

Abstract
Leucine-rich repeat-containing proteins (LRR proteins) are involved in supporting a large number of cellular functions. In this review, we summarize recent advancements in understanding functions of the LRR proteins as signaling scaffolds. In particular, we explore what we have learned about the mechanisms of action of the LRR scaffolds Shoc2 and Erbin and their roles in normal development and disease. We discuss Shoc2 and Erbin in the context of their multiple known interacting partners in various cellular processes and summarize often unexpected functions of these proteins through analysis of their roles in human pathologies. We also review these LRR scaffold proteins as promising therapeutic targets and biomarkers with potential application across various pathologies.
Keywords: cancer, congenital disease, Erbin, Leucine-rich repeats, scaffold proteins, Shoc2, signaling
Introduction
Nonenzymatic signaling scaffolds
Studies over the past two decades have revealed that proteins that act as signaling scaffolds are indispensable for the spatial and temporal regulation of intracellular signaling [1,2]. Scaffolding properties are often attributed to nonenzymatic, structural components within the signaling network that tether core signaling enzymes in order to optimize signal transduction. These proteins serve as molecular platforms that enhance control over the signaling processes and organize signaling activity at a desired site of action [2]. Nonenzymatic signaling scaffolds are a structurally diverse group of proteins. Finding a unifying pattern in molecular mechanisms by which these protein scaffolds exert their control over signal propagation is challenging and often relies on a broad range of methodologies. Yet, identifying common features within a large diverse class of proteins will improve our overall understanding of molecular mechanisms orchestrating signal transmission. In this review, we focus on two signaling scaffolds that use tandem leucine-rich repeats (LRRs) to build extended surfaces that are suitable for protein recognition and assembly of signaling modules.
Leucine-rich repeat proteins
Leucine-rich repeats are apparently simple structural units that are present in over 14 000 proteins and are found in various organisms (e.g., plants, invertebrates, viruses, bacteria, archaea, and eukaryotes). Most LRRs are conserved stretches of 20 to 30 amino acids rich in hydrophobic leucine residues [3]. Individual LRR units are generally divided into highly conserved and variable segments with conserved segments usually containing an 11 (LxxLxLxxNxL) or a 12 (LxxLxLxxCxxL) amino acid sequence where L is Leu, Ile, Val, or Phe; N is Asn, Thr, Ser, or Cys; and C is Cys, Ser, or Asn [4]. Multiple LRRs are often found in tandems of 2 to 30 repeats and form helically twisted solenoid-like curved structures creating convex and concave surfaces [5]. The concave side of the LRR solenoid is lined with a parallel β-sheet in which each LRR contributes one strand. Each strand then is interconnected by various secondary structures that form the convex surface. Structural studies have determined that the hydrophobic core of the first and the last LRR of this solenoid is protected by a flanking N-terminal (LRRNT or N-cap) and C-terminal (LRRCT or C-cap) capping motifs [6]. Several studies that undertook classification of the large repertoire of LRR-containing proteins divided them into eight classes based on different lengths and the consensus sequence of the variable LRR segments [7]. The structural properties of LRR domains are extensively reviewed by Bella et al. [3].
Leucine-rich repeat-containing proteins are remarkably diverse functionally. They are one of the most commonly occurring units in proteins associated with innate and adaptive immunity [7–9]. Beyond immune response, LRR-containing proteins are involved in diverse cellular functions including ubiquitin-related cellular processes, apoptosis, autophagy, nuclear mRNA transport, and neuronal development. To date, more than 60 human diseases have been associated with mutations in LRR-containing proteins [10]. Examples include Crohn’s disease (mutation in NOD2 and TLR4) [11,12], rheumatoid arthritis (CIITA) [13], Legionnaire disease (TLR5) [14], and a number of cancers (LRRC28) [15]. Importantly, despite the large functional diversity, most LRR-containing proteins are involved in protein–ligand and protein–protein interactions thus allowing remarkable scaffolding opportunities. In the present article, we discuss recent advances in our understanding of the two LRR-containing signaling scaffolds: Erbin and Shoc2 and outline the molecular underpinnings connecting these scaffolds.
SHOC2 (SUR-8/SOC2, suppressor of clear): domain organization and its partners
Shoc2 is a case of a signaling scaffold that is almost entirely built of LRRs. All known orthologues preserve a striking sequence conservation throughout evolution and are comprised of two major domains: a short, unstructured N-terminal domain (length from 56 to 145 amino acids in different taxa) that is followed by a long stretch of LRRs [16,17]. Shoc2 lacks apparent intrinsic enzymatic activity, but is capable of partnering with a continuously growing number of proteins with various enzymatic activities (Fig. 1). Both the N terminus and the LRR domain of Shoc2 are involved in protein binding. The N-terminal domain of Shoc2 interacts with several isoforms of canonical RAS GTPases [17–19]. The repertoire of Shoc2 partners recognizing the LRR domain is larger. Shoc2’s LRRs were shown to interact with the catalytic subunits of the protein phosphatase 1 (PP1c) as well as with quite a few proteins of the ubiquitin machinery. Binding to the LRRs are the E3 ligases HUWE1 (also known as LASU1, UREB1, and Mule), FBXW7 (also known as FBX30, SEL-10, and hCdc4), two AAA + ATPase (the ATPase associated with diverse cellular activities) known as PSMC5 (rpt6 or Sug1), and the AAA + ATPase VCP (also known as Cdc48 or p97) (Fig. 1). Other partners of Shoc2 that bind the scaffold indirectly or with yet undetermined binding properties are the serine/threonine-protein kinase RAF-1, the protein phosphatase PP1a (PP1-87B in drosophila), the p110alpha subunit of phosphatidylinositol 3-kinase (PI3K), the MTOR binding protein Raptor, and a transcriptional repressor and pacemaker protein PERIOD (PER). Whether all these partners interact with Shoc2 simultaneously or even in the same cell type is not clear. Data regarding the nature of the interactions within the Shoc2 macromolecular complex, post-translational modification, and specific examples of how the Shoc2 scaffold regulates cellular signaling are discussed in the following sections.
Fig. 1.
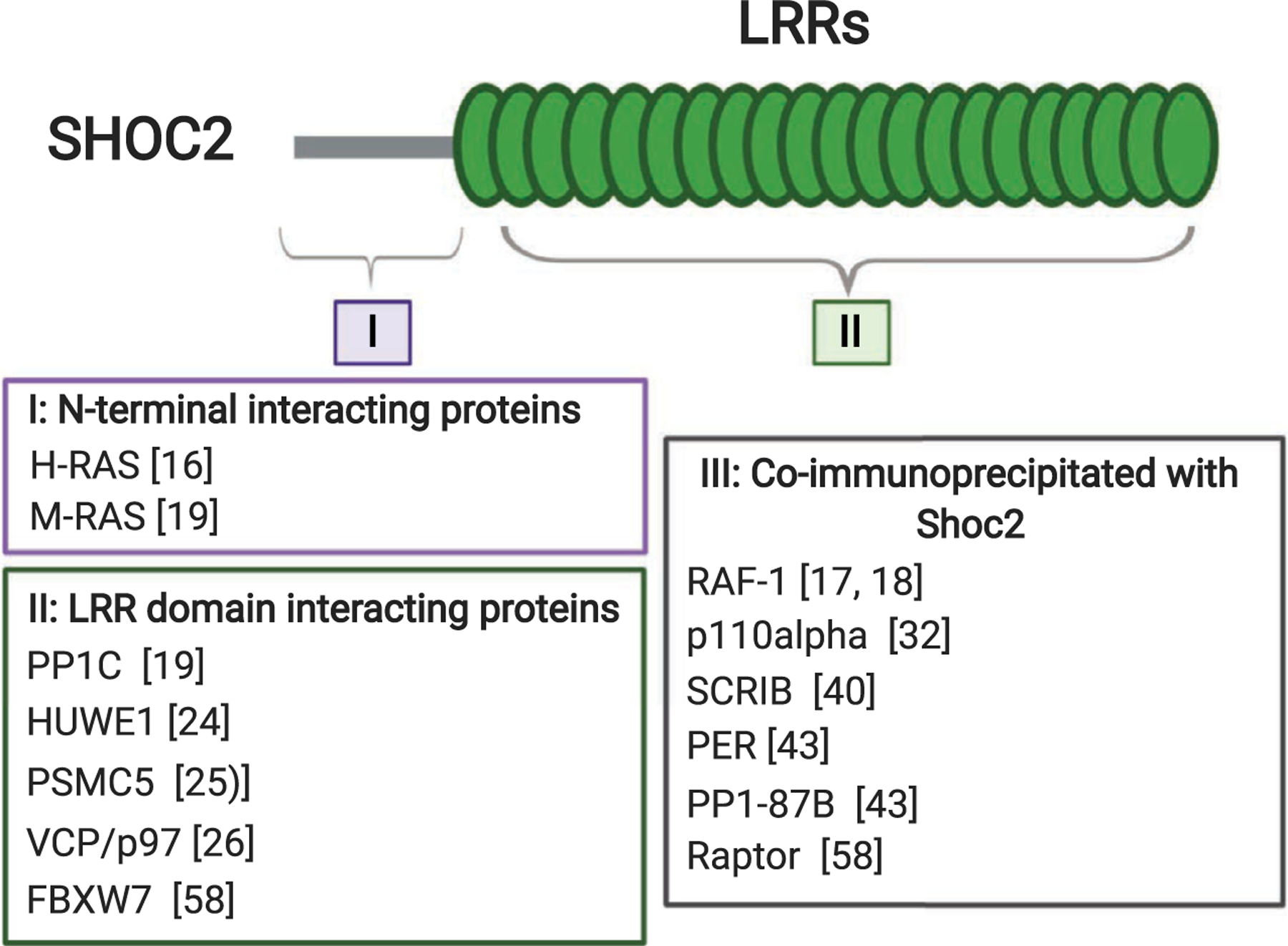
Known Shoc2-interacting proteins, mapped to the N- or C-terminal region of Shoc2. Direct interaction of proteins in I and II with individual domains of Shoc2 was measured by yeast two-hybrid assay, binding assays using recombinantly expressed and purified GST-fusion proteins, or another biochemical strategy. Shoc2 LRR domain contains binding regions for PP1c (LRR4), the E3 ligases HUWE1 (LRR12-14), FBXW7 (LRR18), PSMC5 (LRRC), and VCP/p97 (LRR12-14). Proteins listed in III as ‘coimmunoprecipitated with Shoc2’ have not been shown to bind Shoc2 directly.
Thus far, the best-studied role of Shoc2 is in modulating signals of the extracellular signal-regulated kinase 1/2 (ERK1/2) pathway. Shoc2 tethers RAS and RAF-1 proteins to a close proximity and thus accelerates transmission of signals through the pathway [20,21]. Restricted to its interaction with M-RAS, Shoc2 was reported to form a holoenzyme with the catalytic subunit of protein phosphatase 1 (PP1c) [19]. PP1c, which is known to cooperate with various regulatory proteins, then targets RAF-1 stimulating its kinase activity through dephosphorylation of the RAF-1S259 inhibitory site. The M-RAS/Shoc2/PP1c complex-activated RAF-1 then may be recruited by other RAS proteins or RAS family GTPases [19]. Dephosphorylation of S259 by the M-RAS/Shoc2/PP1c holoenzyme is shown to be critical for RAF heterodimerization—an event essential for the activation of RAF kinase [22,23]. Whether corresponding Shoc2-phosphatase complexes are formed with other RAS homologues (H-RAS, K-RAS, and N-RAS) remains an open question, and future studies are needed to reach a consensus in this regard.
Several of the Shoc2 partners recognizing its LRR domain are involved in fine-tuning of the signals transmitted via the Shoc2 module. We showed that the enzymatic machinery consisting of the HECT-domain E3 ubiquitin ligase HUWE1 and the AAA + ATPases VCP/p97 and PSMC5 allows for a highly coordinated feedback mechanism. In this mechanism, the amplitude of Shoc2-mediated ERK1/2 signals is modified by inducing post-translational modifications [24–26]. Shoc2 ubiquitination mediated by HUWE1 is triggered by growth factor activation of the ERK1/2 pathway and is a prerequisite for the subsequent ubiquitination of the RAF-1 kinase associated with Shoc2 [24]. Current data suggest that these ubiquitin modifications serve as a negative feedback that reduces the amplitude of RAF-ERK1/2 signals. The diversity of ubiquitin chains ligated to Shoc2 by HUWE1 [K63, K48, K6, K11 ([24], unpublished data)] suggests that ubiquitin modifications may play multiple roles in controlling Shoc2 function.
Shoc2 partners and well-known ‘remodelers’, the AAA + ATPases PSMC5 and VCP/p97, make the HUWE1-modulated ubiquitination of Shoc2/RAF highly coordinated and precise. Analogous to its non-proteolytic function as an unfoldase/remodeler of AAA proteins independent of 20S (APIS) complexes [27,28], in the Shoc2 assembly, PSMC5 does not modulate stability of Shoc2 or its known partners. Interestingly, similar to other AAA + ATPase remodelers, PSMC5 recognizes the extreme carboxy terminal residues within the last LRR of Shoc2. This unique stretch of ~ 20 amino acids is also essential for the targeting of Shoc2 to late endosomes [25,29]. However, the mechanistic connection between the remodeling and cellular distribution events is not clear and will require further studies. Nevertheless, PSMC5 was shown to facilitate recruitment of Shoc2 complexes to endosomes where ubiquitin links are recognized by yet another unfoldase: AAA + ATPase VCP/p97 [26]. Experiments using structural mutants of Shoc2 have demonstrated that PSMC5 binding and Shoc2 targeting to endosomes is essential for remodeling of the complex by VCP/p97 [26]. Thus, by incorporating these two mechanoenzymes, the Shoc2 complex acquires another layer of control over the amplitude of the ERK1/2 signals transmitted through the module. Whereas HUWE1-mediated ubiquitination of Shoc2 and RAF-1 fine-tunes the dynamic range of RAF-1 phosphorylation, VCP/p97 controls the levels of ubiquitination by modulating the assembly of molecules in the complex [24,25]. Although some aspects of this feedback mechanism can be explained by existing data, many questions remain. The lack of a clear understanding of how HUWE1 activity in the complex is initiated and the nature of structural changes occurring in the complex upon remodeling makes it difficult to fully appreciate protein dynamics within the module.
Recently, Xie et al. [30] reported crosstalk between Raptor and RAS/RAF signaling via a Shoc2-dependent mechanism that also involves post-translational modifications. In this Shoc2-Raptor axis, Raptor appears to inhibit signals transmission by Shoc2-RASERK1/2. Moreover, the authors show that activation of the ERK1/2 pathway by epidermal growth factor (EGF) results in Shoc2 phosphorylation on Thr-507 by MEK1. Phosphorylated Shoc2 is then ubiquitinated by the E3 ligase FBXW7 leading to Shoc2 degradation. The FBXW7 E3 ligase appears to be involved in balancing the ERK1/2 and MTOR signals for cell proliferation and autophagy. An additional instance of Shoc2 phosphorylation by PKC-alpha and PKC-delta (PKCα/δ) was reported by Lee et al. [31]. It appears that PKCα/δ phosphorylates Shoc2 at Thr-71 and Ser-297 to control the stability of Shoc2 upon FGF2 signaling. This study provides another mechanism for regulation of the stability of Shoc2. Interestingly, the same group reported that Shoc2 interacts with p110α subunit of PI3K and under pathological conditions activates the PI3K-AKT pathways [32]. The connection between these two studies is not clear. As the interplay between ubiquitination and phosphorylation events has become a recurrent theme in the regulation of cell signaling [33], it will not be surprising if future studies uncover an additional crosstalk between the two modifications in terms of mechanisms regulating RAS-Shoc2-ERK1/2 signal propagation.
The role of Shoc2 in normal physiology
Given the critical implications of ERK1/2 signals for a number of cellular processes, several studies have explored the biological significance of Shoc2 in embryonic development. Model organisms, such as Mus musculus (mice) and Danio rerio (zebrafish), have been used to delineate the role of the Shoc2-RAS-ERK1/2 pathway in development. Shoc2 is widely expressed in all tissues. Ablation of Shoc2 in zebrafish results in marked defects in development of facial cartilage, bone, and pigment cells as well as a profound loss of circulating blood cells [34]. Conventional SHOC2−/− (sur-8 in mice) knockout mice died due to an early-stage embryonic lethality and partial absorption of mutant embryos at E8.5 [35]. The conditional disruption of the mouse SHOC2 gene in endothelial cells led to multiple cardiac defects, smaller body size, subcutaneous edema in their dorsal body, fetal lung congestion, and nonsurvival past E14.5. These SHOC2-deficient mice had abnormalities in the transposition of the great arteries, as well as a number of defects in heart morphogenesis [35].
Several studies have utilized cell models to examine the role of Shoc2 in cell proliferation, differentiation, and motility. Shoc2-ERK1/2 signals were shown to stimulate proliferation of neuronal progenitor cells (NPCs), as well as leukemia, pancreatic, and lung cancer cell lines [22,36–38]. These studies connected Shoc2 proliferative functions to the cellular mechanisms controlling the stability of Shoc2 protein. Xie et al. [30] implicated Shoc2 in promoting autophagy via inactivation of the mTORC1 pathway. Several studies also addressed the function of Shoc2 in regulating cell motility [32,39–41]. Shoc2-ERK1/2 signals have been shown to control collective cell migration by modulating turnover of E-cadherin, phosphorylation of p120-catenin, and cell–cell adhesion [39]. Other studies demonstrated that Shoc2-ERK1/2 signals can also regulate cell attachment and motility by controlling expression of a number of proteins of extracellular matrix, including lectin galactoside-binding soluble 3-binding protein (LGALS3BP) [41,42]. This secreted glycoprotein forms oligomers in the extracellular milieu and promotes cell adhesion to matrix proteins.
Interestingly, elevated expression of Shoc2 was reported in human macrophages infected with several different pathogens [7]. Unfortunately, at the moment, we can only speculate in regard to the possible mechanism of this response. An unexpected role of Shoc2 in the regulation of circadian rhythms in Drosophila was recently identified by Xue and co-authors. They showed that Shoc2 is involved in regulation of PER stability through PP1-87B-mediated dephosphorylation [43]. Given that many of the Shoc2 partners are affected in various human pathologies, it is reasonable to expect that cellular signals mediated via the Shoc2 axis are possibly contributing to other pathological conditions. For example, abnormal Shoc2 ubiquitination is found in primary fibroblasts from patients with inclusion body myopathy with Paget’s disease of bone and frontotemporal dementia (IBMPFD) caused by congenital mutation in the Shoc2 partner, VCP/p97 [26]. Altogether, a growing body of evidence emphasizes the key role Shoc2-mediated signals have in maintaining normal physiological functions.
Shoc2 pathologies
Considering the plethora of processes in which Shoc2-ERK1/2 signals are involved potentially, it is not difficult to appreciate the pathological effects upon its dysregulation (Table 1). Germline mutations in the SHOC2 gene (c.4A>G, p.S2G, c.519G>A; p.M173I and c.807_808delinsTT, p.Gln269_His270delinsHisTyr mutation) cause a distinctive hereditary disorder termed Noonan-like syndrome with loose anagen hair (NSLAH) [44–46]. NSLAH belongs to the group of congenital syndromes that are caused by mutations in genes of the ERK1/2 pathway. Cumulatively, these syndromes with overlapping features are called RASopathies. Patients carrying Shoc2 mutations are reported to have an unusual combination of features including reduced growth associated with growth hormone deficiency, cognitive deficits, distinctive hyperactive behavior, and a unique hair anomaly (i.e., loose anagen hair). Over the past several years, characteristics such as variable neurocognitive impairments, brain anomalies, epilepsy, severe hydrops fetalis, Moyamoya syndrome, and even an autoimmune disorder were added to the distinctive craniofacial dysmorphisms and a wide spectrum of congenital heart defects [47–55]. This biological complexity of Shoc2 S2G patients was explored using transcriptome analysis of peripheral blood mononuclear cells [56]. A large transcriptional signature characterized for the Shoc2 S2G mutation further indicated a unique, Shoc2-specific signaling axis. Interestingly, the alterations in the expression of transcription factors (TFs) found in Shoc2 (S2G) patients had very little overlap with the TFs expression pattern affected by the depletion of Shoc2 in cells, emphasizing the selective effect point mutations may have on ERK1/2 activity [41]. Unfortunately, no studies to determine whether other Shoc2 NSLAH mutations affect ERK1/2 signals differently have been performed. Also, recognizing phenotypic variability in patients with NSLAH and apparent phenotypic overlap with patients carrying other mutations in the ERK1/2 pathway, it is likely that individuals with no molecularly confirmed mutations carry different Shoc2 substitutions [57]. Yet, current genetic panels that test for 25 known RASopathy genes only assess the Shoc2 S2G substitution thus preclude us from fully appreciating the frequency of Shoc2 mutations in RASopathy patients.
Table 1.
Cancer subtypes and other clinical pathologies associated with Shoc2 and Erbin.
| Protein | Associated Pathologies | Key reference |
|---|---|---|
| SHOC2 | Noonan-like syndrome with loose anagen hair | [44] |
| Breast cancer | [60] | |
| Colorectal cancer | [31] | |
| Non-small-cell lung cancer | [22] | |
| Pancreatic cancer | [37] | |
| ERBIN | Atopy | [101] |
| Cardiac hypertrophy | [79] | |
| Diabetic peripheral neuropathy | [80] | |
| Inflammatory bowel disease (Crohn’s disease) | [105] | |
| Renal interstitial fibrosis | [104] | |
| Striate palmoplantar keratoderma | [86] | |
| Acute myeloid leukemia | [119] | |
| Breast cancer | [126] | |
| Cervical cancer | [110] | |
| Colorectal cancer | [95] | |
| Head and neck squamous cell cancer | [115] | |
| Hepatocellular carcinoma | [123] |
Shoc2 deregulation is not limited to developmental pathologies, and links have been shown between Shoc2 and cancer. Analysis of publicly available data from the TCGA, COSMIC, and cBioPortal highlights the variability of Shoc2 expression and the presence of Shoc2 mutations or other genomic alterations across different cancer subtypes [58]. Shoc2 is shown to increase ERK1/2 signals in various types of malignant cells (e.g., pancreatic, colon, breast and non-small-cell lung carcinoma, melanoma, neuroblastoma, fibrosarcoma, leukemia, and hepatoma cells) including cells that carry tumorigenic K-Ras, N-Ras, and B-RAF mutations [19,32,41,42]. Importantly, Shoc2 contributes to the acquired drug resistance of cancer cells with K-Ras, N-Ras, and B-RAF oncogenic mutations [23,37,59], possibly by altering signaling connections and re-routing oncogenic signals to other RAF-1 isoforms [59]. Other mechanisms suggested to contribute to Shoc2-mediated RAF and MEK resistance include regulation of contact inhibition, anchorage-independent proliferation and orientation of the microtubule-organizing center of these cells, expression of extracellular matrix proteins [40], and activation of Rac and matrix metalloproteinases (MMPs) through the PI3K pathway [32]. Several recent studies have indicated that Shoc2 might have prognostic value for patients with breast, thyroid, and pancreatic cancers [58,60,61], but further studies are needed to confirm its suitability as a biomarker. With its diverse function in disease progression and prognosis, Shoc2 presents an exciting possibility as a therapeutic target. A recent study by Sulahian et al. in which a search was made for novel targets to sensitize MEK inhibitors highlighted the therapeutic benefit of Shoc2 depletion [37]. Another group has identified Shoc2 as a target of a natural proteasome inhibitor with antitumor activity, Celastrol [62]. Yet, the Shoc2 scaffold remains largely unexplored as a drug target due to the challenges associated with deciphering the organizational complexity of nonenzymatic scaffolds. The lack of available crystal structures for Shoc2 impedes investigation of structural changes as ‘druggable’ targets in these proteins. Such studies investigating the dynamics within scaffold complexes will broaden our understanding of the molecular mechanisms supporting Shoc2’s role in signal transmission and will outline how conformational changes in the Shoc2 structure regulate its function.
Synergy with other LRR scaffolds
An unexpected feature of the Shoc2 scaffold is its cooperativity with other LRR scaffolds. For instance, Young et al. showed that the M-RAS and Shoc2 dimer competes for PP1c binding with Scribbled homolog (SCRIB) [40]. SCRIB, a member of the LAP (LRR And PDZ domain) proteins family, is a known regulator of the ERK1/2 pathway. The study by Young et al. showed that SCRIB antagonizes Shoc2-mediated RAF1 dephosphorylation by competing for PP1 within the same scaffolding complex. Competition of Shoc2 and SCRIB for PP1c binding allows for a multilayered mechanism controlling the frequency and amplitude of Shoc2-transduced ERK1/2 activity and as a consequence affects establishment of cell polarity and tumorigenic growth [40].
In addition, Shoc2 has been shown to compete with Erbin (also known as ERBB2IP), another member of the LAP protein family [63,64], for binding to the RAS/RAF complex. Both SCRIB and Erbin appear to provide an essential mechanism to control the signaling strength of Shoc2-ERK1/2 activation. A comprehensive review of SCRIB, the most studied LAP protein, has recently been published elsewhere [65]. Here, we focus on discussing the role of Erbin in regulating a number of signaling pathways, including its functional interplay with Shoc2 in fine-tuning RAS/RAF/ERK signaling (Fig. 2).
Fig. 2.
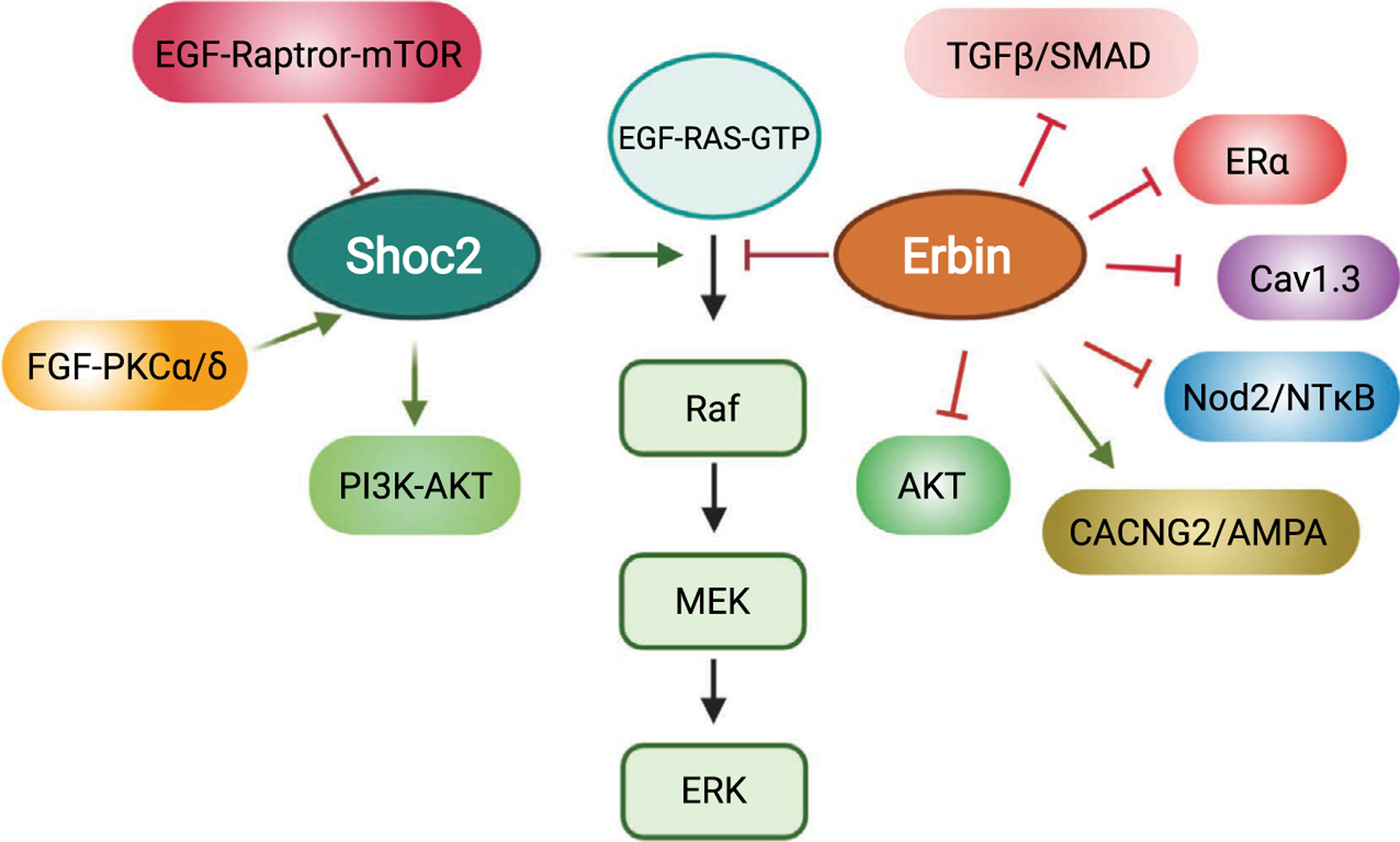
Simplified model for signaling pathways linked to the scaffold proteins Shoc2 and Erbin.
Erbin, a lap family protein that interacts with ERBB2
Building upon the protein–protein interaction platform provided by tandem repeats of LRR domains, members in the LAP protein family contain additional protein–protein interaction modules that expand their capacity for binding diverse groups of ligands. In addition to SCRIB and Erbin, other LAP proteins include vertebrate Densin-180 (LRRC7) and Lano (LRRC1),C. elegans LET-413 and Drosophila Scribble, and Lap1 (Fig. 3). The known LAP proteins contain 16 canonical LRRs located at their amino terminus followed by two conserved LRR-like domains (called LAPSD) and either one or four PDZ domains [66,67]. Despite containing no PDZ domains, Lano has also been classified as a LAP protein as it shares highly conserved LRRs and LAPSDs with other family members [67,68]. The LRR motifs found in Erbin share 38% identity with the LRRs of Shoc2 proteins [63]. However, unlike Shoc2, Erbin is found only in vertebrates. Similar to LRRs, PDZ domains are found in 200–300 proteins within the human genome. These domains are about 80–90 amino acids in length folding into a globular structure that is comprised of six β-strands and often capped by two α-helices [69]. Functionally, PDZ domains mediate protein–protein interactions by binding to specific peptide sequences located at the C-terminal tail of other proteins [70]. Together, LRR and PDZ domains allow Erbin to interact with multiple proteins, possibly simultaneously. Figure 4 summarizes the known binding proteins of Erbin to date.
Fig. 3.
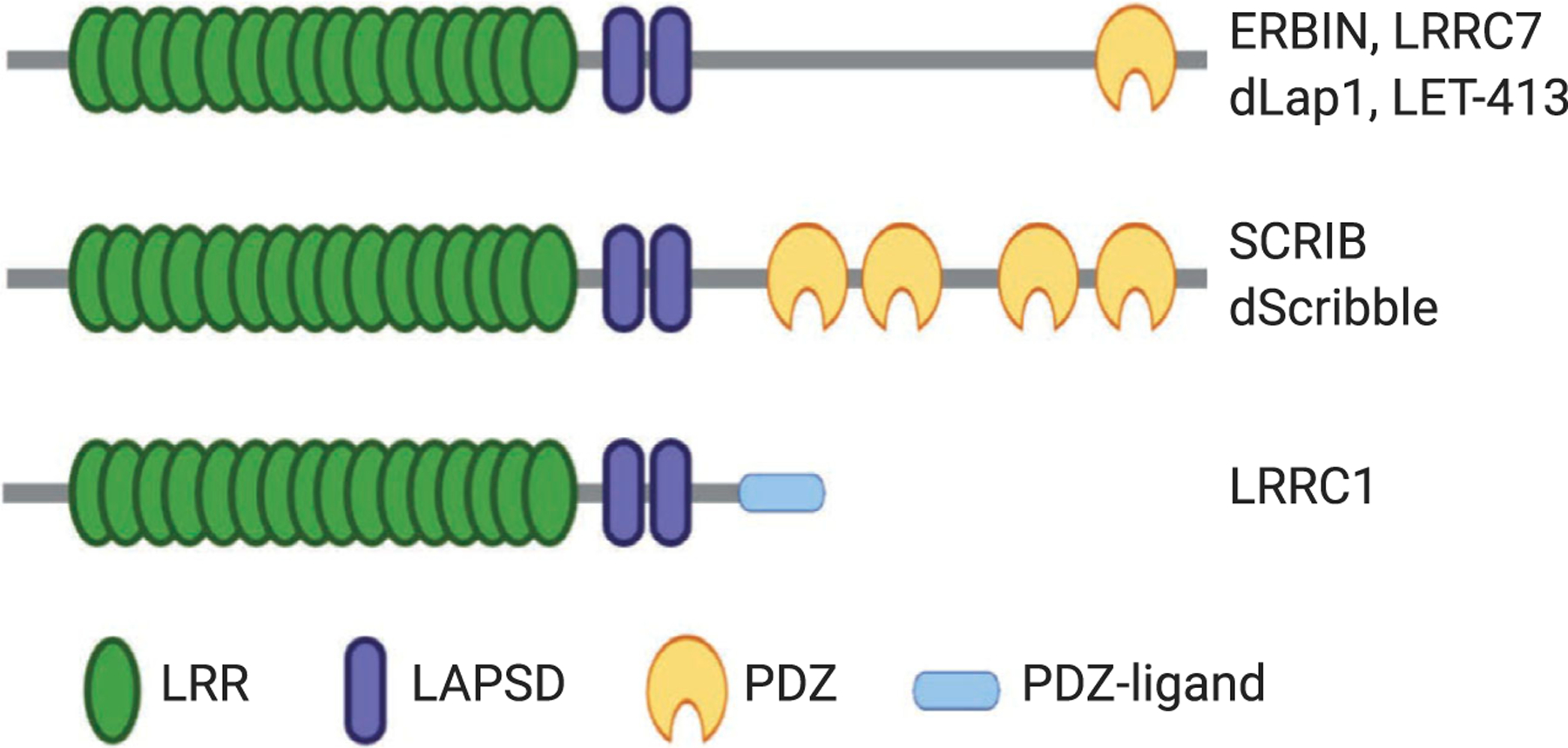
The schematic diagram of the domain composition of LAP family proteins. The LAP family contains vertebrate Erbin, LRRC7, SCRIB, and LRRC1 together with Drosophila Scribble, Lap1, and C. elegans LET-413. All known LAP proteins contain N-terminal LRR domains followed by two conserved LAPSD domains. With the exception of LRRC1, other LAP proteins contain either one or four PDZ domains. The C terminus of LRRC1 contains a PDZ ligand sequence.
Fig. 4.
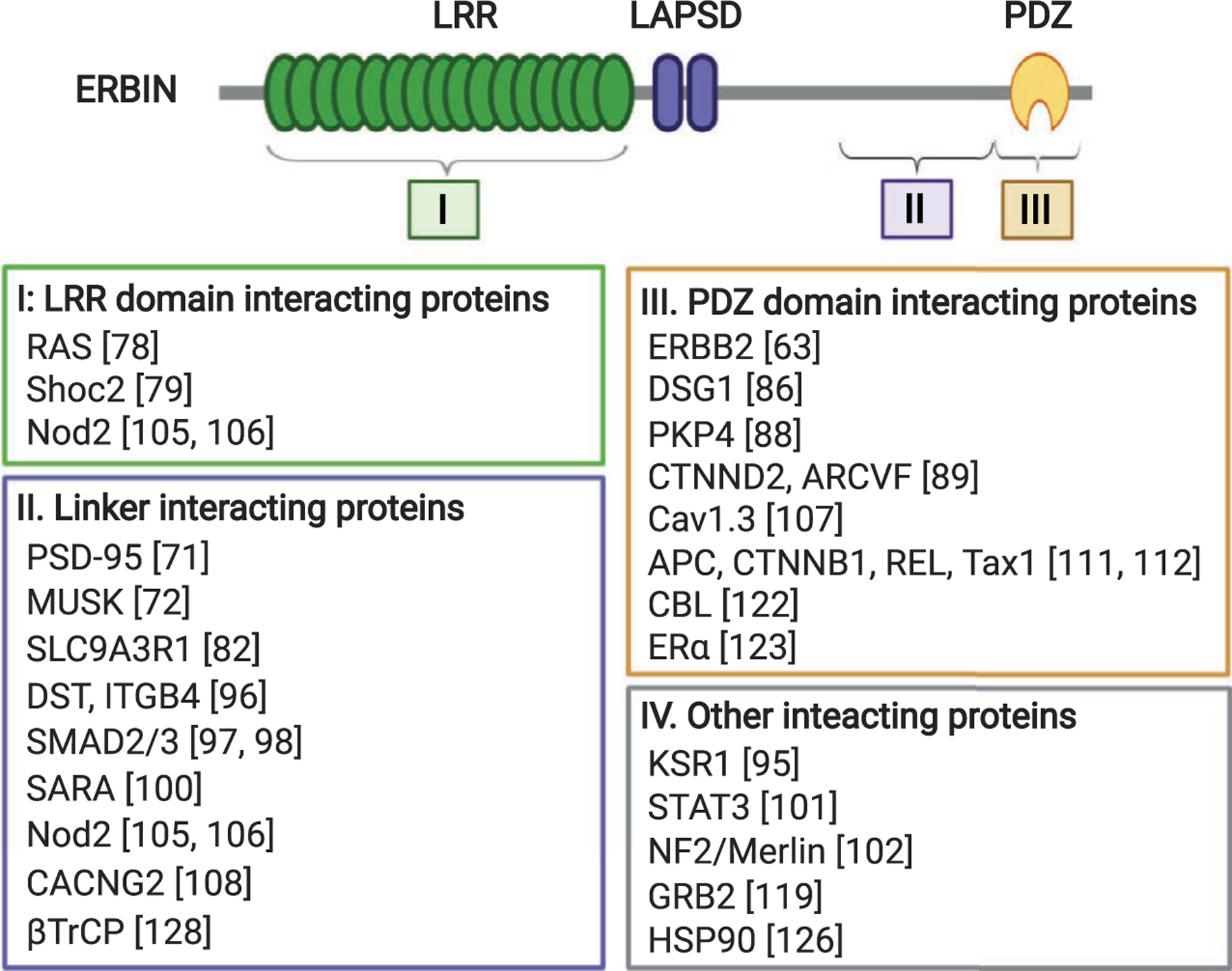
Known Erbin-interacting proteins mapped to the LRR, PDZ, or linker region of Erbin. Erbin-interacting proteins identified to date are subdivided into four groups based on where they bind in Erbin. Proteins listed in group IV were found to interact with Erbin by co-immunoprecipitation experiments but no specific binding sites were identified. Note that Nod2 was found to interact with both the LRR domain and the linker region of Erbin. The interaction site for DSG1 also includes a portion of the linker region upstream of the PDZ domain.
The human Erbin gene is located on the long arm of chromosome 5 and comprised of 26 exons [63]. Splicing variants of Erbin can lead to alterations in the linker region between LRR and PDZ domains. The expression of Erbin mRNA and protein is detected in most human and mouse tissues, including brain, liver, kidney, spleen, intestine, and skeletal muscle [63,71]. Erbin was first identified by Jean-Paul Borg and colleagues in 2000 in the search for ERBB2-interacting proteins that are involved in organizing signaling downstream of the receptor tyrosine kinase [63]. Erbin, specifically its PDZ domain, was found to interact with the C-terminal tail of the ERBB2 receptor, but interestingly not with any other ERBB family members. Conversely, only the PDZ domain of Erbin was able to interact with ERBB2 despite up to 70% sequence identity among all PDZ domains within the LAP family [63]. This binding specificity was confirmed in a similar study in which Erbin was found to interact with ERBB2 and PSD-95 at the postsynaptic membranes [71]. In addition, Erbin concurrently interacts with ERBB2 and another receptor tyrosine kinase, MUSK, at neuromuscular junctions [72].
Although Erbin contains a class I PDZ domain, it preferentially interacts with the C terminus of ERBB2, a class II type PDZ ligand. To better understand the binding specificity between Erbin and ERBB2, several studies have examined the structure of the Erbin PDZ domain bound to the C-terminal peptide of ERBB2. Intriguingly, Erbin PDZ domain has an unusually long β2-β3 loop which partially accounts for its unique binding specificity for PDZ ligands [73,74]. A structural comparison analysis of the Erbin PDZ domain with the first PDZ domain of ZO-1 has provided additional evidence supporting the selectivity of Erbin PDZ [75]. Interestingly, the phosphorylation of ERBB2 at a tyrosine residue at the −7 position of ERBB2 C-terminal peptide (EYLGLDVPV) reduces its interaction with Erbin [63]. The crystal structure of Erbin PDZ and ERBB2 peptide ligand complex revealed that this tyrosine residue fits into the binding pocket formed by the β2-β3 loop of Erbin PDZ; and phosphorylation abolishes its interaction with the pocket without affecting the binding of the last four C-terminal residues of ERBB2 with Erbin PDZ [73]. Since ERBB2 is known to be phosphorylated at this tyrosine residue during activation, the preference of Erbin for binding unphosphorylated ERBB2 potentially provides a regulatory mechanism to control cellular signaling downstream of ERBB2 [63,73].
Following the initial biochemical characterization of the interaction between Erbin and ERBB2, the function of Erbin-mediated regulation of ERBB2 has been investigated using a global Erbin knockout mouse model developed by the Borg group. While the whole-body knockout of SCRIB in mice results in perinatal lethality due to a neural tube closure defect, mice that carry homozygous deletion of both Erbin alleles (Erbin−/− mice) are viable with no apparent developmental defects. Upon closer examinations, Erbin null mice were found to have decreased myelination and aberrant ensheathment of axons in sciatic nerves, which results in decreased nerve conduction velocity [76]. Neuregulin 1 (NRG1) is known to play an important role in myelination by signaling through ERBB family tyrosine kinase receptors. Erbin loss reduces ERBB2 and ERBB3 expression in sciatic nerves and suppresses NRG1 signaling. Interestingly, this Erbin-dependent regulation of myelination and NRG1 signaling relies on its interaction with ERBB2 as mutant ErbinΔC/ΔC mice (the Erbin ΔC allele contains amino acids 1–693 of Erbin fused with β-gal) showed similar phenotypes as Erbin null mice [76]. Moreover, the expression of Erbin has been shown to promote remyelination of regenerating neurons after injury [77].
Erbin-dependent negative regulation of RAS/RAF signaling
The role of Erbin on regulating RAS/RAF signaling was first described in 2003 in a study that discovered the interaction between Erbin and the active form of RAS. The expression of Erbin disrupts the RAS-RAF interaction and inhibits downstream ERK signaling and NGF-induced neuronal differentiation of PC12 cells [78]. Mechanistically, Erbin inhibits the activation of RAS/RAF signaling by directly interacting with Shoc2 via its LRR domain and displacing Shoc2 from the RAS/RAF complex [64]. It has been shown that knockout of Erbin in mice exacerbated isoproterenol-induced cardiac hypertrophy and pressure overload-induced heart failure. An examination of heart tissues from Erbin−/− mice revealed that ERK phosphorylation was increased both basally and upon isoproterenol treatment. The interaction between RAF-1 and Shoc2 was markedly enhanced in Erbin−/− mice, suggesting that Erbin sequesters Shoc2 away from RAF-1 to inhibit RAF-1 phosphorylation. In addition, decreased Erbin expression was observed in mouse models of cardiac hypertrophy and in biopsies of human failing hearts [79]. Similarly, decreased expression of Erbin and corresponding activation of ERK were found in mouse models of diabetic peripheral neuropathy [80]. Furthermore, the expression of Erbin mRNA was shown to be regulated by microphthalmia-associated transcription factor (MITF) and FHL2 under both physiological and pathological conditions in heart [81].
Studies exploring the functional importance of Erbin in modulating RAS/RAF signaling in Schwann cells have shown that silencing Erbin expression disrupts cell–cell contact as a result of increased ERK phosphorylation and cell proliferation [82]. Treating cells with MEK inhibitor reverses the phenotypes induced by decreased Erbin expression. In addition, Erbin forms a complex with NF2 (the protein product encoded by NF2 is often called merlin) and EBP50 (gene name SLC9A3R1) via directly binding to EBP50 in Schwann cells. Given that the phenotypes observed in Erbin knockdown cells are similar as those induced by NF2 loss, it has been suggested that Erbin may regulate NF2 function by targeting NF2 to the adherens junction [82]. Since membrane-localized NF2 inhibits RAS activation [83], it is tempting to speculate that Erbin expression at the adherens junction is required for NF2 to function as a tumor suppressor.
The involvement of Erbin in RASopathies has been demonstrated in studies of desmoglein 1 (DSG1) and a rare autosomal dominant disorder, a striate palmoplantar keratoderma (SPPK), characterized by a thickening of the skin on the palms and soles [84]. SPPK is a RASopathy-like disease where keratinocytes fail to differentiate correctly due to dysregulations in the ERK1/2 pathway. DSG1 is required for maintenances of epidermal tissue integrity by forming cell–cell contact via its extracellular domains. In addition, DSG1 promotes keratinocyte differentiation by suppressing EGFR/ERK1/2 signaling [85]. Loss of DSG1 as a result of mutations in the DSG1 gene (including premature termination codons and frameshift mutations) leads to the development of SPPK. In searching for mechanisms underlying DSG1-mediated inhibition of ERK, Erbin was identified as an interacting protein of DSG1 through a yeast-2-hybrid screen [86]. Colocalization of Erbin and DSG1 was observed at the plasma membrane of cultured epidermal keratinocytes and human skin biopsies. Silencing Erbin in keratinocytes disrupted cell differentiation which recapitulates the phenotypes seen in SPPK patients carrying DSG1 mutations. Co-immunoprecipitation studies showed that Erbin binds Shoc2 and prevents Shoc2-facilitated formation of the RAS/RAF complex. Thus, by providing an anchoring site for Erbin, DSG1 functions to suppress ERK signaling via an Erbin-dependent mechanism [86].
Regulation of membrane localization of Erbin
Multiple studies have demonstrated that Erbin is a plasma membrane-associated protein. The PDZ domain of Erbin recognizes the plasma membrane protein ERBB2 as well as the several members of the p120-catenin/plakophilin subfamily of Armadillo-like proteins, including PKP4, δ-catenin (CTNND2), and ARVCF [87–90], at the adherens junction. Interestingly, the PDZ domain of Erbin is not required for its membrane targeting. Instead, the LRR domain of Erbin has been found as a critical determinant for the membrane localization as mutating a conserved proline residue in the LRR domain to leucine (P315L) results in cytoplasmic expression of Erbin [91,92]. Similarly, LRR domains of other LAP proteins, such as SCRIB, Lano, and LET-413, but not the LRR domain of Shoc2, are required for targeting LAP proteins to the basolateral membrane of epithelial cells [91,92]. In addition, it has been shown that Erbin can be palmitoylated at two Cys residues (Cys14 and Cys16) at its N terminus and the palmitoylation is necessary for its plasma membrane localization [93]. However, since Erbin carrying the LRR domain mutation (P315L) is deficient in palmitoylation, it is likely that both the LRR domain and subsequent palmitoylation are required to maintain a stable membrane pool of Erbin [93]. Moreover, colocalization of Erbin with the adherens junction has been confirmed by a quantitative proteomics study in which Erbin was found to be one of the most abundant proteins associated with E-cadherin containing cell–cell junctions in epithelial cells [94].
It has been well documented that SCRIB plays an evolutionarily conserved role in facilitating the establishment of apical–basolateral polarity in epithelial cells [65]. Given the similar localization of Erbin and SCRIB at the adherens junctions, it is not surprising that recent studies have identified Erbin as another LAP protein that regulates epithelial cell polarity. While a combined knockout of SCRIB, Erbin, and Lano in colon cancer DLD1 cells disrupted the apical–basolateral organization of cell–cell junctions, the expression of Erbin alone was sufficient to maintain normal cell–cell contact suggesting overlapping functions of Erbin and SCRIB [92]. In addition, silencing Erbin expression prevented the formation of acini-like structures in Caco2 cells grown in a 3D matrix indicating a defect in establishing apical–basolateral polarity [95]. Erbin has also been found to interact with cell–cell junction proteins, including bullous pemphigoid antigen 1 (eBPAG1, gene name DST) and the cytoplasmic tail of the β4 integrin (ITGB4) [96]. However, the functional implication of these interactions has not been investigated and the molecular mechanisms by which Erbin regulates epithelial polarity are currently unknown.
The interaction between Erbin and other signaling proteins
In addition to its role in regulating ERBB2 and RAS/RAF signaling, recent studies have identified other signaling molecules that interact with Erbin. Through a yeast two-hybrid screen, Erbin was found to bind the MH2 domain of Smad3. In addition, Erbin interacts with MH2 domains of other Smads, including Smad1, Smad2, Smad4, and Smad7 [97]. A region immediately upstream of the PDZ domain in Erbin has been identified as the Smad-interacting domain (SID) [98]. However, a later study showed that a positively charged residue within the PDZ domain of Erbin may also contribute to an electrostatic interaction with the MH2 domain of Smad3 [99]. Functionally, Erbin negatively regulates TGFβ-dependent transcriptional responses without altering the phosphorylation levels of Smad2 or Smad3; instead, Erbin acts as a sink to sequester Smad2/Smad3 from binding Smad4 and transducing TGFβ signaling [98].
To add another layer of complexity, Erbin has been shown to interact with SARA (Smad anchor for receptor activation) to regulate TGFβ signaling [100]. SARA binds to nonphosphorylated Smad2/Smad3 and recruits them to activated receptors. SARA is localized at the plasma membrane and early endosomes where it was found to interact with Erbin. Interestingly, the previously identified SID region in Erbin is also responsible for binding SARA. SARA competes with Smads for binding Erbin, and the SARA-Erbin interaction attenuates Erbin-mediated inhibitory effects on Smads. Thus, the balance between Erbin-SARA and Erbin-Smad complexes likely dictates the signaling output of TGFβ and activin receptor pathways [100].
Furthermore, the physiological importance of Erbin-mediated regulation of TGFβ signaling has been shown in patients with atopy, a disease associated with increased allergic reactions or immune responses and connective tissue abnormalities. A rare disease-segregating variant in the Erbin gene has been identified in a family with dominantly inherited symptoms that shared both allergic and nonimmunological connective tissue features with patients carrying mutations in the STAT3 gene [101]. Interestingly, this single nucleotide alteration results in a missense mutation (c.1588G>Tp.D530Y) in the nonconserved linker region downstream of the LRR domain in Erbin. The expression of mutant Erbin protein was significantly decreased in fibroblasts and naïve and memory CD4 T cells isolated from patients carrying the c.1588G>T substitution. This loss-of-function mutation in Erbin disrupts the interaction between Erbin and STAT3 and reduces Erbin’s ability to attenuate TGFβ signaling as shown by increased levels of nuclear phosphorylated Smad2/3. This aberrant activation of TGFβ signaling in Erbin mutant lymphocytes leads to elevated levels of T helper type 2 cytokine and IgE production, highlighting the importance of Erbin in mediating signaling crosstalk between STAT3 and Smad in human disease [101].
Studies to define the specificity of signaling activation downstream of TGFβ identified Erbin as a key determinant of epithelial resistance to TGFβ signaling [102]. TGFβ activates PAK2 via a Smad2/3-independent, but Rac1-/Cdc42-dependent mechanism, to stimulate the proliferative and profibrotic response in mesenchymal cells [103]. In a search for epithelialspecific factors preventing TGFβ-induced activation of PAK2, Erbin was found to block the interaction between PAK2 and its upstream activators Cdc42 as part of the Erbin/NF2 complex [102]. In addition, Erbin inhibited TGFβ-induced EMT and ERK signaling in kidney epithelial cells to prevent renal interstitial fibrosis [104]. Taken together, multiple studies have identified Erbin as a negative regulator of TGFβ signaling. Although the binding partners vary under different experimental settings, Erbin functions consistently by sequestering effectors from signaling activation and propagation.
Additionally, Erbin was found to interact with the nucleotide-binding oligomerization domain containing protein (Nod2) in regulating inflammatory responses [105,106]. Both the LRR domain and the C-terminal portion of the linker region in Erbin are responsible for binding Nod2, while the caspase recruitment domains (CARDs) in Nod2 are necessary and sufficient for interaction with Erbin. Improper activation of Nod2 leads to Crohn’s disease and Blau syndrome. Overexpression of Erbin inhibits, whereas Erbin loss increases Nod2-dependent activation of NFκB and proinflammatory cytokine secretion in response to MDP stimulation [105]. The Walker B box mutant of Nod2 (K305R) and a Crohn’s disease-associated frameshift mutant of Nod2 (L1007fs) fail to interact with Erbin [106]. Collectively, these studies identify Erbin as a negative regulator of Nod2-dependent activation of proinflammatory NFκB signaling [105].
Recent findings revealed a novel role of Erbin in regulating Cav1.3 channel activity at the synapse in a PDZ domain-dependent manner. While defining the regulatory mechanism of Ca2+ signaling at excitatory synapses, Calin-Jageman et al., found that Erbin interacts with the L-type voltage-gated Ca2+ channel Cav1.3 subunit (gene name CACNA1D) [107]. The Cav1.3 subunit contains a PDZ ligand sequence (ITTL) at the C terminus of the protein, which is responsible for binding the PDZ domain of Erbin. This interaction is specific as the Cav1.2 subunit of L-type Ca2+ channel and a splicing variance of Cav1.3, a short Cav1.3b lacking the C-terminal PDZ ligand sequence, are unable to bind Erbin. Co-expression of Erbin augments the voltage-dependent facilitation (VDF) of Ca2+ current through Cav1.3 channels.
Following their initial discovery of impaired nerve myelination phenotype in Erbin null mice [76], Mei and co-workers subsequently reported that loss of Erbin results in abnormal locomotive behavior and dysfunction of GABAergic interneurons in mice [108]. The expression of Erbin was found in select GABAergic neurons and the loss of Erbin decreased AMPA receptor (AMPAR)-mediated synaptic transmission. Furthermore, Erbin was found to interact with TARP γ-2 (gene name CACNG2), an auxiliary subunit of AMPAR required for AMPAR membrane trafficking, and the binding domain for γ-2 was mapped to the linker region immediately upstream of the PDZ domain in Erbin. The interaction between Erbin and γ-2 increased γ-2 stability and AMPAR expression at the cell surface. Similar to what was observed in Erbin null mice, the surface expression and function of AMPAR are decreased in ErbinΔC/ΔC mice confirming the importance of the PDZ domain-dependent interaction [108]. Together, these studies provide additional evidence supporting the role of Erbin as a signaling scaffold in neuronal cells.
A complex role of Erbin in cancer
Given its role in the regulating ERBB2 tyrosine kinase receptor and RAS/RAF signaling pathways, it is not surprising that a number of studies have focused on elucidating the functional importance of Erbin in different cancer types. Upon surveying the COSMIC collection of cancer mutations, we found that the frequency of Erbin mutations ranges from no mutations found in several cancer types to up to 8% in liver cancer. Among nonsynonymous mutations, the most common type is a missense substitution followed by a small percentage of nonsense and frameshift mutations. Interestingly, these mutations are evenly distributed throughout the entire coding sequence of Erbin with no particular hotspots (the highest number of a single site mutation found is 4). This type of mutation pattern is often associated with tumor suppressor genes. A number of in vitro and in vivo studies on characterizing the expression and function of Erbin in different cancers have revealed conflicting findings depending on the cancer type and experimental settings. Here, we summarize studies that have implicated Erbin as a tumor suppressor as well as studies showing oncogenic functions of Erbin.
Erbin as a tumor suppressor
Supporting a tumor suppressor role of Erbin, silencing of Erbin expression decreased ERBB2-mediated phosphorylation of Akt and increased the sensitivity of breast cancer MCF7 cells to TRAIL-induced apoptosis [109]. In addition, loss of Erbin rendered cervical cancer cells resistant to cell-detachment-induced anoikis by upregulating nuclear translocation and activation of STAT3 [110]. Moreover, Erbin was found to interact with Tax1, an oncoprotein encoded by human T-cell leukemia virus type I (HTLV-I). Possibly, Tax1 activates RAS/RAF signaling by disrupting Erbin-mediated suppression of this pathway [111–113]. Decreased expression of Erbin protein and mRNA was detected in breast cancer patient samples when compared to normal controls. Silencing of Erbin resulted in increased amplitude and duration of both Akt and ERK1/2 phosphorylation downstream of heregulin stimulation in ERBB2-overexpressing breast cancer cells [114]. More recently, the interaction between Erbin and DSG1 has been shown to inhibit invasion and metastasis of head and neck squamous cell cancer cells by suppressing the formation of invadopodia and EGFR/ERK signaling [115].
Studies on characterizing the expression of Erbin in cancer have identified Erbin as one of the genes that was strongly induced by BRCA1 expression in breast cancer cells using the suppression subtractive hybridization technique [116]. Additionally, it was found that the promoter region of the Erbin gene contains a consensus sequence for binding the transcription factor c-Myb which controls Erbin expression in a cell cycle-dependent manner. Erbin expression peaks in the G2/M phase. The loss of Erbin led to the formation of multipolar spindles and ultimately abnormal chromosome division, suggesting that Erbin is required for the maintenance of chromosomal stability [117]. Moreover, Erbin expression is regulated by the SCFSkp2 ubiquitin ligase. Although an interaction between Erbin and Skp2 was not detected, knockdown of Skp2 increased Erbin protein expression in Hela cells. Functionally, silencing Erbin enhanced signaling through the Akt/Skp2 axis to promote the degradation of p27, a known substrate of Skp2, and cell proliferation. While additional studies are needed to determine whether Erbin is a target of SCF-Skp2, results from this study support a tumor suppressor role for Erbin [118]. More recently, miR-183-5p has been identified as a negative regulator of Erbin mRNA expression in AML cells [119]. Overexpression of miRNA-183 increased Erbin mRNA and protein expression and inhibited cell proliferation, whereas a miRNA-183 mimic had the same effect as knocking down Erbin. In addition, Erbin was found to interact with Grb2, decrease Grb2 stability, and inhibit both PI3K/Akt and RAS/RAF signaling [119]. Taken together, these studies indicate that Erbin expression can be regulated at both mRNA and protein levels in different cancer types and downregulation of Erbin promotes cell growth downstream of oncogenic signaling in these cancers.
To elucidate the functional importance of Erbin in colon cancer in vivo, Stevens et al. showed that the expression of Erbin is significantly downregulated at both mRNA and protein levels in colon cancer patient specimens [95]. While Erbin expression was detected along the epithelial cell–cell junction in normal human colon tissues, the expression of Erbin was markedly reduced and mislocalized to the cytoplasm in tumor tissues. Knockdown of Erbin disrupted epithelial cell polarity and induced EMT. As a consequence, the proliferation in 3D cultures as well as migration and invasion of colon cancer cells were increased. Mechanistically, Erbin interacted with KSR1 and displaced it from the RAF/MEK/ERK complex thereby preventing signaling propagation. Silencing Erbin enhanced the amplitude and duration of signaling through both Akt and RAS/RAF pathways. Furthermore, crossing Erbin−/− mice to the Apc-driven mouse model of colon cancer significantly accelerated tumor progression and reduced survival. Tumor organoids derived from Erbin/Apc double knockout mice acquired increased cancer stem cell properties and tumor initiation potential. Collectively, these studies identify Erbin as a negative regulator of colon cancer tumorigenesis by suppressing Akt and RAS/RAF signaling in vivo.
Erbin as an oncogenic protein
In contrast to what is described above, others showed that increased Erbin expression promotes tumorigenesis. Earlier in vitro studies showed that knockdown of Erbin in HT29 cells reduced the formation of multicellular tumor spheroids in 3D [120]. In addition, increased Erbin has been associated with paclitaxel resistance in gastric cancer cells [121]. Subsequently, Erbin was found to bind the E3 ligase c-Cbl via its PDZ domain and decrease c-Cbl-mediated ubiquitination and degradation of EGFR in colon cancer cells. As a result, knockdown of Erbin led to decreased phosphorylation of Akt and ERK. Moreover, overexpression of Erbin enhanced xenograft tumor growth; and deletion of Erbin C terminus in vivo reduced the average size of colon tumors induced by AOM treatment in ErbinΔC/ΔC mice. These results suggest that Erbin functions to promote tumorigenesis by disrupting c-Cbl-mediating downregulation of EGFR signaling [122].
Consistent with their finding of Erbin as an oncogenic protein, the same research group demonstrated the tumor promoting function of Erbin in hepatocellular carcinoma (HCC) [123]. Analysis of HCC patient samples revealed a stage-dependent increase of Erbin expression in HCC patients. Erbin was found to interact with nuclear receptor ERα, and this interaction enhances the association between ERα and Chip, a known E3 ligase of ERα, and Chip-dependent ubiquitination and degradation of ERα. Since ERα signaling plays a protective role in attenuating HCC development, Erbin attenuates ERα expression and its transactivation activity to drive tumorigenesis [123,124]. Supporting the tumor promoting function of Erbin in HCC, a recent study identified Erbin as a direct target of miR-23c, in that overexpression of miR-23c suppressed Erbin expression to inhibit HCC cell proliferation in vitro and tumor growth in vivo [125].
4324460480706000In an effort to investigate the function of Erbin in breast cancer, Tao et al. [126] showed that deletion of Erbin or its C terminus delays mammary tumor development and prolongs the survival of MMTV-Neu mice. Interestingly, Erbin loss decreased the expression of ERBB2 protein without affecting ERBB2 mRNA in vivo. Erbin was found to form a ternary complex with ERBB2 and HSP90. In this complex, Erbin stabilized the interaction between HSP90 and its protein client ERBB2 and blocked ERBB2 ubiquitination. Silencing of Erbin decreased the proliferation of ERBB2-dependent human breast cancer cells in both 2D and 3D cultures. Nevertheless, crossing Erbin−/− or ErbinΔC/ΔC mice to MMTV-PyVT-driven breast cancer models did not alter tumor development and progression, suggesting that the tumor promoting function of Erbin is unique to ERBB2-driven breast cancer [126]. Based on the finding that Erbin stabilizes ERBB2 expression by disrupting ERBB2-HSP90 interaction, it has been postulated that peptide inhibitors may be developed to treat ERBB2-driven breast cancer by specifically blocking the PDZ domain-dependent interaction between Erbin and ERBB2 [127]. However, given the multifunctional role of Erbin PZD domain beyond its ability to regulate ERBB2, the specificity for such an interference strategy is likely difficult to achieve.
Furthermore, in a study to determine the role of SAG (gene name RNF7), an essential component of the SCF ubiquitin E3 ligase, in KRASG12D-induced papillomagenesis, Erbin was identified as a substrate of SAG-β-TrCP complex [128]. In KRASG12D-expressing primary keratinocytes, SAG deletion significantly accelerates the formation of skin papillomas in vivo. Interestingly, knockout of SAG decreased the activation of RAS/RAF signaling by increasing Erbin expression. The interaction between Erbin and βTrCP was mediated through an evolutionarily conserved consensus binding motif (DSGXXS) at amino acids 958–963 (TSGPQS) in Erbin. In this KRAS-driven tumor model of skin cancer, Erbin functions consistently as a negative regulator of the RAS/RAF pathway. However, because hyperactivation of RAS/RAF signaling induces senescence of keratinocytes, increased Erbin expression downstream of SAG deletion essentially bypasses KRASG12D-induced senescence and promotes papillomagenesis. Indeed, heterozygous deletion of Erbin in KRASG12D/SAG−/− double mutant mice attenuates tumor progression [128].
Collectively, studies on elucidating the role of Erbin in cancer have led to seemingly conflicting conclusions. In seeking for consensus, the following variables in experimental systems need to be taken into consideration. First, Erbin is a membrane-localized protein associated with the adherens junctions in polarized epithelial cells. The function of Erbin likely changes as the epithelial polarity and cell–cell junction are progressively disrupted during tumor progression. This may also explain the functional differences of Erbin in cancer cells of epithelial origins vs. leukemia cells. Second, alterations of Erbin expression or mutations in the Erbin gene are generally not considered drivers of tumorigenesis. Depending on the predominant pathways that drive tumorigenesis, specific phenotypes associated with altered Erbin expression are likely different. For example, Erbin may promote ERBB2-dependent breast cancer by stabilizing ERBB2 expression, whereas it inhibits RAS/RAF signaling to prevent colon cancer progression. In addition, since in vivo tumorigenesis studies have been conducted using whole-body Erbin knockout or mutant mice, the functional contribution of Erbin loss in controlling inflammatory responses has not been integrated into the analysis of tumor phenotypes. Finally, as discussed above Erbin contains multiple protein–protein interacting modules. The overall signaling outcome downstream from Erbin is expected to vary in a cell-type- and cell-context-dependent manner based on the expression of its binding partners.
Conclusions and Closing remarks
Critical roles of scaffolding proteins have been reported in a large number of biological signaling processes [1]. Scaffolds show remarkable diversity in the ways by which they facilitate transmission of intracellular signals. These ways extend beyond a simple concept of bringing signaling proteins to close proximity and increasing an efficiency of their interaction [129]. As discussed above, a collection of functional studies has linked aberrant Shoc2 and Erbin expression with a multitude of human diseases (Table 1). Given the multipotent role of these scaffolding proteins, we anticipate that future studies will continue to dissect the molecular mechanisms underlying the specificity of signaling dynamics.
Studies of Shoc2 over the past decade have exposed the intricacy of the mechanisms regulating assembly of the scaffolding complex and the cellular function of Shoc2-mediated signals. Not only have they have further emphasized the central position of Shoc2 in the ERK1/2 pathway, but also uncovered its role in other intracellular signaling cascades. Nevertheless, our understanding of the multifaceted Shoc2 machinery and biological activities controlled by the module remains incomplete. It would be important to identify all the proteins supporting the process of the Shoc2 scaffold assembly and remodeling. Several gaps remain to be filled in order to understand the stimuli that activate assembly of the scaffolding complex as well as mechanisms that guide subcellular distribution of the Shoc2 scaffolding complex. The final effects of the subcellular redistribution of the complexes and how it affects ERK1/2 signals are yet to be determined. Given the already existing interest in the development of therapeutics agents targeting Shoc2 in cancer, we anticipate to see an expansion of these efforts in identification of novel pharmaceutical agents targeting Shoc2.
Following the first identification of Erbin as a binding partner of ERBB2 two decades ago, more than 20 proteins have been found to interact with Erbin under various experimental conditions (Fig. 3). The structure of the Erbin PDZ domain in complex with the C-terminal PDZ ligand derived from ERBB2 has been solved to explain the binding specificity between the two proteins [73]. However, more studies are needed not only to resolve the controversial role of Erbin in cancer, but also to address other outstanding questions. For example, the molecular mechanisms underlying Erbin-mediated regulation of epithelial cell polarity are currently unknown. It remains an open question if Erbin forms a complex with polarity proteins (such as PAR3 or PAR6) at the adherens junction to facilitate the establishment of apical–basolateral polarity. In addition, structural studies of the LRR domain and linker region of Erbin are needed to better understand how Erbin utilizes different structural elements for interacting with diverse signaling molecules. Moreover, as numerous Erbin mutations are found in various cancer types, studies focusing on determining the functional contribution of these mutants will further advance our understanding of Erbin’s role in cancer.
In summary, by nucleating a large number of protein complexes in cells, Shoc2 and Erbin share an ability to function as signaling scaffolds. Each protein has been shown to regulate multiple signaling pathways and loss-of-function mutations contribute to the development of human disease. Importantly, the intricate interplay between Shoc2 and Erbin in regulating signaling propagation through the RAS/RAF/ERK pathway highlights the importance of balancing signaling outcomes with scaffolding proteins (Fig. 2). Future studies of Shoc2 and Erbin will provide fundamental insights into our understanding of the mechanisms by which other scaffolds may be controlled.
Acknowledgements
We thank Dr. Louis Hersh and members of our laboratories for critical reading of the manuscript. This project was supported by grants from the National Institute of General Medical Sciences (GM113087 and R35 GM136295 to EG), the American Cancer Society (RSG-14-172-01-CSM to EG), and National Cancer Institute (CA133429 and CA208343 to TG). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Health.
Abbreviations
- AAA+
ATPase-associated with diverse cellular activities
- APIS
AAA proteins independent of 20S
- BRCA1
breast cancer 1
- DSG1
Desmoglein 1
- EGF
epidermal growth factor
- Erbin
ERBB2-interacting protein
- EMT
epithelial–mesenchymal transition
- ERK1/2
extracellular signal-regulated kinase
- HNSCC
head and neck squamous cell cancer
- HTLV-I
human T-cell leukemia virus type I
- HCC
hepatocellular carcinoma
- HUWE-1
HECT, UBS, and WWE domain containing 1
- IBMPFD
inclusion body myopathy with Paget’s disease of bone and frontotemporal dementia
- LRR
leucine-rich repeat
- LAP
LRR And PDZ domain
- LGALS3BP
lectin galactoside-binding soluble 3-binding protein
- MITF
microphthalmia-associated transcription factor
- MMPs
matrix metalloproteinases
- NSLAH
Noonan syndrome with loose anagen hair
- NRG1
Neuregulin 1
- PP1C
protein phosphatase 1c
- PSMC5
proteasome 26S subunit ATPase 5
- SARA
SMAD anchor for receptor activation
- SCRIB
Scribbled homolog
- SID
Smad-interacting domain
- SPPK
Striate palmoplantar keratoderma
- TGFβ
Transforming growth factor beta
- VCP
Valosin-containing protein
- VDF
voltage-dependent facilitation
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Langeberg LK & Scott JD (2015) Signalling scaffolds and local organization of cellular behaviour. Nat Rev Mol Cell Biol 16, 232–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Good MC, Zalatan JG & Lim WA (2011) Scaffold proteins: hubs for controlling the flow of cellular information. Science 332, 680–686.21551057 [Google Scholar]
- 3.Bella J, Hindle KL, McEwan PA & Lovell SC (2008) The leucine-rich repeat structure. Cell Mol Life Sci 65, 2307–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kajava AV (1998) Structural diversity of leucine-rich repeat proteins. J Mol Biol 277, 519–527. [DOI] [PubMed] [Google Scholar]
- 5.Kobe B & Deisenhofer J (1993) Crystal structure of porcine ribonuclease inhibitor, a protein with leucine-rich repeats. Nature 366, 751–756. [DOI] [PubMed] [Google Scholar]
- 6.Kobe B & Kajava AV (2001) The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol 11, 725–732. [DOI] [PubMed] [Google Scholar]
- 7.Ng AC, Eisenberg JM, Heath RJ, Huett A, Robinson CM, Nau GJ & Xavier RJ (2011) Human leucine-rich repeat proteins: a genome-wide bioinformatic categorization and functional analysis in innate immunity. Proc Natl Acad Sci USA 108 (Suppl 1), 4631–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ko J & Kim E (2007) Leucine-rich repeat proteins of synapses. J Neurosci Res 85, 2824–2832. [DOI] [PubMed] [Google Scholar]
- 9.Schroeder A & de Wit J (2018) Leucine-rich repeat-containing synaptic adhesion molecules as organizers of synaptic specificity and diversity. Exp Mol Med 50, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matsushima N, Takatsuka S, Miyashita H & Kretsinger RH (2019) Leucine rich repeat proteins: sequences, mutations, structures and diseases. Protein Pept Lett 26, 108–131. [DOI] [PubMed] [Google Scholar]
- 11.Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M et al. (2001) Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603. [DOI] [PubMed] [Google Scholar]
- 12.Cario E & Podolsky DK (2000) Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun 68, 7010–7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swanberg M, Lidman O, Padyukov L, Eriksson P, Akesson E, Jagodic M, Lobell A, Khademi M, Borjesson O, Lindgren CM et al. (2005) MHC2TA is associated with differential MHC molecule expression and susceptibility to rheumatoid arthritis, multiple sclerosis and myocardial infarction. Nat Genet 37, 486–494. [DOI] [PubMed] [Google Scholar]
- 14.Hawn TR, Verbon A, Lettinga KD, Zhao LP, Li SS, Laws RJ, Skerrett SJ, Beutler B, Schroeder L, Nachman A et al. (2003) A common dominant TLR5 stop codon polymorphism abolishes flagellin signaling and is associated with susceptibility to legionnaires’ disease. J Exp Med 198, 1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piepoli A, Palmieri O, Maglietta R, Panza A, Cattaneo E, Latiano A, Laczko E, Gentile A, Carella M, Mazzoccoli G et al. (2012) The expression of leucine-rich repeat gene family members in colorectal cancer. Exp Biol Med (Maywood) 237, 1123–1128. [DOI] [PubMed] [Google Scholar]
- 16.Sieburth DS, Sun Q & Han M (1998) SUR-8, a conserved Ras-binding protein with leucine-rich repeats, positively regulates Ras-mediated signaling in C. elegans. Cell 94, 119–130. [DOI] [PubMed] [Google Scholar]
- 17.Jeoung M, Abdelmoti L, Jang ER, Vander Kooi CW & Galperin E (2013) Functional integration of the conserved domains of Shoc2 scaffold. PLoS One 8, e66067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li W, Han M & Guan KL (2000) The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with Ras and Raf. Genes Dev 14, 895–900. [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M & McCormick F (2006) A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol Cell 22, 217–230. [DOI] [PubMed] [Google Scholar]
- 20.Matsunaga-Udagawa R, Fujita Y, Yoshiki S, Terai K, Kamioka Y, Kiyokawa E, Yugi K, Aoki K & Matsuda M (2010) The scaffold protein Shoc2/SUR-8 accelerates the interaction of Ras and Raf. J Biol Chem 285, 7818–7826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jang ER & Galperin E (2016) The function of Shoc2: A scaffold and beyond. Communicat Int Biol 9, e1188241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jones GG, Del Rio IB, Sari S, Sekerim A, Young LC, Hartig N, Areso Zubiaur I, El-Bahrawy MA, Hynds RE, Lei W et al. (2019) SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nat Commun 10, 2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boned Del Rio I, Young LC, Sari S, Jones GG, Ringham-Terry B, Hartig N, Rejnowicz E, Lei W, Bhamra A, Surinova S & et al. (2019) SHOC2 complex-driven RAF dimerization selectively contributes to ERK pathway dynamics. Proc Natl Acad Sci USA 116, 13330–13339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jang ER, Shi P, Bryant J, Chen J, Dukhante V, Gentry MS, Jang H, Jeoung M & Galperin E (2014) HUWE1 is a molecular link controlling RAF-1 activity supported by the Shoc2 scaffold. Mol Cell Biol 34, 3579–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang ER, Jang H, Shi P, Popa G, Jeoung M & Galperin E (2015) Spatial control of Shoc2-scaffold-mediated ERK1/2 signaling requires remodeling activity of the ATPase PSMC5. J Cell Sci 128, 4428–4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jang H, Jang ER, Wilson PG, Anderson D & Galperin E (2019) VCP/p97 controls signals of the ERK1/2 pathway transmitted via the Shoc2 scaffolding complex: novel insights into IBMPFD pathology. Mol Biol Cell 30, 1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez F, Delahodde A, Kodadek T & Johnston SA (2002) Recruitment of a 19S proteasome subcomplex to an activated promoter. Science 296, 548–550. [DOI] [PubMed] [Google Scholar]
- 28.Sun L, Johnston SA & Kodadek T (2002) Physical association of the APIS complex and general transcription factors. Biochem Biophys Res Commun 296, 991–999. [DOI] [PubMed] [Google Scholar]
- 29.Galperin E, Abdelmoti L & Sorkin A (2012) Shoc2 is targeted to late endosomes and required for Erk1/2 activation in EGF-stimulated cells. PLoS One 7, e36469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie CM & Sun Y (2019) The MTORC1-mediated autophagy is regulated by the FBXW7-SHOC2-RPTOR axis. Autophagy 15, 1470–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee KH, Jeong WJ, Cha PH, Lee SK, Min DS & Choi KY (2017) Stabilization of Sur8 via PKCalpha/delta degradation promotes transformation and migration of colorectal cancer cells. Oncotarget 8, 115596–115608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaduwal S, Jeong WJ, Park JC, Lee KH, Lee YM, Jeon SH, Lim YB, Min DS & Choi KY (2015) Sur8/Shoc2 promotes cell motility and metastasis through activation of Ras-PI3K signaling. Oncotarget 6, 33091–33105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hunter T (2007) The age of crosstalk: phosphorylation, ubiquitination, and beyond. Mol Cell 28, 730–738. [DOI] [PubMed] [Google Scholar]
- 34.Jang H, Oakley E, Forbes-Osborne M, Kesler MV, Norcross R, Morris AC & Galperin E (2019) Hematopoietic and neural crest defects in zebrafish shoc2 mutants: a novel vertebrate model for Noonan-like syndrome. Hum Mol Genet 28, 501–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yi J, Chen M, Wu X, Yang X, Xu T, Zhuang Y, Han M & Xu R (2010) Endothelial SUR-8 acts in an ERK-independent pathway during atrioventricular cushion development. Dev Dyn 239, 2005–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moon BS, Kim HY, Kim MY, Yang DH, Lee JM, Cho KW, Jung HS & Choi KY (2011) Sur8/Shoc2 involves both inhibition of differentiation and maintenance of self-renewal of neural progenitor cells via modulation of extracellular signal-regulated kinase signaling. Stem Cells 29, 320–331. [DOI] [PubMed] [Google Scholar]
- 37.Sulahian R, Kwon JJ, Walsh KH, Pailler E, Bosse TL, Thaker M, Almanza D, Dempster JM, Pan J, Piccioni F et al. (2019) Synthetic Lethal Interaction of SHOC2 depletion with MEK inhibition in RAS-driven cancers. Cell Rep 29, 118–134.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES & Sabatini DM (2017) Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 168, 890–903 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kota P, Terrell EM, Ritt DA, Insinna C, Westlake CJ & Morrison DK (2019) M-Ras/Shoc2 signaling modulates E-cadherin turnover and cell-cell adhesion during collective cell migration. Proc Natl Acad Sci USA 116, 3536–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Young LC, Hartig N, Munoz-Alegre M, Oses-Prieto JA, Durdu S, Bender S, Vijayakumar V, Vietri Rudan M, Gewinner C, Henderson S et al. (2013) An MRAS, SHOC2, and SCRIB complex coordinates ERK pathway activation with polarity and tumorigenic growth. Mol Cell 52, 679–692. [DOI] [PubMed] [Google Scholar]
- 41.Jeoung M, Jang ER, Liu J, Wang C, Rouchka EC, Li X & Galperin E (2016) Shoc2-tranduced ERK1/2 motility signals – novel insights from functional genomics. Cell Signal 28, 448–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rouchka EC, Jeoung M, Jang ER, Liu J, Wang C, Li X & Galperin E (2016) Data set for transcriptional response to depletion of the Shoc2 scaffolding protein. Data Brief 7, 770–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xue Y, Chiu JC & Zhang Y (2019) SUR-8 interacts with PP1-87B to stabilize PERIOD and regulate circadian rhythms in Drosophila. PLoS Genet 15, e1008475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cordeddu V, Di Schiavi E, Pennacchio LA, Ma’ayan A, Sarkozy A, Fodale V, Cecchetti S, Cardinale A, Martin J, Schackwitz W et al. (2009) Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet 41, 1022–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hannig V, Jeoung M, Jang ER, Phillips JA 3rd & Galperin E (2014) A Novel SHOC2 variant in rasopathy. Hum Mutat 35, 1290–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Motta M, Giancotti A, Mastromoro G, Chandramouli B, Pinna V, Pantaleoni F, Di Giosaffatte N, Petrini S, Mazza T, D’Ambrosio V et al. (2019) Clinical and functional characterization of a novel RASopathy-causing SHOC2 mutation associated with prenatal-onset hypertrophic cardiomyopathy. Hum Mutat 40, 1046–1056. [DOI] [PubMed] [Google Scholar]
- 47.Baldassarre G, Mussa A, Banaudi E, Rossi C, Tartaglia M, Silengo M & Ferrero GB (2014) Phenotypic variability associated with the invariant SHOC2 c.4A>G (p.Ser2Gly) missense mutation. Am J Med Genet A 164A, 3120–3125. [DOI] [PubMed] [Google Scholar]
- 48.Gripp KW, Zand DJ, Demmer L, Anderson CE, Dobyns WB, Zackai EH, Denenberg E, Jenny K, Stabley DL & Sol-Church K (2013) Expanding the SHOC2 mutation associated phenotype of Noonan syndrome with loose anagen hair: structural brain anomalies and myelofibrosis. Am J Med Genet A 161, 2420–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoban R, Roberts AE, Demmer L, Jethva R & Shephard B (2012) Noonan syndrome due to a SHOC2 mutation presenting with fetal distress and fatal hypertrophic cardiomyopathy in a premature infant. Am J Med Genet A 158A, 1411–1413. [DOI] [PubMed] [Google Scholar]
- 50.Gargano G, Guidotti I, Balestri E, Vagnarelli F, Rosato S, Comitini G, Wischmeijer A, La Sala GB, Iughetti L, Cordeddu V et al. (2014) Hydrops fetalis in a preterm newborn heterozygous for the c.4A>G SHOC2 mutation. Am J Med Genet A 164A, 1015–1020. [DOI] [PubMed] [Google Scholar]
- 51.Capalbo D, Scala MG, Melis D, Minopoli G, Improda N, Palamaro L, Pignata C & Salerno M (2012) Clinical heterogeneity in two patients with Noonanlike syndrome associated with the same SHOC2 mutation. Italian J Pediat 38, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi JH, Oh MY, Yum MS, Lee BH, Kim GH & Yoo HW (2015) Moyamoya syndrome in a patient with noonan-like syndrome with loose anagen hair. Pediatr Neurol 52, 352–355. [DOI] [PubMed] [Google Scholar]
- 53.Lo FS, Wang CJ, Wong MC & Lee NC (2015) Moyamoya disease in two patients with Noonan-like syndrome with loose anagen hair. Am J Med Genet A 167, 1285–1288. [DOI] [PubMed] [Google Scholar]
- 54.Bader-Meunier B, Cave H, Jeremiah N, Magerus A, Lanzarotti N, Rieux-Laucat F & Cormier-Daire V. (2013) Are RASopathies new monogenic predisposing conditions to the development of systemic lupus erythematosus? Case report and systematic review of the literature. Sem Arthritis Rheumat 43, 217–219. [DOI] [PubMed] [Google Scholar]
- 55.Uehara T, Hosogaya N, Matsuo N & Kosaki K (2018) Systemic lupus erythematosus in a patient with Noonan syndrome-like disorder with loose anagen hair 1: More than a chance association. Am J Med Genet A 176, 1662–1666. [DOI] [PubMed] [Google Scholar]
- 56.Ferrero GB, Picco G, Baldassarre G, Flex E, Isella C, Cantarella D, Cora D, Chiesa N, Crescenzio N, Timeus F et al. (2012) Transcriptional hallmarks of Noonan syndrome and Noonan-like syndrome with loose anagen hair. Hum Mutat 33, 703–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Digilio MC, Romana Lepri F, Dentici ML, Henderson A, Baban A, Roberti MC, Capolino R, Versacci P, Surace C, Angioni A et al. (2013) Atrioventricular canal defect in patients with RASopathies. Europ J Hum Gen 21, 200–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie CM, Tan M, Lin XT, Wu D, Jiang Y, Tan Y, Li H, Ma Y, Xiong X & Sun Y (2019) The FBXW7-SHOC2-raptor axis controls the cross-talks between the RAS-ERK and mTORC1 signaling pathways. Cell Rep 26, 3037–3050.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaplan FM, Kugel CH 3rd, Dadpey N, Shao Y, Abel EV & Aplin AE (2012) SHOC2 and CRAF mediate ERK1/2 reactivation in mutant NRAS-mediated resistance to RAF inhibitor. J Biol Chem 287, 41797–41807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Geng W, Dong K, Pu Q, Lv Y & Gao H (2020) SHOC2 is associated with the survival of breast cancer cells and has prognostic value for patients with breast cancer. Mol Med Rep 21, 867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen X, Qi M, Yang Q & Li JY (2019) MiR-299-3p functions as a tumor suppressor in thyroid cancer by regulating SHOC2. Eur Rev Med Pharmacol Sci 23, 232–240. [DOI] [PubMed] [Google Scholar]
- 62.Xiao-Pei H, Ji-Kuai C, Xue W, Dong YF, Yan L, Xiao-Fang Z, Ya-Min P, Wen-Jun C & Jiang-Bo Z (2018) Systematic identification of Celastrol-binding proteins reveals that Shoc2 is inhibited by Celastrol. Biosci Rep 38, BSR20181233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borg JP, Marchetto S, Le Bivic A, Ollendorff V, Jaulin-Bastard F, Saito H, Fournier E, Adelaide J, Margolis B & Birnbaum D (2000) ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nat Cell Biol 2, 407–414. [DOI] [PubMed] [Google Scholar]
- 64.Dai P, Xiong WC & Mei L (2006) Erbin inhibits RAF activation by disrupting the sur-8-Ras-Raf complex. J Biol Chem 281, 927–933. [DOI] [PubMed] [Google Scholar]
- 65.Bonello TT & Peifer M (2019) Scribble: A master scaffold in polarity, adhesion, synaptogenesis, and proliferation. J Cell Biol 218, 742–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bilder D, Birnbaum D, Borg JP, Bryant P, Huigbretse J, Jansen E, Kennedy MB, Labouesse M, Legouis R, Mechler B et al. (2000) Collective nomenclature for LAP proteins. Nat Cell Biol 2, E114. [DOI] [PubMed] [Google Scholar]
- 67.Santoni MJ, Pontarotti P, Birnbaum D & Borg JP (2002) The LAP family: a phylogenetic point of view. Trends Genet 18, 494–497. [DOI] [PubMed] [Google Scholar]
- 68.Saito H, Santoni MJ, Arsanto JP, Jaulin-Bastard F, Le Bivic A, Marchetto S, Audebert S, Isnardon D, Adelaide J, Birnbaum D et al. (2001) Lano, a novel LAP protein directly connected to MAGUK proteins in epithelial cells. J Biol Chem 276, 32051–32055. [DOI] [PubMed] [Google Scholar]
- 69.Nourry C, Grant SG & Borg JP. (2003) PDZ Domain Proteins: Plug and Play!. Sci Signal 2003, RE7. [DOI] [PubMed] [Google Scholar]
- 70.Harris BZ & Lim WA (2001) Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci 114, 3219–3231. [DOI] [PubMed] [Google Scholar]
- 71.Huang YZ, Wang Q, Xiong WC & Mei L (2001) Erbin is a protein concentrated at postsynaptic membranes that interacts with PSD-95. J Biol Chem 276, 19318–19326. [DOI] [PubMed] [Google Scholar]
- 72.Simeone L, Straubinger M, Khan MA, Nalleweg N, Cheusova T & Hashemolhosseini S (2010) Identification of Erbin interlinking MuSK and ErbB2 and its impact on acetylcholine receptor aggregation at the neuromuscular junction. J Neurosci 30, 6620–6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Birrane G, Chung J & Ladias JA (2003) Novel mode of ligand recognition by the Erbin PDZ domain. J Biol Chem 278, 1399–1402. [DOI] [PubMed] [Google Scholar]
- 74.Skelton NJ, Koehler MF, Zobel K, Wong WL, Yeh S, Pisabarro MT, Yin JP, Lasky LA & Sidhu SS (2003) Origins of PDZ domain ligand specificity. Structure determination and mutagenesis of the Erbin PDZ domain. J Biol Chem 278, 7645–7654. [DOI] [PubMed] [Google Scholar]
- 75.Appleton BA, Zhang Y, Wu P, Yin JP, Hunziker W, Skelton NJ, Sidhu SS & Wiesmann C (2006) Comparative structural analysis of the Erbin PDZ domain and the first PDZ domain of ZO-1. Insights into determinants of PDZ domain specificity. J Biol Chem 281, 22312–22320. [DOI] [PubMed] [Google Scholar]
- 76.Tao Y, Dai P, Liu Y, Marchetto S, Xiong WC, Borg JP & Mei L (2009) Erbin regulates NRG1 signaling and myelination. Proc Natl Acad Sci U S A 106, 9477–9482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liang C, Tao Y, Shen C, Tan Z, Xiong WC & Mei L (2012) Erbin is required for myelination in regenerated axons after injury. J Neurosci. 32, 15169–15180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang YZ, Zang M, Xiong WC, Luo Z & Mei L (2003) Erbin suppresses the MAP kinase pathway. J Biol Chem 278, 1108–1114. [DOI] [PubMed] [Google Scholar]
- 79.Rachmin I, Tshori S, Smith Y, Oppenheim A, Marchetto S, Kay G, Foo RS, Dagan N, Golomb E, Gilon D et al. (2014) Erbin is a negative modulator of cardiac hypertrophy. Proc Natl Acad Sci USA 111, 5902–5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pan P & Dobrowsky RT (2013) Differential expression of neuregulin-1 isoforms and downregulation of erbin are associated with Erb B2 receptor activation in diabetic peripheral neuropathy. Acta Neuropathol Commun 1, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rachmin I, Amsalem E, Golomb E, Beeri R, Gilon D, Fang P, Nechushtan H, Kay G, Guo M, Yiqing PL et al. (2015) FHL2 switches MITF from activator to repressor of Erbin expression during cardiac hypertrophy. Int J Cardiol 195, 85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rangwala R, Banine F, Borg JP & Sherman LS (2005) Erbin regulates mitogen-activated protein (MAP) kinase activation and MAP kinase-dependent interactions between Merlin and adherens junction protein complexes in Schwann cells. J Biol Chem 280, 11790–11797. [DOI] [PubMed] [Google Scholar]
- 83.Petrilli AM & Fernandez-Valle C (2016) Role of Merlin/NF2 inactivation in tumor biology. Oncogene 35, 537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Whittock NV & Bower C (2003) Targetting of desmoglein 1 in inherited and acquired skin diseases. Clin Exp Dermatol 28, 410–415. [DOI] [PubMed] [Google Scholar]
- 85.Getsios S, Simpson CL, Kojima S, Harmon R, Sheu LJ, Dusek RL, Cornwell M & Green KJ (2009) Desmoglein 1-dependent suppression of EGFR signaling promotes epidermal differentiation and morphogenesis. J Cell Biol 185, 1243–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Harmon RM, Simpson CL, Johnson JL, Koetsier JL, Dubash AD, Najor NA, Sarig O, Sprecher E & Green KJ (2013) Desmoglein-1/Erbin interaction suppresses ERK activation to support epidermal differentiation. J Clin Investig 123, 1556–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Izawa I, Nishizawa M, Tomono Y, Ohtakara K, Takahashi T & Inagaki M (2002) ERBIN associates with p0071, an armadillo protein, at cell-cell junctions of epithelial cells. Genes Cells 7, 475–85. [DOI] [PubMed] [Google Scholar]
- 88.Ohno H, Hirabayashi S, Iizuka T, Ohnishi H, Fujita T & Hata Y (2002) Localization of p0071-interacting proteins, plakophilin-related armadillo-repeat protein-interacting protein (PAPIN) and ERBIN, in epithelial cells. Oncogene 21, 7042–9. [DOI] [PubMed] [Google Scholar]
- 89.Laura RP, Witt AS, Held HA, Gerstner R, Deshayes K, Koehler MF, Kosik KS, Sidhu SS & Lasky LA (2002) The Erbin PDZ domain binds with high affinity and specificity to the carboxyl termini of delta-catenin and ARVCF. J Biol Chem 277, 12906–14. [DOI] [PubMed] [Google Scholar]
- 90.Jaulin-Bastard F, Arsanto JP, Le Bivic A, Navarro C, Vely F, Saito H, Marchetto S, Hatzfeld M, Santoni MJ, Birnbaum D et al. (2002) Interaction between Erbin and a Catenin-related protein in epithelial cells. J Biol Chem 277, 2869–75. [DOI] [PubMed] [Google Scholar]
- 91.Legouis R, Jaulin-Bastard F, Schott S, Navarro C, Borg JP & Labouesse M (2003) Basolateral targeting by leucine-rich repeat domains in epithelial cells. Embo Rep 4, 1096–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Choi J, Troyanovsky RB, Indra I, Mitchell BJ & Troyanovsky SM (2019) Scribble, Erbin, and Lano redundantly regulate epithelial polarity and apical adhesion complex. J Cell Biol 218, 2277–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Izawa I, Nishizawa M, Hayashi Y & Inagaki M (2008) Palmitoylation of ERBIN is required for its plasma membrane localization. Genes Cells 13, 691–701. [DOI] [PubMed] [Google Scholar]
- 94.Guo Z, Neilson LJ, Zhong H, Murray PS, Zanivan S & Zaidel-Bar R (2014) E-cadherin interactome complexity and robustness resolved by quantitative proteomics. Sci Signal 7, rs7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stevens PD, Wen YA, Xiong X, Zaytseva YY, Li AT, Wang C, Stevens AT, Farmer TN, Gan T, Weiss HL et al. (2018) Erbin suppresses KSR1-mediated RAS/RAF signaling and tumorigenesis in colorectal cancer. Cancer Res 78, 4839–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Favre B, Fontao L, Koster J, Shafaatian R, Jaunin F, Saurat JH, Sonnenberg A & Borradori L (2001) The hemidesmosomal protein bullous pemphigoid antigen 1 and the integrin beta 4 subunit bind to ERBIN. Molecular cloning of multiple alternative splice variants of ERBIN and analysis of their tissue expression. J Biol Chem. 276, 32427–36. [DOI] [PubMed] [Google Scholar]
- 97.Warner DR, Pisano MM, Roberts EA & Greene RM (2003) Identification of three novel Smad binding proteins involved in cell polarity. FEBS Lett 539, 167–173. [DOI] [PubMed] [Google Scholar]
- 98.Dai F, Chang C, Lin X, Dai P, Mei L & Feng XH (2007) Erbin inhibits transforming growth factor beta signaling through a novel Smad-interacting domain. Mol Cell Biol 27, 6183–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Deliot N, Chavent M, Nourry C, Lecine P, Arnaud C, Hermant A, Maigret B & Borg JP (2009) Biochemical studies and molecular dynamics simulations of Smad3-Erbin interaction identify a non-classical Erbin PDZ binding. Biochem Biophys Res Commun 378, 360–365. [DOI] [PubMed] [Google Scholar]
- 100.Sflomos G, Kostaras E, Panopoulou E, Pappas N, Kyrkou A, Politou AS, Fotsis T & Murphy C (2011) ERBIN is a new SARA-interacting protein: competition between SARA and SMAD2 and SMAD3 for binding to ERBIN. J Cell Sci 124, 3209–3222. [DOI] [PubMed] [Google Scholar]
- 101.Lyons JJ, Liu Y, Ma CA, Yu X, O’Connell MP, Lawrence MG, Zhang Y, Karpe K, Zhao M, Siegel AM et al. (2017) ERBIN deficiency links STAT3 and TGF-beta pathway defects with atopy in humans. J Exp Med 214, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wilkes MC, Repellin CE, Hong M, Bracamonte M, Penheiter SG, Borg JP & Leof EB (2009) Erbin and the NF2 tumor suppressor Merlin cooperatively regulate cell-type-specific activation of PAK2 by TGF-beta. Dev Cell 16, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wilkes MC, Murphy SJ, Garamszegi N & Leof EB (2003) Cell-type-specific activation of PAK2 by transforming growth factor beta independent of Smad2 and Smad3. Mol Cell Biol 23, 8878–8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou Q, Zeng R, Xu C, Liu L, Chen L, Kou P, Pei G, Bai S, Zhang Y, Li C et al. (2012) Erbin inhibits TGF-beta1-induced EMT in renal tubular epithelial cells through an ERK-dependent pathway. J Mol Med (Berl) 90, 563–574. [DOI] [PubMed] [Google Scholar]
- 105.McDonald C, Chen FF, Ollendorff V, Ogura Y, Marchetto S, Lecine P, Borg JP & Nunez G (2005) A role for Erbin in the regulation of Nod2-dependent NF-kappaB signaling. J Biol Chem 280, 40301–40309. [DOI] [PubMed] [Google Scholar]
- 106.Kufer TA, Kremmer E, Banks DJ & Philpott DJ (2006) Role for erbin in bacterial activation of Nod 2. Infect Immun 74, 3115–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Calin-Jageman I, Yu K, Hall RA, Mei L & Lee A (2007) Erbin enhances voltage-dependent facilitation of Ca(v)1.3 Ca2+ channels through relief of an autoinhibitory domain in the Ca(v)1.3 alpha1 subunit. J Neurosci 27, 1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tao Y, Chen YJ, Shen C, Luo Z, Bates CR, Lee D, Marchetto S, Gao TM, Borg JP, Xiong WC & et al. (2013) Erbin interacts with TARP gamma-2 for surface expression of AMPA receptors in cortical interneurons. Nat Neurosci 16, 290–299. [DOI] [PubMed] [Google Scholar]
- 109.Liu N, Zhang J, Zhang J, Liu S, Liu Y & Zheng D (2008) Erbin-regulated sensitivity of MCF-7 breast cancer cells to TRAIL via ErbB2/AKT/NF-kappaB pathway. J Biochem. 143, 793–801. [DOI] [PubMed] [Google Scholar]
- 110.Hu Y, Chen H, Duan C, Liu D, Qian L, Yang Z, Guo L, Song L, Yu M, Hu M et al. (2013) Deficiency of Erbin induces resistance of cervical cancer cells to anoikis in a STAT3-dependent manner. Oncogenesis 2, e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ress A & Moelling K (2006) Interaction partners of the PDZ domain of erbin. Protein Pept Lett 13, 877–881. [DOI] [PubMed] [Google Scholar]
- 112.Song C, Wang W, Li M, Liu Y & Zheng D (2009) Tax1 enhances cancer cell proliferation via Ras-Raf-MEK-ERK signaling pathway. IUBMB Life 61, 685–692. [DOI] [PubMed] [Google Scholar]
- 113.Ress A & Moelling K (2008) The PDZ protein erbin modulates beta-catenin-dependent transcription. Eur Surg Res 41, 284–289. [DOI] [PubMed] [Google Scholar]
- 114.Liu D, Shi M, Duan C, Chen H, Hu Y, Yang Z, Duan H & Guo N (2013) Downregulation of Erbin in Her2-overexpressing breast cancer cells promotes cell migration and induces trastuzumab resistance. Mol Immunol 56, 104–112. [DOI] [PubMed] [Google Scholar]
- 115.Valenzuela-Iglesias A, Burks HE, Arnette CR, Yalamanchili A, Nekrasova O, Godsel LM & Green KJ (2019) Desmoglein 1 regulates invadopodia by suppressing EGFR/Erk signaling in an Erbin-dependent manner. Mol Cancer Res 17, 1195–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Atalay A, Crook T, Ozturk M & Yulug IG (2002) Identification of genes induced by BRCA1 in breast cancer cells. Biochem Biophys Res Commun. 299, 839–846. [DOI] [PubMed] [Google Scholar]
- 117.Liu D, Shi M, Zhang H, Qian L, Yu M, Hu M, Zhang R, Wang T, Han C, Duan H & et al. (2012) c-Myb regulates cell cycle-dependent expression of Erbin: an implication for a novel function of Erbin. PLoS One 7, e42903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huang H, Song Y, Wu Y, Guo N, Ma Y & Qian L (2015) Erbin loss promotes cancer cell proliferation through feedback activation of Akt-Skp2-p27 signaling. Biochem Biophys Res Commun 463, 370–376. [DOI] [PubMed] [Google Scholar]
- 119.Zheng Z, Zheng X, Zhu Y, Gu X, Gu W, Xie X, Hu W & Jiang J (2019) miR-183-5p inhibits occurrence and progression of acute myeloid leukemia via targeting erbin. Mol Ther 27, 542–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dardousis K, Voolstra C, Roengvoraphoj M, Sekandarzad A, Mesghenna S, Winkler J, Ko Y, Hescheler J & Sachinidis A (2007) Identification of differentially expressed genes involved in the formation of multicellular tumor spheroids by HT-29 colon carcinoma cells. Mol Ther 15, 94–102. [DOI] [PubMed] [Google Scholar]
- 121.Murakami H, Ito S, Tanaka H, Kondo E, Kodera Y & Nakanishi H (2013) Establishment of new intraperitoneal paclitaxel-resistant gastric cancer cell lines and comprehensive gene expression analysis. Anticancer Res 33, 4299–4307. [PubMed] [Google Scholar]
- 122.Yao S, Zheng P, Wu H, Song LM, Ying XF, Xing C, Li Y, Xiao ZQ, Zhou XN, Shen T et al. (2015) Erbin interacts with c-Cbl and promotes tumourigenesis and tumour growth in colorectal cancer by preventing c-Cbl-mediated ubiquitination and down-regulation of EGFR. J Pathol 236, 65–77. [DOI] [PubMed] [Google Scholar]
- 123.Wu H, Yao S, Zhang S, Wang JR, Guo PD, Li XM, Gan WJ, Mei L, Gao TM & Li JM (2017) Elevated expression of Erbin destabilizes ERalpha protein and promotes tumorigenesis in hepatocellular carcinoma. J Hepatol 66, 1193–1204. [DOI] [PubMed] [Google Scholar]
- 124.Francavilla A, Polimeno L, DiLeo A, Barone M, Ove P, Coetzee M, Eagon P, Makowka L, Ambrosino G, Mazzaferro V et al. (1989) The effect of estrogen and tamoxifen on hepatocyte proliferation in vivo and in vitro. Hepatology 9, 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zhang L, Wang Y, Wang L, Yin G, Li W, Xian Y, Yang W & Liu Q (2018) miR-23c suppresses tumor growth of human hepatocellular carcinoma by attenuating ERBB2IP. Biomed Pharmacother 107, 424–432. [DOI] [PubMed] [Google Scholar]
- 126.Tao Y, Shen C, Luo S, Traore W, Marchetto S, Santoni MJ, Xu L, Wu B, Shi C, Mei J et al. (2014) Role of Erbin in ErbB2-dependent breast tumor growth. Proc Natl Acad Sci U S A 111, E4429–E4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Mei L & Borg JP (2015) ERBB2 oncogenicity: ERBIN helps to perform the job. Mol Cell Oncol 2, e995033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xie CM, Wei D, Zhao L, Marchetto S, Mei L, Borg JP & Sun Y (2015) Erbin is a novel substrate of the Sag-betaTrCP E3 ligase that regulates KrasG12D-induced skin tumorigenesis. J Cell Biol 209, 721–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Garbett D & Bretscher A (2014) The surprising dynamics of scaffolding proteins. Mol Biol Cell 25, 2315–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]