

Abstract
Nuclear protein 1 (NUPR1) also known as p8 and candidate of metastasis 1 (COM1) functions as a transcriptional regulator, and plays a role in cell cycle, DNA damage response, apoptosis, autophagy, and chromatin remodeling in response to various cellular stressors. Since it was first suggested to contribute to cancer development and progression in 1999, a number of studies have sought to reveal its function. However, NUPR1 and its biological relevance in cancer has proven difficult to pinpoint. Based on evidence of NUPR1 expression in cancer, its function extends from carcinogenesis and tumorigenesis to metastasis and chemotherapeutic resistance. A tumor suppressive function of NUPR1 has also been documented in multiple cancers. By and large, literature involving NUPR1 and cancer is confined to pancreatic and breast cancers, yet significant progress has been made with respect to NUPR1 expression and its function in lung, colorectal, blood, and prostate cancers, among others. Recent evidence strongly supports the notion that NUPR1 is key in chemotherapeutic resistance by mediating both anti-apoptotic activity and autophagy when challenged with anti-cancer compounds. Therefore, it is of significant importance to understand the broad range of molecular functions directed by NUPR1. In this review, NUPR1 expression and its role in breast, lung, and colorectal cancer development and progression will be addressed.
Keywords: Chemotherapeutic resistance, Stress response, Breast cancer, Lung cancer, Colorectal cancer
1. Basic Properties of NUPR1
Nuclear protein 1 (NUPR1) is a stress-response gene upregulated by many biological and chemical stressors such as lipopolysaccharides(Y. F. Jiang, Vaccaro, Fiedler, Calvo, & Iovanna, 1999), TNFα(Goruppi, Patten, Force, & Kyriakis, 2007), amino acid deprivation(Averous et al., 2011), cannabinoids(Carracedo et al., 2006), and heavy metals such as hexavalent chromium (Cr(VI))(D. Chen et al., 2016). NUPR1 was first described by Mallo et al. (1997) in the acute phase of pancreatitis, and was subsequently found to encode an 82 amino-acid monomeric protein (8.8 kDa) that does not share significant homology with other proteins(Mallo et al., 1997; Santofimia-Castaño et al., 2017; Valacco et al., 2006). There are two isoforms of NUPR1; the longer NUPR1 isoform is 100 amino acids in length. The additional 18-amino-acid difference between isoform 1 (isoform b) and isoform 2 (isoform a) corresponds to a flexible loop structure with an unknown functional role to date(Urrutia et al., 2014). NUPR1 also has a paralogue, NUPR1L (NUPR2), which together with NUPR1, have been suggested to encode a family of transcriptional regulators(Urrutia et al., 2014). NUPR1 was initially suggested to function as a transcription factor because of a basic helix-loop-helix structure present at its C-terminal, slight homology with most homeodomains, and potential for phosphorylation by various kinases(Mallo et al., 1997). In addition, NUPR1 binds DNA weakly as shown by electrophoretic mobility shift assay, however, when phosphorylated by protein kinase A, DNA binding properties are significantly enhanced(Encinar et al., 2001). Aside from its primary role as a transcriptional regulator, NUPR1 has been reported to take part in cell-cycle regulation(Malicet, Hoffmeister, et al., 2006), apoptosis regulation(Malicet, Giroux, et al., 2006), DNA-damage response(Aguado-Llera et al., 2013; Gironella et al., 2009), and autophagy(Mu et al., 2018).
At its N-terminal, NUPR1 contains a PEST (Pro/Glu/Ser/Thr-rich) sequence typical of polypeptides subject to modification and degradation by the ubiquitin (Ub)/proteasome system(Goruppi & Kyriakis, 2004). Regulation of NUPR1 stability by the Ub/proteasome system was demonstrated in vitro(Goruppi & Kyriakis, 2004; Kinyamu, Bennett, Bushel, & Archer, 2020), and its proteasomal degradation was further influenced by phosphorylation(Goruppi & Kyriakis, 2004). NUPR1 is also subject to sumoylation(Goruppi et al., 2007). Based on linear motif analyses, NUPR1 was predicted to contain several acetylation and methylation sites, many of which reside within the DNA-binding domain and nuclear location sequence (NLS)(Urrutia et al., 2014). In fact, p300 transcriptional co-activator was shown to specifically acetylate NUPR1, and cytoplasmic accumulation of NUPR1 was reported following inhibition of deacetylation by Trichostatin A, suggesting that acetylation may regulate NUPR1 localization(Hoffmeister et al., 2002; Valacco et al., 2006). NUPR1 contains a canonical bipartite domain of positively charged amino acids, involving protein residues 64–78: typical of a NLS(Urrutia et al., 2014; Valacco et al., 2006). This NLS was determined to be necessary and sufficient for nuclear localization of NUPR1(Valacco et al., 2006). Worth note is that NUPR1 was determined to present nuclear localization in sub-confluent cells, but localizes throughout the whole cell in those grown to high density(Valacco et al., 2006). Two hot-spot regions in NUPR1 involved in ligand binding have also been identified using in silico approaches and subsequently confirmed in vitro(Neira et al., 2017). One region including resides Leu29 and Ala33 (and marginally Gly38) along with a second region including Thr68 (and marginally His61), were determined to have a high probability of ligand binding under favorable conditions through disorder-to-order transition(Neira et al., 2017). However, upon binding both small organic molecules and biomolecules, NUPR1 was determined to remain disordered(Aguado-Llera et al., 2013; Encinar et al., 2001; Malicet, Giroux, et al., 2006; Neira et al., 2017; Neira et al., 2019; Santofimia-Castaño et al., 2017).
NUPR1 is an intrinsically disordered protein (IDP) as it lacks a distinct well-defined secondary and tertiary structure(Encinar et al., 2001). The intrinsically disordered nature of NUPR1 permits the binding of multiple structurally diverse ligands, while retaining some degree of specificity; characteristic of many IDPs(Olsen, Teilum, & Kragelund, 2017). A number of different binding paradigms between IDPs and their ligands have been described (reviewed in (Olsen et al., 2017)). The inability of NUPR1 to undergo ordering following binding to protein partners and small molecules demonstrates that NUPR1 functions primarily through ‘fuzzy’ binding, as suggested by Iovanna and colleagues(Neira et al., 2017). As implied, ‘fuzzy’ ligand-protein binding is ambiguous and dynamic, allowing for ligand-protein complexes to occupy several conformational states(Olsen et al., 2017). This immutable characteristic of NUPR1 may be fundamental for its function, and points toward a far-reaching role in transcriptional regulation and a broad stress response.
NUPR1 has been implicated in a number of cancers including, but not limited to, pancreatic, breast, lung, colorectal, prostate, brain, thyroid, and pituitary (reviewed in (Cano, Hamidi, Sandi, & Iovanna, 2011; Chowdhury, Samant, Fodstad, & Shevde, 2009)). The overwhelming majority of literature pertaining to NUPR1 expression in cancer focuses on pancreatic cancers. However, the relationship between NUPR1 and cancers other than pancreatic is covered in far less detail. The purpose of this review is to provide an update of NUPR1 expression in breast, lung and colorectal cancers, and to elucidate the function of NUPR1 in the context of these particular cancers.
2. NUPR1 Expression in Breast Cancer
In 1999 Ree et al. first identified NUPR1 as highly expressed using an in vivo breast cancer metastasis model and, in vitro, using a panel of metastatic human breast cancer cell lines (Table 1.)(Ree et al., 1999). In tumorigenic, locally aggressive, and aggressive/metastatic panel of breast cancer cell lines, both NUPR1 mRNA and NUPR1 protein levels were determined to be elevated(Clark et al., 2008). In highly metastatic breast carcinoma MDA-LM2–4175 and MDA-BoM-1833 cells derived from lung and bone metastases, respectively, NUPR1 expression was elevated, but not in MDA-BrM-831 cells derived brain metastases(Fish et al., 2018). Interestingly, early passage poorly tumorigenic MCF-7 E cells do not express NUPR1, but late passage tumorigenic MCF-7 L cells do express low levels of NUPR1(Ree et al., 2002). In MCF-7/LCC1 and MCF-7/LCC2, both estrogen-independent derivates of MCF-7 cells that reflect the phenotypes of carcinoma cells observed during the clinical progression of breast cancer, NUPR1 is constitutively expressed(Ree et al., 1999). This prompted the authors to suggest NUPR1 expression might be selected for in long-term culture, and this also provides evidence that NUPR1 expression may correspond with the progression of breast cancers(Ree et al., 2002). NUPR1 was also reported to be induced following treatment with EGF and insulin, which indicates that NUPR1 may be regulated by growth stimulating factors in the breast tumor microenvironment(Ree et al., 2002).
Table 1.
NUPR1 Expression Reported in Breast Cancer Cell Lines and Tissues.
| Cell line/Cancer tissue | Expression (+/−) | Hormone receptor and HER2 status (ER, PR, HER2) | Description | Reference |
|---|---|---|---|---|
| MA-11 | + | ER−, PR−, HER2−(Dai, Cheng, Bai, & Li, 2017) | Adenocarcinoma, derived from metastatic site: bone marrow(Ree et al., 2002) | (Ree et al., 2002; Ree et al., 1999) |
| MT-1 | + | ER−, PR−, HER2 uncharacterized(Naundorf et al., 1992) | Contaminated(MacLeod et al., 1999), originally categorized as large-cell, undifferentiated, medullary(Naundorf et al., 1992) | (Ree et al., 1999) |
| MDA-MB-231 | − | ER−, PR−, HER2−(Dai et al., 2017) | Adenocarcinoma, derived from metastatic site: pleural effusion (ATCC® HTB-26) | (Ree et al., 1999) |
| + | (Clark et al., 2008; W. G. Jiang, Davies, et al., 2005; W. G. Jiang, Watkins, et al., 2005) | |||
| MDA-MB-435 | + | ER−, PR−, HER2−(Dai et al., 2017) | Derived from metastatic site: pericardial effusion (ATCC® HTB-131) | (W. G. Jiang, Watkins, et al., 2005; Ree et al., 1999) |
| MCF-7 | − | ER+, PR+, HER2−(Dai et al., 2017) | Adenocarcinoma, derived from metastatic site: pleural effusion (ATCC® HTB-22) | (Bratland et al., 2000; Ree et al., 1999) |
| + | (W. G. Jiang, Davies, et al., 2005; W. G. Jiang, Watkins, et al., 2005) | |||
| MCF7/LCC1 | + | ER+, PR+, HER2−(Brunner et al., 1993) | Parental MCF-7, derived from metastatic site: pleural effusion(Ree et al., 2002) | (Ree et al., 1999) |
| MCF7/LCC2 | + | ER+, PR+. HER2−(Brunner et al., 1993) | Adenocarcinoma, parental MCF7/LCC1, derived from metastatic site: pleural effusion(Ree et al., 2002) | (Bratland et al., 2000; Ree et al., 2002; Ree et al., 1999) |
| Primary breast tumors | + | Uncharacterized | Infiltrating ductal and lobular or medullar carcinoma | (Ree et al., 2000) |
| PM-1 | + | Uncharacterized | Adenocarcinoma, derived from metastatic site: pleural effusion | (Ree et al., 2002) |
| MCF-7 E | − | ER+, PR+, HER2−(Dai et al., 2017) | MCF-7 early passage breast cancer cells (~150 passages) | |
| MCF-7 L | + | MCF-7 late passage breast cancer cells (>500 passages) | ||
| Primary breast tumors | + | Uncharacterized | Non-invasive ductal breast carcinomas | (Ito et al., 2005) |
| Primary breast tumors | +/− | Uncharacterized | Invasive ductal or lobular breast carcinomas | |
| MDA-MB-157 | + | ER−, PR−, HER2−(Dai et al., 2017) | Medullary carcinoma (ATCC® HTB-24) | (W. G. Jiang, Watkins, et al., 2005) |
| MDA-MB-436 | + | ER−, PR−, HER2−(Dai et al., 2017) | Adenocarcinoma, derived from metastatic site: pleural effusion (ATCC® HTB-130) | |
| MDA-MB-435S | + | Not applicable | Previously described as: ductal carcinoma, derived from metastatic site: pleural effusion. Melanoma, melanocyte (ATCC® HTB-129) | |
| BT-474 | + | ER+, PR+, HER2+(Dai et al., 2017) | Ductal carcinoma (ATCC® HTB-20) | |
| BT-549 | + | ER−, PR−, HER2−(Dai et al., 2017) | Ductal carcinoma (ATCC® HTB-122) | |
| ZR-75–1 | + | ER+, PR+, HER2+(Subik et al., 2010) | Ductal carcinoma, derived from metastatic site: ascites (ATCC® CRL-1500) | |
| Primary breast tumors | +/− | Uncharacterized | Ductal, Lobular, Medullary, Tubular, Mucinous | |
| MCF10AT | + | ER+, PR−, HER2−(H Heppner, R Miller, & Malathy Shekhar, 2000) | Parental MCF10A, Tumorigenic | (Clark et al., 2008; Vincent et al., 2012) |
| MCF10DCIS | + | ER−, PR−, HER2+(Shekhar, Kato, Nangia-Makker, & Tait, 2013) | Locally aggressive | (Clark et al., 2008) |
| MCF10CA | + | Uncharacterized | Aggressive/metastatic | |
| MCF10A | − | ER−, PR−, HER2−(Subik et al., 2010) | Spontaneously immortalized epithelial cells | |
| Primary breast tumors | + | Uncharacterized | Uncharacterized | (Vincent et al., 2012) |
| SUM159 (SUM159PT) | − | ER−, PR−, HER2−(Dai et al., 2017) | Inflammatory breast cancer, p53 loss of function missense mutation | |
| MDA-LM2–4175 | + | ER−, PR−, HER2−(Minn et al., 2005) | Derived from metastatic site: pleural effusion, MDA-MB-231 parental | (Fish et al., 2018) |
| MDA-BoM-1833 | + | ER−, PR−, HER2−(Kang et al., 2003) | Derived from metastatic site: bone, MDA-MB-231 parental | |
| MDA-BrM-831 | − | ER−, PR−, HER2−(Bos et al., 2009) | Derived from metastatic site: brain, MDA-MB-231 parental |
In breast tumor tissues, NUPR1 mRNA was significantly upregulated compared to normal breast tissues(Ree, Pacheco, Tvermyr, Fodstad, & Brentani, 2000; Vincent et al., 2012), and when stratified by breast cancer stage, NUPR1 expression was found increased in advanced breast cancers(Fish et al., 2018). However, no associations were reported between NUPR1 expression and breast cancer subtype(Fish et al., 2018), histological and biochemical characteristics or disease parameters including size, presence of vascular infiltrate or necrosis, steroid receptor status, lymph node status, disease stage at diagnosis, or metastatic development, and survival(Ree et al., 2000). Ito et al. (2005) investigated NUPR1 expression in 50 breast cancer cases of which 60% were classified as high for NUPR1 expression(Ito et al., 2005). In this study, when stratified by breast cancer subtype, NUPR1 was determined to be highly expressed in non-invasive ductal carcinomas, and expressed at a low level in 46.5% of invasive ductal or lobular carcinomas(Ito et al., 2005). The remaining invasive ductal or lobular carcinomas (53.5%) showed high expression of NUPR1(Ito et al., 2005). When evaluated by size and stage, NUPR1 expression was significantly decreased in cases with large tumors and advanced stage(Ito et al., 2005). However, no relationship between NUPR1 expression and age, menopause, histological grade, lymph node metastasis, status of steroid receptors and HER2 expression was reported(Ito et al., 2005). Jung et al. (2012) analyzed copy number alterations in 48 early-stage breast cancers, of which 23 recurrently altered regions (RARs) were identified and possess copy number gains in chromosomal regions containing NUPR1 (16p11.2) and ERBB2 (HER2; 17q12)(Jung et al., 2012). These RARs showed a significant association with poor survival, and patients simultaneously positive for both gains had significantly worse prognosis(Jung et al., 2012). In breast cancer gene expression datasets including The Cancer Genome Atlas, Fish et al. (2018) reported a negative association between NUPR1 expression in breast tumors and patient survival, and they also reported an increase in NUPR1 expression in advanced breast cancers(Fish et al., 2018).
Two studies, in particular, provide evidence supporting the notion that NUPR1 may be downregulated in breast cancer and may possess a tumor suppressive function. Immunohistochemical staining showed significantly reduced nuclear staining of NUPR1 in cells obtained from breast tumor tissues compared to normal epithelial cells(W. G. Jiang, Watkins, et al., 2005). Clinicopathological assessment did not reveal a significant correlation between NUPR1 expression and tumor size(W. G. Jiang, Watkins, et al., 2005). In node positive tumors NUPR1 expression was significantly reduced and its expression was inversely related to prognostic index(W. G. Jiang, Watkins, et al., 2005). In addition, over a median 10-year follow up, patients with metastasis, recurrence, and mortality had lower levels of NUPR1 expression, and in both the overall and overall disease-free survival analyses, there was no significant difference in survival and NUPR1 expression(W. G. Jiang, Watkins, et al., 2005). Interestingly, when stratified by ERα status, low levels of NUPR1 expression were associated with shorter survival in both ERα-positive and ERαnegative patients(W. G. Jiang, Watkins, et al., 2005). In ERβ positive tumors, NUPR1 expression was significantly and inversely correlated with overall survival(W. G. Jiang, Watkins, et al., 2005).
The majority of clinical, in vivo, and in vitro studies on breast cancer show that NUPR1 is upregulated and may be associated with breast cancer progression. Little to no relationship between NUPR1 status and clinicopathological features have been reported, except in one instance with non-invasive ductal, invasive ductal, and lobular breast carcinomas, and in large tumors and advanced stage. This suggests that NUPR1 expression in breast cancers is nonexclusive, and its expression is dynamically regulated during cancer development and progression. Since NUPR1 is a stress-response gene, it is also plausible that its expression is regulated by growth stimulating factors in the tumor microenvironment, including but not limited to, EGF and insulin. Clinically, there has yet to be a definitive association between NUPR1 and hormone receptor status, nor has there been strong evidence showing an association between aberrant NUPR1 expression and hormone receptor status in cell lines (Table 1).
2.1. Upstream Regulators of NUPR1 in Breast Cancer
p23 is a co-chaperone protein best known for its role in aiding steroid hormone receptor folding, its expression increases with breast tumor stage, and is upregulated in metastatic cancers(Rehn & Buchner, 2015; Simpson et al., 2010). Simpson et al. (2010) reported that NUPR1 was downregulated in hormone starved MCF-7 cells upon p23 overexpression(Simpson et al., 2010). This same group previously showed that p23-overexpressing MCF-7 cells exhibit increased invasion without affecting estrogen-dependent cell proliferation(Oxelmark et al., 2006). Together, these studies suggest a regulatory relationship between p23 and NUPR1, although this may be context dependent and has not been elaborated.
E2F1 and E2F2 are classified as transcriptional activators, are well characterized as cell cycle regulators, and are associated with many types of cancer(Hollern, Honeysett, Cardiff, & Andrechek, 2014). Elevated expression levels of E2F1 and E2F2 in breast cancer patients were individually associated with shorter times to distant metastasis than those for patients whose tumors exhibited low levels of expression(Hollern et al., 2014). Using a murine tumor virus (PyMT) model of metastatic breast cancer, tumor onset was reported to be significantly accelerated in E2f1−/− mice(Hollern et al., 2014). In addition, loss of either E2f1 or E2f2 significantly reduced metastatic burden, which was attributed to a reduction in the number of circulating tumor cells(Hollern et al., 2014). Using E2F signature gene expression profiles form published ChIP-seq and ChIP-chip data and filtered using gene sets for metastasis available on MSigDB, NUPR1 was identified as a direct E2F target gene(Hollern et al., 2014). Subsequent validation via RT-qPCR showed that Nupr1 was indeed a target gene of E2f1 and E2f2 and was significantly downregulated in E2f1−/−and E2f2−/− murine breast tumors(Hollern et al., 2014). Altogether, these results suggest that E2f1 and E2f2 play important roles in tumor development and progression as well as metastasis, which may involve Nupr1 given its regulation by E2f1 and E2f2. Furthermore, pathways related to TGF-beta and SMAD activation are impacted by the loss of E2F1 and E2F2, and these pathways have previously been linked to NUPR1(García-Montero et al., 2001).
Lastly, NUPR1 regulation in breast cancer has been tied to epigenetics through orphan noncoding RNA (oncRNA) and micro RNA (miRNA) dysregulation (Figure 1.). NUPR1 expression was shown to be influenced by a cancer-specific oncRNA, T3p, which originates from the 3’ end of TERC and derived from aberrant processing of TERC RNA(Fish et al., 2018). T3p was shown to exert its regulatory effects through interaction with the RNA-induced silencing (RISC) complex, specifically AGO2, and concomitantly modulate miRNA activity of miR-10b and miR-387c, both of which target NUPR1 for degradation(Fish et al., 2018). Increased TERC expression and telomerase activity have been referred to as hallmarks of tumor progression, and oncRNAs as described by Fish et. al (2018) may constitute an evolutionary pathway adopted by cancer cells in order to facilitate tumor progression and metastasis(Fish et al., 2018).
Figure 1.
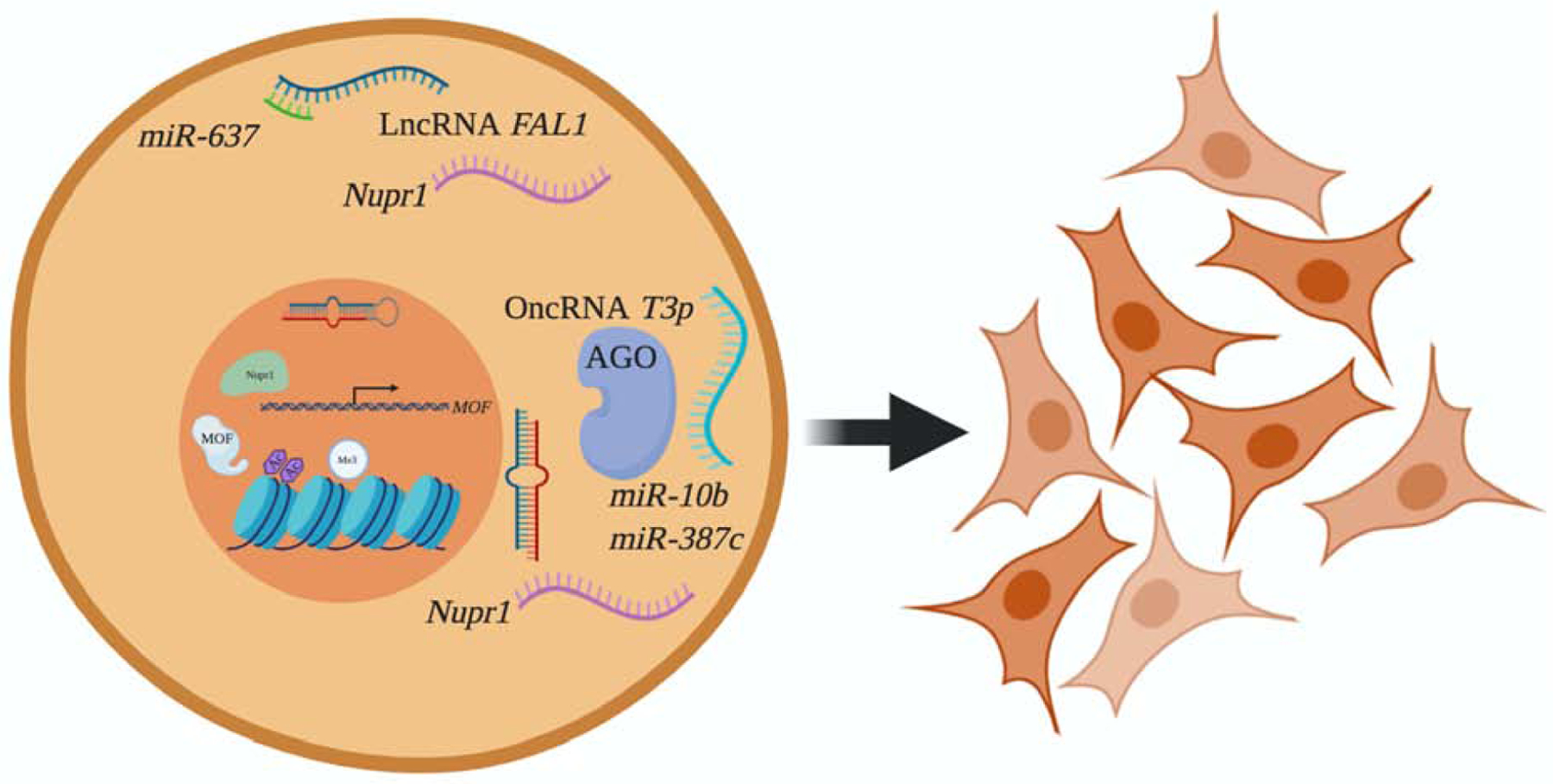
NUPR1 and the Epigenetic Landscape in Breast, Lung, and Colorectal Cancers. LncRNA FAL1 is overexpressed in colorectal cancer, and acts as a ‘sponge’ for miR-637, which negatively regulates NUPR1 expression. Similarly, oncRNA T3p binds to and inhibits miR-10b and miR-387c, which both negatively regulate NUPR1 expression in breast cancer. NUPR1 contributes to Cr(VI)-mediated cell transformation and carcinogenesis in lung cancer by negatively regulating MOF, resulting in reduced levels of H4K16ac and corresponding gene expression. NUPR1 increases H3K4me3, a well-known transcriptional activation mark. Created with BioRender.com.
2.2. Molecular Mechanisms of NUPR1-mediated Chemoresistance and Tumor Suppressive Activity of NUPR1 in Breast Cancer
p21 is considered a cell cycle arrest protein, yet has also been shown to take part in anti-apoptotic signaling. p21 function has been shown to be dependent on its sub-cellular localization such that cytoplasmic p21 enhances cell survival and nuclear p21 acts in a tumor suppressive manner(Vincent et al., 2012). NUPR1 was reported to modulate the cellular localization of p21 by phosphorylation of p21’s NLS in a PI3K/AKT-dependent manner, resulting in cytoplasmic accumulation of p21 in p53-deficient SUM159 triple-negative breast cancer cells(Vincent et al., 2012). In addition, this same study reported that NUPR1 was able to upregulate p21(Vincent et al., 2012). NUPR1’s ability to upregulate p21 was confirmed in immortalized nontumorigenic human breast epithelial MCF10A cells, however, this was determined to require both p53 and p300(Clark et al., 2008). In the context of chemotherapy, doxorubicin induced NUPR1 and resulted in upregulation of p21, which subsequently upregulated anti-apoptotic BCL2L1 (i.e. BCL-XL), a p21-regulated protein(Clark et al., 2008). Upregulation of BCL2L1 (i.e. BCL-XL) bestowed resistance to doxorubicin along with exclusive phosphorylation of RB on Ser807/811, which permits for activation of tyrosine kinase ABL1 (i.e. c-ABL) and cell cycle progression(Clark et al., 2008). Forced expression of NUPR1 in MCF10A cells was also able to confer resistance to Taxol, however the specific molecular mechanisms pertaining to Taxol resistance were not elucidated(Clark et al., 2008). Collectively, these studies provide a mechanistic basis involving p21/BCL2L1/p-RB through which NUPR1 induction by genotoxic agents and chemotherapeutics can confer resistance to chemotherapy in breast cancer (Figure 2.)
Figure 2.
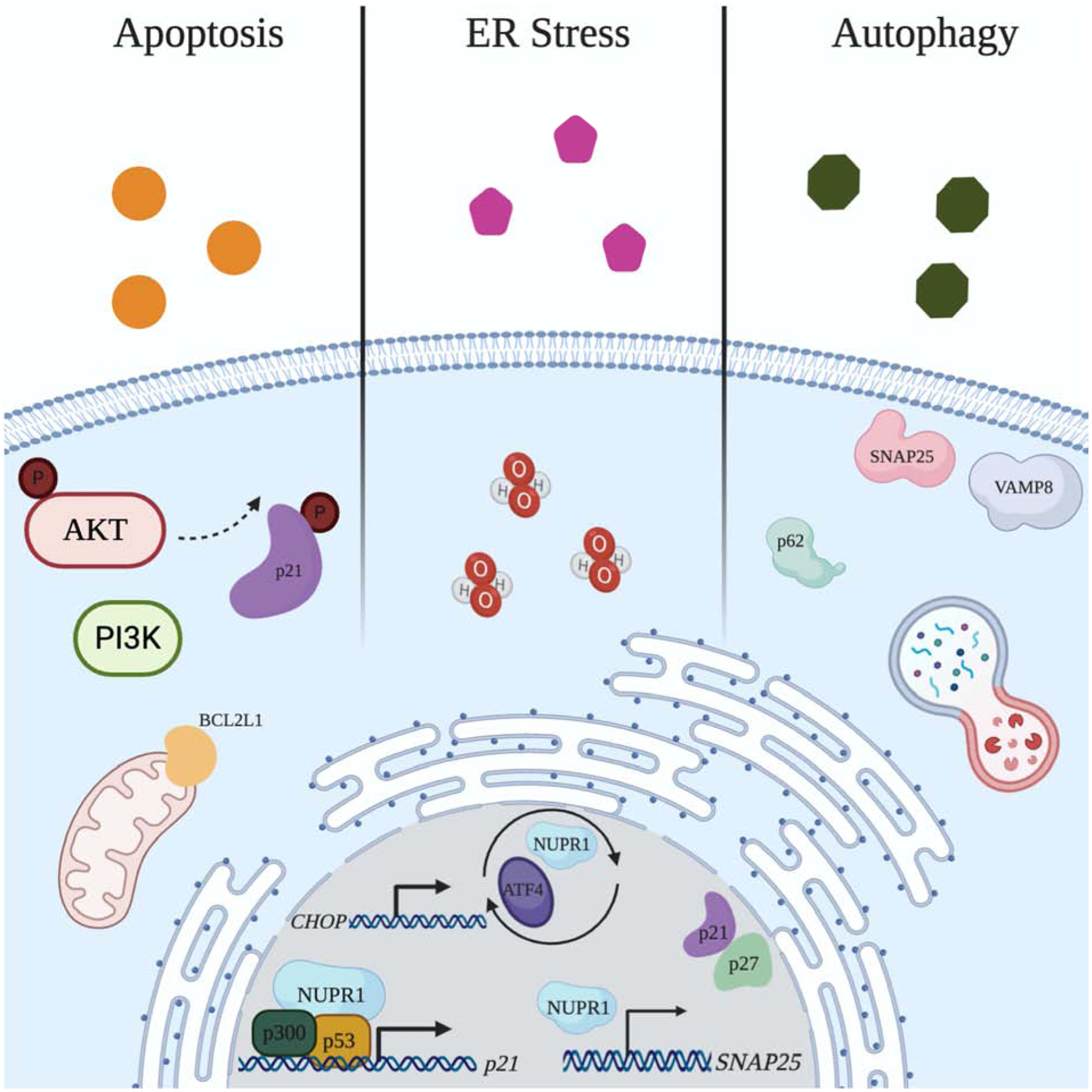
Mechanisms of NUPR1-mediated Chemotherapeutic Resistance in Breast, Lung, and Colorectal Cancers. (left) NUPR1 is induced when challenged with chemotherapeutics doxorubicin and paclitaxel. NUPR1 controls p21 localization by mediating p21 phosphorylation in a PI3K/AKT-dependent manner. NUPR1 upregulates p21, which was shown to require p53 and p300, resulting in the upregulation of p21 target gene, BCL2L1 (i.e. BCL-XL). (middle) NUPR1 is induced by chemotherapeutic oxaliplatin, and induces ER stress, ROS generation, and autophagy. NUPR1 engages in a positive feedback loop with ER stress protein ATF4, and transcriptionally regulates ER stress protein CHOP. (right) NUPR1 mediates autolysosomal processing, and transcriptionally regulates SNAP25, which together with VAMP8, can impact autolysosomal efflux. NUPR1 dysregulation in cancer impacts p62, p21, and p27 expression. Created with BioRender.com.
Bratland et al. (2000) sought to investigate the effects of calcitriol (1,25-dihydroxyvitamin D3) on breast cancer cell growth since it has been previously associated with growth inhibitory effects in breast cancer(Bratland et al., 2000). NUPR1 was found induced following calcitriol treatment in MCF-7/LCC2 cells but not in MCF-7 cells, and following treatment with high concentrations of calcitriol, growth of MCF-7/LCC2 cells in soft agar was inhibited as was cell proliferation(Bratland et al., 2000). At low concentrations, however, soft agar growth was stimulated(Bratland et al., 2000). Moreover, in MCF-7 cells, forced NUPR1 expression using a dexamethasone-inducible NUPR1 construct showed that colony formation was inhibited(Bratland et al., 2000).
Jiang et al. (2005) provided evidence of altered growth and invasion in breast cancer cells aberrantly expressing NUPR1 with and without 17,β-estradiol and reported a connection between ERβ and NUPR1(W. G. Jiang, Davies, & Fodstad, 2005). In MDA-MB-231 (ERα−, ERβ+) cells treated with 17,β-estradiol both NUPR1 knockdown and overexpression resulted in an increase in growth rate, albeit the growth rate was increased to a higher degree in NUPR1 knockdown compared to overexpression(W. G. Jiang, Davies, et al., 2005). However, knockdown or overexpression of NUPR1 in MCF-7 (ERα+, ERβ+) treated with 17-β-estradiol had no impact on growth rate(W. G. Jiang, Davies, et al., 2005). In the absence of 17-β-estradiol treatment, in both MDA-MB-231 and MCF-7 cells, NUPR1 knockdown cells exhibited a faster growth rate compared to WT and vector controls(W. G. Jiang, Davies, et al., 2005). NUPR1 overexpression in both cell lines resulted in a slower growth rate(W. G. Jiang, Davies, et al., 2005). In addition, NUPR1 knockdown in MDA-MB-231 cells increased invasiveness, but there was no difference upon NUPR1 knockdown in MCF-7 cells(W. G. Jiang, Davies, et al., 2005). Overexpression of NUPR1 in both cell lines decreased invasiveness, but the difference was not significant(W. G. Jiang, Davies, et al., 2005).
Treatment with 17-β-estradiol in MDA-MB-231 (ERα−, ERβ+) cells showed a loss of nuclear NUPR1 and unchanged cytoplasmic immunocytochemical staining(W. G. Jiang, Davies, et al., 2005). Similar patterns were observed with ERβ immunocytochemical staining(W. G. Jiang, Davies, et al., 2005). This prompted investigation into the connection between NUPR1 and ERβ. In fact, ERβ but not ERα was determined to coimmunoprecipitate with NUPR1(W. G. Jiang, Davies, et al., 2005). Furthermore, in order to investigate the reason for loss of nuclear NUPR1, cells were co-treated with a ubiquitin inhibitor and 17-β-estradiol, and the loss of NUPR1 was reversed(W. G. Jiang, Davies, et al., 2005). Treatment with a proteasome inhibitor showed similar results(W. G. Jiang, Davies, et al., 2005). Collectively, this indicates that estradiol may facilitate the depletion of nuclear NUPR1 through the Ub/proteasome system.
3. NUPR1 Expression in Lung Cancer
In vitro, NUPR1 expression was found to be upregulated in a panel of non-small cell lung cancer (NSCLC) cell lines (Table 2.)(Guo et al., 2012). In lung tumor tissues, NUPR1 expression was determined to be variable, but was unexpressed in cancer-adjacent tissues and in human bronchial epithelial cells(Mu et al., 2018). Survival analysis showed that high NUPR1 expression correlates significantly with poor survival, and low expression corresponds with longer survival times(Mu et al., 2018). However, no correlation between primary tumor, regional lymph nodes, and distant metastasis (TNM) status, smoking history, age, or gender was evident(Mu et al., 2018). Guo et al. (2012) assessed NUPR1 expression in NSCLC tissue samples, and consistently found that NUPR1 was upregulated in adenocarcinoma, squamous carcinoma, and adenosquamous carcinomas compared to peritumor lung tissues(Guo et al., 2012). Therefore, current evidence suggests that NUPR1 is indeed upregulated in lung cancer tissues and cell lines, and likely plays a role in lung cancer development and progression.
Table 2.
NUPR1 Expression Reported in Lung Cancer Cell Lines and Tissues
| Cell line/Cancer tissue | Expression (+/−) | Description | Reference |
|---|---|---|---|
| Non-small cell lung cancer tumors | + | Squamous cell carcinoma | (Guo et al., 2012) |
| Non-small cell lung cancer tumors | + | Adenocarcinoma | |
| Non-small cell lung cancer tumors | + | Adenosquamous carcinoma | |
| A549 | + | Carcinoma (ATCC® CCL-185) | (Y. Li et al., 2020; Mu et al., 2018) |
| SK-MES-1 | + | Derived from metastatic site: pleural effusion, squamous cell carcinoma (ATCC® HTB-58) | (Guo et al., 2012) |
| 95-D (PLA-801D) | + | Non-small Cell Lung Cancer | |
| NCI-H460 | + | Pleural effusion, large cell lung cancer (ATCC® HTB-177) | (Guo et al., 2012; Mu et al., 2018) |
| NCI-H1650 | + | Adenocarcinoma, bronchoalveolar carcinoma, derived from metastatic site: pleural effusion (ATCC® CRL-5883) | (Guo et al., 2012) |
| NCI-H1299 | + | Derived from metastatic site: lymph node, non-small cell lung cancer (ATCC® CRL-5803) | (Guo et al., 2012; Mu et al., 2018) |
| BEAS-2B | + | Virus transformed bronchial epithelial cells | (D. Chen et al., 2016) |
| - | (Mu et al., 2018) | ||
| Non-small cell lung cancer tumors | + | Uncharacterized | (Mu et al., 2018) |
| NCI-H209 | - | Derived from metastatic site: bone marrow, small cell lung cancer (ATCC® HTB-172) | |
| NCI-H441 | + | Papillary adenocarcinoma (ATCC® HTB-174) | |
| NCI-H446 | + | Derived from metastatic site: pleural effusion, small cell lung cancer (ATCC® HTB-171) | |
| NCI-H385 | - | Bronchiole; derived from metastatic site: alveolus, bronchioalveolar carcinoma, non-small cell lung cancer (ATCC® CRL-5807) | |
| NCI-H1155 | + | Derived from metastatic site: lymph node, non-small cell lung cancer (ATCC® CRL-5818) | |
| NHBE | - | Normal bronchial epithelial cells |
3.1. Molecular Mechanisms Downstream of NUPR1 and Upstream Regulators of NUPR1 in Lung Cancers
NUPR1’s function in autophagy and senescence was investigated in NSCLC. Mu et al. (2018) investigated NUPR1 and its control over autolysosomal dynamics(Mu et al., 2018). First, they showed that upon NUPR1 knockdown, perinuclear vacuole accumulation occurred in A549, H460, and H1155 cells(Mu et al., 2018). Knockdown of NUPR1 increased LC3 puncta in a time-dependent manner, and impaired the subcellular localization of LC3(Mu et al., 2018). Movement of vacuoles in the NUPR1 knockdown cells was also less active(Mu et al., 2018). Altogether, this provides evidence that NUPR1 affects both autolysosomal clearance and trafficking of intracellular components(Mu et al., 2018)(Figure 2.). Consistent with this scenario, NUPR1 knockdown showed increased processing of LC3B-I to LC3B-II and accumulation of SQSTM1 (i.e p62)(Mu et al., 2018).
Using an inhibitor of autolysosomal and lysosomal fusion, autolysosomal vacuole formation brought on by NUPR1 knockdown was reversed, while LC3B puncta accumulation and LC3B-I to LC3B-II conversion was maintained (Mu et al., 2018). From this, it was concluded that NUPR1 is required for a critical step in late-stage autolysosomal processing, and that accumulation of LC3B-II and SQSTM1 upon NUPR1 knockdown is due to impaired autolysosomal processing, presumably through autophagic flux and decreased autolysosomal efflux(Mu et al., 2018)(Figure 2.). Interestingly, using both a Tet-on inducible shRNA against NUPR1 and re-expression using a flag-tagged NUPR1 construct, the phenotype of autolysosomal vacuolization, LC3B turnover, or SQSTM1 accumulation was not rescued, meaning that the autolysosomal processes is irreversibly impaired by NUPR1 depletion(Mu et al., 2018).
Taking this a step further, the authors showed that NUPR1 directly transcriptionally activates SNAP25, which was noted to be important for lysosomal trafficking and fusion(Mu et al., 2018)(Figure 2.). They also reported that the effects of SNAP25 on autolysosomal efflux are dictated, in part, through one of its binding partners, VAMP8 (Figure 2.). In order to rule out an association between NUPR1 depletion and activation of autolysosomal degradation enzymes, cathepsin processing was evaluated. Processing of lysosomal proteases cathepsins B and D was not significantly changed in NUPR1 or SNAP25 knockdown cells.
Due to the association between cytoplasmic vacuolization and senescence induction, the authors investigated and showed that NUPR1 knockdown, in vitro, induced premature senescence(Mu et al., 2018). Tumorigenesis and metastases-related phenotypes for NUPR1 knockdown were also evaluated. NUPR1 knockdown mitigated cell migration in vitro, and when xenografted into nude mice, subcutaneously injected NUPR1-depleted cells delayed tumor growth and significantly decreased tumor weights(Mu et al., 2018). NUPR1 expression also positively correlated with SNAP25 expression in NSCLS tissues(Mu et al., 2018). In a follow-up study, the same group confirmed that upon knocking down NUPR1, autophagy is impaired and premature senescence is induced(Y. Li et al., 2020). p62 and SNAP25 were increased and decreased, respectively, following NUPR1 knockdown, and cell cycle inhibitors p21 and p27 were both upregulated(Y. Li et al., 2020)(Figure 2.). Moreover, this study demonstrated the effects of NUPR1 knockdown on autophagy and premature senescence in vivo, and confirmed significantly reduced tumor volumes, weight, and size upon NUPR1 knockdown(Y. Li et al., 2020). The relationship between NUPR1 and tumorigenesis in vivo was also investigated by Guo et al. (2012), who reported that NUPR1 knockdown in H1299 and SK-MES-1 cells formed smaller tumors and with lower weights in a murine xenograft model(Guo et al., 2012). In addition, NUPR1 knockdown reduced proliferation and colony formation ability, and resulted in G0/G1 arrest as well as increased apoptosis(Guo et al., 2012).
Grasso et al. (2015) also showed an association between NUPR1 and senescence in vivo. Using a constitutively active mutant Kras murine model, Nupr1 inactivation was shown to induce senescence when Kras was expressed, whereas Nupr1 expression resulted in the development of significantly more lung adenomas compared to Nupr1−/− mice(Grasso et al., 2015). Since Kras is partially linked to the induction of premature senescence and Nupr1+/+-Kras mutant mice failed to induce senescence, it’s likely that Nupr1 modifies Kras-induced senescence to facilitate oncogenic transformation(Grasso et al., 2015).
Our group previously showed that NUPR1 was epigenetically regulated because its mRNA and protein expression was increased following treatment with 5-azaC and sodium butyrate, inhibitors of DNA methylation and histone acetylation, respectively(D. Chen et al., 2016). In addition, NUPR1 overexpression reduced histone 4 lysine 16 acetylation (H4K16as) and histone acetyltransferase MOF expression, the former considered a ‘hallmark’ of cancer(D. Chen et al., 2016)(Figure 1.). More specifically, H4K16ac levels were reduced in the promoters of TRIM42S and IAP and D4Z4 repeat array in subtelomeric regions(D. Chen et al., 2016)(Figure 1.). Histone 3 lysine 4 trimethylation (H3K4me3) levels were also altered by NUPR1 overexpression(D. Chen et al., 2016)(Figure 1.). Furthermore, NUPR1 overexpression in human bronchial epithelial cells (BEAS-2B) resulted in cell transformation(D. Chen et al., 2016). Since NUPR1 was observed to be induced by the known human carcinogen Cr(VI), NUPR1 was knocked down in BEAS-2B cells treated with Cr(VI) to determine if it plays a role in Cr(VI)-induced carcinogenesis. Indeed, upon knockdown, Cr(VI)-induced cell transformation was prevented(D. Chen et al., 2016).
4. NUPR1 Expression in Colorectal Cancer
NUPR1 was first characterized in colorectal cancers in 2010(Davies, Parr, Sanders, Fodstad, & Jiang, 2010). In normal and tumor colorectal tissue samples, NUPR1 was expressed in 22.8% and 43.6% of samples tested, respectively(Davies et al., 2010). NUPR1 was significantly overexpressed in tumor tissues; however, there was a significant decrease in percentage of tumors overexpressing NUPR1 relative to their matched normal counterpart with increasing primary tumor stage(Davies et al., 2010). With regards to regional lymph nodes and distant metastasis, there was a decrease in tumors overexpressing NUPR1, but neither decrease was significant(Davies et al., 2010). Wang et. al (2019) reported NUPR1 was highly expressed in colorectal cancer tissues(Wang, Jiang, Xia, & Zhang, 2019). Histological grade analysis showed that levels of NUPR1 overexpression decreased with worsening degree of tumor differentiation, but again this difference was not significant(Davies et al., 2010). Interestingly, analysis of IHC staining for NUPR1 in normal tissues showed strong nuclear and peri-nuclear staining with little or no cytoplasmic staining(Davies et al., 2010). In tumor tissues, overall NUPR1 staining was greater, and there was a higher degree of cytoplasmic staining but little nuclear or peri-nuclear staining(Davies et al., 2010). Finally, when early stage and advanced stage was considered, there was a greater degree of staining in early stage and a lower degree of overall NUPR1 staining in advanced stage compared to matched normal tissues(Davies et al., 2010).
The same authors subsequently investigated the impact of NUPR1 knockdown on cell growth, migration, and apoptosis. NUPR1 knockdown in RKO and CaCO2 human colorectal cancer cell lines showed significantly reduced growth(X. Li, Martin, & Jiang, 2012). NUPR1 knockdown in both RKO and CaCO2 cells did not impact migration using both ECIS and wound healing assays(X. Li et al., 2012). In both RKO and CaCO2 cells with NUPR1 knocked down, an increase in apoptosis was observed, which suggests that NUPR1 may function as an anti-apoptotic gene and promote cell growth in RKO and CaCO2 cells(X. Li et al., 2012). In summary, based on all available evidence, NUPR1 is variably expressed in colorectal cancers and plays an unclear role in cancer progression (Table 3).
Table 3.
NUPR1 Expression Reported in Colorectal Cancer Cell Lines and Tissues.
| Cell line/Cancer tissue | Expression (+/−) | Description | References |
|---|---|---|---|
| Primary colon tumors | + | Uncharacterized | (Davies et al., 2010) |
| RKO | + | Colon carcinoma (ATCC® CRL-2577) | (X. Li et al., 2012) |
| CaCO-2 | + | Colorectal adenocarcinoma (ATCC® HTB-37) | |
| HRT-18 (HCT-8) | + | Ileocecal colorectal adenocarcinoma (ATCC® CCL-224) | |
| HCT116 | + | Colorectal carcinoma (ATCC® CCL-247) | (S. S. Chen et al., 2015) |
| Primary colorectal tumors | + | Uncharacterized | (Wang et al., 2019) |
4.1. Molecular Mechanisms Downstream of NUPR1 and Chemoresistance in Colorectal Cancer
Oxaliplatin is a chemotherapeutic used in combination with 5-fluorouracil and leucovorin for metastatic colorectal cancer(Shi et al., 2012). Autophagy, which plays an important role in therapeutic efficacy, was shown to be induced following oxaliplatin treatment in CaCO2 cells, and was determined to protect against oxaliplatin-induced cell death(Shi et al., 2012). This study further showed that autophagy induced by oxaliplatin depends on reactive oxygen species (ROS) generation and that autophagy decreases oxaliplatin-induced ROS, thereby acting as a cell survival mechanism(Shi et al., 2012). NUPR1 was revealed to play a part in autophagy induction by oxaliplatin since upon NUPR1 silencing, ROS production was increased and autophagy decreased(Shi et al., 2012). It was previously reported that NUPR1 engages in a positive feedback loop with ER stress-related gene, ATF4, and can influence CHOP and TRB3 mRNA expression, both ATF4 target genes and associated with the ER stress response(Jin et al., 2009). Shi et al., (2012) concluded that because both NUPR1 and CHOP silencing increased ROS and decreased autophagy, ER stress lies upstream of autophagy and ROS generation in oxaliplatin-treated CaCO2 cells(Shi et al., 2012). This indicates that NUPR1’s role in mediating autophagy is likely linked to the ER stress response (Figure 2.). NUPR1’s function as a mediator of autophagy can facilitate cell survival in the face of oxaliplatin treatment, and may inadvertently facilitate chemotherapeutic resistance. Moreover, NUPR1’s control over autophagy in the face of a toxic insult may allow cells that have undergone extensive intracellular damage to survive and potentially contribute to cell transformation and drive carcinogenesis.
Dihydroartemisinin (DHA) is an antimalaria compound that has also been shown to possess anticancer activity, and NUPR1 is upregulated by DHA treatment in HCT116 human colorectal cancer cells(S. S. Chen, Hu, Wang, Lou, & Zhou, 2015). ATF4 and CHOP were also found induced by DHA, and more importantly, their induction was dependent on NUPR1 since its silencing blocked their induction(S. S. Chen et al., 2015)(Figure 2.). Furthermore, the NUPR1-ATF4-CHOP axis was reported to be involved in DHA-induced autophagy in HCT116 cells(S. S. Chen et al., 2015). NUPR1 induction by DHA decreases sensitivity of HCT116 cells to DHA(S. S. Chen et al., 2015). This again demonstrates a chemotherapeutic resistance mechanism involving NUPR1 and its control over autophagy-mediated cell survival. Similar to DHA and oxaliplatin, combination treatment of HCT116 spheroids with chloroquine and irinotecan was associated with autophagy induction and likely involved NUPR1; however, NUPR1 was predicted to be inactivated based on Ingenuity Pathway Analysis (IPA) of upstream regulators(Schroll, LaBonia, Ludwig, & Hummon, 2017).
4.2. Upstream Regulators of NUPR1 in Colorectal Cancer
ZYX is a potential oncogene in colorectal cancer that has been shown to facilitate anchorage-independent growth, migration and invasion, in vitro, and is pro-tumorigenic in vivo(Zhong et al., 2019). NUPR1 was suggested to be downstream of ZYX based on IPA analysis of ZYX silenced HCT116 cells, and functional enrichment suggested that genes associated with ZYX are related to cell motility, angiogenesis, cell proliferation, growth, and adhesion(Zhong et al., 2019).
Like oncRNAs, NUPR1 is regulated by long noncoding RNAs (LncRNAs)(Wang et al., 2019)(Figure 1.). In HCT16 and HT29 colorectal cancer cell lines, LncRNA FAL1 was shown to regulate mRNA and protein expression of NUPR1 and downstream targets HIF1A and LASP1, by binding to and inhibiting miR-637, which targets and downregulates NUPR1(Wang et al., 2019)(Figure 1.). By binding to and inhibiting miR-637, FAL1 is capable to functioning as an oncogene in colorectal cancer. Phenotype analysis showed that FAL1 increased cell viability, colony formation, migration and invasion, and reduced cadherin 1 but increased vimentin expression, epithelial and mesenchymal markers, respectively(Wang et al., 2019). NUPR1 was also shown to act as an oncogene by affecting viability, colony formation, migration and invasion, and by directly regulating cadherin 1 and vimentin(Wang et al., 2019). Conversely, miR-637 acts as a tumor suppressor, and was determined to inversely regulate these same oncogenic properties(Wang et al., 2019). FAL1 was also evaluated in colorectal cancer tissues and cells, and determined to be upregulated in 90% of tumor tissues and was expressed to a higher degree in colorectal cancer cell lines compared to normal human colon mucosal epithelial cells(Wang et al., 2019). Expression levels of FAL1 were also significantly correlated with tumor size, TNM stage, lymph node metastasis, and high expression was associated with worse overall survival(Wang et al., 2019).
5. NUPR1: A New Target for Cancer Treatment
An in vitro molecular screening approach was applied to characterize the interactions of 1120 FDA-approved drugs with NUPR1 to identify potential chemotherapeutics that act by targeting NUPR1. Fifteen compounds were determined to significantly bind to and interact with NUPR1, and the antipsychotic drug trifluoperazine (TFP) demonstrated the highest dissociation constant among those tested(Neira et al., 2017). Phenotypic analysis of pancreatic ductal adenocarcinoma (PDAC)-derived cells treated with TFP indicated that TFP is effective in reducing cell proliferation, metastasis, and in vivo reports show that TFP is capable of preventing tumor growth, especially when co-administered with NUPR1 gene silencing therapies in both pancreatic and lung cancer xenograft models(Y. Li et al., 2020; Neira et al., 2017). TFP treatment was also reported to substantially reduce the IC50 of gemcitabine and oxaliplatin, induce cellular senescence, and hamper the interaction between NUPR1 and one of its binding partners, MSL1, which is involved in DNA damage repair and H4K16 acetylation together with MOF(Neira et al., 2017). Mechanistically, it was determined that combination therapy of NUPR1 silencing and TFP administration, in vivo, reduced tumorigenesis by inducing premature senescence and dysregulating autophagy, consistent with studies showing that NUPR1 mediates autophagy in vitro(Y. Li et al., 2020). Prior to the knowledge that TFP likely acts as a small molecule inhibitor of NUPR1, the anticancer activity of TFP in colorectal, breast, and lung cancer was investigated thoroughly in vitro and in vivo.
Qian et al. (2018) reported that TFP suppresses colorectal cancer cell proliferation, promotes apoptosis through autophagy, inhibits migration and invasion by regulating cadherin 2, SNAI1, and SNAI2 expression, and suppresses tumorigenesis in vivo(Qian et al., 2019). In addition, TFP inhibits cancer stem cell (CSC) growth, which is particularly important because CSCs contribute to cancer relapse, chemotherapeutic resistance, and cancer initiation(Yeh et al., 2012). In a TNBC tumor xenograft model using both murine 4T1 cells and metastatic human MDA-MB-436 cells, TFP administration dose-dependently and significantly attenuated tumor growth(Feng et al., 2018). Notably, TFP inhibited brain metastasis growth and prolonged the survival of mice bearing metastatic TNBC brain tumors(Feng et al., 2018). As pointed out by the investigators, metastatic TNBC is known to present a high risk of brain metastases, and because TFP demonstrates good bioavailability in the brain as an antipsychotic, it may be that TFP is particularly effective against brain metastases derived from TNBC(Feng et al., 2018). In TNBC patient tumors (n=58), activation of NUPR1 was observed after chemotherapy intervention, and it was confirmed, in vitro, that chemotherapeutic intervention in six TNBC cell lines upregulated NUPR1(Solzak, Wang, Hancock, & Radovich, 2020). Combination therapy of NUPR1 inhibition using TFP and either targeted therapies or paclitaxel showed synergy, and this was mimicked by silencing NUPR1 using an siRNA-based approach(Solzak et al., 2020).
Contrary to the promising results that TFP has shown in vivo and in vitro, efficient doses of TFP needed for anticancer activity were reported to cause neurological effects in mice, and may preclude TFP as a treatment option(Santofimia-Castaño et al., 2019). A TFP-derived compound, ZZW-115, was subsequently developed using an in silico ligand design approach in lieu of this limitation. ZZW-115 mechanistically mimics NUPR1 inactivation, and induces cell death by necroptosis and apoptosis, with concomitant ER stress-mediated mitochondrial metabolism failure that triggers lower production of ATP and overproduction of ROS(Santofimia-Castaño et al., 2018; Santofimia-Castaño et al., 2019). ZZW-115 was also shown to be capable of preventing tumor growth and decreasing tumor size until disappearance (Santofimia-Castaño et al., 2019). Collectively, TFP and ZZW-115 demonstrate that NUPR1 inhibition using a small molecule-based approach is certainly an option for chemotherapeutic intervention, yet more studies and clinical trials are needed to confirm that NUPR1 inhibition and/or gene silencing is effective.
Conclusion
NUPR1 is overexpressed in breast, lung, and colorectal cancers based on all available evidence of its expression in tissues and cell lines (Tables 1., 2., and 3.). In breast cancer, early studies support a metastatic role for NUPR1; however, the site of metastasis seems to be an important factor in its expression based on in vitro analyses. However, clinical evidence supporting a metastatic role is lacking, and is greatly needed in order to come to a definitive conclusion. In lung cancers, particularly NSCLC, in vitro and in vivo evidence supports a carcinogenic and tumorigenic role for NUPR1 based on its ability to facilitate cell transformation and its association with reduced tumor growth following knockdown. In colorectal cancer, in vitro evidence points toward increased tumorigenic capacity for cells overexpressing NUPR1 but not increased potential for migration and invasion. However, additional in vitro and, particularly, in vivo, studies are needed to fully understand the impact that NUPR1 overexpression has on colorectal cancer. NUPR1 appears to be epigenetically regulated across all cancer types covered herein based on the impact that oncRNAs, lncRNAs, miRNAs, and HDAC and DNMT inhibitors have on its expression (Figure 1.). Yet, much still remains to be elaborated on in this regard such as its interaction with and influence over epigenetic machinery proteins like histone acetyltransferase MOF.
In breast cancer there is strong evidence showing that NUPR1 is regulated by E2F1 and E2F2, which is important because E2F genes are major transcriptional regulators and are highly expressed in virtually all cancers (reviewed in (Kent & Leone, 2019)). Downstream molecular mechanisms describing how NUPR1 imparts its oncogenic effects are understudied and are greatly needed. The only studies to date that provide comprehensive mechanistic details of NUPR1 in breast cancer concern p21/BCL2L1 (i.e. BCL-XL)/p-RB. NUPR1 functions as an anti-apoptotic gene by upregulating p21 and BCL2L1 (i.e. BCL-XL) under conditions of genotoxic and chemotherapeutic stress (Figure 2.). While NUPR1 is acting to enhance cell survival when challenged with anti-cancer agents, it is likely that NUPR1 is inadvertently permitting cells that have incurred extensive damage to survive, which may later prove to be oncogenic.
There is meaningful evidence showing that NUPR1 regulates autophagy in lung cancers by transcriptionally activating snare protein SNAP25 (Figure 2.). NUPR1 depletion in lung cancer cells deregulates autophagic flux and impairs autolysosomal clearance, resulting in premature senescence. This is an important finding because autophagy is capable of conferring a survival advantage to cancer cells by mitigating various cellular stressors encountered by either chemotherapeutic intervention or in the natural progression of cancer cells in unfavorable growth conditions. In recent years autophagy has been recognized as a double-edge sword in cancer such that it may promote drug resistance and tumor cell adaptation to stress (reviewed in (Glick, Barth, & Macleod, 2010; Sui et al., 2013)). This may be an important consideration for NUPR1’s role in breast cancer and other cancers, given some of the inconsistencies and tumor suppressive functions reported. beclin-1, which is central component in autophagy, is mono-allelically deleted in breast, ovarian, and prostate cancer (reviewed in (Glick et al., 2010)). Therefore, the influence that NUPR1 has on autophagy should be taken into consideration when discussing its tumor suppressive or oncogenic properties, and is a lucrative avenue for future studies. Similar to breast cancer, in lung cancer NUPR1 was also confirmed to upregulate p21 in vitro, and modify Kras-induced senescence in vivo. Due to the impact that histone posttranslational modifications have on global gene expression and their association with cancers, it is important that NUPR1 was determined to reduce H4K16ac. H4K16ac is localized to enhancers and promoters of active genes, is involved in chromatin decondensation, and is associated with many cancers, such as lung cancers(D. Chen et al., 2016). Additional studies on the impact that NUPR1 has on the epigenetic status of cancer cells would be invaluable.
In colorectal cancer, there is strong evidence that NUPR1 operates by inducing autophagy in a pro-survival manner when challenged with anticancer agents (Figure 2.). Furthermore, a connection between ER stress and NUPR1 has been shown and involves transcriptional activation of ATF4 and CHOP (Figure 2.). Like autophagy, ER stress acts as both a friend and foe in cancer; under normal conditions ER stress compensates for cellular damage through initiating the unfolded protein response, but in cancer can trigger cell transformation, enhance survival, and adjust the metabolic status of cells (reviewed in (Urra, Dufey, Avril, Chevet, & Hetz, 2016)). In summary, NUPR1 is overexpressed in breast, lung, and colorectal cancers, and acts oncogenically, in part, by regulating autophagy, influencing ER stress response, and acting as an anti-apoptotic gene. Through these mechanisms, cells are able to adapt to chemical and biological stressors, and evade programmed cell death, permitting cell survival under harsh conditions and leading to the acquisition of malignant characteristics. NUPR1 can be targeted by small molecule inhibitors, namely TFP and ZZW-115, and inhibition by these compounds prevents cancer cell growth and acquisition of malignant characteristics. NUPR1 silencing mimics inhibition by small molecule drugs, and because of the general specificity of gene silencing techniques, this may be preferable to avoid or minimize off-target effects. In combination with traditional chemotherapeutics, NUPR1 inhibition or silencing is the most probable approach for cancer treatment due to the synergy this exhibits along with the observed dose reduction of primary therapeutics needed to maintain a sufficient response. NUPR1 and its functions are challenging to unmask given that NUPR1 has a disordered protein structure, is expressed differentially during early and late passage, is localized differentially based on cell density, is induced by a myriad of chemical and biological stressors, and is generally not expressed in normal tissues and cells. Aside from those factors, experimental conditions such as routine medium change may impact the basal and induced levels of NUPR1 and should be recognized to avoid potential discrepancies(Garcia-Montero et al., 2001). However, much more remains to be uncovered with regards to NUPR1 expression and its functions, as it seems to be key in cellular adaptation to unfavorable conditions and chemotherapeutic resistance in cancer.
Highlights.
NUPR1 is often overexpressed in breast, lung, and colorectal cancers, among other cancers
NUPR1 overexpression contributes to carcinogenesis, tumorigenesis, metastasis, and chemotherapeutic resistance
By mediating autophagy, ER stress response, and anti-apoptotic mechanisms, NUPR1 is capable of conferring chemotherapeutic resistance
Genetic or pharmacological inhibition of NUPR1 is a novel and promising new strategy to treat cancers
Funding:
This research was funded by the following NIH grants: ES000260, ES022935, ES023174, ES0261.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: There are no conflicts of interest
References
- Aguado-Llera D, Hamidi T, Doménech R, Pantoja-Uceda D, Gironella M, Santoro J, … Iovanna JL (2013). Deciphering the binding between Nupr1 and MSL1 and their DNA-repairing activity. PLoS One, 8(10), e78101. doi: 10.1371/journal.pone.0078101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Averous J, Lambert-Langlais S, Cherasse Y, Carraro V, Parry L, B’Chir W, … Fafournoux P (2011). Amino acid deprivation regulates the stress-inducible gene p8 via the GCN2/ATF4 pathway. Biochem Biophys Res Commun, 413(1), 24–29. doi: 10.1016/j.bbrc.2011.08.028 [DOI] [PubMed] [Google Scholar]
- Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, … Massagué J (2009). Genes that mediate breast cancer metastasis to the brain. Nature, 459(7249), 1005–1009. doi: 10.1038/nature08021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratland A, Risberg K, Maelandsmo GM, Gützkow KB, Olsen OE, Moghaddam A, … Ree AH (2000). Expression of a novel factor, com1, is regulated by 1,25-dihydroxyvitamin D3 in breast cancer cells. Cancer Res, 60(19), 5578–5583. Retrieved from https://cancerres.aacrjournals.org/content/canres/60/19/5578.full.pdf [PubMed] [Google Scholar]
- Brünner N, Frandsen TL, Holst-Hansen C, Bei M, Thompson EW, Wakeling AE, … Clarke R (1993). MCF7/LCC2: a 4-hydroxytamoxifen resistant human breast cancer variant that retains sensitivity to the steroidal antiestrogen ICI 182,780. Cancer Res, 53(14), 3229–3232. Retrieved from https://cancerres.aacrjournals.org/content/canres/53/14/3229.full.pdf [PubMed] [Google Scholar]
- Cano CE, Hamidi T, Sandi MJ, & Iovanna JL (2011). Nupr1: the Swiss-knife of cancer. J Cell Physiol, 226(6), 1439–1443. doi: 10.1002/jcp.22324 [DOI] [PubMed] [Google Scholar]
- Carracedo A, Lorente M, Egia A, Blázquez C, García S, Giroux V, … Velasco G (2006). The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell, 9(4), 301–312. doi: 10.1016/j.ccr.2006.03.005 [DOI] [PubMed] [Google Scholar]
- Chen D, Kluz T, Fang L, Zhang X, Sun H, Jin C, & Costa M (2016). Hexavalent Chromium (Cr(VI)) Down-Regulates Acetylation of Histone H4 at Lysine 16 through Induction of Stressor Protein Nupr1. PLoS One, 11(6), e0157317. doi: 10.1371/journal.pone.0157317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SS, Hu W, Wang Z, Lou XE, & Zhou HJ (2015). p8 attenuates the apoptosis induced by dihydroartemisinin in cancer cells through promoting autophagy. Cancer Biol Ther, 16(5), 770–779. doi: 10.1080/15384047.2015.1026477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury UR, Samant RS, Fodstad O, & Shevde LA (2009). Emerging role of nuclear protein 1 (NUPR1) in cancer biology. Cancer Metastasis Rev, 28(1–2), 225–232. doi: 10.1007/s10555-009-9183-x [DOI] [PubMed] [Google Scholar]
- Clark DW, Mitra A, Fillmore RA, Jiang WG, Samant RS, Fodstad O, & Shevde LA (2008). NUPR1 interacts with p53, transcriptionally regulates p21 and rescues breast epithelial cells from doxorubicin-induced genotoxic stress. Curr Cancer Drug Targets, 8(5), 421–430. doi: 10.2174/156800908785133196 [DOI] [PubMed] [Google Scholar]
- Dai X, Cheng H, Bai Z, & Li J (2017). Breast Cancer Cell Line Classification and Its Relevance with Breast Tumor Subtyping. Journal of Cancer, 8(16), 3131–3141. doi: 10.7150/jca.18457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies ML, Parr C, Sanders AJ, Fodstad O, & Jiang WG (2010). The transcript expression and protein distribution pattern in human colorectal carcinoma reveal a pivotal role of COM-1/p8 as a tumour suppressor. Cancer Genomics Proteomics, 7(2), 75–80. Retrieved from http://cgp.iiarjournals.org/content/7/2/75.full.pdf [PubMed] [Google Scholar]
- Encinar JA, Mallo GV, Mizyrycki C, Giono L, Gonzalez-Ros JM, Rico M, … Iovanna JL (2001). Human p8 is a HMG-I/Y-like protein with DNA binding activity enhanced by phosphorylation. J Biol Chem, 276(4), 2742–2751. doi: 10.1074/jbc.M008594200 [DOI] [PubMed] [Google Scholar]
- Feng Z, Xia Y, Gao T, Xu F, Lei Q, Peng C, … Yu L (2018). The antipsychotic agent trifluoperazine hydrochloride suppresses triple-negative breast cancer tumor growth and brain metastasis by inducing G0/G1 arrest and apoptosis. Cell Death & Disease, 9(10), 1006. doi: 10.1038/s41419-018-1046-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish L, Zhang S, Yu JX, Culbertson B, Zhou AY, Goga A, & Goodarzi H (2018). Cancer cells exploit an orphan RNA to drive metastatic progression. Nat Med, 24(11), 1743–1751. doi: 10.1038/s41591-018-0230-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Montero A, Vasseur S, Mallo GV, Soubeyran P, Dagorn JC, & Iovanna JL (2001). Expression of the stress-induced p8 mRNA is transiently activated after culture medium change. European Journal of Cell Biology, 80(11), 720–725. doi: 10.1078/0171-9335-00209 [DOI] [PubMed] [Google Scholar]
- García-Montero AC, Vasseur S, Giono LE, Canepa E, Moreno S, Dagorn JC, & Iovanna JL (2001). Transforming growth factor beta-1 enhances Smad transcriptional activity through activation of p8 gene expression. Biochem J, 357(Pt 1), 249–253. doi: 10.1042/0264-6021:3570249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gironella M, Malicet C, Cano C, Sandi MJ, Hamidi T, Tauil RM, … Iovanna JL (2009). p8/nupr1 regulates DNA-repair activity after double-strand gamma irradiation-induced DNA damage. J Cell Physiol, 221(3), 594–602. doi: 10.1002/jcp.21889 [DOI] [PubMed] [Google Scholar]
- Glick D, Barth S, & Macleod KF (2010). Autophagy: cellular and molecular mechanisms. J Pathol, 221(1), 3–12. doi: 10.1002/path.2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goruppi S, & Kyriakis JM (2004). The pro-hypertrophic basic helix-loop-helix protein p8 is degraded by the ubiquitin/proteasome system in a protein kinase B/Akt- and glycogen synthase kinase-3-dependent manner, whereas endothelin induction of p8 mRNA and renal mesangial cell hypertrophy require NFAT4. J Biol Chem, 279(20), 20950–20958. doi: 10.1074/jbc.M312401200 [DOI] [PubMed] [Google Scholar]
- Goruppi S, Patten RD, Force T, & Kyriakis JM (2007). Helix-loop-helix protein p8, a transcriptional regulator required for cardiomyocyte hypertrophy and cardiac fibroblast matrix metalloprotease induction. Mol Cell Biol, 27(3), 993–1006. doi: 10.1128/mcb.00996-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso D, Bintz J, Lomberk G, Molejon MI, Loncle C, Garcia MN, … Iovanna JL (2015). Pivotal Role of the Chromatin Protein Nupr1 in Kras-Induced Senescence and Transformation. Sci Rep, 5, 17549. doi: 10.1038/srep17549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Wang W, Hu J, Feng K, Pan Y, Zhang L, & Feng Y (2012). Lentivirus-mediated RNAi knockdown of NUPR1 inhibits human nonsmall cell lung cancer growth in vitro and in vivo. Anat Rec (Hoboken), 295(12), 2114–2121. doi: 10.1002/ar.22571 [DOI] [PubMed] [Google Scholar]
- H Heppner G, R Miller F, & Malathy Shekhar PV (2000). Nontransgenic models of breast cancer. Breast Cancer Research, 2(5), 331. doi: 10.1186/bcr77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmeister A, Ropolo A, Vasseur S, Mallo GV, Bodeker H, Ritz-Laser B, … Iovanna JL (2002). The HMG-I/Y-related protein p8 binds to p300 and Pax2 trans-activation domain-interacting protein to regulate the trans-activation activity of the Pax2A and Pax2B transcription factors on the glucagon gene promoter. J Biol Chem, 277(25), 22314–22319. doi: 10.1074/jbc.M201657200 [DOI] [PubMed] [Google Scholar]
- Hollern DP, Honeysett J, Cardiff RD, & Andrechek ER (2014). The E2F transcription factors regulate tumor development and metastasis in a mouse model of metastatic breast cancer. Mol Cell Biol, 34(17), 3229–3243. doi: 10.1128/mcb.00737-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y, Yoshida H, Motoo Y, Iovanna JL, Nakamura Y, Kakudo K, … Miyauchi A (2005). Expression of p8 protein in breast carcinoma; an inverse relationship with apoptosis. Anticancer Res, 25(2a), 833–837. Retrieved from http://ar.iiarjournals.org/content/25/2A/833.full.pdf [PubMed] [Google Scholar]
- Jiang WG, Davies G, & Fodstad O (2005). Com-1/P8 in oestrogen regulated growth of breast cancer cells, the ER-beta connection. Biochem Biophys Res Commun, 330(1), 253–262. doi: 10.1016/j.bbrc.2005.02.157 [DOI] [PubMed] [Google Scholar]
- Jiang WG, Watkins G, Douglas-Jones A, Mokbel K, Mansel RE, & Fodstad O (2005). Expression of Com-1/P8 in human breast cancer and its relevance to clinical outcome and ER status. Int J Cancer, 117(5), 730–737. doi: 10.1002/ijc.21221 [DOI] [PubMed] [Google Scholar]
- Jiang YF, Vaccaro MI, Fiedler F, Calvo EL, & Iovanna JL (1999). Lipopolysaccharides induce p8 mRNA expression in vivo and in vitro. Biochem Biophys Res Commun, 260(3), 686–690. doi: 10.1006/bbrc.1999.0953 [DOI] [PubMed] [Google Scholar]
- Jin HO, Seo SK, Woo SH, Choe TB, Hong SI, Kim JI, & Park IC (2009). Nuclear protein 1 induced by ATF4 in response to various stressors acts as a positive regulator on the transcriptional activation of ATF4. IUBMB Life, 61(12), 1153–1158. doi: 10.1002/iub.271 [DOI] [PubMed] [Google Scholar]
- Jung SH, Lee A, Yim SH, Hu HJ, Choe C, & Chung YJ (2012). Simultaneous copy number gains of NUPR1 and ERBB2 predicting poor prognosis in early-stage breast cancer. BMC Cancer, 12, 382. doi: 10.1186/1471-2407-12-382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordón-Cardo C, … Massagué J (2003). A multigenic program mediating breast cancer metastasis to bone. Cancer Cell, 3(6), 537–549. doi: 10.1016/s1535-6108(03)00132-6 [DOI] [PubMed] [Google Scholar]
- Kent LN, & Leone G (2019). The broken cycle: E2F dysfunction in cancer. Nature Reviews Cancer, 19(6), 326–338. doi: 10.1038/s41568-019-0143-7 [DOI] [PubMed] [Google Scholar]
- Kinyamu HK, Bennett BD, Bushel PR, & Archer TK (2020). Proteasome inhibition creates a chromatin landscape favorable to RNA Pol II processivity. J Biol Chem, 295(5), 1271–1287. doi: 10.1074/jbc.RA119.011174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Martin TA, & Jiang WG (2012). COM-1/p8 acts as a tumour growth enhancer in colorectal cancer cell lines. Anticancer Res, 32(4), 1229–1237. Retrieved from http://ar.iiarjournals.org/content/32/4/1229.full.pdf [PubMed] [Google Scholar]
- Li Y, Yin Y, Ma J, Sun Y, Zhou R, Cui B, … Ma Z (2020). Combination of AAV-mediated NUPR1 knockdown and trifluoperazine induces premature senescence in human lung adenocarcinoma A549 cells in nude mice. Oncol Rep, 43(2), 681–688. doi: 10.3892/or.2020.7455 [DOI] [PubMed] [Google Scholar]
- MacLeod RA, Dirks WG, Matsuo Y, Kaufmann M, Milch H, & Drexler HG (1999). Widespread intraspecies cross-contamination of human tumor cell lines arising at source. Int J Cancer, 83(4), 555–563. doi: [DOI] [PubMed] [Google Scholar]
- Malicet C, Giroux V, Vasseur S, Dagorn JC, Neira JL, & Iovanna JL (2006). Regulation of apoptosis by the p8/prothymosin alpha complex. Proc Natl Acad Sci U S A, 103(8), 2671–2676. doi: 10.1073/pnas.0508955103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malicet C, Hoffmeister A, Moreno S, Closa D, Dagorn JC, Vasseur S, & Iovanna JL (2006). Interaction of the stress protein p8 with Jab1 is required for Jab1-dependent p27 nuclear-to-cytoplasm translocation. Biochem Biophys Res Commun, 339(1), 284–289. doi: 10.1016/j.bbrc.2005.11.018 [DOI] [PubMed] [Google Scholar]
- Mallo GV, Fiedler F, Calvo EL, Ortiz EM, Vasseur S, Keim V, … Iovanna JL (1997). Cloning and expression of the rat p8 cDNA, a new gene activated in pancreas during the acute phase of pancreatitis, pancreatic development, and regeneration, and which promotes cellular growth. J Biol Chem, 272(51), 32360–32369. doi: 10.1074/jbc.272.51.32360 [DOI] [PubMed] [Google Scholar]
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, … Massagué J (2005). Genes that mediate breast cancer metastasis to lung. Nature, 436(7050), 518–524. doi: 10.1038/nature03799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y, Yan X, Li D, Zhao D, Wang L, Wang X, … Liu Z (2018). NUPR1 maintains autolysosomal efflux by activating SNAP25 transcription in cancer cells. Autophagy, 14(4), 654–670. doi: 10.1080/15548627.2017.1338556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naundorf H, Rewasowa EC, Fichtner I, Büttner B, Becker M, & Görlich M (1992). Characterization of two human mammary carcinomas, MT-1 and MT-3, suitable forin vivo testing of ether lipids and their derivatives. Breast Cancer Research and Treatment, 23(1), 87–95. doi: 10.1007/BF01831480 [DOI] [PubMed] [Google Scholar]
- Neira JL, Bintz J, Arruebo M, Rizzuti B, Bonacci T, Vega S, … Abián O (2017). Identification of a Drug Targeting an Intrinsically Disordered Protein Involved in Pancreatic Adenocarcinoma. Sci Rep, 7, 39732. doi: 10.1038/srep39732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neira JL, Palomino-Schätzlein M, Ricci C, Ortore MG, Rizzuti B, & Iovanna JL (2019). Dynamics of the intrinsically disordered protein NUPR1 in isolation and in its fuzzy complexes with DNA and prothymosin α. Biochim Biophys Acta Proteins Proteom, 1867(11), 140252. doi: 10.1016/j.bbapap.2019.07.005 [DOI] [PubMed] [Google Scholar]
- Olsen JG, Teilum K, & Kragelund BB (2017). Behaviour of intrinsically disordered proteins in protein-protein complexes with an emphasis on fuzziness. Cell Mol Life Sci, 74(17), 3175–3183. doi: 10.1007/s00018-017-2560-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxelmark E, Roth JM, Brooks PC, Braunstein SE, Schneider RJ, & Garabedian MJ (2006). The cochaperone p23 differentially regulates estrogen receptor target genes and promotes tumor cell adhesion and invasion. Mol Cell Biol, 26(14), 5205–5213. doi: 10.1128/mcb.00009-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian K, Sun L, Zhou G, Ge H, Meng Y, Li J, … Fang X (2019). Trifluoperazine as an alternative strategy for the inhibition of tumor growth of colorectal cancer. J Cell Biochem, 120(9), 15756–15765. doi: 10.1002/jcb.28845 [DOI] [PubMed] [Google Scholar]
- Ree AH, Bratland A, Kroes RA, Aasheim HC, Flørenes VA, Moskal JR, … Maelandsmo GM (2002). Clinical and cell line specific expression profiles of a human gene identified in experimental central nervous system metastases. Anticancer Res, 22(4), 1949–1957. [PubMed] [Google Scholar]
- Ree AH, Pacheco MM, Tvermyr M, Fodstad O, & Brentani MM (2000). Expression of a novel factor, com1, in early tumor progression of breast cancer. Clin Cancer Res, 6(5), 1778–1783. Retrieved from https://clincancerres.aacrjournals.org/content/clincanres/6/5/1778.full.pdf [PubMed] [Google Scholar]
- Ree AH, Tvermyr M, Engebraaten O, Rooman M, Røsok O, Hovig E, … Fodstad O (1999). Expression of a novel factor in human breast cancer cells with metastatic potential. Cancer Res, 59(18), 4675–4680. Retrieved from https://cancerres.aacrjournals.org/content/canres/59/18/4675.full.pdf [PubMed] [Google Scholar]
- Rehn AB, & Buchner J (2015). p23 and Aha1. Subcell Biochem, 78, 113–131. doi: 10.1007/978-3-319-11731-7_6 [DOI] [PubMed] [Google Scholar]
- Santofimia-Castaño P, Lan W, Bintz J, Gayet O, Carrier A, Lomberk G, … Iovanna J (2018). Inactivation of NUPR1 promotes cell death by coupling ER-stress responses with necrosis. Sci Rep, 8(1), 16999. doi: 10.1038/s41598-018-35020-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santofimia-Castaño P, Rizzuti B, Pey Á L, Soubeyran P, Vidal M, Urrutia R, … Neira JL (2017). Intrinsically disordered chromatin protein NUPR1 binds to the C-terminal region of Polycomb RING1B. Proc Natl Acad Sci U S A, 114(31), E6332–e6341. doi: 10.1073/pnas.1619932114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santofimia-Castaño P, Xia Y, Lan W, Zhou Z, Huang C, Peng L, … Iovanna J (2019). Ligand-based design identifies a potent NUPR1 inhibitor exerting anticancer activity via necroptosis. J Clin Invest, 129(6), 2500–2513. doi: 10.1172/jci127223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroll MM, LaBonia GJ, Ludwig KR, & Hummon AB (2017). Glucose Restriction Combined with Autophagy Inhibition and Chemotherapy in HCT 116 Spheroids Decreases Cell Clonogenicity and Viability Regulated by Tumor Suppressor Genes. J Proteome Res, 16(8), 3009–3018. doi: 10.1021/acs.jproteome.7b00293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar MPV, Kato I, Nangia-Makker P, & Tait L (2013). Comedo-DCIS is a precursor lesion for basal-like breast carcinoma: identification of a novel p63/Her2/neu expressing subgroup. Oncotarget, 4(2), 231–241. doi: 10.18632/oncotarget.818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Tang B, Yu PW, Tang B, Hao YX, Lei X, … Zeng DZ (2012). Autophagy protects against oxaliplatin-induced cell death via ER stress and ROS in Caco-2 cells. PLoS One, 7(11), e51076. doi: 10.1371/journal.pone.0051076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson NE, Lambert WM, Watkins R, Giashuddin S, Huang SJ, Oxelmark E, … Garabedian MJ (2010). High levels of Hsp90 cochaperone p23 promote tumor progression and poor prognosis in breast cancer by increasing lymph node metastases and drug resistance. Cancer Res, 70(21), 8446–8456. doi: 10.1158/0008-5472.Can-10-1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solzak JP, Wang C, Hancock B, & Radovich M (2020). Abstract P3-10-12: Targeting a common drug compensation pathway using NUPR1 inhibition in triple negative breast cancer. Cancer Research, 80(4 Supplement), P3–10–12. doi: 10.1158/1538-7445.SABCS19-P3-10-12 [DOI] [Google Scholar]
- Subik K, Lee J-F, Baxter L, Strzepek T, Costello D, Crowley P, … Tang P (2010). The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast cancer : basic and clinical research, 4, 35–41. Retrieved from https://pubmed.ncbi.nlm.nih.gov/20697531https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2914277/ [PMC free article] [PubMed] [Google Scholar]
- Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, … Pan H (2013). Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death & Disease, 4(10), e838–e838. doi: 10.1038/cddis.2013.350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urra H, Dufey E, Avril T, Chevet E, & Hetz C (2016). Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer, 2(5), 252–262. doi: 10.1016/j.trecan.2016.03.007 [DOI] [PubMed] [Google Scholar]
- Urrutia R, Velez G, Lin M, Lomberk G, Neira JL, & Iovanna J (2014). Evidence supporting the existence of a NUPR1-like family of helix-loop-helix chromatin proteins related to, yet distinct from, AT hook-containing HMG proteins. J Mol Model, 20(8), 2357. doi: 10.1007/s00894-014-2357-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valacco MP, Varone C, Malicet C, Cánepa E, Iovanna JL, & Moreno S (2006). Cell growth-dependent subcellular localization of p8. J Cell Biochem, 97(5), 1066–1079. doi: 10.1002/jcb.20682 [DOI] [PubMed] [Google Scholar]
- Vincent AJ, Ren S, Harris LG, Devine DJ, Samant RS, Fodstad O, & Shevde LA (2012). Cytoplasmic translocation of p21 mediates NUPR1-induced chemoresistance: NUPR1 and p21 in chemoresistance. FEBS Lett, 586(19), 3429–3434. doi: 10.1016/j.febslet.2012.07.063 [DOI] [PubMed] [Google Scholar]
- Wang L, Jiang F, Xia X, & Zhang B (2019). LncRNA FAL1 promotes carcinogenesis by regulation of miR-637/NUPR1 pathway in colorectal cancer. Int J Biochem Cell Biol, 106, 46–56. doi: 10.1016/j.biocel.2018.09.015 [DOI] [PubMed] [Google Scholar]
- Yeh CT, Wu AT, Chang PM, Chen KY, Yang CN, Yang SC, … Huang CY (2012). Trifluoperazine, an antipsychotic agent, inhibits cancer stem cell growth and overcomes drug resistance of lung cancer. Am J Respir Crit Care Med, 186(11), 1180–1188. doi: 10.1164/rccm.201207-1180OC [DOI] [PubMed] [Google Scholar]
- Zhong C, Yu J, Li D, Jiang K, Tang Y, Yang M, … Yuan Y (2019). Zyxin as a potential cancer prognostic marker promotes the proliferation and metastasis of colorectal cancer cells. J Cell Physiol. doi: 10.1002/jcp.28236 [DOI] [PubMed] [Google Scholar]