

Abstract
Rationale: Event-driven primary endpoints are increasingly used in pulmonary arterial hypertension clinical trials, substantially increasing required sample sizes and trial lengths. The U.S. Food and Drug Administration advocates the use of prognostic enrichment of clinical trials by preselecting a patient population with increased likelihood of experiencing the trial’s primary endpoint.
Objectives: This study compares validated clinical scales of risk (Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension, the French score, and Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management [REVEAL] 2.0) to identify patients who are likely to experience a clinical worsening event for trial enrichment.
Methods: Baseline data from three pulmonary arterial hypertension clinical trials (AMBITION [a Study of First-Line Ambrisentan and Tadalafil Combination Therapy in Subjects with Pulmonary Arterial Hypertension], SERAPHIN [Study of Macitentan on Morbidity and Mortality in Patients with Symptomatic Pulmonary Arterial Hypertension], and GRIPHON [Selexipag in Pulmonary Arterial Hypertension]) were pooled and standardized. Receiver operating curves were used to measure each algorithm’s performance in predicting clinical worsening within the pooled placebo cohort. Power simulations were conducted to determine sample size and treatment time reductions for multiple enrichment strategies. A cost analysis was performed to illustrate potential financial savings by applying enrichment to GRIPHON.
Measurements and Main Results: All risk algorithms were compared using area under the receiver operating curve and substantially outperformed prediction per New York Heart Association Functional Class. The REVEAL 2.0’s risk grouping provided the greatest time and sample size savings in AMBITION and GRIPHON for all enrichment strategies but lacked appropriate inputs (i.e., N-terminal-proB-type natriuretic peptide) to perform as well in SERAPHIN. Cost analysis applied to GRIPHON demonstrated the greatest financial benefit by enrolling patients with a REVEAL score ≥8.
Conclusions: This preliminary study demonstrates the feasibility of risk algorithms for pulmonary arterial hypertension trial enrichment and a need for further investigation.
Keywords: pulmonary arterial hypertension, clinical trial enrichment, risk score calculator
At a Glance Commentary
Scientific Knowledge on the Subject
The U.S. Food and Drug Administration encourages the use of prognostic enrichment of phase III drug efficacy trials to increase the odds of completing a successful trial in a timely manner. Traditional means of enriching clinical trials specifically for cardiovascular products typically was performed by New York Heart Association (NYHA) Classification. Enrolling patients with greatest disease severity (i.e., higher NYHA class) allows event-driven trials to observe more events in a shorter time period and achieve adequate statistical power more efficiently.
What This Study Adds to the Field
This study demonstrates that for pulmonary arterial hypertension (PAH) trials, the use of registry risk calculators (specifically, the Registry to Evaluate Early and Long-Term PAH Disease Management 2.0, French score, and Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension score) offer better prognostication of clinical worsening than NYHA class or other individual biomarkers and therefore are better suited for PAH clinical trial enrichment. Our study shows, for the first time, that risk-based enrichment not only reduces sample sizes and treatment time in trials but also that screening rates can be kept modest and that there is significant potential for financial benefit in trial enrichment.
Pulmonary arterial hypertension (PAH) is a collection of rare and progressive disorders of the pulmonary vasculature with no known cure (1). Our investigations focused on PAH in World Health Organization (WHO) group I, in which most of the experience lies. Although there has been an explosion of clinical trials seeking new therapeutic options and approaches in PAH in the last decade, endpoints to demonstrate drug efficacy efficiently and effectively in PAH trials are lacking (2, 3). Early-era PAH trials were primarily required to show a statistically significant increase in 6-minute-walk distance (6MWD) for demonstrating drug efficacy. However, it is now known that improvement in 6MWD is only weakly associated with reductions in clinical events. Moreover, demonstrated improvements in 6MWD are typically small (average 30 m), with debatable clinical relevance (4). Contemporary PAH clinical trials switched focus to complex determinants of therapeutic efficacy, such as time to clinical worsening, which is a composite endpoint of death, hospitalization, and other measures of disease progression. Although such event-driven endpoints demonstrate a benefit with clear clinical relevance to patients, clinical worsening is relatively infrequent. Hence, successful PAH trials end up requiring large-scale patient enrollment for lengthy durations, with substantial economic expenditure (4).
The U.S. Food and Drug Administration (FDA) recently suggested the use of prognostic enrichment strategies to improve clinical trial efficiency (5). This includes strategies that leverage enrollment based on the probability of an individual patient experiencing a disease-related endpoint for event-driven studies. In a PAH drug efficacy trial, this would emphasize enrollment of patients who are deemed to be at an intermediate or high risk for clinical worsening.
In the current study, we hypothesized that existing validated risk-prediction algorithms derived from PAH registry data could be used to identify patients at intermediate and high risk of clinical worsening using baseline clinical trial data. The goals of the present study were to demonstrate that these algorithms are prognostic of a clinical worsening event and that, in simulated scenarios, a patient cohort enriched with higher-risk patients (as identified by risk algorithms) can demonstrate a significant treatment benefit with a substantially smaller sample size compared with other, more simplistic measures or biomarkers of PAH prognosis.
Methods
The following three contemporary PAH risk-prediction algorithms were used to stratify patient risk of clinical worsening at baseline: Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) (6), the French pulmonary hypertension registry score (French) (7), and the U.S. Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL 2.0) (8). A description of the design of all these scores, including inclusion and exclusion criteria, has been published previously.
Three contemporary PAH trials, AMBITION (A Study of First-Line Ambrisentan and Tadalafil Combination Therapy in Subjects with PAH) (9), GRIPHON (Selexipag in PAH) (10), and SERAPHIN (Study of Macitentan on Morbidity and Mortality in Patients with Symptomatic PAH) (11) were chosen for analysis because they had time to clinical worsening as their primary endpoint. Definitions of clinical worsening for each trial are provided in Table 1. Data across trials were unified and standardized for testing the investigational risk algorithms. The variables used in each algorithm and the method for computing their final scores are shown in Table 2. All risk algorithms were applied as intended in the original publication except for COMPERA, which was left as a continuous variable and not rounded to the nearest integer after averaging. The following variables (underlined in Table 2) were unavailable across all clinical trials: diffusing lung capacity and hospitalization 6 months before randomization for REVEAL 2.0 and mixed venous oxygen saturation for COMPERA. Estimated glomerular filtration rate for REVEAL 2.0 was calculated using the Levey and colleagues equation, which considers race, age, and sex (12).
Table 1.
Components and Definitions of the Primary Endpoint for Each Clinical Trial
| Clinical Trial | Definition of Clinical Worsening |
|---|---|
| AMBITION |
|
| SERAPHIN |
|
| GRIPHON |
|
Definition of abbreviations: 6MWD = 6-minute-walk distance; AMBITION = A Study of First-Line Ambrisentan and Tadalafil Combination Therapy in Subjects with Pulmonary Arterial Hypertension; FC = functional class; GRIPHON = Selexipag in Pulmonary Arterial Hypertension; PAH = pulmonary arterial hypertension; SERAPHIN = Study of Macitentan on Morbidity and Mortality in Patients with Symptomatic Pulmonary Arterial Hypertension; WHO = World Health Organization.
Table 2.
Clinical Variables and Risk Algorithm Calculation
| Algorithm Clinical Variables | French Score | COMPERA | REVEAL 2.0 |
|---|---|---|---|
| BNP/NT-proBNP, pg/ml | × | +1 if <300 | −2 if <300 |
| +2 if ≤1,400 | +2 if ≥1,100 | ||
| +3 if >1,400 | |||
| NYHA class | +1 if ≤II | +1 if <III | −1 if I |
| +2 if =III | +1 if III | ||
| +3 if >III | +2 if IV | ||
| 6-minute-walk distance, m | +1 if >440 | +1 if >440 | −2 if ≥440 |
| +2 if ≥165 | −1 if >320 | ||
| +3 <165 | +1 if <165 | ||
| Right atrial pressure, mm Hg | +1 if <8 | +1 if <8 | +1 if >20 |
| +2 if ≤14 | |||
| +3 if >14 | |||
| Sex | × | × | +2 if male and >60 yr |
| Age, yr | × | × | +2 if male and >60 |
| Etiology | × | × | +1 if connective, +3 if portal, +2 if familial |
| Systolic blood pressure, mm Hg | × | × | +1 if <110 |
| Heart rate, bpm | × | × | +1 if >96 |
| Pulmonary vascular resistance, Wood units | × | × | −1 if <5 |
| Renal function, ml/min/1.73 m2 | × | × | +1 if <60 |
| Recent hospitalizations | × | × | +1 if within 6 mo |
| Diffusing lung capacity, %DlCO | × | × | +1 if <40% |
| Pericardial effusion | × | × | +1 if present |
| Mixed venous oxygen saturation, % | × | +1 if >65 | × |
| +2 if ≥60 | |||
| +3 if <60 | |||
| Cardiac index, L/min/m2 | +1 if ≥ 2.5 | +1 if ≥2.5 | × |
| +2 if ≥2 | |||
| +3 if <2 | |||
| Final score | Sum total | Average of total available measurements | Sum total + 6 |
Definition of abbreviations: BNP = B-type natriuretic peptide; bpm = beats per minute; COMPERA = Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension; NT-proBNP = N-terminal-proB-type natriuretic peptide; NYHA = New York Heart Association; REVEAL = Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management.
Clinical variables not included in the algorithm are marked with ×; clinical variables that were not available in any trial are underlined.
For statistical analysis, receiver operating characteristic (ROC) curves were generated for each algorithm to determine their ability to predict clinical worsening as defined by the trial’s original primary endpoint. Algorithms were benchmarked against a traditional clinical means of patient risk stratification (New York Heart Association [NYHA] functional class used in isolation) via nonparametric statistical analysis (i.e., bootstrapping) to determine statistical significance of the difference in the areas under the curve (AUCs). Algorithms were further benchmarked against 6MWD, NT-proBNP (N-terminal-proB-type natriuretic peptide), and the three hemodynamics used for risk calculation (listed in Table 2) to determine whether commonly used single clinical variables could provide the same degree of predictive performance as a multivariable risk score (see online supplement). Patients who were censored early from the primary endpoint were imputed as event-free, and a sensitivity analysis was conducted to determine the impact of this assumption (see online supplement). To avoid confounding baseline risk and treatment effects, only the placebo populations in each trial were used for ROC analysis.
Because both COMPERA and REVEAL 2.0 generate risk scores on a near continuous scale, we were interested in finding a consistent means to define cut points that allowed for simplified patient groupings for enrichment (low risk vs. intermediate risk vs. high risk, with the possibility for very low and/or very high depending on algorithm precision). We were also interested in optimizing cut points such that the high-risk patients not only had more clinical worsening events but also a faster time to clinical worsening. To that end, a survival tree analysis was applied to the pooled placebo population to determine such cut points for each algorithm (applied via rpart R package) (13, 14). Each survival tree was optimized to find the largest number of cut points such that each group had a statistically significantly different survival curve (per exponential regression) using 10-fold cross-validation. Our aim in using the largest number of cut points was to identify an optimal high-risk patient group from one candidate algorithm that could be recommended for prognostic enrichment without a trial sponsor conducting their own analysis.
Identified cut points were applied to the pooled treatment population to determine whether each simplified risk group saw a benefit in treatment, as determined by Cox proportional hazards. This analysis is necessary to support the goals of prognostic enrichment as a means to bridge therapy established in higher-risk groups to lower-risk patient populations. P < 0.05 was used to determine a statistical significance. The incidence rate of clinical worsening for each group and each treatment arm was calculated as events per 100 patient-years.
Sample size estimates for each trial were recalculated by resampling from patients with no missing algorithm data and employing the method originally proposed by Freeman in 1982 to estimate probability of clinically worsening events in each treatment arm (applied via the powerSurvEpi R package) (15, 16). Resampling was conducted to reflect the following multiple enrichment strategies: 1) selecting only intermediate- and high-risk patients (intermediate–high strategy); 2) selecting 50% of patients from the high-risk–only group and 50% from all other risk groups (high-other strategy); and 3) selecting 100% of patients from the high-risk group only (high-risk strategy). A nonparametric bootstrap analysis was used to generate 95% confidence intervals (CIs) for each estimated sample size determined from an event-driven power analysis. The parameters of the power analysis, namely the confidence level, anticipated effect size, and power to detect the hypothesized treatment effect, were kept as published in the original trial’s statistical design (see Table E1 in the online supplement). Although no attrition rate is specified in this analysis, resampling from the original trial populations allows results to reflect sample size estimations in the presence of early withdrawals. Mean times in trial (i.e., treatment time) were also calculated for each enrichment strategy per algorithm. Sample size and time in trial from simulations are presented as a percentage reduction from simulations using the nonenriched subpopulation, in which higher reduction indicates lower sample size, shorter treatment time, and, therefore, improved trial efficiency. For further details on the procedure to perform the nonparametric power analysis, see the online supplement.
As patient screening can become burdensome for trial enrichment, we aimed to estimate the likelihood of finding a patient identified per each algorithm’s risk groupings by calculating the ratio of total patients screened to patients enrolled per risk category in the pooled trial dataset. Ratios are presented as the number of patients who must be screened to enroll 100 patients, in which a higher ratio is indicative of higher screening numbers and therefore lesser screening efficiency.
Finally, a hypothetical cost savings exercise was conducted with the GRIPHON trial to demonstrate the benefit of enrichment in balancing the increased cost of screening by reducing the cost of treating enrolled patients (i.e., research costs) and to determine the enrichment strategy optimal for balancing these costs. Minimum estimated costs per patient were based on figures reported by Ryan and colleagues and other clinical procedure price estimations. Costs of treatment are provided in Table 3 (3, 17, 18). Mean time to clinical worsening or censor, as calculated from power simulations, was used in the cost analysis as the average treatment time per patient receiving the study drug. For this analysis, we assumed that every patient, regardless of risk algorithm, required a right heart catherization procedure in screening to confirm PAH group I diagnosis. Although this study used fewer variables to stratify patients, final estimated screening cost for each algorithm reflected the cost of collecting all data required for each algorithm. To account for the worst case of screened patients failing because of selection criteria outside of the matching risk level (such as very low 6MWD or prostacyclin analog background therapy), the number of patients required for screening was calculated using the following equation:
Table 3.
Cost Analysis for Pulmonary Arterial Hypertension Clinical Trial
| Study Element | Baseline Cost (USD) | Iterations per Study per ITT Patient | REVEAL 2.0 Screening | COMPERA Screening | French Screening | Nonenrichment Screening |
|---|---|---|---|---|---|---|
| Informed consent processing | 150 | 0 | 1 | 1 | 1 | 1 |
| History and PE | 500 | 0+ | 1 | 1 | 1 | 1 |
| Vital sign assessment | 50 | 3+ | 1 | 1 | 1 | 1 |
| Right heart catheterization | 3,500–5,000 | 0+ | 1 | 1 | 1 | 1 |
| 6-minute-walk test | 550 | 3+ | 1 | 1 | 1 | 1 |
| NT-proBNP | 140 | 0+ | 1 | 1 | — | — |
| Lung capacity test | 500 | — | 1 | — | — | — |
| Mixed venous O2 saturation | 200 | — | 1 | 1 | — | — |
| Creatinine | 50 | 0+ | 1 | — | — | — |
| IRB fees | 4,000 | 1 | — | — | — | — |
| Study drugs | 12,100 | 1 | — | — | — | — |
| Total | $13,900+ per treated patient | $5,640 per patient | $5,090 per patient | $4,750 per patient | $4,750 per patient | |
| $1,800+ per placebo patient | ||||||
Definition of abbreviations: COMPERA = Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension; IRB = institutional review board; ITT = intention to treat; NT-proBNP = N-terminal-proB-type natriuretic peptide; PE = physical examination; REVEAL = Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management; USD = U.S. dollars.
Costs per ITT patient reflects cost for every 12-week period of enrollment in a trial. Costs per ITT patient can vary considerably because of trial design; estimates reflect a minimum, with (+) indicating an anticipated increase.
Cost saving percentage was calculated as the percentage difference between total cost for the given enrichment strategy and the estimated cost with no enrichment.
Results
From a total of 1,769 patients, we identified n = 976 (55%) and n = 793 (45%) patients with all required variables from the treatment and placebo groups, respectively. As shown in Figure 1, all algorithms performed similarly in their ability to predict clinical worsening (COMPERA AUC, 0.70; 95% CI, 0.66–0.73; French score AUC, 0.66; 95% CI, 0.63–0.70; and REVEAL 2.0 AUC, 0.70; 95% CI, 0.66–0.73). Each investigational algorithm outperformed NYHA functional class (AUC, 0.61; 95% CI, 0.57–0.64) at predicting clinical worsening prognosis (COMPERA P = 2.26 × 10−6, French P = 6.5 × 10−4, and REVEAL P = 1.63 × 10−6). Furthermore, all risk algorithms had better performance in predicting clinical worsening than singular clinical variables (see Table E2). In our sensitivity analyses to test the assumption that early censored patients were event-free, we note that all algorithms perform worse if patients with a censor time of less than 3 years are removed from the analysis, though excluding patients censored before 1 year largely did not change the AUC of each algorithm (Figure E1). This indicates that algorithms may be somewhat optimistic in their prediction. It is worth noting that REVEAL 2.0 was, however, the most robust against this assumption and largely maintained its prognostic performance.
Figure 1.

Receiver operating curves (ROCs) for each investigational algorithm. The population used for ROCs included pooled placebo patients from each clinical trial with all available values listed in Table 2, excluding variables not available for any trial. N = 793. Area under curve (AUC) comparable for all risk algorithms for prediction of clinical worsening at end of study (COMPERA AUC, 0.70; 95% confidence interval [CI], 0.66–0.73; French score AUC, 0.66 95% CI, 0.63–0.70; and REVEAL 2.0 AUC, 0.70; 95% CI, 0.66–0.73. All investigational algorithms outperformed NYHA Classification (AUC, 0.61; 95% CI, 0.57–0.64) in nonparametric statistical analysis (COMPERA P = 2.26 × 10−6; French P = 6.5 × 10−4; and REVEAL P = 1.63 × 10−6). COMPERA = Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension; French = French score; NYHA = New York Heart Association; REVEAL = Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management.
Application of Survival Tree Analysis to Identify Risk Groups
Each investigational algorithm identified at least three unique risk groups with statistically significantly different time to clinical worsening rates using survival tree analysis. Cut points and incidence rates (as number of events per 100 patient-years) for each risk group are shown in Table 4. REVEAL 2.0 was the most precise and identified four statistically significantly different ranked groups for clinical worsening (P < 2 × 10−16; its full survival tree is shown as an example in Figure 2), specifically identifying an additional very low–risk group, and its high-risk group had a much higher incidence rate than those of COMPERA or French score. Only three different groups were identified using either COMPERA (P < 2 × 10−16) or the French score (P = 8.98 × 10−16). When used in isolation, NYHA functional class identified only two statistically significantly different ranked groups (P = 1.18 × 10−9). Hazard ratios between the pooled treatment and pooled placebo groups for reduction in clinical worsening rate were statistically significant for all risk groups identified, irrespective of the risk algorithm used (P < 0.05 for all), demonstrating that even lower-risk patients saw a treatment benefit and that bridging efficacy to these groups would be appropriate. Treatment effects were not significantly different between groups (i.e., there were no interactive effects between baseline risk and placebo versus treatment), although we note that this retrospective study is not powered to determine interactive effects.
Table 4.
Incidence Rate and Treatment Effect per Risk Group as Defined by Risk Algorithm and Its Survival Tree Cut Points
| Algorithm | Risk Group | Pooled Placebo Incidence Rate of Clinical Worsening (Events per 100 Patient-Years) | Pooled Treatment Incidence Rate of Clinical Worsening (Events per 100 Patient-Years) | Hazard Ratio for Reduction of Clinical Worsening Rate (95% Confidence Interval) |
|---|---|---|---|---|
| French score | Low (2, 3) | 17.10 | 10.47 | 0.61 (0.46–0.81) |
| Intermediate (1) | 29.75 | 18.19 | 0.60 (0.46–0.79) | |
| High (0) | 51.44 | 25.08 | 0.50 (0.38–0.67) | |
| COMPERA | Low (≤1.7) | 13.90 | 8.27 | 0.59 (0.42–0.85) |
| Intermediate (>1.7–2.1) | 27.37 | 16.36 | 0.59 (0.46–0.76) | |
| High (>2.1) | 52.52 | 24.81 | 0.48 (0.37–0.62) | |
| REVEAL 2.0 | Very Low (≤5) | 11.00 | 6.61 | 0.60 (0.36–0.99) |
| Low (6–8) | 21.19 | 11.97 | 0.56 (0.42–0.74) | |
| Intermediate (9, 10) | 36.90 | 23.07 | 0.63 (0.48–0.83) | |
| High (>10) | 72.32 | 34.52 | 0.48 (0.34–0.67) | |
| NYHA | Low (≤ II) | 16.69 | 9.79 | 0.58 (0.43–0.78) |
| Intermediate (>II) | 35.02 | 20.95 | 0.60 (0.50–0.73) |
Definition of abbreviations: COMPERA = Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension; NYHA = New York Heart Association; REVEAL = Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management.
Figure 2.
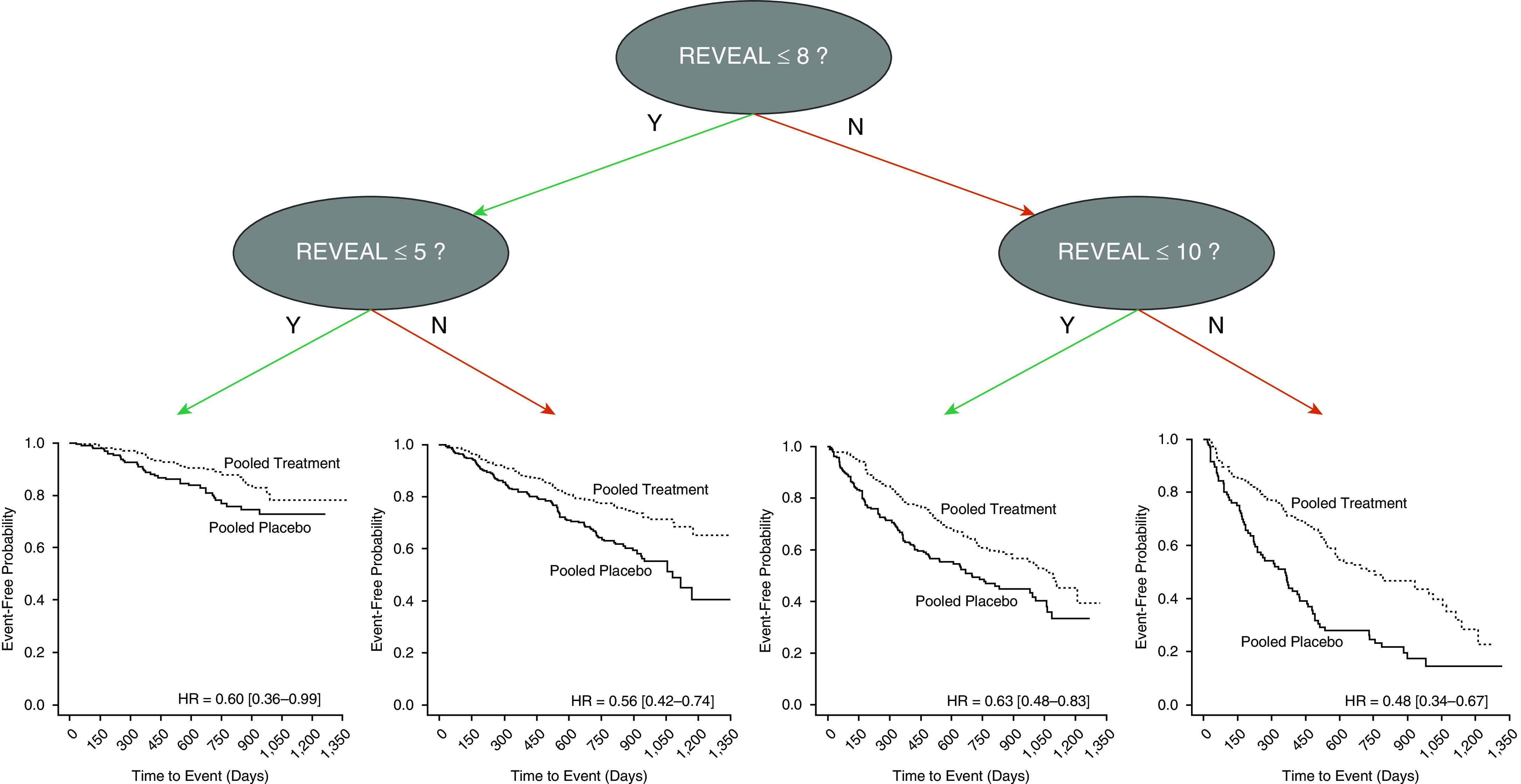
Survival tree analysis applied with REVEAL 2.0 risk stratification. Example of splitting conducted by survival tree is shown. Cut points to define risk groups were calculated by maximizing the statistical difference in time to clinical worsening between each risk group. Cut points were optimized on pooled placebo data before applying to treatment group. Once applied to the treatment group, Kaplan-Meier curves were generated, and hazard ratios (HRs) between pooled placebo and pooled treatment group were calculated. HRs and 95% confidence intervals are shown. REVEAL = Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management.
Impact on Sample Size and Treatment Time
Shown in Figure 3 are the results for sample size reduction (bar graph, y-axis, left) and average treatment time reduction (superimposed line graph, y-axis, right) for multiple enrichment strategies. REVEAL 2.0 performed best for both reducing the total number of patients needed for enrollment and the average treatment time for all enrichment methods in AMBITION and GRIPHON. However, the French score, on average, outperformed both COMPERA and REVEAL 2.0 for all enrichment methods in the SERAPHIN trial. This discrepancy is likely due to the use of a nonstandard assay for measuring NT-proBNP in the SERAPHIN trial, which would compromise the accuracy of both COMPERA and REVEAL 2.0 for risk stratification in this trial but not of the French score, as it does not use NT-proBNP to estimate risk.
Figure 3.
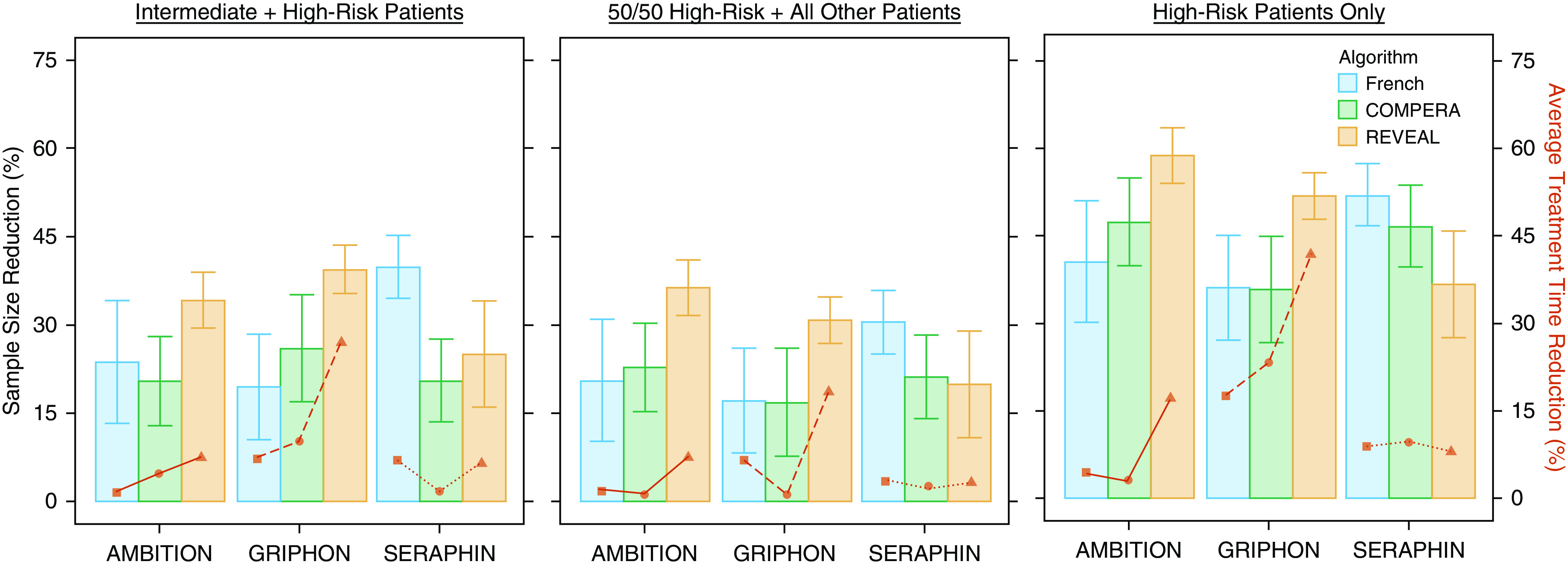
Estimated sample size and treatment time reduction. Sample size reduction (shown in bars, y-axis, left) is presented as percentage of the average sample size with the nonenriched population used for power analysis. Error bars represent 95% confidence intervals. Lines (y-axis, right) show estimated reduction in average treatment time as a percentage of the average treatment time with nonenriched population used for power analysis. AMBITION = A Study of First-Line Ambrisentan and Tadalafil Combination Therapy in Subjects with Pulmonary Arterial Hypertension; COMPERA = Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension; French = French score; GRIPHON = Selexipag in Pulmonary Arterial Hypertension; REVEAL = Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management; SERAPHIN = Study of Macitentan on Morbidity and Mortality in Patients with Symptomatic Pulmonary Arterial Hypertension.
Estimated Screen-to-Enroll Ratios
Table 5 provides the estimated screen-to-enroll ratios of each risk group per algorithm, as determined by the pooled dataset. REVEAL’s screen-to-enroll ratio was highest (i.e., worst screening efficiency) for all enrichment methods. The French score using an intermediate–high enrichment strategy could achieve a comparable screen-to-enroll ratio to those of the original trials.
Table 5.
Estimated Number Screened to Enroll 100 Patients Based on Risk Proportions in Pooled Dataset
| Enrichment Method | Risk Algorithm | Number Screened to Enroll 100 Patients |
|---|---|---|
| Intermediate and high risk | COMPERA | 170 |
| French | 127 | |
| REVEAL | 263 | |
| 50% high risk/50% all other | COMPERA | 214 |
| French | 259 | |
| REVEAL | 417 | |
| High risk only | COMPERA | 427 |
| French | 518 | |
| REVEAL | 833 | |
| None (average of original trials) | 124 | |
Definition of abbreviations: COMPERA = Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension; French = French score; REVEAL = Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management.
Impact on Cost Savings
Figure 4 shows the potential cost savings that GRIPHON may have benefited from if enrichment strategies had been used as well as the ratio of screening cost to research (treatment) costs per enrichment strategy. Although all enrichment strategies reduced cost on average, the high-other strategy provided minimal net savings. The intermediate–high strategy provided the greatest financial benefit under REVEAL 2.0 (by reducing the total trial cost by 40%), and the high-risk–only strategy reduced overall cost but substantially increased screening costs.
Figure 4.
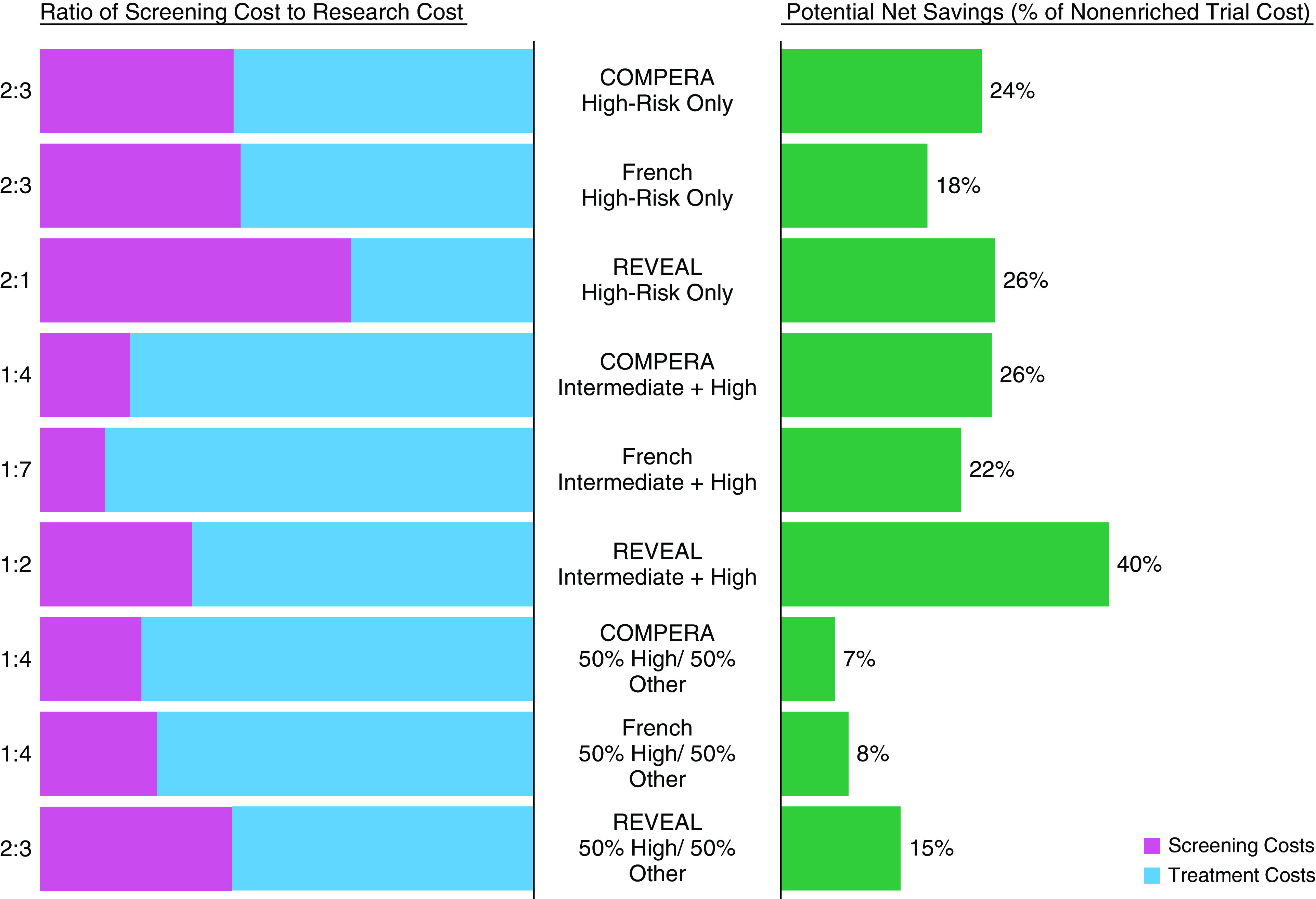
Cost analysis for applying enrichment to GRIPHON (Selexipag in Pulmonary Arterial Hypertension). Each enrichment strategy was applied to GRIPHON, and estimated ratios of total cost of screening versus total cost of conducting trial (i.e., research cost) as well as net savings as a percentage of the cost of a nonenriched trial were calculated. Cost of screening and cost of research were estimated per Table 3. COMPERA = Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension; French = French score; REVEAL = Registry to Evaluate Early and Long-Term Pulmonary Arterial Hypertension Disease Management.
Discussion
Clinical trial design in PAH has evolved into large, placebo-controlled studies focusing on a composite endpoint of clinical outcomes to determine therapeutic efficacy. However, such approaches are cumbersome and costly, and trial durations extend over many years in hopes of achieving the desired statistical power.
The FDA supports using clinical trial enrichment, advocating that the prospective use of patient characteristics to select a study population in which detection of a drug effect (benefit or lack thereof) is more likely than in a broad patient population (5). For any given desired power in an event-based study, the appropriate sample size depends on effect size and the event rate in the control group. Prognostic enrichment strategies are encouraged, not to affect the relative risk reduction but to increase the proportion of patients likely to experience a disease-related endpoint, allowing for a higher number of events in a shorter time period, hence reducing overall sample size requirements. In our analysis, all risk algorithms met the guidance criteria for prognostic enrichment; 1) they were shown to be prognostic of clinical worsening by ROC analysis, and 2) when applied using multiple enrichment strategies, they reduced the average estimated sample size compared with estimations made with the nonenriched population. Furthermore, all risk groups were determined to have the same proportional pooled treatment benefit, supporting the FDA’s statement that relative risk reduction is not affected with a prognostic enrichment strategy.
Currently, the FDA has no pharmacological concerns with bridging treatment efficacy established in a higher-risk PAH group to treat lower-risk patients with PAH. The current understanding of PAH disease state and pathophysiology points to maintenance of a treatment effect regardless of a patient’s individual risk of morbidity or mortality. However, as stated in the FDA guidance document for trial enrichment, there is no absolute guarantee that prognostic enrichment and predictive enrichment are mutually exclusive. It is possible that risk-based prognostic enrichment also accomplishes the goal of predictive enrichment by selecting for a patient population that experiences an effect that would not be present in an unselected population. Such a result was not supported by our analysis but cannot be ruled out for PAH drugs of differing mechanisms, as all investigated trials tested vasodilators.
An enriched patient cohort could experience a lack of treatment efficacy rather than experiencing a greater treatment response. The latter is possible specifically when a treatment cannot have a therapeutic effect quickly enough to slow the deterioration of a high-risk patient. Many cardiovascular drugs not expected to rapidly improve heart function still achieved approval by demonstrating efficacy in very ill patients with rapid deterioration and high mortality (5). Therefore, it is almost never clear when it is too late to clinically intervene, especially with the limited pilot data that precede a phase III trial, and prognostic trial enrichment should still be considered.
If there is a truly well-captured reason that the drug must be used as an early intervention, such a trial would still benefit from risk-stratified enrichment. A trial sponsor could also consider an adaptive clinical trial, for which the cut point for an appropriate risk group is determined through interim analyses rather than initially determined and static throughout the trial. The use of adaptive clinical trials for prognostic and/or predictive enrichment is well supported by the FDA, and several adaptive designs have been previously published (19). In fact, one of the greatest challenges in adaptive enrichment is finding the correct biomarker among some number of candidates that provides prognosis of the desired endpoint (20). Our analysis clearly demonstrates that risk stratification is prognostic for clinical worsening, avoiding the need for multiple hypothesis testing that “spends the alpha” used to control the family-wise error rate during interim analysis.
As shown in Figure 3, REVEAL 2.0 outperformed COMPERA and the French score in two of the three clinical trials for all enrichment methods but appeared less informative in SERAPHIN. One possible explanation for this variation is that REVEAL 2.0 and COMPERA were both optimized for a different NT-proBNP analytical assay from the one used in the SERAPHIN trial (21). Specifically, the SERAPHIN NT-proBNP measurements were determined from an enzyme immunoassay rather than a chemiluminescence immunoassay, meaning that its range and scale do not translate to the cut points used in COMPERA and REVEAL 2.0. This leads to all SERAPHIN patients assessed appearing to have a high-risk NT-proBNP level per these two algorithms. By comparison, the invasive French score does not consider NT-proBNP for its algorithm and therefore could not be skewed by this value, which may be why it performed better than both COMPERA and REVEAL 2.0 at reducing sample size in simulations for SERAPHIN. This demonstrates the importance of using clinical variables measured with the appropriate scales when applying risk stratification, especially for trial enrichment. Moreover, it emphasizes the need for standardization of choice of biomarker tested and its range of values in risk assessment tools.
This result also leads into an important discussion about ease of use and data collection for risk-driven screening at trial baseline. REVEAL 2.0 requires 13 clinical variables (with a minimum of seven), COMPERA requires five, and the French score requires four. Although the number of variables for each algorithm differs, the overall cost of each algorithm as a screening tool was similar because right heart catherization was the biggest contributing factor to cost. Further investigations are warranted to explore whether REVEAL Lite (22) or the French noninvasive score (6) can identify the intermediate–high and/or high-risk–only groups with considerably fewer variables and no hemodynamic variables.
Although the ideal enrichment strategy is a single, inexpensive biomarker, as shown in our additional analyses, hemodynamic variables, 6MWD, and NT-proBNP used in isolation were not as predictive as any of the three multivariable risk algorithms. Use of these singular biomarkers was unlikely to substantially reduce costs versus REVEAL 2.0 because confirmation of PAH diagnosis by right heart catheterization would still be required. Furthermore, although our results demonstrate that REVEAL 2.0 performed best in terms of cost savings and precise prediction, if all variables considered in our analysis cannot be measured, we recommend the use of a risk algorithm that best fits the available data to avoid inaccurate prognosis of screened patients.
The cost savings exercise conducted with the GRIPHON data showed that intermediate-high–risk enrollment as determined by a REVEAL score of 8 or more provides the greatest financial gain. This enrichment strategy had the triple benefit of 1) reducing total number of enrolled patients, 2) reducing average treatment time per enrolled patient, and 3) keeping screening costs modest. For our analysis, most of the cost per enrolled patient stemmed from distributing study drugs over a long period of time. Our estimation of other research costs was conservative, as we did not account, for example, for regularly scheduled lab tests, multiple right heart catheterizations, and the cost of personnel, all of which can vary considerably per trial and geographic location (16). For a comparison of scale, our economic analysis estimates an average cost of $17,000 (U.S. dollars) per enrolled patient for 26 weeks of treatment, whereas current estimations for costs per enrolled patient in international trials with a median trial time of 26 weeks were $31,802 per patient as a low estimate (23). This further illustrates the importance of keeping trial sizes small and treatment time short, even when requiring a more involved screening process.
PAH clinical trials tend to be international, multisite endeavors to reach necessary sample sizes. Economic research cited by the FDA established that international trials can benefit from lower costs of clinical procedures found abroad, although benefits in terms of the cost of study drugs is uncertain (24, 25). Our estimates of the studied drug (selexipag) are based on previously published literature specific to its 2017 market price submitted by the manufacturer in the United States and Canada. Specific selexipag cost data in countries where costs of clinical trials are far lower, such as China and Russia, could not be found. Although our estimates may not be entirely reflective of total clinical trial cost, especially for sites outside the United States, our analysis illustrates a proportional reduction in cost that would translate to international sites because of a reduction in treated patients and treatment time.
The rarity of algorithm-identified risk groups is a serious challenge for enrichment. Although a high risk–only strategy provides the biggest reduction in sample size, the rarity of these patients makes screening prohibitive not only financially but also in the sheer number of available patients to screen. We found that our screen-to-enrollment ratios were reflective of the real world, as they matched the availability of an enriched population in the REVEAL registry, although rounding of the COMPERA score affected the apparent availability of its high-risk population (6). These estimates are primarily meant to illustrate the relative difficulty of identifying a subpopulation within an already rare disease population. As stated above, PAH clinical trials typically require multiple sites across several countries to achieve adequate sample sizes, and this is expected to still be necessary with an enrichment strategy. At this time, the need for additional sites to increase screening numbers is not expected with an intermediate-high–risk strategy, but the possibility should be considered. We recommend the use of feasibility surveys that consider availability of intermediate- and high-risk patients before site recruitment to mitigate costs.
Our estimation of screen-to-enrollment ratios assumed no prescreening. Prescreening via electronic health records may allow a high-risk–only enrichment strategy to become more viable (23). The feasibility of a high-risk–only enrichment strategy will become increasingly important, as the sophistication and treatment costs of PAH clinical trials are increasing (4). Trials with particularly high costs per enrolled patient, such as those studying dual or triple combination therapies, will substantially benefit from a high-risk–only enrichment strategy, specifically using REVEAL 2.0, which provided the greatest sample size and treatment time reductions with appropriate inputs.
Finally, the overall availability of intermediate- and high-risk patients to be enrolled per year also contributes to overall trial length. Although PAH trials typically expect to enroll roughly 200–350 patients per year, intermediate- and high-risk patients only compose an estimated 35–50% of the total current registry population (8). Therefore, enrollment efforts are likely to slow and not be entirely offset by the reduction in the total number of patients needed for enrollment. However, an enriched study will still stand to benefit from more primary endpoint events occurring at a quicker pace, as shown in our analysis with reduced treatment time. Therefore, trials may still benefit from a reduction in needed patient treatment-years. Future studies will investigate how to identify an enrichment strategy more precisely based on clinical trial simulations that account for the availability of patients.
Our findings address the limitations of current PAH clinical trials by demonstrating the benefit of risk stratification of patients with validated scales in PAH at baseline for optimizing enrollment. These data demonstrate, for the first time, the efficacy of established PAH risk-prediction algorithms in selecting patients most likely to experience clinical events. When applied retrospectively to contemporary PAH clinical trials, our patient enrichment strategy reduced the enrolled/intent-to-treat population size required to detect a statistically significant treatment effect. Thus, results from this study establish that a priori risk stratification maximizes the likelihood of observing a statistically significant treatment effect with a smaller study population and should be considered in future study designs. Accordingly, the application of our approach to clinical study design is, in turn, expected to increase efficiency of successful PAH randomized clinical trials.
Limitations
This study had several limitations. Because of missing clinical variables at baseline, COMPERA and REVEAL 2.0 were not evaluated as originally intended. Furthermore, all risk algorithms were optimized for predicting 1-year mortality rather than clinical worsening.
COMPERA was applied without employing the rounding methods used with the algorithm applied to clinical risk stratification. Overall, not rounding has little effect on the overall functionality of the algorithm, as all inputs and calculations are the same. However, rounding could profoundly affect COMPERA’s prognostic performance, as ranking accuracy almost always suffers when scales become less precise.
Relative algorithm accuracy was determined on the basis of a pooled placebo group with differing definitions for clinical worsening. Given the nature of event adjudication, namely the lack of available source data surrounding worsening symptoms, it is infeasible to create a common clinical worsening endpoint to be used in all trials. This limitation motivates the understanding of how different risk algorithms perform for different definitions of clinical worsening given all the proper inputs for calculations, which were not available for this study.
Although sampling from a trial population with early withdrawal, power simulations did not force a specific attrition rate. Sample size reduction estimates were therefore controlled by comparing with a nonenriched population without attrition rather than the original trial’s study size. We expect the percentage change to be similar overall. Random dropout, drug intolerability, or lack of satisfactory clinical progress (but not an explicit adjudicated event) can all contribute to an underestimation of drug effect, which could substantially increase the required sample sizes. However, use of a high-risk enrichment strategy is less likely to be affected by attrition rates, as events occur earlier in the trial. It may, in fact, increase the likelihood of patients experiencing a clinically worsening endpoint before any other factor, leading to an early withdrawal.
In addition, by assuming that all patients with early withdrawal were event-free and thereby reducing the estimated event rate in each risk group, estimations of sample size reduction may be conservative. Our sensitivity analysis of the ROC curves demonstrates that this assumption produces a modest bias in the performance of each algorithm. However, underestimating sample size reduction is desirable for providing appropriate recommendations versus providing inflated estimations that later prove to provide no significant reduction in sample size.
In terms of bridging treatment efficacy, given the broad range of etiologies even for WHO group I PAH, it is important to balance a trial’s patient cohort appropriately so that it is representative of the intended drug indication. Our analysis demonstrated a treatment effect in all REVEAL risk populations even though etiology contributed to risk stratification, providing preliminary evidence that there are no concerns about bridging efficacy between risk groups of different etiological proportion. However, this concept warrants further investigation.
There are two major limitations to our economic analysis. First, increase in clinical trial costs because of the need for additional sites for screening was not considered. Next, all costs were based on estimates of clinical procedures and cost of the study drug (selexipag) in the United States. Although a proportional reduction in cost is still expected, it is speculative.
Lastly, the studied population may not be representative of the risk in the entire trial population. Missing clinical variables for baseline risk assessment are assumed to be missing at random, but there may be some underlying cause.
Conclusions
Use of risk-prediction algorithms as a prognostic enrichment tool must be validated in prospective clinical trials. This preliminary retrospective study demonstrates that such enrichment would reduce needed enrollment size and the duration of treatment and observation. This has many significant patient benefits, such as reducing the duration of treatment with placebo and improving time-to-market for potentially life-saving medications. Furthermore, the financial burden of future PAH clinical trials can be reduced by improving trial efficiency, allowing drug developers to reinvest savings into research and drug innovation.
Supplementary Material
Acknowledgments
Acknowledgment
The authors thank Dr. Mardi Gomberg-Maitland (Department of Cardiology, George Washington School of Medicine and Health Sciences, Washington, D.C.), Dr. Charles G. Elliot (Pulmonary Division, Department of Medicine, Intermountain Medical Center, Salt Lake City, Utah), and Dr. Bradley A. Maron (Cardiovascular Division, Department of Medicine, Brigham and Women’s Hospital, Boston, Massachusetts) for commenting on and editing of the manuscript and contributing important interpretation to the results.
Footnotes
Supported by the NIH NHLBI grant R01 HL134673, Pulmonary Hypertension Outcomes Risk Assessment, Oak Ridge Institute For Science and Education, and U.S. Department of Energy. This article reflects the views of the authors and should not be construed to represent U.S. Food and Drug Administration’s views or policies.
Author Contributions: C.E.G., M.K.K., N.L.S., and R.L.B. designed the study. J.V.S., C.E.G., N.L.S., and R.L.B. designed the statistical analyses, and J.V.S. executed statistical and data analyses. All authors interpreted the data. J.V.S., C.E.G., M.K.K., and R.L.B. drafted and revised the manuscript critically for important intellectual content. N.L.S. revised the manuscript critically for important intellectual content. All authors gave final approval of this version to be submitted.
This article has an online supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1164/rccm.202002-0357OC on September 16, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol. 2015;65:1976–1997. doi: 10.1016/j.jacc.2015.03.540. [DOI] [PubMed] [Google Scholar]
- 2.Kanwar MK, Thenappan T, Vachiéry JL. Update in treatment options in pulmonary hypertension. J Heart Lung Transplant. 2016;35:695–703. doi: 10.1016/j.healun.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 3.Ryan JJ, Rich JD, Maron BA. Building the case for novel clinical trials in pulmonary arterial hypertension. Circ Cardiovasc Qual Outcomes. 2015;8:114–123. doi: 10.1161/CIRCOUTCOMES.114.001319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sitbon O, Gomberg-Maitland M, Granton J, Lewis MI, Mathai SC, Rainisio M, et al. Clinical trial design and new therapies for pulmonary arterial hypertension. Eur Respir J. 2019;53:1801908. doi: 10.1183/13993003.01908-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.U.S. Food and Drug Administration Enrichment strategies for clinical trials to support approval of human drugs and biological products - guidance for industry Silver Spring, MD: U.S. Food and Drug Administration; 2019 [accessed 2019 Sep]. Available from: https://www.fda.gov/media/121320/download [Google Scholar]
- 6.Hoeper MM, Kramer T, Pan Z, Eichstaedt CA, Spiesshoefer J, Benjamin N, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J. 2017;50:1700740. doi: 10.1183/13993003.00740-2017. [DOI] [PubMed] [Google Scholar]
- 7.Boucly A, Weatherald J, Savale L, Jaïs X, Cottin V, Prevot G, et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J. 2017;50:1700889. doi: 10.1183/13993003.00889-2017. [DOI] [PubMed] [Google Scholar]
- 8.Benza RL, Gomberg-Maitland M, Elliott CG, Farber HW, Foreman AJ, Frost AE, et al. Predicting survival in patients with pulmonary arterial hypertension: the REVEAL risk score calculator 2.0 and comparison with ESC/ERS-based risk assessment strategies. Chest. 2019;156:323–337. doi: 10.1016/j.chest.2019.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, McLaughlin VV, et al. AMBITION Investigators. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373:834–844. doi: 10.1056/NEJMoa1413687. [DOI] [PubMed] [Google Scholar]
- 10.Sitbon O, Channick R, Chin KM, Frey A, Gaine S, Galiè N, et al. GRIPHON Investigators. Selexipag for the treatment of pulmonary arterial hypertension. N Engl J Med. 2015;373:2522–2533. doi: 10.1056/NEJMoa1503184. [DOI] [PubMed] [Google Scholar]
- 11.Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, Ghofrani HA, et al. SERAPHIN Investigators. Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med. 2013;369:809–818. doi: 10.1056/NEJMoa1213917. [DOI] [PubMed] [Google Scholar]
- 12.Levey AS, Stevens LA, Schmid CH, Zhang YL, Castro AF, III, Feldman HI, et al. CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150:604–612. doi: 10.7326/0003-4819-150-9-200905050-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Y, McArdle JJ. Rationale and applications of survival tree and survival ensemble methods. Psychometrika. 2015;80:811–833. doi: 10.1007/s11336-014-9413-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Therneau TAB, Ripley B.rpart: recursive partitioning and regression trees 2019 [accessed 2019 Sep]. Available from: https://cran.r-project.org/package=rpart
- 15.Freedman LS. Tables of the number of patients required in clinical trials using the logrank test. Stat Med. 1982;1:121–129. doi: 10.1002/sim.4780010204. [DOI] [PubMed] [Google Scholar]
- 16.Qiu W, Chavarro J, Lazarus R, Rosner B, Ma J.powerSurvEpi: power and sample size calculation for survival analysis of epidemiological studies 2018 [accessed 2019 Nov]. Available from: https://cran.r-project.org/package=powerSurvEpi
- 17.Boston Scientific Procedural payment guide: hospital inpatient and outpatient, ASC and physician reimbursement information Chicago, IL: American Medical Association2019 [accessed 2019 Nov]. Available from: https://www.bostonscientific.com/content/dam/bostonscientific/Reimbursement/RhythmManagement/assets/2019_Procedural_Payment_Guide.pdf [Google Scholar]
- 18.Ottawa, Canada: Canadian Agency for Drugs and Technologies in Health; 2017. Pharmacoeconomic review report: selexipeg (Uptravi) [PubMed] [Google Scholar]
- 19.Pallmann P, Bedding AW, Choodari-Oskooei B, Dimairo M, Flight L, Hampson LV, et al. Adaptive designs in clinical trials: why use them, and how to run and report them. BMC Med. 2018;16:29. doi: 10.1186/s12916-018-1017-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon N, Simon R. Adaptive enrichment designs for clinical trials. Biostatistics. 2013;14:613–625. doi: 10.1093/biostatistics/kxt010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mueller T, Gegenhuber A, Poelz W, Haltmayer M. Comparison of the Biomedica NT-proBNP enzyme immunoassay and the Roche NT-proBNP chemiluminescence immunoassay: implications for the prediction of symptomatic and asymptomatic structural heart disease. Clin Chem. 2003;49:976–979. doi: 10.1373/49.6.976. [DOI] [PubMed] [Google Scholar]
- 22.Benza RL, Kanwar M, Raina A, Lohmueller LC, Pasta DJ, La R, et al. Comparison of risk discrimination between the REVEAL 2.0 calculators, the French Pulmonary Registry algorithm and the Bologna method in patients with Pulmonary Arterial Hypertension (PAH) [abstract] Am J Respir Crit Care Med. 2020;201:A2512. [Google Scholar]
- 23.Moore TJ, Zhang H, Anderson G, Alexander GC. Estimated costs of pivotal trials for novel therapeutic agents approved by the US Food and drug administration, 2015-2016. JAMA Intern Med. 2018;178:1451–1457. doi: 10.1001/jamainternmed.2018.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang YT, Chen B, Bennett CL. Offshore pharmaceutical trials: evidence, economics, and ethics. Mayo Clin Proc Innov Qual Outcomes. 2018;2:226–228. doi: 10.1016/j.mayocpiqo.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sertkaya A, Wong HH, Jessup A, Beleche T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin Trials. 2016;13:117–126. doi: 10.1177/1740774515625964. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.