

Abstract
Stem rust is one of the most important diseases, threatening global wheat production. Identifying genomic regions associated with resistance to stem rust at the seedling stage may contribute wheat breeders to introduce durably resistant varieties. Genome‐wide association study (GWAS) approach was applied to detect stem rust (Sr) resistance genes/QTLs in a set of 282 Iranian bread wheat varieties and landraces. Germplasms evaluated for infection type and latent period in four races of Puccinia graminis f. sp. tritici (Pgt). A total of 3 QTLs for infection type (R2 values from 9.54% to 10.76%) and 4 QTLs for the latent period (R2 values from 8.97% to 12.24%) of studied Pgt races were identified in the original dataset. However, using the imputed SNPs dataset, the number of QTLs for infection type increased to 10 QTLs (R2 values from 8.12% to 11.19%), and for the latent period increased to 44 QTLs (R2 values from 9.47% to 26.70%). According to the results, the Iranian wheat germplasms are a valuable source of resistance to stem rust which can be used in wheat breeding programs. Furthermore, new information for the selection of resistant genotypes against the disease through improving marker‐assisted selection efficiency has been suggested.
Keywords: association mapping, Puccinia graminis f. sp. tritici, Sr genes, wheat
The results obtained from this study can be important to facilitate and accelerate breeding programs through marker‐assisted selection.

1. INTRODUCTION
Wheat (Triticum aestivum L.) is a major staple food crop in the world. It is predicted that the cereal yield should increase by 50% to meet the food demand of the human population by 2050 (Godfray et al., 2010). Wheat production and yield are constantly challenged by various biotic and abiotic stresses. Diseases and pests are among the most important biotic stresses resulting in approximately 20% losses of the global wheat yield annually (Oerke, 2006). Rusts are among the most damaging cereal diseases which have coexisted and evolved during the domestication of cereals (Schafer et al., 1984). Black stem rust of wheat caused by Puccinia graminis Pers. f. sp. tritici (Pgt) is considered a very serious threat in major wheat‐growing regions around the world. This disease, which rapidly develops under humid conditions and high temperatures (15–35ºC), is the most damaging rust among wheat rusts and the extent of damage may result in the large‐scale destruction across wheat fields (Roelfs et al., 1992; Singh et al., 2015).
Fungicide application and stem rust‐resistant wheat cultivars are two important strategies to combat Pgt. However, the fungicide application may not always be feasible due to the high cost, risk of fungicide resistance, and environmental contamination. Using resistant wheat cultivars is the most effective and economical way to control the disease. In this regard, having sufficient knowledge on population genetics of the disease causal agent and identifying effective resistance genes in the selected wheat genotypes are of great importance.
Plant disease resistance is generally divided into monogenic and polygenic categories based on the mode of inheritance in plants. In the monogenic type, a gene is responsible for the expression of resistance, and the type of resistance, dominance, and codominance, as well as discrete variation, can be obtained in differentiating generations and can be identified using the Mendelian laws and the monosomic technique of the genes. In the polygenic type, resistance is governed by several genes, so monosomic techniques and Mendelian laws cannot be used to identify resistance genes. Linkage mapping to detect quantitative traits loci (QTLs) is used as a methodology to understand the genetic control of polygenic traits. The major limitations of this methodology are a small number of crossovers, resulting in low genetic mapping resolution (10–20 cM) and the high cost of population replication to reach sufficient crossovers. Besides, the created populations are only useful for limited traits and studies (Gupta et al., 2005; Holland, 2007). An alternative methodology with the advantage of using natural populations is association mapping.
Association mapping seeks to identify continuous markers linked to one or more quantitative traits. It is based on the assumption that the observed phenotypic variation is related to genetic variation. In this method, a large and diverse set of individuals/lines drawn from a population is randomly collected and mapping is done based on linkage disequilibrium. Association mapping has been used for both whole‐genome scanning and candidate gene analysis (Kraakman et al., 2004; Pasam et al., 2012; Rafalski & Morgante, 2004; Rostoks et al., 2006; Thornsberry et al., 2001). Association mapping is much more accurate than linkage mapping due to its many recombinations (Moose & Mumm, 2008). It is also more compatible with genetically diverse germplasm and allows the mapping of several traits simultaneously. Therefore, there is no need to create two‐parental populations for each trait, which in turn incurs additional costs for genotypic and phenotypic evaluation.
The objectives of this study were to carry out a genome‐wide search in Iranian wheat genotypes for resistance loci to Pgt races at the seedling stage, and the identification of genomic regions suitable for marker‐assisted selection and further genetic dissection.
2. MATERIALS AND METHODS
2.1. Plant materials
A set of 282 Iranian bread wheat genotypes, including 193 landraces collected between 1931 and 1968 and 89 commercial varieties released between 1942 and 2014 were selected to form a stem rust AM panel. Landraces were obtained from the Gene Bank of the University of Tehran and commercial varieties from Seed and Plant Improvement Institute (SPII), Karaj, Alborz, Iran, and Dryland Agricultural Research Institute (DARI), Maragheh, East Azarbaijan, Iran. A detailed description of the AM panel is attached as the additional file 1, Table S1, and S2.
2.2. Phenotypic evaluation
Phenotypic evaluation of AM panel against Pgt was carried out at the Cereal Research Department, Seed and Plant Improvement Institute (SPII), Karaj, Alborz, Iran, in 2019. The AM panel was evaluated under greenhouse conditions using a randomized complete block design with two replications for each of the four Pgt races. Seeds of each genotype were planted in pots with 10 cm height and diameter containing 1:1:2 ratio of common soil, peat moss, and leaf mold. After 8–10 days, when the first leaf of the seedlings well developed (12 stage of phenological development based on the Zadoks et al. (1974) scale), the spores of four stem rust races were collected from different regions of Iran (Table 1) were inoculated separately. The inoculated plants were kept in a dark chamber for 24 hr at 18 ± 2°C and near saturation moisture and then transferred to a 22 ± 2°C greenhouse with a 16‐hr photoperiod. Inoculated plants were examined from the second day to record the latent period (LP). Fourteen days after inoculation, seedling infection type (IT) was recorded using the 0 to 4 scale introduced by Stakman et al. (1962) and modified by McIntosh et al. (1995) where ITs of 0, 1, and 2 were considered as low ITs, and ITs of 3, and 4 were considered as high ITs.
TABLE 1.
Detailed description of the Pgt races used to evaluate the wheat genotypes
| Isolate | Location | Race | Stem rust resistance (Sr) genes | |
|---|---|---|---|---|
| Ineffective | Effective | |||
| 94–8 | Boroujerd, Lorestan, Iran | TTTTF | Sr5, Sr6, Sr7a, Sr7b, Sr8a, Sr8b, Sr9a, Sr9b, Sr9d, Sr9e, Sr9g, Sr10, Sr11, Sr12, Sr13, Sr14, Sr15, Sr16, Sr17, Sr18, Sr19, Sr20, Sr21, Sr22, Sr23, Sr25, Sr28, Sr29, Sr30, Sr34, Sr35, Sr36, Sr37, Sr38, Sr40, SrTmp, and SrMcN | Sr24, Sr26, Sr27, Sr31, Sr33, and Sr39 |
| 94–15 | Kelardasht, Mazandaran, Iran | PTRTF | Sr5, Sr6, Sr7b, Sr8a, Sr8b, Sr9a, Sr9b, Sr9d, Sr9e, Sr9g, Sr10, Sr11, Sr12, Sr13, Sr14, Sr15, Sr16, Sr17, Sr18, Sr19, Sr20, Sr25, Sr27, Sr28, Sr29, Sr34, Sr35, Sr36, Sr37, Sr38, Sr39, SrTmp, and SrMcN | Sr7a, Sr21, Sr22, Sr23, Sr24, Sr26, Sr30, Sr31, Sr32, Sr33, and Sr40 |
| 95–2 | Shavour, Khouzestan, Iran | TTKTK | Sr5, Sr6, Sr7a, Sr7b, Sr8a, Sr8b, Sr9a, Sr9b, Sr9d, Sr9e, Sr9g, Sr10, Sr11, Sr12, Sr14, Sr15, Sr16, Sr17, Sr18, Sr19, Sr20, Sr21, Sr23, Sr28, Sr29, Sr30, Sr31, Sr33, Sr34, Sr37, Sr38, SrTmp, and SrMcN | Sr13, Sr22, Sr24, Sr25, Sr26, Sr27, Sr32, Sr35, Sr36, Sr39, and Sr40 |
| 95–31 | Kelardasht, Mazandaran, Iran | TKTTF | Sr5, Sr6, Sr7a, Sr7b, Sr8a, Sr8b, Sr9a, Sr9b, Sr9d, Sr9e, Sr9g, Sr10, Sr11, Sr12, Sr14, Sr15, Sr16, Sr17, Sr18, Sr19, Sr20, Sr21, Sr23, Sr28, Sr29, Sr30, Sr34, Sr35, Sr36, Sr37, Sr38, SrTmp, and SrMcN | Sr22, Sr24, Sr25, Sr26, Sr27, Sr31, Sr32, Sr33, Sr39, and Sr40 |
2.3. Phenotypic data analysis
Before performing the analysis of variance, the hypotheses of variance analysis were examined and the results confirmed the hypotheses. To calculate genetic parameters, analysis of variance was performed for the IT and LP of the studied Pgt races. Genetic, environmental, and phenotypic variances were calculated using the Comstock & Robinson (1952) method, using Equations 1, 2, and 3, respectively:
| (1) |
| (2) |
| (3) |
In these equations, MSg is genotype mean square, MSe is mean square error and r is the number of experimental replications. Heritability was estimated by the Falconer (1989) method through the 4 equation:
| (4) |
where is the genetic variance and is the phenotypic variance obtained from the variance analysis table according to Comstock & Robinson (1952) method. To estimate the coefficient of genetic and phenotypic variation, Singh and Chaudhary (1985) method was used based on 5 and 6 equations:
| (5) |
| (6) |
where is the average of trait. Genetic efficiency was also calculated according to the Allard (1960) and Singh and Chaudhary (1985) methods using the (7) equation:
| (7) |
where k is 10% of the selection pressure (1.75), is the phenotypic standard deviation and h 2 is the heritability.
A combined analysis of variance was performed using SAS software 9.3, and genetic parameters were calculated using Excel. The race grouping was performed based on Ward's method using SPSS software.
2.4. Genotyping by sequencing and imputation method
The development and sequencing of a GBS library for the Iranian wheat have previously been described by Alipour et al. (2017). The genomic library was constructed according to the method of Poland et al. (2012). For this purpose, first, the extracted DNA was normalized and then enzymatic digestion was performed with two enzymes PstI and MspI. Then, barcode adapters were attached to each sample, so that each sample has a different barcode. To remove extras except for barcoded genomic DNA, purification was performed by QIAquick (Qiagen) PCR purification kits. Finally, the amplified fragments were selected on an E‐gel system with a size between 300–250 bp and sent for sequencing by the Ion Proton System. Sequencing data were treated for 64 bp, and the same reads were grouped into tags. Identical sequence tags were aligned to identify the SNPs within the tags and the SNPs were summoned using the UNEAK (Universal Network Enabled Analysis Kit) GBS pipeline (Lu et al., 2013) as part of the TASSEL 4.0 bioinformatics package (Bradbury et al., 2007). SNPs with heterozygosity > 10%, minor allele frequency (MAF) <5%, and missing data > 20% were eliminated to reduce false‐positive error results. After specifying the haplotype phase for all individuals, the data were subjected to imputation using BEAGLE v3.3.2 (Browning & Browning, 2009) based on available allele frequencies obtained. During imputation, four different reference genomes were assessed, among which the W7984 reference genome was shown to have the greatest imputation accuracy (Alipour et al., 2019). The different chromosomes LD decay was obtained using the ggplot2 package in RStudio (Team, 2015) based on LOESS regression.
2.5. Population structure and kinship matrix
The population structure of the AM panel was investigated using 8,959 SNP markers for the original dataset, and 43,921 SNP markers for the imputed dataset, which were distributed across the 21 wheat chromosomes, using the software STRUCTURE v2.3.3 (Pritchard et al., 2000). The optimal number of K was determined by an admixture model and with a burn‐in and simulation phase consisting of 10,000 steps for values of K = 1 to 10. The ΔK statistic was used to determine the most desirable subpopulation number, and its plot was plotted for consecutive K values. The observed and expected values allele frequencies were used to estimate the linkage disequilibrium between markers in TASSEL v.5 (Bradbury et al., 2007). Then, population structure matrix Q (n × p), where n is the number of assayed genotypes and p is the number of defined subpopulations, was used in association studies. Cluster analysis and principal component analysis were also performed to determine the genetic relationships between the AM panel.
2.6. Genome‐wide association study
A genome‐wide association study for loci governing Pgt resistance was conducted using phenotypic data converted to a linear scale. To use the modified Stakman et al. (1962) ITs in the genome‐wide association studies (GWAS), the 0 to 4 scale was converted to a 1 to 13 linear. Both the general linear model (GLM) and mixed linear model (MLM) were used to examine the accurate association between marker and trait in TASSEL v.5 (Bradbury et al., 2007). The GAPIT package (Lipka et al., 2012) was also used to perform association mapping in both GLM and MLM methods in RStudio (Team, 2015). The results of TASSEL and GAPIT were investigated using a t test. According to the results, it was found that the general linear model derived from TASSEL provides more accurate information about the marker–trait association.
2.7. Gene annotation
Sequences surrounding of significant SNP markers were obtained from the wheat 90 K SNP database (Wang et al., 2014) and used for assessing gene annotation using Gramene (http://www.gramene.org/) by aligning them to the IWGSC RefSeq v1.0 annotation (https://wheat‐urgi.versailles. inra.fr/Seq‐Repository/Annotations). The function of putative genes was explored by investigating the pathways in which the encoded enzymes were involved in. After aligning SNPs sequences to the reference genome, overlapping genes with the highest identity percentage and blast score were selected for further processing. The ontology of each adjacent genes with Triticum aestivum, including molecular function and biological process, and also orthologous genes in related species were extracted from the ensemble‐gramene database (http://ensembl.gramene.org).
3. RESULTS
3.1. Phenotypic data analysis
The result of the combined analysis of variance for IT and the LP of wheat genotypes to four Pgt races is presented in Table 2. According to the results, the effects of race, genotype, and genotype × race interaction were highly significant for all race's IT and LP.
TABLE 2.
Combined analysis of variance for infection types and the latent period of wheat genotypes to four Pgt races
| Source of variation | df | Mean squares | Explained variance (%) | ||
|---|---|---|---|---|---|
| Infection type | Latent period | Infection type | Latent period | ||
| Race | 3 | 145.69** | 169.59** | 3.97 | 8.80 |
| Block (race) | 4 | 18.78 | 3.67 | 0.68 | 0.25 |
| Genotype | 281 | 21.20** | 8.93** | 54.12 | 43.39 |
| Genotype × race interaction | 843 | 4.74** | 2.53** | 36.33 | 36.91 |
| Error | 1,124 | 0.48 | 0.55 | 4.90 | 10.69 |
| Coefficient of variation (%) | 6.10 | 9.58 | |||
Significant at the 1% probability level.
Genetic parameters including variance components, genetic coefficients, and phenotypic variations, heritability, and genetic efficiency for IT and LP of wheat genotypes against four Pgt races were calculated (Table 3). The results showed high genetic and phenotypic variances for IT and LP of wheat genotypes to all races except TKTTF IT and LP. The highest genetic and phenotypic coefficients of variation were obtained for TTKTK IT and LP; the lowest values of these coefficients were detected for TKTTF IT and LP. The heritability and genetic efficiency of the ITs varied from 84.02% to 97.80% and 25.33 to 38.04%, respectively, and for the LPs ranged from 67.68% to 95.34% and 24.85 to 33.55%, respectively.
TABLE 3.
Amounts of genetic parameters for infection types and latent period collected from the response of wheat genotypes to four Pgt races
| Genetic parameters | TTTTF | PTRTF | TTKTK | TKTTF | ||||
|---|---|---|---|---|---|---|---|---|
| IT | LP | IT | LP | IT | LP | IT | LP | |
| Genetic variance | 4.131 | 2.050 | 4.872 | 2.050 | 4.862 | 2.870 | 3.380 | 1.525 |
| Genetic coefficient of variation (%) | 17.33 | 19.087 | 19.47 | 19.632 | 20.66 | 19.826 | 15.66 | 16.034 |
| Phenotypic variance | 4.375 | 2.250 | 4.981 | 2.150 | 5.786 | 4.240 | 3.958 | 1.945 |
| Phenotypic coefficient of variation (%) | 17.84 | 19.987 | 19.68 | 20.105 | 22.54 | 24.097 | 16.95 | 18.107 |
| Environmental variance | 0.244 | 0.200 | 0.109 | 0.100 | 0.924 | 1.370 | 0.578 | 0.420 |
| Heritability (%) | 94.43 | 91.11 | 97.80 | 95.34 | 84.02 | 67.68 | 85.40 | 78.40 |
| Genetic efficiency (%) | 29.48 | 31.87 | 33.69 | 33.55 | 33.14 | 28.54 | 25.33 | 24.85 |
IT: Infection type, LP: Latent period.
The frequency of genotypes was calculated based on the 0 to 4 IT scales (McIntosh et al., 1995; Stakman et al., 1962) for each race, and the results were presented in Table 4. Genotypes with seedling response between 0 and 2 (0–0;‐ ;‐ ;1– 1– 1+‐ 2‐‐2C‐ 2 and 2+) as resistance reaction and genotypes with IT between 3 and 4 (3– 3+ and 4) were considered as susceptible reaction (Letta et al., 2014). The results showed that in TTTTF, 16 varieties (17.98%) and 33 landraces (17.10%), in PTRTF, 18 varieties (20.22%) and 40 landraces (20.73%), in TTKTK, 34 varieties (38.20%) and 64 landraces (33.16%), and in TKTTF, 16 varieties (17.98%) and 35 landraces (18.13%) had resistance reaction.
TABLE 4.
Classification of wheat genotypes based on the reaction to four Pgt races
| Infection type | Cultivar/ landrace | TTTTF | PTRTF | TTKTK | TKTTF | ||||
|---|---|---|---|---|---|---|---|---|---|
| Number | % | Number | % | Number | % | Number | % | ||
| Resistance reactions | |||||||||
| 0, 0; and ; | Cultivar | – | – | ‐ | ‐ | 6 | 6.74 | 1 | 1.12 |
| landrace | 2 | 1.04 | 3 | 1.55 | 5 | 2.59 | 1 | 0.52 | |
| ;1, 1 and 1+ | Cultivar | 4 | 4.49 | 5 | 6.74 | 11 | 12.36 | 6 | 6.74 |
| landrace | 12 | 6.22 | 13 | 6.73 | 17 | 8.81 | 10 | 5.18 | |
| 2‐, 2C, 2 and 2+ | Cultivar | 12 | 13.48 | 12 | 13.48 | 17 | 19.10 | 9 | 10.11 |
| landrace | 19 | 9.84 | 25 | 12.95 | 41 | 21.24 | 24 | 12.44 | |
| Total | Cultivar | 16 | 17.98 | 18 | 20.22 | 34 | 38.20 | 16 | 17.98 |
| landrace | 33 | 17.10 | 40 | 20.73 | 64 | 33.16 | 35 | 18.13 | |
| Susceptible reactions | |||||||||
| 3 and 3+ | Cultivar | 48 | 53.93 | 48 | 53.93 | 46 | 51.68 | 30 | 33.71 |
| landrace | 63 | 32.64 | 82 | 42.49 | 106 | 54.92 | 58 | 30.05 | |
| 4 | Cultivar | 25 | 28.09 | 23 | 25.84 | 9 | 10.11 | 43 | 48.41 |
| landrace | 97 | 50.26 | 71 | 36.79 | 23 | 11.92 | 100 | 51.81 | |
| Total | Cultivar | 73 | 82.02 | 71 | 79.77 | 55 | 61.80 | 73 | 82.02 |
| landrace | 160 | 82.90 | 153 | 79.27 | 129 | 66.84 | 158 | 81.87 | |
The pattern of diversity among the four Pgt races based on Ward's method was shown in Figure 1. The dendrogram obtained from the analysis classified the races into three well‐distinct groups. The TTTTF and TKTTF clustered together while PTRTF and TTKTK showed independent virulence patterns.
FIGURE 1.
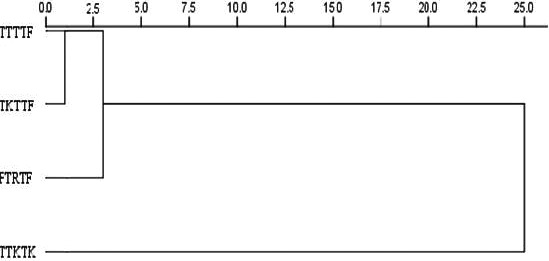
Dendrogram of cluster analysis of four Pgt races based on infection types and latent periods of 297 wheat genotypes using the Ward's method
3.2. Population structure and kinship analysis
To prevent false‐positive results, the AM panel was studied in terms of structure and kinship, and K and ∆K were extracted and their 2D graph was drawn and shown in Figure 2. The graph of ∆K at K = 3 represents the highest value in perfectly specified fracture, dividing the present population into three subpopulations. The allocation of each individual to either group was carried out based on the 70% membership threshold. Details of the subpopulation structure for each of the wheat genotypes are shown in Figure 3. Out of a total of 282 genotypes, 51 genotypes (18.09%) were in the first subpopulation, 103 genotypes (36.52%) were in the second subpopulation and 61 genotypes (21.63%) were in the third subpopulation. The first subpopulation contained 49 landraces and two varieties, the second subpopulation contained 97 landraces and six varieties and the third subpopulation contained 59 varieties and two landraces. When used the imputed SNPs, the studied germplasm has also divided into three subpopulations such as the original dataset that the first subpopulation consisted of 51 genotypes containing 49 landraces and two varieties, the second subpopulation contained 103 genotypes consisting of 97 landraces and six varieties and the third subpopulation contained 61 genotypes consisting of 59 varieties and two landraces.
FIGURE 2.
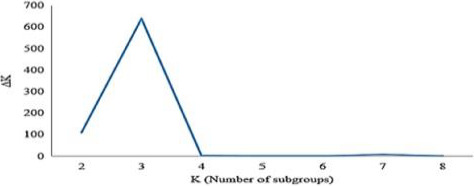
Determination of subpopulation number in wheat genotypes based on ΔK values
FIGURE 3.
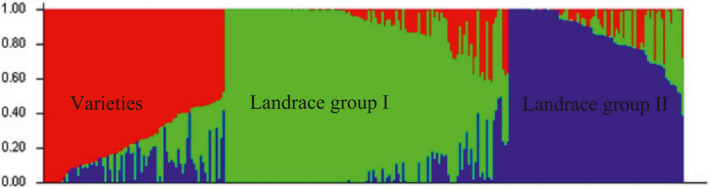
A structure plot of the 282 wheat genotypes and landraces determined by K = 3
The principal component analysis was performed on the matrix derived from both original and imputed SNPs to further evaluation of population structure and investigation of genetic relationships among wheat genotypes. According to genotypic data, the 2D plot of the original (Figure 4a) and imputed SNPs (Figure 4b) showed almost distinct grouping into three groups. Genotyping variance was explained by the first two main principal components that were 12.70% and 5.95%, respectively, for the original SNPs, and 17.20% and 6.10%, for the imputed SNPs.
FIGURE 4.
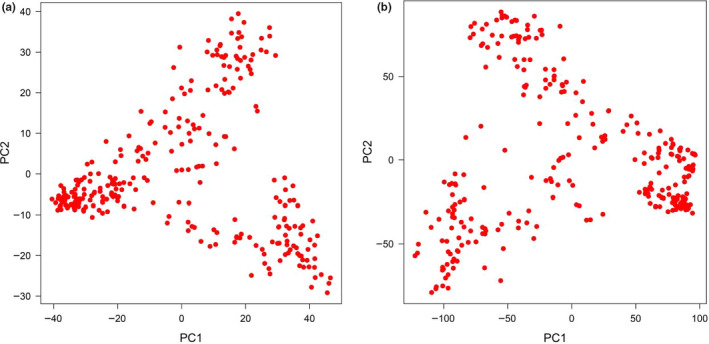
Principal component analysis of the wheat genotypes using original (A) and imputed SNPs (B)
Group I contains 129 genotypes with 113 landraces and 16 varieties; Group II contains 82 genotypes with 77 landraces and five varieties, and Group III contains 68 varieties and three landraces (Figure 5a). Genotypes also clustered into three main groups when used the imputed SNPs, where Group I contains 111 genotypes with 105 landraces and six varieties; Group II contains 99 genotypes with 85 landraces and 14 varieties; and Group III contains 72 genotypes with 68 varieties and three landraces (Figure 5b). According to the original SNP dataset, 21 varieties appear to be admixed with the two landrace groups, while for the imputed SNP dataset, only 20 such admixed varieties were identified. The admixed varieties originated from Iranian landraces and varieties.
FIGURE 5.
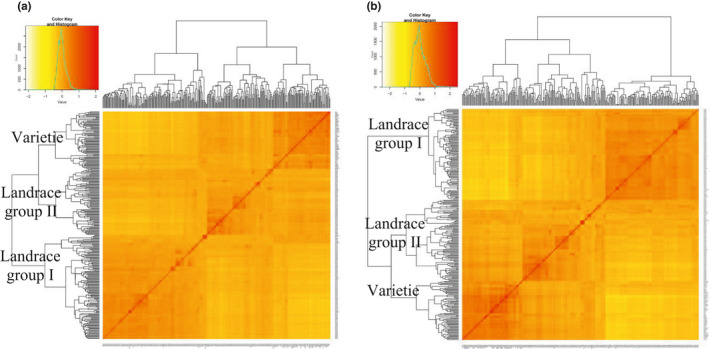
Cluster analysis using Kinship matrix of original (A) and imputed dataset (B) for the wheat genotypes
3.3. Linkage disequilibrium analysis
Linkage disequilibrium (LD) was examined for wheat varieties and landraces by 404,825 and 40,824 pairwise SNPs, respectively, in the original dataset, and by 2,152,925 SNPs for both wheat varieties and landraces in the imputed dataset. The SNPs were distributed across 21 chromosomes. The range of LD was estimated with a square of allele frequencies (r2) between the pairwise markers. LD analysis using the original dataset showed that the average genetic distance for the wheat varieties and landraces was 221.7440 and 221.7461 cM, respectively, and the mean of squared allele‐frequency correlations for them was 0.130 and 0.096, respectively (Table 5). Using an imputed dataset, the average genetic distance was equal for both wheat varieties and landraces (52.82 cM), but their mean of squared allele‐frequency correlation was 0.1924 and 0.1623, respectively (Table 6). The markers were not evenly distributed over different genomes. In the original dataset, the number of markers located on A and B genomes in varieties was equal to the number of markers located on A and B genomes in landraces, but in the D genome, landraces had one less marker than varieties. In the imputed dataset, there was no difference between the number of markers placed on each of the three genomes (A, B, and D) in the varieties with those in the landraces. The B genome had the highest number of markers, whereas the D genome showed the lowest number of markers in the original and imputed dataset. The proportion of each A, B, and D genomes from total pairwise original SNP markers in varieties and landraces were estimated at almost 37, 46, and 17%, respectively, and in the imputed SNP markers approximately 36, 50, and 13%, respectively. Out of 404,825 pairwise SNP markers, 86,415 pairwise markers (21.35%) in varieties and out of 404,824 pairwise SNP markers, 107,976 pairwise SNP markers (26.67%) in landraces showed significant linkage at 1% probability level in the original dataset. According to the imputed dataset, out of 2,152,925 pairwise SNP markers in varieties, 842,785 pairwise SNP markers (39.15%) and out of 2,152,925 pairwise SNP markers in landraces, and 968,270 pairwise SNP markers (44.98%) had significant linkage at 1% probability level.
TABLE 5.
A summary of observed linkage disequilibrium among pairwise markers and the number of significant pairwise markers per chromosome and genome using the original dataset
| Chromosome | Cultivars | Landraces | ||||||
|---|---|---|---|---|---|---|---|---|
| NSP | Distance (cM) | R2 | SSP | NSP | Distance (cM) | R2 | SSP | |
| 1A | 20,325 | 6.5208 | 0.1095 | 4,019 | 20,325 | 6.5208 | 0.0731 | 4,762 |
| 1B | 26,450 | 4.5786 | 0.1373 | 5,867 | 26,450 | 4.5786 | 0.0955 | 8,673 |
| 1D | 13,350 | 11.9226 | 0.1803 | 3,453 | 13,350 | 11.9226 | 0.1183 | 4,137 |
| 2A | 23,950 | 4.8296 | 0.1315 | 5,427 | 23,950 | 4.8296 | 0.1276 | 8,259 |
| 2B | 33,350 | 4.2026 | 0.1288 | 8,539 | 33,350 | 4.2026 | 0.0966 | 9,575 |
| 2D | 17,050 | 6.5433 | 0.2647 | 4,553 | 17,050 | 6.5433 | 0.1999 | 5,078 |
| 3A | 21,500 | 9.5170 | 0.0970 | 3,613 | 21,500 | 9.5170 | 0.0706 | 4,114 |
| 3B | 33,050 | 4.5077 | 0.1281 | 7,928 | 33,050 | 4.5077 | 0.0932 | 9,166 |
| 3D | 7,550 | 22.2324 | 0.0933 | 701 | 7,550 | 22.2324 | 0.0927 | 1547 |
| 4A | 17,450 | 8.0520 | 0.1312 | 3,752 | 17,450 | 8.0520 | 0.1106 | 4,245 |
| 4B | 10,600 | 9.9421 | 0.0995 | 1847 | 10,600 | 9.9421 | 0.0527 | 1,386 |
| 4D | 3,700 | 25.9186 | 0.1359 | 588 | 3,700 | 25.9186 | 0.1116 | 1,010 |
| 5A | 18,250 | 8.0378 | 0.1085 | 3,289 | 18,250 | 8.0378 | 0.0816 | 4,711 |
| 5B | 28,850 | 6.5358 | 0.1314 | 7,721 | 28,850 | 6.5358 | 0.0726 | 6,691 |
| 5D | 8,350 | 27.4992 | 0.0936 | 805 | 8,350 | 27.4992 | 0.0696 | 1,394 |
| 6A | 17,850 | 7.3937 | 0.1057 | 3,279 | 17,850 | 7.3937 | 0.1210 | 6,715 |
| 6B | 27,850 | 4.3847 | 0.1409 | 6,565 | 27,850 | 4.3847 | 0.0753 | 6,593 |
| 6D | 9,250 | 18.1532 | 0.1055 | 1,397 | 9,250 | 18.1532 | 0.0802 | 2059 |
| 7A | 28,550 | 6.1499 | 0.1520 | 5,580 | 28,550 | 6.1499 | 0.1123 | 8,528 |
| 7B | 26,800 | 4.7642 | 0.1118 | 5,429 | 26,800 | 4.7642 | 0.0837 | 6,846 |
| 7D | 10,750 | 20.0585 | 0.1618 | 2063 | 10,749 | 20.0606 | 0.0932 | 2,487 |
| A genome | 147,875 | 50.5007 | 0.1194 | 28,959 | 147,875 | 50.5007 | 0.0995 | 41,334 |
| B genome | 186,950 | 38.9156 | 0.1254 | 43,896 | 186,950 | 38.9156 | 0.0814 | 48,930 |
| D genome | 70,000 | 132.3277 | 0.1479 | 13,560 | 69,999 | 132.3298 | 0.1094 | 17,712 |
| Total | 404,825 | 221.7440 | 0.1309 | 86,415 | 404,824 | 221.7461 | 0.0968 | 107,976 |
NSP: Number of SNP pairwise, SSP: Significant pairwise SNP (p < .01).
TABLE 6.
A summary of observed linkage disequilibrium among pairwise markers and the number of significant pairwise markers per chromosome and genome using the imputed dataset
| Chromosome | Cultivars | Landraces | ||||||
|---|---|---|---|---|---|---|---|---|
| NSP | Distance (cM) | R2 | SSP | NSP | Distance (cM) | R2 | SSP | |
| 1A | 110,675 | 1.3446 | 0.1392 | 33,200 | 110,675 | 1.3446 | 0.1079 | 41,497 |
| 1B | 149,000 | 0.9442 | 0.1923 | 59,280 | 149,000 | 0.9442 | 0.1497 | 74,142 |
| 1D | 47,950 | 3.5143 | 0.3028 | 20,528 | 47,950 | 3.5143 | 0.2257 | 24,767 |
| 2A | 136,250 | 0.8628 | 0.2672 | 64,307 | 136,250 | 0.8628 | 0.2766 | 76,493 |
| 2B | 185,800 | 0.7705 | 0.1841 | 77,464 | 185,800 | 0.7705 | 0.1554 | 86,029 |
| 2D | 67,250 | 1.6415 | 0.2201 | 21,080 | 67,250 | 1.6415 | 0.1777 | 26,828 |
| 3A | 95,500 | 2.3004 | 0.1487 | 32,745 | 95,500 | 2.3004 | 0.1212 | 33,346 |
| 3B | 199,900 | 0.7787 | 0.2307 | 92,545 | 199,900 | 0.7787 | 0.2029 | 103,406 |
| 3D | 35,850 | 4.6331 | 0.1521 | 7,560 | 35,850 | 4.6331 | 0.1547 | 12,763 |
| 4A | 129,850 | 1.3855 | 0.3447 | 66,704 | 129,850 | 1.3855 | 0.3293 | 69,407 |
| 4B | 59,800 | 2.2142 | 0.1378 | 18,268 | 59,800 | 2.2142 | 0.0968 | 16,220 |
| 4D | 13,200 | 9.3335 | 0.1573 | 2,876 | 13,200 | 9.3335 | 0.1239 | 4,212 |
| 5A | 71,350 | 2.0200 | 0.1620 | 24,756 | 71,350 | 2.0200 | 0.1344 | 28,504 |
| 5B | 151,500 | 1.3002 | 0.1888 | 66,026 | 151,500 | 1.3002 | 0.1377 | 68,944 |
| 5D | 31,450 | 6.9574 | 0.1470 | 7,085 | 31,450 | 6.9574 | 0.1286 | 9,923 |
| 6A | 98,550 | 1.3027 | 0.1721 | 37,415 | 98,550 | 1.3027 | 0.1688 | 48,695 |
| 6B | 188,750 | 0.6656 | 0.1834 | 75,156 | 188,750 | 0.6656 | 0.1275 | 82,520 |
| 6D | 37,800 | 4.1908 | 0.1357 | 9,535 | 37,800 | 4.1908 | 0.1358 | 14,802 |
| 7A | 147,900 | 1.1793 | 0.2230 | 58,549 | 147,900 | 1.1793 | 0.1951 | 71,306 |
| 7B | 148,700 | 0.9965 | 0.1464 | 53,428 | 148,700 | 0.9965 | 0.1154 | 57,384 |
| 7D | 45,900 | 4.4855 | 0.2044 | 14,278 | 45,900 | 4.4855 | 0.1436 | 17,082 |
| A genome | 790,075 | 10.39523 | 0.2081 | 317,676 | 790,075 | 10.39523 | 0.1905 | 369,248 |
| B genome | 1,083,450 | 7.669932 | 0.1805 | 442,167 | 1,083,450 | 7.669932 | 0.1408 | 488,645 |
| D genome | 279,400 | 34.75617 | 0.1885 | 82,942 | 279,400 | 34.75617 | 0.1557 | 110,377 |
| Total | 2,152,925 | 52.82133 | 0.1924 | 842,785 | 2,152,925 | 52.82133 | 0.1623 | 968,270 |
NSP: Number of Pairwise SNP, SSP: Significant Pairwise SNP (p < .01).
3.4. Marker–trait association
Table 7 summarized the results of the marker–trait association for the two traits of IT and the LP for each studied Pgt race. Totally, 69 and 62 marker–trait associations were identified for IT and the LP of the used races, respectively, in the original dataset with a probability level of 0.1% (p ≤ .001). In the imputed dataset, the number of marker–trait associations increased to 504 and 454 for IT and the LP, respectively (p ≤ .001). The IT corresponding to all races in the original and imputed dataset had the highest number of the marker–trait association on the B genomes of chromosomes. The TTKTK IT had the same number of the marker–trait association on all three genomes in the original dataset. In the TTTTF, the lowest marker–trait association in original and imputed SNPs in A genome and the other races in the D genome were observed. Regarding the TTTTF, PTRTF, and TKTTF LPs, it was found that the highest number of marker–trait association of original SNPs was located on the B genome and for TTKTK on the A genome. These results were a little different in the imputed SNPs, as the highest number of the marker–trait association was observed for PTRTF and TTKTK in A genome and B genome, respectively. Both the TTTTF and TKTTF had the highest number of marker–trait association located on the B genome in imputed SNPs similar to the results of the original SNPs. Among the ITs of races, TTTTF had 29 and 208 marker–trait associations in the original and imputed SNPs, respectively, and among the LPs of races, the TTKTK race had 19 and 160 marker–trait associations in the original and imputed SNPs, respectively, in the 0.1% probability, which were the highest among the other races.
TABLE 7.
A summary of marker–trait associations for infection type and the latent period of studied races in Iranian wheat genotypes
| Genome | TTTTF | PTRTF | TTKTK | TKTTF | ||||
|---|---|---|---|---|---|---|---|---|
| IT | LP | IT | LP | IT | LP | IT | LP | |
| Original data | ||||||||
| Marker–trait association | 29 | 15 | 15 | 18 | 10 | 19 | 15 | 10 |
| Genome A | 2 | 2 | 2 | 5 | 3 | 6 | 4 | 3 |
| Genome B | 17 | 8 | 9 | 6 | 3 | 2 | 8 | 5 |
| Genome D | 8 | 2 | 1 | 3 | 3 | 8 | 2 | 1 |
| Unassembled Chromosomes | 2 | 3 | 2 | 4 | 1 | 3 | 1 | 1 |
| Imputed data | ||||||||
| Marker–trait association | 208 | 128 | 109 | 106 | 83 | 160 | 104 | 60 |
| Genome A | 55 | 37 | 27 | 44 | 25 | 53 | 37 | 10 |
| Genome B | 95 | 69 | 67 | 35 | 39 | 75 | 54 | 33 |
| Genome D | 56 | 19 | 13 | 23 | 18 | 29 | 12 | 16 |
| Unassembled Chromosomes | 2 | 3 | 2 | 4 | 1 | 3 | 1 | 1 |
IT: Infection type, LP: Latent period
The results of the Bonferroni correction at the 5% probability level of the association analysis of original SNPs are presented in Table 8. For Bonferroni correction of the original dataset, 0.05 was divided by the number of the employed markers (8,959 markers) and finally, it was identified that markers were significant at the probability level of 5.58098E‐06. Two QTLs for the TTTTF LP, one QTL for the IT and four QTLs for the LP of PTRTF, one QTL for the IT, and one QTL for the LP of TTKTK, and one QTL for the IT of TKTTF were identified according to the results of Bonferroni correction. The identified QTLs associated with the LP of the studied races, except for one QTL related to the LP of PTTTF located on chromosome 7A with a distance of 133.92 cM, the rest were associated with markers whose genomic location was unknown. The QTL identified for the PTRTF IT was located on chromosome 7A at 132.92 cM. QTL of TTKTK IT was on chromosome 6B at 43.284 cM and QTL of TKTTF IT was on chromosome 1D at 64.821 cM.
TABLE 8.
Most significant original SNP markers associated with quantitative trait loci for resistance to stem rust races of TTTTF, PTRTF, TTKTK, and TKTTF
| Marker | Chr | Pos | P‐value | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TTTTF | PTRTF | TTKTK | TKTTF | |||||||
| IT | LP | IT | LP | IT | LP | IT | LP | |||
| rs46629 | 1D | 64.821 | — | — | — | — | — | — | 1.91E‐6 | — |
| rs23510 | 6B | 43.284 | — | — | — | — | 3.56E‐6 | — | — | — |
| rs22132 | 7A | 133.92 | — | — | 3.23E‐6 | 2.81E‐6 | — | — | — | — |
| rs13588 | Un | 0 | — | 1.56E‐6 | — | 3.38E‐8 | — | — | — | — |
| rs18901 | Un | 0 | — | 1.56E‐6 | — | 3.38E‐8 | — | — | — | — |
| rs27487 | Un | 0 | — | — | — | 5.18E‐6 | — | 2.75E‐7 | — | — |
Chr: Chromosome, Pos: Position, IT: Infection type, LP: Latent period, Un: Unassembled Chromosome
Bonferroni correction of the imputed SNPs was performed as with the original SNPs, but with this difference in the number of markers used in this dataset (43,921 markers), 0.05 was divided into these markers number (43,921) and the level of significance was considered to be 1.1384E‐06 and the results are shown in Table 9. In this regard, the TTTTF IT was controlled by six QTLs, which one, two, and one QTLs were located on chromosome 1, chromosome 3, and chromosome 5 of the B genome, respectively; and the other two QTLs were located on chromosome 2D. For the LP of this race (TTTTF), there were 26 QTLs that eight QTLs were on the A genome, 12 QTLs were on the B genome and six QTLs were on the D genome. Chromosomes three, six, and two of the A genomes, chromosomes one, six, three, and two of the B genome, and chromosomes four, seven, and one of the D genome carried QTLs identified in the TTTTF LP. For the IT and LP of PTRTF, 1 and 31 QTLs were, respectively, observed. The only QTL of the IT was present at 4.558 cM of chromosome 3A. From the 31 identified QTLs for the LP, 3, 3, 1, 1, 1, 4, 3, 3, 6, 1, and 3 QTLs were located on chromosomes of 1A, 1B, 1D, 2A, 2B, 3A, 3B, 4D, 6A, 6B, and 7D, respectively; two QTLs were also associated with markers with unknown genomic loci. For the LP of TTKTK, 14 QTLs were detectable, but no QTL was recorded for the IT. The identified QTLs of the LP was located on the A (chromosomes six, three, five, and seven, respectively) and B genomes (chromosomes one and three, respectively), and a QTL was also associated with markers with unknown genomic loci. In the TKTTF, only three QTLs associated with the IT were identified, two QTLs were located on chromosome 1B at 11.376 cM and one QTL was located on chromosome 1D at 64.821 cM.
TABLE 9.
Most significant imputed SNP markers associated with quantitative trait loci for resistance to stem rust races of TTTTF, PTRTF, TTKTK, and TKTTF
| Marker | Chr | Pos | P‐value | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TTTTF | PTRTF | TTKTK | TKTTF | |||||||
| IT | LP | IT | LP | IT | LP | IT | LP | |||
| rs63550 | 1A | 44.512 | — | — | — | 1.15E‐7 | — | — | — | — |
| rs63551 | 1A | 44.512 | — | — | — | 1.15E‐7 | — | — | — | — |
| rs63552 | 1A | 44.512 | — | — | — | 1.15E‐7 | — | — | — | — |
| rs12408 | 1B | 54.669 | — | 1.98E‐7 | — | — | — | — | — | — |
| rs21674 | 1B | 58.08 | — | 3.47E‐18 | — | 7.11E‐19 | — | 1.35E‐10 | — | ‐ |
| rs45874 | 1B | 11.376 | — | — | — | — | — | 7.55E‐7 | 2.11E‐7 | — |
| rs45875 | 1B | 11.376 | — | — | — | — | — | 7.55E‐7 | 2.11E‐7 | — |
| rs47978 | 1B | 54.669 | — | 1.36E‐7 | — | 2.69E‐7 | — | — | — | — |
| rs51316 | 1B | 25.027 | — | 8.32E‐9 | — | 1.12E‐10 | — | — | — | — |
| rs5923 | 1B | 106.992 | 1.06E‐6 | — | — | — | — | — | — | — |
| rs33527 | 1D | 47.767 | — | 3.89E‐8 | — | 8.69E‐8 | — | — | — | — |
| rs46629 | 1D | 64.821 | — | — | — | — | — | — | 5.21E‐7 | — |
| rs17477 | 2A | 92.517 | — | 4.31E‐8 | — | 1.31E‐8 | — | — | — | — |
| rs25281 | 2B | 40.98 | — | 8.93E‐7 | — | 2.62E‐8 | — | — | — | — |
| rs46339 | 2D | 79.911 | 1.00E‐6 | — | — | — | — | — | — | — |
| rs46340 | 2D | 79.911 | 1.00E‐6 | — | — | — | — | — | — | — |
| rs13329 | 3A | 43.437 | — | — | — | — | — | 6.96E‐8 | — | — |
| rs50704 | 3A | 4.558 | — | 5.03E‐8 | — | 7.21E‐8 | — | — | — | — |
| rs50705 | 3A | 4.558 | — | 5.03E‐8 | — | 7.21E‐8 | — | — | — | — |
| rs50706 | 3A | 4.558 | — | 5.03E‐8 | — | 7.21E‐8 | — | — | — | — |
| rs62417 | 3A | 4.558 | — | — | — | — | — | 1.68E‐7 | — | — |
| rs7318 | 3A | 4.558 | — | 2.55E‐10 | 6.70E‐8 | 2.92E‐13 | — | — | — | — |
| rs20274 | 3B | 31.882 | 5.93E‐7 | 3.72E‐18 | — | 1.77E‐19 | — | 1.11E‐10 | — | — |
| rs20275 | 3B | 31.882 | 5.93E‐7 | 3.72E‐18 | — | 1.77E‐19 | — | 1.11E‐10 | — | — |
| rs20294 | 3B | 31.882 | — | 2.75E‐8 | — | 3.82E‐9 | — | — | — | — |
| rs33960 | 3B | 62.576 | — | — | — | — | — | 1.78E‐7 | — | — |
| rs65252 | 4D | 54.756 | — | 2.61E‐8 | — | 7.93E‐9 | — | — | — | — |
| rs65253 | 4D | 54.756 | — | 2.61E‐8 | — | 7.93E‐9 | — | — | — | — |
| rs65254 | 4D | 54.756 | — | 2.61E‐8 | — | 7.93E‐9 | — | — | — | — |
| rs51204 | 5A | 93.664 | — | — | — | — | — | 1.53E‐7 | — | — |
| rs22086 | 5B | 86.610 | 4.63E‐7 | — | — | — | — | — | — | — |
| rs17868 | 6A | 90.292 | — | — | — | 2.62E‐7 | — | — | — | — |
| rs34668 | 6A | 50.208 | — | 2.56E‐18 | — | 4.91E‐19 | — | 1.91E‐10 | — | — |
| rs35063 | 6A | 50.208 | — | 2.21E‐18 | — | 4.78E‐19 | — | 1.91E‐10 | — | — |
| rs35074 | 6A | 50.208 | — | 2.74E‐18 | — | 4.73E‐19 | — | 1.95E‐10 | — | — |
| rs4500 | 6A | 10.247 | — | — | — | 6.97E‐7 | — | — | — | — |
| rs53936 | 6A | 99.391 | — | — | — | 1.80E‐7 | — | — | — | — |
| rs14828 | 6B | 58.062 | — | 1.00E‐6 | — | — | — | — | — | — |
| rs14829 | 6B | 58.062 | — | 1.00E‐6 | — | — | — | — | — | — |
| rs14830 | 6B | 58.062 | — | 1.00E‐6 | — | — | — | — | — | — |
| rs25306 | 6B | 47.831 | — | — | — | 6.48E‐8 | — | — | — | — |
| rs32944 | 6B | 47.831 | — | 4.11E‐8 | — | — | — | — | — | — |
| rs48710 | 7A | 71.904 | — | — | — | — | — | 8.54E‐8 | — | — |
| rs16240 | 7D | 83.31 | — | 7.72E‐9 | — | 4.73E‐9 | — | — | — | — |
| rs20930 | 7D | 83.31 | — | 1.21E‐8 | — | 3.81E‐9 | — | — | — | — |
| rs40699 | 7D | 82.173 | — | — | — | 4.09E‐7 | — | — | — | — |
| rs13588 | Un | 0 | — | — | — | 3.38E‐8 | — | — | — | — |
| rs18901 | Un | 0 | — | — | — | 3.38E‐8 | — | — | — | — |
| rs27487 | Un | 0 | — | — | — | — | — | 2.75E‐7 | — | — |
Chr: Chromosome, Pos: Position, IT: Infection type, LP: Latent period, Un: Unassembled Chromosome
Highly significant markers associated with IT and the LP of Pgt races, their chromosomal sequence and position, the closest wheat gene or genes to them, orthologous genes (with the highest percentage of identity), identity percentage of the wheat gene or genes that match to the ortholog, molecular function and biological processes of the wheat gene or genes associated with the markers and the extent of phenotype variance explain (R2) are reported in Table 10. Out of the 51 highly significant, identified SNP markers, only 9 markers had the adjacent wheat gene or genes with the same chromosomal position. The Ensembl (https://asia.ensembl.org/index.html) site was used to gain further information about adjacent genes beyond the genetic position of the markers. The genes with the highest identity percentage and the lowest E‐value with significant markers were reported. The amount of phenotypic variance explained by SNP markers ranged between 9% and 25%. The rs21674 and rs51316 markers on the 1B justified the high variance explained for the associated traits to other markers. These markers are in the closest genetic position to the TraesCS1B02G208400 and TraesCS1B02G037100 genes, respectively, and each of these genes has a genetic identity with the orthologous genes of AET1Gv20503500 (96.74%) in Aegilops tauschii and TRIDC1AG003150 (95) in Triticum dicoccoides. The wheat TraesCS1D02G315800 gene, with an rs46629 marker located on the chromosome 1D, is incomplete identity (100%) with the AET1Gv20751400 ortholog gene of Aegilops tauschii.
TABLE 10.
Basic local alignment search tool results of significant markers for seedling resistance to Pgt races
| R2 | 0.09 | 0.10 |
0.25 0.15 0.25 |
0.09 0.11 |
0.10 | 0.09 | 0.09 | 0.09 |
0.12 0.15 |
|---|---|---|---|---|---|---|---|---|---|
| Trait | PTRTF (LP) | TTTTF (LP) |
TTTTF (LP) PTRTF (LP) TTKTK (LP) |
TTTTF (LP) PTRTF (LP) |
TTKTK (LP) | TTTTF (IT) | TTTTF (IT) | TKTTF (IT) |
TTTTF (LP) PTRTF (LP) |
| Biological process | — | protein phosphorylation, phosphorylation | regulation of transcription, DNA‐templated | — | protein phosphorylation | — | – | protein processing, oxidation‐reduction process | defense response to bacterium |
| Molecular function | protein binding | nucleotide‐binding, protein kinase activity, protein serine/threonine kinase activity, protein binding, ATP binding, kinase activity, transferase activity | DNA binding, lipid binding, sequence‐specific DNA binding | protein binding | protein kinase activity, MAP kinase activity, ATP binding | ADP binding | ADP binding | serine‐type endopeptidase activity, superoxide dismutase activity | copper ion binding |
| Identity | 99.70 | 85.81 | 96.74 | 96.94 | 96.53 | 74.78 | 74.78 | 100 | 95.00 |
| Orthologous gene | TRIDC6AG012980 a | TRIDC1BG053570 a | AET1Gv20503500 b | AET2Gv20533600 b | TRIDC3BG039070 a | TRIDC6AG061710 a | TRIDC6AG061710 a | AET1Gv20751400 b | TRIDC1AG003150 a |
| Position (bp) | 67,642,014‐67,642,038 | 555,948,792‐555,952,831 | 377,964,396‐377,970,306 | 367,657,356‐367,709,290 | 418,887,290‐418,898,340 | 571,193,736‐571,202,194 | 571,193,736‐571,202,194 | 410,793,556‐410,798,235 | 17,730,527‐17,739,883 |
| Adjacent T. aestivum gene | TraesCS6A02G100400 | TraesCS1B02G330100 | TraesCS1B02G208400 | TraesCS2B02G269700 | TraesCS3B02G260900 | TraesCS2D02G465300 | TraesCS2D02G465300 | TraesCS1D02G315800 | TraesCS1B02G037100 |
| Chromosome | 6A | 1B | 1B | 2B | 3B | 2D | 2D | 1D | 1B |
| Allele | C/G | A/G | C/T | A/G | G/T | A/G | C/A | C/T | A/G |
| Sequence | TGCAGACGGCGGCGGCGCGAACAAGCATGGTGGCGAAGCCGACACGGAGGAGGATCTCGGCGAG | TGCAGCAAGCTAAACCAAGTCTTGAAACTGGAGCAGCGAACAATTCTAAAGTCGCTTGAGGCTC | TGCAGCCAGAGCTCAAGAGGCCCCCTCGGCGGCATCTCAAAGGAGAAGGAAGGAGGAAGGCGAA | TGCAGCCGCCCGTCCACCCCCTTCGTCTTCGTCCAGCACCAACCCGTCGCCAGCGTCGCCGTCA | TGCAGCGTGTAGATTACCATGCTATTCACACAGAAGTTATCGGGTGTATATGTCATCAACTTTT | TGCAGGCAGCTGACTGTACTGGAGGGCCACACATTTATCGAAACCCTGCTGCCTGCCCTGGTTC | TGCAGGCAGCTGACTGTACTGGAGGGCCACACATTTATCGAAACCCTGCTGCCTGCCCTGGTTC | TGCAGGCATTCCATCAAGCAACCAGCAAGTCAGCACCAGAATCACCCCAATCAGACATGAGTGA | TGCAGGGGGTAGCCCTGAGCGCCGCCAAATCAGATCAAACGATGCCCTTCACTACATAAGCCAG |
| Marker | rs4500 | rs12408 | rs21674 | rs25281 | rs33960 | rs46339 | rs46340 | rs46629 | rs51316 |
a: Triticum dicoccoides, b: Aegilops tauschii, IT: Infection type, LP: Latent period.
4. DISCUSSION
The characteristics of the races used to evaluate the seedling stage and the effective and ineffective genes for each race are presented in Table 1. These races are known as the dominant black rust races in the country collected during 2015 and 2016 and with inoculated on 20 lines and differential cultivar of North American (Singh et al., 2008), the race of those has been determined. Isolate 95–2 was pathogenic to the Sr31 resistance gene, which caused resistance to Pgt for a very long time. This isolate belonged to the Ug99 race group and was named TTKTK.
The results of the combined analysis of variance showed that in both traits of IT and LP, the highest total sum of squares was explained by the genotype effect (54.12% and 43.39%, respectively); therefore, it is inferred that genotypes have had a significant effect on data variation. After that, the highest justified variance of IT (36.33%) and LP (36.91%) was due to genotype–race interaction. The effect of the race for IT and LP explained 3.97% and 8.80% of the total variation, respectively. The small effect of the race indicates a relatively low diversity among the races under study.
The values of the genetic and phenotypic variation coefficients for ITs and LPs of all races were very close to each other. This indicates the high impact of genes on creating diversity among genotypes. Each of the phenotypic, genetic, and environmental diversity coefficients provides useful information about the observed genetic or environmental diversity. As seen in Table 3, the coefficient of phenotypic variation was higher than the genetic variation coefficient in all studied ITs and LPs. This is due to the influence of environmental factors. Since the phenotypic variance is derived from the sum of environmental and genetic variance, if the genetic variance is assumed constant, what causes one trait to differ in phenotypic and genetic variation would be environmental variance; that is, if the trait has high phenotypic variation and low genetic diversity, this indicates the effect of the environment. The higher the variation ratio of genetic to the environment, the greater the efficiency of selection, and therefore, favorable genotypes will be identified and selected more accurately from unfavorable ones.
The genetic variation coefficient reflects the variation among genotypes in terms of specificity understudy and cannot lonelily determine the extent of inheritance of this variation. This index, along with heritability, provides a good estimate of genetic progress in phenotypic selection (Burton & Devane, 1953). Simultaneous application of two very important parameters of heritability and genetic efficiency plays an important role in the development of varieties and genotypes. The high rate of genetic efficiency indicates additive gene action, and its low level represents nonadditive gene action. Genetic efficiency will not necessarily be high when estimated a high heritability for a trait. If high heritability and high genetic efficiency coincide, it will reflect the additive effects of genes; however, if high heritability is associated with low genetic function, it would indicate epistatic effects or dominance (Johnson et al., 1955). If trait heritability is < 0.2 (20%), it indicates low heritability, if it is between 0.2 (20%) to 0.5 (50%), it has moderate heritability, and if it is > 0.5 (50%) it has high heritability (Stansfield, 1991). High heritability with favorable genetic efficiency was observed in the PTRTF IT and LP, confirming the additive effects of the gene, and indicating that a large proportion of phenotypic variation explains the genotypic variation. High inheritance along with high genetic efficiency is a very important factor in predicting the effects of selecting the best individuals in a population.
Population structure has been used in genetic studies to explain the relationship among individuals "within populations" and "between populations" and shows a perspective on the evolutionary relationship of individuals in a population. Besides the existence of structure in a population that studied for association mapping, it is a deterrent to achieving reliable and positive results in a population that is not considered by the effects of population structure factors and relationships (Breseghello & Sorrells, 2006). Out of 282 genotypes in both groups of the original and imputed datasets, 215 genotypes (76.24%) belonged to three identified subpopulations and the remaining 67 genotypes (23.76%), including 22 variety and 45 landraces, were excluded from the mixed genotypes. The number of these genotypes in the mixed group was small indicating low mixing in the studied population. Allelic incorporation in landraces of more than one single gene cohort could have caused a slight mixing due to the wheat distribution in more than one ancestral population (Oliveira et al., 2012). The presence of gene flow from the introduction of new genotypes into farms previously can be another reason for mixing. In this regard, germplasm exchange between different Mediterranean regions due to the development of ancient empires is considered as another possible reason for mixing (Moragues et al., 2007). Geographical origin of genotypes, selection, and genetic drift cause subpopulation within a large population (Buckler IV & Thornsberry, 2002; Flint‐Garcia et al., 2003). The reason some varieties fall into associated groups of landrace is that some of the varieties have been selected and introduced based on indices from landrace and thus have a high genetic similarity to landrace (Alipour et al., 2017).
The extent of LD determines the number of markers required to identify marker–trait association as well as mapping resolution (Flint‐Garcia et al., 2003). On the other hand, in genome‐wide association mapping, locating QTLs is based on the extent of LD and is therefore of particular importance (Al‐Maskri et al., 2012). Generally, LD decreased with increasing genetic distance. The range of LD varied across chromosomes as well as from chromosomes to other chromosomes. The share of each of the A, B, and D genomes of the total original SNP markers was estimated to be in varieties and landraces, approximately 37, 46, and 17%, respectively, and in the imputed SNP markers, approximately 36, 50, and 13%, respectively. Nearly 87% of markers pairwise and 90% of significant markers pairwise in varieties and in landraces nearly 87% of markers pairwise and 88% of significant markers pairwise in original SNPs had genetic distance < 10 cM. In the imputed SNPs, all the pairwise markers and the significant pairwise markers related to the varieties and landraces were located < 10 cM. This issue indicants a high impact of LD on population, but LD in the population decreased (but not disappeared) with increasing distance between pairwise markers according to the results; this is due to other factors such as population structure, genetic drift, migration, selection, and mutation. As well as, the results showed higher LD values in varieties than landraces. This increase in LD in varieties can be attributed to the selection, whether natural or synthetic, which causes an LD among the selected and associated genes. Also, the selection of the ascending (or descending) trait controlled by two or more nonlinkage genes, despite the high genetic (physical) distance among the genes, increases the LD (Ataei et al., 2017). The relatively high level of LD in many chromosomal regions in the population indicates the usefulness and effectiveness of association mapping for identifying and confirming QTLs in these regions (Zhang et al., 2010).
Since association mapping was introduced in plants (Thornsberry et al., 2001), interest in the identification of new genes has gained popularity due to significant advances in DNA sequencing technology. The application of this method in plant populations to identify loci responsible for genetic variation including disease resistance is increasing (Hall et al., 2010; Kollers et al., 2013; Reich et al., 2001). Knowledge of the genetic diversity of wheat to accelerate genetic and breeding studies is essential to produce disease‐resistant genotypes because understanding the genetic nature of disease resistance can play a key role in the development of resistant genotypes. The use of genetic diversity of wheat germplasm to understand the genetic mechanisms of disease resistance through GWAS can be very effective in breeding programs aimed at producing resistant varieties of Pgt.
Association mapping introduced a total of 69 QTLs for the IT and 62 QTLs for the LP in the original dataset and the imputed dataset, 504 QTLs for the IT, and 454 QTLs for the LP with a 0.1% probability level (p ≤ .001). The highest number of QTLs identified for the IT of studied races in the original and imputed dataset was nonuniformly distributed on the B genome chromosomes.
After Bonferroni correction aimed to identify markers highly correlated with the target trait, the number of identified QTLs reduced in the original dataset, so the total of the QTLs for the studied races IT was 3 and for the LP 7 QTLs remained. In the imputed dataset, the number of QTLs of the IT was reduced to 10 QTLs and the number of QTLs of the LP decreased to 71 QTLs. To answer the question of whether QTLs identified with specific genomic regions in this study have been previously introduced or not, the results of this study were compared with previous findings. Some of the QTLs identified in this study corresponded to the genomic regions of the Sr genes and the QTLs reported in previous studies (Yu et al., 2011, 2012). B chromosomes were fined chromosomes carrying rust resistance genes in wheat, which has been reported in various studies (Aoun et al., 2016; Crossa et al., 2007; Letta et al., 2014; Yu et al., 2011). In the present study, only one marker (rs23510) was identified based on the results of the B genome located on chromosome 6 and was associated with the genomic locus that caused resistance to TTKTK. The number of markers in the B genome associated with the IT and the LP was significantly higher in the imputed dataset than in the original dataset and they were distributed on chromosomes 1B, 6B, 3B, 2B, and 5B, respectively. Letta et al. [19] identified a QTL on chromosome 1B for resistance to the JRCQC; this disease‐resistant genomic region has also been mentioned. The rs51316 marker associated with the TTTTF and PTRTF IT was largely close to the QTL identified by Letta et al. (2014). On the other hand, in this chromosomal position, the Sr14 gene is resistant to some Pgt races in wheat (Singh et al., 2006), but a significant effect on the IT of each race was observed in this study. A specific study on TTKTK infection type belonging to the Ug99 group and reported in the study of Jin et al. (2007) did not show any significant effect at 106.99 cM located on chromosome 1B marker rs5923. This marker is related to the genomic region that caused seedling resistance to the TTTTF and was reported in the study by Letta et al. (2014). Not only the Pgt resistance gene is located in this region, but also the Lr46/Yr29/Pm39 gene block and the unknown resistance genes are in the adult plant stage (Bhavani et al., 2011; Singh et al., 2013). Several Pgt resistance genes, including Sr9, Sr16, and Sr28 (McIntosh et al., 1995) and SrWeb, are located on the long arm of chromosome 2B. The SrWeb gene causes resistance to Ug99, while none of the four Sr9 alleles has this ability (Jin et al., 2007). No resistance to TTKTK was found on chromosome 2B; the SrWeb gene may not be present in the studied germplasm, or there is no marker associated with this gene. After the B genome, the A genome had the highest marker–trait association. In the original dataset after Bonferroni correction, only one marker (rs22132) was identified in the A genome, located on chromosome 7 at 133.92 cM, and was associated with both PTRTF IT and LP characteristics. There were no QTLs in the A genome in the imputed SNPs for the IT of any of the studied races, but 8 QTL (1 QTL on 2A, 4 QTL on 3A, and 3 QTL on 6A) in TTTTF, 14 QTL (3 QTL on 1A, 1 QTL on 2A, 4 QTL on 3A and 6 QTL on 6A) in PTRTF, and 7 QTL (2 QTL on 3A, 1 QTL on 5A, 3 QTL on 6A and 1 QTL on 7a) in TTKTK were identified for the LP. TKTTF was not associated with genomic A chromosomes. The Sr34 and Sr38 Pgt genes are located on chromosome 2A (Letta et al., 2014) and are somewhat close to the rs17477 marker in genomic position, but because the resistance of these two genes is broken by the studied races and as a result, they were inactivated against rust disease with no QTL associated region with the IT. Therefore, this marker can be used in marker‐assisted selection to identify Sr34 and Sr38. The coexistence of two markers cfa2201 and wPt‐5839 with two Sr34 and Sr38 Pgt resistance genes was reported in a study (Letta et al., 2014). These two genes are ineffective against the Ug99 race group, one of which (TTKTK) has been investigated in the present study (Jin et al., 2007; Singh et al., 2011). The Sr7 and SrND643 resistance genes (probably the Sr7 allele) are located on the long arm of chromosome 4A (Basnet et al., 2015; McIntosh et al., 1995). This gene caused resistance to the PTRTF race, but no marker–trait association was found in this study. Other studies have also mapped genomic regions associated with resistance to black rust disease in the A genome such as a marker wpt‐734078 (Bhavani et al., 2011) and wpt‐6869 (Rouse et al., 2014) on chromosome 1AS.
The D genome had the lowest number of marker–trait associations. In the original dataset between the IT and the LP of all the studied races, only one QTL was identified for the TKTTF IT associated with the rs46629 marker on the chromosome 1D. In the imputed dataset, this marker retained its association with the TKTTF IT, and the only marker had the marker–trait association on all TKTTF D genome chromosomes. The TTKTK had no marker–trait association with the D genome. For the TTTTF IT, 2 QTLs were on the 2D and for the LP, 1, 3, and 2 QTLs were on 1D, 4D, and 7D, respectively. In PTRTF, 1, 3, and 3 QTLs were observed on 1D, 4D, and 7D, respectively. Edae et al. (2018) in their study reported QTHJC resistance‐related IWB17135 marker and TPMKC resistance‐related IWA2415 marker on the short arm of chromosome 2D. Tsilo et al. (2009) mapped a QTL on the chromosome 2D at 78.50 cM, which was 1.1 cm from the Sr6 Pgt resistance gene. In the original dataset, there were 6 QTLs on unknown chromosomes, 2 QTLs for TTTTF LP, 3 QTLs for PTRTF LP, and 1 QTL for TTKTK LP. In the imputed dataset, 2 QTLs associated with PTRTF LP and 1 QTL associated with TTKTK LP overlapped with the original dataset were identified.
The markers with the highest marker–trait association were blasted at the Ensembl database to identify overlapping genes, their molecular function, and biological processes, as well as to identify orthologous genes. The results indicated that the genes adjacent to the markers play an important role in biosynthetic pathways such as ion transport, redox, protein processing, phosphorylation, and so on. In general, environmental stresses cause significant changes in the expression levels of many of these genes in plants, so such changes result in the accumulation or reduction of important metabolites, changes in enzyme activity and protein synthesis rates, as well as the production of new proteins (Zhu, 2016) that deal with environmental stress in various ways. ROS, phospholipid‐derived signals, and cyclic nucleotide‐dependent signals, as well as some plant hormones, are involved in stress signaling (Ali et al., 2018; Jamei et al., 2018). In response to the pathogen attack, ROS is produced by the attacked plants, acts as an important sign of stress, and reduces the amount of stress damage possible by activating defense mechanisms (Ali et al., 2018; Jamei et al., 2018; Lehmann et al., 2015). Membrane lipids by regulating membrane fluidity and other physicochemical properties cause secondary signaling molecules in response to stress. Biosynthesis of lipids and their degrading enzymes plays several roles such as direct or indirect regulation or effect on stress signaling and tolerance (Jamei et al., 2018). In plants, a variety of biotic and abiotic stresses such as pathogens, drought, salinity, high temperature, intense light, hormones, and nodulation factors cause changes in cytosolic calcium levels and lead to stress transmission (Monihan, 2011; Pandey et al., 2015). In many transcriptional signaling pathways, the major form of signal transduction is reversible protein phosphorylation. Protein kinases such as mitogen‐activated protein kinase (MAPK) are essential in stress developmental, hormonal, biotic, and abiotic signaling. Protein phosphatases are responsible for dephosphorylating the phospho‐proteins. Protein phosphatases are subdivided into serine/threonine phosphatase, tyrosine phosphatase, and dual phosphatases through their specific substrates, which play an important role in serine/threonine phosphatase transcriptional stress transcription (Jamei et al., 2018). Eventually, protein kinases are likely to target transcription factors and bind to the stress‐responsive gene's promoter and consequently activate transcription (Jamei et al., 2018).
After identifying the nearest wheat gene or genes to the markers, orthologous genes with these genes were examined. Numerous orthologous genes were observed, but only genes with a high percentage of identity were selected. The identified orthologous genes were generally related to two species of Triticum dicoccoides and Aegilops tauschii, the ancestors of wheat, and they have introduced to it during the evolutionary stages of wheat by hybridization (Dvorak et al., 1998; Kerber, 1964; Kihara, 1944; Kislev, 1979; Matsuoka & Nasuda, 2004; McFadden & Sears, 1946). These orthologous genes had a high percentage of identity with the genes identified in wheat, which involved in stress resistance mechanisms.
5. CONCLUSION
Association mapping of resistance of Iranian bread wheat varieties and landraces to four Pgt races in the seedling stage in terms of LP and IT. The studied germplasm was grouped into three subpopulations. According to the results of the original and imputed dataset, the highest number of the pairwise marker in varieties and landraces was observed on the B genome. In the original dataset, the highest LD was observed in the D genome of varieties and landraces but in an imputed dataset, it was observed in the A genome of varieties and landraces. The results showed a significant difference in the LD value on different chromosomes. However, the varieties had higher LD compared with the landraces, which could be attributed to the selection made during the production and release stages of the varieties. Genome‐wide association study based on the original dataset revealed 69 and 62 marker–trait associations for races IT and their LP, respectively. The number of marker–trait associations in the imputed dataset was 504 for IT and 454 for the LP. When Bonferroni correction was performed, in the original dataset, two QTLs for the TTTTF LP, one QTL for the IT and four QTLs for the LP of PTRTF, one QTL for the IT, and one QTL for the LP of TTKTK, and one QTL for the TKTTF IT remained. In the imputed dataset, the TTTTF IT by six QTLs and its LP by 26 QTLs, the PTRTF IT, and the LP by 1 and 31 QTLs, respectively, the TTKTK LP by 14 QTLs, and the TKTTF IT by 3 QTLs was controlled. No marker–trait association was detected in the TTKTK IT and TKTTF LP. Nine markers out of 51 SNP markers (very significant) had a chromosomal position similar to the adjacent Triticum aestivum gene or genes. By studying the molecular function and biological processes of these genes, it was found that these genes have different mechanisms that cause stress resistance. The results obtained from this study can be important to facilitate and accelerate breeding programs through marker‐assisted selection.
CONFLICT OF INTEREST
The authors declare that they do not have conflict of interest. The funding body was involved in the material creation, designing the study, data analysis, and writing the manuscript.
Ethical Approval
Not applicable.
Saremirad A, Bihamta MR, Malihipour A, Mostafavi K, Alipour H. Genome‐wide association study in diverse Iranian wheat germplasms detected several putative genomic regions associated with stem rust resistance. Food Sci Nutr. 2021;9:1357–1374. 10.1002/fsn3.2082
Contributor Information
Ali Saremirad, Email: Asaremirad@gmail.com.
Mohammad Reza Bihamta, Email: mrghanad@ut.ac.ir.
REFERENCES
- Ali, M. , Cheng, Z. , Ahmad, H. , & Hayat, S. (2018). Reactive oxygen species (ROS) as defenses against a broad range of plant fungal infections and case study on ROS employed by crops against Verticillium dahliae wilts. Journal of Plant Interactions, 13(1), 353–363. 10.1080/17429145.2018.1484188 [DOI] [Google Scholar]
- Alipour, H. , Bai, G. , Zhang, G. , Bihamta, M. R. , Mohammadi, V. , & Peyghambari, S. A. (2019). Imputation accuracy of wheat genotyping‐by‐sequencing (GBS) data using barley and wheat genome references. PLoS One, 14(1), 1–20. 10.1371/journal.pone.0208614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alipour, H. , Bihamta, M. R. , Mohammadi, V. , Peyghambari, S. A. , Bai, G. , & Zhang, G. (2017). Genotyping‐by‐sequencing (GBS) revealed molecular genetic diversity of Iranian wheat landraces and cultivars. Frontiers in Plant Science, 8(1), 1–14. 10.3389/fpls.2017.01293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allard, R. (1960). Principies of plant breeding, 2, (p. 36). USA: John Wiley & Sons, INC. [Google Scholar]
- Al‐Maskri, A. Y. , Sajjad, M. , & Khan, S. H. (2012). Association mapping: A step forward to discovering new alleles for crop improvement. International Journal of Agriculture & Biology, 14(1), 153–160. [Google Scholar]
- Aoun, M. , Breiland, M. , Kathryn Turner, M. , Loladze, A. , Chao, S. , Xu, S. S. , Ammar, K. , Anderson, J. A. , Kolmer, J. A. , & Acevedo, M. (2016). Genome‐wide association mapping of leaf rust response in a durum wheat worldwide germplasm collection. The Plant Genome, 9(3), 1–24. 10.3835/plantgenome2016.01.0008 [DOI] [PubMed] [Google Scholar]
- Ataei, R. , Gholamhoseini, M. , & Mohammadi, V. (2017). Association mapping in plants. Crop Biotechnology, 18(1), 28–37. [Google Scholar]
- Basnet, B. R. , Singh, S. , Lopez‐Vera, E. E. , Huerta‐Espino, J. , Bhavani, S. , Jin, Y. , Rouse, M. N. , & Singh, R. P. (2015). Molecular mapping and validation of SrND643: A new wheat gene for resistance to the stem rust pathogen Ug99 race group. Phytopathology, 105(4), 470–476. 10.1094/PHYTO-01-14-0016-R [DOI] [PubMed] [Google Scholar]
- Bhavani, S. , Singh, R. P. , Argillier, O. , Huerta‐Espino, J. , Singh, S. , Njau, P. , Brun, S. , Lacam, S. , Desmouceaux, N. , & McIntosh, R. A. (2011). Mapping durable adult plant stem rust resistance to the race Ug99 group in six CIMMYT wheats, 1(42). Minnesota, USA: In Borlaug Global Rust Initiative 2011 Technical Workshop June 13‐16 Saint Paul. [Google Scholar]
- Bradbury, P. J. , Zhang, Z. , Kroon, D. E. , Casstevens, T. M. , Ramdoss, Y. , & Buckler, E. S. (2007). TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics, 23(19), 2633–2635. 10.1093/bioinformatics/btm308 [DOI] [PubMed] [Google Scholar]
- Breseghello, F. , & Sorrells, M. E. (2006). Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics, 172(2), 1165–1177. 10.1534/genetics.105.044586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning, B. L. , & Browning, S. R. (2009). A unified approach to genotype imputation and haplotype‐phase inference for large data sets of trios and unrelated individuals. The American Journal of Human Genetics, 84(2), 210–223. 10.1016/j.ajhg.2009.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler, E. S. IV , & Thornsberry, J. M. (2002). Plant molecular diversity and applications to genomics. Current Opinion in Plant Biology, 5(2), 107–111. 10.1016/S1369-5266(02)00238-8 [DOI] [PubMed] [Google Scholar]
- Burton, G. W. , & Devane, D. E. (1953). Estimating heritability in tall fescue (Festuca arundinacea) from replicated clonal material 1. Agronomy Journal, 45(10), 478–481. 10.2134/agronj1953.00021962004500100005x [DOI] [Google Scholar]
- Comstock, R. E. , & Robinson, H. F. (1952). Genetic parameters, their estimation, and significance. In Proceedings of the 6th international Grassland congress, 1, 248–291. [Google Scholar]
- Crossa, J. , Burgueño, J. , Dreisigacker, S. , Vargas, M. , Herrera‐Foessel, S. A. , Lillemo, M. , Singh, R. P. , Trethowan, R. , Warburton, M. , Franco, J. , Reynolds, M. , Crouch, J. H. , & Ortiz, R. (2007). Association analysis of historical bread wheat germplasm using additive genetic covariance of relatives and population structure. Genetics, 177(3), 1889–1913. 10.1534/genetics.107.078659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak, J. , Luo, M.‐C. , Yang, Z.‐L. , & Zhang, H.‐B. (1998). The structure of the Aegilops tauschii genepool and the evolution of hexaploid wheat. Theoretical and Applied Genetics, 97(4), 657–670. 10.1007/s001220050942 [DOI] [Google Scholar]
- Edae, E. A. , Pumphrey, M. O. , & Rouse, M. N. (2018). A genome‐wide association study of field and seedling response to individual stem rust pathogen races reveals combinations of race‐specific genes in North American spring wheat. Frontiers in Plant Science, 9(1), 1–18. 10.3389/fpls.2018.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer, D. S. (1989). IntrInoduction to quantitative genetics, 1, 3rd ed, Harlow: Longman Scientific & Technica. [Google Scholar]
- Flint‐Garcia, S. A. , Thornsberry, J. M. , & Buckler, E. S. IV (2003). Structure of linkage disequilibrium in plants. Annual Review of Plant Biology, 54(1), 357–374. 10.1146/annurev.arplant.54.031902.134907 [DOI] [PubMed] [Google Scholar]
- Godfray, H. C. J. , Beddington, J. R. , Crute, I. R. , Haddad, L. , Lawrence, D. , Muir, J. F. , Pretty, J. , Robinson, S. , Thomas, S. M. , & Toulmin, C. (2010). Food security: The challenge of feeding 9 billion people. Science, 327(5967), 812–818. 10.1126/science.1185383 [DOI] [PubMed] [Google Scholar]
- Gupta, S. K. , Charpe, A. , Koul, S. , Prabhu, K. V. , & Haq, Q. M. R. (2005). Development and validation of molecular markers linked to an Aegilops umbellulata–derived leaf‐rust‐resistance gene, Lr9, for marker‐assisted selection in bread wheat. Genome, 48(5), 823–830. 10.1139/g05-051 [DOI] [PubMed] [Google Scholar]
- Hall, D. , Tegström, C. , & Ingvarsson, P. K. (2010). Using association mapping to dissect the genetic basis of complex traits in plants. Briefings in Functional Genomics, 9(2), 157–165. 10.1093/bfgp/elp048 [DOI] [PubMed] [Google Scholar]
- Holland, J. B. (2007). Genetic architecture of complex traits in plants. Current Opinion in Plant Biology, 10(2), 156–161. 10.1016/j.pbi.2007.01.003 [DOI] [PubMed] [Google Scholar]
- Jamei, R. , Kargar‐Khorami, S. , & Zanganeh, R. (2018). Plant cellular and molecular mechanism against biotic stress, 1(166). Iran: Urmia University Press. [Google Scholar]
- Jin, Y. , Singh, R. , Ward, R. , Wanyera, R. , Kinyua, M. , Njau, P. , Fetch, T. , Pretorius, Z. , & Yahyaoui, A. (2007). Characterization of seedling infection types and adult plant infection responses of monogenic Sr gene lines to race TTKS of Puccinia graminis f. sp. tritici. Plant Disease, 91(9), 1096–1099. 10.1094/PDIS-91-9-1096 [DOI] [PubMed] [Google Scholar]
- Johnson, H. W. , Robinson, H. , & Comstock, R. (1955). Estimates of genetic and environmental variability in soybeans 1. Agronomy Journal, 47(7), 314–318. 10.2134/agronj1955.00021962004700070009x [DOI] [Google Scholar]
- Kerber, E. (1964). Wheat: Reconstitution of the tetraploid component (AABB) of hexaploids. Science, 143(3603), 253–255. 10.1126/science.143.3603.253 [DOI] [PubMed] [Google Scholar]
- Kihara, H. (1944). Discovery of the DD–analyzer, one of the analyzers of Triticum vulgare . Agriculture and Horticulture, 19(1), 889–890. [Google Scholar]
- Kislev, M. E. (1979). Triticum parvicoccum sp. Nov., The oldest naked wheat. Israel Journal of Botany, 28(2), 95–107. 10.1080/0021213X.1979.10676861 [DOI] [Google Scholar]
- Kollers, S. , Rodemann, B. , Ling, J. , Korzun, V. , Ebmeyer, E. , Argillier, O. , Hinze, M. , Plieske, J. , Kulosa, D. , Ganal, M. W. , & Röder, M. S. (2013). Whole genome association mapping of fusarium head blight resistance in European winter wheat (Triticum aestivum L.). PLoS One, 8(2), 1–10. 10.1371/journal.pone.0057500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraakman, A. T. W. , Niks, R. E. , Van den Berg, P. M. M. , Stam, P. , & Van Eeuwijk, F. A. (2004). Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics, 168(1), 435–446. 10.1534/genetics.104.026831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann, S. , Serrano, M. , L’Haridon, F. , Tjamos, S. E. , & Metraux, J.‐P. (2015). Reactive oxygen species and plant resistance to fungal pathogens. Phytochemistry, 112(1), 54–62. 10.1016/j.phytochem.2014.08.027 [DOI] [PubMed] [Google Scholar]
- Letta, T. , Olivera, P. , Maccaferri, M. , Jin, Y. , Ammar, K. , Badebo, A. , Salvi, S. , Noli, E. , Crossa, J. , & Tuberosa, R. (2014). Association mapping reveals novel stem rust resistance loci in durum wheat at the seedling stage. Plant Genome, 7(1), 1–13. 10.3835/plantgenome2013.08.0026 [DOI] [Google Scholar]
- Lipka, A. E. , Tian, F. , Wang, Q. , Peiffer, J. , Li, M. , Bradbury, P. J. , Gore, M. A. , Buckler, E. S. , & Zhang, Z. (2012). GAPIT: Genome association and prediction integrated tool. Bioinformatics, 28(18), 2397–2399. 10.1093/bioinformatics/bts444 [DOI] [PubMed] [Google Scholar]
- Lu, F. , Lipka, A. E. , Glaubitz, J. , Elshire, R. , Cherney, J. H. , Casler, M. D. , Buckler, E. S. , & Costich, D. E. (2013). Switchgrass genomic diversity, ploidy, and evolution: Novel insights from a network‐based SNP discovery protocol. PLoS One, 9(1), 1–14. 10.1371/journal.pgen.1003215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka, Y. , & Nasuda, S. (2004). Durum wheat as a candidate for the unknown female progenitor of bread wheat: An empirical study with a highly fertile F 1 hybrid with Aegilops tauschii Coss. Theoretical and Applied Genetics, 109(8), 1710–1717. 10.1007/s00122-004-1806-6 [DOI] [PubMed] [Google Scholar]
- McFadden, E. S. , & Sears, E. R. (1946). The origin of Triticum spelta and its free‐threshing hexaploid relatives. Journal of Heredity, 37(3), 81–89. 10.1093/oxfordjournals.jhered.a105590 [DOI] [PubMed] [Google Scholar]
- McIntosh, R. A. , Wellings, C. R. , & Park, R. F. (1995). Cloud‐Guest A. Wheat rusts: An atlas of resistance genes, 1(81). Australia: CSIRO Publications. [Google Scholar]
- Monihan, S. (2011). The Arabidopsis Calcineurin B‐Like10 Calcium Sensor Couples Environmental Signals to Developmental Responses. The University of Arizona, (201)Tucson, United States. http://hdl.handle.net/10150/202993.
- Moose, S. P. , & Mumm, R. H. (2008). Molecular plant breeding as the foundation for 21st century crop improvement. Plant Physiology, 147(1), 969–977. 10.1104/pp.108.118232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moragues, M. , Moralejo, M. , Sorrells, M. E. , & Royo, C. (2007). Dispersal of durum wheat [Triticum turgidum L. ssp. turgidum convar. durum (Desf.) MacKey] landraces across the Mediterranean basin assessed by AFLPs and microsatellites. Genetic Resources and Crop Evolution, 54(5), 1133–1144. 10.1007/s10722-006-9005-8 [DOI] [Google Scholar]
- Oerke, E. C. (2006). Crop losses to pests. Journal of Agricultural Science, 144(1), 31–43. 10.1017/S0021859605005708 [DOI] [Google Scholar]
- Oliveira, H. R. , Campana, M. G. , Jones, H. , Hunt, H. V. , Leigh, F. , Redhouse, D. I. , Lister, D. L. , & Jones, M. K. (2012). Tetraploid wheat landraces in the Mediterranean basin: Taxonomy, evolution and genetic diversity. PLoS One, 7(5), 1–13. 10.1371/journal.pone.0037063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey, G. K. , Kanwar, P. , Singh, A. , Steinhorst, L. , Pandey, A. , Yadav, A. K. , Tokas, I. , Sanyal, S. K. , Kim, B.‐G. , Lee, S.‐C. , Cheong, Y.‐H. , Kudla, J. , & Luan, S. (2015). Calcineurin B‐like protein‐interacting protein kinase CIPK21 regulates osmotic and salt stress responses in Arabidopsis. Plant Physiology, 169(1), 780–792. 10.1104/pp.15.00623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasam, R. K. , Sharma, R. , Malosetti, M. , Eeuwijk, F. , Haseneyer, G. , Kilian, B. , & Graner, A.. (2012). Genome wide association studies for agronomical traits in a worldwide spring barely collection. BMC Plant Biology, 12(16), 1–16. 10.1186/1471-2229-12-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poland, J. A. , Brown, P. J. , Sorrells, M. E. , & Jannink, J. L. (2012). Development of high‐density genetic maps for barley and wheat using a novel two‐enzyme genotyping‐by‐sequencing approach. PLoS One, 7(2), 1–8. 10.1371/journal.pone.0032253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafalski, A. , & Morgante, M. (2004). Corn and humans: Recombination and linkage disequilibrium in two genomes of similar size. TRENDS in Genetics, 20(2), 103–111. 10.1016/j.tig.2003.12.002 [DOI] [PubMed] [Google Scholar]
- Reich, D. E. , Cargill, M. , Bolk, S. , Ireland, J. , Sabeti, P. C. , Richter, D. J. , Lavery, T. , Kouyoumjian, R. , Farhadian, S. F. , Ward, R. , & Lander, E. S. (2001). Linkage disequilibrium in the human genome. Nature, 411(1), 199–204. 10.1038/35075590 [DOI] [PubMed] [Google Scholar]
- Roelfs, A. P. , Singh, R. P. , & Saari, E. E. (1992). Hettel G. P. Rust Diseases of Wheat: Concepts and Methods of Disease Management, 1, (81). Mexico: CIMMYT. [Google Scholar]
- Rostoks, N. , Ramsay, L. , MacKenzie, K. , Cardle, L. , Bhat, P. R. , Roose, M. L. , Svensson, J. T. , Stein, N. , Varshney, R. K. , Marshall, D. F. , Graner, A. , Close, T. J. , & Waugh, R. (2006). Recent history of artificial outcrossing facilitates whole‐genome association mapping in elite inbred crop varieties. Proceedings of the National Academy of Sciences, 103(49), 18656–18661. 10.1073/pnas.0606133103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouse, M. N. , Talbert, L. E. , Singh, D. , & Sherman, J. D. (2014). Complementary epistasis involving Sr12 explains adult plant resistance to stem rust in Thatcher wheat (Triticum aestivum L.). Theoretical and Applied Genetics, 127(7), 1549–1559. 10.1007/s00122-014-2319-6 [DOI] [PubMed] [Google Scholar]
- Schafer, J. , Roelfs, A. , & Bushnell, W. (1984). Contributions of early scientists to knowledge of cereal rusts. The Cereal Rusts, 2(1), 3–38. 10.1086/329514 [DOI] [Google Scholar]
- Singh, A. , Pandey, M. P. , Singh, A. K. , Knox, R. E. , Ammar, K. , Clarke, J. M. , Clarke, F. R. , Singh, R. P. , Pozniak, C. J. , DePauw, R. M. , McCallum, B. D. , Cuthbert, R. D. , Randhawa, H. S. , & Fetch, T. G. (2013). Identification and mapping of leaf, stem and stripe rust resistance quantitative trait loci and their interactions in durum wheat. Molecular Breeding, 31(2), 405–418. 10.1007/s11032-012-9798-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, R. K. , & Chaudhary, B. D. (1985). Biometrical methods in quantitative analysis, 1, (304). Ludhiana, India: Kalyani. [Google Scholar]
- Singh, R. P. , Hodson, D. P. , Huerta‐Espino, J. , Jin, Y. , Bhavani, S. , Njau, P. , Herrera‐Foessel, S. , Singh, P. K. , Singh, S. , & Govindan, V. (2011). The emergence of Ug99 races of the stem rust fungus is a threat to world wheat production. Annual Review of Phytopathology, 49(1), 465–481. 10.1146/annurev-phyto-072910-095423 [DOI] [PubMed] [Google Scholar]
- Singh, R. P. , Hodson, D. P. , Huerta‐Espino, J. , Jin, Y. , Njau, P. , Wanyera, R. , Herrera‐Foessel, S. A. , & Ward, R. W. (2008). Will stem rust destroy the world's wheat crop? Advances in Agronomy, 98(1), 271–309. 10.1016/S0065-2113(08)00205-8 [DOI] [Google Scholar]
- Singh, R. P. , Hodson, D. P. , Jin, Y. , Huerta‐Espino, J. , Kinyua, M. G. , Wanyera, R. , Njau, P. , & Ward, R. W. (2006). Current status, likely migration and strategies to mitigate the threat to wheat production from race Ug99 (TTKS) of stem rust pathogen. CAB Reviews: Perspectives in Agriculture, Veterinary Science, Nutrition and Natural Resources, 1(54), 1–13. 10.1079/PAVSNNR20061054 [DOI] [Google Scholar]
- Singh, R. P. , Hodson, D. P. , Jin, Y. , Lagudah, E. S. , Ayliffe, M. A. , Bhavani, S. , Rouse, M. N. , Pretorius, Z. A. , Szabo, L. J. , Huerta‐Espino, J. , Basnet, B. R. , Lan, C. , & Hovmøller, M. S. (2015). Emergence and spread of new races of wheat stem rust fungus: Continued threat to food security and prospects of genetic control. Phytopathology, 105(7), 872–884. 10.1094/PHYTO-01-15-0030-FI [DOI] [PubMed] [Google Scholar]
- Stakman, E. C. , Stewart, D. , & Loegering, W. (1962). Identification of physiologic races of Puccinia graminis var. tritici, 1(54). Washington, USA: USDA. [Google Scholar]
- Stansfield, W. (1991). Molecular Genetics. Theory and Problems of Genetics, 3rd ed., Schaum's Outline Series, McGraw‐Hill, 1(1), 356. [Google Scholar]
- Team, R. (2015). RStudio: integrated development for R. RStudio, Inc., Boston, MA, 42(1), 1–14. http://www.rstudio.com [Google Scholar]
- Thornsberry, J. M. , Goodman, M. M. , Doebley, J. , Kresovich, S. , Nielsen, D. , & Buckler, E. S. (2001). Dwarf8 polymorphisms associate with variation in flowering time. Nature Genetics, 28(3), 286–289. 10.1038/90135 [DOI] [PubMed] [Google Scholar]
- Tsilo, T. J. , Chao, S. , Jin, Y. , & Anderson, J. A. (2009). Identification and validation of SSR markers linked to the stem rust resistance gene Sr6 on the short arm of chromosome 2D in wheat. Theoretical and Applied Genetics, 118(3), 515–524. 10.1007/s00122-008-0917-x [DOI] [PubMed] [Google Scholar]
- Wang, S. , Wong, D. , Forrest, K. , Allen, A. , Chao, S. , Huang, B. E. , Maccaferri, M. , Salvi, S. , Milner, S. G. , & Cattivelli, L. (2014). Characterization of polyploid wheat genomic diversity using a high‐density 90 000 single nucleotide polymorphism array. Plant Biotechnology Journal, 12(6), 787–796. 10.1111/pbi.12183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L.‐X. , Lorenz, A. , Rutkoski, J. , Singh, R. P. , Bhavani, S. , Huerta‐Espino, J. , & Sorrells, M. E. (2011). Association mapping and gene–gene interaction for stem rust resistance in CIMMYT spring wheat germplasm. Theoretical and Applied Genetics, 123(8), 1257–1268. 10.1007/s00122-011-1664-y [DOI] [PubMed] [Google Scholar]
- Yu, L.‐X. , Morgounov, A. , Wanyera, R. , Keser, M. , Singh, S. K. , & Sorrells, M. (2012). Identification of Ug99 stem rust resistance loci in winter wheat germplasm using genome‐wide association analysis. Theoretical and Applied Genetics, 125(4), 749–758. 10.1007/s00122-012-1867-x [DOI] [PubMed] [Google Scholar]
- Zadoks, J. C. , Chang, T. T. , & Konzak, C. F. (1974). A decimal code for the growth stages of cereals. Weed Research, 14(6), 415–421. 10.1111/j.1365-3180.1974.tb01084.x [DOI] [Google Scholar]
- Zhang, D. , Bai, G. , Zhu, C. , Yu, J. , & Carver, B. F. (2010). Genetic diversity, population structure, and linkage disequilibrium in US elite winter wheat. The Plant Genome, 3(2), 117–127. 10.3835/plantgenome2010.03.0004 [DOI] [Google Scholar]
- Zhu, J.‐K. (2016). Abiotic stress signaling and responses in plants. Cell, 167(2), 313–324. 10.1016/j.cell.2016.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]