

Abstract
Objective
Ligelizumab is a humanised IgG1 anti‐IgE antibody that binds IgE with higher affinity than omalizumab. Ligelizumab had greater efficacy than omalizumab on inhaled and skin allergen provocation responses in mild allergic asthma. This multi‐centre, randomised, double‐blind study was designed to test ligelizumab in severe asthma patients not adequately controlled with high‐dose inhaled corticoids plus long‐acting β2‐agonist.
Methods
Patients received 16 weeks ligelizumab (240 mg q2w), omalizumab or placebo subcutaneously, and ACQ‐7 was measured as primary outcome at Week 16. In addition, the study generated dose‐ranging data of ligelizumab and safety data.
Results
A total of 471 patients, age 47.4 ± 13.36 years, were included in the study. Treatment with ligelizumab did not significantly improve asthma control (ACQ‐7) and exacerbation rates compared to omalizumab and placebo. Therefore, primary and secondary objectives of the study were not met. The compound was well tolerated, and the safety profile showed no new safety findings. Pharmacokinetic data demonstrated faster clearance and lower serum concentrations of ligelizumab than historical omalizumab data, and exploratory in vitro data showed differential IgE blocking properties relative to FcεRI and FcεRII/CD23 between the two compounds.
Conclusion
Ligelizumab failed to demonstrate superiority over placebo or omalizumab. Although ligelizumab is more potent than omalizumab at inhibiting IgE binding to the high‐affinity FcεRI, there is differential IgE blocking properties relative to FcεRI and FcεRII/CD23 between the two compounds. Therefore, the data suggest that different anti‐IgE antibodies might be selectively efficacious for different IgE‐mediated diseases.
Keywords: asthma, biologics, CD23, ligelizumab, omalizumab
In this multi‐centre, randomised, double‐blind study, 471 severe asthmatics were treated with the high‐affinity anti‐IgE antibody ligelizumab. Treatment did not significantly improve asthma control and exacerbations compared to omalizumab and placebo. Although ligelizumab is more potent than omalizumab at inhibiting IgE binding to the high‐affinity FcεRI, there is differential IgE blocking properties relative to FcεRI and FcεRII/CD23 between the two compounds, potentially blunting the effect in asthma.

Introduction
Despite improved treatment regimens and pharmaceutical advancements, severe asthma remains a global burden. It affects approximately 5% of the overall asthma population, 15 million patients worldwide (ATS Workshop on refractory asthma 2000, GINA 2011). New potential monoclonal antibody therapies focus on severe asthma not only because of high morbidity and mortality among this subgroup, but also because it accounts for more than 80% of disease costs. 1
Omalizumab is the only licensed anti‐IgE antibody and is approved for severe, inadequately controlled asthma and chronic spontaneous urticaria (CSU). It has been established that lowering free IgE levels translates into significant clinical benefit. 2 , 3 , 4 Omalizumab has a moderate affinity for IgE (K D = 6.8 nm) and needs to be used in molar excess of IgE, limiting utilisation in patients with high body weight and/or high IgE levels (> 1500 IU mL−1). 5 Since an estimated 75% of severe asthmatics have IgE‐mediated asthma, the development of new anti‐IgE antibodies with improved properties remains important. 6 , 7 Moreover, a retrospective analysis of patients treated with omalizumab indicates that higher suppression of free IgE seems to be linked to better outcomes. 8
Ligelizumab (QGE031) is a humanised IgG1 monoclonal anti‐IgE antibody with high affinity (K D = 0.13 nm), which potently inhibits human IgE binding to the high‐affinity IgE receptor (FcεRI). Preclinical studies have shown an about 50‐fold increased affinity for human IgE compared to omalizumab. 9 First clinical studies confirmed that ligelizumab is more potent than omalizumab in suppressing free IgE and skin prick wheal responses in atopic patients. 9 Furthermore, ligelizumab showed greater efficacy than omalizumab on early responses to inhaled allergen provocations in mild allergic asthmatics. 10 , 11 , 12 Therefore, the purpose of this study was to evaluate whether the pharmacodynamic effects of ligelizumab translated into improved clinical outcomes for patients with severe uncontrolled asthma. Efficacy and safety data were collected in a randomised, multi‐centre phase‐2 dose‐ranging study comparing multiple doses of ligelizumab to placebo and omalizumab. The primary objective was efficacy of ligelizumab treatment compared to placebo and determined as responder rates. Response was defined to be improvement of ≥ 0.5 points of the ACQ‐7 score from baseline. A key secondary objective was the comparison of responder rates between ligelizumab and omalizumab. Since in this study ligelizumab failed to demonstrate superiority over placebo or omalizumab, further mechanistically in vitro investigations were conducted.
Results
A total of 1377 patients were screened (beginning 14 December 2012), of which 471 patients entered the treatment phase and were randomised to receive the study medication. Five patients did not receive any study drug and were, therefore, excluded from the full analysis set (FAS: 466 patients). Of all randomised patients, 92.4% (n = 435) completed the treatment phase of the study (until 21 January 2016). The main reasons for not completing the treatment phase were patient/guardian decision (2.5%), followed by protocol deviation (2.3%) and adverse event (1.9%). For complete information, see trial profile in Figure 1b. The patient groups were well matched, with no significant differences between the groups (Table 1).
Figure 1.

(a) Schematic study design, the main comparative component framed in red. (b) Trial profile.
Table 1.
Patient characteristics (FAS). The High‐dose ligelizumab group is pooled from ligelizumab 240 mg q2w, ligelizumab 240 mg q4w, ligelizumab 180 mg q2w, and ligelizumab 120 mg q2w treatment arms. The Low‐dose ligelizumab group is pooled from ligelizumab 36 mg q2w and ligelizumab 12 mg q2w treatment arms. The Placebo group is pooled from all ligelizumab placebo and omalizumab placebo.
| Ligelizumab | Omalizumab | Placebo | ||
|---|---|---|---|---|
| Low dose | High dose | |||
| Number | N = 199 | N = 40 | N = 131 | N = 96 |
|
Age Years – mean (range) |
47.6 (17–75) | 46 (22–69) | 46.8 (18–73) | 47.5 (25–75) |
|
Female sex No. (%) |
113 (56.8) | 20 (50) | 90 (68.7) | 62 (64.4) |
|
BMI kg m−2 – mean ± SD |
27.5 ± 4.70 | 27.4 ± 4.98 | 26.8 ± 4.55 | 27.4 ± 4.86 |
|
Duration of asthma Years – mean ± SD |
21.4 ± 15.37 | 18.2 ± 12.02 | 21.8 ± 13.56 | 22.9 ± 16.15 |
|
Asthma exacerbation (last 24 months) No. – mean ± SD |
2.5 ± 2.04 | 2.2 ± 1.70 | 2.2 ± 1.05 | 2.4 ± 1.61 |
|
Baseline IgE IU mL−1 – mean ± SD |
291.1 ± 287.21 | 302.0 ± 268.01 | 307.9 ± 253.06 | 292.1 ± 245.46 |
| FEV1 | ||||
|
FEV1 baseline Litres – mean ± SD |
1.86 ± 0.57 | 1.86 ± 0.54 | 1.80 ± 0.56 | 1.76 ± 0.52 |
|
Percent pred. FEV1 % – mean ± SD |
61.44 ± 10.12 | 60.54 ± 10.47 | 60.89 ± 11.10 | 59.44 ± 11.70 |
|
Increase in FEV1 after inhalation % – mean ± SD |
23.19 ± 14.78 | 23.18 ± 18.55 | 24.81 ± 17.43 | 25.31 ± 18.13 |
|
Former smokers No. (%) |
41 (20.6) | 8 (20.0) | 15 (11.5) | 17 (17.7) |
|
Baseline ICS category No. (%) | ||||
| Low | 12 ( 6.0) | 3 ( 7.5) | 7 ( 5.3) | 9 ( 9.4) |
| Medium | 48 (24.1) | 13 (32.5) | 29 (22.1) | 24 (25.0) |
| High | 127 (63.8) | 23 (57.5) | 84 (64.1) | 54 (56.3) |
|
Baseline ACQ‐7 Mean ± SD |
2.4 ± 0.65 | 2.5 ± 0.56 | 2.5 ± 0.69 | 2.4 ± 0.59 |
At Week 16, there was no statistical difference in the ACQ‐7 score among the ligelizumab, omalizumab and placebo groups. A numerically lower proportion of patients in the ligelizumab 240 mg q2w group (63.16%) achieved an improvement of ≤ −0.5 in ACQ‐7 score from baseline compared to the placebo group (70.21%) or to the omalizumab group (69.17%). Therefore, the responder rate as the primary objective was not met. The odds ratio for comparison between the responder rates of ligelizumab 240 mg q2w and the placebo group was not in favor of the ligelizumab treatment (OR: 0.79) while the comparison was not statistically significant (P = 0.58).
At Week 16, the mean change in ACQ‐7 score from baseline for ligelizumab 240 mg q2w, the placebo and omalizumab groups was −0.73, −0.78 and −0.76, respectively, as shown in Figure 2.
Figure 2.

ACQ‐7 change from baseline: There was no notable difference in the change from baseline in the ACQ‐7 score between the ligelizumab 240 mg q2w group (n = 120) and placebo (n = 49). Similarly for other ligelizumab dose groups, no notable differences could be seen compared to placebo.
The rate of moderate or severe asthma exacerbations per year was 0.64 for the ligelizumab high‐dose group versus 1.01 for the placebo group and 0.28 for the omalizumab group. Comparing ligelizumab 240 mg q2w to its placebo (Ratio of rates 1.04, 95% CI 0.417–2.621, P‐values 0.925) showed no difference as well as compared to omalizumab (Ratio of rates 2.02, 95% CI 0.865–4.713, P‐value 0.104). Kaplan–Meier estimates of time‐to‐first asthma exacerbation show a non‐significant improvement of ligelizumab treatment over placebo, but less pronounced than omalizumab treatment (Figure 3) in a post hoc analysis. The results of the omalizumab group were compatible with previous studies, although the study was not designed to measure the effect of omalizumab.
Figure 3.
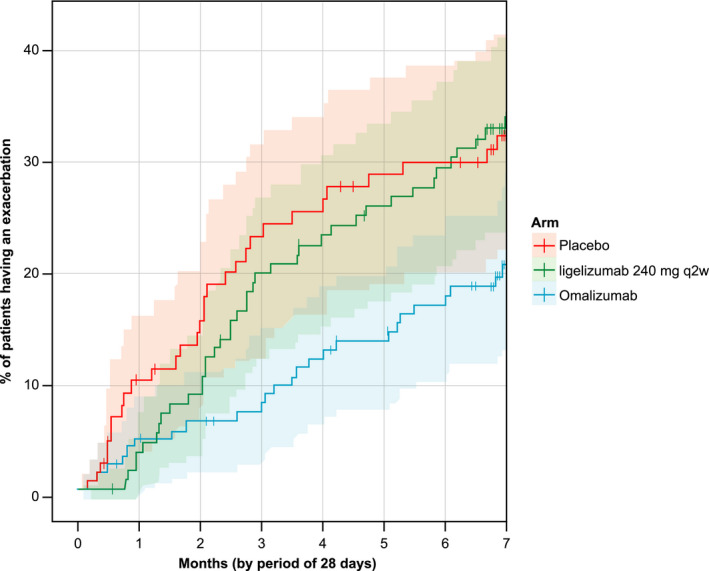
Time‐to‐first exacerbation: No difference in time‐to‐first exacerbation between ligelizumab (n = 120) and placebo (n = 96) 16 weeks after completion of treatment period in post hoc analysis (combined placebo groups).
Ligelizumab showed the expected pharmacokinetic behaviour with an increase in serum concentration during the initial dosing period approaching steady state after 6–12 weeks, with the exception of the Week 14 time point of the 120 mg q2w treatment group, which was biased by a single patient showing very high ligelizumab levels (Figure 4a).
Figure 4.

(a) Steady state of ligelizumab serum concentration was reached after 6–12 weeks, peaks differing according to dosing. (b) At 12 weeks, total IgE was increased in no apparent dose‐related manner by forming ligelizumab‐IgE complexes with reduced clearance. Mean free IgE was suppressed below the lower level of quantification at all concentrations above 120 mg q2w.
The ligelizumab levels measured at the follow‐up visit on Week 28, that is 84 days after the last administration, were only a small fraction of their corresponding end‐of‐treatment levels.
As expected, by forming ligelizumab‐IgE complexes with reduced clearance compared to free IgE, total serum IgE levels increased upon treatment with ligelizumab. There was no obvious correlation of IgE increase with dose. This was in line with analysis of free IgE, which indicated that already at the lowest dose of ligelizumab the majority of free serum IgE was bound as IgE complexes. Free serum IgE levels returned back to almost baseline levels on Week 28, 12 weeks after the last dose of ligelizumab. For q2w doses of ligelizumab of 12 and 36 mg, there were free IgE levels detectable throughout the period of active treatment, while at the 120 mg q2w dose level free serum IgE was temporarily suppressed below the lower level of quantification (LLOQ 2.50 ng mL−1). At doses higher than 120 q2w, free serum IgE was suppressed below LLOQ at almost all time points after treatment initiation (Figure 4b).
Following SC injection, the apparent clearance of ligelizumab (CL/F) and apparent volume of distribution (V/F) were estimated as 0.903 L day−1 (±3% standard error for parameter estimate) and 21.7 L (±3% standard error for parameter estimate) in a reference patient (body weight of 70 kg, baseline IgE level of 365 ng mL−1) using one‐compartment PK modelling. Therefore, ligelizumab exhibited faster apparent clearance and a larger apparent volume than omalizumab in the same reference patient (0.205 L day−1, 8.31 L, as estimated by one‐compartment PK modelling integrated in a PK‐IgE binding model 13 ).
There were no significant differences in overall adverse events (AE) between the study groups (61.8%, 65.0%, 52.7% and 56.3% of the patients in the ligelizumab high‐dose, ligelizumab low‐dose, omalizumab and placebo treatment groups). Overall, the most commonly affected system organ classes (SOCs) (≥ 10% in total) were infections and infestations (30.9%); respiratory, thoracic and mediastinal disorders (26.2%); and general disorders and administration site conditions (21.2%). One death was reported from the study which was from the ligelizumab placebo group.
Due to the negative outcome of the trial, further mechanistic in vitro investigations were conducted.
IgE‐mediated degranulation of mast cells was performed using a hapten‐specific IgE (JW8) and triggered with hapten‐conjugated BSA (see Methods section). Ligelizumab and omalizumab inhibited this IgE‐mediated degranulation of mast cell with IC50 values of 0.14 and 4.52 nm, respectively (Figure 5a). Thus, in this assay system, ligelizumab was about 32‐fold more potent than omalizumab to inhibit FcεRI‐induced degranulation of mast cells.
Figure 5.

(a) Inhibition of FcεRI‐induced hexosaminidase release from mast cells by ligelizumab (black symbols) and omalizumab (blue symbols). (b) Inhibition of IgE binding to FcεRI (left) or CD23 (right) by omalizumab (blue, n = 5) or ligelizumab (ligelizumab, black, n = 5). Individual IC50 values from repeat experiments and medians (black lines) are shown. CD23 binding data are for RPMI 8866 (circles, n = 4), IM9 (triangles n = 2) and EBV B cells (squares, n = 2). Ligelizumab inhibits IgE binding to FcεRI bearing mast cells more potently than omalizumab, whereas omalizumab is more potent at inhibiting IgE binding to CD23. P‐values from t‐tests for the respective groups are indicated on top.
The two antibodies were analysed for their relative potency to inhibit IgE binding to FcεRI and FcεRII/CD23 bearing cells (Figure 5b). Ligelizumab inhibited IgE binding to FcεRI‐expressing mast cells more potently than omalizumab (statistically not significant). However, this was not the case for CD23‐expressing cells, where ligelizumab was less potent than omalizumab. These data show that the two therapeutic anti‐IgE antibodies show important qualitative differences in the interference of the two IgE‐mediated pathways.
Discussion
This randomised multi‐centre phase‐2 study was designed to assess the efficacy and safety of ligelizumab, a humanised, high‐affinity IgG1 anti‐IgE antibody and to compare its effect to omalizumab and placebo in a population of severe and uncontrolled asthmatics. In previous studies, ligelizumab had greater efficacy than omalizumab on inhaled and skin allergen provocation responses in mild allergic asthmatics, which provided a plausible rationale to investigate ligelizumab in severe asthmatics. Surprisingly, the primary objective of the study, improvement of the ACQ‐7, was not met.
In order to achieve significant outcomes, the ACQ‐7 was chosen as the primary endpoint. The ACQ‐7 is used extensively in clinical trials to measure clinically meaningful change in asthma control. 14 Still, the determination of primary endpoints remains an inherent problem of asthma research, especially since lung function testing often does not correlate with clinical benefit. 1 The ACQ‐7 scoring system consists of patient questions, β‐agonist use and lung function testing. 15 Hence, the establishment of superiority of a new drug for this composite endpoint might be more difficult. Comparable studies for omalizumab utilised exacerbation rates as primary endpoint and were able to show a significant decrease. 2 , 16 In addition, several ‘real life’ studies have shown better asthma control and a significant decrease in the exacerbations in patients treated with omalizumab. 17 , 18 The analysis of exacerbations, exacerbation rate and time‐to‐first exacerbation did not show a significant benefit of ligelizumab over placebo. It is possible though that the duration of the study was too short to address exacerbations as outcome, since even in the placebo group only less than a quarter of the patients experienced an exacerbation within the 16‐week treatment period. However, in a post hoc analysis, the ligelizumab 240 mg q2w group did show favorable findings related to reductions in severe asthma exacerbations. Nevertheless, it needs to be noted that the presented study was not powered for exacerbations and findings, therefore, need to be interpreted with caution.
A recent meta‐analysis has shown that a 16‐week treatment with omalizumab has an average impact on the ACQ of −41%, whereas exacerbations are reduced by 81%. 19 We, therefore, conclude that the lower magnitude of change of the ACQ compared to exacerbations combined with utilisation of the ACQ‐7 might explain part of the missing effect of ligelizumab on the primary endpoint.
Additional mechanistic data were generated in response to the initially somehow unexpected clinical observations of this study. The data suggested that ligelizumab is more potent than omalizumab at inhibiting IgE signalling through FcεRI, and it is less potent at inhibiting IgE signalling mediated by FcεRII/CD23. These findings have been supported by recently published additional mechanistic data describing different profiles of the two compounds relative to their ability to inhibit the IgE signalling through FcεRI and CD23 20 as a consequence of the different epitopes on the IgE molecule they bind to. These data are in alignment with additional prior research showing experimental antibodies having the ability to differentially influence IgE binding to these two receptors; that is, anti‐IgE antibodies have been described that only modulate IgE binding to FcεRII/CD23. 21 , 22
Therefore, the outcomes of this study might reflect the clinical translation of the unique mechanistic profiles of ligelizumab and omalizumab in a complex disease like asthma, where the relative contribution of the two IgE receptors to its pathophysiology might require further investigations. Although ligelizumab showed faster systemic clearance than omalizumab, ligelizumab effectively suppressed free IgE in serum and showed longer and higher IgE inhibition than omalizumab, consistent with its higher binding affinity to IgE. 9 However, PK exposure and IgE data at local pulmonary tissue were not available to assess any interactions in the target tissues (e.g. levels of local IgE concentrations and relative inhibition).
Nevertheless, in prior studies, ligelizumab was consistently more effective than omalizumab in inhibiting clinical pharmacodynamics endpoints strongly associated with the FcεRI (e.g. skin prick test wheal size), 9 , 10 hence supporting its further development in indications where IgE and its high‐affinity receptor potentially play a more pronounced role like CSU, seasonal rhinitis and food allergies. 23 The recent report of strong clinical results of a Phase 2b dose range finding study comparing ligelizumab and omalizumab efficacy in CSU patients supports this concept of an anti‐IgE antibody more potently and selectively blocking IgE action at the FcεRI to be sufficient for a larger clinical benefit in this indication. 24
These results raise the question about the role of the CD23 receptor in asthma. A potential key role of CD23 in allergic asthma was demonstrated in animal models, where allergen‐induced pulmonary eosinophilic inflammation was shown to be a CD23 pathway‐dependent event and could be prevented by appropriate anti‐IgE treatment. 25 , 26 CD23 on B cells may also participate in activating allergen‐specific T cells 27 and consecutively maintaining the eosinophilic inflammation through cytokine release. Moreover, Assayag et al. 28 could detect a receptor modulation of the CD23 after treatment of severe asthmatics with omalizumab. Therefore, a plausible hypothesis is that CD23 may play an important role in the inflammatory process associated with asthma and that ligelizumab's less potent inhibition (compared to omalizumab) might have contributed to the findings of this study.
In conclusion, this study hints to a more complex pathophysiology of IgE‐driven diseases, where anti‐IgE antibodies with different modes of action seem to be selectively efficacious for different IgE‐mediated diseases. FcεRI‐predominant diseases like chronic urticaria, seasonal rhinitis or food allergies might, therefore, benefit from ligelizumab, whereas the concomitant inhibition of FcεRII/CD23 seems to be important in allergic asthma.
Methods
Study design
This was a randomised, multi‐centre, parallel‐group, 14‐arm, double‐blind study design, where ligelizumab, omalizumab or matched placebo (s.c. injection) were added to high‐dose inhalative corticosteroids (ICS) plus long‐acting β‐agonist (LABA) according to GINA 2011 guideline step 4 asthma therapy. Placebo was administered according to the volume and frequency of each active component; ligelizumab 240 mg every 2 weeks and matched placebo and omalizumab and matched placebo as per locally approved dosing table (treatment arms A and B). This study also included an active comparative component, with comparisons of different doses of ligelizumab ranging from 12 mg biweekly to 240 mg every 4 weeks (Arms C to G, see Figure 1 for detail). Treatment period encompassed 16 weeks with treatment‐free follow‐up of additional 12 weeks. For a graphical presentation of the study design, see Figure 1a. The primary objective was efficacy of ligelizumab treatment compared to placebo and determined as responder rates. The response was defined to be improvement of ≥ 0.5 points of the ACQ‐7 score from baseline. The key secondary objective was the comparison of responder rates between ligelizumab and omalizumab. Other secondary and explorative objectives were AQLQ (Asthma Quality of Life Questionnaire) scores, Trough FEV1 and exacerbation rates.
For specific post hoc analyses, treatment groups were pooled into a high‐dose group (ligelizumab 240 mg q2w, 240 mg q4w, 180 mg q2w, 120 mg q2w), a low‐dose group (ligelizumab 36 mg q2w, 12 mg q2w) and a placebo group (all ligelizumab placebo and omalizumab placebo). The study was approved by local ethics committee and registered with ClinicalTrials.gov as NCT01716754.
Study population
Adult patients aged ≥ 18 to ≤ 75 years (BMI 18–40 kg m−2) with a diagnosis of asthma (according to GINA 2011 guidelines) for a period of at least 24 months that had signed written informed consent were eligible to participate in the study. A documented history of at least one asthma exacerbation in the previous 12 months that had a documented treatment with systemic corticosteroids for at least 3 days or a depot‐injectable dose of corticosteroids or that had required hospitalisation or emergency room (ER) was mandatory. Pulmonary function testing had to exhibit a FEV1 of ≥ 40% and ≤ 80% of the predicted normal value for the patient, after withholding bronchodilators and demonstrate an increase in FEV1 of ≥ 12% and 200 mL within 30 min after administration of 400 µg salbutamol/albuterol (or equivalent dose) prior to randomisation. Daily treatment at Visit 1 had to include a medium‐ or high‐dose ICS plus a LABA (b.i.d. or equivalent) for ≥ 3 months prior and demonstrate inadequately controlled asthma on current treatment by an Asthma Control Questionnaire (ACQ) score of ≥ 1.5 15 at Visit 1 and prior to randomisation. Patients had to be allergic or atopic to a perennial aeroallergen (house dust mite, cockroach, mould, cat/dog dander or other perennial allergens), as diagnosed by either a skin prick test (≥ 3 mm diameter above background) or a positive in vitro specific IgE (e.g. RAST/CAP) test.
Exclusion criteria included active smoking within the 6‐month period prior to Visit 1, or a smoking history of greater than 10 pack years, asthma attack/exacerbation requiring a short burst of systemic corticosteroids for at least 3 days within 6 weeks prior to Visit 1, a respiratory tract infection or asthma worsening within 4 weeks prior to Visit 1.
Randomisation and masking
Randomisation was achieved via Interactive Response Technology (IRT). The independent and unblinded study pharmacist or other appropriately trained personnel contacted the IRT system after the investigator or his/her delegate confirmed that the patient fulfilled all the inclusion and exclusion criteria. The IRT assigned a randomisation number to the patient.
Randomisation was stratified by prior history of asthma exacerbations in the 24 months prior to screening (> 2; yes/no), and by baseline serum IgE (≤ 700 IU mL−1, > 700 IU mL−1).
The appearance of placebo medication was identical to that of active drug to maintain the blind. Patients, investigator staff, persons performing the study assessments, and data analysts remained blinded to the identity of the treatment from the time of randomisation until database lock.
Asthma Control Questionnaire (ACQ‐7)
The Asthma Control Questionnaire 15 , 29 consists of five items to assess symptoms and activity limitations, one item on airway calibre (FEV1 % predicted) and one item on rescue β2‐agonist use. Patients complete items 1–6 and clinical staff score item 7 on FEV1 % predicted. The recall time was 1 week. All seven items were scored on a 7‐point Likert scale.
Spirometry (Trough FEV1)
Equipment for spirometry assessments was provided to all study sites by a Central Spirometry vendor, and all measurements were reviewed by trained spirometry technicians at the central vendor.
Exacerbations
The start date of an asthma exacerbation was defined as the first day that required the use of ‘rescue’ treatment, defined as systemic corticosteroids for at least 3 days or hospitalisation or emergency department visit. 30
At the end of an exacerbation, the patient was required to attend the clinic, where possible, for an assessment of the episode. Clinical significant exacerbations were defined as either a severe asthma exacerbation (treatment with ‘rescue’ systemic corticosteroids for greater than or equal to 3 days, and hospitalisation or emergency department visit (greater than 24 h) or death due to asthma) or a moderate asthma exacerbation (‘rescue’ systemic corticosteroids for greater than or equal to 3 days either as an outpatient or in emergency department visits).
Ligelizumab, total and free IgE concentrations
A single blood collection accounted for detection of ligelizumab, total IgE and free IgE analytes. Concentrations were assessed from blood samples taken predose on weeks 0, 2, 4, 6, 8, 10, 12, 14 and 16. An additional blood sample was collected on the follow‐up visit at Week 28 (Day 196), that is 12 weeks (84 days) after the last administration of study drug. Ligelizumab concentrations were determined in serum by an ELISA assay with a lower limit of quantification (LLOQ) of 0.500 µg·mL−1. Total IgE in serum was determined on Phadia 100 platform by ImmunoCAP® Total IgE assay (Phadia AB, Uppsala, Sweden) with LLOQ of 2.0 IU mL−1 in human serum (1 IU = 2.42 ng mL−1). Free IgE in serum was determined using a sandwich ELISA with a lower limit of quantification (LLOQ) of 2.50 ng mL−1 and an upper limit of quantification (ULOQ) of 250 ng mL−1 in human serum.
Clearance
Clearance of ligelizumab was calculated using a nonlinear mixed effect pharmacokinetic model specifying one distribution compartment, a single clearance process and first‐order absorption from the injection site. It was run using the MONOLIX software system, version 4.3.2 (Lixoft, Paris, France).
Preparation of human mast cells
Human peripheral blood‐derived mast cells were generated from CD34+ progenitor cells according to Schmetzer et al. 31 with modifications. Peripheral blood‐derived CD34+ cells were obtained from mononuclear cells by magnetic separation using anti‐CD34 antibody (Miltenyi, Bergisch‐Gladbach, Germany) and LS columns (Miltenyi). The cells were cultured for 6 weeks in a methylcellulose‐based medium (MethoCult, STEMCELL Technologies, Vancouver, BC, Canada) supplemented with 1 µg mL−1 SCF, 0.25 µg mL−1 IL‐6 and 0.025 µg mL−1 IL‐3 to promote differentiation into mast cells. After 6 weeks, the cells were transferred to liquid culture in IMDM media containing 1% Pen Strep, 50 µm 2‐mercaptoethanol, 0.1 µg mL−1 SCF, 0.05 µg mL−1 IL‐6, 1:100 dilution of Insulin‐Transferrin‐Selenium and 0.1% BSA. Cell characterisation and quantification of FcεRI expression confirmed that cells at this point are considered mature mast cells. In order to further upregulate FcεRI expression and, thus, to increase sensitivity to IgE, mast cells were treated with IL‐4 at 10 ng mL−1 for 2 days before use.
Mast cell degranulation assay (hexosaminidase release)
On the day of the assay, 2.5 × 104 mast cells (in HBSS with Ca2+/Mg2+ plus 0.1% BSA) were plated in 50 µL per well into 96‐well round bottom polypropylene plates. Ligelizumab or omalizumab was serially diluted at 3× the final concentration, and 50 µL was added to the cells and incubated at 37°C for 30 min. Thereafter, 50 µL of a mixture of NIP‐specific IgE (clone JW8) or non‐specific IgE (Abbiotec, Escondido, CA, USA) was added to a final concentration of 20 or 80 ng mL−1, respectively. The cells were incubated for 2 h, then centrifuged and washed in the same buffer. Cells were resuspended in 110 µL of buffer containing 20 ng mL−1 NIP5‐BSA, which cross‐links cell‐bound JW8. For the total amount of hexosaminidase, cells were lysed with 110 µL 0.1% Triton X‐100. Further controls assessed spontaneous hexosaminidase release and stimulation without anti‐IgE. Cells were incubated for 30 min at 37°C before the plates were placed on ice, centrifuged and supernatants harvested and tested for β‐hexosaminidase using 50 µL of supernatant and an equal volume of β‐hexosaminidase substrate solution (192.5 mL of 0.1 m sodium acetate pH 4.4 plus 250 mg p‐nitrophenyl N‐acetyl, β‐d‐glucosaminide). After incubation for 1.5 h at 37°C, the reaction was quenched by adding 50 µL of 1 m Tris‐base. The plates were then read with excitation at 355 nm and emission at 460 nm on the Envision plate reader (Perkin Elmer, Waltham, MA, USA). Data were analysed as a % release of total content taking spontaneous release into account and expressed as per cent inhibition.
Inhibition of IgE binding to FcεRI and FcεRII bearing cells
For B cell lines, RPMI 8866 (Sigma‐Aldrich, St. Louis, MO, USA), IM9 cells (ATCC CCL‐159) and the EBV‐transformed B cells HD2 (received from Dr Heiko Sic, Novartis) were used. In brief, for each condition, 10 µL of human IgE clone B11/SUS‐11 (final concentration of 5.26 nm) was mixed with 10 µL of pre‐diluted omalizumab or ligelizumab and incubated at 37°C for 30 min. The concentration range of omalizumab and ligelizumab tested was between 0.02 and 337 nm.
Mast cells or B cells were resuspended in incubation buffer at a cell density of 1.25 × 106 cells mL−1. Of this cell suspension, 105 cells per well were added to the pre‐mixed IgE/anti‐IgE and incubated for 60 min at 37°C. After the incubation, the plates were cooled on ice and centrifuged to pellet the cells. The cells were then incubated with 20 µL per well of Privigen (human IgG, 1 mg mL−1, CSL Behring, King of Prussia, USA) to block Fc receptors after which 20 µL of anti‐IgE‐PE (Ige21; eBioscience, San Diego, CA, USA) was added to detect surface‐bound IgE. In some experiments, double stainings were performed with anti‐IgE‐PE and anti CD23‐APC (BD Bioscience, Franklin Lakes, NJ, USA) or anti‐IgE‐PE and anti FcεRI‐APC (BioLegend, San Diego, CA, USA). After incubation for 30 min at 4°C, cells were washed twice with 100 µL of ice‐cold FACS buffer, resuspended in 50 µL of FACS buffer and measured in a CyAn flow cytometer (Beckman Coulter, Brea, CA, USA). Inhibition of IgE binding was calculated using ExcelFit and the one‐site dose‐response model. Binding of IgE to CD23 and inhibition by omalizumab and ligelizumab represent combined data obtained with RPMI8866, IM9 and EBV HD2 B cells.
Statistical analysis
The sample size of 104 patients per treatment arm had 85% power with a two‐sided type I error of 0.05, to detect a difference of 20% in responder rates of ligelizumab and omalizumab based on Fisher's exact test. Assuming 10% drop out during the study, the sample size was rounded to 120 patients per ligelizumab and omalizumab arms.
Data were analysed by Novartis according to the planned data analyses. The full analysis set (FAS) included all randomised patients who received at least one dose of the study drug. Following the intent‐to‐treat principle, patients were analysed according to the treatment they were assigned to at random. FAS was used for all efficacy variables, unless otherwise stated. The patients who were randomised but not diagnosed with asthma were excluded from the full analysis set. The proportion of patients with a change from baseline in ACQ‐7 of ≤ –0.5 was summarised by treatment and analysed using a repeated measures logistic regression.
Conflict of interest
JT has nothing to disclose. AG is an employee of Novartis. AP is an employee of Novartis Pharmaceuticals Corporation. HGZ is an employee of Novartis, during the conduct of the study. IB is an employee of Novartis Pharma AG. RW reports personal fees from Novartis Pharma AG, during the conduct of the study, and personal fees from Novartis Pharma AG, outside the submitted work. XJ is an employee of Novartis. CH has nothing to disclose. SZ reports grants and personal fees from bene‐Arzneimittel GmbH, grants and personal fees from Biotest GmbH, grants from Vifor Pharma Deutschland GmbH, grants from ALK Arzneimittel, personal fees from Novartis GmbH, personal fees from Böhringer Ingelheim, personal fees from Lofarma GmbH, personal fees from IMS HEALTH GmbH & Co. OHG, personal fees from GSK, personal fees from Stallergen, personal fees from Procter and Gamble, personal fees from Allergopharma GmbH, grants and personal fees from Allergy Therapeutics, personal fees from Engelhard Arzneimittel, personal fees from Sanofi‐Pasteur, personal fees from AstraZeneca, personal fees from EryDel, and personal fees from Bionorica GmbH, outside the submitted work.
Author contributions
Jordis Trischler: Data curation; Writing‐original draft. Ivan Bottoli: Conceptualization; Writing‐review & editing. Reinhold Janocha: Conceptualization; Writing‐review & editing. Christoph Heusser: Investigation; Writing‐review & editing. Xavier Jaumont: Conceptualization. Philip John Lowe: Conceptualization; Formal analysis. Aurelie Gautier: Conceptualization; Formal analysis; Writing‐review & editing. Abhijit Pethe: Conceptualization; Formal analysis; Writing‐review & editing. Ralph Woessner: Conceptualization. Hans‐Günter Zerwes: Investigation. Stefan Zielen: Data curation; Writing‐review & editing.
Acknowledgments
The contribution of A Hauchard for the IgE binding assays is acknowledged.
ClinicalTrials.gov Identifier: NCT01716754.
References
- 1. Normansell R, Walker S, Milan SJ, Walters EH, Nair P. Omalizumab for asthma in adults and children. Cochrane Database Syst Rev 2014; (1): CD003559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Busse W, Corren J, Lanier BQ et al. Omalizumab, anti‐IgE recombinant humanized monoclonal antibody, for the treatment of severe allergic asthma. J Allergy Clin Immunol 2001; 108: 184–190. [DOI] [PubMed] [Google Scholar]
- 3. Humbert M, Beasley R, Ayres J et al. Benefits of omalizumab as add‐on therapy in patients with severe persistent asthma who are inadequately controlled despite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 2005; 60: 309–316. [DOI] [PubMed] [Google Scholar]
- 4. Soler M, Matz J, Townley R et al. The anti‐IgE antibody omalizumab reduces exacerbations and steroid requirement in allergic asthmatics. Eur Respir J 2001; 18: 254–261. [DOI] [PubMed] [Google Scholar]
- 5. Zielen S, Lieb A, De La Motte S et al. Omalizumab protects against allergen‐ induced bronchoconstriction in allergic (immunoglobulin E‐mediated) asthma. Int Arch Allergy Immunol 2013; 160: 102–110. [DOI] [PubMed] [Google Scholar]
- 6. Moore WC, Bleecker ER, Curran‐Everett D et al. Characterization of the severe asthma phenotype by the National Heart, Lung, and Blood Institute's Severe Asthma Research Program. J Allergy Clin Immunol 2007; 119: 405–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. The ENFUMOSA cross‐sectional European multicentre study of the clinical phenotype of chronic severe asthma. European Network for Understanding Mechanisms of Severe Asthma. Eur Respir J 2003; 22: 470–477. [DOI] [PubMed] [Google Scholar]
- 8. Lowe PJ, Tannenbaum S, Gautier A, Jimenez P. Relationship between omalizumab pharmacokinetics, IgE pharmacodynamics and symptoms in patients with severe persistent allergic (IgE‐mediated) asthma. Br J Clin Pharmacol 2009; 68: 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arm JP, Bottoli I, Skerjanec A et al. Pharmacokinetics, pharmacodynamics and safety of QGE031 (ligelizumab), a novel high‐affinity anti‐IgE antibody, in atopic subjects. Clin Exp Allergy 2014; 44: 1371–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gauvreau GM, Arm JP, Boulet LP et al. Efficacy and safety of multiple doses of QGE031 (ligelizumab) versus omalizumab and placebo in inhibiting allergen‐induced early asthmatic responses. J Allergy Clin Immunol 2016; 138: 1051–1059. [DOI] [PubMed] [Google Scholar]
- 11. Galli SJ, Tsai M. IgE and mast cells in allergic disease. Nat Med 2012; 18: 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hide M, Francis DM, Grattan CE, Hakimi J, Kochan JP, Greaves MW. Autoantibodies against the high‐affinity IgE receptor as a cause of histamine release in chronic urticaria. N Engl J Med 1993; 328: 1599–1604. [DOI] [PubMed] [Google Scholar]
- 13. Honma W, Gautier A, Paule I, Yamaguchi M, Lowe PJ. Ethnic sensitivity assessment of pharmacokinetics and pharmacodynamics of omalizumab with dosing table expansion. Drug Metab Pharmacokinet 2016; 31: 173–184. [DOI] [PubMed] [Google Scholar]
- 14. Barnes PJ, Casale TB, Dahl R, Pavord ID, Wechsler ME. The Asthma Control Questionnaire as a clinical trial endpoint: past experience and recommendations for future use. Allergy 2014; 69: 1119–1140. [DOI] [PubMed] [Google Scholar]
- 15. Juniper EF, Bousquet J, Abetz L, Bateman ED, Committee G. Identifying ‘well‐controlled’ and ‘not well‐controlled’ asthma using the Asthma Control Questionnaire. Respir Med 2006; 100: 616–621. [DOI] [PubMed] [Google Scholar]
- 16. Soler M. Omalizumab, a monoclonal antibody against IgE for the treatment of allergic diseases. Int J Clin Pract 2001; 55: 480–483. [PubMed] [Google Scholar]
- 17. Barnes N, Menzies‐Gow A, Mansur AH et al. Effectiveness of omalizumab in severe allergic asthma: a retrospective UK real‐world study. J Asthma 2013; 50: 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Deschildre A, Marguet C, Salleron J et al. Add‐on omalizumab in children with severe allergic asthma: a 1‐year real life survey. Eur Respir J 2013; 42: 1224–1233. [DOI] [PubMed] [Google Scholar]
- 19. MacDonald KM, Kavati A, Ortiz B, Alhossan A, Lee CS, Abraham I. Short‐ and long‐term real‐world effectiveness of omalizumab in severe allergic asthma: systematic review of 42 studies published 2008–2018. Expert Rev Clin Immunol 2019; 15: 553–569. [DOI] [PubMed] [Google Scholar]
- 20. Gasser P, Tarchevskaya SS, Guntern P et al. The mechanistic and functional profile of the therapeutic anti‐IgE antibody ligelizumab differs from omalizumab. Nat Commun 2020; 11: 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Davies AM, Allan EG, Keeble AH et al. Allosteric mechanism of action of the therapeutic anti‐IgE antibody omalizumab. J Biol Chem 2017; 292: 9975–9987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shiung YY, Chiang CY, Chen JB et al. An anti‐IgE monoclonal antibody that binds to IgE on CD23 but not on high‐affinity IgE.Fc receptors. Immunobiology 2012; 217: 676–683. [DOI] [PubMed] [Google Scholar]
- 23. Zellweger F, Eggel A. IgE‐associated allergic disorders: recent advances in etiology, diagnosis, and treatment. Allergy 2016; 71: 1652–1661. [DOI] [PubMed] [Google Scholar]
- 24. Maurer M, Gimenez‐Arnau AM, Sussman G et al. Ligelizumab for chronic spontaneous urticaria. N Engl J Med 2019; 381: 1321–1332. [DOI] [PubMed] [Google Scholar]
- 25. Coyle AJ, Wagner K, Bertrand C, Tsuyuki S, Bews J, Heusser C. Central role of immunoglobulin (Ig) E in the induction of lung eosinophil infiltration and T helper 2 cell cytokine production: inhibition by a non‐anaphylactogenic anti‐IgE antibody. J Exp Med 1996; 183: 1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Palaniyandi S, Liu X, Periasamy S et al. Inhibition of CD23‐mediated IgE transcytosis suppresses the initiation and development of allergic airway inflammation. Mucosal Immunol 2015; 8: 1262–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Selb R, Eckl‐Dorna J, Neunkirchner A et al. CD23 surface density on B cells is associated with IgE levels and determines IgE‐facilitated allergen uptake, as well as activation of allergen‐specific T cells. J Allergy Clin Immunol 2017; 139: 290–299 e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Assayag M, Moshel S, Kohan M, Berkman N. The effect of omalizumab treatment on the low affinity immunoglobulin E receptor (CD23/fc epsilon RII) in patients with severe allergic asthma. Allergy Asthma Proc 2018; 39: 36–42. [DOI] [PubMed] [Google Scholar]
- 29. Juniper EF, O'Byrne PM, Guyatt GH, Ferrie PJ, King DR. Development and validation of a questionnaire to measure asthma control. Eur Respir J 1999; 14: 902–907. [DOI] [PubMed] [Google Scholar]
- 30. Reddel HK, Taylor DR, Bateman ED et al. An official American Thoracic Society/European Respiratory Society statement: asthma control and exacerbations: standardizing endpoints for clinical asthma trials and clinical practice. Am J Respir Crit Care Med 2009; 180: 59–99. [DOI] [PubMed] [Google Scholar]
- 31. Schmetzer O, Valentin P, Smorodchenko A, et al. A novel method to generate and culture human mast cells: Peripheral CD34+ stem cell‐derived mast cells (PSCMCs). J Immunol Methods 2014; 413: 62–68. [DOI] [PubMed] [Google Scholar]