

Abstract
Aggregated amyloid beta (Aβ) is widely reported to cause neuronal dystrophy and toxicity through multiple pathways: oxidative stress, disrupting calcium homeostasis, and cytoskeletal dysregulation. The neuro-cytoskeleton is a dynamic structure that reorganizes to maintain cell homeostasis in response to varying soluble and physical cues presented from the extracellular matrix (ECM). Due this relationship between cell health and the ECM, we hypothesize that amyloid toxicity may be directly influenced by physical changes to the ECM (stiffness and dimensionality) through mechanosensitive pathways, and while previous studies demonstrated that Aβ can distort focal adhesion signaling with pathological consequences, these studies do not address the physical contribution from a physiologically relevant matrix. To test our hypothesis that physical cues can adjust Aβ toxicity, SH-SY5Y human neuroblastoma and primary human cortical neurons were plated on soft and stiff, 2D polyacrylamide matrices or suspended in 3D collagen gels. Each cell culture was exposed to escalating concentrations of oligomeric or fibrillated Aβ(1–42) with MTS viability and lactate dehydrogenase toxicity assessed. Actin restructuring was further monitored in live cells by atomic force microscopy nanoindentation, and our results demonstrate that increasing either matrix stiffness or exposure to oligomeric Aβ promotes F-actin polymerization and cell stiffening, while mature Aβ fibrils yielded no apparent cell stiffening and minor toxicity. Moreover, the rounded, softer mechanical phenotype displayed by cells plated onto a compliant matrix also demonstrated a resilience to oligomeric Aβ as noted by a significant recovery of viability when compared to same-dosed cells plated on traditional tissue culture plastic. This recovery was reproduced pharmacologically through inhibiting actin polymerization with cytochalasin D prior to Aβ exposure. These studies indicate that the cell–ECM interface can modify amyloid toxicity in neurons and the matrix-mediated pathways that promote this protection may offer unique targets in amyloid pathologies like Alzheimer’s disease.
Keywords: Alzheimer’s disease, extracellular matrix, mechanical properties
Graphical Abstract
INTRODUCTION
In healthy brains, the low nanomolar concentration of amyloid beta (Aβ) has been reported to modulate neurogenesis and help maintain synaptic plasticity.1 However, excess Aβ production and the resulting aggregates are associated with numerous pathways of neuronal dysfunction and toxicity.2 In an early study to demonstrate Aβ toxicity, Yankner et al. reported that toxicity was observed in hippocampal neurons at concentrations of 20 μM after 24 h.3 However, the same dose of Aβ did not have any observed toxicity to glial cells. Recently, a shift in the amyloid cascade hypothesis has moved the primary focus from total Aβ concentration to the small oligomeric, intermediate species.4 Associated with this toxicity, Aβ has been shown to dysregulate the actin cytoskeleton, with subsequent synaptic and dendritic malfunction, which generally precedes neuronal toxicity, loss and amyloid-induced dementia.5
Abnormalities in cytoskeletal organization are a common feature of many neurodegenerative disorders, including Alzheimer’s disease,6,7 Parkinson’s disease,8 and Huntington’s disease.9 Neurons are polarized cells, with two distinct functional compartments, and the neuro-cytoskeleton is crucial in maintaining shape and intercompartment polarity differences for efficient signal transmission.10 Thus, an appropriately organized cytoskeleton is necessary during nervous system development, maintenance, and recovery after injury. Cytoskeletal defects including alterations in microtubule stability, stalled actin dynamics, and prolonged/fragmented F-actin bundles have all been observed in several neurodegenerative conditions.11 For example, Alzheimer’s disease is associated with both neurofibrillary tangles and microtubule defects caused by the aberrant hyperphosphorylation of the tau protein.12
A key regulator of cytoskeletal remodeling and neuronal function is the extracellular matrix (ECM). Physical characteristics of the ECM—stiffness, composition, and topology—are tissue specific, and resident cells have evolved to be optimally functional within a specific ECM. An ECM that is excessively stiff may prompt actin polymerization and stress fiber formation, an increase in RhoA-mediated intracellular tension, and/or an increase in the release of enzymes that degrade the ECM. Alternatively, a matrix that is too soft may trigger actin depolymerization, a decrease in cell spreading, and increased collagen secretion to potentially rebuild and fortify the surrounding ECM.13 Neuropotentiation has been further shown to increase overall brain tissue stiffness, while age, injury, and disease state have been associated with a decrease overall brain tissue stiffness. Brain tissue is soft with reported Young’s moduli ranging from 0.1 to 16 kPa and is significantly more compliant than other tissues in the body such as endothelial, muscle, or bone. Moreover, the brain ECM is not constant, and elastography imaging has shown that brain tissue softness decreases with age, and for patients suffering from Alzheimer’s disease (AD) this tissue softening is enhanced.14 It is unclear, however, how a changing brain ECM may contribute to the pathology of AD.
Herein we explore the ECM’s potential role in aggregated Aβ toxicity in both SH-SY5Y neuroblastoma and primary neurons. ECM stiffness helps regulate cell shape, adhesion, differentiation, and survival, among numerous other cellular behaviors. The ECM also affects the endocytosis of both nanoparticles and peptides, including our previous work that displays an increased uptake for both the 40- and 42-residue isoforms of Aβ on soft polyacrylamide (PA) matrices relative to Aβ uptake into cells plated on tissue culture plastic.15 Notably, this increase in uptake on soft matrices is enhanced for the 42-residue isoform, suggesting that cell–matrix interactions could influence uptake in a mechanism-specific manner.
In addition to mechanosensitive endocytosis, it has previously been reported that cells can be more susceptible to certain toxins, such as acrylamide or acetaminophen, when they are plated in soft matrices compared to plastic substrates.16,17 This susceptibility in soft alginate substrates was argued to be a result of increased Rho GTPase activity, a key regulator of the actin cytoskeleton. For AD, it has been shown that the involvement of Rho GTPases led to an increased level of RhoA and decreased neurite length in dystrophic neurons located at the periphery Aβ plaque deposits. Song et al. determined that the overproduction of Aβ in a septal neuronal cell line induced the formation of actin stress fibers.18 In addition, Aβ(1—42) has been shown to stimulate actin polymerization in these neurons through the activation of the Rho GTPases Rac1 and Cdc42.19
The cytoskeleton provides mechanical stiffness to the cell, and thus changing cell stiffness (whether due to soluble or physical factors) may serve as a marker of cytoskeletal status. Ungureanu et al. observed a decrease in cell stiffness when the cells were dosed with 10 μM Aβ for 2 h in artificially aged primary neurons.20 This decrease in cell stiffness was not observed for cells that were cultured for 1 week, and they concluded that membrane elasticity changes are induced with a “sublethal” concentration of preaggregated Aβ. Additionally, aged neurons were found to be more susceptible to amyloid toxicity than nonaged or “young” neurons.
To better understand the influence of cytoskeletal dysregulation and AD, Furukawa and Mattson observed that pretreatment with cytochalasin D rescued primary hippocampal neurons from fibrillated Aβ toxicity in a dose dependent manner.21 Cytochalasin D, a pharmacological agent that depolymerizes actin, was further reported to decrease intracellular calcium levels, even after exposure to Aβ. Cells dosed with Aβ displayed an increase in intracellular calcium, while this increase was not observed in cells that were treated with cytochalasin D prior to exposure. In addition, actin remodeling was considered the key contributor to this neuroprotection, because destabilization of microtubules did not prevent Aβ induced toxicity. The destabilization of actin limited the amount of intracellular calcium in the cells and decreased excitotoxicity.21–23
Although these studies show varying consequences of actin and microtubule remodeling, they collectively confirm that there is a definitive, cytoskeletal-mediated, cellular response to Aβ(1—42). As cytoskeletal dysregulation has been implicated in multiple neurodegenerative disorders, this work aims to determine whether cytoskeletal remodeling, prompted by physical modifications to the ECM, can adjust Aβ toxicity. If successful, we anticipate that detailing the specific, mechano-biochemical pathways that orchestrate this actin response would offer novel therapeutic targets in the treatment of Alzheimer’s disease.
RESULTS AND DISCUSSION
Characterization of Oligomeric and Fibrillated Aβ.
To confirm the preparation protocol generated two different aggregated states of Aβ each preparation was analyzed using atomic force microscopy (AFM). Previous studies have analyzed Aβ aggregation using SDS-PAGE electrophoresis; however, these techniques may lead to apparent aggregates that arise from technical artifacts due to matrix effects.24 Additionally, AFM imaging permits identification of the aggregate conformation, either globular or fibrillated. As shown in Figure 1, when our aggregate preparations were analyzed using AFM, distinct fibrils were observed for samples prepared in 10 mM HCl at 37 °C. Alternatively, when the sample was prepared in cell culture media at 4 °C, globular aggregates, which were considerably smaller in size, appeared. This is consistent with the results described in the protocol reported by Stine et al.24 Note that in Figure 1 some differences were observed for Aβ preparations using material sourced to different vendors.
Figure 1.

AFM images of oligomeric and fibrillated Aβ(1–42) preparations from California Peptide and American Peptide Company. Scale bar for the fibrillated species of CA peptide and APC are 750 and 200 nm, respectively. The scale bar for the oligomeric species is 1 μm for both samples. Height data indicate the fibrillated species of each are 2.8 nm (CA) and 4.7 nm (APC), and the height of the oligomeric species is approximately 12.6 nm.
Baseline Studies: Oligomeric Aβ Reduces Cell Viability More than Fibrillated Aβ Does.
Prior to exploring the potential role of the ECM in amyloid-mediated toxicity, it is important to first establish baseline viability data using traditional, tissue culture plastic. Monomeric Aβ is considered benign, particularly at low micromolar concentrations, and indeed recent studies have reported that low nanomolar concentrations of monomeric Aβ exert a positive influence on synaptic plasticity, neurogenesis, and memory formation.25 An early study reported by Lorenzo and Yankner tested the toxicity of monomeric and fibrillated Aβ, and they observed no toxicity when primary rat hippocampal neurons were treated with 20 μM of monomeric Aβ(1–40) or Aβ(1–42) over 72 h.26 However, significant toxicity was observed for neurons dosed with fibrillated Aβ. Pretreatment with Congo red, a commonly used inhibitor of amyloidogenesis, restored viability to control levels, and thus, it was concluded that aggregation was requisite for Aβ toxicity.
For our study, SH-SY5Y cells and primary neurons were treated with 1, 5, and 10 μM concentrations of oligomeric or fibrillated Aβ. Note that, in Figure 2, each of these aggregated species reduced cell viability in a concentration-dependent fashion. However, exposure to the oligomeric species is consistently more toxic than that to fibrillated Aβ for both cell types, and at all concentrations. A modest reduction in cell viability was observed for fibrillated Aβ only at the highest in-well concentration, and for both cell lines cell viability remained at near 60–70% (relative to untreated control cells) after a 48 h exposure to fibrillated Aβ. Our results are similar to those reported by Cecchi et al. whereby undifferentiated SH-SY5Y cells exposed to 1 and 10 μM doses of small (ca. 50 nm diameter), globular aggregates of Aβ(1—42) reduced MTT viability to near 55% after 24 h.27 In the same study, retinoic acid differentiated SH-SY5Y cells displayed a significant resistance to Aβ injury, suggesting a strong, phenotype-specific, toxicity response to Aβ. The relationship between chemical differentiation of SH-SY5Y and sensitivity to amyloid toxicity was further explored Krishtal et al.28 At a 10 μM concentration of HFIP-disaggregated Aβ (1–42), undifferentiated SH-SY5Y cells displayed no reduction in WST-1 viability relative to untreated control cells. Upon differentiation, however, WST-1 viability reduced significantly and to varying extents, depending upon the specific differentiation protocol. One clear exception is SH-SY5Y cells differentiated through retinoic acid followed by tetradecanoylphorbol acetate to a dopaminergic phenotype that was completely resistant to Aβ at either 10 or 20 μM concentrations.
Figure 2.
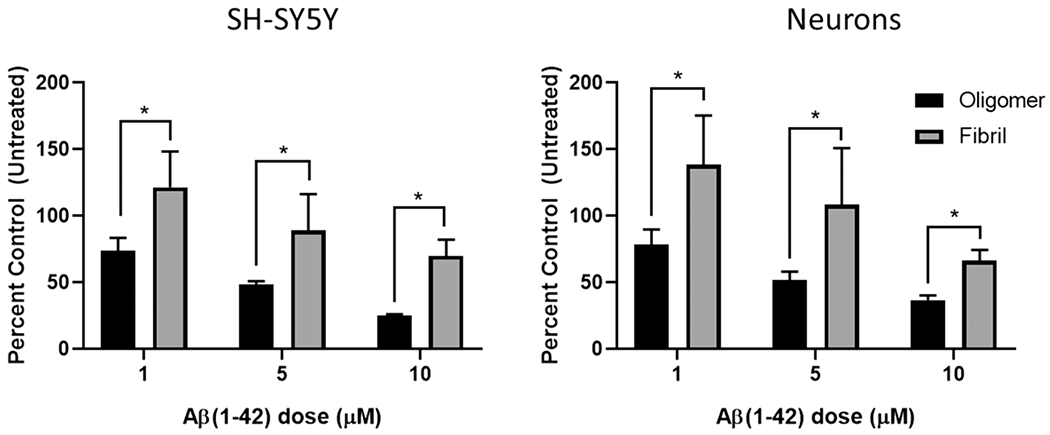
MTS viability results for SH-SY5Y cells and primary cortical neurons after exposure to increasing concentrations of oligomeric and fibrillated Aβ species for 48 h. Oligomeric Aβ is significantly more toxic than fibrillated Aβ to both cell types (two-way ANOVA, p < 0.05).
The mechanism of amyloid-mediated toxicity is also sensitive to the specific aggregation state of Aβ. Gharibyan et al. studied amyloidogenic protein aggregation and its associated toxicity using lysozyme as a model protein.29 Early stage aggregates of lysozyme formed oligomers that induced apoptosis in SH-SY5Y cells, while late-stage aggregates formed mature fibrils that induced a necrotic cell death primarily through disruption of the plasma membrane. Consistent with our studies, Gharibyan et al. found the oligomeric species to be significantly more toxic to the neuroblastoma than either the monomeric or fibrillated species; however, the specific mechanism(s) that distinguish the pronounced toxicity of oligomeric lysozyme remain unclear. Recent treatment strategies for Alzheimer’s disease have been developed to exclusively target oligomeric Aβ, rather than the monomers or mature fibrils. For example, Limbocker et al. treated SH-SY5Y cells with trodusquemine, a compound that facilitates nucleation and Aβ fibrillation, which led to a significant reduction in toxicity, despite a significant increase in overall plaque load.30
Toxicity to Oligomeric Aβ(1–42) is Matrix-Dependent.
Neuronal differentiation is not restricted to chemical cues only, but the physical influence of the ECM—topography, dimensionality, stiffness, microstructure, and pore size—all play a critical role in directing the terminal fate of progenitor cells.31–35 Analogous to the chemical differentiation studies described above, cells exposed to different extracellular matrices may express a distinct, “mechanical” phenotype. Cancer cells are well-known to display a mechanical phenotype often markedly softer than that for corresponding normal tissue cells. As reported by Lin et al., this mechanical transformation to a soft phenotype occurs regardless of cancer type—breast, bladder, cervix, or pancreas—and is driven largely by suppressing caveolin-1 expression to reduce stiffness sensing and traction force at cell-ECM interface.36 The authors suggest that this loss of stiffness sensing may confer this soft phenotype with an antiapoptotic character that permits the anchorage-independent growth which is characteristic of many cancer types.
The potential for matrix-dependent apoptosis has significant bearing for Alzheimer’s disease. The brain ECM changes in composition and stiffness with age, and the alternative adhesion signaling in resident neurons results in cytoskeletal restructuring. To test the potential matrix-dependency of oligomeric Aβ(1–42) toxicity to SH-SY5Y cells and primary neurons, each cell type was plated on (1) soft and stiff 2D PA matrices, (2) 3D collagen gels, or (3) tissue culture plastic and the cell viability was assessed with an MTS proliferation assay. Note that, for our experiments, the “soft” PA matrix had a Young’s modulus of near 3 kPa and the “stiff” PA matrix had a Young’s modulus of near 24 kPa. The shear modulus of the 3D collagen gel (2 mg/mL) was determined to be 54.7 Pa, and we estimate the Young’s modulus to be 0.156 kPa assuming a Poisson’s ratio of 0.5. This approximation is reasonable because a hydrated gel is mostly composed of water, an incompressible fluid. Specific details about the rheological measurements are provided in the Supporting Information; however, the composition and stiffness data for all matrices used in this study are collected for reference in Table 1. Our studies using the 2D matrices were performed using collagen type IV as an ECM protein because it is similar to the nonfibrous proteins found in vivo in the brain ECM. This linker protein is the primary point of integrin-mediated adhesion and helps communicate matrix stiffness to the cell; thus, changing this linker protein would have definite implications in modifying cell behavior. In our work, additional studies were completed using fibronectin and laminin. However, for each of these ECM proteins, sufficient cell attachment was not achieved on the softest matrices (data not shown), which led to poor cell counts and challenged follow-on analyses by flow cytometry.
Table 1.
Composition and Stiffness Data for Each Model Matrix
| composition (w/v) | Young’s modulus, E (kPa) | |
|---|---|---|
| collagen (3D) | 0.2% rat tail collagen type I | 0.156 |
| soft (2D) | 8% acrylamide, 0.03% BIS | 3.53 |
| stiff (2D) | 10% acrylamide, 0.30% BIS | 23.6 |
| plastic/glass | ~106 |
Each sample was exposed to 10 μM oligomeric Aβ(1–42) for 48 h. Note that SH-SY5Y cell and primary neuron proliferation are matrix dependent, and thus, MTS results could not be directly compared across the different matrices because the raw cell counts were different for different matrices. To address this challenge, each sample is compared to an untreated control of cells plated on the same corresponding matrix. As displayed in Figure 3, for the 2D PA matrices, both cell types displayed a significant recovery in MTS viability when compared to same-dosed cells on traditional tissue culture plastic. When comparing the viability of SH-SY5Y cells on soft and stiff PA matrices, no significant difference was observed at α = 0.05; however, at the same significance level, the human primary neurons on the soft PA matrix displayed a higher viability than that observed for cells on the stiff PA matrix. Further shown in Figure 3, both SH-SY5Y cells and primary neurons suspended in the soft, 3D collagen gel yielded the highest overall increase in viability relative to both PA matrices and tissue culture plastic. Overall, both ECM stiffness and dimensionality may play a significant role in regulating amyloid toxicity.
Figure 3.
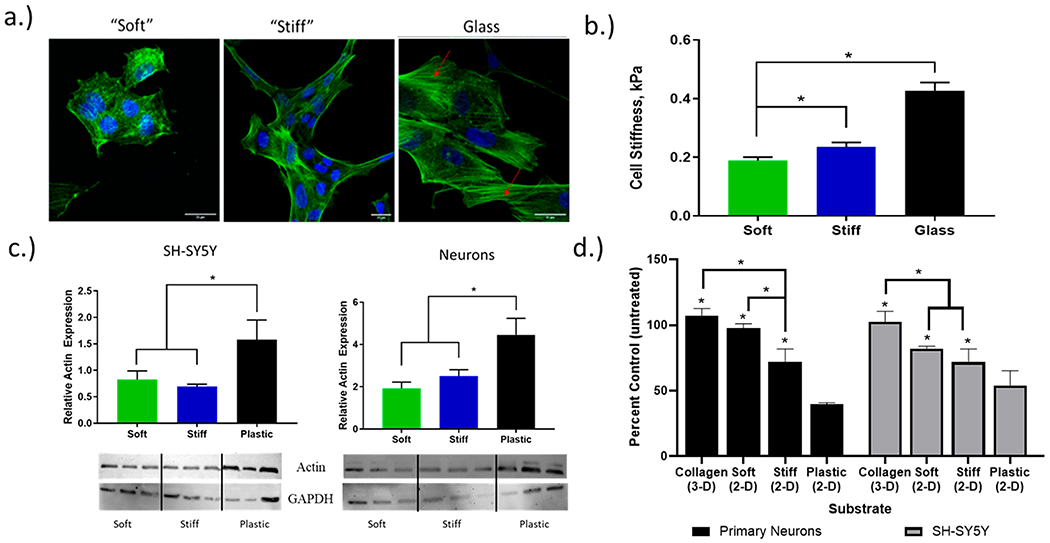
Cell behavior changes dramatically when exposed to varying environments. (a) Confocal images of human primary cortical neurons display an increase in cell spreading and F-actin polymerization on stiffer substrates. Red arrows point to F-actin stress fibers formed in the cells plated on the glass substrate. (b) AFM nanoindentation measurements of cell stiffness indicate an increase in matrix stiffness results in an increase in cell stiffness, when plated on PA gels. (c) Total actin expression for SH-SY5Y cells and primary neurons plated on each PA matrix. Actin expression increases with higher matrix stiffness. All results reported relative to GAPDH expression (one-way ANOVA, p < 0.05). (d) MTS viability of SH-SY5Y and primary neurons on each substrate, when exposed to 10 μM Aβ(1–42). This data suggests a neuroprotective effect, with decreased matrix stiffness resulting in higher cell viability when exposed to the same treatment (two-way ANOVA, p < 0.05).
Actin Polymerization and Cell Stiffness Adjust in Response to Both Matrix Stiffness and Aggregated Aβ.
Characterizing the mechanical properties of individual cells is sensitive to cell type, sample preparation (e.g., live/fixed), and method of measurement. Note that cell stiffness is generally reported as a Young’s modulus (E) and an increasing E indicates lower compressibility and higher cell stiffening. Consistent with our previous studies, live human primary neurons were plated on the soft and stiff PA matrices, as well as a glass coverslip, prior to indentation measurements. Because AFM indentation requires physical contact between the AFM probe and the sample, cell stiffness could not be obtained for cells suspended in the 3D collagen gel. Previous indentation measurements of cell stiffness were found to be 0.23–0.52 kPa for cortical neurons,37,38 2–11 kPa for human fibroblasts, and 1.2–2.8 kPa for human lung cells.39 As shown in Figure 3, our AFM nanoindentation results are consistent with these previous studies and confirm that primary human neurons are sensitive to changes in matrix stiffness. Neurons adapting to the softest PA matrix displayed a Young’s modulus of 0.18 kPa. On the stiffest PA matrix, a modest, yet significant, increase to 0.22 kPa was observed. On glass, a material like traditional tissue culture plastic with GPa stiffness, neurons display a stiff phenotype with a spread morphology and a Young’s modulus of 0.42 kPa, which is nearly double that observed on the softer PA matrices. Our stiffness results for live neurons plated on a glass substrate are comparable to previous results reported in the literature.39
Further shown in Figure 3, the primary neurons display a clear, matrix-dependent morphology similar to that observed previously for SH-SY5Y cells. On the softest matrix, neurons display a rounded morphology, yet when plated onto the stiffest matrix a spread morphology is adopted. An ImageJ analysis of neuronal spread area is provided in the Supporting Information. Cells adherent to a stiff matrix generally adopt a spread morphology with an increased apical cell area. A stiff matrix can better resist cell-generated traction forces leading to an increase in actomyosin activation, stress fiber formation, and intracellular tension. Qualitative confocal fluorescence images of primary neurons plated on the stiff PA matrices displayed a clear increase in phallodin-stained F-actin and stress fiber formation, while the rounded neurons on the softest matrix stained weakly and thus qualitatively display the lowest F-actin fraction. In no instance for the 2D matrices did we see a traditional neuronal morphology with long neurites extending from a well-defined soma. Only in the 3D collagen matrix was this traditional, neuronal morphology observed.
To better quantify the extent of matrix-dependent actin reorganization, total actin expression was measured for cells that were plated on the each of the 2D PA matrices. As displayed in Figure 3, both neuroblastoma and primary neurons significantly increased actin production when plated on traditional tissue culture plastic. However, when comparing total actin levels from cells plated on either soft and stiff PA matrices, no significant differences were observed. Attempts to isolate G- and F-actin pools separately were unsuccessful, and therefore, total actin is reported (normalized to GADPH levels). However, we suspect that if F-actin were isolated, its fraction would decrease as matrix stiffness decreased. The large increase in actin production from cells plated on plastic is directly in line with the large increase in cell stiffness observed. Overall, for neurons and SH-SY5Y neuroblastoma, the morphological changes in cell spreading clearly correspond to reorganization of the actin cytoskeleton that subsequently manifests as a change in cell stiffness. This inter-relationship between cell area, F-actin, and cell stiffness had previously been reported by Solon et al. for NIH 3T3 fibroblasts.40 Using a similar series of PA matrices, fibroblasts preferentially adopted a spread morphology (higher cell area) on a stiff matrix and a rounded morphology (lower cell area) on softer matrices. Plotting fibroblast cell stiffness against cell area displayed a positive correlation and furthermore displayed an increase in F-actin polymerization.
Apart from physical stimuli, chemical cues presented to the cell membrane from the extracellular space can also modify cell stiffness.41,42 To investigate the potential of aggregated Aβ inducing changes in cell stiffness, each cell type was exposed to 1 μM of oligomeric or fibrillated Aβ for 24 h. All stiffness experiments were performed at 37 °C with live cells cultured onto a glass coverslip. We selected a subtoxic concentration of aggregated Aβ because our interest was whether cell-Aβ interactions may equally potentiate changes in cell stiffness. If toxic doses were used, then cytoskeletal restructuring that results from either apoptotic blebbing or necrotic membrane damage would confound any stiffness changes introduced by cell-Aβ interactions alone. As seen in Figure 4, for both cell types, an increase in cell stiffness was observed when exposed to the oligomeric Aβ; however, exposure to the same concentration of fibrillated Aβ produced no change in stiffness for either cell type. Referring to Figure 2, this concentration and exposure would result in a minor (if any) reduction in viability, and therefore, the aggregation dependent increase in cell stiffness is not likely a result of actin restructuring due toxicity, but rather it indicates that oligomeric and fibrillated Aβ interact with neuronal cells in distinctly separate fashions.
Figure 4.
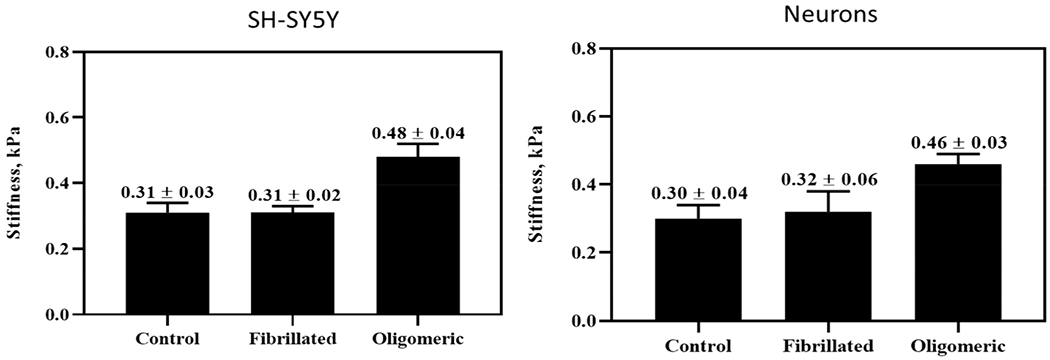
AFM cell stiffness measurements of SH-SY5Y cells and human primary cortical neurons after treatment with 1 μM Aβ(1—42) for 24 h. Treatment with oligomeric species induces cell stiffening, while this was not observed after treatment with the fibrillated species (one-way ANOVA, p < 0.05).
Multiple studies have explored cell—Aβ interactions using cell stiffness to better detail the mechanism of amyloid-mediated toxicity.43–45 Lulevich et al. reported that untreated N2a neuroblastoma have a Young’s modulus of 0.9 MPa and after exposure to oligomeric Aβ(1—42) the Young’s modulus increased to 1.85 MPa.44 Exposure to monomeric or fibrillated Aβ yielded either no change (monomeric) or a modest increase (fibrillated) in cell stiffness. An even more pronounced change in stiffness was observed for HT22 hippocampal neurons, whereby exposure to oligomeric Aβ prompted a greater than 3-fold increase in cell stiffness from 1.73 MPa (untreated) to 5.5 MPa (treated). In a separate study, the stiffness of primary hippocampal neurons, exposed to oligomeric Aβ, was further studied Ungureanu et al.; however, depending upon the “age” of the neuron, exposure to Aβ(1–42) resulted in a significant decrease in cell stiffness.20 For cortical neurons harvested from 17 day old Sprague—Dawley rats, the viscoelastic response was monitored as a function of oligomeric Aβ(1—42) exposure using microrheology. Using a 1 μM Aβ(1—42) dose, the scaled shear modulus (Go) of cortical neurons was found to increase from control values (~42 Pa) to ~55 kPa after a 6 h exposure. With continued exposure up to 24 h, this increased rigidity displayed no further change. Similarly, the viscosity increased from 3.0 to ~4.7 Pa·s after a 3 h exposure and remained constant thereafter for 24 h.
Potential Physical Mechanisms That Regulate Neuronal Survival.
The relationship between cytoskeletal structure and survival suggests that amyloid toxicity is inherently sensitive to changes in the ECM. ECM stiffness has previously been shown to affect the survival of neurons and cancer cells exposed to various toxins, nanoparticles, and chemotherapeutics. As reported by Senthebane et al., the ECM proteins of the tumor microenvironment are an essential component to understanding chemoresistance in esophageal squamous cancer cells (ESCCs).46 A PCR analysis of the decellularized ESCC biopsies revealed increased mRNA expression for multiple ECM proteins: collagen, laminin, and fibronectin. Moreover, cells seeded into these fortified matrices displayed an enhanced resistance to common chemotherapies relative to cells grown on plastic or in collagen- and fibronectin-lacking matrices. Ramamoorthi and co-workers furthermore reported that glioblastoma exposed to acrylamide, acetaminophen, quinidine, or cadmium chloride displayed a clear matrix-dependent cell viability.17 Glioblastoma cells were plated on traditional tissue culture plastic or suspended in two different 3D alginate matrices designated as soft and stiff, and cells grown in the soft alginate matrix were found to be more susceptible to each of the four toxins. In a separate study, Wang et al. found when two breast cancer cell lines were plated on 2D substrates with stiffness values ranging from 1—25 kPa, the cells grown on the softest matrix were resistant to paclitaxel treatment.47 Combined these studies confirm cell survival is sensitive to the ECM; however, the specific mechanism that promotes this sensitivity is unknown.
An initial consideration in mechanosensitive toxicity is endocytosis. In general, when the physical properties of a cell change, it would be anticipated that endocytic pathways could be affected. This uptake sensitivity to the ECM would, however, be pathway-specific, because different endocytic pathways require a specific membrane deformation, actin organization, and/or protein recruitment. From our previous studies on monomeric Aβ endocytosis, undifferentiated SH-SY5Y cells plated on progressively softer PA matrices displayed a clear reduction in spread area and cell stiffness.15 This soft, rounded phenotype of SH-SY5Y cells also displayed an increased uptake of monomeric Aβ. This finding was consistent with a recently published theoretical model that describes the energetics and kinetics of membrane deformation and wrapping.48 Further support is found from the experimental study that detailed the substrate dependent uptake of carboxylated polystyrene particles into bovine endothelial cells.49
While the expectation may be that softening promotes endocytosis, monomeric and oligomeric Aβ display completely different physicochemical properties and thus are not expected to be internalized through the same endocytic path. As shown in the Supporting Information, our initial uptake studies using oligomeric Aβ (prepared as described earlier but doped with fluorescently labeled monomeric Aβ) indicate that SH-SY5Y cells on soft and stiff matrices internalize less oligomeric Aβ than SH-SY5Y cells grown on traditional cell culture plastic. Therefore, the improved viability of SH-SY5Y cells exposed to oligomeric Aβ may be partly attributed to the reduced uptake. The internalization of oligomeric Aβ into primary neurons, however, displayed no significant sensitivity to the underlying substrate. Therefore, the improved viability observed on the softest matrix does not stem from differences in endocytic uptake. Further attempts to assess uptake of oligomeric Aβ in cells suspended in the 3D matrix were unsuccessful due to a limited cell number. Note that, despite a micrometer-sized pore structure for 2 mg/mL collagen gels, the lower diffusivity of oligomeric Aβ into the 3D matrix may also contribute to the improved viability for both SH-SY5Y cells and primary neurons.50
Oligomeric Aβ(1–42) exerts toxicity through multiple mechanisms, some of which—oxidative stress and calcium dysregulation—are known to be sensitive to the actin cytoskeleton and intracellular tension.51–56 It has previously been shown that primary hippocampal neurons, when pretreated with cytochalasin D for 1 h, display a dose-dependent increase in viability when challenged with 50 μM Aβ(25–35) after 48 h.21 Cytochalasins are a class of fungal metabolites that act to destabilize the cytoskeleton by binding to the plus-end of actin microfilaments and preventing further actin fiber elongation and polymerization. However, when the microtubules were destabilized using a 100 nM colchicine pretreatment, there was no recovery in viability, suggesting that specifically actin depolymerization contributed to the improved viability. To investigate the role of actin turnover in our studies, both SH-SY5Y cells and primary neurons were pretreated with 100 nM cytochalasin D, prior to a 10 μM challenge of oligomeric Aβ(1–42) for 48 h. As shown in Figure 5, cells pretreated with cytochalasin D demonstrated improved MTS viability and reduced LDH toxicity when compared to cells treated with oligomeric Aβ alone. Thus, both the pharmacological and matrix-mediated depolymerization of the actin cytoskeleton can provide protection against oligomeric Aβ.
Figure 5.
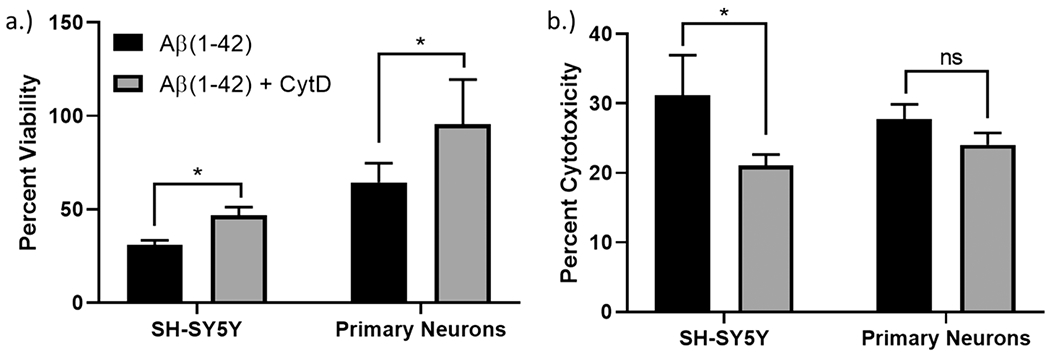
(a) MTS viability and (b) LDH toxicity studies for SH-SY5Y cells and primary neurons when pretreated with cytochalasin D and followed by exposure to 10 μM oligomeric Aβ(1—42) for 48 h (one-way ANOVA, p < 0.05).
Actin dynamics and cell—ECM adhesion signaling are interrelated.57–59 Adhesion to the ECM is predominantly integrin-mediated, and depending upon the specific integrin activation different scaffolding and adaptor proteins are recruited to the intracellular focal adhesion. This focal adhesion furthermore connects to the actin cytoskeleton and through its composition and structure may signal secondary messengers that modulate actin turnover. An early, upstream regulator of cytoskeletal status is paxillin; its recruitment and phosphorylation state helps regulate cell adhesion and cytoskeletal structure. Both SH-SY5Y cells and primary neurons display a significant decrease in paxillin expression (normalized to the GAPDH band) when plated on the soft PA matrices, indicating that paxillin expression is highly matrix-dependent. as shown in Figure 6. The decrease of total paxillin is consistent with decreased cell adhesion and actin polymerization, and this correlates with improved viability; however, further studies are underway to identify whether paxillin (or its phosphorylation status) directly participates in matrix-mediated, neuronal survival. As shown in the Supporting Information, our initial attempts to agonize integrin activation using MnCl2 resulted in reduced viability for SH-SY5Y cells exposed to 5 μM oligomeric Aβ, but no significant effect was observed for the primary neurons. Given that paxillin and its phosphorylation state are associated with amyloid-mediated cytoskeletal dysfunction in Alzheimer’s disease,60–62 understanding how adhesion dynamics may protect against neuronal loss in an aging and/or pathological ECM would offer multiple untapped targets in the search for effective, new therapies in neurodegenerative disorders.
Figure 6.
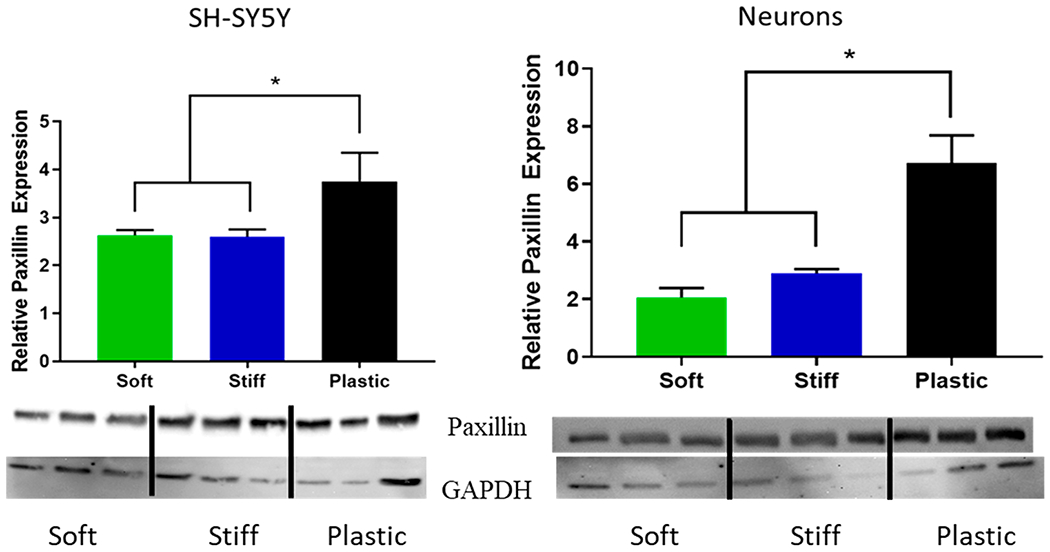
Relative paxillin expression is matrix dependent for SH-SY5Y cells and human primary cortical neurons on PA and tissue culture plastic substrates (one-way ANOVA, p < 0.05).
In conclusion, Aβ aggregates are a key component of Alzheimer’s disease, and their mechanism of toxicity is a primary focus when developing next-generation therapeutic options. Consistent with previous studies in alternative cell lines, oligomeric Aβ is significantly more toxic to both SH-SY5Y neuroblastoma and primary cortical neurons than fibrillated Aβ. We find that this toxicity, however, is matrix-dependent and cytoskeletal remodeling may directly influence amyloid toxicity. Cells cultured on soft 2D or 3D matrices and exposed to aggregated Aβ display a significant recovery in MTS viability compared to same-dosed cells plated on conventional tissue culture plastic. For cells adherent to stiff matrices, there is an increase in actin polymerization and stress fiber formation, which translates to an increase in cell stiffness. For both cell types, treatment with a subtoxic concentration of oligomeric Aβ further prompted significant cell stiffening. Both SY-SY5Y cells and primary neurons grown on the softer matrices displayed a rounded, soft mechanical phenotype that weakly stained for F-actin. We suggest this is due to reduced cell adhesion to the soft ECM, and this is consistent with the reduced paxillin levels observed. We anticipate that cells grown in a matrix that more closely mimics the mechanical environment of the brain are buffered against cell stiffening introduced by oligomeric Aβ. The precise mechanism that confers this matrix-dependent protection is unclear, and future studies are underway, but our results suggest that actin depolymerization can protect against amyloid toxicity. Our focus on the actin cytoskeleton is furthermore supported by pharmacological depolymerization of actin using cytochalasin D, which improves the viability for both cell types when challenged with oligomeric Aβ. Overall, recognizing that physical cues presented from the ECM may regulate cell survival introduces a variety of new sites for potential therapeutic intervention in Alzheimer’s disease.
MATERIALS AND METHODS
Preparation of Oligomeric and Fibrillated Aβ.
Amyloid beta used for AFM characterization was purchased from California Peptide and American Peptide Company. Further toxicity and AFM cell stiffness analyses were done using only Aβ(1–42) purchased from California Peptide. Aβ(1–42) oligomers and fibrils were prepared by following the protocol reported by Stine et al.24 Any pre-existing aggregates were removed by dissolving the Aβ to 5 mM in hexafluoro-2-propanol (HFIP), aliquoted into microcentrifuge tubes, and the allowed to evaporate in a fume hood overnight. These samples were stored at −20 °C in a desiccant jar until further use. For sample preparation, Aβ(1—42) was dissolved to 5 mM in dimethyl sulfoxide (DMSO). For oligomeric samples, Aβ(1–42) was further diluted to 100 μM in serum-free,phenol-free cell culture medium and incubated at 4 °C for 24–48 h. For fibrillated samples, Aβ(1–42) was diluted to 100 μM in 10 mM HCl and incubated at 37 °C in a heat block for 24 h prior to AFM characterization or dosing.
Structural Characterization of Aβ(1–42) Preparations via AFM.
Height images were recorded using a Molecular Force Probe 3D (Asylum Research) atomic force microscope. Fibrillated and oligomeric Aβ samples were prepared by drop casting Aβ solutions (100 μM) onto a freshly cleaved atomically flat mica (V–I grade, SPI Supplies) substrates. AFM height images of deposited Aβ samples were collected in air using the AC mode with silicon nitride AFM probes, a nominal spring constant of 0.3 N/m, and a typical tip radius of curvature of 8 nm (Mikromasch, CSC37). In this study, images were collected with a spatial resolution of 5 nm and 1 Hz scan rate. Raw AFM data and a detailed experimental analysis are provided in the Supporting Information.
2D Polyacrylamide Matrix Preparation.
Polyacrylamide (PA) matrices were prepared exactly as described in our previous publication.15 A detailed protocol can be found in the Supporting Information. Note that, consistent with our previous studies, we selected a “soft” and “stiff” PA matrix to contrast with results provided from cells plated on traditional, tissue culture plastic. Our “soft” and “stiff” PA matrices have Young’s moduli of approximately 3 and 24 kPa, respectively. Rat tail collagen type IV was selected as an ECM linker protein to promote cell adhesion. This protein was covalently bound to the surface of each PA matrix using standard sulfo-SANPAH chemistry. Note that all traditional cell culture plates (plastic) also had a thin layer of collagen type IV deposited onto the surface to assist adhesion.
Cell Culture.
SH-SY5Y human neuroblastoma cells were obtained from American Type Culture Collection. Cells were maintained at 37 °C and 5% CO2 and were grown in Opti-MEM supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, MEM nonessential amino acids (Gibco), and 1% penicillin/streptomycin. All uptake experiments used Opti-MEM without phenol in the media. SH-SY5Y cells would not be used when the passage number exceeded 30. Primary human cortical neurons were obtained from Neuromics (Edina, MN) and grown in neuronal culture medium, also purchased from Neuromics. These neurons were cultured strictly according to the culture conditions advised by Neuromics. Primary neurons were passaged 2–3 times before beginning anew with frozen neurons.
Confocal Microscopy.
After primary neurons were plated onto the 2D polyacrylamide matrices, the cells were fixed in ice cold, 4% paraformaldehyde. Cells were then permeabilized with 0.1% Triton-X, and stained with FITC-phalloidin (green). The cells were mounted on microscope slides with a drop of VectaShield mounting media containing DAPI for staining cell nuclei blue. The cells were imaged using a Zeiss 710 confocal microscope located in the Central Microscopy Research Facility at the University of Iowa.
3D Collagen Matrix Preparation and Rheological Characterization.
Collagen matrices were prepared according to the method reported by Baker et al.63 Briefly, the gels were prepared by first combining 100 mM HEPES in DPBS with high concentration collagen I (Corning) in equal proportions. This solution was mixed with cell culture media and cells to get a final concentration of 2 mg/mL and 1 × 105 cells/mL. The mixture was plated into 96-well plates at 100 μL per well. The plate was incubated at 37 °C, 5% CO2 for 2 h while the collagen matrix spontaneously self-assembled. After the matrix had fully formed, 100 μL of complete medium was added above each gel and the cells were grown for 24 h before staining or treatment.
The viscoelastic properties of these collagen gels were determined using a RheoStress One rheometer with a 35 mm stainless steel, 4 ° cone. Collagen mixtures of different concentrations were prepared and allowed to polymerize on the rheometer for 2 h at 37 °C. Frequency and stress sweeps were first performed to determine the linear viscoelastic range of each matrix using an oscillatory stress of 250 Pa while varying the frequency from 0.1 to 10 Hz and measuring 8 points per decade. Then, using a frequency of 1 Hz, the oscillatory stress was varied between 0.04 and 10 Pa. Each frequency and stress sweep were performed at 37 °C and is the average of three independently prepared samples.
Cell Viability and Toxicity Assays.
MTS Proliferation Assay.
SH-SY5Y cells or primary neurons were plated in 12-well plates on 2D PA matrices or tissue culture plastic at 1.0 × 106 cells per well. Otherwise SH-SY5Y cells or primary neurons were plated in 2 mg/mL 3D collagen matrices or on tissue culture plastic in 96-well plates at 1.0 × 105 cells per well as described earlier. After 24 h of growth, cells were treated with either oligomeric or fibrillated Aβ at various concentrations for 48 h in serum-free and phenol-free media. After this exposure period, the media was aspirated and the cells were gently washed with DPBS. MTS reagent was added at 1 mL reagent plus 10 mL of Hank’s balanced salt solution with 1 mg/mL glucose. The plate was incubated at 37 °C for an additional 4 h, and then the absorbance was measured at 490 nm using a plate reader. Viability for all samples was compared to that of untreated control cells that were plated on the same corresponding matrix.
Lactate Dehydrogenase Release Assay.
SH-SY5Y cells or primary neurons were plated in 12-well plates on 2D PA matrices or tissue culture plastic at 1.0 × 106 cells per well. After 24 h of growth, cells were treated with either oligomeric or fibrillated Aβ at various concentrations for 48 h in serum-free, phenol-free media. Thirty minutes before analysis, 10 μL of 10× lysis buffer was added to positive control wells for each cell line and on each substrate. Lactate dehydrogenase (LDH) release was measured using a LDH release assay kit (Thermo Scientific), according to the manufacturer’s instructions. After 50 μL of sample media was transferred to a 96-well plate, 50 μL of the working reagent was added to each well. The plate was stored in the dark at room temperature for 1 h. Then 50 μL of a stop solution was added to each well. The absorbance of the plate was read at 490 nm on a plate reader. Toxicity is reported as the percentage of LDH release relative to the completely lysed sample.
Nanoindentation to Measure Cell Stiffness.
The AFM nanoindentation for cells grown on either the 2D PA matrices or glass was done exactly as described in our previous paper. A detailed experimental protocol is further provided in the Supporting Information.
Western Blot Analysis to Measure Total Actin Expression.
To measure actin expression for each of the 2D PA substrates, PA gels were prepared as previously mentioned. SH-SY5Y cells or primary neurons were plated on each substrate at 1.5 × 106 cells per well on the softest substrate and at 1.0 × 106 cells per well on the stiff and plastic substrates and were grown for 3 days in complete culture medium. Cells were lysed and prepared for a Western blot. Next, 20 μg/well of each sample was loaded onto a 10% SDS-PAGE gel and run at 90 V. The samples were transferred electrophoretically onto a nitrocellulose membrane and then blocked in 5% BSA in TBST for 2 h. The membrane was then probed with an actin primary antibody (Cytoskeleton, Inc.) and a GAPDH housekeeping control (University of Iowa Hybridoma Bank) according to the vendors’ instructions. The membrane was washed twice with TBST and then probed with a rabbit secondary antibody (Santa Cruz, 1:20,000) for 1 h. The membrane was washed twice with TBST and then analyzed using a chemiluminescent substrate according to the manufacturer’s instructions (ThermoScientific). Each sample is presented relative to the amount of GAPDH expression.
Supplementary Material
ACKNOWLEDGMENTS
This work utilized the Zeiss LSM710 confocal microscope in the University of Iowa Central Microscopy Research Facilities that was purchased with funding from the NIH SIG grant RR025439-01.
Funding
We would like to acknowledge the Dale & June Wurster Pharmaceutics Graduate Student Fellowship for the financial support of T.M.K. Research reported in this publication was in part supported by the Environmental Health Sciences Research Center of the National Institutes of Health under award number ES005605.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.9b00401.
Experimental and fitting details for AFM nanoindentation on live cells; AFM images of oligomeric and fibrillated; Aβ fitted force—indentation plots for cells dosed with monomeric, oligomeric, and fibrillated Ab; fitted force—indentation plots for primary neurons grown on glass and different stiffness PA matrices; fitted force—indentation plots for bare soft and stiff PA matrices; cell spread analysis for primary neurons grown on different substrates; shear moduli for 3D collagen gels; experimental details about oligomeric Aβ uptake studies; uptake results from flow cytometry; manganese chloride pretreatment prior to oligomeric Aβ challenge; collected references (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acschemneuro.9b00401
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
Contributor Information
Terra M. Kruger, Department of Pharmaceutical Sciences and Experimental Therapeutics, College of Pharmacy, The University of Iowa, Iowa City, Iowa 52242, United States
Kendra J. Bell, Department of Pharmaceutical Sciences and Experimental Therapeutics, College of Pharmacy, The University of Iowa, Iowa City, Iowa 52242, United States
Thiranjeewa I. Lansakara, Department of Chemistry, The University of Iowa, Iowa City, Iowa 52242, United States.
Alexei V. Tivanski, Department of Chemistry, The University of Iowa, Iowa City, Iowa 52242, United States.
Jonathan A. Doorn, Department of Pharmaceutical Sciences and Experimental Therapeutics, College of Pharmacy, The University of Iowa, Iowa City, Iowa 52242, United States
Lewis L. Stevens, Department of Pharmaceutical Sciences and Experimental Therapeutics, College of Pharmacy, The University of Iowa, Iowa City, Iowa 52242, United States.
REFERENCES
- (1).Parihar MS, and Brewer GJ (2010) Amyloid-beta as a modulator of synaptic plasticity. J. Alzheimer’s Dis 22, 741–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Rajasekhar K, Chakrabarti M, and Govindaraju T (2015) Function and toxicity of amyloid beta and recent therapeutic interventions targeting amyloid beta in Alzheimer’s disease. Chem. Commun 51, 13434–13450. [DOI] [PubMed] [Google Scholar]
- (3).Yankner BA, Duffy LK, and Kirschner DA (1990) Neurotrophic and neurotoxic effects of amyloid beta protein: reversal of tachykinin neuropeptides. Science 250, 279–282. [DOI] [PubMed] [Google Scholar]
- (4).Sengupta U, Nilson AN, and Kayed R (2016) The role of amyloid-beta oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 6, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Henriques AG, Oliveira JM, Carvalho LP, and da Cruz e Silva OAB (2015) Abeta influences cytoskeletal signaling cascades with consequences to Alzheimer’s disease. Mol. Neurobiol 52, 1391–1407. [DOI] [PubMed] [Google Scholar]
- (6).Ishii T, Haga S, and Tokutake S (1979) Presence of neurofilament protein in Alzheimer’s neurofibrillary tangles (ANT). An immunofluorescent study. Acta Neuropathol. 48, 105–112. [DOI] [PubMed] [Google Scholar]
- (7).Nukina N, Kosik KS, and Selkoe DJ (1987) Recognition of Alzheimer paired helical filaments by monoclonal neurofilament antibodies is due to cross reaction with tau protein. Proc. Natl. Acad. Sci. U. S. A 84, 3415–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lavedan C, Buchholtz S, Nussbaum RL, Albin RL, and Polymeropoulos MH (2002) A mutation in the human neurofilament M gene in Parkinson’s disease that suggests a role for the cytoskeleton in neuronal degeneration. Neurosci. Lett 322, 57–61. [DOI] [PubMed] [Google Scholar]
- (9).Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, and Saudou F (2007) Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci 27, 3571–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Craig AM, and Banker G (1994) Neuronal polarity. Annu. Rev. Neurosci 17, 267–310. [DOI] [PubMed] [Google Scholar]
- (11).Eira J, Silva CS, Sousa MM, and Liz MA (2016) The cytoskeleton as a novel therapeutic target for old neurodegenerative disorders. Prog. Neurobiol 141, 61–82. [DOI] [PubMed] [Google Scholar]
- (12).Cairns NJ, Lee VM, and Trojanowski JQ (2004) The cytoskeleton in neurodegenerative diseases. J. Pathol 204, 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bonneh-Barkay D, and Wiley CA (2009) Brain extracellular matrix in neurodegeneration. Brain Pathol. 19, 573–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Murphy MC, Huston J, Jack CR III, Glaser KJ, Manduca A, Felmlee JP, and Ehman RL (2011) Decreased brain stiffness in Alzheimer’s disease determined by magentic resonance elastography. J. Magn. Reson. Imaging 34, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kruger TM, Bell KJ, Lansakara TI, Tivanski AV, Doorn JA, and Stevens LL (2019) Reduced extracellular matrix stiffness prompts SH-SY5Y cell softening and actin turnover to selectively increase Abeta(1–42) endocytosis. ACS Chem. Neurosci 10, 1284–1293. [DOI] [PubMed] [Google Scholar]
- (16).Zustiak SP, Dadhwal S, Medina C, Steczina S, Chehreghanianzabi Y, Ashraf A, and Asuri P (2016) Three-dimensional matrix stiffness and adhesive ligands affect cancer cell response to toxins. Biotechnol. Bioeng 113, 443–452. [DOI] [PubMed] [Google Scholar]
- (17).Ramamoorthi K, Hara J, Ito C, and Asuri P (2014) Role of three-dimensional matrix stiffness in regulating the response of human neural cells to toxins. Cell. Mol. Bioeng 7, 278–284. [Google Scholar]
- (18).Song C, Perides G, Wang D, and Liu YF (2002) Beta-amyloid peptide induces formation of actin stress fibers through p38 mitogen-activated protein kinase. J. Neurochem 83, 828–836. [DOI] [PubMed] [Google Scholar]
- (19).Mendoza-Naranjo A, Gonzalez-Billault C, and Maccioni RB (2007) Abeta1–42 stimulates actin polymerization in hippocampal neurons through Rac1 and Cdc42 Rho GTPases. J. Cell Sci 120, 279–288. [DOI] [PubMed] [Google Scholar]
- (20).Ungureanu AA, Benilova I, Krylychkina O, Braeken D, De Strooper B, Van Haesendonck C, Dotti CG, and Bartic C (2016) Amyloid beta oligomers induce neuronal elasticity changes in age-dependent manner: a force spectroscopy study on living hippocampal neurons. Sci. Rep 6, 25841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Furukawa K, and Mattson MP (1995) Cytochalasins protect hippocampal neurons against amyloid beta peptide toxicity - evidence that actin depolymerization suppresses Ca2+ influx. J. Neurochem 65, 1061–1068. [DOI] [PubMed] [Google Scholar]
- (22).Mattson MP (2010) ER calcium and Alzheimer’s disease: in a state of flux. Sci. Signaling 3, p e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dutta R, and Trapp BD (2011) Mechanisms of neuronal dysfunction and degeneration in multiple sclerosis. Prog. Neurobiol 93, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Stine WB, Jungbauer L, Yu C, and LaDu MJ (2010) Preparing syntheitc Abeta in different aggregation states. Methods Mol. Biol 670, 13–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Puzzo D, and Arancio O (2012) Amyloid-beta: Dr. Jekyll or Mr. Hyde? J. Alzheimer’s Dis. 33, S111–S120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Lorenzo A, and Yankner BA (1994) Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proe. Natl. Acad. Sci. U. S. A 91, 12243–12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cecchi C, Pensalfini A, Liguri G, Baglioni S, Fiorillo C, Guadagna S, Zampagni M, Formigli L, Nosi D, and Stefani M (2008) Differentiation increases the resistance of neuronal cells to amyloid toxicity. Neurochem. Res 33, 2516–2531. [DOI] [PubMed] [Google Scholar]
- (28).Krishtal J, Metsla K, Bragina O, Tougu V, and Palumaa P (2019) Toxicity of amyloid-beta peptides varies depending upon differentiation route of SH-SY5Y cells. J. Alzheimer’s Dis 71, 879–887. [DOI] [PubMed] [Google Scholar]
- (29).Gharibyan AL, Zamotin V, Yanamandra K, Moskaleva OS, Margulis BA, Kostanyan IA, and Morozova-Roche LA (2007) Lysozyme amyloid oligomers and fibrils induce cellular death via different apoptotic/necrotic pathways. J. Mol. Biol 365, 1337–1349. [DOI] [PubMed] [Google Scholar]
- (30).Limbocker R, Chia S, Ruggeri FS, Perni M, Cascella R, Heller GT, Meisl G, Mannini B, Habchi J, Michaels TCT, Challa PK, Ahn M, Casford ST, Fernando N, Xu CK, Kloss ND, Dobson CM, et al. (2019) Trodusquemine enhances Abeta42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat. Commun 10, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Li Y, Liu M, Yan Y, and Yang S-T (2014) Neural differentiation from pluripotent stem cells: The role of natural and synthetic extracellular matrix. World J. Stem Cells 6, 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Sart S, Yan Y, Li Y, Lochner E, Zeng C, Ma T, and Li Y (2016) Crosslinking of extracellular matrix scaffolds derived from pluripotent stem cell aggregates modulates neural differentiation. Acta Biomater. 30, 222–232. [DOI] [PubMed] [Google Scholar]
- (33).Yan Y, Martin LM, Bosco DB, Bundy JL, Nowakowski RS, Sang QX, and Li Y (2015) Differential effects of acellular embryonic matrics on pluripotent stems cell expansion and neural differentiation. Biomaterials 73, 231–242. [DOI] [PubMed] [Google Scholar]
- (34).Long KR, and Huttner WB (2019) How the extracellular matrix shapes neural development. Open Biol. 9, 180216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Farrukh A, Zhao S, and del Campo A (2018) Micro-environments designed to support growth and function of neuronal cells. Front. Mater 5, 1–22. [Google Scholar]
- (36).Lin H-H, Lin H-K, Lin I, Chiou Y-W, Chen H, Liu C, Harn HI, Chiu W, Wang Y, Shen M, and Tang M (2015) Mechanical phenotype of cancer cells: cell softening and loss of stiffness sensing. Oncotarget 6, 20946–20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bernick KB, Prevost TP, Suresh S, and Socrate S (2011) Biomechanics of single cortical neurons. Acta Biomater. 7, 1210–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Spedden E, White JD, Naumova EN, Kaplan DL, and Staii C (2012) Elasticity maps of living neurons measured by combined fluorescence and atomic force microscopy. Biophys. J 103, 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Luo Q, Kuang D, Zhang B, and Song G (2016) Cell stiffness determined by atomic force microscopy and its correlation with cell motility. Biochim. Biophys. Acta, Gen. Subj 1860, 1953–1960. [DOI] [PubMed] [Google Scholar]
- (40).Solon J, Levental I, Sengupta K, Georges PC, and Janmey PA (2007) Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys. J 93, 4453–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Raudenska M, Kratochvilova M, Vicar T, Gumulec J, Balvan J, Polanska H, Pribyl J, and Masarik M (2019) Cisplatin enhances cell stiffness and decreases invasiveness rate in prostate cancer cells by actin accumulation. Sci. Rep 9, 1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Williams LM, and Ridley AJ (2000) Lipopolysaccharide induces actin reorganization and tyrosine phosphorylation of Pyk2 and paxillin in monocytes and macrophages. J. Immunol 164, 2028–2036. [DOI] [PubMed] [Google Scholar]
- (43).Lu Z, Li H, Hou C, Peng Y, Long J, and Liu J (2017) Endogenously generated amyloid-beta increases stiffness in human neuroblastoma cells. Eur. Biophys. J 46, 415–424. [DOI] [PubMed] [Google Scholar]
- (44).Lulevich V, Zimmer CC, Hong HS, Jin LW, and Liu GY (2010) Single-cell mechanics provides a sensitive and quantitative means for probing amyloid-beta peptide and neuronal cell interactions. Proc. Natl. Acad. Sci. U. S. A 107, 13872–13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Jazvinscak Jembrek M, Simic G, Hof PR, and Segota S (2015) Atomic force microscopy as an advanced tool in neuroscience. Transl. Neurosci 6, 117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Senthebane DA, Jonker T, Rowe A, Thomford NE, Munro D, Dandara C, Wonkam A, Govender D, Calder B, Soares NC, Blackburn JM, Parker MI, and Dzobo K (2018) The role of tumor microenvironment in chemoresistance: 3D extracellular matrices as accomplices. Int. J. Mol. Sci 19, No. e2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wang Y, Gong T, Zhang ZR, and Fu Y (2017) Matrix stiffness differentially regulates cellular uptake behavior of nano-particles in two breast cancer cell lines. ACS Appl. Mater. Interfaces 9, 25915–25928. [DOI] [PubMed] [Google Scholar]
- (48).Yi X, and Gao H (2017) Kinetics of receptor-mediated endocytosis of elastic nanoparticles. Nanoscale 9, 454–463. [DOI] [PubMed] [Google Scholar]
- (49).Huang C, Butler P, Tong S, Muddana H, Bao G, and Zhang S (2013) Substrate stiffness regulates cellular uptake of nanoparticles. Nano Lett 13, 1611–1615. [DOI] [PubMed] [Google Scholar]
- (50).Yang Y, Motte S, and Kaufman LJ (2010) Pore size variable type I collagen gels and their interaction with glioma cells. Biomaterials 31, 5678–5688. [DOI] [PubMed] [Google Scholar]
- (51).Wang Y, Liu Q, Xu Y, Zhang Y, Lv Y, Tan Y, Jiang N, Cao G, Ma X, Wang J, Cao Z, Yu B, and Kou J (2016) Ginsenoside Rg1 protects against oxidative stress-induced neuronal apoptosis through myosin IIa-actin related cytoskeletal reorganization. Int. J. Biol. Sci 12, 1341–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Fiaschi T, Cozzi G, Raugei G, Formigli L, Ramponi G, and Chiarugi P (2006) Redox regulation of beta-actin during integrin-mediated cell adhesion. J. Biol. Chem 281, 22983–22991. [DOI] [PubMed] [Google Scholar]
- (53).Farah ME, Sirotkin V, Haarer B, Kakhniashvili D, and Amberg DC (2011) Diverse protective roles of the actin cytoskeleton during oxidative stress. Cytoskeleton 68, 340–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gardiner J, Overall R, and Marc J (2013) The nervous system cytoskeleton under oxidative stress. Diseases 1, 36–50. [Google Scholar]
- (55).Takeshita N, Evangelinos M, Zhou L, Serizawa T, Somera-Fajardo RA, Lu L, Takaya N, Nienhaus GU, and Fischer R (2017) Pulses of Ca2+ coordinate actin assembly and exocytosis for stepwise cell extension. Proc. Natl. Acad. Sci. U. S. A 114, 5701–5706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Rosenmund C, and Westbrook GL (1993) Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron 10, 805–814. [DOI] [PubMed] [Google Scholar]
- (57).Parsons JT, Horwitz AR, and Schwartz MA (2010) Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol 11, 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Gardel ML, Schneider IC, Aratyn-Schaus Y, and Waterman CM (2010) Mechanical integration of actin and adhesion dynamics in cell migration. Annu. Rev. Cell Dev. Biol 26, 315–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Case L, and Waterman C (2015) Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat. Cell Biol 17, 955–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Caltagarone J, Jing Z, and Bowser R (2007) Focal adhesions regulate Abeta signaling and cell death in Alzheimer’s disease. Biochim. Biophys. Acta, Mol. Basis Dis 1772, 438–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Berg MM, Krafft GA, and Klein WL (1997) Rapid impact of beta-amyloid on paxillin in a neural cell line. J. Neurosci. Res 50, 979–989. [DOI] [PubMed] [Google Scholar]
- (62).Caltagarone J, Hamilton RL, Murdoch G, Jing Z, DeFranco DB, and Bowser R (2010) Paxillin and hydrogen peroxide-inducible-clone 5 (Hic-5) expression and distribution in control and Alzheimer disease hippocampus. J. Neuropathol. Exp. Neurol 69, 356–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Baker EL, Lu J, Yu D, Bonnecaze RT, and Zaman MH (2010) Cancer cell stiffness: integrated roles of three-dimensional matrix stiffness and transforming potential. Biophys. J 99, 2048–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.