

CONSPECTUS:
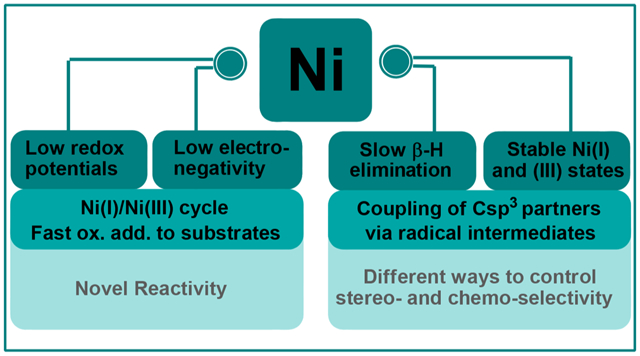
Nickel complexes exhibit distinct properties from other group 10 metals, including a small nuclear radius, high paring energy, low electronegativity, and low redox potentials. These properties enable Ni catalysts to accommodate and stabilize paramagnetic intermediates, access radical pathways, and undergo slow β-H elimination. Our research program investigates how each of these fundamental attributes impact the catalytic properties of Ni, in particular in the context of alkene functionalization.
Alkenes are versatile functional groups, but stereoselective carbofunctionalization reactions of alkenes have been under-developed. This challenge may derive from the difficulty of controlling selectivity via traditional two-electron migratory insertion pathways. Ni catalysts could lead to different stereo-determining steps via radical mechanisms, allowing access to molecular scaffolds that are otherwise difficult to prepare. For example, an asymmetric alkene diarylation reaction developed by our group relies upon the radical properties of Ni(III) intermediates to control the enantioselectivity and give access to a library of chiral α,α,β-triarylethane molecules with biological activity.
Mechanistic studies on a two-component reductive 1,2-difunctionalization reaction have shed light on the origin of the cross-electrophile selectivity, as C sp2 and C sp3 electrophiles are independently activated at Ni(I) via two-electron and radical pathways, respectively. Catalyst reduction has been identified to be the turnover-limiting step in this system. A closer investigation of the radical formation step using a (Xantphos)Ni(I)Ar model complex reveals that Ni(I) initiates radical formation via a concerted halogen-abstraction pathway.
The low redox potentials of Ni have allowed us to develop a reductive, trans-selective diene cyclization, wherein a classic two-electron mechanism operates on a Ni(I)/Ni(III) platform, accounting for the chemo- and stereoselectivity. This reaction has found applications in the efficient synthesis of pharmaceutically relevant molecules, such as 3,4-dimethylgababutin.
The tendency of Ni to undergo one-electron redox processes prompted us to explore dinuclear Ni-mediated bond formations. These studies provide insight into Ni–Ni bonding and how two metal centers react cooperatively to promote C–C, C–X, and N–N bond forming reductive elimination.
Finally, isolation of β-agostic Ni and Pd complexes has allowed for X-ray and neutron diffraction characterization of these highly reactive molecules. The bonding parameters serve as unambiguous evidence for β-agostic interactions and help rationalize the slower β-H elimination at Ni relative to Pd. Overall, our research has elucidated the fundamental properties of Ni complexes in several contexts. Greater mechanistic understanding facilitates catalyst design and helps rationalize the reactivity and selectivity in Ni-catalyzed alkene functionalization reactions.
Graphical Abstract
INTRODUCTION
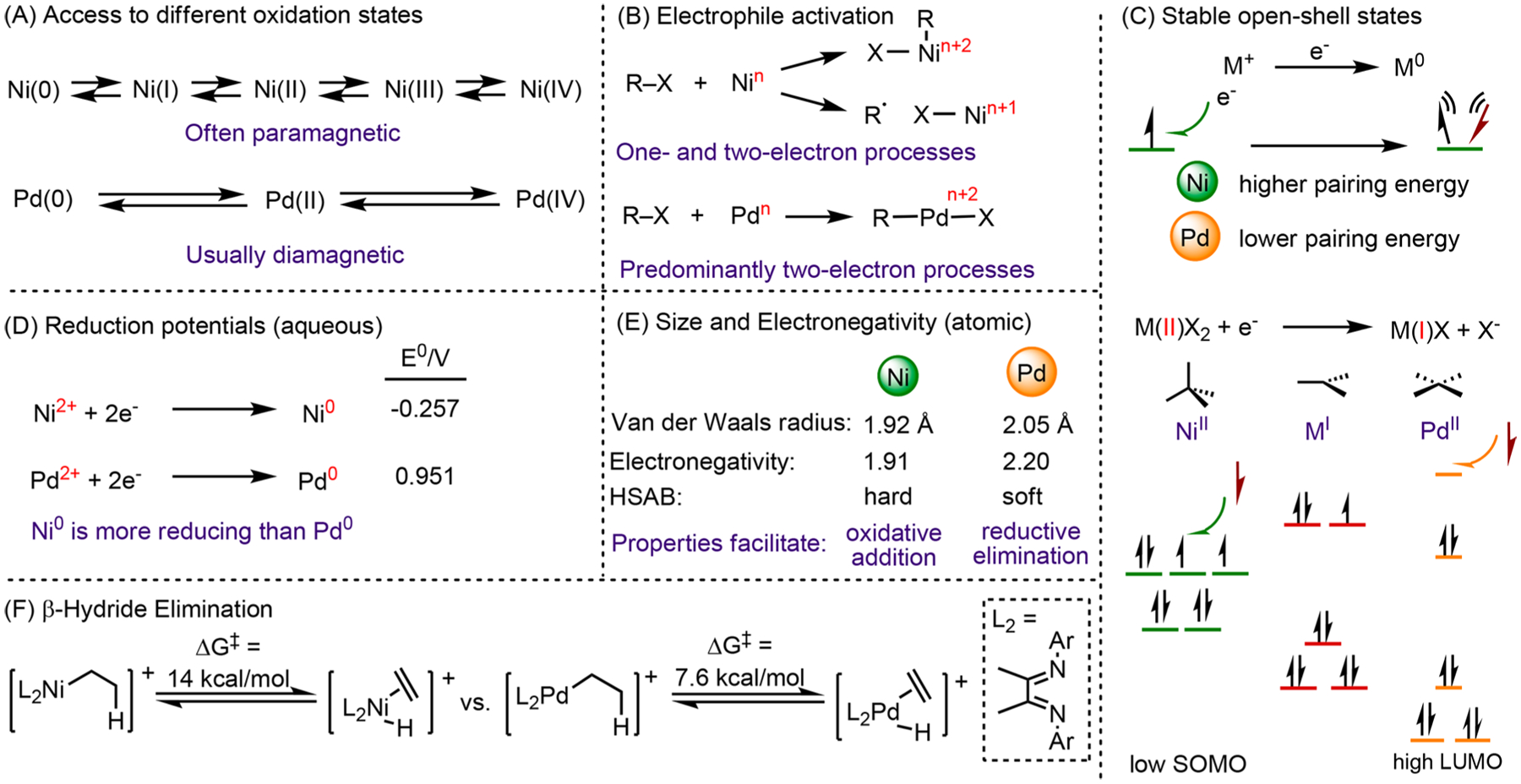
Nickel (Ni) catalysts are an inexpensive and earth-abundant alternative to precious metal catalysts, such as Pd, for cross-coupling reactions. In addition, Ni exhibits unique reactivity, leading to different mechanisms.1 First, Ni complexes can adopt both high- and low-spin configurations and are stable in a variety of oxidation states ranging from Ni(0) to Ni(IV).2 In comparison, Pd compounds are primarily low-spin and are most commonly encountered as diamagnetic Pd(0), Pd(II), and Pd(IV) compounds (Scheme 1A).3 The propensity of Ni to adopt open-shell electronic configurations, such as Ni(I) and Ni(III), is likely a result of the higher pairing energy of Ni compared to Pd. The nucleus radius of Ni is smaller than Pd, hence the electron distribution of the valence orbitals is smaller. When two electrons occupy the same orbital, they experience higher repulsion (Scheme 1C).4 Moreover, the geometry of high-spin Ni species could lead to a lower barrier for single electron transfer. For example, one electron reduction of a tetrahedral Ni(II) complex is easier than that of a square planar Pd(II) complex by introducing the electron to a lower SOMO (Scheme 1C). As a consequence of the stable open-shell electronic configuration, activation of electrophiles by Ni can proceed either by two-electron oxidative addition to give organonickel intermediates or by single-electron pathways to afford radicals (Scheme 1B).5
Scheme 1.
Overview of Fundamental Properties of Ni and Comparison with Pd
The reduction potential,6 atomic radius,7 and electronegativity8 of Ni are lower than those of Pd (Scheme 1D,E). This leads to facile oxidative addition of Ni(0) to bonds that traditionally do not undergo oxidative addition with Pd(0), such as ether C–O9 and amide C–N bonds.10 Finally, Ni–alkyl compounds undergo β-hydride (β-H) elimination more slowly than Pd compounds, when compared in the same ligand framework (Scheme 1F).11 As a result, alkyl coupling partners that decompose via β-H elimination with Pd are compatible with Ni.
The different reactivity and selectivity of Ni in alkene functionalization, compared to Pd, highlights the distinct properties outlined above. Alkenes are versatile functional groups. The Pd-catalyzed Heck reaction proceeds through migratory insertion of Pd–carbyl intermediates to alkenes (Scheme 2A, step ii), followed by β-H elimination to restore unsaturation (Scheme 2 A, step iii).12 In contrast, Ni catalysts have been shown to perform olefin insertions without subsequent β-H elimination. In addition, Ni can induce radical formation from electrophiles, which then add to alkenes. This reactivity has been leveraged to develop hydroarylation,13 hydroalkylation,14 and dicarbofunctionalization15 reactions. Ni typically activates C sp2 electrophiles through a classic two-electron oxidative addition pathway to afford organonickel intermediates, which then undergo migratory insertion to alkenes (Scheme 2B, step iv).16 In contrast, activation of C sp3 electrophiles by Ni can proceed by halide abstraction to form radicals, which then add to alkenes (step v).14,17 Organic radical intermediates can be trapped by Ni to afford Ni–alkyl 1 (step iv). Due to a slow β-H elimination, relative to Pd compounds, 1 can undergo further transformations to form new C–C bonds and afford difunctionalization or hydrofunctionalization product 2 (step vii). These reactions enable the rapid construction of complex motifs starting from simple and readily available coupling partners (Scheme 2C).15 The necessity of stoichiometric and often air-sensitive, organometallic reagents can be avoided through the use of reductive strategies for olefin functionalization.18
Scheme 2.
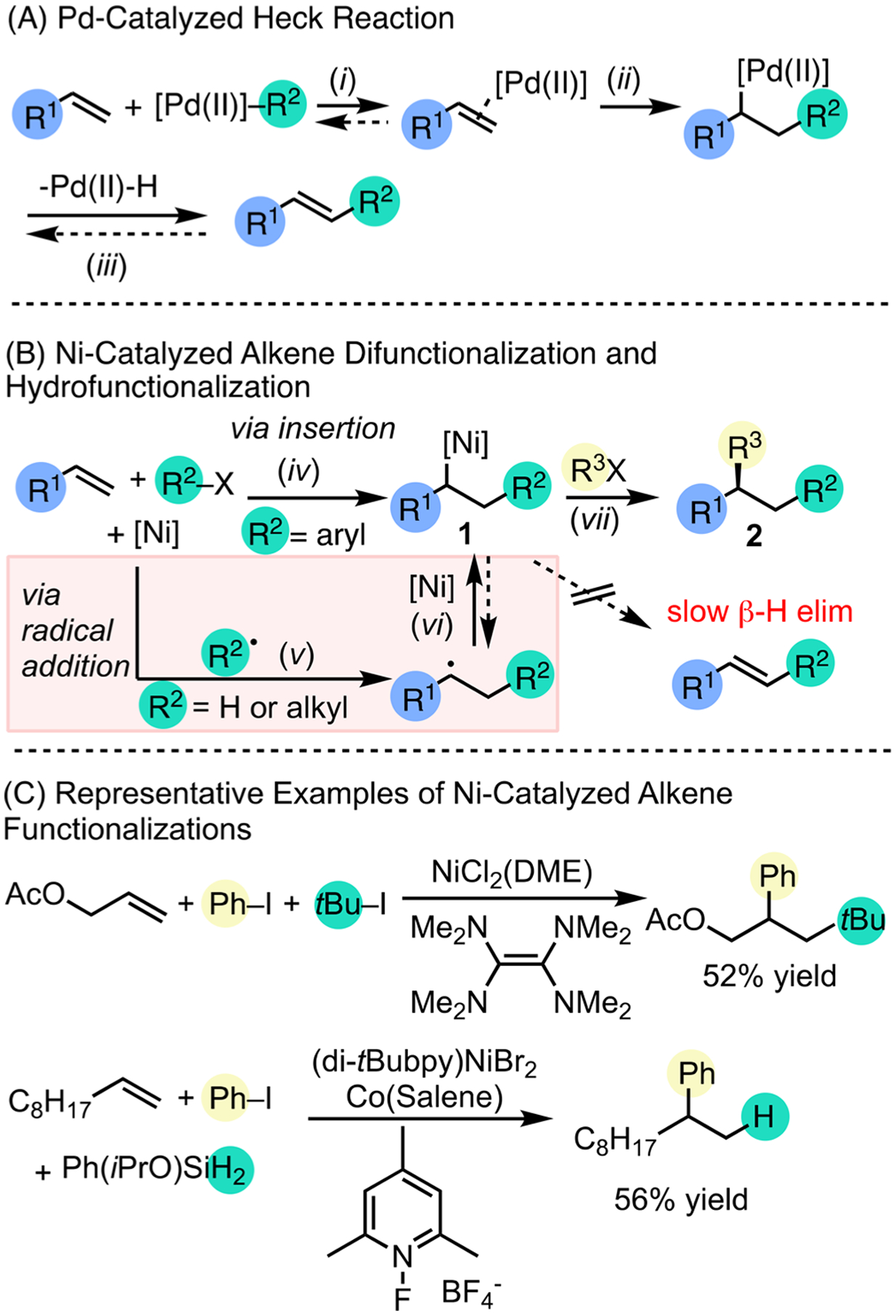
Ni-Catalyzed Difunctionalization and Hydrofunctionalization, in Comparison to the Pd-Catalyzed Heck Reaction
In addition to diversifying the methods of alkene functionalization, Ni catalysts could provide new ways to control stereochemistry. The Pd-catalyzed asymmetric Heck reaction has been limited to electronically biased or engineered substrates.19 The constraints may derive from the difficulty of controlling selectivity via traditional alkene coordination and migratory insertion (Scheme 2A, step i or ii) or the reversible β-H elimination.12 Ni-catalyzed asymmetric alkene functionalizations with C sp2 coupling partners mostly proceed through two-electron migratory insertion (Scheme 2B, step iv). Intramolecular examples include the difunctionalization of allyl ethers with aryl boronates20 and the reductive difunctionalization of acrylamides with aryl halides.21 Ni-catalyzed intermolecular asymmetric diarylation reactions are rare,22 and asymmetric difunctionalizations with C sp3 coupling partners have yet to be discovered. Both intramolecular and intermolecular asymmetric hydrofunctionalization reactions have been developed with Ni catalysts. Intramolecular hydroarylation reactions provide access to indoline derivatives.23 The intermolecular hydroarylation of styrenes with boronic acids has led to enantioselective formation of α,α-diarylethanes.13 In contrast to two-electron pathways in hydroarylation, Ni-catalyzed hydroalkylation reactions of olefins with α-bromoamides may proceed through radical pathways with different enantio-determining steps.24
This Account summarizes our efforts to probe the mechanistic details of Ni-mediated bond activation and formation processes, including radical formation, reduction of Ni intermediates, reductive elimination, and β-H elimination (Scheme 3). The stoichiometric studies have been performed on isolated complexes, which represent different conditions compared to the catalytic reactions. Nevertheless, modest catalytic reactivity of the isolated model molecules demonstrates their catalytic relevance. The stoichiometric studies are complemented by kinetic and spectroscopic characterizations of catalytic alkene functionalization reactions. The combined organometallic and catalytic studies allowed us to draw conclusions on the mechanisms and helped us develop stereoselective alkene functionalization reactions for synthesizing biologically active molecules.
Scheme 3.
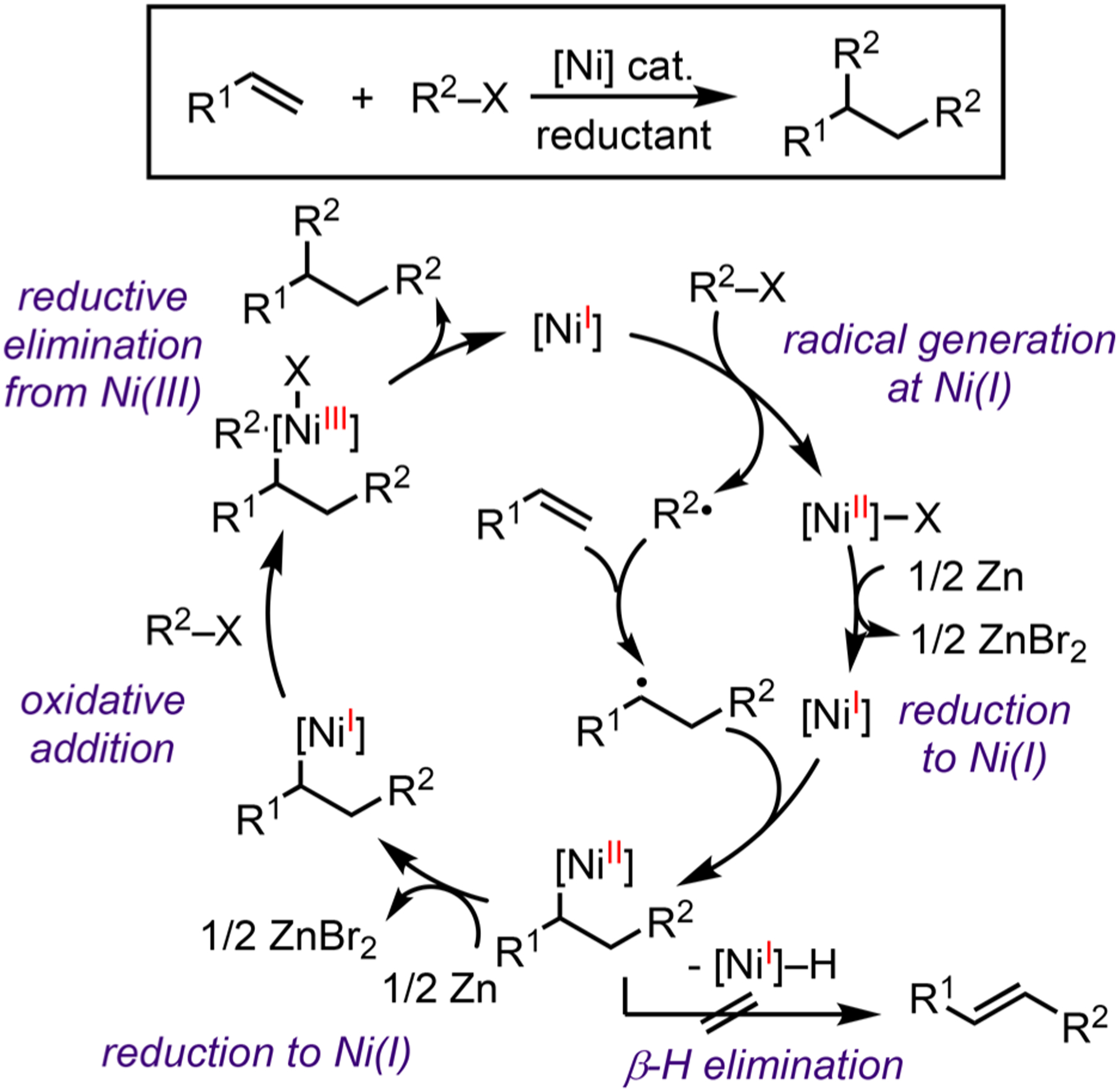
Plausible Fundamental Steps in Alkene Difunctionalization
REDUCTIVE ASYMMETRIC ALKENE DIARYLATION
The α,α,β-triarylated ethane motif is present in a number of important pharmaceutical compounds and biologically active molecules. Accessing enantioenriched compounds would facilitate the assessment of their efficacy in biological studies. Previous methods to prepare these structures, including asymmetric hydrogenation25 and stereospecific and stereoconvergent cross-coupling,26 require multistep synthesis of the substrates. We developed an asymmetric 1,2-diarylation of vinylarenes to assemble chiral α,α,β-triarylated ethane structures with simple vinylarene and bromoarene building blocks (Scheme 4).27 The use of reductive conditions, in combination with aryl bromide electrophiles, avoids stoichiometric air-sensitive organometallic reagents.
Scheme 4.
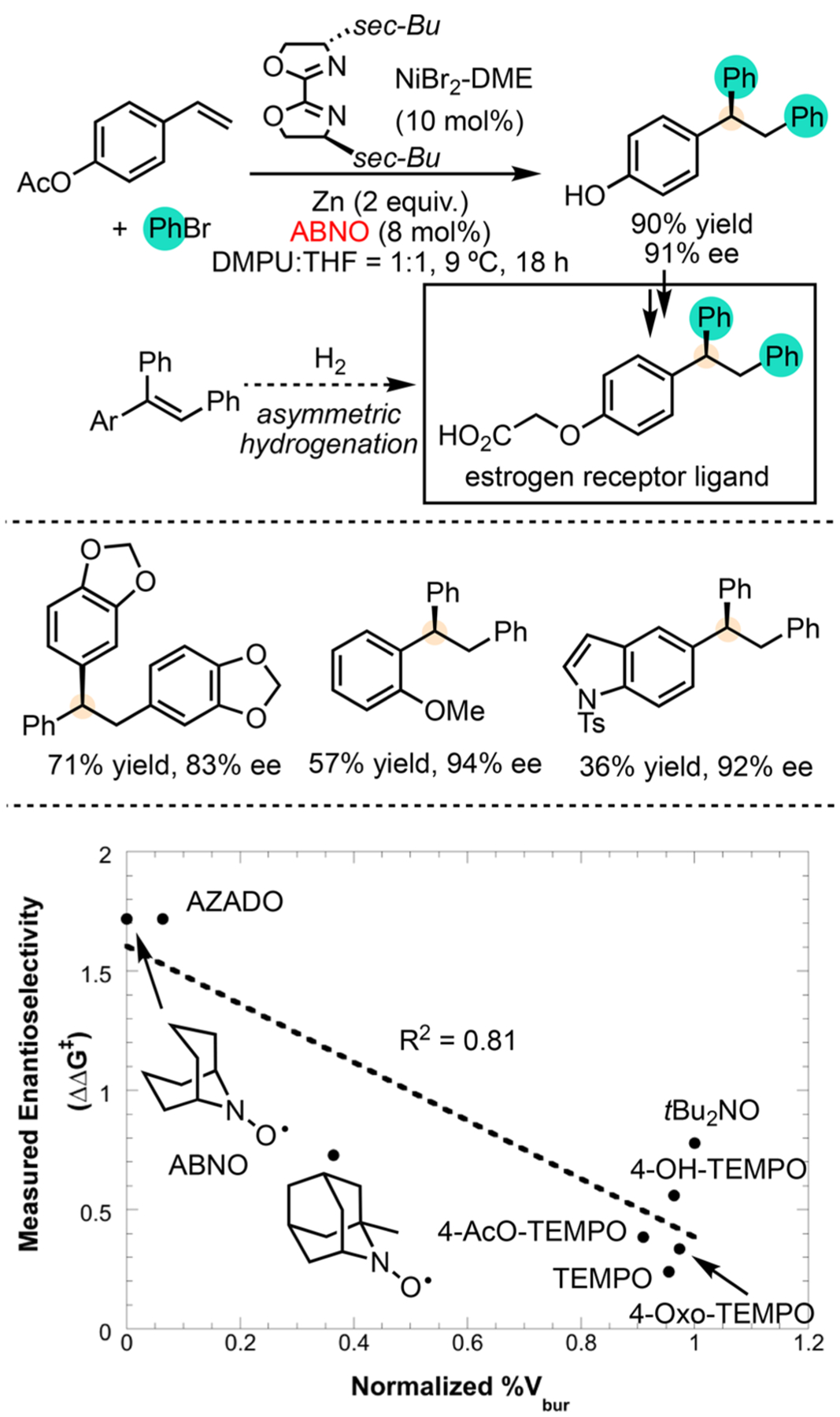
Ni-Catalyzed Asymmetric Diarylation of Vinylarenes
During catalyst optimization, we noticed that changing the substituents on the biOx (bioxazoline) ligands had a relatively small effect on the ee of the products. The use of vinylarene that was purified by distillation reduced the ee compared to substrate used directly from the commercial bottle, in which radical inhibitors are present to prevent polymerization. This observation prompted us to systematically evaluate radical inhibitors, which did not drastically improve the ee. Given that radical inhibitors form stabilized radicals after H-atom abstraction, we reasoned that stabilized radicals may be important to the enantioselectivity. Indeed, the addition of TEMPO in catalytic amounts allowed us to obtain consistent enantioselectivities. Screening stabilized radicals revealed that the ee of the reaction exhibits a correlation with the steric bulk of the N-oxyl radicals (Scheme 4). The least hindered N-oxyl radicals, ABNO and AZADO, gave the highest ee. Although further mechanistic investigation is underway, the different reaction colors in the presence and absence of N-oxyl radicals suggest that they may serve as a ligand to Ni.28
Preliminary mechanistic observations reveal the formation of benzylic radicals as intermediates (Scheme 5). A plausible mechanism begins with a Ni(I)–Ph-mediated migratory insertion (step iii), followed by oxidative addition to activate another molecule of PhBr (step iv). The resulting Ni(III) intermediate undergoes reversible radical ejection that scrambles the benzylic stereocenter (step v). This process has been proposed by Kozlowski, Molander, and co-workers based on computational studies29 and accounts for the observed trans-diphenylation of indene. The formation of dimers at the benzylic position of the monoarylation intermediate, the lack of a Hammett correlation between para-substituents on vinylarenes, and er implies formation of organic radicals rather than polar intermediates. The subsequent reductive elimination (step vi) may serve as the enantio-determining step. We are carrying out a more detailed study to precisely identify the enantio-determining step, build the stereochemical model for predicting the enantioselectivity, resolve the sequence of reagent activation, and elucidate the role of N-oxyl radical additives.
Scheme 5.
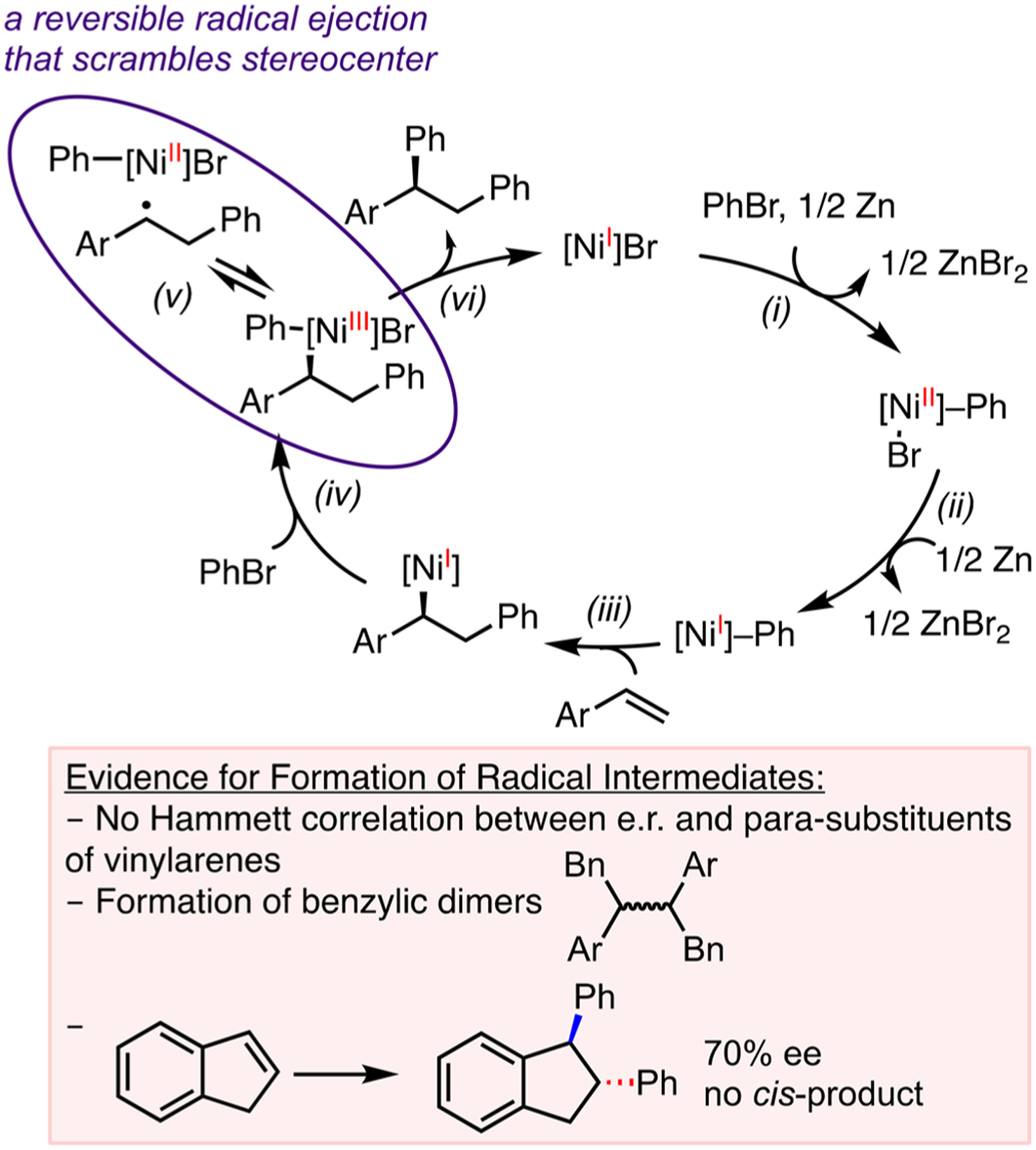
Proposed Mechanism for Ni-Catalyzed Asymmetric Diarylation of Vinylarenes
REDUCTIVE TWO-COMPONENT ALKENE DICARBOFUNCTIONALIZATION
Inspired by previous reports on radical cyclizations and followed by reductive cross-coupling reactions,30 we developed a two-component alkene difunctionalization reaction in which the C sp3-electrophile is tethered to the alkene (Scheme 6).31 The use of reductive conditions with electrophiles bypasses the need for pregeneration of organometallic reagents, which broadens the substrate scope to aryl, heteroaryl, and alkyl bromide electrophiles, while improving tolerance to functional groups such as chlorides, ketones, aldehydes, pyridines, and indoles. This combinatorial approach gives access to a variety of cyclopentane, pyrrolidine, piperidine, and tetrahydrofuran derivatives that are of pharmaceutical interest. We envision that this methodology can be used to rapidly build a library of structurally diverse molecules for medicinal chemistry screening.
Scheme 6.
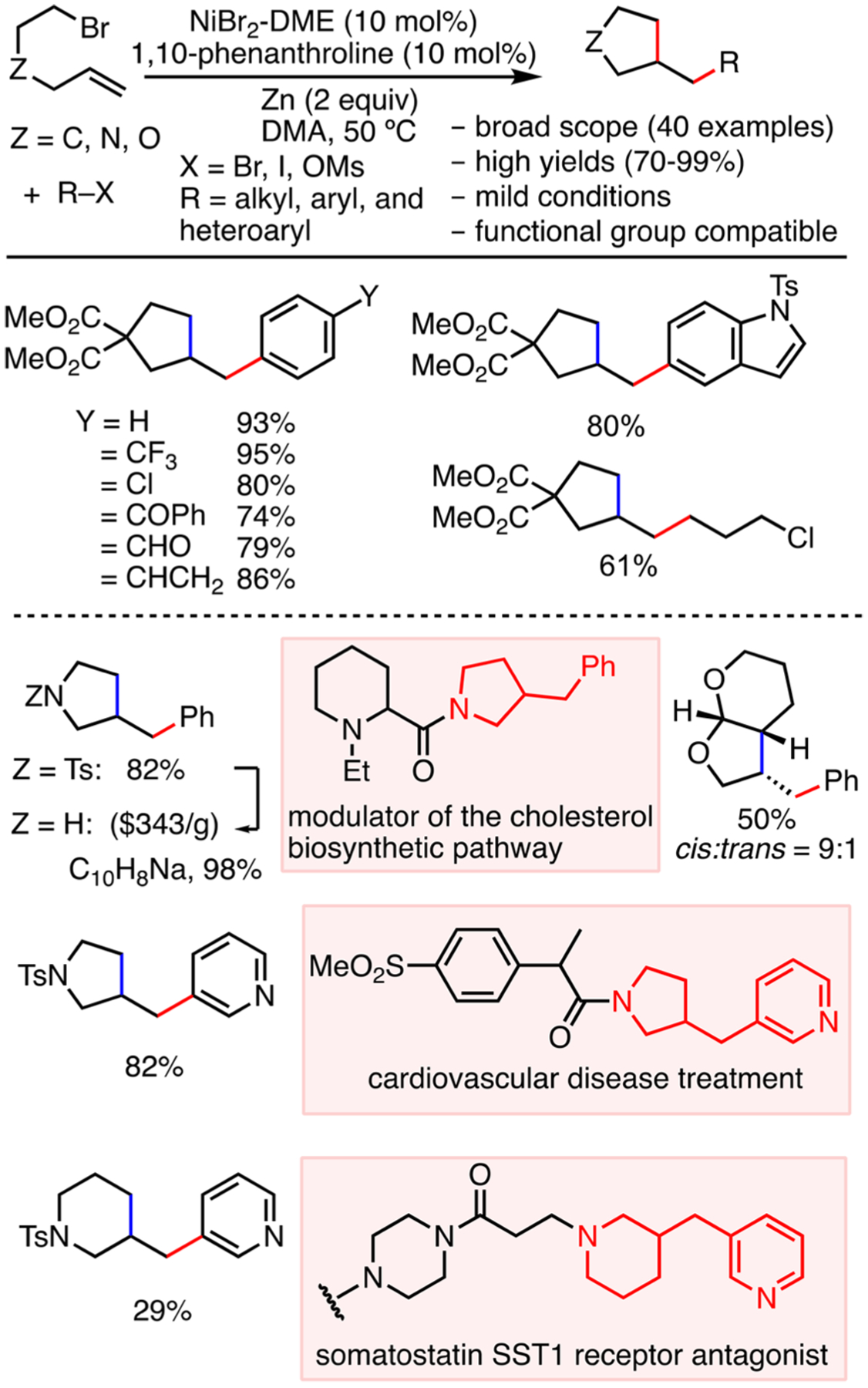
Two-Component Reductive 1,2-Dicarbofunctionalization of Alkenes
MECHANISM OF Ni-CATALYZED REDUCTIVE ALKENE DICARBOFUNCTIONALIZATION
The aforementioned reductive 1,2-dicarbofunctionalization provides us with a platform to investigate the mechanism of Ni-catalyzed cross-electrophile coupling reactions. In general, two types of mechanisms have been proposed for cross-electrophile coupling reactions, “radical chain”32 and “sequential reduction” pathways33 (Scheme 7). The main distinguishing feature of these pathways is the sequence of electrophile activation. In the radical chain mechanism, C sp3 electrophiles are activated by Ni(I)–X (X = halide) to form radicals prior to the oxidative addition of C sp2 electrophiles to Ni(0) (pathways A and B). The radical can either combine with Ni(0) (step iii, pathway A) or Ni(II) (step vii, pathway B). This pathway is kinetically feasible when the Ni species that combines with the radical is the catalyst resting state. In the sequential reduction pathway, oxidative addition of the C sp2 electrophile precedes reduction to afford a Ni(I)–aryl intermediate, which then activates the C sp3 electrophile and immediately combines with the radical (pathway C). The sequence and mechanism of the activation of C sp2 and C sp3 electrophiles are critical to achieving chemoselectivity in cross-electrophile coupling.
Scheme 7.
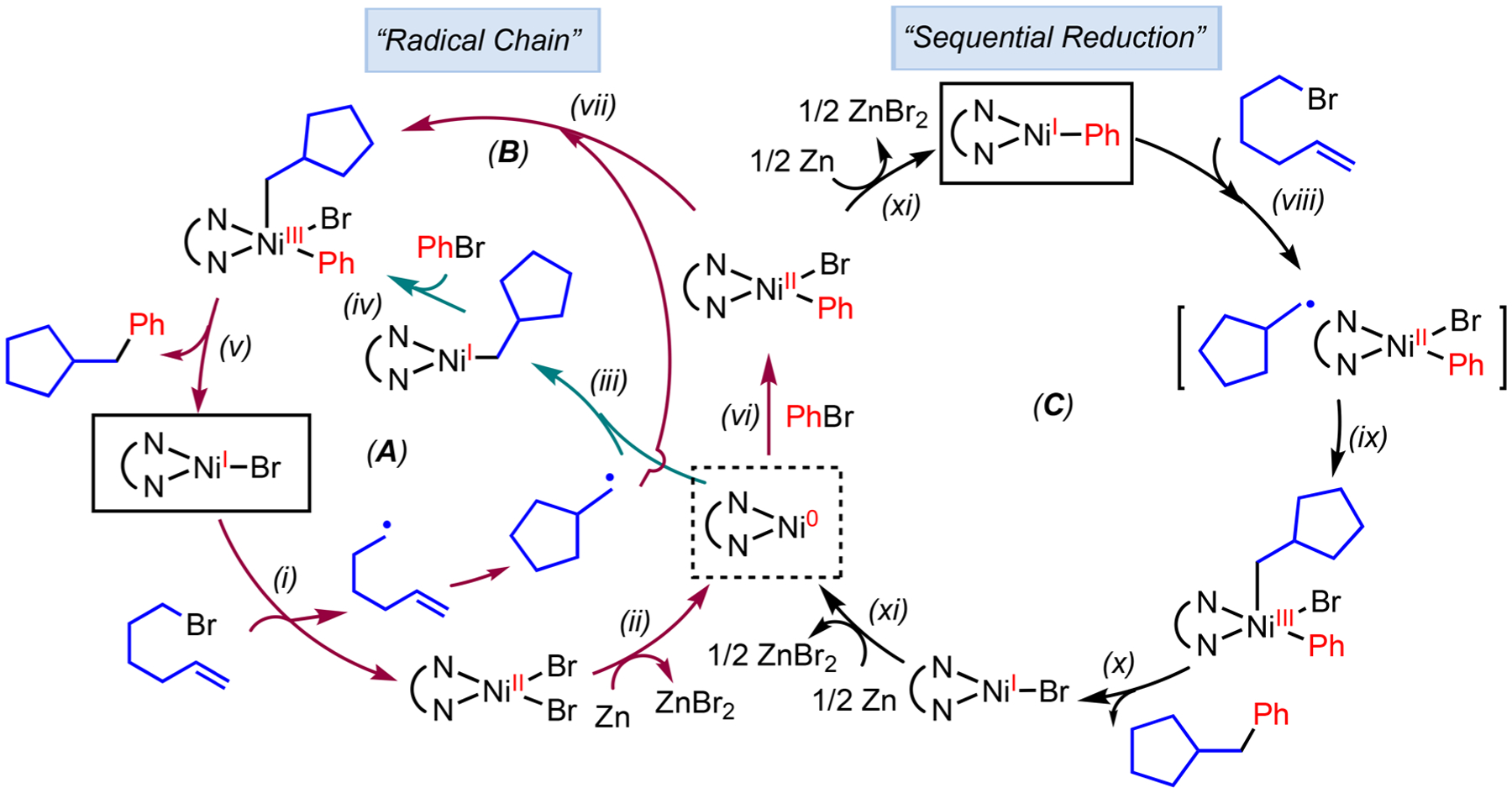
Possible Mechanisms of Ni-Catalyzed Reductive 1,2-Dicarbofunctionalization of Alkenes
We carried out kinetic, spectroscopic, and organometallic studies on the 1,2-dicarbofunctionalization of alkenes (Scheme 6).31 The sequential reduction pathway is most consistent with the data (Scheme 8).34 The lack of rate dependence on olefin and PhBr concentrations, in combination with the coexistence of (phen)NiBr2 3 and (phen)Ni(Ar)(Br) 4 as the catalyst resting state, suggests that the reduction of Ni by Zn is the turnover-limiting step, which is consistent with previous observations.35 This result highlights the importance of catalyst reduction, suggesting that it is critical to ensure efficient mixing of the heterogeneous reductants in order to maintain the reaction rate for large scale production. The preference of (phen)Ni(I)Br to activate ArBr relative to alkyl-Br (step i, Scheme 7) suggested that the radical chain mechanism was not operative, since step i in Scheme 7 would be outcompeted by the reaction of (phen)Ni(I)Br with ArBr. A closer investigation of the catalyst reduction step by cyclic voltammetry, NMR, and EPR spectroscopy, as well as stoichiometric studies, reveals that Zn is only sufficient to reduce (phen)Ni(II) to (phen)Ni(I) rather than (phen)Ni(0).
Scheme 8.
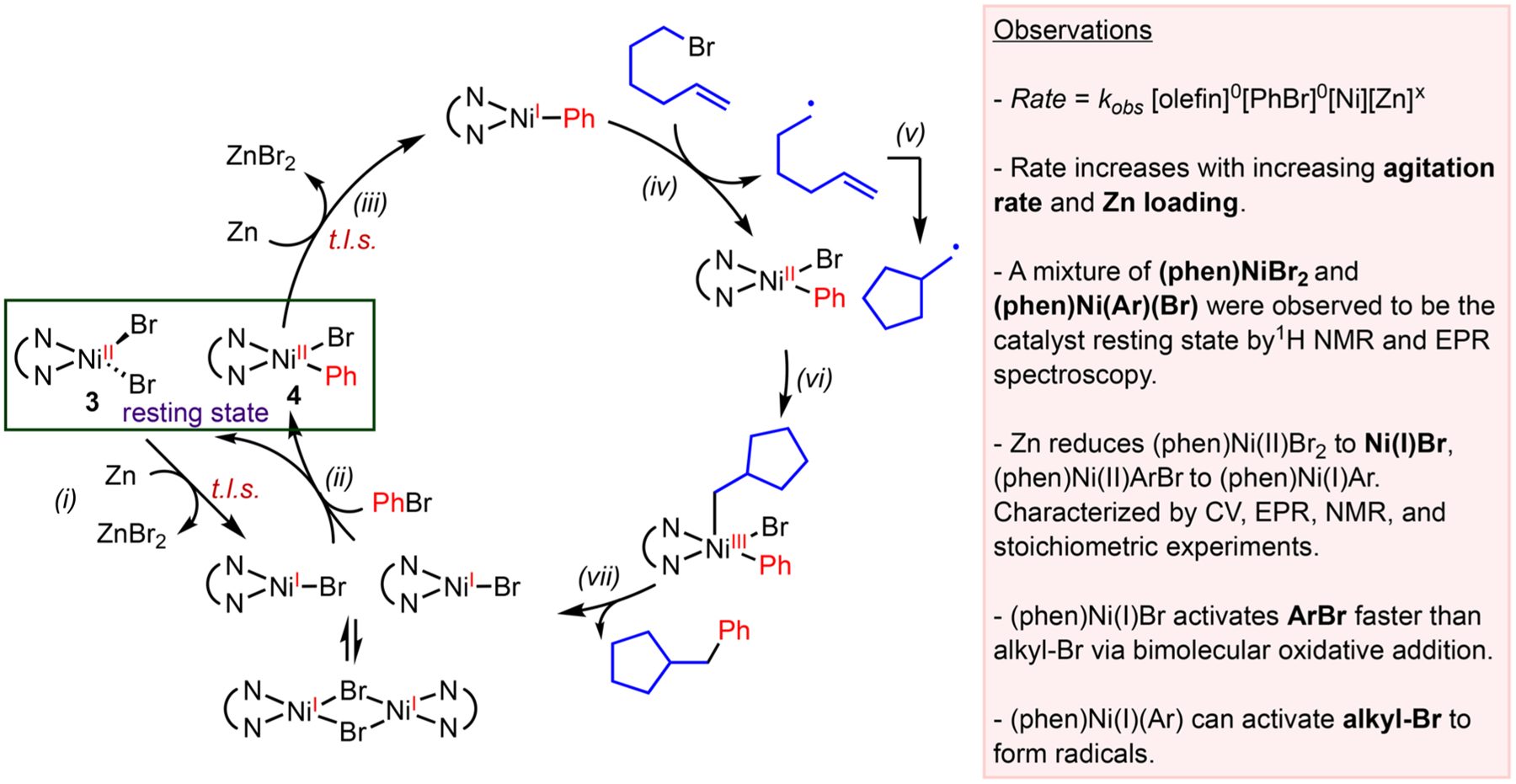
Ni-Catalyzed 1,2-Dicarbofunctionalization of Alkenes by a Proposed “Sequential Reduction” Mechanism and Experimental Evidence
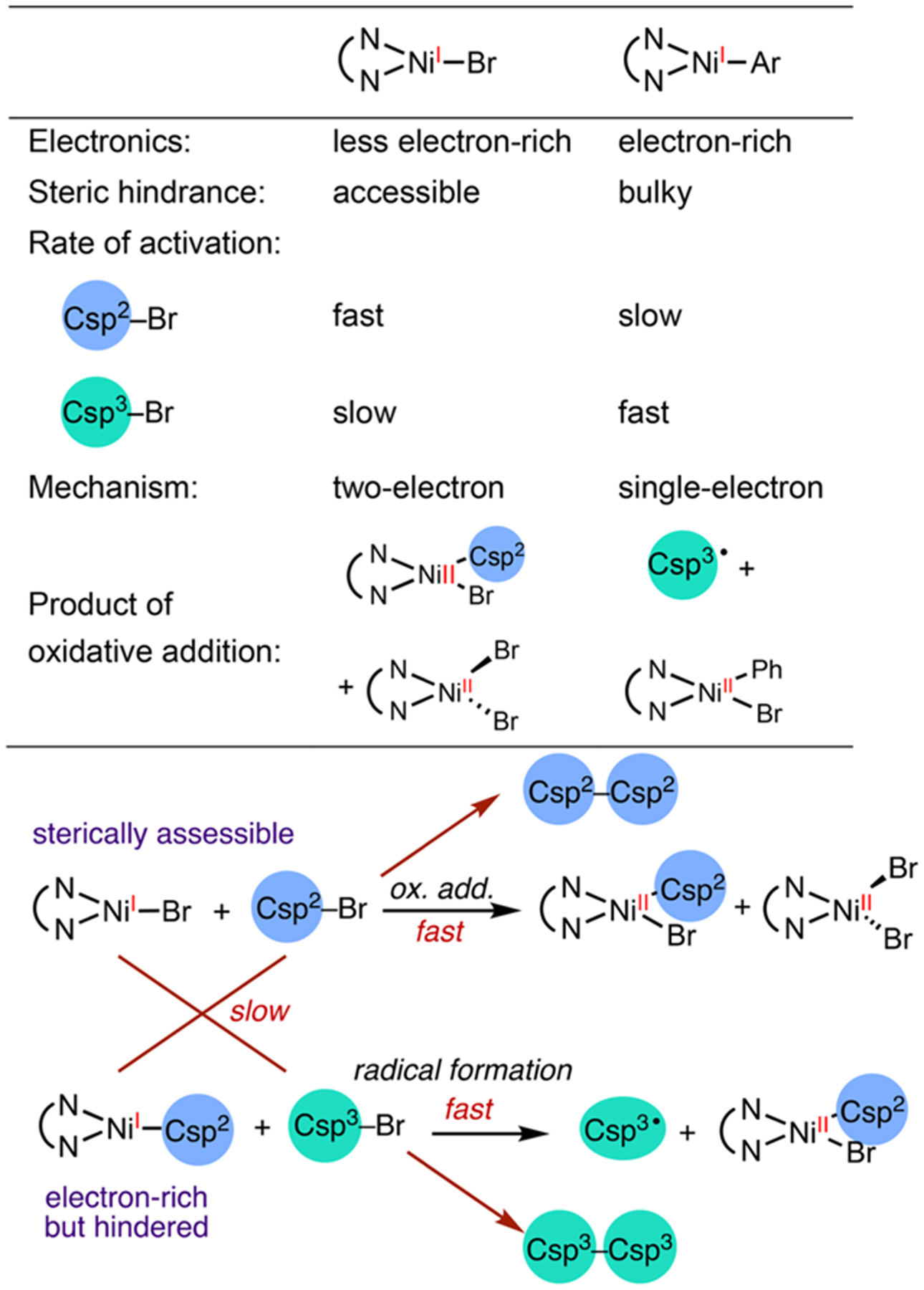
Our data shed light on the selectivity of cross-electrophile coupling reactions and can be explained on the basis of steric and electronic properties of the Ni intermediates (Table 1). Oxidative addition of the C sp2–Br most likely proceeds through a two-electron oxidative addition pathway, due to the instability of C sp2 radicals and the high C sp2–Br bond strength. Stoichiometric studies reveal that two molecules of (phen)Ni(I) Br react with one molecule of Ar–Br to afford (phen)NiBr2 and (phen)Ni(Ar)Br in a 1:1 ratio. While the precise mechanism of this step is still under investigation, it is conceivable that a two-electron oxidative addition on (phen)Ni(I)Br affords (phen)-Ni(III)(Ar)Br2, which undergoes rapid comproportionation with an equivalent of (phen)Ni(I)Br. The two-electron activation of the C sp2–Br by different Ni species is therefore primarily influenced by steric effects. (Phen)Ni(I)Br is sterically more accessible than (phen)Ni(I)Ar; thus it preferentially reacts with the C sp2–Br. The activation of the C sp3–Br proceeds through a one-electron pathway to generate radicals and is largely influenced by electronic effects. The more electron-rich, yet sterically hindered, (phen)Ni(I)Ar therefore reacts with the C sp3–Br faster than the C sp2–Br.
Table 1.
Selectivity of Different Electrophiles
|
MECHANISM OF Ni(I)-MEDIATED RADICAL FORMATION FROM ALKYL BROMIDES
Formation of radicals from the single-electron oxidative activation of alkyl halides by Ni(I) has been extensively proposed in Ni-catalyzed cross-coupling reactions (Scheme 8, step iv).36 Radical intermediates erase the stereochemical information of racemic electrophiles in stereoconvergent cross-coupling,37 enable selectivity in reductive cross-electrophile coupling,18 and accommodate new reactivity via Ni–photoredox dual catalysis.38 Four pathways are possible for Ni(I)-mediated radical formation from alkyl halides (Scheme 9). Two-electron oxidative addition at a Ni(I) center gives a Ni(III) intermediate, which can eject a radical and form a Ni(II) species (Scheme 9A). An outer-sphere single-electron transfer (SET) from Ni(I) to alkyl halides can form a radical anion, which fragments to generate a C-centered radical and a halide anion (Scheme 9B). Alternatively, SET could take place through an inner-sphere pathway via an encounter complex (Scheme 9C). Finally, the concerted halogen atom abstraction has been proposed on the basis of recent DFT calculations (Scheme 9D).39 Prior to our work, there was no experimental study to distinguish these pathways, which was, in part, due to the fact that Ni(I) complexes, with a d9 configuration, are difficult to synthesize and isolate.2a
Scheme 9.
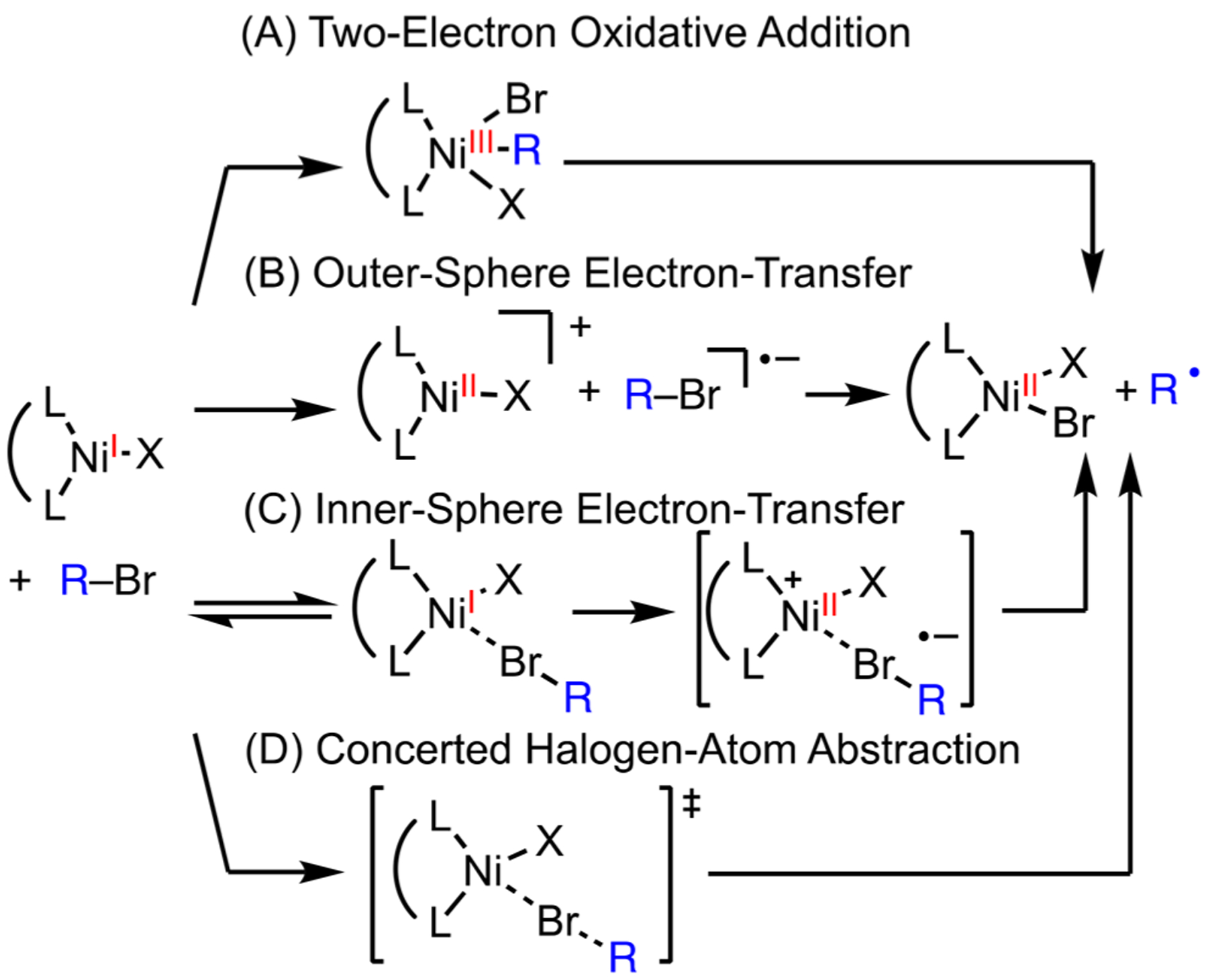
Possible Mechanisms for Ni(I)-Mediated Halide Abstraction
Bulky bidentate phosphine ligands, such as di-tBu-phosphinoethane (dtbpe) and 1,1′-bis(diphenylphosphino)ferrocene (dppf), have been employed to stabilize Ni(I)–halide complexes,40 but Ni(I)–carbyl molecules are more challenging to isolate. We discovered that t-Bu-Xantphos enabled the isolation of a series of Ni(I)–Ar compounds, which can be stored as solids in the glovebox at room temperature for weeks.5 The large bite angle and the secondary interaction of the O-atom on Xantphos to Ni may contribute to this stabilization. Typically, Ni(I) complexes are prepared by reduction of Ni(II),36d comproportionation of Ni(0) and Ni(II),36e or oxidation of Ni(0).41 In our experience, the former method gives the cleanest Ni(I)-halides with mild reductants, such as Zn and cobaltocene, being optimal. Ni(I)–aryl 5 was inert toward PhBr, but reacted with alkyl bromides to give radicals and Ni(II) 6, as verified by radical clocks and trapping experiments (eq 1). The selective activation of alkyl halides over aryl halides is consistent with our observations in the mechanistic study of a reductive 1,2-dicarbofunctionalization (Scheme 8).
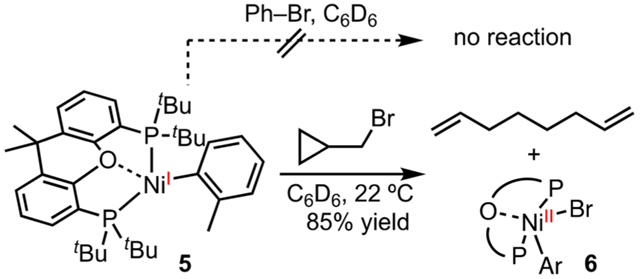 |
(1) |
Kinetic studies, in combination with DFT calculations, allowed us to differentiate the four pathways (Table 2). Using in situ NMR measurements, we found that secondary R–Br reacts faster than primary R–Br, which is inconsistent with the oxidative addition pathway (Scheme 9A). Bulky aryl groups on the Ni(I) compound reduce the rate of halide abstraction, which is inconsistent with the outer-sphere electron transfer pathway (Scheme 9B). The faster rates with electron-rich Ni–Ar complexes are consistent with all four pathways, but the slope of ΔG‡ vs ΔG deviates from the prediction by Marcus theory. The difference between the inner-sphere electron transfer (Scheme 9C) and the concerted mechanism (Scheme 9D) is the formation of an ionic intermediate. An ionic intermediate formed from inner-sphere electron transfer should be stabilized by polar solvents, and the lack of kinetic correlation to solvent polarity rules out this pathway. DFT calculations identified concerted halogen-atom abstraction (Scheme 9D) as the pathway with the lowest activation barrier. In transition state 7, the Ar group, Ni, Br, and the R group organize in a nearly linear geometry, where the unpaired electron is donated to the σ* orbital of the C–Br bond.
Table 2.
Evidence for and against the Possible Pathways of Ni(I)-Mediated Alkyl Halide Activation
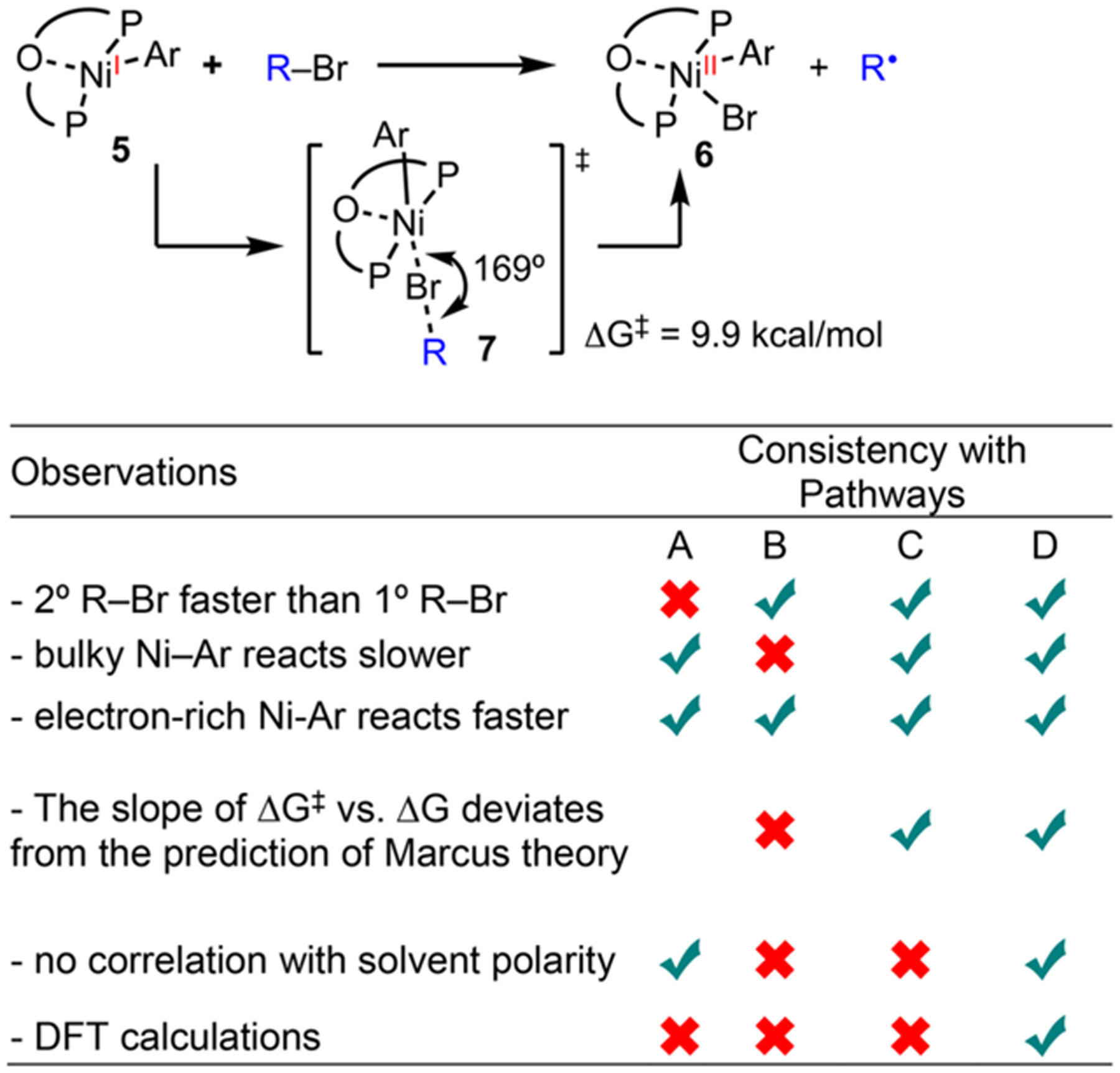
|
Based on this mechanism, we developed a reductive coupling of α-oxyacrylates with alkyl halides to access lactic acid derivatives (eq 2).42 1,1′-Bis(diphenylphosphino)ferrocene exhibits the best reactivity in combination with Ni and Zn to generate a radical, which adds to α-oxyacrylates, an excellent radical acceptor due to the captodative effect. This method represents a concise and efficient way to access α-oxyacrylates, as building blocks for pharmaceutically relevant molecules.
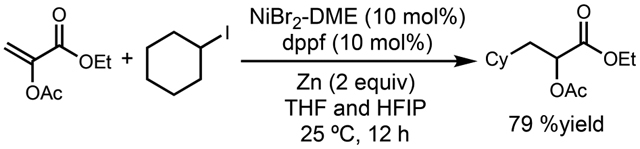 |
(2) |
Bidentate and tridentate N-ligands, such as bipyridine (bpy), bioxazoline (biOx), terpyridine (terpy), and pyridine-bioxazo-line (pybox), are more frequently utilized than phosphine ligands in Ni-catalyzed cross-coupling reactions. These stronger donors, relative to phosphine ligands,43 lead to higher field splitting. The redox activity of these chelating ligands may stabilize the open-shell Ni intermediates. The fundamental advantages of N-ligands over phosphine ligands are worth more in-depth investigation. Our ongoing studies focus on exploring the mechanism of Ni-mediated electrophile activation with N-ligands, as well as understanding the effect of their redox activity on catalysis.
Ni(I)-MEDIATED CO2 INSERTION
Ni-catalyzed functionalization of carbon dioxide (CO2) has become an efficient and sustainable approach to synthesize carboxylate molecules.44 These reactions are proposed to proceed through a pathway consisting of substrate activation by Ni, reduction of Ni(II) to Ni(I) species, and carboxylation of the Ni(I)–alkyl intermediates to form carboxylates (Scheme 10A).45 Previous experimental46 and computational studies39b show that Ni(II) species are not nucleophilic enough to insert into CO2, but once reduced to Ni(I), CO2 insertion is facile. In order to provide experimental evidence for the reduction of Ni(II) to Ni(I) and the insertion of CO2 at a Ni(I)–alkyl center, we prepared (t-Bu-Xantphos)Ni(II)Br2 8, which is reduced by excess Zn to afford (t-Bu-Xantphos)Ni(I)Br 9.47 NMR and XRD experiments show that ZnBr2 is coordinated to the bromide of Ni(I) 9. (t-Bu-Xantphos)NiMe 10 and (t-Bu-Xantphos)NiEt 11 were obtained by transmetalation of Ni(I)Br 9 with lithium reagents (Scheme 10B). These (t-Bu-Xantphos)-Ni(I)–alkyl complexes are much more sensitive than their aryl counterparts, decomposing at room temperature within 30 min and requiring handling at low temperature in the glovebox.
Scheme 10.
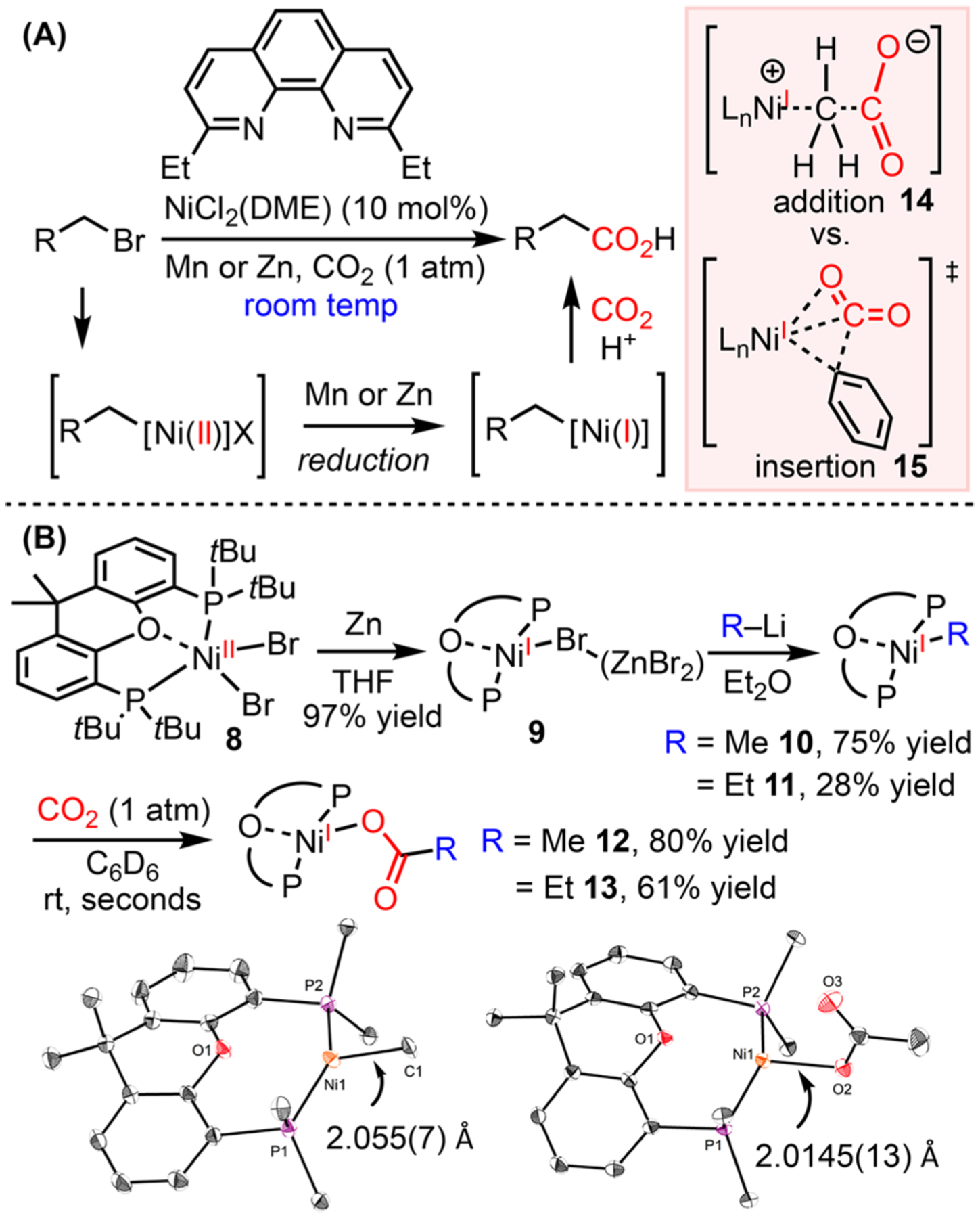
Reduction of Ni(II) to Ni(I) by Zn and Carboxylation of Ni(I)–Me
Rapid insertion of CO2 at 10 and 11 afforded the corresponding carboxylates 12 and 13 at room temperature in seconds. In contrast, (t-Bu-Xantphos)NiPh and (t-Bu-Xantphos)Ni-acetylide are unreactive toward CO2. This contrasting reactivity between sp, sp2, and sp3 Ni(I) complexes sheds light on the mechanism of CO2 insertion. Ni(I)–C sp3 complexes could undergo insertion via nucleophilic attack of the alkyl groups to CO2 (intermediate 14), where sp and sp2 nucleophiles can only undergo migratory insertion (TS 15), but the approach of CO2 to these (tBu-Xantphos)Ni(I) complexes is inhibited by the bulky substituents on the ligand.
REDUCTIVE INTRAMOLECULAR ALKENE COUPLING VIA TWO-ELECTRON PATHWAYS MEDIATED BY Ni(I)/Ni(III)
The low oxidation potentials of Ni complexes (Scheme 1E) motivated us to pursue reactions going through two-electron redox pathways mediated by Ni(I)/Ni(III) intermediates. The redox potentials of Ni(I)/Ni(III) couples may resemble those of Pd(0)/Pd(II), but the radical properties of Ni(I) and Ni(III) intermediates can lead to different selectivity. Cycloisomerization reactions of 1,5-dienes typically give unsaturated products (Scheme 11).48 Employing d8 transition metal catalysts, these reactions commence with hydride insertion to one alkene, followed by insertion to the other alkene, and β-H elimination, which restores the unsaturation. In light of the prevalence of trans-vicinal disubstituents in carbocycles49 and heterocycles50 of biological relevance, we developed a reductive diene coupling reaction where formation of the C–C bond is accompanied by the installation of trans-stereocenters (Scheme 11).51 This reaction is accomplished by simply changing the electronic structure of the catalyst from d8 to d9 18 and using Et2SiH2 as the reductant. The utility of this method was demonstrated in the preparation of 3,4-dimethyl pyrrolidine 17 and cyclopentane derivative 19 that are of pharmaceutical interest. The (R, R) enantiomer of 3,4-dimethylgababutin derivative 19 exhibits excellent efficacy toward neuropathic pain and anxiety. In contrast to the previous synthesis,49 which requires 13 steps for a 14% overall yield, the route going through the reductive diene coupling requires 6 steps with an overall yield of 42%.
Scheme 11.
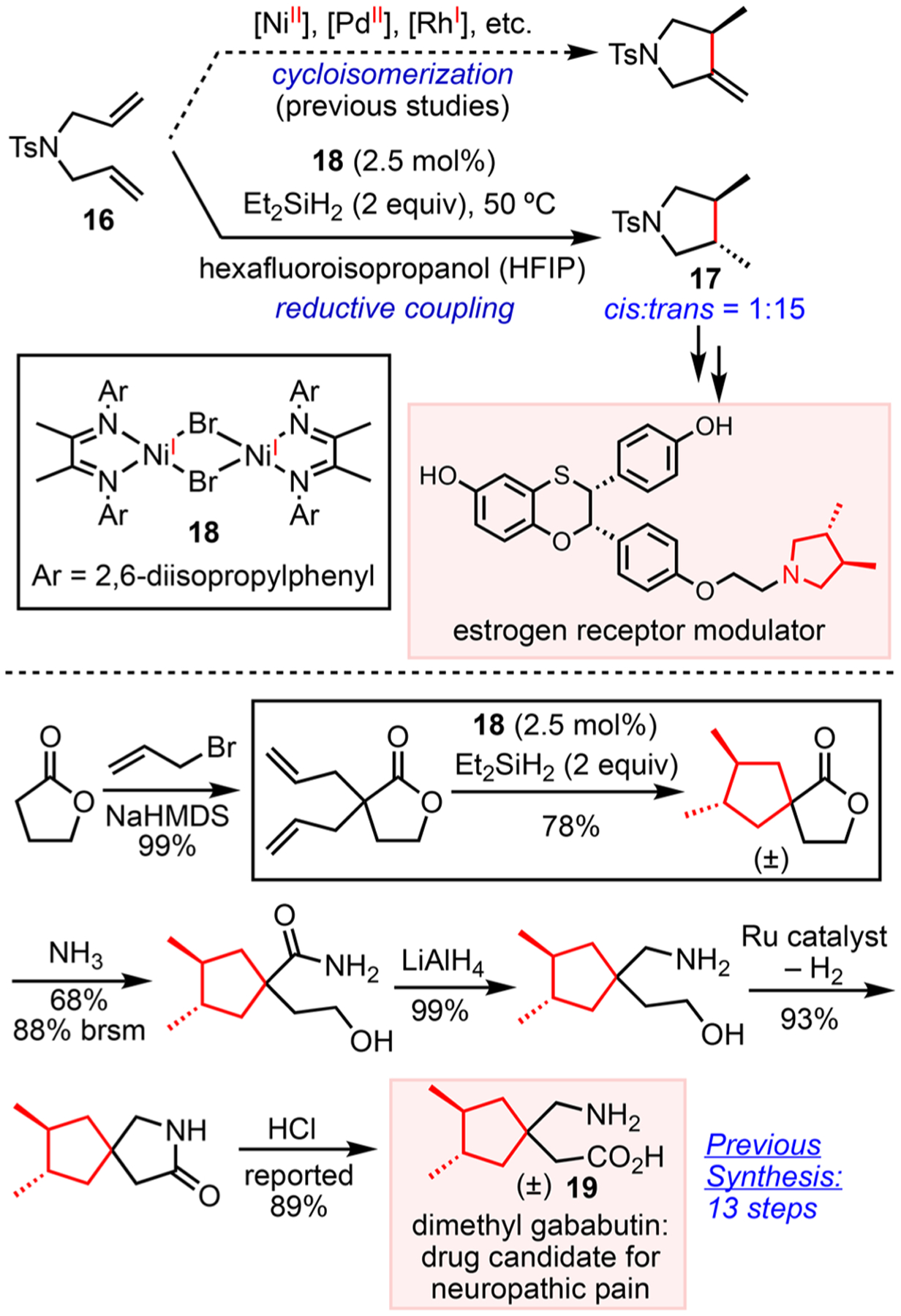
Reductive Diene Coupling and Applications
This reductive diene coupling reaction differs from several recent olefin functionalization reactions in its scope and mechanism. Fe- and Co-catalyzed diene coupling reactions developed by Baran and Shenvi are selective for activated or substituted alkenes,52 going through a H-atom transfer (HAT) pathway to form radical intermediates. Our Ni-catalyzed intramolecular reductive diene coupling is effective with unactivated alkenes, but the reaction is drastically inhibited by substituents on the olefin. When a radical clock was built into the diene substrate, no ring opening was observed, suggesting that HAT is not occurring in this system. We characterized its mechanism by kinetic and spectroscopic studies, which support a classic two-electron pathway mediated by paramagnetic Ni(I) and Ni(III) intermediates (Scheme 12). This pathway accounts for the chemoselectivity for reductive coupling relative to redox-neutral cycloisomerization. Ni(I) 22 is stabilized by the redox-active diimine ligand and is capable of undergoing oxidative addition with silanes to form Ni(III) 24, whereas the previous Ni(II) catalyst affords unsaturated products via β-H elimination (22 → 26). The trans-diastereoselectivity is determined by the second olefin insertion step, 21 → 22. This cyclization step is responsible for the limited scope in cycloisomerization reactions, as substituents on the olefin inhibit the cyclization.48
Scheme 12.
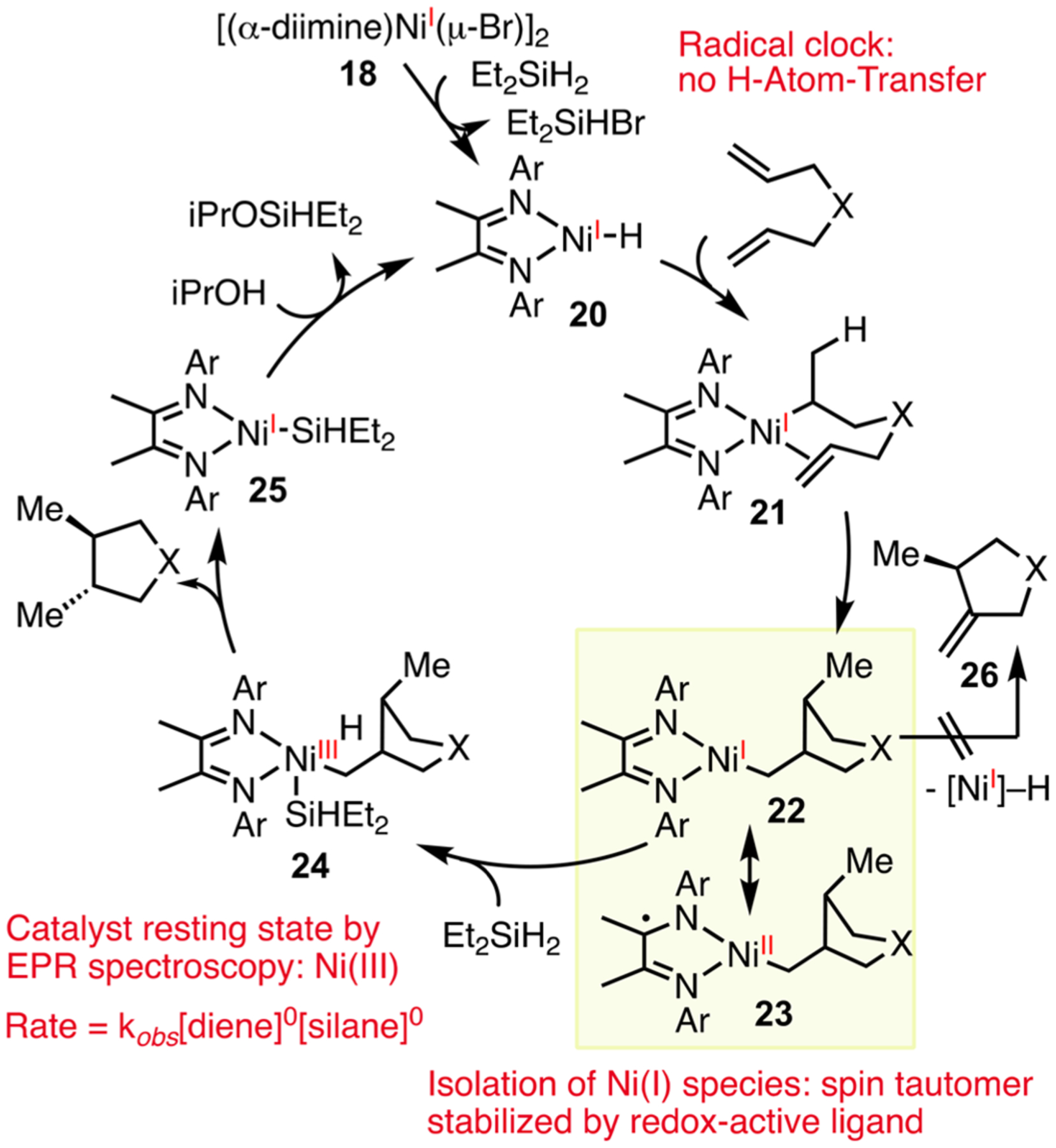
Proposed Mechanism to Account for Chemo- and Diastereoselectivity
DINUCLEAR Ni(III)⋯Ni(III)-MEDIATED C–C BOND FORMING REDUCTIVE ELIMINATION
The formation and activation of a chemical bond usually involves two-electron transformations. For example, oxidative addition of Rh(I) to C–H bonds affords Rh(III) hydride compounds.53 An illustrative example reveals that the C–H activation can be facilitated by engaging two Rh(II)-porphyrin molecules with each Rh(II) center offering one electron.54 The one-electron redox process at each Rh center is expected to lower the kinetic barrier relative to two-electron redox chemistry. Since Ni has the tendency to undergo one-electron redox chemistry, we sought to facilitate bond formation processes by utilizing dinuclear Ni-centers.
Our investigations began with preparation of (py-Mepyrr)-NiMe(2,4-lutidine) complex 27 (py-Mepyrr = 3,5-dimethyl-2-(2-pyridyl)pyrrole) from transmetalation of (py-Mepyrr)NiMe-(acac) (acac = acetylacetonate) with MeMgBr (Scheme 13).55 Alternatively, addition of py-Mepyrr to (TMEDA)NiMe2 in the presence of 3,5-lutidine can cleanly afford 28 in high yield. 2,4-Lutidine has the right amount of steric protection for stabilizing the complex. Bulkier lutidines dissociate from Ni and less bulky pyridine derivatives are insufficient at stabilizing the Ni compounds. Oxidation of 27 by I2 or O2 liberates ethane. A combination of 1H NMR and EPR studies revealed a rapid conversion of 27 to Ni(III) 28 upon oxidation, followed by dimerization to generate 29. The subsequent dinuclear reductive elimination is slow and exhibits a first-order dependence on 29. This result establishes the first dinuclear Ni-mediated C–C bond formation, where each Ni center reacts cooperatively to accept one electron.
Scheme 13.
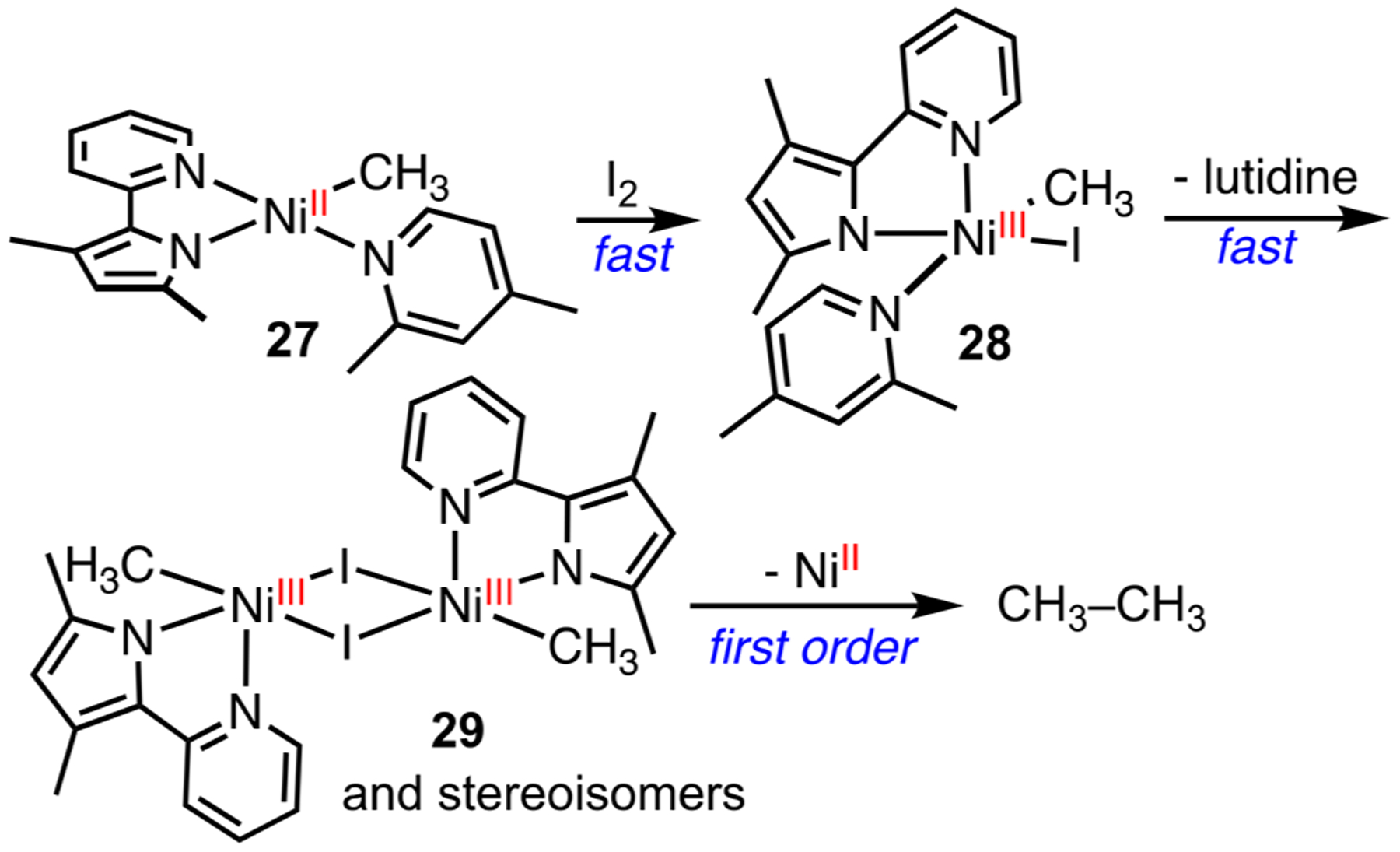
Dinuclear Ni(III)-Mediated Ethane Formation
DINUCLEAR Ni(III)⋯Ni(III)-MEDIATED CARBON–HALOGEN BOND FORMING REDUCTIVE ELIMINATION
Dinuclear transition metal compounds and metal–metal interactions are ubiquitous in biological and catalytic systems.56 Multimetallic cofactors are commonly involved in redox reactions. Hydrogenase enzymes featuring a [NiFe] core catalyze the oxidation of H2, as well as the reduction of protons to H2, and represent well-characterized examples of metal–metal bonding in biology.56 Synthetically, bimetallic intermediates have been observed in Ni-catalyzed vinylidene transfer reactions57 as well as many others.58 First-row transition metals are expected to form weaker metal–metal bonds than second-and third-row metals, which contributes to the lack of examples of isolated high-valent Ni complexes with metal–metal bonding.
Ritter and co-workers have shown that a Pd(III)–Pd(III) dimer, 30, is stabilized by a metal–metal bond and can undergo synergistic reductive elimination to afford bromobenzo[h]quinoline in 95% yield (theoretical yield is 200%) and Pd(II) (Scheme 14A).59 We prepared an analogous Ni(II)⋯Ni(II) dimer, 31.60 Oxidation of 31 with Umemoto’s reagent, 5-(trifluoromethyl)dibenzothiophenium triflate (TDTT), afforded the mixed-valent Ni(2.5)–Ni(2.5), 32. Fractional oxidation states are common in multimetallic Ni complexes61 but are rare with Pd.62 This one-electron oxidation, however, is insufficient to induce reductive elimination. Further oxidation is required to form carbon–halogen bonds. The use of PhNMe3Br3 led to a Ni(III)⋯Ni(III) dimer 34, a geometric isomer of 33, which underwent rapid reductive elimination at −80 °C to form bromo-benzo[h]quinoline in 190% yield and Ni(I).
Scheme 14.
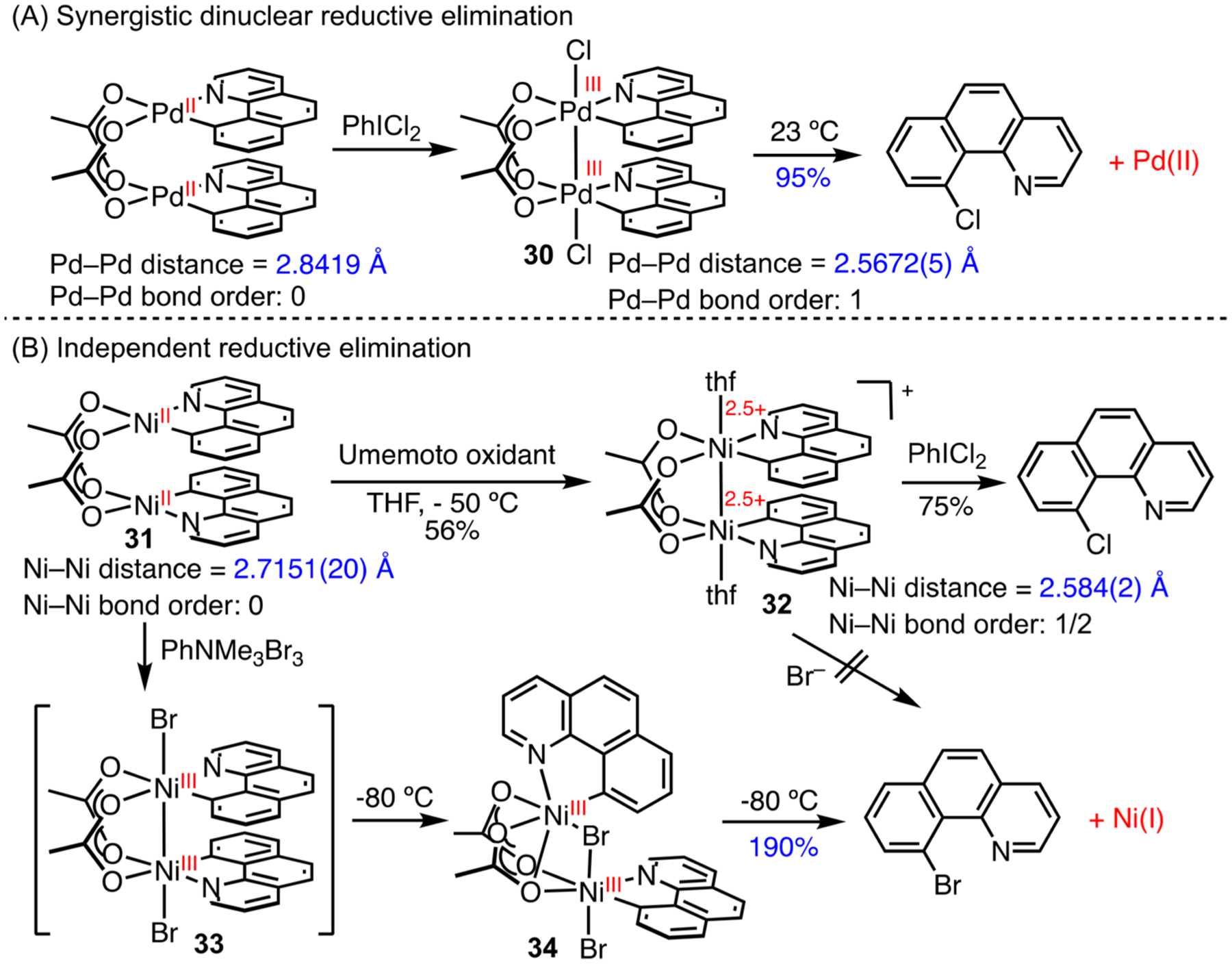
Comparison of Dinuclear Ni and Pd in Metal–Metal Bonding and Reductive Elimination
These results suggest that the Ni(III)⋯Ni(III) dimer 33, if ever formed, has a weak metal–metal bond and prefers to rearrange to 34. This contrasts with Pd and is consistent with weaker metal–metal bonding for first-row transition metals. It is speculated that the more constrained electron-density of first-row transition metals restricts sufficient orbital overlap for metal–metal bonding interactions. The facile reductive elimination of Ni(III) 34 to form Ni(I) occurs independently at each metal center. This reactivity differs from that of Pd(III)–Pd(III) dimer 30, which undergoes cooperative reductive elimination to eject half of the benzo[h]quinoline ligand as bromo-benzo[h]quinoline and forms Pd(II). The different reactivity likely results from a stable Ni(I) oxidation state.
DINUCLEAR Ni(III)⋯Ni(II)-MEDIATED N–N BOND FORMING REDUCTIVE ELIMINATION
Our investigation of Ni–Ni bonds was extended to a “paddle-wheel” system.63 (TBD)4Pd2 35 (TBD = triazabicyclodecene) can be oxidized to Pd(III)–Pd(III) 36, which does not undergo reductive elimination (Scheme 15A).64 Compared to 35, the shorter Pd⋯Pd distance of 36 indicates a Pd–Pd bond order of 1. A Ni2(TBD)4 dimer has been electrocatalytically oxidized to form [Ni2(TBD)4]+, but the structure was not characterized.65 In order to compare Ni–Ni bonding with Pd and probe the intrinsic reactivity of the high-valent Ni system, we oxidized Ni2(TBD)4 37 with 0.5 equiv of PhICl2 to afford the mixed-valent complex 38 (Scheme 15B). The reduction of 38 is facile and can be accomplished with Tl(I) compounds. Further oxidation of 38 with excess PhICl2 promotes N–N bond forming reductive elimination to give Ni(II) 39, which represents the first example of such reactivity. Treating Ni(II) 39 with Ni(cod)2 and Li(TBD) causes N–N bond cleavage, reforming the paddle-wheel Ni(II)–Ni(II) complex 37.
Scheme 15.
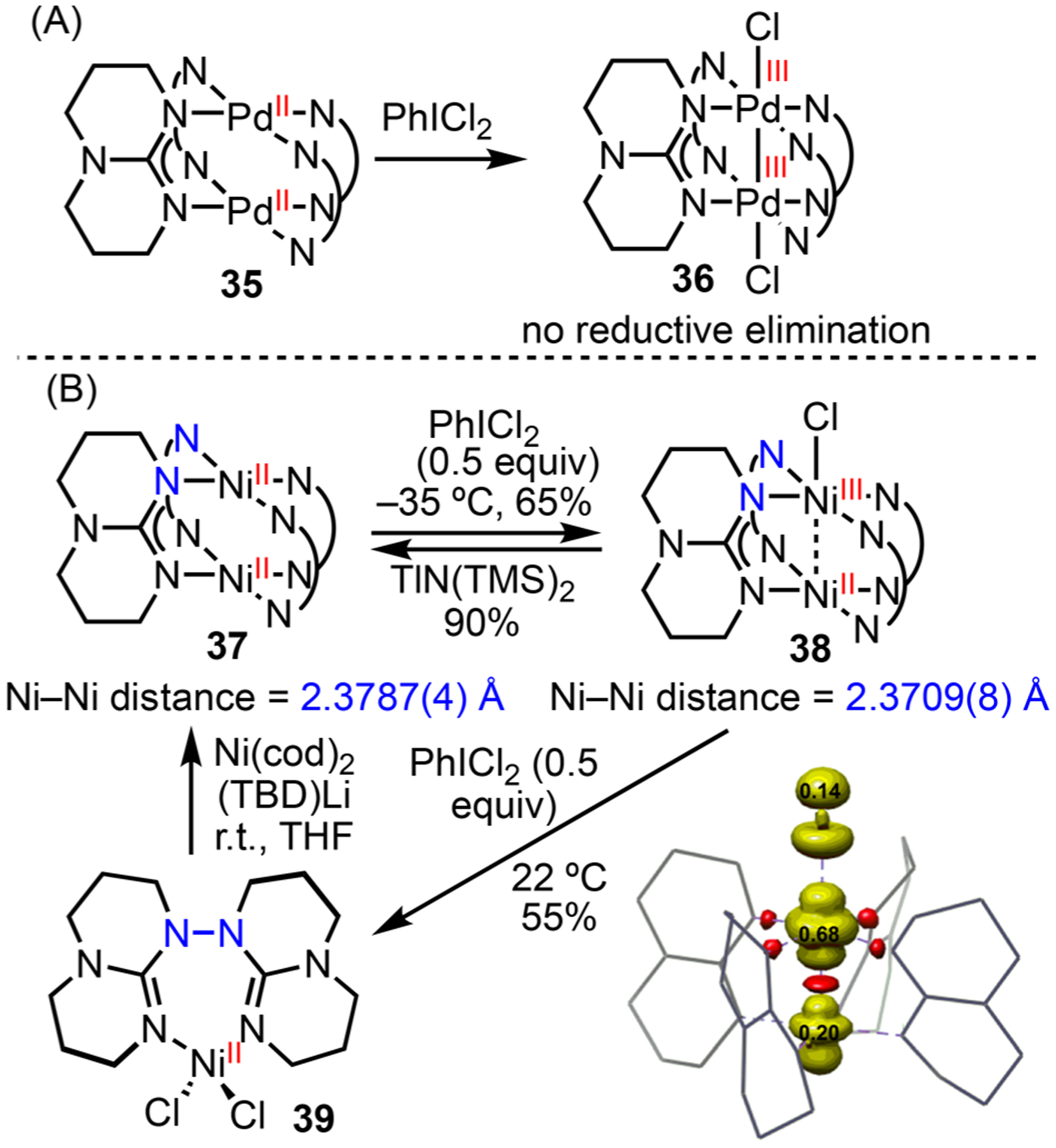
Comparison of Ni and Pd in Metal–Metal Bonding and Reductive Elimination with Paddle-wheel Complexes
Assignment of Ni–Ni bonds in this system requires careful consideration. The σ and σ* orbitals along the metal–metal axis of the Ni(II) dimer 37 are filled; however, mixing of the 4s, 4p, and 3d orbitals reduces the energy of the σ* orbital, resulting in partial bonding character.66 Complex 38 forms a 3c/3e bond. The EPR spectrum of 38 shows a Ni-centered radical. DFT calculations suggest that the unpaired electron has the highest density on Ni(III). The electronic structure of 38 is best described as discrete Ni(II) and Ni(III) centers with weak bonding interactions between the two Ni atoms. This assignment is corroborated by the similar bond lengths between 37 and 38. Although bond lengths may be constrained by the paddle-wheel geometry and may not accurately reflect the bonding interactions, contractions of bond lengths have been observed and serve as indications of increases in bond orders in previous Ni2 paddle-wheel complexes.66
STRUCTURES OF (α-DIIMINE)Ni AND Pd AGOSTIC COMPLEXES AND IMPLICATIONS IN β-H ELIMINATION
Agostic interactions are three-center, two-electron bonds formed from the donation of electron density from the C–H bond of an alkyl ligand to the empty d-orbital of a transition metal.67 Ni and Pd agostic complexes are commonly proposed intermediates in catalytic reactions going through C–H activation and β-H elimination steps. Cationic (α-diimine)Ni and Pd complexes display intriguing reactivity and properties in olefin polymerization (Scheme 16).68 The rate of β-H elimination (step i) relative to olefin coordination (step iii) determines the polymer structure and thus its morphology and properties. Structural characterization of Ni and Pd agostic complexes, however, remains difficult, owing to their instability. Warren reported the isolation of a neutral Ni-agostic molecule,69 but its reactivity is drastically lower than the cationic catalysts.
Scheme 16.
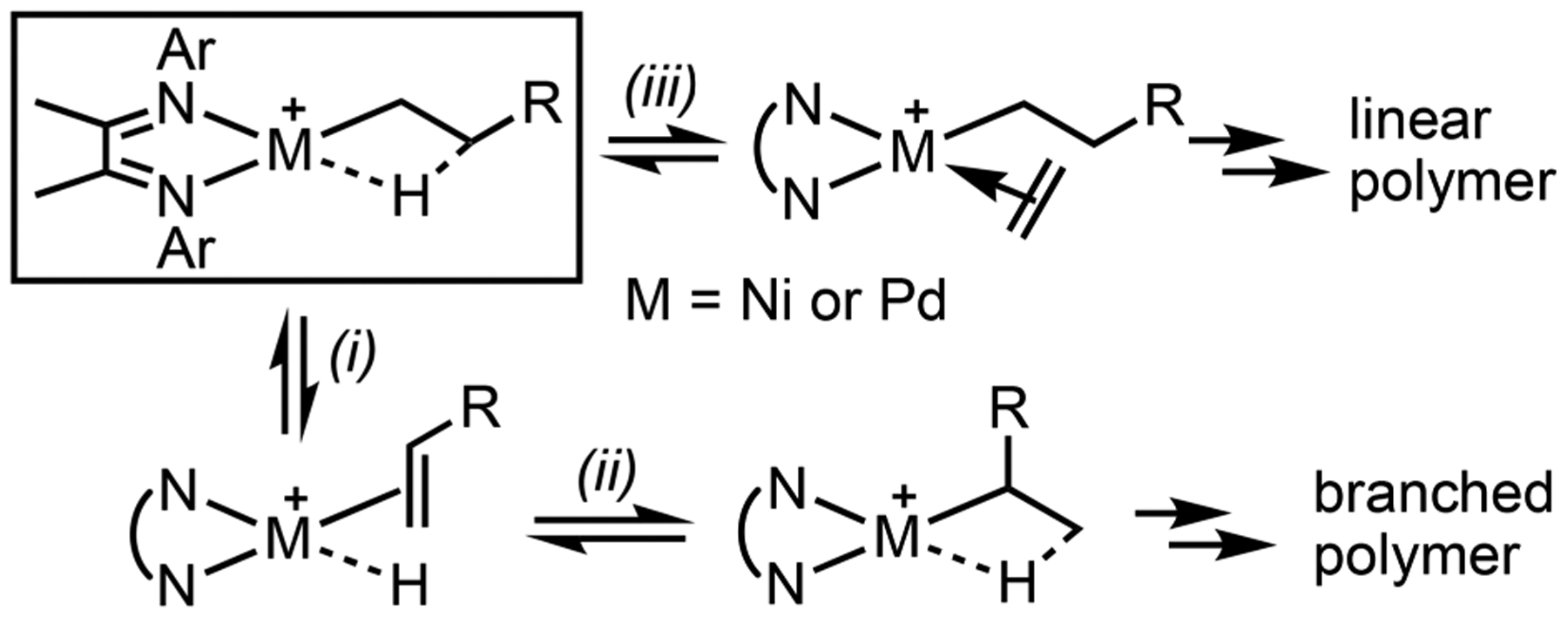
(α-Diimine)Ni and Pd-Catalyzed Olefin Polymerization
We discovered that a cyclohexane backbone on the α-diimine ligand can increase the catalyst stability, while maintaining its catalytic reactivity. This modification allowed us to obtain single crystals of β-agostic [(iPrα-diimine)Ni-Et]+[BArF4]− 40 and [(iPrα-diimine)Pd-Et]+[BArF]− 41 that are suitable for characterization by X-ray diffraction and neutron diffraction (Figure 1).70 The structures feature short M–H distances (M = Ni and Pd), short Cα–Cβ distances, and acute M–Cα–Cβ bond angles (Table 3). The bond lengths of Cα–Cβ (C31–C32) for 40 and 41 are 1.468(7) and 1.469(6) Å, which are shorter than that of a nonagostic Pd–Et complex (1.528(3) Å), suggesting greater sp2 character of the Cα and Cβ atoms as a C=C bond partially forms in the agostic structure. The M–Cα–Cβ bond angles are substantially smaller than the ideal angle of 109° for a sp3 hybridized carbon, consistent with the attraction between Pd and Cβ. These properties combined serve as unambiguous evidence for the β-agostic interaction.
Figure 1.
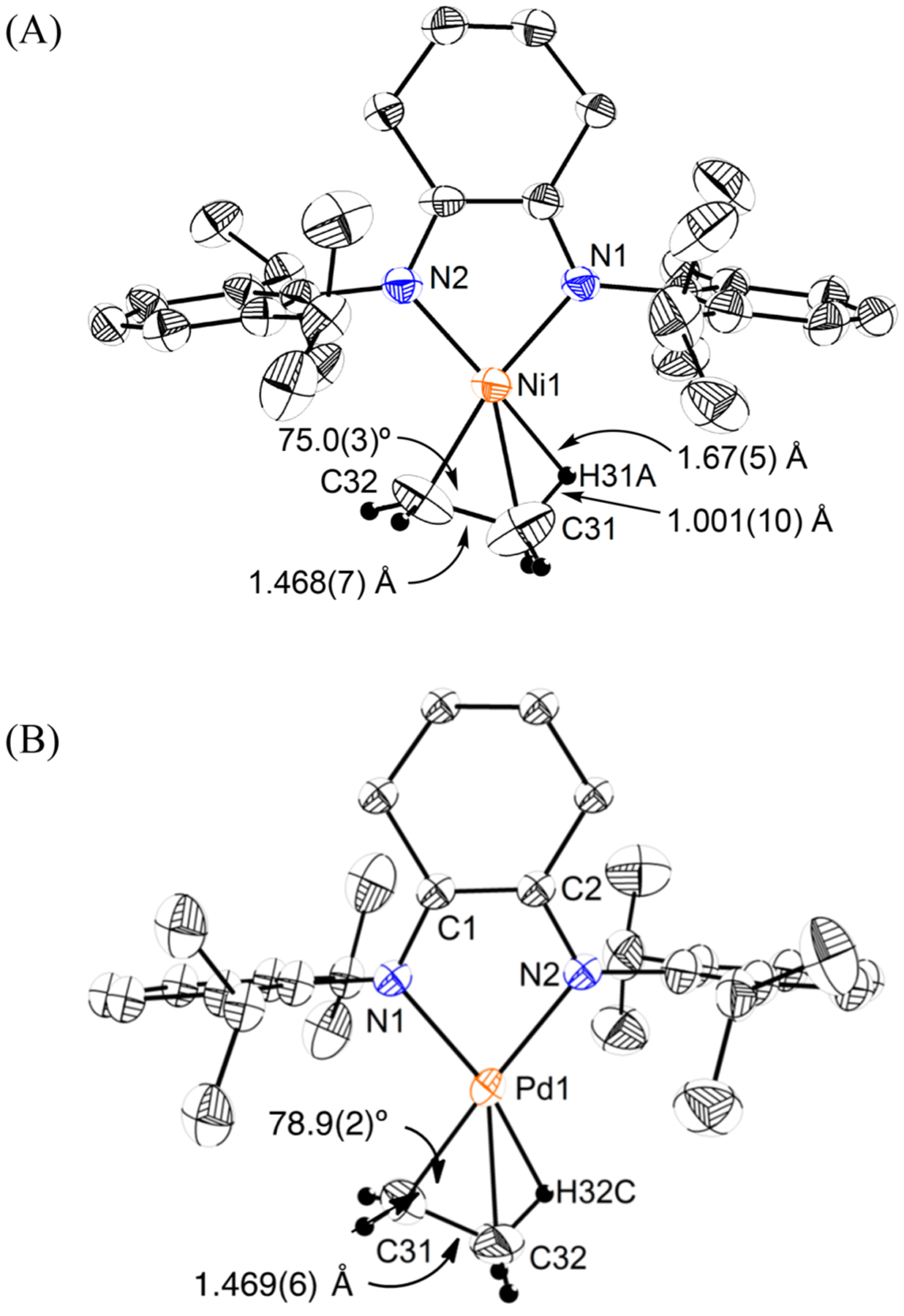
X-ray structure of [(iPrα-diimine)Ni-Et]+[BArF4]− (40) and [(iPrα-diimine)Pd-Et]+[BArF]− (41). Atom thermal ellipsoids are shown at the 50% probability level. The hydrogen atoms on the α-diimine ligands and [BArF4]− are omitted for clarity.
Table 3.
Comparison of Bond Parameters for β-Agostic (α-Diimine)Ni and Pd Complexes
| 40 (Ni) | 41 (Pd) | |
|---|---|---|
| M–C31 (Å) | 1.901(4) | 2.030(4) |
| M–C32 (Å) | 2.081(5) | 2.264(4) |
| C31–C32 (Å) | 1.468(7) | 1.469(6) |
| M–C31–C32 (deg) | 75.0(3) | 78.9(2) |
| M–H34A (Å) | 1.67(5) | 1.60(4)a |
| C34–H34A (Å) | 1.001(10) | 1.20(5)a |
Bond lengths determined by neutron diffraction for (iPr,para‑MeOα-diimine)Pd.
Complexes 40 and 41 with the cyclohexyl backbone are as competent as the parent catalysts in olefin polymerization. We measured kinetic barriers for β-H elimination (Scheme 17). The slower β-H elimination from Ni, relative to Pd, is reminiscent of previous studies by Brookhart.11 Multiple factors influence this rate. The nature of the agostic bond is the donation of electron density from the C–H σ orbital to the metal d orbital. The lower electronegativity of Ni results in a weaker agostic interaction relative to Pd. Second, in the metal hydrides, 42 and 43, the alkene resides in a conformation that is perpendicular to the square-planar complex, representing a twisted geometry in the late transition state. Ni has a smaller atom radius and thus a shorter Ni–C bond length and a more acute Ni–C–C bond angle. As a result, the transition state for Ni has a more strained geometry, which should contribute to a higher transition state energy.
Scheme 17.
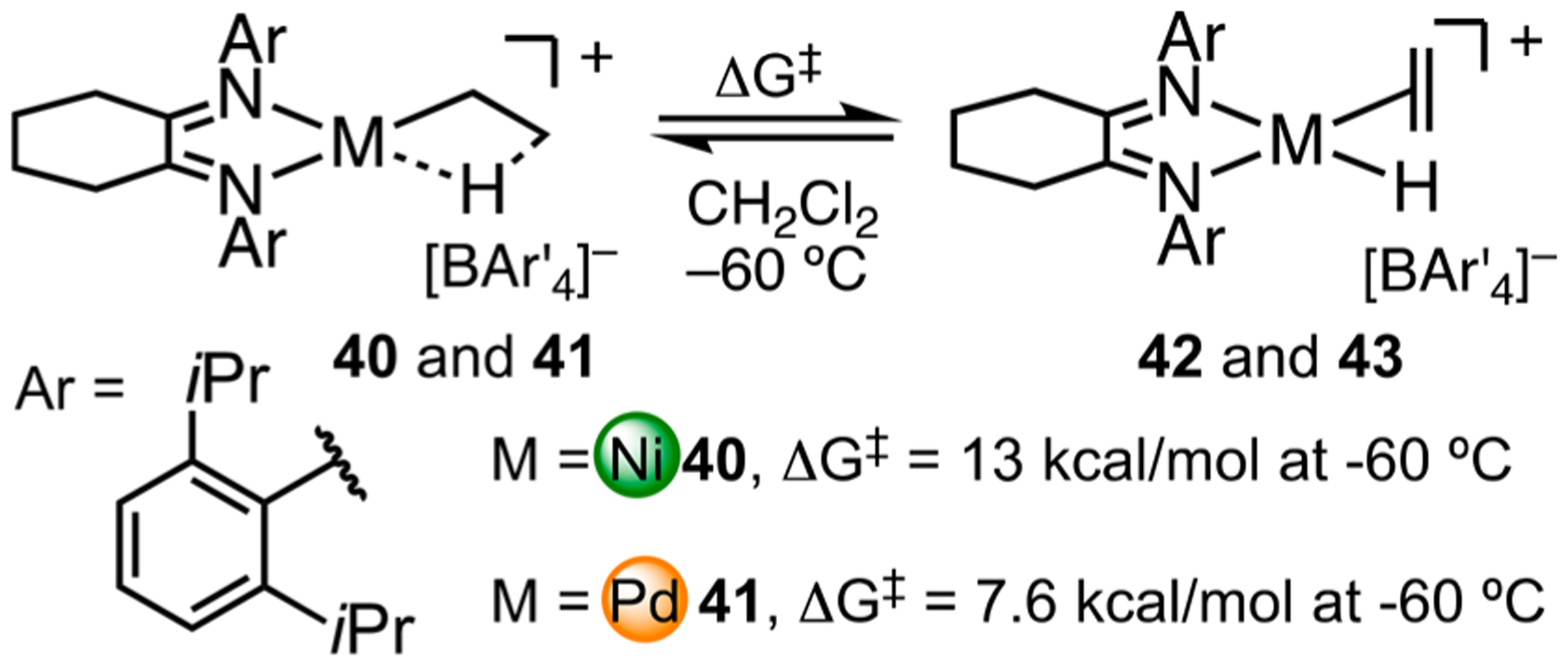
Kinetic Barriers for β-H Elimination
CONCLUSION
We investigated the fundamental properties and the consequent reactivity of nickel catalysts in various aspects. The single-electron redox pathways led us to develop an asymmetric 1,2-diarylation reaction of alkenes via a radical pathway. Mechanistic studies of the reductive 1,2-difunctionalization reaction of alkenes shed light on the mechanistic origin for selectivity in cross-electrophile coupling reactions. C sp2 electrophiles are activated by Ni(I)–Br via two-electron pathways, whereas C sp3 electrophiles are activated by Ni(I)–Ar via radical processes. Activation of the C sp3 electrophile by (Xantphos)Ni(I) has been characterized to proceed through a concerted halogen atom abstraction pathway. The low redox potentials of Ni catalysts allowed us to develop a trans-selective reductive diene coupling, going through a two-electron pathway mediated by a Ni(I)/Ni(III) cycle. The tendency of Ni to undergo single-electron pathways prompted us to explore dinuclear Ni-mediated C–C, C–halogen, and N–N bond formation processes. These stoichiometric studies contribute to fundamental understanding of metal–metal bonding. Finally, we obtained the X-ray crystal structures of the long-sought-after Ni and Pd β-agostic complexes, which help rationalize slower β-H elimination for Ni complexes.
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation (CHE-1654483) and the National Institutes of Health (R01 GM127778). T.D. is a recipient of the Alfred P. Sloan Research Fellowship (FG-2018-10354) and the Camille-Dreyfus Teacher-Scholar Award (TC-19-019).
Biographies
Justin B. Diccianni received his B.S. in chemistry from Stony Brook University in 2014 and his Ph.D. in chemistry from NYU in 2019 under the supervision of Prof. Tianning Diao. His thesis is entitled “Synthetic and Mechanistic Studies of Nickel-Mediated Reactions.” Justin is now a medicinal chemist at Janssen.
Qiao Lin received her B.S. in chemistry from Sichuan University in 2016. She is currently a graduate student in Professor Diao’s group. Her research focuses on understanding the mechanisms of Ni-catalyzed cross-coupling reactions.
Tianning Diao received her Ph.D. in chemistry from the University of Wisconsin–Madison in 2012 under the supervision of Prof. Shannon Stahl. She conducted her postdoctoral research with Prof. Paul Chirik at Princeton University. In 2014, she joined the faculty of NYU, where she is currently an Assistant Professor of Chemistry.
Footnotes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.accounts.0c00032
REFERENCES
- (1).(a) Tasker SZ; Standley EA; Jamison TF Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fu GC Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes. ACS Cent. Sci 2017, 3, 692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Lin C-Y; Power PP Complexes of Ni(I): a “rare” oxidation state of growing importance. Chem. Soc. Rev 2017, 46, 5347–5399. [DOI] [PubMed] [Google Scholar]; (aa) Zheng B; Tang F; Luo J; Schultz JW; Rath NP; Mirica LM Organometallic Nickel(III) Complexes Relevant to Cross-Coupling and Carbon–Heteroatom Bond Formation Reactions. J. Am. Chem. Soc 2014, 136, 6499–6504. [DOI] [PubMed] [Google Scholar]; (c) Camasso NM; Sanford MS Design, synthesis, and carbon-heteroatom coupling reactions of organometallic nickel(IV) complexes. Science 2015, 347, 1218–1220. [DOI] [PubMed] [Google Scholar]
- (3). Dinuclear Pd(III) complexes are common and are stabilized by Pd–Pd bonds, giving closed-shell electron configurations. For an example, see ref 58.
- (4).Poli R; Cacelli I Orbital Splitting and Pairing Energy in Open-Shell Organometallics: A Study of Two Families of 16-Electron Complexes [Cp2M] (M = Cr, Mo, W) and [CpM(PH3)] (M = Co, Rh, Ir). Eur. J. Inorg. Chem 2005, 2005, 2324–2331. [Google Scholar]
- (5).Diccianni JB; Katigbak J; Hu C; Diao T Mechanistic Characterization of (Xantphos)Ni(I)-Mediated Alkyl Bromide Activation: Oxidative Addition, Electron Transfer, or Halogen-Atom Abstraction. J. Am. Chem. Soc 2019, 141, 1788–1796. [DOI] [PubMed] [Google Scholar]
- (6).Lide DR CRC handbook of chemistry and physics, 87th ed.; CRC Press: 2008. [Google Scholar]
- (7).Batsanov SS Van der Waals Radii of Elements. Inorg. Mater 2001, 37, 871–885. [Google Scholar]
- (8).Mann JB; Meek TL; Knight ET; Capitani JF; Allen LC Configuration Energies of the d-Block Elements. J. Am. Chem. Soc 2000, 122, 5132–5137. [Google Scholar]
- (9).Rosen BM; Quasdorf KW; Wilson DA; Zhang N; Resmerita A-M; Garg NK; Percec V Nickel-Catalyzed Cross-Couplings Involving Carbon-Oxygen Bonds. Chem. Rev 2011, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Weires NA; Baker EL; Garg NK Nickel-catalysed Suzuki–Miyaura coupling of amides. Nat. Chem 2016, 8, 75. [DOI] [PubMed] [Google Scholar]
- (11).Leatherman MD; Svejda SA; Johnson LK; Brookhart M Mechanistic Studies of Nickel(II) Alkyl Agostic Cations and Alkyl Ethylene Complexes: Investigations of Chain Propagation and Isomerization in (α-diimine)Ni(II)-Catalyzed Ethylene Polymerization. J. Am. Chem. Soc 2003, 125, 3068–3081. [DOI] [PubMed] [Google Scholar]
- (12).Ashimori A; Bachand B; Calter MA; Govek SP; Overman LE; Poon DJ Catalytic Asymmetric Synthesis of Quaternary Carbon Centers. Exploratory Studies of Intramolecular Heck Reactions of (Z)-α,β-Unsaturated Anilides and Mechanistic Investigations of Asymmetric Heck Reactions Proceeding via Neutral Intermediates. J. Am. Chem. Soc 1998, 120, 6488–6499. [Google Scholar]
- (13).(a) Chen Y-G; Shuai B; Xu X-T; Li Y-Q; Yang Q-L; Qiu H; Zhang K; Fang P; Mei T-S Nickel-catalyzed Enantioselective Hydroarylation and Hydroalkenylation of Styrenes. J. Am. Chem. Soc 2019, 141, 3395–3399. [DOI] [PubMed] [Google Scholar]; (b) Shevick SL; Obradors C; Shenvi RA Mechanistic Interrogation of Co/Ni-Dual Catalyzed Hydroarylation. J. Am. Chem. Soc 2018, 140, 12056–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS A general alkyl-alkyl cross-coupling enabled by redox-active esters and alkylzinc reagents. Science 2016, 352, 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Dhungana RK; Kc S; Basnet P; Giri R Transition Metal-Catalyzed Dicarbofunctionalization of Unactivated Olefins. Chem. Rec 2018, 18, 1314–1340. [DOI] [PubMed] [Google Scholar]
- (16).Nguyen J; Chong A; Lalic G Nickel-catalyzed anti-Markovnikov hydroarylation of alkenes. Chem. Sci 2019, 10, 3231–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).García-Domínguez A; Li Z; Nevado C Nickel-Catalyzed Reductive Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 2017, 139, 6835–6838. [DOI] [PubMed] [Google Scholar]
- (18).(a) Knappke CEI; Grupe S; Gärtner D; Corpet M; Gosmini C; Jacobi von Wangelin A Reductive Cross-Coupling Reactions between Two Electrophiles. Chem. - Eur. J 2014, 20, 6828–6842. [DOI] [PubMed] [Google Scholar]; (b) Everson DA; Weix DJ Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem 2014, 79, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Weix DJ Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res 2015, 48, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gu J; Wang X; Xue W; Gong H Nickel-catalyzed reductive coupling of alkyl halides with other electrophiles: concept and mechanistic considerations. Org. Chem. Front 2015, 2, 1411–1421. [Google Scholar]
- (19).Werner EW; Mei T-S; Burckle AJ; Sigman MS Enantioselective Heck Arylations of Acyclic Alkenyl Alcohols Using a Redox-Relay Strategy. Science 2012, 338, 1455–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cong H; Fu GC Catalytic Enantioselective Cyclization/Cross-Coupling with Alkyl Electrophiles. J. Am. Chem. Soc 2014, 136, 3788–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Wang K; Ding Z; Zhou Z; Kong W Ni-Catalyzed Enantioselective Reductive Diarylation of Activated Alkenes by Domino Cyclization/Cross-Coupling. J. Am. Chem. Soc 2018, 140, 12364–12368. [DOI] [PubMed] [Google Scholar]; (b) Jin Y; Wang C Nickel-Catalyzed Asymmetric Reductive Arylalkylation of Unactivated Alkenes. Angew. Chem., Int. Ed 2019, 58, 6722–6726. [DOI] [PubMed] [Google Scholar]
- (22).Wang F; Chen P; Liu G Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations. Acc. Chem. Res 2018, 51, 2036–2046. [DOI] [PubMed] [Google Scholar]
- (23).(a) Qin X; Lee MWY; Zhou JS Nickel-Catalyzed Asymmetric Reductive Heck Cyclization of Aryl Halides to Afford Indolines. Angew. Chem., Int. Ed 2017, 56, 12723–12726. [DOI] [PubMed] [Google Scholar]; (b) Tian Z-X; Qiao J-B; Xu G-L; Pang X; Qi L; Ma W-Y; Zhao Z-Z; Duan J; Du Y-F; Su P; Liu X-Y; Shu X-Z Highly Enantioselective Cross-Electrophile Aryl-Alkenylation of Unactivated Alkenes. J. Am. Chem. Soc 2019, 141, 7637–7643. [DOI] [PubMed] [Google Scholar]
- (24).(a) Wang Z; Yin H; Fu GC Catalytic enantioconvergent coupling of secondary and tertiary electrophiles with olefins. Nature 2018, 563, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhou F; Zhang Y; Xu X; Zhu S NiH-Catalyzed Remote Asymmetric Hydroalkylation of Alkenes with Racemic α-Bromo Amides. Angew. Chem., Int. Ed 2019, 58, 1754–1758. [DOI] [PubMed] [Google Scholar]
- (25).Tolstoy P; Engman M; Paptchikhine A; Bergquist J; Church TL; Leung AWM; Andersson PG Iridium-Catalyzed Asymmetric Hydrogenation Yielding Chiral Diarylmethines with Weakly Coordinating or Noncoordinating Substituents. J. Am. Chem. Soc 2009, 131, 8855–8860. [DOI] [PubMed] [Google Scholar]
- (26).Do H-Q; Chandrashekar ERR; Fu GC Nickel/Bis(oxazoline)-Catalyzed Asymmetric Negishi Arylations of Racemic Secondary Benzylic Electrophiles to Generate Enantioenriched 1,1-Diarylalkanes. J. Am. Chem. Soc 2013, 135, 16288–16291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Anthony D; Lin Q; Baudet J; Diao T Nickel-Catalyzed Asymmetric Reductive Diarylation of Vinylarenes. Angew. Chem., Int. Ed 2019, 58, 3198–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Isrow D; Captain B Synthesis and Reactivity of a Transition Metal Complex Containing Exclusively TEMPO Ligands: Ni(η2-TEMPO)2. Inorg. Chem 2011, 50, 5864–5866. [DOI] [PubMed] [Google Scholar]
- (29).Gutierrez O; Tellis JC; Primer DN; Molander GA; Kozlowski MC Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereo-convergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc 2015, 137, 4896–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).(a) Yu X; Yang T; Wang S; Xu H; Gong H Nickel-Catalyzed Reductive Cross-Coupling of Unactivated Alkyl Halides. Org. Lett 2011, 13, 2138–2141. [DOI] [PubMed] [Google Scholar]; (b) Peng Y; Xiao J; Xu X-B; Duan S-M; Ren L; Shao Y-L; Wang Y-W Stereospecific Synthesis of Tetrahydronaphtho[2,3-b]furans Enabled by a Nickel-Promoted Tandem Reductive Cyclization. Org. Lett 2016, 18, 5170–5173. [DOI] [PubMed] [Google Scholar]
- (31).Kuang Y; Wang X; Anthony D; Diao T Ni-catalyzed two-component reductive dicarbofunctionalization of alkenes via radical cyclization. Chem. Commun 2018, 54, 2558–2561. [DOI] [PubMed] [Google Scholar]
- (32).Biswas S; Weix DJ Mechanism and selectivity in nickel-catalyzed cross-electrophile coupling of aryl halides with alkyl halides. J. Am. Chem. Soc 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).(a) Dai Y; Wu F; Zang Z; You H; Gong H Ni-Catalyzed Reductive Allylation of Unactivated Alkyl Halides with Allylic Carbonates. Chem. - Eur. J 2012, 18, 808–812. [DOI] [PubMed] [Google Scholar]; (b) Cherney AH; Kadunce NT; Reisman SE Catalytic Asymmetric Reductive Acyl Cross-Coupling: Synthesis of Enantioenriched Acyclic α,α-Disubstituted Ketones. J. Am. Chem. Soc 2013, 135, 7442–7445. [DOI] [PubMed] [Google Scholar]
- (34).Lin Q; Diao T Mechanism of Ni-Catalyzed Reductive 1,2-Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc 2019, 141, 17937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).(a) Everson DA; Jones BA; Weix DJ Replacing Conventional Carbon Nucleophiles with Electrophiles: Nickel-Catalyzed Reductive Alkylation of Aryl Bromides and Chlorides. J. Am. Chem. Soc 2012, 134, 6146–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang X; Ma G; Peng Y; Pitsch CE; Moll BJ; Ly TD; Wang X; Gong H Ni-Catalyzed Reductive Coupling of Electron-Rich Aryl Iodides with Tertiary Alkyl Halides. J. Am. Chem. Soc 2018, 140, 14490–14497. [DOI] [PubMed] [Google Scholar]
- (36).(a) Jones GD; Martin JL; McFarland C; Allen OR; Hall RE; Haley AD; Brandon RJ; Konovalova T; Desrochers PJ; Pulay P; Vicic DA Ligand Redox Effects in the Synthesis, Electronic Structure, and Reactivity of an Alkyl-Alkyl Cross-Coupling Catalyst. J. Am. Chem. Soc 2006, 128, 13175–13183. [DOI] [PubMed] [Google Scholar]; (b) Cornella J; Gómez-Bengoa E; Martin R Combined Experimental and Theoretical Study on the Reductive Cleavage of Inert C–O Bonds with Silanes: Ruling out a Classical Ni(0)/Ni(II) Catalytic Couple and Evidence for Ni(I) Intermediates. J. Am. Chem. Soc 2013, 135, 1997–2009. [DOI] [PubMed] [Google Scholar]; (c) Breitenfeld J; Ruiz J; Wodrich MD; Hu X Bimetallic oxidative addition involving radical intermediates in nickel-catalyzed alkyl-alkyl Kumada coupling reactions. J. Am. Chem. Soc 2013, 135, 12004–12012. [DOI] [PubMed] [Google Scholar]; (d) Schley ND; Fu GC Nickel-catalyzed Negishi arylations of propargylic bromides: a mechanistic investigation. J. Am. Chem. Soc 2014, 136, 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mohadjer Beromi M; Nova A; Balcells D; Brasacchio AM; Brudvig GW; Guard LM; Hazari N; Vinyard DJ Mechanistic Study of an Improved Ni Precatalyst for Suzuki-Miyaura Reactions of Aryl Sulfamates: Understanding the Role of Ni(I) Species. J. Am. Chem. Soc 2017, 139, 922–936. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Manzoor A; Wienefeld P; Baird MC; Budzelaar PHM Catalysis of Cross-Coupling and Homocoupling Reactions of Aryl Halides Utilizing Ni(0), Ni(I), and Ni(II) Precursors; Ni(0) Compounds as the Probable Catalytic Species but Ni(I) Compounds as Intermediates and Products. Organometallics 2017, 36, 3508–3519. [Google Scholar]
- (37).Lucas EL; Jarvo ER Stereospecific and stereoconvergent cross-couplings between alkyl electrophiles. Nat. Rev. Chem 2017, 1, 0065. [Google Scholar]
- (38).Milligan JA; Phelan JP; Badir SO; Molander GA Alkyl Carbon–Carbon Bond Formation by Nickel/Photoredox Cross-Coupling. Angew. Chem., Int. Ed 2019, 58, 6152–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).(a) Kehoe R; Mahadevan M; Manzoor A; McMurray G; Wienefeld P; Baird MC; Budzelaar PHM Reactions of the Ni(0) Compound Ni(PPh3)4 with Unactivated Alkyl Halides: Oxidative Addition Reactions Involving Radical Processes and Nickel(I) Intermediates. Organometallics 2018, 37, 2450–2467. [Google Scholar]; (b) Yoo C; Ajitha MJ; Jung Y; Lee Y Mechanistic Study on C–C Bond Formation of a Nickel(I) Monocarbonyl Species with Alkyl Iodides: Experimental and Computational Investigations. Organometallics 2015, 34, 4305–4311. [Google Scholar]
- (40).Mohadjer Beromi M; Brudvig GW; Hazari N; Lant HMC; Mercado BQ Synthesis and Reactivity of Paramagnetic Nickel Polypyridyl Complexes Relevant to C(sp2)–C(sp3)Coupling Reactions. Angew. Chem., Int. Ed 2019, 58, 6094–6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhang K; Conda-Sheridan M; Cooke R, S.; Louie J NHeterocyclic Carbene Bound Nickel(I) Complexes and Their Roles in Catalysis. Organometallics 2011, 30, 2546–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Diccianni JB; Chin M; Diao T Synthesis of lactate derivatives via reductive radical addition to α-oxyacrylates. Tetrahedron 2019, 75, 4180–4185. [Google Scholar]
- (43).Van Hecke GR; Horrocks WD Approximate Force Constants for Tetrahedral Metal Carbonyls and Nitrosyls. Inorg. Chem 1966, 5, 1960–1968. [Google Scholar]
- (44).Tortajada A; Juliá-Hernández F; Börjesson M; Moragas T; Martin R Transition-Metal-Catalyzed Carboxylation Reactions with Carbon Dioxide. Angew. Chem., Int. Ed 2018, 57, 15948–15982. [DOI] [PubMed] [Google Scholar]
- (45).Sayyed FB; Tsuji Y; Sakaki S The crucial role of a Ni(I) intermediate in Ni-catalyzed carboxylation of aryl chloride with CO2: a theoretical study. Chem. Commun 2013, 49, 10715–10717. [DOI] [PubMed] [Google Scholar]
- (46).(a) Schmeier TJ; Hazari N; Incarvito CD; Raskatov JA Exploring the reactions of CO2 with PCP supported nickel complexes. Chem. Commun 2011, 47, 1824–1826. [DOI] [PubMed] [Google Scholar]; (b) Jonasson KJ; Wendt OF Synthesis and Characterization of a Family of POCOP Pincer Complexes with Nickel: Reactivity Towards CO2 and Phenylacetylene. Chem. - Eur. J 2014, 20, 11894–11902. [DOI] [PubMed] [Google Scholar]; (c) Charboneau DJ; Brudvig GW; Hazari N; Lant HMC; Saydjari AK Development of an Improved System for the Carboxylation of Aryl Halides through Mechanistic Studies. ACS Catal 2019, 9, 3228–3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Diccianni JB; Hu CT; Diao T Insertion of CO2 Mediated by a (Xantphos)NiI–Alkyl Species. Angew. Chem., Int. Ed 2019, 58, 13865–13868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Widenhoefer RA Synthetic and Mechanistic Studies of the Cycloisomerization and Cyclization/Hydrosilylation of Functionalized Dienes Catalyzed by Cationic Palladium(II) Complexes. Acc. Chem. Res 2002, 35, 905–913. [DOI] [PubMed] [Google Scholar]
- (49).Blakemore DC; Bryans JS; Carnell P; Field MJ; Kinsella N; Kinsora JK; Meltzer LT; Osborne SA; Thompson LR; Williams SC Synthesis and in vivo evaluation of 3,4-disubstituted gababutins. Bioorg. Med. Chem. Lett 2010, 20, 248–251. [DOI] [PubMed] [Google Scholar]
- (50).Blizzard TA; DiNinno F; Morgan JD; Chen HY; Wu JY; Kim S; Chan W; Birzin ET; Yang YT; Pai L-Y; Fitzgerald PMD; Sharma N; Li Y; Zhang Z; Hayes EC; DaSilva CA; Tang W; Rohrer SP; Schaeffer JM; Hammond ML Estrogen receptor ligands. Part 9: Dihydrobenzoxathiin SERAMs with alkyl substituted pyrrolidine side chains and linkers. Bioorg. Med. Chem. Lett 2005, 15, 107–113. [DOI] [PubMed] [Google Scholar]
- (51).Kuang Y; Anthony D; Katigbak J; Marrucci F; Humagain S; Diao T Ni(I)-Catalyzed Reductive Cyclization of 1,6-Dienes: Mechanism-Controlled trans-Selectivity. Chem 2017, 3, 268–280. [Google Scholar]
- (52).(a) Lo JC; Gui J; Yabe Y; Pan C-M; Baran PS Functionalized olefin cross-coupling to construct carbon–carbon bonds. Nature 2014, 516, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crossley SWM; Barabé F; Shenvi RA Simple, Chemoselective, Catalytic Olefin Isomerization. J. Am. Chem. Soc 2014, 136, 16788–16791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Weiller BH; Wasserman EP; Bergman RG; Moore CB; Pimentel GC Time-resolved IR spectroscopy in liquid rare gases: direct rate measurement of an intermolecular alkane carbon-hydrogen oxidative addition reaction. J. Am. Chem. Soc 1989, 111, 8288–8290. [Google Scholar]
- (54).Zhang X-X; Wayland BB Rhodium(II) Porphyrin Bimetalloradical Complexes: Preparation and Enhanced Reactivity with CH4 and H2. J. Am. Chem. Soc 1994, 116, 7897–7898. [Google Scholar]
- (55).Xu H; Diccianni JB; Katigbak J; Hu C; Zhang Y; Diao T Bimetallic C–C Bond-Forming Reductive Elimination from Nickel. J. Am. Chem. Soc 2016, 138, 4779–4786. [DOI] [PubMed] [Google Scholar]
- (56).Lindahl PA Metal–metal bonds in biology. J. Inorg. Biochem 2012, 106, 172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Zhou Y-Y; Uyeda C Catalytic reductive [4 + 1]-cycloadditions of vinylidenes and dienes. Science 2019, 363, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Powers IG; Uyeda C Metal–Metal Bonds in Catalysis. ACS Catal 2017, 7, 936–958. [Google Scholar]
- (59).Powers DC; Ritter T Bimetallic Pd(III) complexes in palladium-catalysed carbon–heteroatom bond formation. Nat. Chem 2009, 1, 302. [DOI] [PubMed] [Google Scholar]
- (60).Diccianni JB; Hu C; Diao T Binuclear, High-Valent Nickel Complexes: Ni-Ni Bonds in Aryl–Halogen Bond Formation. Angew. Chem., Int. Ed 2017, 56, 3635–3639. [DOI] [PubMed] [Google Scholar]
- (61).Berry JF; Cotton FA; Daniels LM; Murillo CA; Wang X Oxidation of Ni3(dpa)4Cl2 and Cu3(dpa)4Cl2: Nickel-Nickel Bonding Interaction, but No Copper-Copper Bonds. Inorg. Chem 2003, 42, 2418–2427. [DOI] [PubMed] [Google Scholar]
- (62).Cotton FA; Matusz M; Poli R; Feng X Dinuclear formamidinato complexes of nickel and palladium. J. Am. Chem. Soc 1988, 110, 1144–1154. [Google Scholar]
- (63).Diccianni JB; Hu C; Diao T N-N Bond Forming Reductive Elimination via a Mixed-Valent Nickel(II)–Nickel(III) Intermediate. Angew. Chem., Int. Ed 2016, 55, 7534–7538. [DOI] [PubMed] [Google Scholar]
- (64).Cotton FA; Gu J; Murillo CA; Timmons DJ The First Dinuclear Complex of Palladium(III). J. Am. Chem. Soc 1998, 120, 13280–13281. [Google Scholar]
- (65).Berry JF; Bothe E; Cotton FA; Ibragimov SA; Murillo CA; Villagrán D; Wang X Metal-Metal Bonding in Mixed Valence Ni25+ Complexes and Spectroscopic Evidence for a Ni26+ Species. Inorg. Chem 2006, 45, 4396–4406. [DOI] [PubMed] [Google Scholar]
- (66).Berry JF Two-Center/Three-Electron Sigma Half-Bonds in Main Group and Transition Metal Chemistry. Acc. Chem. Res 2016, 49, 27–34. [DOI] [PubMed] [Google Scholar]
- (67).Brookhart M; Green MLH; Parkin G Agostic interactions in transition metal compounds. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 6908–6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Ittel SD; Johnson LK; Brookhart M Late-Metal Catalysts for Ethylene Homo- and Copolymerization. Chem. Rev 2000, 100, 1169–1204. [DOI] [PubMed] [Google Scholar]
- (69).Kogut E; Zeller A; Waren TH; Strassner TJ Am. Chem. Soc 2004, 126 (38), 11984–11994. [DOI] [PubMed] [Google Scholar]
- (70).(a) Xu H; White PB; Hu C; Diao T Structure and Isotope Effects of the β-H Agostic (α-Diimine)Nickel Cation as a Polymerization Intermediate. Angew. Chem., Int. Ed 2017, 56, 1535–1538. [DOI] [PubMed] [Google Scholar]; (b) Xu H; Hu CT; Wang X; Diao T Structural Characterization of β-Agostic Bonds in Pd-Catalyzed Polymerization. Organometallics 2017, 36, 4099–4102. [Google Scholar]