of inhibition compared with DC113. Surprisingly, a 5-quinolinyl group in compound 3 abolished the inhibition to NTMT1. Furthermore, we introduced halogens onto the naphthyl ring of DC113 to obtain 4 and 5, also displaying about seven- to eight-fold loss compared with DC113. Thus DC113 was used as the lead compound for the subsequent investigation at the C-terminal region.
C-Terminal Region. To understand the contribution of Arg4 to the interaction with NTMT1, we replaced Arg with other amino acids, such as Lys, Gly, Ala, Glu, His, Phe, Trp, and Tyr, to yield 6–13. Among them, Lys was chosen because it is also a basic amino acid and has been observed in NTMT1 protein substrates like DNA damage binding domain (DDB2) and kelch-like protein 31 (KLH31).2,3,8 Gly and Ala were designed to explore the role of the guanidine group of Arg. Glu served as a negative control, as it is an acidic amino acid. Aromatic amino acids His, Phe, Trp, and Tyr were designed to explore the size of this binding pocket. Moreover, the deletion of the fourth position Arg produced 14 to further explore the contribution of the interaction of the Arg4 backbone with NTMT1. Compounds 6–14 showed inhibitory activities with IC50 values ranging from 0.34 to 4.2 μM, which were 3–40 times higher than that of DC113 (Table 2). Compound 9 containing Ala showed a comparable IC50 value as DC113, suggesting a trivial contribution from the guanidine group of Arg4 to the interaction of NTMT1. This result is consistent with the information from the cocrystal structure, as the side chain of Arg4 orients to the solvent.10 However, the introduction of an acidic residue like Glu at the C-terminus is unfavorable, exhibiting a ∼40-fold loss in inhibition. Except for the acidic residue, other substitutions including aromatic residues Trp, Tyr, and Phe are reasonably tolerant at the C-terminal region.
Table 2. Modifications at C-Terminal Regiona.
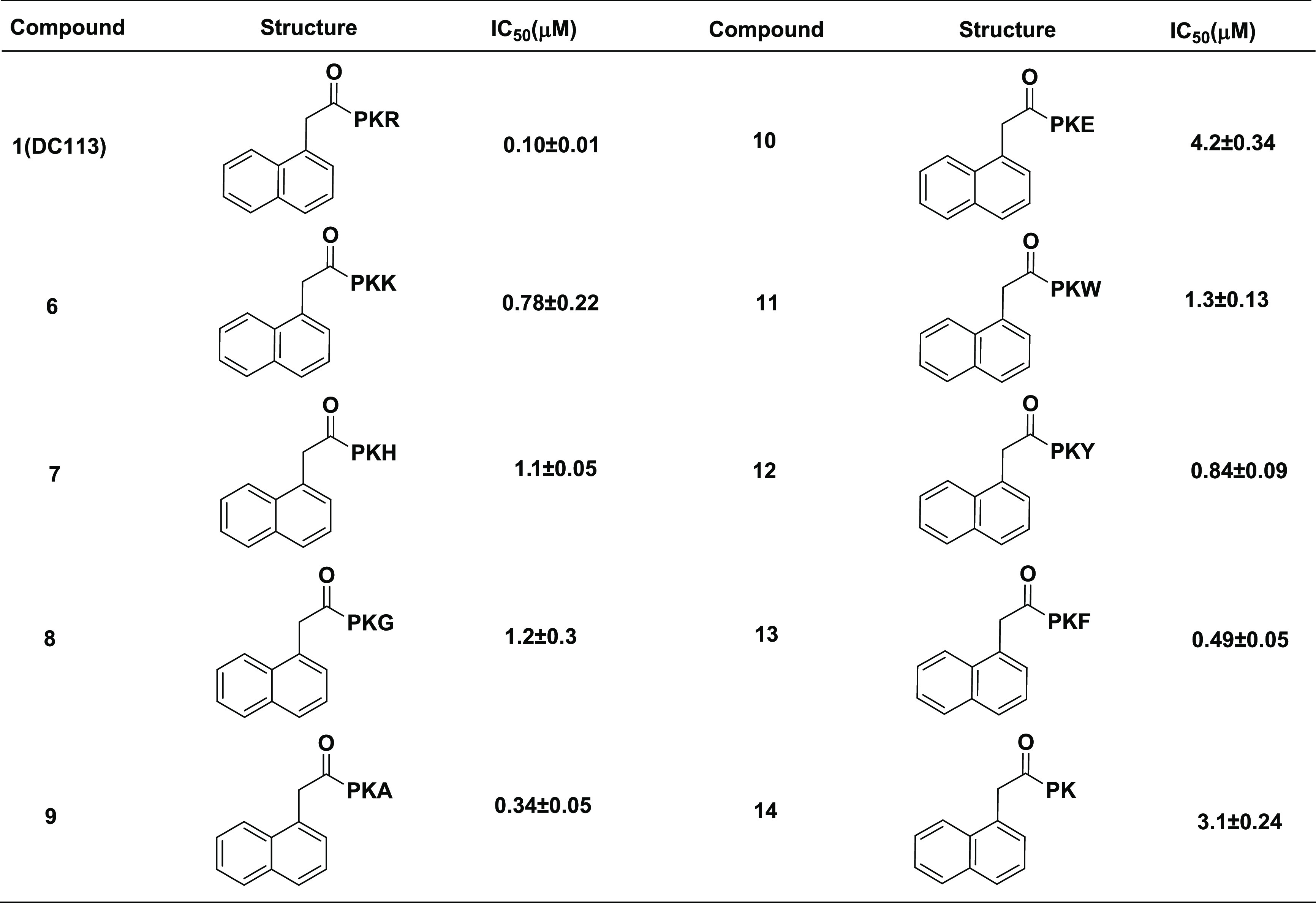
IC50 values were determined in triplicate (n = 3) and are presented as the mean ± standard deviation (SD).
The inhibition mechanism of DC113 was examined to check if it similarly interacted with NTMT1 compared with BM30. As shown in Figure 3, the IC50 values of DC113 increased proportionally to the rising concentrations of the peptide substrate, which indicated that the compound interacted with the peptide substrate-binding site. Conversely, the IC50 values of DC113 were almost unaffected when the concentration of SAM increased, exhibiting a noncompetitive inhibition pattern with the cofactor SAM. These results suggested that DC113 retained a similar binding mode as BM30.
Figure 3.

Inhibition mechanism of DC113. (A) IC50 curves of DC113 at varying concentrations of the peptide (GPKRIA) with a fixed concentration of SAM. (B) Linear regression plot of IC50 values with corresponding concentrations of the peptide (GPKRIA). (C) IC50 curves of DC113 at varying concentrations of SAM with a fixed concentration of the peptide (GPKRIA). (D) Linear regression plot of IC50 values with corresponding concentrations of SAM.
The selectivity of DC113 for NTMT1 was evaluated against a panel of methyltransferases (MTs) including two representative PKMTs (G9a and SETD7), three PRMTs (PRMT1, PRMT3, and TbPRMT7), nicotinamide N-methyltransferase (NNMT), and the coupling enzyme SAHH. Our results showed that DC113 inhibited <50% of the activities of those enzymes at 100 μM, demonstrating the >1000-fold selectivity of DC113 for NTMT1 over other MTs (Figure 4).
Figure 4.

Selectivity study of DC113 (n = 2).
To elucidate the interaction between DC113 and NTMT1 at the molecular level, we obtained the X-ray cocrystal structure of NTMT1–DC113–SAH (PDB ID: 7K3D) (Figure 5). The results showed the DC113 only occupied the substrate-binding site of NTMT1, similarly to the lead compound BM30.18 The structure alignment of the NTMT1–DC113–SAH complex with the NTMT1–BM30–SAH structure further revealed a similar binding interaction (Figure 5D,E). For example, the carbonyl oxygen of the first residue forms a H bond with Asn 168. The amino group of Lys interacts with the carboxylate groups of Asp177 and Asp180 through electrostatic interactions. Importantly, the naphthyl ring appeared to insert more deeply into the binding pocket compared with the phenyl ring, as we hypothesized, which enabled it to have a π–π interaction with Trp20. Compared with BM30, DC113 lost the H bond between the 4-hydroxyl of BM30 and Asp180. However, an extra phenyl ring of DC113 was able to push one water molecule in the binding pocket, and the naphthyl ring restricted the rotation to decrease the entropy loss, which may contribute to its increased inhibitory activity.
Figure 5.

X-ray cocrystal structure of NTMT1 (gray cartoon)–DC113 (green stick)–SAH (green stick) (PDB ID: 7K3D). (A) Fo – Fc omit density map of ligands in the cocrystal structure. (B) Interactions of DC113 with NTMT1. H-bond interactions are shown as yellow dotted lines. (C) DC113 2D interaction diagram (Schrödinger Maestro) with NTMT1. (D) Structural alignment of NTMT1 (gray cartoon)–DC113 (green stick)–SAH (green stick) and NTMT1 (yellow cartoon)–BM30 (yellow stick)–SAH (yellow stick) (PDB ID: 6WH8). (E) Zoomed view of structural alignment.
NTMT1 can catalyze the methylation of its substrate, starting with an SPK motif. Therefore, the NTMT1 cell-permeable inhibitor is expected to decrease the N-terminal methylation level. Compared with BM30 (cLogP = −2.33), we proposed that DC113 (cLogP = −0.49) would enhance the cell penetration due to its increased hydrophobicity. The mass spectrometry (MS) study confirmed that DC113 displayed enhanced cell permeability compared with BM30, as DC113 but not BM30 could be detected in the cells at 100 μM (Figure S1).18 Then, we proceeded to assess the inhibitory effects of DC113 on the N-terminal trimethylation level via Western blotting with the specific antibody to detect the N-terminal trimethylation level, as it is predominant in HCT116 cells. However, DC113 decreased by only ∼50% of the me3-RCC1 and SET levels at 1 mM in HCT116 cells (Table 3).
Table 3. Optimizations at C-Terminal Region to Increase Cellular Potency.
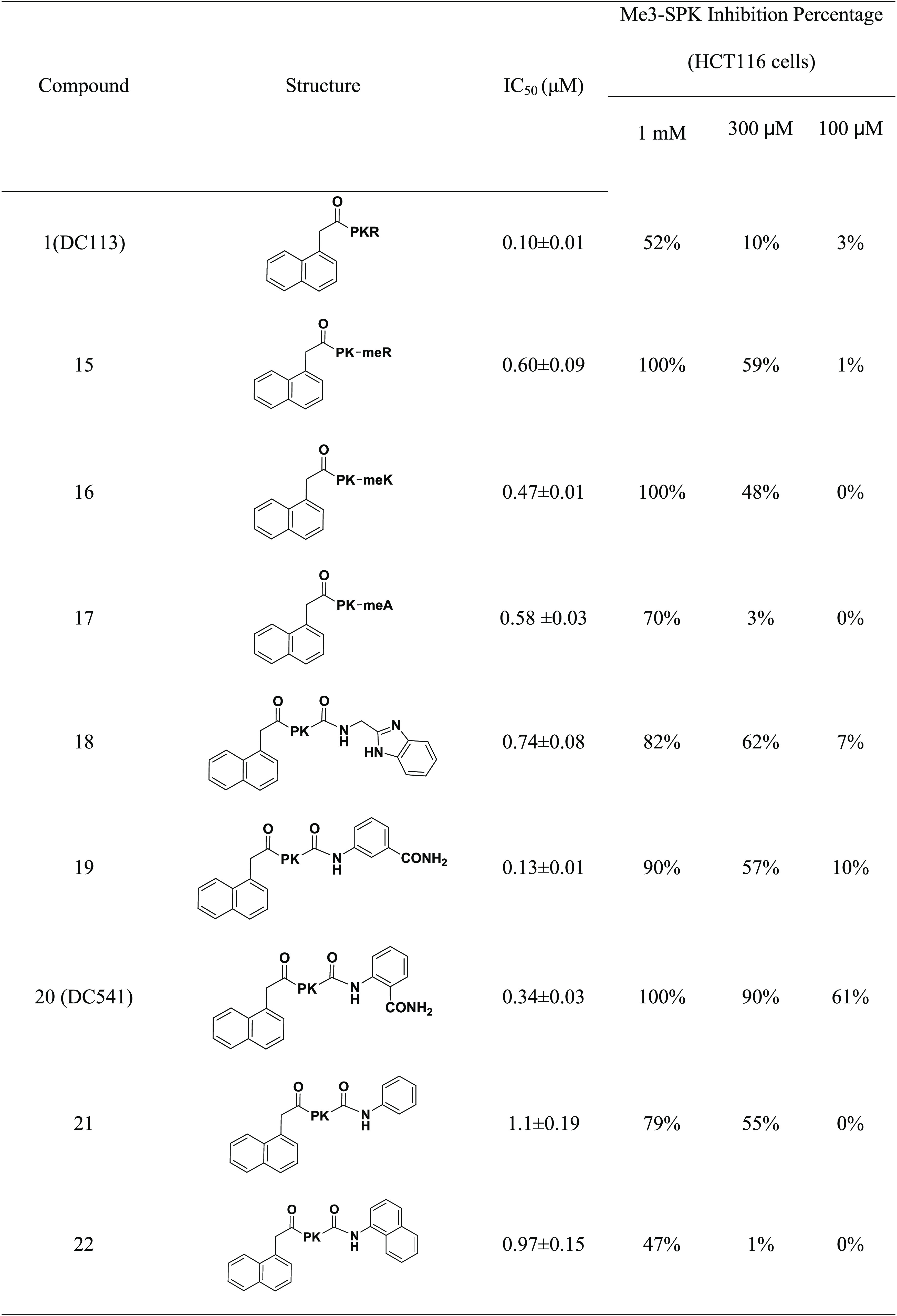
To increase the cellular potency, we proceeded to introduce an N-methylation amino acid at the fourth position to generate compounds 15–17 to increase the stability of compounds against proteases. Although the inhibitory activity decreased four to six times compared with that of DC113 (Table 3), 15 and 16 demonstrated three-fold increased cellular potency compared with DC113 in HCT116 cells based on the trimethylation levels. After incubation with trypsin for 1 h, peaks of cleaved products of DC113 can be detected in the MS (Figure S2); however, the incubation of 15–17 with trypsin for 1 h confirmed their improved stability, as no fragment peaks were detected in the MS (Figure S3). Compounds 18–20 were designed and synthesized to replace Arg4 with a non-amino-acid moiety, including benzimidazole and ortho- and meta-aminobenzoic amide groups. Among them, 19 demonstrated the most potent inhibition on NTMT1, with an IC50 value of 0.13 μM, which was comparable to that of DC113; however, its positional isomer 20 led to a three-fold loss of inhibitory activity. To explore the importance of the amide group, 21 was prepared and led to ten- and three-fold decreased inhibition compared with that of 19 and 20, respectively. Meanwhile, a C-terminal naphthyl group was introduced to produce 22 to further increase the hydrophobicity, yielding a comparable activity to that of 21. Then, cellular N-terminal methylation indicated that compound 20 (DC541) exhibited the best cellular inhibition among all tested compounds. Thus DC541 was chosen to evaluate its cellular inhibition on two colorectal cancer cell lines, HT29 and HCT116 (Figure 6). DC541 significantly inhibited me3-RCC1 at 100 μM and showed an approximate IC50 value of 30 μM on the me3-RCC1 level in HT29 cells (Figure 6A). On the basis of the results, the inhibitory effects of the compound were more sensitive in HT29 cells than in HCT116 cells, as the IC50 value of DC541 was ∼100 μM in HCT116 cells (Figure 6B), supporting the importance of simultaneously assessing both the biochemical and cellular activities. Interestingly, different inhibitory effects were also observed on the N-terminal methylation between RCC1 and the SET protein, with an me3-RCC1 level about ten times more sensitive to DC541 inhibition. One possible reason is that the endogenous level of me3-SET is higher than that of me3-RCC1.
Figure 6.

Inhibition of cellular N-terminal methylation by DC541 in HT29 and HCT116 cells. Representative Western blot results of the effects of DC541 (0–300 μM) on the cellular methylation level (n = 2) of (A) HT29 and (B) HCT116 cells. RCC1, SET, and Lamin B1 blots are shown as loading controls.
To further validate the selectivity of DC541 for NTMT1, we evaluated the inhibitory activity of DC541 against a panel of methyltransferases and the coupling enzyme SAHH, similarly to DC113. The results showed that DC541 did not inhibit 50% of those enzyme activities up to 100 μM, indicating more than 300-fold selectivity against those methyltransferases and SAHH (Figure 7).
Figure 7.

Selectivity study of DC541 (n = 2).
The cytotoxicity of DC541 was evaluated on both HT29 and HCT116 cells through the alamarBlue assay (Figure 8). Cells were treated with DC541 for 24, 48, and 72 h and tested in the alamarBlue assay. No significant toxicity was observed in the viability of either cell line up to 1 mM at all three time points.
Figure 8.

Cytotoxicity studies of DC541 on (A) HT29 and (B) HCT116 cells via alamarBlue assay (n = 3).
In summary, we developed a series of cell-potent NTMT1 peptidomimetic inhibitors that target the substrate-binding pocket. The inhibition mechanism and cocrystal structure of NTMT1–DC113–SAH confirmed that naphthyl substitution at the N-terminal region can retain the interaction at the peptide substrate-binding site. Furthermore, it demonstrated high selectivity for NTMT1 by exhibiting over 1000-fold potency for NTMT1 compared with other MTs. Although DC113 showed better inhibitory activity and cell permeability than BM30, the compound decreased the me3-SPK level with an IC50 value of ∼1 mM in HCT116 cells. Further modification at the C-terminal region to increase the hydrophobicity and stability yielded DC541, showing an approximate IC50 value of 30 μM in HT29 and 100 μM in HCT116 cells. The 100- to 300-fold differences between the biochemical inhibition and the cellular potency need to be examined in more detail by quantifying its cell permeability in the future. Nevertheless, this is the first report of cell-permeable tetrapeptidomimetic inhibitors for NTMT1 to our knowledge. Therefore, this study provides a valuable tool for the research community to study the functions of NTMT1. Future investigation will focus on its application in the cell-based study to understand the biological function of NTMT1 as well as to improve the cell potency of NTMT1 inhibitors by exploring the C-terminal region.
Acknowledgments
We thank Mr. Congyu Zhang for his assistance with the synthesis of several peptidomimetic analogues. We also thank Mr. Yuanrui Zhao for his assistance with the purification and NMR spectrum of compounds. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. We also acknowledge support from the Department of Medicinal Chemistry and Molecular Pharmacology (R.H.) and the Department of Biological Sciences (N.N.) at Purdue University.
Glossary
Abbreviations
- NTMT
protein N-terminal methyltransferase
- SAM
S-5′-adenosyl-l-methionine
- SAH
S-5′-adenosyl-l-homocysteine
- SAHH
SAH hydrolase
- MT
methyltransferase
- PKMT
protein lysine methyltransferase
- PRMT
protein arginine methyltransferase
- PRMT1
protein arginine methyltransferase 1
- G9a
euchromatic histone–lysine N-methyltransferase 2
- SETD7
SET domain-containing protein 7
- PRMT3
protein arginine methyltransferase 3
- TbPRMT7
Trypanosoma brucei protein arginine methyltransferase 7
- rt
room temperature
- TFA
trifluoroacetic acid
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00012.
Experimental section. MS characterization of compounds 1–22. MS and HPLC analysis of compounds 1–22. NMR spectra of compounds 18 and 20–23. Figure S1. Cell permeability evaluation of DC113. Figure S2. Stability evaluation of compound DC113. Figure S3. Stability evaluation of compounds 15–17. Figure S4. Inhibition of cellular N-terminal methylation by DC541 in HT29 and HCT116 cells. Table S1. Crystallography data and refinement statistics (PDB ID: 7K3D) (PDF)
Accession Codes
The coordinates for the structure of human NTMT1 in complex with compound DC113 (PDB ID: 7K3D) have been deposited in Protein Data Bank.
Author Contributions
The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.
We acknowledge the support from NIH grants R01GM117275 (R.H.), 1R01GM127896 (N.N.), 1R01AI127793 (N.N.), and P30 CA023168 (Purdue University Center for Cancer Research). GM/CA@APS has been funded in whole or in part with federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006).
The authors declare no competing financial interest.
Supplementary Material
References
- Chen T.; Muratore T. L.; Schaner-Tooley C. E.; Shabanowitz J.; Hunt D. F.; Macara I. G. N-Terminal Alpha-Methylation of RCC1 Is Necessary for Stable Chromatin Association and Normal Mitosis. Nat. Cell Biol. 2007, 9, 596–603. 10.1038/ncb1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q.; Fu L.; Wang Z.; Gan N.; Dai X.; Wang Y. α-N-Methylation of Damaged DNA-Binding Protein 2 (DDB2) and Its Function in Nucleotide Excision Repair. J. Biol. Chem. 2014, 289, 16046–16056. 10.1074/jbc.M114.558510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R. Chemical Biology of Protein N-Terminal Methyltransferases. ChemBioChem 2019, 20, 976–984. 10.1002/cbic.201800615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann-Liebold B.; Pannenbecker R. Primary Structure of Protein L33 from the Large Subunit of the Escherichia Coli Ribosome. FEBS Lett. 1976, 68, 115–118. 10.1016/0014-5793(76)80417-6. [DOI] [PubMed] [Google Scholar]
- Sathyan K. M.; Fachinetti D.; Foltz D. R. α-Amino Trimethylation of CENP-A by NRMT Is Required for Full Recruitment of the Centromere. Nat. Commun. 2017, 8, 14678. 10.1038/ncomms14678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X.; Rulten S. L.; You C.; Caldecott K. W.; Wang Y. Identification and Functional Characterizations of N-Terminal α-N-Methylation and Phosphorylation of Serine 461 in Human Poly(ADP-Ribose) Polymerase 3. J. Proteome Res. 2015, 14, 2575–2582. 10.1021/acs.jproteome.5b00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X.; Otake K.; You C.; Cai Q.; Wang Z.; Masumoto H.; Wang Y. Identification of Novel A-N-Methylation of CENP-B That Regulates Its Binding to the Centromeric DNA. J. Proteome Res. 2013, 12, 4167–4175. 10.1021/pr400498y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaner Tooley C. E.; Petkowski J. J.; Muratore-Schroeder T. L.; Balsbaugh J. L.; Shabanowitz J.; Sabat M.; Minor W.; Hunt D. F.; Macara I. G. NRMT Is an Alpha-N-Methyltransferase That Methylates RCC1 and Retinoblastoma Protein. Nature 2010, 466, 1125–1128. 10.1038/nature09343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkowski J. J.; Bonsignore L. A.; Tooley J. G.; Wilkey D. W.; Merchant M. L.; Macara I. G.; Schaner Tooley C. E. NRMT2 Is an N-Terminal Monomethylase That Primes for Its Homologue NRMT1. Biochem. J. 2013, 456, 453–462. 10.1042/BJ20131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C.; Mao Y.; Tempel W.; Qin S.; Li L.; Loppnau P.; Huang R.; Min J. Structural Basis for Substrate Recognition by the Human N-Terminal Methyltransferase 1. Genes Dev. 2015, 29, 2343–2348. 10.1101/gad.270611.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C.; Dong G.; Li L.; Zhu L.; Tempel W.; Liu Y.; Huang R.; Min J. An Asparagine/Glycine Switch Governs Product Specificity of Human N-Terminal Methyltransferase NTMT2. Commun. Biol. 2018, 1, 183. 10.1038/s42003-018-0196-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsignore L. A.; Butler J. S.; Klinge C. M.; Tooley C. E. S. Loss of the N-Terminal Methyltransferase NRMT1 Increases Sensitivity to DNA Damage and Promotes Mammary Oncogenesis. Oncotarget 2015, 6, 12248–12263. 10.18632/oncotarget.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsignore L. A.; Tooley J. G.; Van Hoose P. M.; Wang E.; Cheng A.; Cole M. P.; Schaner Tooley C. E. NRMT1 Knockout Mice Exhibit Phenotypes Associated with Impaired DNA Repair and Premature Aging. Mech. Ageing Dev. 2015, 146, 42–52. 10.1016/j.mad.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.; Dong G.; Noinaj N.; Huang R. Discovery of Bisubstrate Inhibitors for Protein N-Terminal Methyltransferase 1. J. Med. Chem. 2019, 62, 3773–3779. 10.1021/acs.jmedchem.9b00206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.; Dong C.; Dong G.; Srinivasan K.; Min J.; Noinaj N.; Huang R. Probing the Plasticity in the Active Site of Protein N-Terminal Methyltransferase 1 Using Bisubstrate Analogs. J. Med. Chem. 2020, 63, 8419–8431. 10.1021/acs.jmedchem.0c00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Richardson S. L.; Mao Y.; Huang R. Design, Synthesis, and Kinetic Analysis of Potent Protein N-Terminal Methyltransferase 1 Inhibitors. Org. Biomol. Chem. 2015, 13, 4149–4154. 10.1039/C5OB00120J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Huang R. Facile Synthesis of SAM-Peptide Conjugates through Alkyl Linkers Targeting Protein N-Terminal Methyltransferase 1. RSC Adv. 2016, 6, 6768–6771. 10.1039/C5RA20625A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie B. D.; Chen D.; Dong G.; Dong C.; Parker H.; Schaner Tooley C. E.; Noinaj N.; Min J.; Huang R. Selective Peptidomimetic Inhibitors of NTMT1/2: Rational Design, Synthesis, Characterization, and Crystallographic Studies. J. Med. Chem. 2020, 63, 9512–9522. 10.1021/acs.jmedchem.0c00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson S. L.; Mao Y.; Zhang G.; Hanjra P.; Peterson D. L.; Huang R. Kinetic Mechanism of Protein N-Terminal Methyltransferase 1. J. Biol. Chem. 2015, 290, 11601–11610. 10.1074/jbc.M114.626846. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.