

Abstract

Indoleamine 2,3-dioxygenase 1 (IDO1) has been identified as a target for small-molecule immunotherapy for the treatment of a variety of cancers including renal cell carcinoma and metastatic melanoma. This work focuses on the identification of IDO1 inhibitors containing replacements or isosteres for the amide found in BMS-986205, an amide-containing, IDO1-selective inhibitor currently in phase III clinical trials. Detailed subsequently are efforts to identify a structurally differentiated IDO1 inhibitor via the pursuit of a variety of heterocyclic isosteres, leading to the discovery of highly potent, imidazopyridine-containing IDO1 inhibitors.
Keywords: Indoleamine 2,3-dioxygenase 1; IDO1; immuno-oncology; amide isostere
The clinical success of monoclonal antibody checkpoint inhibitors such as Yervoy (Bristol Myers Squibb, BMS), Keytruda (Merck), and Opdivo (BMS) has received much attention in the field of immuno-oncology (IO). These IO therapies leverage a patient’s native immune system to reverse tumor-induced immune suppression and enhance immune response toward cancer.1 More recently, the effect of metabolism, including amino acid catabolism, on immune response in the tumor microenvironment (TME) has provided a basis for exploring IO targets capable of perturbation by small molecules. One such strategy for small-molecule cancer immunotherapy involves the inhibition of indoleamine 2,3-dioxygenase 1 (IDO1) to decrease local kynurenine levels in the TME and restore cancer immunity.1
IDO1 is a monomeric, heme-containing dioxygenase enzyme that degrades tryptophan by catalyzing the initial, rate-limiting step of tryptophan metabolism. This step involves the oxidative cleavage of the indole 2,3-double bond of tryptophan to give N-formyl kynurenine. Subsequent hydrolysis of N-formyl kynurenine yields kynurenine.2 IDO1 promotes tumoral immune escape from host immune surveillance and plays an important role in tumor-associated immunosuppression, leading to tolerance toward tumors. Local depletion of tryptophan as well as accumulation of kynurenine have been shown to elicit immunomodulatory activity. Effects include suppression of the T effector (Teff) cell immune response, induction of naïve T cell differentiation into regulatory T (Treg) cells, and both activation of as well as a decrease in dendritic cell (DC) function.3 IDO1 is widely expressed in antigen-presenting cells (DCs, macrophages) and tumor cells, as well as epithelial and vascular endothelial cells. Furthermore, IDO1 can be induced in the tumor microenvironment in response to inflammation and T cell activation following immunotherapy, radiotherapy, or chemotherapy.4 This observation is indicative of the potential for IDO1 inhibitors in the context of combination therapies. High IDO1 expression in the tumor or tumor-draining lymph nodes is generally associated with poor prognosis and reduced survival in patients.5 Therefore, inhibition of IDO1 is a promising strategy for the re-establishment of immunogenic responses to cancer.
Several IDO1 inhibitors have entered clinical trials for the treatment of cancer (Figure 1).6 Epacadostat (INCB24360) (1), an orally active, hydroxyamidine-containing small molecule developed by Incyte, entered Phase III clinical trials in 2017.7 Preclinically, epacadostat selectively inhibits IDO1 enzymatic activity and effectively regulates the functions of various immune cells including T cells, natural killer (NK) cells, and DCs.8,9In vivo, epacadostat (1) suppressed IDO1 activity in mouse and dog plasma and inhibited tumor growth in a lymphocyte-dependent manner in mice.9,10 Results from a phase I/II combination study with epacadostat and ipilimumab in melanoma patients showed a 56% overall response rate among 54 efficacy-evaluable patients and a median progression-free survival of 12.4 months.11a However, a phase III study evaluating the combination of epacadostat and keytruda for the treatment of metastatic melanoma demonstrated no significant improvement in progression-free survival.11b,11c Navoximod (2), an imidazoisoindoline compound, was discovered by NewLink Genetics and later licensed to Genentech as GDC-0919. Rights to this molecule were later returned to NewLink after phase I clinical trial results were disclosed.12 The third candidate, iTeos/Pfizer’s compound PF-06840003 (3), was dosed as a single agent, once daily, in a phase I study in patients with malignant gliomas.13 In January 2018, Pfizer stopped the development of the compound due to a lack of efficacy.13
Figure 1.

IDO1 inhibitors evaluated in clinical trials.
In February 2015, BMS expanded its immuno-oncology pipeline in an agreement with Flexus Biosciences and acquired linrodostat (4, BMS-986205). Preclinically, compound 4 exhibits potent inhibitory activity against IDO1 (human HeLa cellular IC50 = 2 nM and murine M109 cellular IC50 = 5 nM), with a human whole blood (hWB) IC50 potency ranging between 2 and 42 nM depending on the donor. It also enhances the proliferation of Teff cells with an EC50 range of 2–7 nM in a human T cell + DC mixed lymphocyte reaction (MLR) assay. Linrodostat was advanced into phase I clinical studies in 2016.14a Currently, linrodostat is in several clinical trials including a phase III study in bladder cancer in combination with nivolumab.14b
More recently, we disclosed the structure of a closely related clinical candidate, BMS-986242 (5), which entered phase I clinical trials in 2017.14c,14d Preclinical biotransformation studies of BMS-986242 in hepatocytes across several species revealed the formation of metabolites resulting from quinoline oxidation and amide bond cleavage. For this reason, we pursued structurally differentiated IDO1 inhibitors that no longer contained an amide (this work) and/or contained a less metabolically labile replacement for the quinoline. Studies around the quinoline portion of the molecule will be disclosed in due course. Notably, other groups have pursued similar approaches to optimize this chemotype by replacing the amide in linrodostat with an oxalamide.15a,15b
A docking pose of compound 4 (Figure 2, shown in magenta) in hIDO1 was modeled based on an X-ray cocrystal structure of compound (6) (a closely related analogue is shown in green).14e This model reveals that the chlorophenyl moiety makes an edge-to-face pi-stacking interaction with Tyr126. The amide NH forms a key H-bond interaction with Ser167. The cyclohexyl core serves as a rigid scaffold that correctly positions the quinoline and phenyl group in the preferred cis-configuration. The quinoline moiety occupies a hydrophobic pocket, and the quinoline nitrogen could form a H-bond with Arg343. These observations provided key insight into the design of new analogues.
Figure 2.

Putative binding mode of 4 (magenta) in hIDO1, based on an X-ray cocrystal structure of compound 6 (green).
Herein, we describe our efforts toward identifying a nonamide-containing IDO1 inhibitor while maintaining potency and improving pharmacologic properties. Heterocyclic amide isosteres have been extensively studied in the literature.16 Previous examples have shown that five-membered heterocycles can be effective amide isosteres.17a,17b,18 The success of a given isostere will depend on whether the amide is simply a spacer or if it is taking part in key interactions that are critical to molecular recognition. Given the critical nature of the H-bonding event between Ser167 and compound 6, it was important to select and tailor an isosteric replacement that would maintain this interaction.
All isosteres discussed can be synthesized via the intermediacy of a carboxylic acid such as previously reported acid 7 (Scheme 1).19 To obtain oxazole 8 or imidazole 9, intermediate 7 is coupled with 2-amino-1-(4-chlorophenyl)ethan-1-one. To yield 8, the resulting amide can be heated in POCl3 to affect dehydration/cyclization. Alternatively, this amide can be heated with ammonium acetate in the presence of acetic acid to give imidazole 9. In route to oxadiazole 10 or triazole 11, intermediate 7 can be treated with t-butyl carbazide followed by treatment with acid to give a hydrazide (not shown). This hydrazide can be coupled with 4-chlorobenzoic acid and cyclized with POCl3 to give 10. Additionally, the hydrazide can be treated with 4-chlorobenzamidine and heated to furnish triazole 11. Alternatively, the hydrazide can be reacted with 4-methoxyphenylisocyanate followed by cyclization, again with POCl3, to provide amino-oxadiazole 12. Amino-oxadiazole 13 could be made in a three-step sequence employing a Curtius rearrangement followed by trapping of the isocyanate intermediate with 4-methoxybenzohydrazide and finally heating in POCl3 to affect dehydration/cyclization to give 13. Benzimidazole 14 could be synthesized from 7 by first converting the acid to an acyl chloride and treating with 4-fluoro-1,2-phenylenediamine. Heating the resulting amide with methanesulfonic acid provided benzimidazole 14.
Scheme 1. Synthesis of Heterocyclic Amide Isosteres and Replacements Starting from Common Intermediate Carboxylic Acid 7.

Reagents and conditions: (a) HATU, NMM, 2-amino-1-(4-chlorophenyl)ethan-1-one (64%); (b) POCl3, 100 °C (56%); (c) NH4OAc, HOAc, EtOH, 100 °C (17%); (d) (i) HATU, NMM, tert-butyl carbazate; (ii) HCl (89% over 2 steps); (e) HATU, NMM, 4-chlorobenzoic acid (83%); (f) POCl3, 90 °C (55%); (g) pyridine, 4-chlorobenzamidine·HCl salt, 110 °C (31%); (h) DIPEA, 1-isocyanato-4-methoxybenzene (93%); (i) POCl3, 90 °C (26%); (j) DPPA, TEA, toluene, 70 °C; (k) 4-methoxybenzohydrazide, DIPEA (54% over 2 steps); (l) POCl3, 100 °C (52%); (m) (i) SOCl2, DMF (ii) CH2Cl2, TEA, 4-fluoro-1,2-phenylenediamine, (n) MsOH, 90 °C (50% over 3 steps).
These various amide isosteres and replacements were evaluated in HeLa (human) and M109 (murine) cellular IDO1 inhibition assays where IDO1 activity can be assessed upon induction with IFNγ (see SI for details). Several five-membered heterocycles were found to be effective replacements for the amide in compound 4 (Table 1). Both imidazole 9 and triazole 11 displayed more potent IDO1 inhibitory activity compared to oxazole 8. One hypothesis that would account for the greater potency of 9 and 11 is that they maintain a key H-bond donor NH to interact with Ser167. This hypothesis is consistent with the modest potency of oxadiazole 10. It is of note that 10 is more potent than 8 possibly due to either stronger H-bond acceptor ability or containing two potential H-bond-accepting nitrogens as opposed to one. Analogue 12, which has an amino linkage between the oxadiazole and aryl ring, displayed a significant loss of hIDO1 cellular activity compared to 4. This may be due to the unfavorable orientation of the amino group NH. The “flipped” amino-oxadiazole 13 seems to reorient the NH favorably. Both amino-oxadiazole 13 and benzimidazole 14 demonstrated single-digit nanomolar cellular activity; however, 14 showed inferior metabolic stability.
Table 1. Effects of Heterocyclic Amide Replacements on IDO1 Inhibition Potency and Metabolic Stability.


Data reported as average test results (N = 2, unless otherwise noted). See Supporting Information for a description of assay conditions.
Fraction of the parent compound (0.5 μM) remaining after a 10 min incubation with 1 mg/mL of human, mouse, and rat liver microsomes (HLM, MsLM, RLM).
Based on the cellular potency of amino-oxadiazole 13 and benzimidazole 14, both were further pursued. Modular synthetic approaches were developed to allow for modification of R1 and quinoline R2 substitution (see Scheme 2). A vinyl boronic acid with R1 groups preinstalled like 15(19−21) (Scheme 2a) could be joined to quinolinyl halides (16) with Suzuki coupling. The resulting styrenyl olefin in 17 could be reduced under either transfer or standard hydrogenation conditions and hydrolyzed to give 18 as a mixture of cis- and trans-racemates. Alternatively, the R1 group could be installed later in the route by first coupling 16 (Scheme 2b) to unsubstituted boronic acid 19(19) to give 20. The position alpha to the ester could then be alkylated either before or after reduction of the olefin. Hydrolysis would again give intermediates of type 18. O-Linked quinolines could be made from trans-alcohol 21(19,21) employing SNAr on 4-bromoquinoline 22. Hydrolysis provides racemic acid 24. Acids of type 18 and 24 can be converted to amino-oxadiazoles, benzimidazoles, or closely related imidazopyridines as outlined in Scheme 1. All compounds synthesized by the above general methods and discussed in Tables 2 and 3 have been chirally resolved to obtain enantiopure material. For all final compounds discussed in Tables 2 and 3, only the most potent enantiomer is shown. To elucidate the relative cis-/trans-stereochemistry of final compounds or synthetic precursors, 1H NMR, 13C NMR, 2D COSY, 2D NOESY, and 1H–13C–Dept-HSQC were performed (see SI for details). Absolute stereochemistry was not confirmed unless specifically noted.
Scheme 2. General Schemes for the Synthesis of Acid Intermediates with Modified R1 and Quinoline R2 Substitution.

Reagents and conditions: (a) Pd(PPh3)4, K2CO3, dioxane, H2O, 100 °C; (b) Pd–C, MeOH, HCOONH4, 80 °C or Pd–C, MeOH, H2; (c) LiOH, H2O, THF, rt–70 °C; (d) 1,3-dimethyl-3,4,5,6-tetrahydro-2(1H)-pyrimidinone, lithium diisopropylamide, THF, R1X, – 78 °C–rt; (e) NaH, DMSO, 60 °C, 4 h.
Table 2. Effects of R1 Substitution and Quinoline Modifications to Amino-oxadiazoles on IDO1 Inhibitory Activity, in Vitro Metabolic Stability, and PXR Activation.

Data reported as average of test results (N = 2 unless otherwise noted). See Supporting Information for a description of assay conditions.
Fraction of the parent compound (0.5 μM) remaining after a 10 min incubation with 1 mg/mL of human, mouse, and rat liver microsomes (HLM, MsLM, RLM).
The pregnane X receptor (PXR) transactivation activity was measured by comparing to activation with rifampicin to assess the potential for induction of cytochrome P450 (CYP) 3A4 (Et = ethyl, MOM = methoxymethyl, EOM = ethoxymethyl).
Table 3. Effects of R1 Substitution and Quinoline Modification of Benzimidazoles and Imidazopyridines on IDO1 Inhibitory Activity and in Vitro Metabolic Stability.
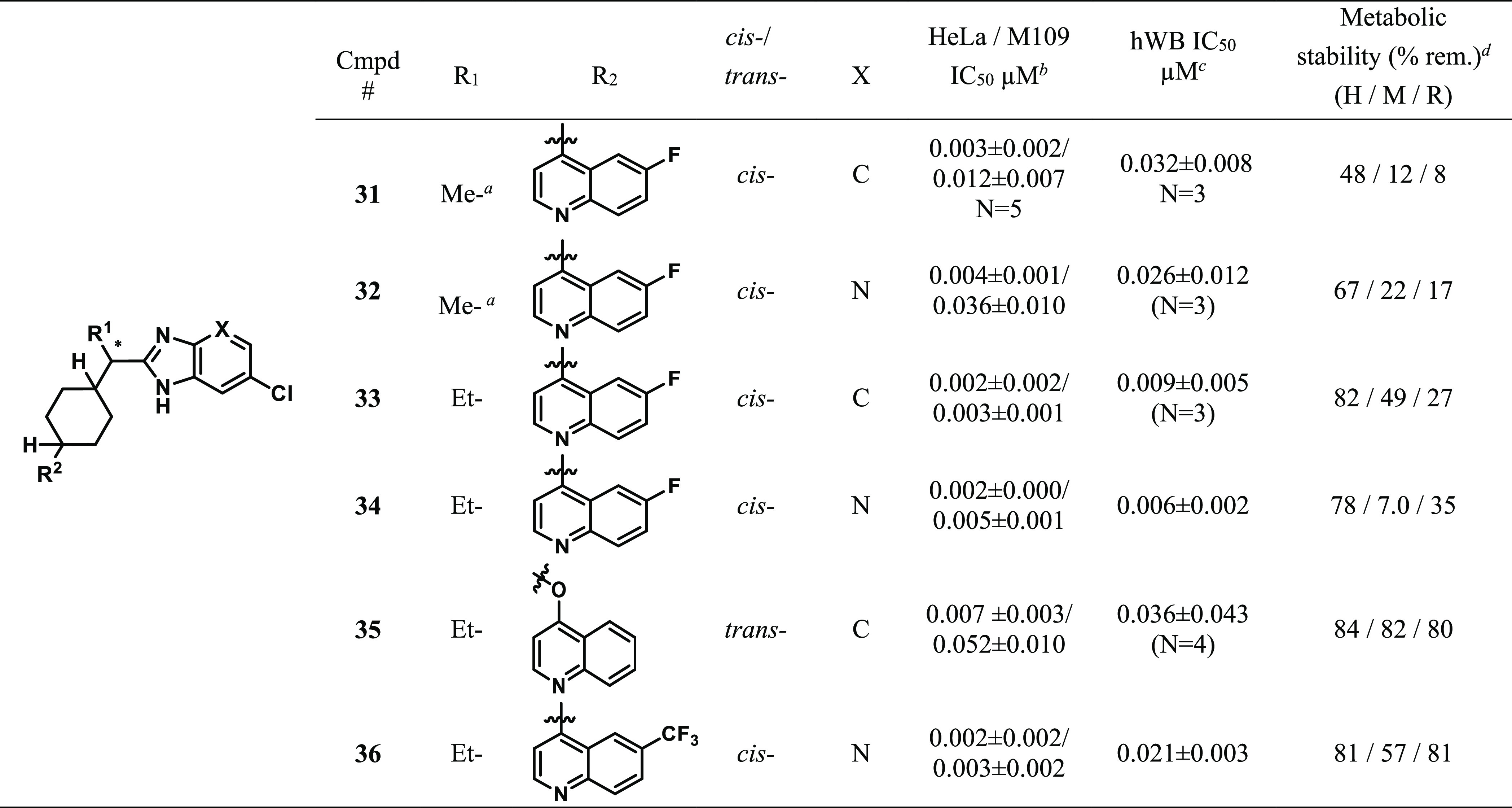
Absolute stereochemistry is R-.
Data reported as average test results (N = 3, unless otherwise noted). See Supporting Information for a description of assay conditions.
hWB = human whole blood; data reported as average test results (N = 2, unless otherwise noted). See Supporting Information for a description of assay conditions.
Fractions of the parent compound (0.5 μM) remaining after a 10 min incubation with 1 mg/mL of human, mouse, and rat liver microsomes (HLM, MsLM, RLM).
Amino-oxadiazoles related to 13 are discussed in Table 2. Introducing an ethyl substituent at the position alpha to the ester (25) maintained potency and proved to be beneficial in terms of metabolic stability. Analogue 25 also showed potent hWB activity. Since quinolines are a known potential site of metabolic oxidation,22a we turned our attention to modeling substituent effects on the oxidation potentials of quinolines.22b Reduction potentials of nitrogen-containing compounds have been reported to correlate with the lowest unoccupied molecular orbital (LUMO) energies.23,24 Modeling calculations suggested that electron-withdrawing group(s) on the quinoline ring could reduce N-oxidation potential. For example, 6-CF3 quinoline displays higher predicted reduction potential Ered = 2.12 than that of 6-F-quinoline Ered = 1.93.22,23 Gratifyingly, the 6-CF3-substituted quinoline 26 offered potent cellular activity as well as improved metabolic stability but had a 3–4-fold loss of hWB potency compared to analogue 25. Analogue 27, with a 6,8-difluoro-quinoline moiety, unexpectedly showed less stability, especially in mouse LMs. Further profiling revealed that all three cis-isomers (25–27) led to PXR activation. As a consequence of this finding, efforts were focused on improving PXR activity. We were pleased to find that O-linked trans-isomer 28 did not activate PXR, but unfortunately it showed not only a 10-fold disparity between human and mouse IDO1 activity, which would hinder in vivo studies, but also a significant drop in hWB potency. It is worth noting that the trans-isomer of C-linked compound 25 (not shown) also did not activate PXR; however, IDO1 inhibitory activity was very poor. We then turned our attention to alpha-substituent modification and found that α-MOM-substituted analogue 29 and α-EOM-substituted analogue 30 both demonstrated significant improvement in PXR activity compared to compounds 25–27. While 29 and 30 maintained good hIDO1 activity in cells, they had more modest hWB potency (IC50 = 0.031 μM and 0.096 μM, respectively, vs 0.002–0.042 μM for 4) and metabolic stability compared to lead compound 4 (see Table 1). Since PXR activation could not be remedied while maintaining suitable hWB and mouse cellular activity, this series was not progressed.
We then focused on the benzimidazole series. Although benzimidazole 14 displayed poor metabolic stability, it had very potent cellular activity (see Table 1). Therefore, identification of a more stable benzimidazole was of primary interest. Addition of a chloro substituent on the phenyl ring of the benzimidazole in combination with incorporation of a nitrogen atom into the ring yielded imidazopyridine 32 (Table 3), which exhibited a significant improvement in stability compared to 31 (H/M/R = 67/22/17) while maintaining hWB activity (IC50 = 0.039 μM). As was previously observed in the amino-oxadiazole series, introduction of ethyl substitution at the alpha-position generally increased potency and improved metabolic stability. Analogue 33 displayed potent hWB activity and improved stability, while imidazopyridine 34 revealed unexpectedly poor mouse metabolic stability (H/M/R = 78/7/35). Consistent with trends observed in the amino-oxadiazole series (see 28, Table 2), incorporating an O-linked trans-isomer (35) was found to increase metabolic stability. Once again, however, mouse cellular activity suffered. Combining the more potent alpha-ethyl group with the more stable imidazopyridine and the 6-CF3-quinoline (see 26, Table 2, vide supra) led to compound 36. Docking models of 36, based on the X-ray cocrystal of compound 6 and hIDO1 in Figure 2, indicated that the imidazopyridine likely maintains the same key interactions observed for the amide (Figure 3). The NH of the imidazopyridine nicely overlays with the NH of the amide bond in 6 which would correctly position it to make the key hydrogen bond with Ser167. While the pyridine portion of the imidazopyridine is positioned a little lower than the phenyl ring of 6, it still is in a position to make a productive edge-to-face pi-stacking interaction with Tyr126. Imidazopyridine 36 possessed the best overall in vitro profile in terms of potency and stability for this series and was selected for more extensive profiling.
Figure 3.

Putative binding mode of compound 36 in hIDO1, based on an X-ray cocrystal structure of compound 6 (green).
Further in vitro profiling (Table 4) showed the compound had modest oxidative metabolic stability; however, this metabolic stability profile was superior to both linrodostat (4) (T1/2 human = 53, mouse = 4, rat = 20, dog = 17, cyno = 7) and BMS-986242 (5) (T1/2 human = 14, mouse = 4, rat = 10, dog = 10, cyno = 2). Compound 36 had good intrinsic permeability in a PAMPA assay and Caco-2 cells (a-b = 113 nm/s; efflux ratio = 0.6). Unfortunately, compound 36 showed modest PXR activation (PXR EC50 = 1.2 μM (30% Ymax)) and CYP inhibition in several human isoforms including potent inhibition of CYP 2C9.
Table 4. Compound 36in Vitro Profiling.
| parameter | compound 36 |
|---|---|
| met. stability CYP (T1/2 min) | 54 (H), 17 (M), 60 (R), 41 (D), 21 (C) |
| met. stability UGT (T1/2 min) | 95 (H), >120 (M), 107 (R), 105 (D), >120 (C) |
| PAMPA cosolvent (pH 7.4) | pH 5.5: 2380 nm/s |
| pH 7.4: 2630 nm/s | |
| caco (a-b:b-a) | 113: 66 nm/s |
| PXR-TA EC50 (μM) | 1.19 (32% Ymax) |
| human rCYP panel IC50 (μM) | 1A2: 2.55 |
| 2D6: 4.28 | |
| 2C8: 1.47 | |
| 2C9: 0.242 | |
| 2C19: 1.13 | |
| 3A4: 1.93 |
Compound 36 was further examined in the SKOV3 human ovarian carcinoma xenograft model (Table 5). In this model, immune-compromised nu/nu nude mice were implanted with SKOV3 cells, and the resulting tumors were allowed to grow for 2 weeks. On day 14, tumor-bearing mice were dosed with imidazopyridine 36 QD at 20 mg/kg for 5 days. On day 18 at 2, 6, and 24 h time points, tumor kynurenine concentration (PD) and compound 36 concentration (PK) were measured in the tumor. The reduction of kynurenine levels, when compared to a vehicle control, was used as a pharmacodynamic marker. Imidazopyridine 36 demonstrated a robust profile. At a 20 mg/kg dose, it achieved 56% reduction in tumor kynurenine levels and tumor exposure of 43.6 μM*h AUC. This profile compares well with linrodostat (4): at a 60 mg/kg dose, 4 achieved a 61% reduction in kynurenine levels and a tumor AUC of 34.9 μM*h.
Table 5. PK–PD Study of Imidazopyridine 36 vs Linrodostat (4) in a Human SKOV3 Xenograft Tumor Mouse Model.
| treatment | dose (QDX5) (mg/kg) | PKa AUC0–24h | PDb AUEC0–24h, %[Kyn]↓ |
|---|---|---|---|
| linrodostat 4 | 60 | 34.9 | 61 |
| compound 36 | 20 | 43.6 | 56 |
PK is AUC (0–24 h) in tumor μM*h.
PD is percent kynurenine AUEC (0–24 h) reduction. % Kyn reduction was measured at a steady state after the 5th dose in the tumor, calculated as the area under the Kyn concentration–time curve from 0 to 24 h and compared with that of vehicle control.
In summary, structurally differentiated IDO1 inhibitors were identified. Heterocyclic amide isosteres and replacements were investigated. Amino-oxadiazoles, such as compound 25, demonstrated potent cellular and hWB potency but led to PXR activation. Optimization of benzimidazole 14 led to the identification of imidazopyridine 36. Lead compound 36 possessed potent cellular and hWB activity as well as improved metabolic stability (T1/2). Additionally, it had a suitable permeability profile. Based on these findings, compound 36 was advanced into an in vivo human SKOV3 xenograft tumor model in mice. Compound 36 demonstrated a robust PK/PD profile with improved exposure and comparable PD effects to linrodostat (4). In contrast to linrodostat (4), which showed less PXR activation (PXR EC50 > 50 μM (13% Ymax)) and a cleaner rCYP panel profile, compound 36 demonstrated more significant PXR activation and CYP inhibition across several isoforms; therefore, compound 36 was not investigated further.
Acknowledgments
We thank Jay Markwalder, Weifang Shan, and the Richard Rampulla/BBRC group for providing intermediates. We thank Dr. Shana Posy for assistance with Figure 3.
Glossary
ABBREVIATIONS
- HATU
1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate
- NMM
N-methylmorpholine
- DIPEA
N,N-diisopropylethylamine
- DMF
N,N-dimethylformamide
- TEA
triethylamine
- DPPA
diphenylphosphorylazide
- MsOH
methanesulfonic acid
- H
human
- M
mouse
- R
rat
- D
dog
- C
cyno
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00014.
Biological assay protocols, in vivo pharmacokinetic–pharmacodynamic study protocols, experimental procedures, and analytical data for all final compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Adams J. L.; Smothers J.; Srinivasan R.; Hoos A. Big opportunities for small molecules in immuno-oncology. Nat. Rev. Drug Discovery 2015, 14, 603–622. 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- Austin C. J. D.; Rendina L. M. Targeting key dioxygenases in tryptophan-kynurenine metabolism for immunomodulation and cancer chemotherapy. Drug Discovery Today 2015, 20, 609–617. 10.1016/j.drudis.2014.11.007. [DOI] [PubMed] [Google Scholar]
- Munn D. H.; Mellor A. L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Invest. 2007, 117, 1147–1154. 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn D. H.; Mellor A. L. Indoleamine 2,3-dioxygenase and metabolic control of immune responses. Trends Immunol. 2013, 34, 137–143. 10.1016/j.it.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn D. H.; Mellor A. L. IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 2016, 37, 193–207. 10.1016/j.it.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prendergast G. C.; Malachowski W. P.; DuHadaway J. B.; Muller A. J. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 2017, 77, 6795–6811. 10.1158/0008-5472.CAN-17-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue E. W.; Sparks R.; Polam P.; Modi D.; Douty B.; Wayland B.; Glass B.; Takvorian A.; Glenn J.; Zhu W.; Bower M.; Liu X.; Leffet L.; Wang Q.; Bowman K. J.; Hansbury M. J.; Wei M.; Li Y.; Wynn R.; Burn T. C.; Koblish H. K.; Fridman J. S.; Emm T.; Scherle P. A.; Metcalf B.; Combs A. P. INCB24360 (epacadostat), a highly potent and selective indoleamine-2,3-dioxygenase 1 (IDO1) inhibitor for immunooncology. ACS Med. Chem. Lett. 2017, 8, 486–491. 10.1021/acsmedchemlett.6b00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs A. P.; Yue E. W.. Preparation of 4-amino-N’-hydroxy-1,2,5-oxadiazole-3-carboximidamides and related compounds as modulators of indoleamine 2,3-dioxygenase for inhibiting immunosuppression and treating various disorders. WO 2006122150 A1, November 16, 2006.
- Liu X.; Shin N.; Koblish H. K.; Yang G.; Wang Q.; Wang K.; Leffet L.; Hansbury M. J.; Thomas B.; Rupar M.; Waeltz P.; Bowman K. J.; Polam P.; Sparks R. R.; Yue E. W.; Li Y.; Wynn R.; Fridman J. S.; Burn T. C.; Combs A. P.; Newton R. C.; Scherle P. A. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010, 115, 3520–3530. 10.1182/blood-2009-09-246124. [DOI] [PubMed] [Google Scholar]
- Koblish H. K.; Hansbury M. J.; Bowman K. J.; Yang G.; Neilan C. L.; Haley P. J.; Burn T. C.; Waeltz P.; Sparks R. B.; Yue E. W.; Combs A. P.; Scherle P. A.; Vaddi K.; Fridman J. S. Hydroxyamidine inhibitors of indoleamine-2,3-dioxygenase potently suppress systemic tryptophan catabolism and the growth of IDO-expressing tumors. Mol. Cancer Ther. 2010, 9, 489–498. 10.1158/1535-7163.MCT-09-0628. [DOI] [PubMed] [Google Scholar]
- a Hamid O.; Gajewski T. F.; Frankel A. E.; Bauer T. M.; Olszanski A. J.; Luke J. J.; Balmanoukian A. S.; Schmidt E. V.; Sharkey B.; Maleski J.; Jones M. J.; Gangadhar T. C. Epacadostat plus pembrolizumab in patients with advanced melanoma: phase 1 and 2 efficacy and safety results from ECHO-202/KEYNOTE-037. Ann.Oncol 2017, 28, v428–v448. 10.1093/annonc/mdx377.001. [DOI] [Google Scholar]; b NCT 02752074, ECHO-301/KEYNOTE-252. [Google Scholar]; c Mullard A. IDO takes a blow. Nat. Rev. Drug Discovery 2018, 17, 307. 10.1038/nrd.2018.67. [DOI] [PubMed] [Google Scholar]
- Burris H. A.; Gordon M. S.; Hellmann M. D.; LoRusso P.; Emens L. A.; Hodi F. S.; Lieu C. H.; Infante J. R.; Tsai F. Y.-C.; Eder J. P.; Cleary J. M.; Jelovac D.; Tsuhako A. L.; Mueller L.; Lin R.; Morrissey K.; Mahrus S.; Morley R.; Pirzkall A.; Davis S. L. A phase Ib dose escalation study of combined inhibition of IDO1 (GDC-0919) and PD-L1 (atezolizumab) in patients with locally advanced or metastatic solid tumors. J. Clin. Oncol. 2017, 35, 105–105A. 10.1200/JCO.2017.35.15_suppl.105. [DOI] [Google Scholar]
- ClinicalTrials.gov Identifier: NCT 02764151: First in patient study for PF-06840003 in Malignant gliomas:; http://www.iteostherapeutics.com/news/20180104-iteos-therapeutics-regains-worldwide-rights-to-clinical-stage-IDO1-inhibitor.
- a Balog A.; Lin T.-A.; Maley E.; Gullo-Brown J.; Hamza Kandoussi E.; Zeng J.; Hunt J. T. Preclinical characterization of Linrodostat mesylate, a novel, potent, and selective oral indoleamine 2,3-dioxygenase 1 inhibitor. Mol. Cancer Ther. 2020, molcanther.0251.2020. 10.1158/1535-7163.MCT-20-0251. [DOI] [PubMed] [Google Scholar]; b A Study of chemo only versus chemo plus nivo with or without BMS-986205, followed by post-surgery therapy with nivo or nivo and BMS-986205 in patients with MIBC. ClinicalTrials.gov. Identifier: NCT03661320. [Google Scholar]; c Cherney E. C.; Zhang L.; Nara S.; Zhu X.; Gullo-Brown J.; Maley D.; Lin T.; Hunt J.; Huang C.; Yang Z.; D́Arienzo C.; Discenza L.; Ranasinghe A.; Grubb M.; Ziemba T.; Traeger S.; Li X.; Johnston K.; Kopcho L.; Fereshteh M.; Foster K. A.; Stefanski K.; Fargnoli J.; Swanson J.; Brown J.; Delpy D.; Seitz S.; Borzilleri R.; Vite G.; Balog A. Discovery and preclinical evaluation of BMS-986242, a potent, selective inhibitor of indoleamine-2,3-dioxygenase 1. ACS Med. Chem. Lett. 2021, 12, 288. 10.1021/acsmedchemlett.0c00668. [DOI] [PMC free article] [PubMed] [Google Scholar]; d An investigational immunotherapy study of experimental medication BMS-986242 given in combination with nivolumab in patients with advanced cancer. ClinicalTrials.gov. Identifier: NCT03351231. [Google Scholar]; e Nelp M. T.; Kates P. A.; Hunt J. T.; Newitt J. A.; Balog A.; Maley D.; Zhu X.; Abell L.; Allentoff A.; Borzilleri R.; Lewis H. A.; Lin Z.; Seitz S. P.; Yan C.; Groves J. T. Immune-modulating enzyme indoleamine 2,3-dioxygenase is effectively inhibited by targeting its apo-form. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 3249–3254. 10.1073/pnas.1719190115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Steeneck C.; Kinzel O.; Anderhub S.; Hornberger M.; Pinto S.; Morschhaeuser B.; Albers M.; Sonnek C.; Czekanska M.; Hoffmann T. Discovery and optimization of substituted oxalamides as novel heme-displacing IDO1 inhibitors. Bioorg. Med. Chem. Lett. 2021, 33, 127744. 10.1016/j.bmcl.2020.127744. [DOI] [PubMed] [Google Scholar]; b Kinzel O.; Steeneck C.; Anderhub S.; Hornberger M.; Pinto S.; Morschhaeuser B.; Albers M.; Sonnek C.; Wang Y.; Mallinger A.; Czekańska M.; Hoffmann T. Discovery of highly potent heme-displacing IDO1 inhibitors based on a spirofused bicyclic scaffold. Bioorg. Med. Chem. Lett. 2021, 33, 127738. 10.1016/j.bmcl.2020.127738. [DOI] [PubMed] [Google Scholar]
- Meanwell N. A. Synopsis of some recent tactical application of bioisosteres in drug design. J. Med. Chem. 2011, 54, 2529–2591. 10.1021/jm1013693. [DOI] [PubMed] [Google Scholar]
- a Kumari S.; Carmona A. V.; Tiwari A. K.; Trippier P. C. Amide bond bioisosteres: strategies, synthesis, and successes. J. Med. Chem. 2020, 63, 12290–12358. 10.1021/acs.jmedchem.0c00530. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Mohammed I.; Kummetha I. R.; Singh G.; Sharova N.; Lichinchi G.; Dang J.; Stevenson M.; Rana T. M. 1,2,3-triazoles as amide bioisosteres: discovery of new class of potent HIV-1 vif antagonists. J. Med. Chem. 2016, 59, 7677–7682. 10.1021/acs.jmedchem.6b00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Francesco M. E.; Dessole G.; Nizi E.; Pace P.; Koch U.; Fiore F.; Pesci S.; Di Muzio J.; Monteagudo E.; Rowley M.; Summa V. Novel macrocyclic inhibitors of hepatitis C NS3/4A protease featuring a 2-amino-1,3-thiazole as a P4 carbamate replacement. J. Med. Chem. 2009, 52, 7014–7028. 10.1021/jm900524b. [DOI] [PubMed] [Google Scholar]
- Beck H. P.; Jaen J. C.; Osipov M.; Powers J. P.; Reilly M. K.; Shunatona H. P.; Walker J. R.; Zibinsky M.; Balog J. A.; Williams D. K.; Markwalder J. A.; Seitz S. P.; Cherney E. C.; Zhang L.; Shan W.; Guo W.; Huang A.. Preparation of immunoregulatory agents. WO 2016073774, May 12, 2016.
- Beck H. P.; Jaen J. C.; Osipov M.; Powers J. P.; Reilly M. K.; Shunatona H. P.; Walker J. R.; Zibinsky M.; Balog J. A.; Williams D. K.; Guo W.. Immunoregulatory agents. WO 2016073738, A2, May 12, 2016.
- Beck H. P.; Jaen J. C.; Osipov M.; Powers J. P.; Reilly M. K.; Shunatona H. P.; Walker J. R.; Zibinsky M.; Balog J. A.; Williams D. K.; Markwalder J. A.; Cherney E. C.; Shan W.; Huang A.. Immunoregulatory agents. WO 2016073770, A1, May 12, 2016.
- a Bai Q.; Yang L.; Li R.; Chen B.; Zhang L.; Zhang Y.; Rittmann B. E. Rittmann. Accelerating quinoline biodegradation and oxidation with endogenous electron donors. Environ. Sci. Technol. 2015, 49, 11536–11542. 10.1021/acs.est.5b03293. [DOI] [PubMed] [Google Scholar]; b Modeling methods for calculating N-oxidation potential: Molecular structures of each model compound are constructed using the Maestro v2015-4 software (Schrodinger, 2015). QM calculations are performed using the Jaguar software (Bochevarov, 2013). Each model compound is subjected to geometry optimization at the B3LYP/6-31G** level of theory and single-point energy calculation at the B3LYP/cc-pVTZ(-f) level of theory, from which the LUMO energy term is extracted. LUMO energy is converted to unscaled reduction potential (eV) via Ered = −27.2107 * LUMO (hartree) (ref (23)) such that higher Ered signifies a lower propensity for oxidation.
- Assary R. S.; Brushett F. R.; Curtiss L. A. Reduction potential predictions of some aromatic nitrogen-containing molecules. RCS Advances 2014, 4, 57442–57451. [Google Scholar]
- Bochevarov A. D.; Harder E.; Hughes T. F.; Greenwood J. R.; Braden D. A.; Philipp D. M.; Rinaldo D.; Halls M. D.; Zhang J.; Friesner R. A. Jaguar A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. 10.1002/qua.24481. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.