

Significance
Host-adapted bacteria, both pathogenic and commensal, have developed multimodular enzymatic systems to cope with the complex glycans found in the host environment, such as the highly competitive gut niche. Among these deployed enzymatic arsenals are O-glycopeptidases, which uniquely target proteins with O-linked glycan modification. Through meticulous structural and functional analyses, we probe the molecular basis of glycan recognition and illuminate the complete structure of an ultramultimodular O-glycopeptidase. This provides extraordinary insight into glycan recognition in the catalytic site of this unique class of peptidase. It also reveals how noncatalytic carbohydrate binding by ancillary modules and substrate recognition in the O-glycopeptidase active site can be specifically coordinated in three-dimensional space.
Keywords: Clostridium, mucin, structure, multimodular, O-glycan
Abstract
A challenge faced by peptidases is the recognition of highly diverse substrates. A feature of some peptidase families is the capacity to specifically use post-translationally added glycans present on their protein substrates as a recognition determinant. This is ultimately critical to enabling peptide bond hydrolysis. This class of enzyme is also frequently large and architecturally sophisticated. However, the molecular details underpinning glycan recognition by these O-glycopeptidases, the importance of these interactions, and the functional roles of their ancillary domains remain unclear. Here, using the Clostridium perfringens ZmpA, ZmpB, and ZmpC M60 peptidases as model proteins, we provide structural and functional insight into how these intricate proteins recognize glycans as part of catalytic and noncatalytic substrate recognition. Structural, kinetic, and mutagenic analyses support the key role of glycan recognition within the M60 domain catalytic site, though they point to ZmpA as an apparently inactive enzyme. Wider examination of the Zmp domain content reveals noncatalytic carbohydrate binding as a feature of these proteins. The complete three-dimensional structure of ZmpB provides rare insight into the overall molecular organization of a highly multimodular enzyme and reveals how the interplay of individual domain function may influence biological activity. O-glycopeptidases frequently occur in host-adapted microbes that inhabit or attack mucus layers. Therefore, we anticipate that these results will be fundamental to informing more detailed models of how the glycoproteins that are abundant in mucus are destroyed as part of pathogenic processes or liberated as energy sources during normal commensal lifestyles.
The turnover of proteins in normal or pathogenic processes requires hydrolysis of the peptide bond, a reaction catalyzed by the peptidase enzyme superfamily. The concept of peptidase specificity is captured in the universally adopted nomenclature defining subsites (S sites) in the enzyme active site that accommodate specific amino acids of the protein or peptide substrate (P residues) (1). As potential protease substrates, however, proteins are often post-translationally modified, and, as such, peptidases must also contend with these modifications to their substrates. Glycosylation is thought to be the most frequent modification of proteins with an estimated one-half of all proteins in nature bearing covalently linked glycans (2). The glycans on glycoproteins are most frequently attached to either asparagine (N-linked) or serine/threonine (O-linked) sidechains. They are structurally diverse and contribute to the complexity of the so-called glycome of an organism, where the key roles glycans play in intercellular and intracellular processes are integral to multicellularity (3). Individual glycoproteins can be simple, bearing a single defined glycan, to having heavy modifications with varied glycans. Mucins are an example of the latter and can be up to 80% by mass of primarily O-linked glycans (4). Glycosylation is frequently credited for the increased stability of these glycoproteins, yet these glycoconjugates remain substrates for peptidases. Indeed, some peptidases target only mucins or glycoproteins having mucin-type glycosylation, indicating that they tolerate glycosylation. In the case of some peptidases, they even rely on glycosylation as a key substrate recognition determinant (5–10).
Pfam 13402 or the “peptidase_M60 family” was classified through characterization of a founding member combined with bioinformatics analysis (9). This revealed a mucin-active peptidase with the defining feature of relatedness to insect enhancins, the presence of a zinc-metallopeptidase gluzincin motif (HEXXHX(8,28)E), and multimodularity (i.e., the occurrence of noncatalytic carbohydrate-binding modules; CBMs) (11, 12). The peptidase_M60 family presently has nearly 900 sequence entries from bacteria, plants, fungi, and animals. Recently, proteins from Bacteroides thetaiotaomicron (BT4244), Pseudomonas aeruginosa (IMPa), Clostridium perfringens (ZmpB), and Akkermansia muciniphila (Amuc_0627, Amuc_1514, Amuc_0908), which all contain peptidase_M60 domains, were demonstrated to have a high specificity for the glycans of O-glycoslyated substrates. These enzymes hydrolyzed the peptide bond immediately N terminal to the O-glycan bearing residue in a zinc-dependent reaction (5, 8). Thus, for these “O-glycopeptidases” the active site nomenclature was modified to include the presence of G-sites that accommodate the glycan components. Notably, BT4244 and ZmpB are classified in the MEROPS peptidase database (13) as family M60 peptidases, IMPa is classified as M88, and A. muciniphila Amuc_1514 is classified as an M98 peptidase, yet all fall into the peptidase_M60 family. This indicates that the peptidase_M60 family (i.e., Pfam 13402) is a complex superfamily of multiple individual MEROPS classified metallopeptidase families which may share glycan recognition as another defining feature.
The human gut is host to trillions of bacteria where the interaction of this microbial community with the host is mediated largely by a mucous layer. As competition for nutrients in this environment is intense, and glycans decorating the glycoconjugates in the mucous layer (e.g., glycoproteins like mucins) represent a consistent carbohydrate source, many species of bacteria have adapted to utilize these substrates (14). Consistent with this, genes encoding peptidase_M60 domains are frequently found in members of the dominant gut phyla: Bacteroidetes, Verrucomicrobia, and Firmicutes (15). For example, genes encoding proteins with peptidase_M60 domains are noted to be expanded in human gut Bacteroides sp., and the expression of these genes are up-regulated under in vivo conditions where metabolism of the host mucin layer is promoted (16). Similarly, three of the four peptidase_M60 proteins of A. muciniphila are up-regulated during growth on mucin; two of these have been demonstrated as functional O-glycopeptidases (8, 17). Interpreted in the context of the biochemical activity of characterized peptidase_M60 domain containing proteins, this has bolstered the concept that the peptidase_M60 family domains play a role in the processing of glycoproteins in the mucous layer by host-adapted microbes (5, 11).
C. perfringens is a Firmicute that is widely distributed in nature but is most commonly considered an opportunistic pathogen that is native to the flora of human and animal gastrointestinal tracts (18–20). In addition to an arsenal of glycan processing enzymes, strains of C. perfringens frequently have one to three genes encoding proteins containing peptidase_M60 domains that specifically fall into MEROPS family M60 (5, 21). Two are typically found as genomically encoded: a “large” M60 protein of over 2,100 amino acids, which we call ZmpA, and a “small” version that is typically 1,400 to 1,700 amino acids and is referred to as ZmpB. A third M60 peptidase, ZmpC, is found on virulence plasmids (22). ZmpB has been detected as an expressed extracellular protein, and deletion of the genes encoding ZmpB and ZmpC (the latter referred to as ZmpA in the previous report) in C. perfringens strains revealed a profound contribution of these proteins to the development of necrotic enteritis in an avian model system (23, 24). This directly demonstrates the importance of this class of enzyme in host–microbe interactions. Peptidase_M60 domain containing proteins and O-glycopeptidases in general are proving to have important roles in biological processes and as glycobiology tools (8, 25). Yet, our understanding of how glycan recognition impacts their activity and particularly how their complex molecular architectures might influence their functions is only nascent.
Here, we study ZmpA and ZmpB (locus tags CPF_1489 and CPF_1073, respectively) from C. perfringens strain ATCC13124, which is regarded as a human commensal and opportunistic pathogen, and ZmpC (locus tag CP4_3468) from the virulence plasmid of the avian C. perfringens strain CP4. The goal is to examine their functions with a particular focus on the molecular basis of glycoprotein recognition and processing. Overall, combined with examination of the activities and structures of the ZmpA and ZmpC M60 domains, the complete structural and functional analysis of the 1,687 amino acid ZmpB protein provides unique structure–activity relationships for these architecturally complex proteins. This in turn provides insights into how these proteins may participate in the host–microbe interaction.
Results
Modularity of the C. perfringens M60 Peptidases.
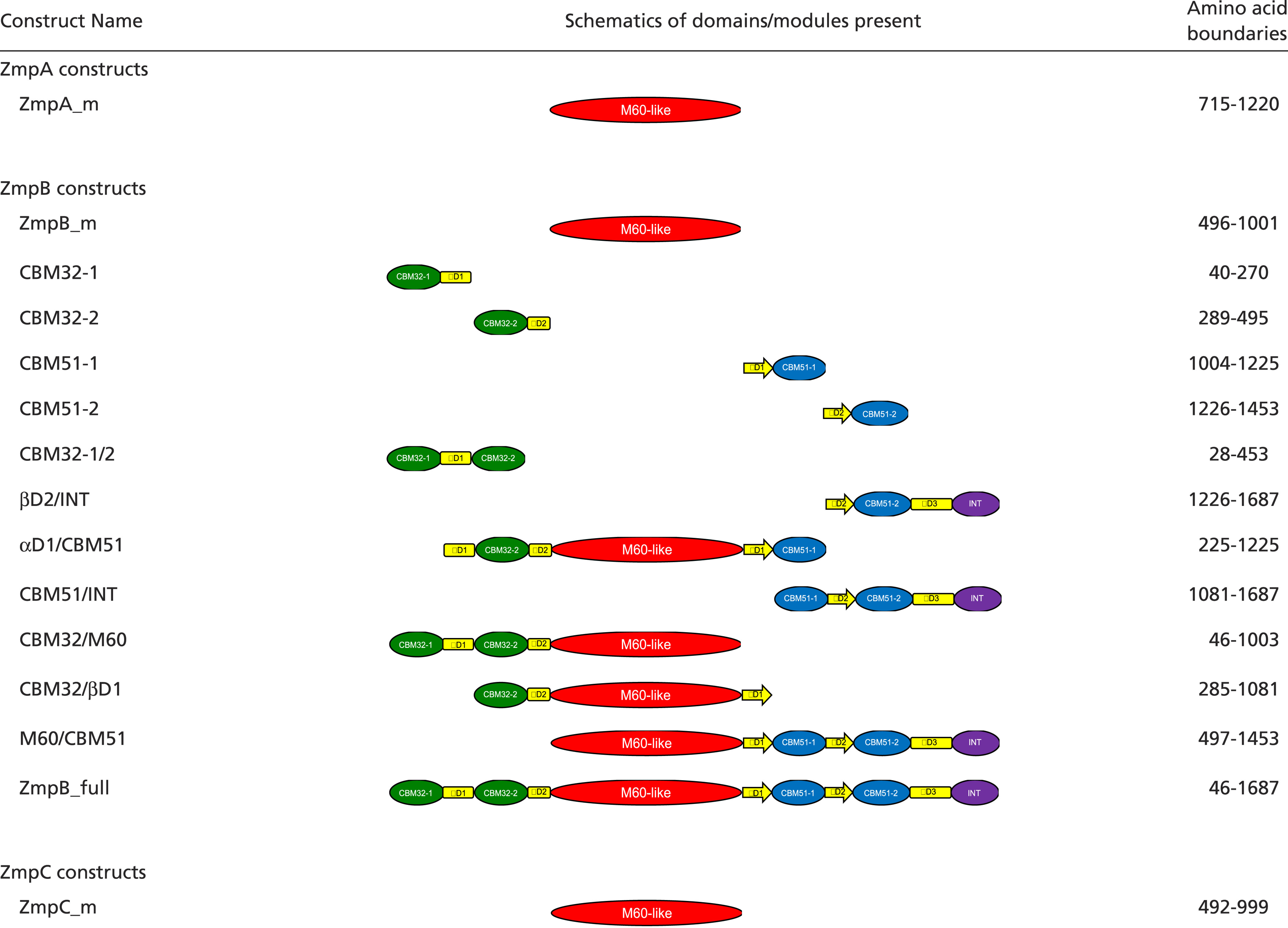
With prior knowledge of the domain boundaries of the M60 catalytic domain in ZmpB (5), we used a combination of sequence similarity searches and fold-recognition with Phyre2 (26) to predict the extensive domain/module organization of ZmpB (Fig. 1). Using this domain dissection as a guide, we also annotated the putative organizations of ZmpA and ZmpC, which display obvious similarities, including the conserved key catalytic signature sequence in the M60 domain (Fig. 1). Key differences are the additional CBM32/αD and βD/CBM51 tandem domains in ZmpA and the unidentified domain of ZmpC that comprises ∼200 amino acids at its C terminus. This C-terminal domain in ZmpC does not show significant amino acid sequence identity to proteins of known function but it is predicted to have an all α-helical structure. Using this annotation of domain architecture, and with particular focus on ZmpB as a model, we generated recombinant constructs that would enable us to probe the various domain functions and the molecular basis thereof (Table 1).
Fig. 1.

Modular schematics of the Clostridium perfringens Zmps. The respective Zmps are labeled and indicated whether their genes reside in the genome or a plasmid. The schematics of ZmpA and ZmpB are based on the enzymes from C. perfringens ATCC 13124, while ZmpC is from C. perfringens CP4. Module/domain labels are as follows: SP, a likely secretion signal peptide of 27 residues; CBM32, family 32 carbohydrate-binding modules (also classified as PF18344) of ∼160 amino acids; αD, all-α helical domains of ∼60 to 70 amino acids; M60, the MEROPS family M60 catalytic domain of ∼500 amino acids (also classified into the PF13402 or peptidase_M60 superfamily); CBM51, family 51 carbohydrate-binding modules of ∼150 amino acids; βD, all-β strand domains of ∼75 amino acids (also annotated as DUF5011/PF16403); INT, an internalin-like domain of ∼170 amino acids. Shaded regions indicate the amino acid sequence identity of the modules between the paired Zmps. The orange bar in the M60 domain indicates the gluzincin metallopeptidase motif in the M60 domain, the sequence alignment of which is indicated in the inset with the catalytic glutamic acid shown with an arrow.
Table 1.
Details of Zmp constructs (see SI Appendix, Table S1 for additional details)
 |
See SI Appendix, Table S1 for additional details.
Activity of the C. perfringens M60 Peptidase Catalytic Modules.
To assess the potential peptidase activity of recombinant ZmpA_m and ZmpC_m relative to the previously characterized ZmpB_m, we screened their activities against various glycoproteins by sodium dodecyl sulfatate polyacrylamide gel electrophoresis (SDS-PAGE). This failed to show any activity for ZmpA_m but revealed a largely similar, but not identical, activity profile for ZmpC_m compared with ZmpB_m, including inhibition by ethylenediaminetetraacetic acid (EDTA) (Table 2 and SI Appendix, Fig. S1).
Table 2.
Summary of Zmp glycoprotein digestion
| Glycoprotein substrate | ZmpA_m | ZmpA_m + EDTA | ZmpB_m | ZmpB_m + EDTA | ZmpC_m | ZmpC_m + EDTA |
| BSM | — | — | +++ | — | +++ | — |
| Fetuin | — | — | ++ | — | — | — |
| Asialofetuin | — | — | — | — | — | — |
| TIM1 | — | — | +++ | — | +++ | — |
| TIM4 | — | — | ++ | — | + | — |
| G1/C1 inhibitor | — | — | + | — | — | — |
| CD55 | — | — | +/− | — | +/− | — |
| IgA1 | — | — | — | — | — | — |
+, ++, and +++ qualitatively indicate low, medium, and high activity, respectively. +/− indicates possible activity. Data are reproduced from ref. 5 for comparison. See SI Appendix, Fig. S1 for additional details.
Given the high amino acid sequence identity of 75% between ZmpB_m and ZmpC_m, and their action on similar proteins, we sought to identify a quantitative substrate for these enzymes. Informed by our previous analysis of ZmpB_m activity on synthetic glycopeptides (5), we chemoenzymatically generated a fluorescence resonance energy transfer (FRET)-based substrate bearing sialyl-T-antigen whereby the core 1 glycan is α-2,6-sialylated on the N-acetylgalactosamine (GalNAc) residue (SI Appendix and Fig. 2A). This substrate could be cleaved by ZmpB_m and displayed a linear response of fluorescence dequenching with time upon peptide cleavage (SI Appendix, Fig. S2 B–E). A kinetic analysis of ZmpB_m activity on this substrate yielded a KM of 532.5 (±81.8) μM and a kcat of 0.026 (±0.003) s−1 (SI Appendix, Fig. S3A). The kcat was ∼3 orders of magnitude less than the “average” enzyme (27) and therefore quite poor, which we attribute to either the FRET pairs influencing activity or a nonoptimized peptide sequence in the substrate. ZmpA_m had no discernible activity on this substrate (SI Appendix, Fig. S3B). The rates of substrate hydrolysis by ZmpC_m proved to have a linear relationship up to maximum accessible substrate concentrations, thereby preventing a full kinetic determination. However, estimation of the kcat/KM value revealed its efficiency on this substrate to be ∼10-fold lower compared with ZmpB_m (Table 3 and SI Appendix, Fig. S3B).
Fig. 2.

Structure analysis of the ZmpA and ZmpC M60 domains. (A) A cartoon representation of ZmpA_m with the putative catalytic center surrounded by a σa-weighted Fo-Fc omit map contoured at 3σ. (B) Overlay of the catalytic center of ZmpB (gray with bound the zinc ion as a sphere) with the putative catalytic center of ZmpA_m (purple). (C) A cartoon representation of ZmpC_m with the putative catalytic center surrounded by a σa-weighted Fo-Fc omit map contoured at 3σ. A bound zinc ion with its coordinating waters are shown as gray and red spheres, respectively. (D) The complex of ZmpC_m with Galβ1–3[Neu5Acα2–6]GalNAcα1-Ser shown from two angles. The blue mesh shows the electron density of the glycosylated amino acid (shown as green sticks) as a σa-weighted Fo-Fc omit map contoured at 3σ. Relevant amino acids in the active site that interact with the ligand are shown as sticks and hydrogen bonds as dashed lines. The catalytic glutamic acid residue is E752. The bound zinc ion is shown as a gray sphere. (E) Comparison of the ZmpB and ZmpC O-glycopeptidase catalytic sites. The structure of ZmpB_m in complex with Galβ1–3(Neu5Acα2–6)GalNAcα1-Ser (blue; PDB ID 1KDU) overlapped with the structure of ZmpC_m in complex with Galβ1–3(Neu5Acα2–6)GalNAcα1-Ser (gray and the ligand in green sticks). Individual residues in the ligands and the G subsites they occupy are labeled. All residues in the vicinity of the ligand are shown as sticks, revealing very high sequence and structure identity in the active sites.
Table 3.
Kinetic analysis of the Zmp catalytic domains and mutants
| Protein | kcat/Km (s−1 ⋅ M−1) | Relative to ZmpB_m |
| ZmpA_m | ND* | ND* |
| ZmpB_m | 48.9 (±8.9)† | 1 |
| ZmpC_m | 4.5 (±0.1) | 0.09 |
| ZmpB_m_W752F | 10.2 (±0.1) | 0.21 |
| ZmpB_m_N749A | 9.5 (±0.1) | 0.19 |
| ZmpB_m_N775A | 4.6 (±0.1) | 0.09 |
| ZmpB_m_F727A | 1.3 (±0.2) | 0.03 |
| ZmpB_m_R790A | 1.1 (±0.1) | 0.02 |
ND indicates activity was not detectable.
Calculated from full kinetic analysis.
With the FRET substrate in hand, we extended this analysis by performing structure-guided mutagenesis to quantitatively assess the importance of interactions between the ZmpB_m active site and the glycan moiety on the substrate. Five residues in the three specific glycan binding subsites were chosen based on their interactions with the glycan moiety: F727, N749, W752, N775, and R790 (SI Appendix, Fig. S3C). As with ZmpC_m, the ZmpB_m mutants did not show saturation kinetics, so kcat/KM values were determined (Table 3 and SI Appendix, Fig. S3D). The effect of the mutations in the glycan-binding subsites ranged from modest (approximately fivefold) to more substantive (∼50-fold), supporting the importance of the protein–glycan interaction (Table 3).
Structural Analysis of the M60 Catalytic Domains.
Given that ZmpA_m displayed no activity in our assays and that ZmpC_m had reduced activity relative to ZmpB_m, we solved their crystal structures to provide potential insight into these observations. The structure of ZmpA_ determined to 2.1 revealed its fold to be the same as that previously described for ZmpB_m (5) (Fig. 2A). The rmsd of ZmpA_m with ZmpB_m was 1.41 Å. Examination of the ZmpA_m structure most obviously exposes its lack of a zinc ion bound in the metal binding site at what is the catalytic center of other M60 peptidases (Fig. 2A). This is unusual, as multiple other M60 peptidase structures have a bound ion in this position despite the proteins not being intentionally precomplexed with the ion or having it as a part of the crystallization condition. Nevertheless, we attempted soaks with zinc and cocrystallization with the ion to trap a zinc complexed protein. This did not yield occupancy of the metal binding site. Furthermore, the addition of zinc to the ZmpA_m activity assays did not restore activity, thus leading us to conclude that the metal binding site in this recombinant protein is nonfunctional. Indeed, the loop that harbors the distal glutamate of the HEXXH(8,24)E gluzincin motif is pulled down and away from the active site, disengaging this residue from metal binding site (Fig. 2B). The incomplete valency of the metal binding likely makes it unable to bind zinc, which would otherwise be a key element of the catalytic cycle, rendering the recombinant protein catalytically inactive. Though many residues in the putative ZmpA_m glycan binding site are conserved with ZmpB, disorder in some sidechains and a loop bordering the active site makes it unclear if this region of the protein is able to recognize glycan (SI Appendix, Fig. S4).
The structure of ZmpC_m at 2.25 Å resolution also showed its fold to be typical of M60 peptidases (Fig. 2C). It had rmsds of 1.57 Å and 0.72 Å with ZmpA_m and ZmpB_m, respectively. The structural identity between ZmpB_m with ZmpC_m suggested their catalytic and glycan binding sites to be essentially indistinguishable. Based on this, we also determined the structure of ZmpC_m in complex with the α2,6-sialylated core-1 O-glycan [Galβ1–3(Neu5Acα2–6)GalNAcα1-Ser] to 2.5 Å resolution (Fig. 2D). This confirmed a set of interactions between ZmpC_m and the glycosylated amino acid that were very similar to that previously observed in ZmpB_m (Fig. 2 D and E). This suggests that the glycan specificity of ZmpC_m and ZmpB_m is the same via protein–carbohydrate interactions in essentially identical G subsites [using the previously proposed O-glycopeptidase subsite nomenclature (5)].
Function of the Carbohydrate-Binding Modules.
An evident feature of the three clostridial Zmp proteins is their content of multiple CBMs (Fig. 1). To interrogate the potential carbohydrate binding function of the CBMs in ZmpB, we used isothermal titration calorimetry to examine the binding of the isolated recombinant CBM32-1, CBM32-2, CBM51-1, and CBM51-2 proteins to monosaccharides typically found in N- and O-glycans. Through this screen, we found that only CBM32-1 and CBM51-2 bound to GalNAc and N-acetylglucosamine (GlcNAc) with measurable dissociation constants (Kd) of 2.9 mM and 2.6 mM, respectively (SI Appendix, Table S2 and Fig. S5). Though the Kd values were low, these are in the range typically observed for type C CBM32 and CBM51 proteins (28–33). The individual CBM constructs resisted crystallization; however, diffraction quality crystals of the larger multimodular CBM-containing constructs of CBM32-1/2 and βD2/INT were obtained.
Structures of the CBM32 Modules.
Structures of the CBM32-1/2 construct were determined without ligand and in complex with GalNAc and in two different space groups. The unliganded structure determined at 2.0 Å resolution revealed its bilobed structure with the two calcium-complexed CBM32 modules separated by a three α-helix bundle (αD1) that resembles the Found In Various ARchetectures (FIVAR) modules found in other C. perfringens carbohydrate-active enzymes (Fig. 3A) (34). The GalNAc bound structure was determined at 2.0 Å resolution in a different space group and showed the same calcium-bound bilobed structure. However, an overlap of the two structures indicated that the CBM modules in the GalNAc complex were closed in toward one another by ∼4 to 5 Å (overall rmsd of 2.0 Å), suggesting only slight flexibility in this polypeptide (Fig. 3B). In the GalNAc complex, a fragment of the N terminus of the polypeptide, which was not visible in the unbound structure, was modeled at the interface of CBM32-1 and αD1 where it potentially contributed to the more compact conformation (Fig. 3B).
Fig. 3.

Structures of the family 32 CBMs. (A) Modular schematic of the crystallized and modeled CBM32-1/2 protein according to Fig. 1 shown with a cartoon representation of the CBM32-1/2 crystal structure. The N and C termini are indicated and bound calcium ions are shown as blue spheres. (B) Cartoon representation of the CBM32-1/2 structure in complex with GalNAc (shown as green sticks). The electron density for the GalNAc residue is shown as a σa-weighted Fo-Fc omit map contoured at 3σ. A peptide derived from the N terminus of the protein that was bound at an interface of CBM32-1 and αD1 is shown as a cyan ribbon. This structure was overlayed via the CBM32-1 modules with the CBM32-1/2 structure determined in the absence of GalNAc; the unbound structure is shown in transparent blue. (C) Overlap of the CBM32-1 module (blue with GalNAc as green sticks) with the CBM32-2 (orange) from the complexed structure. (D) Close-up of the CBM32-1 binding site showing specific interactions and the solvent accessible surface as transparent gray. (E) An overlap of the CBM32-1 binding site (blue) with the analogous region of CBM32-2 (orange with orange labels). The surface of CBM32-2 is shown as transparent gray.
Clear electron density for an α-GalNAc was observed at the apex of the CBM32-1 β-sandwich, which is a typical binding site in CBM32s (35), but no electron density consistent with a bound sugar was observed near CBM32-2 (Fig. 3B). The two separate CBMs overlap with an rmsd of 1.1 Å and have their bound calcium ions present in a conserved binding site (Fig. 3C). CBM32-1 bound GalNAc through a limited series of hydrogen bonds that include selectivity for the axial O4 that defines galacto-configured sugars (Fig. 3D). An aromatic corner is formed by the perpendicular positioning of the Y86 and W200 sidechains; the ring of Y86 makes CH-π interactions with the C3-C4-C5 plane of the GalNAc ring and the C6 hydroxymethyl group sits in the aromatic corner. The acetamido group makes only a single water mediated hydrogen bond with the protein but lies packed along the protein surface, which appears in a sufficient set of interactions to make the CBM selective for this monosaccharide.
The binding sites of CBM32-1 and CBM32-2 are largely conserved, except for the W200 residue present in CBM32-1, which is replaced by a serine (S422) in CBM32-2 (Fig. 3E). The loss of the aromatic corner and the potential interaction with carbohydrate C6 hydroxymethyl group seems sufficient to lose monosaccharide binding, but potentially opens this side of the binding site to accommodate another monosaccharide. We therefore tested binding to sialylated ligands but still found no binding (SI Appendix, Table S2); thus, it remains unclear if CBM32-2 is nonfunctional or if it requires a different and/or more complex carbohydrate ligand.
Structures of CBM51-2 and INT.
Structures of the βD2/INT construct, comprising βD2, CBM51-2, αD3, and INT, were determined in unbound and GalNAc bound forms. The overall structure displays an elongated conformation comprising four domains/modules (Fig. 4A). The CBM51-2 module is preceded by an elongated all β-strand domain (βD2; also annotated as DUF5011/PF16403) and followed by a three-α helix bundle (αD3), though this is more extended than αD1. αD3 in turn is trailed by the mostly β-structure of the INT domain. Electron density for a well-defined GlcNAc residue was found at the apex of the CBM51-2 β-sandwich, which like the CBM32s is also a typical binding site in CBM51s (Fig. 4 A and B) (28). The orientation of the GlcNAc indicates specific recognition of the nonreducing end, while the pattern of hydrogen bonding is consistent with specific binding of an equatorial O4 (i.e., gluco-configured) (Fig. 4B). W1438 provides an aromatic platform against which the pyranose ring packs in a parallel fashion. The lack of glucose binding to this CBM indicates that the acetamido group of GlcNAc is a key recognition determinant through a similar set of interactions with the CBM as was observed for GalNAc binding to CBM32-1.
Fig. 4.

Structure of the C terminus of ZmpB. (A) Modular schematic of the crystallized and modeled βD2/INT protein according to Fig. 1 shown with a cartoon representation of the unliganded βD2/INT crystal structure. The N and C termini are indicated, and a bound calcium ion is shown as a blue sphere. The carbohydrate binding region of the protein is indicated by the orange amino acid side chains that are shown as sticks. (B) A close-up of the CBM51-2 carbohydrate binding site in the βD2/INT GlcNAc complex. The electron density of the bound GlcNAc residue (green sticks) is shown as a blue mesh σa-weighted Fo-Fc omit map contoured at 3σ. Residues involved in the interaction are shown as an orange stick, hydrogen bonds as dashed lines, and a coordinated water as a red sphere. (C) Comparison of the INT domain, and its subdomains, with Internalin B (InlB) from Listeria monocytogenes (PDB ID 1H6T). The cap (CAP) subdomain is shown in green, the leucine-rich repeat (LRR) in red, and the Ig-like fold (IR) in blue. The overlay was generated by separately overlapping the CAP/LRR and IR fragments with InlB. (D) Conservation of the INT surface residues with ∼163 nonredundant INT homologs with greater than 30% amino acid sequence identity and 75% sequence coverage. The conservation was mapped onto the INT structure using CONSURF (72). (E) Structure of the INT domain showing distinctive solvent-exposed aromatic amino acid residues.
The 165 amino acids at the C terminus of ZmpB comprise a module that we refer to as INT and whose composite domains show structural similarity to the internalin family of adhesins (Fig. 4C) (36). The INT module is made up of a truncated α-helical EF-hand domain, referred to as the CAP, followed by three leucine-rich repeats (LRR) and a C-terminal domain with an Ig-like fold (IR region). Proteins with the internalin-like architecture are typically associated with mediating protein–protein interactions, as represented by the interaction of the canonical internalin A (InlA) from Listeria monocytogenes with E cadherin on the surface of gastrointestinal epithelial cells (37). Though the potential ligand for INT from ZmpB is unknown it displays an arrangement of two solvent exposed phenylalanine side chains and a solvent exposed tyrosine that create a distinctive hydrophobic surface suggestive of a binding functionality (Fig. 4D).
ZmpB Displays an Architecturally Complex and Extended Conformation.
To relate the structures and functions of the individual modules in ZmpB to the context of the complete enzyme, we purused a model of the complete enzyme. Though we were unable to crystallize the intact protein, we were able to crystallize the CBM32/M60 and M60/CBM51 constructs and collect diffraction data to 4.6 Å resolution data for both proteins. The low-resolution structures were solved taking advantage of our existing high-resolution structures, leading to high confidence in the resulting models. Both structures revealed extended conformations somewhat resembling “beads on a string” (Fig. 5 A and B). These structures, in combination with the other high-resolution structures determined in this study and previously, were used to generate a model of full-length ZmpB (Fig. 5 C and D).
Fig. 5.

Structure full-length ZmpB. Cartoon representations of the 4.6 Å crystal structures of (A) CBM32/M60 and (B) M60/CBM51. Modular schematics of the crystallized and modeled protein according to Fig. 1 are shown for each protein. (C) The full-length composite structure of ZmpB. The CBM32/M60 and M60/CBM51 were overlapped via their M60 domains, resulting in a model of CBM32/CBM51. The CBM51-2 module of this intermediate structure was overlapped with the CBM52-2 module of the βD2/INT structure to generate an initial full-length model of ZmpB. Finally, where possible, the modules/domains were replaced with the high-resolution structures of relevant complexes to generate the final full-length model, which is shown as a solvent-accessible surface. The complex of the M60 domain with an α-2,6-sialylated core 3 pentapeptide (PDB ID 5KDS) was used for this domain (5). D shows an alternate orientation of ZmpB rotated by 90°. In C and D, the surfaces contributing to known or putative carbohydrate binding sites are colored purple. The glycopeptidase active site is shown as a yellow surface. The inset in D shows the distinctive apolar solvent-exposed surface in the INT domain as orange. GalNAc in the CBM32-1 and GlcNAc in the CBM51-2 binding sites are shown as green sticks. The α-2,6-sialylated core 3 pentapeptide is shown in the glycopeptidase active site as green and orange sticks (only the glycosylated threonine of the pentapeptide is shown). Relevant dimensions are indicated. In all panels, the modules/domains are labeled according to Fig. 1.
Given the extended nature of the ZmpB model, also interrogated its potential flexibility. The CBM51-1/2 tandem was captured in three different crystal structures, all of which showed the CBMs to be in quite similar relative orientations (Figs. 3 A and B and 5A), indicating this region of the protein displays only subtle changes in conformation. We also used small-angle X-ray scattering (SAXS) to assess the solution conformations of a set of unique ZmpB constructs: αD1/CBM51, CBM51/INT, and CBM32/βD1. General analysis of the scattering data revealed molecular weights and extended dimensions (Rg and Dmax) consistent with the analogous fragments from the reconstructed X-ray crystal structure–based model (SI Appendix, Table S3). Kratky and Porod-Debye analysis of the scattering data showed properties indicative of folded proteins having an absence of significant flexibility/disorder (SI Appendix, Fig. S6) (38). Finally, the ab initio envelopes generated from the SAXS data show an excellent match to the αD1/CBM51, CBM51/INT, and CBM32/βD1 coordinates extracted from the full-length model (SI Appendix, Fig. S6). The χ2 values for these models fit to the scattering data were 5.4, 4.5, and 2.6, respectively, again revealing good agreement between the models constructed from X-ray–derived coordinates and solution scattering data. Furthermore, normal mode analysis of the full-length ZmpB model with both ElNemo (39) and iMod (40) suggest largely insignificant conformational heterogeneity that is limited to some flexing along the spine of the structure. On this basis, we argue that the model of full-length ZmpB constructed from the X-ray structures is a reliable representation of a likely dominant conformation of the protein.
Discussion
A defining feature of the peptidase_M60 domain containing proteins that have been characterized to date is the recognition of an O-glycan on the amino acid residue immediately following the hydrolyzed peptide bond. Recent work has provided excellent qualitative insight into substrate consensus structures of some glycopeptidases as well as the structural basis of glycan recognition (5, 8). Here, we complement this by using a quantitative kinetic approach to demonstrate the key importance of the specific protein–glycan interactions in the G-sites. This has revealed that two closely related M60 domains, ZmpB_m and ZmpC_m, display quite different kinetic properties, despite having identical G-sites. We suggest that this points to the importance of how variations in the peptide portion of the substrate(s) may be recognized by the two peptidases, which is an observation also made with immunomodulating metalloprotease of Pseudomonas aeruginosa (IMPa) (5). Indeed, some structural variations exist between ZmpB_m and ZmpC_m in a cleft adjacent to the catalytic machinery that may interact with the peptide portion substrates and result in slightly different capacities to recognize the peptide backbone. However, it is notable at this point that, to date, specific S subsites, other than that bearing the glycosylated amino acid, have not been identified in a M60 peptidase, making it difficult to specifically assess the contribution of peptide recognition to catalytic efficiency in this class of peptidase.
The frequent possession of CBMs is a notable feature of peptidase_M60 proteins (11). A survey of the clostridial M60 peptidases indicates variety in the number of putative CBMs present with the pattern of 1 to 3 CBM32s and 2 to 3 CBM51s that are N and C terminal, respectively, to the M60 domain. Biochemical analysis of the four putative CBMs present in ZmpB and high-resolution structural analysis of all but CBM51-1 revealed the ability of CBM32-1 and CBM51-2 to bind carbohydrates. CBM32-1 displayed a mode of GalNAc binding that is characteristic of nonreducing terminal galacto-configured sugar recognition by family 32 carbohydrate-binding modules (30, 32, 41). The acetamido group makes only a single water mediated hydrogen bond with the protein but lies packed along the protein surface, which appears in a sufficient set of interactions to make the CBM selective for this monosaccharide. CBM32-5 from the C. perfringens GH89 (AgnC) utilizes a highly similar set of interactions to achieve selectivity for GalNAc even though the two CBMs only share ∼20% amino acid sequence identity (SI Appendix, Fig. S7) (30). Though CBM32-2 displayed considerable structural identity with CBM32-1, we could not find a carbohydrate-binding function for this module. This suggests it is either nonfunctional or, more likely, the carbohydrate ligand is something other than the typical candidates that we explored, such as a more complex glycan.
To date, structurally and functionally characterized CBM51 modules have displayed two modes of ligand recognition. One is typified by the terminal galactose binding CBM51 from C. perfringens GH95 (referred to as GH95CBM51), and the other mode is represented by the blood group antigen binding CBM51 from C. perfringens GH98 (referred to as GH98CBM51) (28). These two modes of carbohydrate recognition by related CBMs are dissimilar via nonconserved binding sites (28) (SI Appendix, Fig. S8 A and B). The recognition of GlcNAc by CBM52-2, which is a novel selectivity for a CBM51 module, represents a third mode of binding, as its binding site is poorly conserved with the other representative CBM51s. CBM51-1, however, shows ∼50% amino acid sequence identity to GH95CBM51, including relatively well-conserved binding site residues with GH95CBM51, and therefore has a potentially competent galactose binding site (SI Appendix, Fig. S8C). Nevertheless, much like with CBM32-2, we were unable to detect binding of CBM51-1 to a monosaccharide, suggesting subtle differences in the binding site that renders it inactive or generates a different specificity for an as yet unidentified carbohydrate ligand.
A comparison of the putative CBMs in the other C. perfringens Zmps to the ZmpB CBMs provides some insights into the possible functions of the CBMs in the other Zmps. The CBM32 tandem in ZmpC shows high amino acid sequence identity and conservation of key amino acids, suggesting similar binding functions for these two modules (SI Appendix, Fig. S7A). Consistent with the CBM32 pattern, the tandem of CBM51 modules in ZmpC also displays the features of functions that likely parallel those of the CBM51 tandem in ZmpB (SI Appendix, Fig. S8C). Thus, it is probable that the CBMs in ZmpC have identical functions to those in ZmpB. In ZmpA, however, which appears to be a noncatalytic protein, it is notable that the three CBM32 modules in ZmpA show better conservation with the binding site residues of ZmpB CBM32-1 than with ZmpB CBM32-2 (SI Appendix, Fig. S7A). The two key aromatic amino acids are conserved and, for the most part, hydrogen bond donor/acceptors are functionally conserved. This suggests that all of the CBM32s may be functional in ZmpA and would target terminal galacto-configured sugars. Like CBM51-1, the three CBM51 modules of ZmpA show good conservation of the functional residues in GH95CBM51, suggesting these modules may also recognize galacto-configured sugars (SI Appendix, Fig. S8C). On this basis, we speculate that the CBMs in ZmpA may compensate for its apparent lack of catalytic activity by potentially giving the protein an adhesive role where the expansion of the CBMs would provide potential for significant binding avidity to the termini of glycans presented on mucins and other glycoproteins.
The C termini of the clostridial Zmps typically comprise one of two types of domains: a predicted all α-helical bundle domain, as in the case of ZmpC, or more commonly the INT domain observed in ZmpB and ZmpA. Sequence similarity searches using the ZmpB INT amino acid sequence (using 30% amino acid sequence identity and 75% coverage cutoffs) yielded over 400 results. Of these entries, over 95% originate from Firmicutes. Additionally, most were found in putative M60 peptidases possessing multimodular architectures, including the presence of a preponderance of possible CBMs. The coassociation of INT-like domains with M60 peptidase domains suggests a possibly conserved function for the INT-like domains. However, mapping conserved residues among homologs revealed very little conservation of surface residues (Fig. 4C). More stringent amino acid sequence identity cutoffs of 40% and 60% also failed to yield clear patterns of surface residue conservation, though above 60% amino acid sequence identity the putative hydrophobic functional surface observed in the ZmpB INT structure (Fig. 4D) was largely conserved. The lack of conserved surface residues among the INT-like domains suggests either a general lack of function and corresponding absence of a selective driving force, which appears unlikely, or the targeting of ligands that are not conserved between the different INTs. Indeed, even the INT domains from ZmpA and ZmpB display quite low amino acid sequence identity at 33% (Fig. 1). Given the association of internalin-like domains with protein–protein interactions, we suggest that the poor conservation of surface residues among the INT-like domains may reflect the diversity of possible protein ligands. Potential likely ligands may either be within the mucosal layer of the host, other secreted C. perfringens effectors, or components on the surface of the bacterium. In the latter case, the INT domains would mediate bacterial surface presentation of the enzymes, though there is evidence that ZmpB is a secreted protein (24), making the former options more likely.
The catalytic activity of the C. perfringens Zmps and their role in necrotic enteritis caused by this bacterium is consistent with the biological target of the proteins being the mucosal layer and the mucins that comprise it. Heavily O-glycosylated mucins can be membrane bound or secreted. The structures adopted by mucin, however, are not well-understood, though evidence is consistent with the O-glycosylated mucin repeats adopting extended structures, sometimes referred to as a “bottle brush” architecture (42). Recent low-resolution cryo-electron microscopy analysis has refined this concept by showing the structure of highly O-glycosylated mucin to present as a repeat of roughly 45 to 50 nM diameter spherical domains, whereby the domains are presumed to be highly glycosylated and the intervening regions less so (43). Remarkably, the composite domains and modules of ZmpB and their arrangement in three-dimensional space appears to uniquely tailor the enzyme to the recognition and degradation of mucin. Overall, the protein adopts an extended conformation, which measures ∼280 Å end to end (Fig. 5D). The protein comprises 13 modules and subdomains with individual folds [the M60 domain comprises three discrete folds (5) along with the additional 10 described here], yet it displays a surprising lack of significant conformational heterogeneity. The most salient feature of the architecture is the directionality of the active sites: All of the CBM binding sites and the M60 active site are arranged along a straight line and are oriented in the same direction (Fig. 5 C and D). The organization of the CBM binding sites and the M60 active site are, therefore, consistent with the coordinated recognition of the extended conformation of highly glycosylated mucin or mucin-like substrates. Additionally, the CBM binding sites in ZmpB are on roughly 45 to 50 nM centers, despite employing two structurally distinct types of linker domains (i.e., the αD1 and βD2 linkers), suggesting an organization tailored to accommodate and interact with the spacing of highly glycosylated mucin domains (Fig. 5D). This would place the catalytic domains near the regions separating mucin domains, potentially promoting access to more exposed regions of the peptide backbone. Thus, one surface of ZmpB appears to display features that mirror the known characteristics of mucin. This is reminiscent of the M88 O-glycopeptidase from Pseudomonas aeruginosa, IMPa, that appears to incorporate a separate proline/glycan binding ancillary domain that is in line with the catalytic site (44). However, the putative binding site on the ZmpB INT domain points in a direction opposite to that of the mucin-active surface of ZmpB and is thus consistent with a role not directly associated with mucin recognition or degradation, such as interaction with other C. perfringens enzymes or the bacterial cell surface.
The members of the peptidase_M60 superfamily are widely distributed in nature, and these proteins, which are often very large, frequently display the property of multimodularity. Presently characterized members are all glycopeptidases that recognize and require an O-linked glycan but cleave the peptide bond in the polypeptide backbone. The presence of functional CBMs in these proteins, such as the C. perfringens Zmps, reveals that in addition to direct glycan recognition in the catalytic site, the CBMs can mediate more general binding to distal glycans on glycoprotein substrates. Moreover, in the case of ZmpB, the remarkable elongated architecture of the protein is directionally arranged in three-dimensional space such that it appears tailored to the recognition of mucin and/or mucin-like substrates and potentially provide avid recognition of multivalent glycosylated substrates. This sophisticated mode of substrate recognition would then provide a significant advantage in specifically targeting and maintaining proximity to glycoproteins in the mucous layers of a host, thereby aiding in its destruction to either enable access to underlying epithelial layers, as in the case of toxin-deploying C. perfringens, or in liberating an energy source, such as in commensal Bacteroides species.
Materials and Methods
All reagents and chemicals were purchased from Sigma, unless otherwise stated.
Gene Cloning, Protein Production, and Purification.
The gene and fragment genes encoding all ZmpA and ZmpB constructs (see Table 1 for construct name and boundaries) were amplified from C. perfringens strain ATCC13124 genomic DNA using the oligonucleotide primers listed in SI Appendix, Table S3. The gene fragment encoding for the ZmpC construct was amplified from an Escherichia coli codon optimized gene (synthesized by GenScript) based on the sequence from C. perfringens strain CP4. PCR-amplified gene and fragment genes were cloned into pET28a (Novagen) using NheI and XhoI restriction sites or into a PCR linearized pET28a (see SI Appendix, Table S4 for oligonucleotide primer sequences) using the In Fusion HD cloning kit (Clontech). The recombinant plasmids encoded the desired polypeptide fused to an N-terminal six-histidine tag by a thrombin protease cleavage site. Mutagenesis of previously cloned pET28a-ZmpB_m (5) to introduce point mutations (F727A, N749A, W752A, N775A or R790A) was performed using the In Fusion HD cloning kit. All mutagenic primers are listed in SI Appendix, Table S4. The fidelity of all constructs was confirmed by bidirectional sequencing.
All recombinant expression vectors were transformed into E. coli BL21 Star(DE3) cells (Invitrogen), and proteins were produced using 2xYT medium containing kanamycin (50 μg/mL) supplemented at an OD600nm with 0.5 mM isopropyl β-D-1-thiogalactopyranoside for 18 h at 16 °C. Bacterial cells were harvested by centrifugation and disrupted by chemical lysis using previously described procedures (45). The recombinant proteins were purified from the cleared cell-lysate by immobilized metal-affinity chromatography and size-exclusion chromatography using either an S100, S200, or S300 HiPrep 16/60 Sephacryl column (GE Healthcare) as appropriate. Protein purity was assessed by SDS-PAGE, and protein concentrations were determined using extinction coefficients calculated by ProtParam (46) (SI Appendix, Table S1).
Activity Assays.
Glycopeptidase activities were assessed by using bovine submaxillary mucin (BSM) type I-S, bovine fetuin, and bovine asialofetuin (ASF) as substrates in the absence or presence of 50 mM EDTA as described (5). Briefly, enzymes were incubated with BSM in a 1:175 (wt/wt) ratio for 20 h and with fetuin or ASF in a 1:25 (wt/wt) ratio for 3 h, in 20 mM Tris HCl pH 7.5 at 37 °C. Reaction products were then analyzed on 10 to 15% SDS/PAGE and stained for specific glycoprotein detection with the periodic acid-Schiff (PAS) stain (47). TIM1, TIM4, CD55, G1/C1 (R&D Systems), and IgA1 (Abcam) were also tested as substrates in the absence or presence of 50 mM EDTA using conditions identical to those described previously for ZmpB_m and using standard Coomassie blue to stain the gels (5).
Kinetic Analyses.
The synthesis of the FRET-glycopeptidase substrate is schematically shown in SI Appendix, Fig. S2A. An N-terminal 6-FAM/C-terminal DABCYL–labeled peptide derived from bovine fetuin with the sequence AEAPSAVPDK was prepared synthetically by Bio Basic Inc. The Galβ1–3[Neu5Acα2–6]GalNAcα-O-Ser glycan was constructed via sequential glycosyltransferase reactions of 1 mL total volume incubated at 30 °C. First, α-GalNAc addition to form the Tn antigen was performed in a coupled epimerase/transferase reaction containing 50 mM Hepes pH 7.0, 10 mM MnCl2, 10 mg/mL peptide, 0.5 mg/mL human ppGalNAcT2 (48), 0.5 mg/mL UDP-GlcNAc-4 epimerase (49), and 12.5 mM UDP-GlcNAc. The α-GalNAc (Tn antigen) was then α-2,6-sialylated in a reaction mixture of 20 mM Bis-Tris pH 6.5, 2 mM EDTA, 1 mM DTT, 10 mg/mL Tn-peptide, 0.25 mg/mL EGFP-ST6GalNAc1 fusion protein (50), and 11 mM CMP-Neu5Ac. Lastly, β-1,3-galactose was added in a reaction mixture of 50 mM Hepes pH 7.0, 10 mM MnCl2, 10 mg/mL SiaTn-peptide, 0.25 mg/mL Core 1 GalT, and 10 mM UDP-Gal. The Core 1 GalT was produced as an N-terminal E. coli maltose binding proteins fusion to a Δ46 amino acid N-terminal truncation of C1GalTA (sequence tag CG9520) from Drosophila melanogaster. The maltose binding protein fusion expression plasmid was described previously (51). All reactions were evaluated for completion by thin layer chromatography on silica plates (Supelco) in a solvent mixture of ethyl acetate, methanol, water, and acetic acid (40:20:10:1) followed by p-anisaldehyde staining. The product of each reaction was purified on C18 derivatized silica (Supelco) between synthesis steps with elution in 100% MeOH elution and then drying before the next step was performed.
All steady state kinetics were performed at room temperature on a SpectraMax M5 plate reader in 384-well microtiter plates using SoftMax Pro-6.2.1 software. Standard reaction mixtures were done in 20 mM Tris HCl pH 7.0 and 100 μM zinc chloride containing 0.5 to 2 μM enzyme and 0 to 400 μM F9F. Fluorescence resulting from enzyme activity was measured at 25 °C using the wavelength of 492 and 530 nm for excitation and emission, respectively. The 5-FAM-AEAP peptide product of hydrolysis (purchased from Bio Basic Inc.) was used to generate a standard curve. The measured fluorescence for the activity assays was corrected for inner filter effects for each substrate concentration as previously described (52). Briefly, the fluorescence of the FRET substrate at each concentration is measured with and without the 5-FAM-AEAP fluorescent product and adjusted based on the fluorescence of the product only. Kinetic values for ZmpB_m were determined by fitting the Michaelis–Menton equation to the rate data. For all other kinetic experiments, kcat/KM values were determined from the slope of linear fits to the rate data, which were normalized to the enzyme concentration.
Crystallization, Data Collection, and Data Processing.
All crystals were grown using sitting-drop vapor diffusion for screening and hanging drop vapor diffusion for optimization at 18 °C.
Crystals of selenomethionine-labeled CBM32-1/2 (20 mg/mL) and unlabeled CBM32-1/2 were grown in 1.6 M tribasic ammonium citrate pH 7.0, supplemented with 3% glycerol and 5 mM N-acetylgalactosamine. An additional crystal form of unlabeled CBM32-1/2 (15 mg/mL) was obtained in 0.18 M calcium chloride, 20% polyethylene glycol (PEG) 3350, 0.1 M Hepes pH 7.5. Crystals were cryoprotected in crystallization solution containing 20% glycerol and flash cooled in a liquid nitrogen (N2).
βD2/INT (20 mg/mL) in the absence of ligand and precomplexed with 5 mM N-acetylglucosamine was crystallized in 0.15 M sodium phosphate monobasic, 18% PEG 3350, and 0.1 M Tris HCl pH 7.0. Crystals were cryoprotected in crystallization solution containing 15% ethylene glycol and flash cooled in N2.
Crystals of ZmpA_m (35 mg/mL) were grown in 0.1 M MES:NaOH pH 6.5 and 12% PEG 20000 and cryoprotected by addition of 25% ethylene glycol prior to flash cooling in N2.
ZmpC_m crystals (21 mg/mL) were produced in 7.3 to 7.6% Tascimate pH 5.8 and 17 and 19% PEG 3350. To achieve ZmpC_m crystal complex, crystals obtained were soaked in the crystallization solution supplemented with an excess of α2,6-sialylated core-1 O-glycan (Galβ1–3[Neu5Acα2–6[GalNAcα1-Ser) (STAg) for 2 min. Both complexed and uncomplexed crystals were cryoprotected in crystallization solution supplemented with 30% ethylene glycol prior to flash cooling in N2.
CBM32/M60 (25 mg/mL) crystallized in 1 to 2% Tascimate pH 6.0, 0.1 M BisTris, and 15% Lax-A Day laxative (PEG 3350). Crystals were cryoprotected in crystallization solution containing 30% ethylene glycol and flash cooled in N2.
M60/CBM51 was prepared for crystallization by thrombin treatment to remove the N-terminal six histidine tag followed by separation on and S300 HiPrep 16/60 Sephacryl column (GE Healthcare) and concentration. M60/CBM51 (25 mg/mL) crystallized at pH 8.3 in 0.2 M potassium citrate tribasic monohydrate and 20% PEG 3350. Crystals were cryoprotected in crystallization solution containing 20% ethylene glycol prior to flash cooling in N2.
X-ray diffraction data were collected at the Stanford Synchrotron Radiation Laboratories (Stanford) on beamlines BL7-1 or BL9-2 or on an “in house” instrument comprising a Pilatus 200K 2D detector coupled to a MicroMax-007HF X-ray generator with a VariMaxTM-HF ArcSec Confocal Optical System and an Oxford Cryostream 800, as indicated in SI Appendix, Table S4. Diffraction data for selenomethionine-labeled CBM32-1/2 crystals were collected on beamline BL9-2 at the peak wavelength for optimized selenium determined by fluorescence scans. Diffraction data were processed with iMOSFLM/SCALA (53, 54) and HKL2000 (55) for SSRL and “in house” collected data, respectively. Data processing statistics are given in SI Appendix, Table S5.
Structure Solution.
The structure of selenomethionine-labeled CBM32-1/2 was solved using the single wavelength anomalous dispersion method. ShelxC/D (56) was used with data extending to 3.0 Å to find four of the six expected selenium atoms present in the single molecule of CBM32-1/2 in the asymmetric unit. Initial phasing and refinement of selenium atom sites was performed with SHARP (57). Two additional selenium sites were added upon inspection of residual maps, and final phasing was performed using the six selenium sites. Phase improvement was achieved by solvent flattening using DM (58). Automated model building with ref. 59 resulted in a model that was easily completed with rounds of model building with COOT (60) and refinement with REFMAC (61). This model was used as a starting point to determine the structures of the native CBM32-1/2 in both of the obtained space groups.
The structure of βD2/INT was solved by molecular replacement. The coordinates of the family 51 carbohydrate binding module from Clostridium perfringens [Protein Data Bank (PDB) code 2VMG (28)], which has 40% amino acid sequence identity over ∼35% of βD2/INT, was used as a search model in PHASER (62). Initial phases generated by refinement with REFMAC were improved by 10 cycles of solvent flattening with automated phase extension using DM. The resulting phases were of sufficient quality for ARP/WARP to build a complete model of βD2/INT. The model was corrected with COOT followed by refinement with REFMAC.
The structures of ZmpA_m and ZmpC_m in complex with sialylated T-antigen (STAg) or not were solved by molecular replacement using the coordinates of ZmpB_m [PDB code 5KDN (5)], which shares at least 50% sequence identity, as a search model in PHASER. The generated models were further built using BUCCANEER (63) and corrected with COOT followed by refinement with REFMAC.
The structure of CBM32/M60 was solved by molecular replacement using PHASER with the structures of CBM32-1/2 and ZmpB_m (PDB code 5KDN) as search models. The initial model was further built using COOT and improved through a combination of LORESTR (64) and Phenix.Rosetta refinements (65).
The structure of M60/CBM51 was solved by molecular replacement using PHASER with the structure of βD2/CBM51-2 extracted from the βD2/INT structure and ZmpB_m (PDB code 5KDN) as search models. Homology models of the CBM51-1 and βD1 domains were generated with Phyre2 threading (26) using the βD2 and CBM51-2 coordinates, respectively, as templates. These were individually placed using PHASER, the positions of which were verified by examination of the difference electron density maps generated after refinement of the model comprising only the M60, βD2, and CBM51-2 modules/domains. The initial model was corrected and further built using COOT and improved through a combination of LORESTR (64) and Phenix.Rosetta refinements (65).
In all cases, 5% of the reflections were flagged as “free” and used to monitor model building and refinement procedures (66). Waters were added using FINDWATERS in COOT and inspected manually. All models were validated using MOLPROBITY (67). Model quality statistics are given in SI Appendix, Table S5.
SAXS.
SAXS data were collected at the SSRL beamline 4-2 using a MarCCD 165 detector. Diffraction data were measured with an exposure time of 1 min at 288 K with a wavelength of 1.127 Å. The sample-to-detector distance was set at 2.5 m, leading to scattering vectors q (defined as q = 4π/λsinθ, where 2θ is the scattering angle) ranging from 0.01 to 0.4 Å−1. Three concentrations of the protein samples in 50 mM Tris pH 8.0 and 150 mM NaCl were examined (SI Appendix, Table S3). SAXS data processing and determination of the radii of gyration (Rg), maximum particle size (Dmax), and Porod–Debye parameters were performed in accordance with procedures described previously (68). The ab initio low-resolution envelopes of all proteins were generated with DAMMIF (69), using 10 independent runs with no shape constraints. The solutions were then aligned and averaged using the DAMAVER suite of programs (70). SAXS data are summarized in SI Appendix, Table S3.
Carbohydrate-Binding Assays.
Isothermal titration calorimetry was performed as described previously (71) using a VP-ITC (MicroCal) in 50 mM potassium phosphate buffer (pH 7.0) at 22 °C using 100 μM CBM32-1, CBM32-2, CBM51-1, or CBM51-2 in the reaction cell and 20 mM monosaccharide in the syringe. All carbohydrate solutions were prepared using buffer saved from the last step of extensive dialysis of the protein solutions. All solutions were filtered and degassed immediately before use. Integrated data were fit with a one-site binding model assuming a stoichiometry (n) of one. This approach was chosen because of the low affinity, resulting in low C-values, which prevented accurate fitting of the n value, but is justified based on the 1:1 binding observed in the crystal structures.
Supplementary Material
Acknowledgments
This research was supported by operating grants to A.B.B. from the Canadian Institutes of Health Research (MOP 130305) and the GlycoNet National Centre of Excellence (AM-1). We thank the beamline staff at the Stanford Synchrotron Radiation Lightsource (SSRL). Use of the SSRL, SLAC National Accelerator Laboratory, is supported by the US Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the NIH, National Institute of General Medical Sciences (NIGMS; P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
The authors declare no competing interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at https://www.pnas.org/lookup/suppl/doi:10.1073/pnas.2019220118/-/DCSupplemental.
Data Availability
X-ray crystal structures data have been deposited in PDB. Coordinates and structure factors have been deposited with the following accession numbers: 6XSX for ZmpA_m, 6XSZ for ZmpC_m, 6XT1 for ZmpC_m in complex with STAg, 7JNB for selenomethionine-labeled CBM32-1/2 in complex with GalNAc, 7JND for CBM32-1/2, 7JNF for CBM32-1/2 in complex with GalNAc, 7JRM for CBM51/INT, 7JRL for CBM51/INT in complex with GlcNAc, 7JFS for CBM32/M60, and 7JS4 for M60/CBM51.
References
- 1.Hooper N. M., Proteases: A primer. Essays Biochem. 38, 1–8 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Apweiler R., Hermjakob H., Sharon N., On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim. Biophys. Acta 1473, 4–8 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Varki A., Biological roles of glycans. Glycobiology 27, 3–49 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brockhausen I., Stanley P., “O-GalNAc glycans” in Essentials of Glycobiology, 3rd edition, Varki A., Ed. et al. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 2015), pp. 113–123. [Google Scholar]
- 5.Noach I., et al., Recognition of protein-linked glycans as a determinant of peptidase activity. Proc. Natl. Acad. Sci. U.S.A. 114, E679–E688 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu A. C. Y., Worrall L. J., Strynadka N. C. J., Structural insight into the bacterial mucinase StcE essential to adhesion and immune evasion during enterohemorrhagic E. coli infection. Structure 20, 707–717 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Malaker S. A., et al., The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc. Natl. Acad. Sci. U.S.A. 116, 7278–7287 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shon D. J., et al., An enzymatic toolkit for selective proteolysis, detection, and visualization of mucin-domain glycoproteins. Proc. Natl. Acad. Sci. U.S.A. 117, 21299–21307 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haurat M. F., et al., The glycoprotease CpaA secreted by medically relevant acinetobacter species targets multiple O-linked host glycoproteins. MBio 11, e02033-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trastoy B., Naegeli A., Anso I., Sjögren J., Guerin M. E., Structural basis of mammalian mucin processing by the human gut O-glycopeptidase OgpA from Akkermansia muciniphila. Nat. Commun. 11, 4844 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakjang S., Ndeh D. A., Wipat A., Bolam D. N., Hirt R. P., A novel extracellular metallopeptidase domain shared by animal host-associated mutualistic and pathogenic microbes. PLoS One 7, e30287 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang P., Granados R. R., An intestinal mucin is the target substrate for a baculovirus enhancin. Proc. Natl. Acad. Sci. U.S.A. 94, 6977–6982 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rawlings N. D., Barrett A. J., Finn R., Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 44, D343–D350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cockburn D. W., Koropatkin N. M., Polysaccharide degradation by the intestinal microbiota and its influence on human Health and disease. J. Mol. Biol. 428, 3230–3252 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Arumugam M.et al.; MetaHIT Consortium , Enterotypes of the human gut microbiome. Nature 473, 174–180 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desai M. S., et al., A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167, 1339–1353.e21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ottman N., et al., Genome-scale model and omics analysis of metabolic capacities of Akkermansia muciniphila reveal a preferential mucin-degrading lifestyle. Appl. Environ. Microbiol. 83, e01014-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuda K., Tsuji H., Asahara T., Kado Y., Nomoto K., Sensitive quantitative detection of commensal bacteria by rRNA-targeted reverse transcription-PCR. Appl. Environ. Microbiol. 73, 32–39 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Minamoto Y., Dhanani N., Markel M. E., Steiner J. M., Suchodolski J. S., Prevalence of Clostridium perfringens, Clostridium perfringens enterotoxin and dysbiosis in fecal samples of dogs with diarrhea. Vet. Microbiol. 174, 463–473 (2014). [DOI] [PubMed] [Google Scholar]
- 20.Vince A., Dyer N. H., O’Grady F. W., Dawson A. M., Bacteriological studies in Crohn’s disease. J. Med. Microbiol. 5, 219–229 (1972). [DOI] [PubMed] [Google Scholar]
- 21.Low K. E., Smith S. P., Abbott D. W., Boraston A. B., The glycoconjugate-degrading enzymes of Clostridium perfringens: Tailored catalysts for breaching the intestinal mucus barrier. Glycobiology, 10.1093/glycob/cwaa050 (2020). [DOI] [PubMed] [Google Scholar]
- 22.Mehdizadeh Gohari I., et al., Plasmid characterization and chromosome analysis of two netF+ Clostridium perfringens isolates associated with foal and canine necrotizing enteritis. PLoS One 11, e0148344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wade B., et al., Two putative zinc metalloproteases contribute to the virulence of Clostridium perfringens strains that cause avian necrotic enteritis. J. Vet. Diagn. Invest. 32, 259–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kulkarni R. R., Parreira V. R., Sharif S., Prescott J. F., Clostridium perfringens antigens recognized by broiler chickens immune to necrotic enteritis. Clin. Vaccine Immunol. 13, 1358–1362 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang S., et al., Deciphering protein O-glycosylation: Solid-phase chemoenzymatic cleavage and enrichment. Anal. Chem. 90, 8261–8269 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Kelley L. A., Sternberg M. J. E., Protein structure prediction on the web: A case study using the Phyre server. Nat. Protoc. 4, 363–371 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Bar-Even A., et al., The moderately efficient enzyme: Evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 50, 4402–4410 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Gregg K. J., Finn R., Abbott D. W., Boraston A. B., Divergent modes of glycan recognition by a new family of carbohydrate-binding modules. J. Biol. Chem. 283, 12604–12613 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Higgins M. A., Ficko-Blean E., Meloncelli P. J., Lowary T. L., Boraston A. B., The overall architecture and receptor binding of pneumococcal carbohydrate-antigen-hydrolyzing enzymes. J. Mol. Biol. 411, 1017–1036 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Ficko-Blean E., et al., Carbohydrate recognition by an architecturally complex α-N-acetylglucosaminidase from Clostridium perfringens. PLoS One 7, e33524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boraston A. B., Ficko-Blean E., Healey M., Carbohydrate recognition by a large sialidase toxin from Clostridium perfringens. Biochemistry 46, 11352–11360 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Ficko-Blean E., Boraston A. B., The interaction of a carbohydrate-binding module from a Clostridium perfringens N-acetyl-β-hexosaminidase with its carbohydrate receptor. J. Biol. Chem. 281, 37748–37757 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Gilbert H. J., Knox J. P., Boraston A. B., Advances in understanding the molecular basis of plant cell wall polysaccharide recognition by carbohydrate-binding modules. Curr. Opin. Struct. Biol. 23, 669–677 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Adams J. J., Gregg K., Bayer E. A., Boraston A. B., Smith S. P., Structural basis of Clostridium perfringens toxin complex formation. Proc. Natl. Acad. Sci. U.S.A. 105, 12194–12199 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abbott D. W., Eirín-López J. M., Boraston A. B., Insight into ligand diversity and novel biological roles for family 32 carbohydrate-binding modules. Mol. Biol. Evol. 25, 155–167 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Schubert W.-D., et al., Internalins from the human pathogen Listeria monocytogenes combine three distinct folds into a contiguous internalin domain. J. Mol. Biol. 312, 783–794 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Schubert W.-D., et al., Structure of internalin, a major invasion protein of Listeria monocytogenes, in complex with its human receptor E-cadherin. Cell 111, 825–836 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Rambo R. P., Tainer J. A., Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Suhre K., Sanejouand Y.-H., ElNemo: A normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 32, W610–W614 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lopéz-Blanco J. R., Garzón J. I., Chacón P., iMod: multipurpose normal mode analysis in internal coordinates. Bioinformatics 27, 2843–2850 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Grondin J. M., et al., Diverse modes of galacto-specific carbohydrate recognition by a family 31 glycoside hydrolase from Clostridium perfringens. PLoS One 12, e0171606 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hansson G. C., Mucins and the microbiome. Annu. Rev. Biochem. 89, 769–793 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hughes G. W., et al., The MUC5B mucin polymer is dominated by repeating structural motifs and its topology is regulated by calcium and pH. Sci. Rep. 9, 17350 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Noach I., Boraston A. B., Structural evidence for a proline-specific glycopeptide recognition domain in an O-glycopeptidase. Glycobiology, 10.1093/glycob/cwaa095 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pluvinage B., Hehemann J. H., Boraston A. B., Substrate recognition and hydrolysis by a family 50 exo-β-agarase, Aga50D, from the marine bacterium Saccharophagus degradans. J. Biol. Chem. 288, 28078–28088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gasteiger E., et al., ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doerner K. C., White B. A., Detection of glycoproteins separated by nondenaturing polyacrylamide gel electrophoresis using the periodic acid-Schiff stain. Anal. Biochem. 187, 147–150 (1990). [DOI] [PubMed] [Google Scholar]
- 48.Du T., et al., A bacterial expression platform for production of therapeutic proteins containing human-like O-linked glycans. Cell Chem. Biol. 26, 203–212.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Bernatchez S., et al., A single bifunctional UDP-GlcNAc/Glc 4-epimerase supports the synthesis of three cell surface glycoconjugates in Campylobacter jejuni. J. Biol. Chem. 280, 4792–4802 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Moremen K. W., et al., Expression system for structural and functional studies of human glycosylation enzymes. Nat. Chem. Biol. 14, 156–162 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bernatchez S., et al., Variants of the β 1,3-galactosyltransferase CgtB from the bacterium Campylobacter jejuni have distinct acceptor specificities. Glycobiology 17, 1333–1343 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Liu Y., et al., Use of a fluorescence plate reader for measuring kinetic parameters with inner filter effect correction. Anal. Biochem. 267, 331–335 (1999). [DOI] [PubMed] [Google Scholar]
- 53.Powell H. R., The Rossmann Fourier autoindexing algorithm in MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 55, 1690–1695 (1999). [DOI] [PubMed] [Google Scholar]
- 54.Evans P., Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Otwinowski Z., Minor W., Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 56.Sheldrick G. M., Experimental phasing with SHELXC/D/E: Combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vonrhein C., Blanc E., Roversi P., Bricogne G., “Automated structure solution with autoSHARP” in Macromolecular Crystallography Protocols (Methods in Molecular Biology, 2007) (Humana Press, 2007), vol. 2, pp. 215–230. [DOI] [PubMed] [Google Scholar]
- 58.Cowtan K. D., Zhang K. Y. J., Main P., “DM/DMMULTI software for phase improvement by density modification” in International Tables for Crystallography (2012), Volume F. Crystallography of Biological molecules (John Wiley & Sons, 2012), pp. 407–412. [Google Scholar]
- 59.Langer G., Cohen S. X., Lamzin V. S., Perrakis A., Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murshudov G. N., et al., REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCoy A. J., et al., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cowtan K., The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Kovalevskiy O., Nicholls R. A., Murshudov G. N., Automated refinement of macromolecular structures at low resolution using prior information. Acta Crystallogr. D Struct. Biol. 72, 1149–1161 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DiMaio F., et al., Improved low-resolution crystallographic refinement with Phenix and Rosetta. Nat. Methods 10, 1102–1104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brünger A. T., Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 355, 472–475 (1992). [DOI] [PubMed] [Google Scholar]
- 67.Chen V. B., et al., MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pluvinage B., et al., Conformational analysis of StrH, the surface-attached exo-β-D-N-acetylglucosaminidase from Streptococcus pneumoniae. J. Mol. Biol. 425, 334–349 (2013). [DOI] [PubMed] [Google Scholar]
- 69.Franke D., Svergun D. I., DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 42, 342–346 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Petoukhov M. V., Svergun D. I., Ambiguity assessment of small-angle scattering curves from monodisperse systems. Acta Crystallogr. D Biol. Crystallogr. 71, 1051–1058 (2015). [DOI] [PubMed] [Google Scholar]
- 71.van Bueren A. L., Morland C., Gilbert H. J., Boraston A. B., Family 6 carbohydrate binding modules recognize the non-reducing end of β-1,3-linked glucans by presenting a unique ligand binding surface. J. Biol. Chem. 280, 530–537 (2005). [DOI] [PubMed] [Google Scholar]
- 72.Ashkenazy H., et al., ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 44, W344–W350 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
X-ray crystal structures data have been deposited in PDB. Coordinates and structure factors have been deposited with the following accession numbers: 6XSX for ZmpA_m, 6XSZ for ZmpC_m, 6XT1 for ZmpC_m in complex with STAg, 7JNB for selenomethionine-labeled CBM32-1/2 in complex with GalNAc, 7JND for CBM32-1/2, 7JNF for CBM32-1/2 in complex with GalNAc, 7JRM for CBM51/INT, 7JRL for CBM51/INT in complex with GlcNAc, 7JFS for CBM32/M60, and 7JS4 for M60/CBM51.