

Abstract
Application of a microfluidic CE device for CZE-MS allows for fast, rapid, and in-depth analysis of large sample sets. The microfluidic CZE-MS device, the 908 Devices ZipChip, involves minimal sample preparation and is ideal for small cation analytes, such as alkaloids. Here, we evaluated the microfluidic device for the analysis of alkaloids from Lobelia cardinalis hairy root cultures. Extracts from wild-type, transgenic, and selected mutant plant cultures were analyzed and data batch processed using the mass spectral processing software MZmine2 and the statistical software Prism 8. In total 139 features were detected as baseline resolved peaks via the MZmine2 software optimized for the electrophoretic separations. Statistically significant differences in the relative abundance of the primary alkaloid lobinaline (C27H34N2), along with several putative “lobinaline-like” molecules were observed utilizing this approach. Additionally, a method for performing both targeted and untargeted tandem mass spectrometry experiments using the microfluidic device was developed and evaluated. Coupling data-processing software with CZE-MS data acquisition has enabled comprehensive metabolomic profiles from plant cell cultures to be constructed within a single working day.
Introduction
The development of a sensitive, high-throughput analytical screening for plant alkaloids and metabolites is crucial for natural pharmaceutical product discovery and plant metabolomic profiling. Mass spectrometry for the identification of metabolites has traditionally focused on using HPLC-MS or GC-MS [1–4]. While both are viable options, the screenings can be lengthy, involve extensive sample preparation, or may not be suitable for all types of analytes (i.e. derivatization requirements for GC-MS samples). The separation of structural isomers can also be a challenge with these methods [1,5]. Here, a microfluidic capillary zone electrophoresis (CZE) chip, first designed in 2008 and later commercially produced in 2016 for research use, has been employed to attempt to address these issues [6,7]. The microfluidic device consists of a small, etched glass chip capable of efficient and fast electrophoretic separations (Figure S1). The chip is enclosed in a polymer casing to establish “wells” and is inserted into the device interface mounted directly to the inlet of the mass spectrometer.
The ZipChip is marketed primarily for analysis of intact antibodies, proteins, and amino acids. A variety of uses for this microfluidic CZE-MS device have been reported in the literature. Glycomic and glycoproteomic applications have been reported by Zaia et al. and Zang et al [8,9]. In utilization of the HR variety chip, intact antibodies have been analyzed as well as viral capsid proteins by Chen et al. and Zhang et al. respectively [10,11]. In early 2018, the first small-molecule metabolomics application was published by Bereman et al [5]. Currently, the only real limitation observed with this device is that its application is limited to positively charged molecules due to the nature of the separation mechanism employed in the device [7].
The alkaloids in question for this application of the microfluidic device are those found in the cardinal flower, Lobelia cardinalis. L. cardinalis produces a binitrogenous alkaloid, lobinaline (decahydro-1-methyl-5,7-diphenyl-6-(3,4,5,6-tetrahydro-2-pyridinyl)-quinoline - C27H34N2 – Figure S2), recently shown to have inhibitory activity on the dopamine transporter (DAT) [12–16]. In order to find novel metabolites with similar activity, Naprogenix Inc. (Lexington, KY) successfully transformed L. cardinalis to produce hairy root cultures expressing the human dopamine transporter (hDAT). The cultures were then further mutated to induce random gain-of-function mutations and these transgenic mutants were selected on media containing MPP+ (which is accumulated intracellularly by activity of the hDAT) [12]. Surviving mutants are predicted to contain up-regulated secondary metabolites with hDAT inhibitory activity [12]. The aim of this study was to develop an analytical method to identify those specific metabolites that are increased in MPP+-resistant mutants relative to wild type (WT).
Here, samples of the surviving hairy root cultures were propagated, harvested, extracted, and then analyzed using the microfluidic device for the pharmacological screening of alkaloids in plant material. Due to the complexity of the sample, the software program, MZmine2 was used to enhance the screening procedure [17]. Additionally, a method was developed for the utilization of capillary zone electrophoresis as a separation method prior to both targeted (parallel reaction monitoring – PRM) and untargeted (data-dependent analysis – DDA) tandem mass spectrometry (MS/MS) analyses. This is the first reported use of the ZipChip for alkaloid analysis.
Materials and Methods
HCl, optima H2O, chloroform, and sodium hydroxide were purchased from Sigma Aldrich (St. Louis, MO, USA). Metabolites diluent (H2O, MeOH, ammonium acetate), metabolites BGE (H2O, MeOH, formic acid), and ZipChip HS microfluidic chips were purchased from 908 Devices (Boston, MA, USA). MeOH extracts of L. Cardinalis hairy roots were provided by Naprogenix Inc. (Lexington, KY, USA).
Methanolic extraction of hairy roots
Methanolic extracts of L. cardinalis hairy roots were produced by Naprogenix. Briefly, hairy root cultures were collected, rinsed to remove media, and lyophilized. The lyophilized roots were ground to a fine powder and transferred to amber glass vials with phthalate-free caps. Powdered roots were transferred to a tared glass vial, 5mL of MeOH per 100mg was added, and the vials were agitated for 24 hours. The methanolic extraction was repeated three times. The combined extracts were centrifuged at 20,000xg. The supernatant was collected, transferred to a 3mL amber vial, and the MeOH was removed under a stream of dry nitrogen.
Sample preparation
Dried methanolic extracts of wild type (WT) hairy roots, virally transformed hDAT mutants (DAT+) hairy roots, and induced gain of function DAT+ mutants (MUT) of L. cardinalis hairy roots were obtained by Naprogenix Inc. Aliquots of the dried extracts were weighed and reconstituted in MeOH to produce 1 mg/mL solutions. A 250 μL aliquot of each sample was diluted in 200 μL of optima H2O and acidified with 50 mM HCl to pH 2. The acidified extracts were extracted with 500 μL of CHCl3 and the organic layer was removed. The pH of the aqueous layer was increased to pH 10 with 100 mM NaOH and extracted again with 500 μL of CHCl3. The organic layer was collected and reduced to dryness under a stream of dry nitrogen. The dried CHCl3 extracts were reconstituted in 250 μL of the 908 Devices metabolites diluent and transferred into autosampler vials with 300 μL inserts for analysis. Each sample was analyzed in triplicate.
Microfluidic CZE-MS analysis
The samples were delivered to an HS chip housed within the ZipChip Interface (ZCI) via an attached autosampler. Each sample was delivered to the sample well and loaded into the sample channel for 24 seconds (5.00 nL - default HS Metabolites method). The sample set was analyzed using the predefined standard HS Metabolites method, field strength: 1000 V/cm, pressure assist start time: 2.00 minutes. All separations were performed for 3.00 minutes with approximately three minutes of pre-run time to transfer the sample from the autosampler to the ZCI, totaling approximately 55 minutes of total run time for all nine samples. BGE refreshes were performed between every three samples. The ZCI was coupled to a Q Exactive Orbitrap Mass Spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Acquisition parameters included a full scan range from m/z 70 to 600, mass resolution of 17,500, microscans set to 1 (~8 scans per second), sheath gas flow set to 2.0 psi, and an AGC target of 3e6. The current version of the ZipChip can only be utilized for cation separations, therefore only positive ion mode acquisitions were performed.
Analytical method validation
A purified lobinaline standard was obtained after multiple passes through a phenomenex Luna pHPLC column (10 um C18, 100 A, 100 × 10 mm) on a Hewlett Packard 1100 series HPLC binary pump (G1312A) coupled to a UV detector (G1314A). An isocratic method consisting of 3 mL/min of 70% ACN in water with 0.1% formic acid was utilized for the separation. UV peaks were monitored at 260 nm and 280 nm. The subsequent purified lobinaline standard was used to create a calibration curve for quantification of lobinaline in the plant extracts. Additionally, percent recovery and the method detection limit (MDL) were assessed using this standard. For percent recovery, following assessment of the lobinaline concentration naturally found in the plant extracts, a known amount of lobinaline was spiked into six samples of dry plant material. Spiked material was extracted and analyzed as stated above. The determined concentration of the spiked extracts was compared to the theoretical concentration in order to assess the recovery percentage. For the MDL determination, the procedure established by the Environmental Protection Agency (EPA), EPA 821-R-16-006, was followed. Briefly, seven replicates of lobinaline spiked into surrogate matrices (sedum acre dried plant extracts) at ten times (100 nM) the suspected limit of detection (10 nM) were compared to seven blank surrogate extracts. This procedure was repeated on three separate days. The resulting concentrations of lobinaline in the spiked population was calculated and the standard deviation (s) of the concentrations was used in the following equation.
Data processing via MZmine2 software
MZmine2 data processing was performed as reported by Olivon et al. with modifications [18]. Data files (.RAW) were converted to .mzXML files using the open source MSConvert software prior to MZmine2 processing. Masses were detected from 0.70–2.50 minutes with an exact mass detector and a noise level of 1.0e5. The mass list was then filtered with an FTMS shoulder peak filter using the Gaussian function and a mass resolution of 17,500. Chromatograms were built from the filtered mass lists from 0.70–2.50 minutes with a minimum duration of 0.00 minutes, a minimum height of 1.5e6, and an m/z tolerance of 0.002. The resulting chromatogram lists were then deconvoluted using a local minimum search with the following parameters: a chromatographic threshold of 10.0%, a minimum RT of 0.01 minutes, a minimum relative intensity of 15.0%, a minimum absolute height of 1.0e6, a minimum ratio of peak top/edge of 5, and a peak duration range of 0.01 to 0.10. Following deconvolution, the electropherograms were then deisotoped with an m/z tolerance of 0.002, RT tolerance of 0.1 absolutes minutes, and represented by the most intense ion. Alignment for comparison and data export was performed using the RANSAC alignment tool with tolerances consistent with previous processes along with a threshold value of 1 and the minimum number of points set to 10%. Aligned lists were then gap-filled for peaks with a 25% intensity tolerance and again with the m/z and RT range gap filler. Three separate peak lists were then produced corresponding to 1) all features detected, 2) features present in at least two of the replicates within the population, and 3) features only found in all three replicates for each population.
Targeted and untargeted CZE-MS/MS analysis
For microfluidic CZE separations coupled to MS/MS experiments, the separation times were increased to 6.00 minutes and the initial applied field strength was reduced to 300 V/cm with a linearly increasing field strength gradient from 300 V/cm to 1000 V/cm over the 6.00 minutes. Pressure assist was initiated at the 3.00 minute mark. Untargeted MS/MS analyses were performed via the Xcalibur data-dependent acquisition (DDA, Full MS/dd-MS2) module. Briefly, full scan acquisitions were acquired as listed above. For the dd-MS2 parameters: a Top3 loop count with a 0.5 s dynamic exclusion was applied at 17,500 resolution with a 1e5 AGC target, 8e3 AGC minimum, 50 ms maximum IT, 2.0 m/z isolation window, and a fixed NCE of 30. Due to the ionization character of the alkaloids of interest, doubly charged ions and isotopes were also excluded. An exclusion list was also developed based on a blank full-scan analysis to prevent background ions or those attributed to contaminants in the BGE or diluent from being selected by the Top3 experiment. Targeted MS/MS analyses were performed via the Xcalibur Parallel Reaction Monitoring (PRM) module. An inclusion list formulated based on the average (n=6) retention times and durations for seven of the ions of interest was applied. All applicable MS/MS parameters applied for the DDA were also applied for the PRM experiment. Fragmentation was induced with either 35 or 40 NCE. All analyses were performed in positive ion mode only, as stated previously.
Results and Discussion
The goal of this work was to assess microfluidic CZE-MS as an analytical tool for alkaloid metabolomics screening. Triplicate extracts from WT Lobelia cardinalis hairy roots were analyzed and compared with transformed hDAT-expressing mutant culture extracts and extracts from an hDAT mutant culture that was induced to overproduce protective alkaloids. Example base peak electropherograms for each strain (WT, DAT+, and Mutant) are shown in Figure 1.
Figure 1:
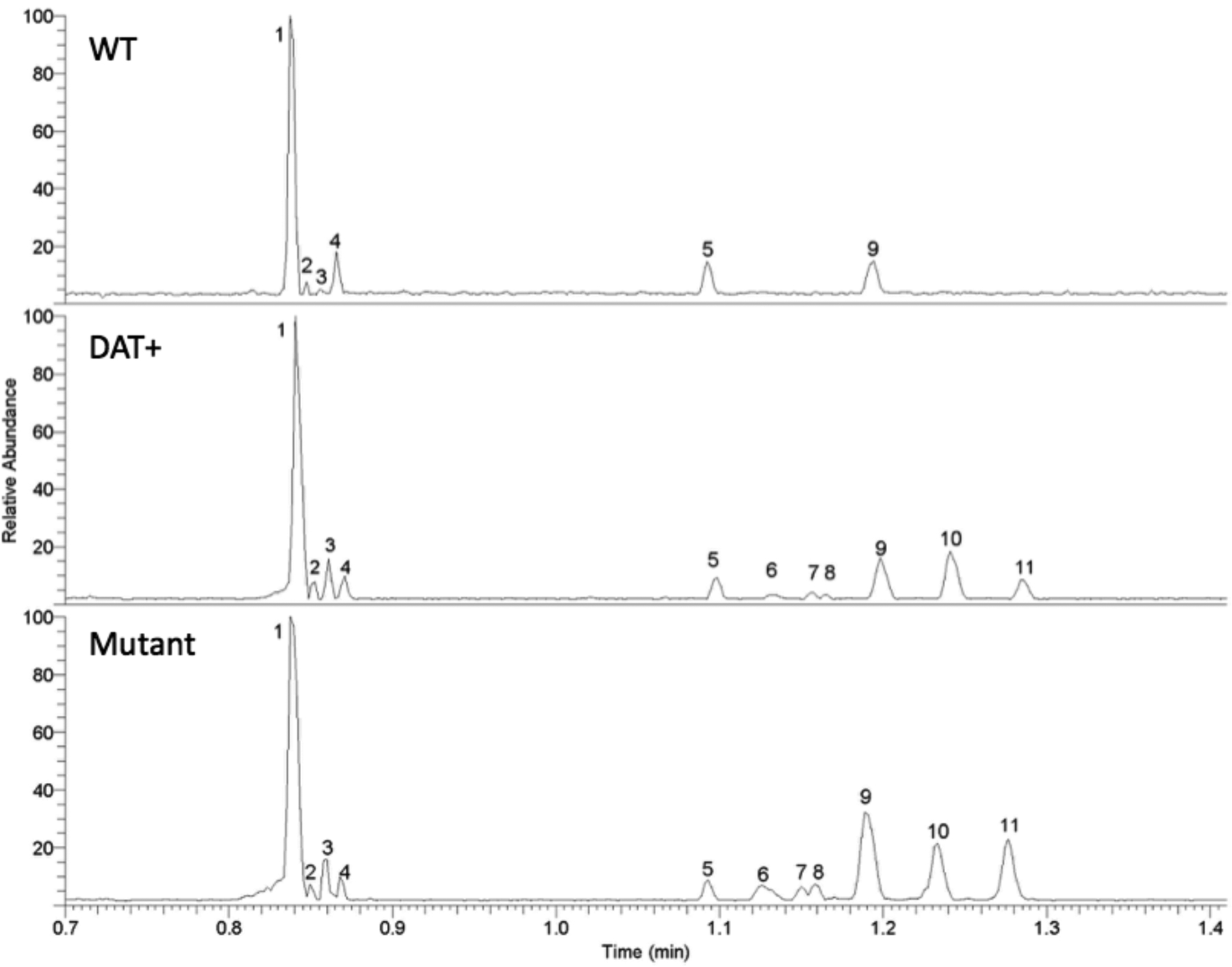
Base peak electropherograms for WT (top), DAT+ (middle), and the mutant (bottom) L. cardinalis extracts resulting from the microfluidic CZE-MS method. Peak numbers correspond to structures in Supporting Information Figure S5 and the data in Table 1.
Prior to any data processing, notable differences were observed between the WT sample compared with the DAT+ and mutant samples. Five additional peaks for the DAT+ and mutant were observable in the base peak electropherogram. It is also noteworthy that all of the observed peaks elute between the 0.7 and 1.5-minute marks making the entire separation approximately ten-times faster than the time required for a standard HPLC separation of similar material.
The intra-day precision of the microfluidic CZE separation method was also evaluated. A WT extract was analyzed in triplicate once immediately preceding the first blank analysis of the day (t=0) and then again in triplicate approximately twelve hours later (t=12) with ~60 analyses in-between on the same HS chip. Twelve hours was chosen as the limit due to the manufacturer stating that the BGE and diluents expire after twelve hours due to pH instability. For this assessment, the area count and retention time of the lobinaline (m/z 387.2795 – C27H35N2) peak was compared. At t=0, the average lobinaline area count was 2.4e10 with an RSD of 14% and at t=12, the average area count was 2.5e10 with an RSD of 3%. Using a paired, two-there was no statistical difference between these two means (p=0.70). A similar trend was observed in the average retention times of the lobinaline peak at 0.76 min (RSD = 1%) and 0.75 min (RSD = 2%) for the t=0 and t=12, respectively. Here, again, no statistical difference was observed via the t-test (p=0.48). This result would suggest that the microfluidic device is similarly comparable to traditional HPLC in terms of reproducibility.
The .RAW data files were converted to .mzXML files and processed using the MZmine2 software following the workflow visualized in Figure S3 to increase the depth of metabolite coverage. Using MZmine2 as a data-mining software, peak lists were generated to allow for direct comparison between the strains both in the number of features detected and in area counts corresponding to each of those features. Prior to peak list generation, mass detection and chromatogram building must be performed.
For this data set, a low noise level was set in order to ensure that low intensity signals corresponding to potential analytes in the samples were not discarded during processing. Any features associated with background noise that were included in the mass list due to the low noise level were processed out in a later step based on their peak duration. Masses were also only detected during the elution window starting with the baseline increase brought about by the elution of the leading electrolyte from the separation channels, approximately 0.70 minutes. Following mass detection and noise filtering, the resulting mass lists were converted into chromatogram lists for processing. Analyte band compaction during the separation and focusing process in the separation channels resulted in sharp, narrow electropherographic peaks with durations less than 0.1 minutes, therefore, the duration parameter in the chromatogram builder module was set to 0.0 minutes. The resulting peak lists were then deconvoluted and deisotoped to remove the aforementioned background and noise-related features and any features that are part of an isotopic distribution. The filtered electropherograms were then aligned using the RANSAC aligner tool to allow for direct comparison of each feature based on common retention times and peak durations. Any individual feature discarded in a prior process was then reclaimed using the gap-filling modules. Lastly, any feature not present in at least two replicates was filtered out using a peak list row filter.
Using MZmine2, 139 unique, resolved peaks were detected that appeared in at least two replicates of the same sample. This strict filtering approach was utilized to reduce noise/spike related peaks and possible laboratory contaminants that were not common to all the samples, especially those within each triplicate set. The WT, DAT+, and mutant sample sets contained 136, 139, and 137 peaks, respectively observed in at least two of the triplicate samples (full feature list with associated area counts in supplementary material - S6). This master peak list was then used to select several known or putatively identified molecules of interest for a statistical comparison list. Lobinaline and lobinaline-like derivatives, henceforth referred to as “lobinalines” were initially set as top priority molecules due to the hDAT inhibitory activity of lobinaline reported previously [13]. Lobinalines all share a common capability to become doubly charged molecules during electrospray ionization experiments. Figure 2 shows an example mass spectrum of lobinaline (Figure 2 (top) – C27H34N2 MW=386.2722, m/z for z: +1 = 387.2795, m/z for z: +2 = 194.1438) and the N-oxide version of lobinaline (Figure 2 (bottom) - C27H34N2O, m/z for z: +1 = 403.2740, m/z for z: +2 = 202.1410) displaying this common charge state motif. Additionally, features considered to share similar biosynthetic precursors to lobinaline, such as putative sedamine-like derivatives, and those found in relatively high abundance within the samples (i.e., visible in the base peak) were also added to the comparison list.
Figure 2:
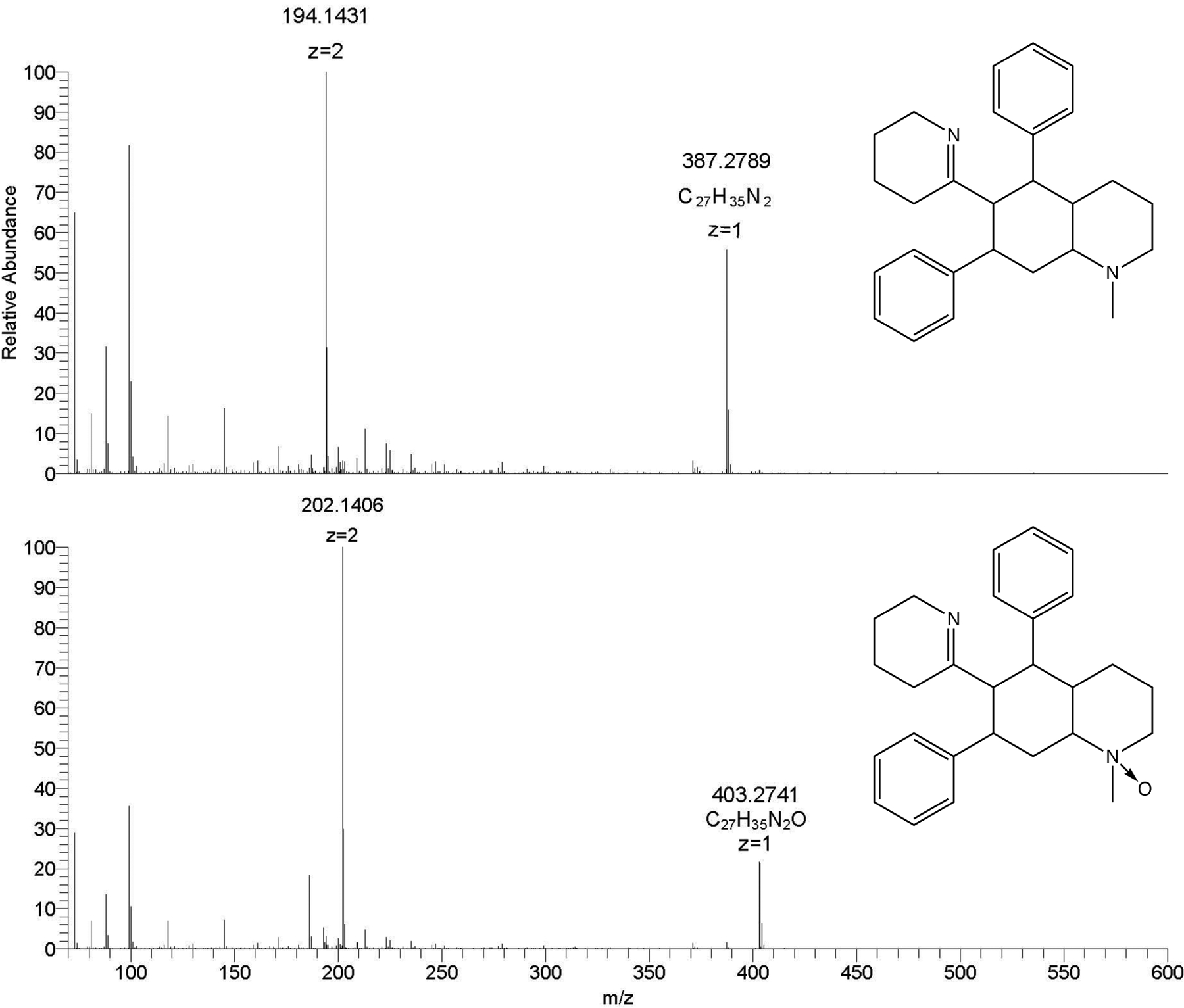
Mass spectra for lobinaline (top) and the lobinaline N-oxide (bottom) displaying the doubly charged properties of bi-nitrogenous alkaloids.
Table S4 displays the final comparison list of all the selected features with the corresponding average area count and relative standard deviation. Correlating putative structures can be observed in Supporting Information Figure S5.
Average area counts for the lobinalines generally display an increasing trend from the WT to the mutant sample sets. This observation fits with the overarching hypothesis that lobinalines are protective agents against MPP+ toxicity used in L. cardinalis mutant selection. The observed abundance increases for the N-oxide (m/z 403.2740 – peak 3) and bi N-oxide (m/z 419.2693) versions of lobinaline are very promising since N-oxides tend to increase hydrophilicity, making both molecules excellent targets for further study.
Structures for both m/z 130.1595 (peak 3) and m/z 200.2362 (peak 4) are putative and drawn as primary amines for simplicity. Alternatively, secondary or tertiary structures would be equally probable. It should be noted that m/z 129 [M+•] has been previously reported in a methanolic plant extract analyzed by GC-MS therefore our observation of m/z 130.1595 [M+H+] was not deemed a laboratory contaminant [19]. Lastly, the lobeline-like molecules, peaks 5–11 (see Table 1 and Supporting Information Figure S5), also displayed a generally increasing trend. Though lobeline (8,10-diphenyllobelionol - C22H27NO2) itself was not observed in these samples and has not been found in extracts of intact plant material of L. cardinalis in the past, both lobinaline and lobeline derive from similar precursors [13]. Molecules very similar to these have been reported in the literature to have activity both on the DAT and on nicotinic acetylcholine receptors (nAChRs) [13].
Table 1:
Average area counts and %RSD for each of the most abundant features corresponding to the peaks in figure 1.
| Peak | Primary Feature m/z (charge) | Putative EF | Avg Area Count - WT | Avg Area Count - DAT | Avg Area Count - Mutant |
|---|---|---|---|---|---|
| 1 | 387.2795 (z=1), 194.1415 (z=2) | C27H35N2 | 1.75E+09 | 3.45E+09 | 5.23E+09 |
| 2 | 399.2822 (z=1), 200.1446 (z=2) | C27H31N2O | 7.28E+07 | 1.57E+08 | 1.60E+08 |
| 3 | 403.2740 (z=1), 202.1410 (z=2) | C27H35N2O | 8.38E+07 | 3.84E+08 | 5.31E+08 |
| 4 | 130.1595 (z=1) | C8H20N | 1.43E+08 | 1.51E+08 | 1.72E+08 |
| 5 | 200.2363 (z=1) | C13H30N | 1.78E+08 | 2.03E+08 | 2.19E+08 |
| 6 | 242.2115 (z=1) isomer 1 | C14H28NO2 | 8.49E+07 | 1.69E+08 | 4.09E+08 |
| 7 | 228.1967 (z=1) | C13H26NO2 | 7.17E+07 | 1.77E+08 | 3.03E+08 |
| 8 | 242.2115 (z=1) isomer 2 | C14H28NO2 | 5.60E+07 | 1.00E+08 | 2.58E+08 |
| 9 | 242.2115 (z=1) isomer 3 | C14H28NO2 | 3.24E+08 | 5.22E+08 | 1.43E+09 |
| 10 | 258.2437 (z=1) | C15H32NO2 | 1.03E+08 | 8.22E+08 | 1.28E+09 |
| 11 | 272.2574 (z=1) | C16H34NO2 | 6.82E+07 | 3.82E+08 | 1.15E+09 |
Preparative HPLC was used to isolate and purify a sample of lobinaline that was used to develop calibration standards for analysis using the microfluidic device (Figure 3). Based on the resulting calibration curve, the WT, DAT, and mutant extract samples were found to contain 83±13 μM, 168±13 μM, and 256±15 μM lobinaline, respectively (calculations for error in concentration values can be found in the supplementary material S7). To translate these concentrations into a whole plant basis, 1.64 g (wet weight) of hairy roots yielded 68 mg of methanol extractable material or 4.14% of the total mass of extractables. Lobinaline comprised 3.2% of the methanol extractable material for WT or 0.13% of the plant (wet weight basis). Similarly, lobinaline comprised 6.5% of the methanol extractable material for DAT (0.27% plant basis), and 9.8% of the methanol extractable material for the mutant (0.41% plant basis).
Figure 3:
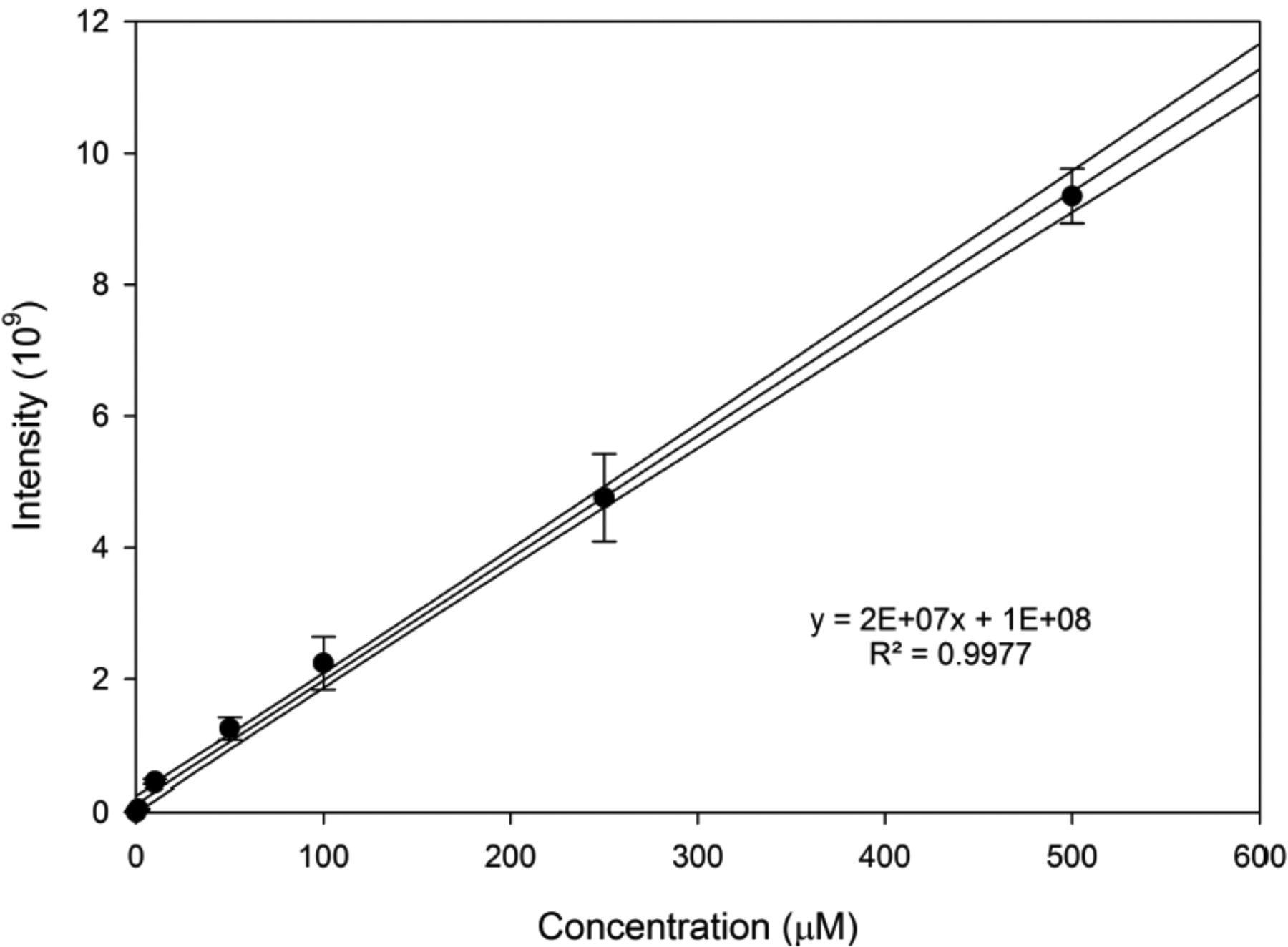
Calibration curve for lobinaline analysis and quantification.
Additionally, the purified standard was also used to assess both the percent recovery of the extraction method and the method detection limit. The percent recovery was determined to be 99±22% based on the recovery of lobinaline spiked into three separate WT samples and three separate mutant samples (6 total samples). The variation observed in the recovery is most likely due to percent moisture (not determined) in the plant material. For the MDL determination, lobinaline was spiked into powdered Sedum acre plant material at a concentration of 100 nM. Following the EPA guidelines for MDL determination, the MDL for this method was determined to be 74 nM.
To make a global comparison with WT, variations in isolation, extraction, and analysis of all components must be controlled. Borrowing from quantitative western blot analysis, we evaluated the use of a “loading control” to normalize our data. All nine data sets (3 each from WT, DAT+, and mutant) were compiled and the variance for each of the detected peaks was calculated. The putative monoamine (m/z 200.2363 – peak 5) was found to have the lowest variance and was used as a normalization standard. Several bar graphs displaying normalized area counts for the prioritized features can be seen in Figure 4 (all sixteen graphs can be found in the supplementary data S8). Normalized area counts for all 139 features can also be seen in the supplementary material (Table S9). Statistical analyses were performed on the prioritized list using the GraphPad Prism 8 software. Tukey’s multiple comparison test was utilized as a post-hoc test for the selected features of interest. Statistically significant differences were observed for pairwise comparisons in fourteen of the sixteen prioritized features. Figure 4 (supplementary data Figure S8) also displays the post-hoc test result in significant differences denoted by stars (★) under the corresponding bar graphs.
Figure 4: Normalized area counts for three of the sixteen prioritized peaks.
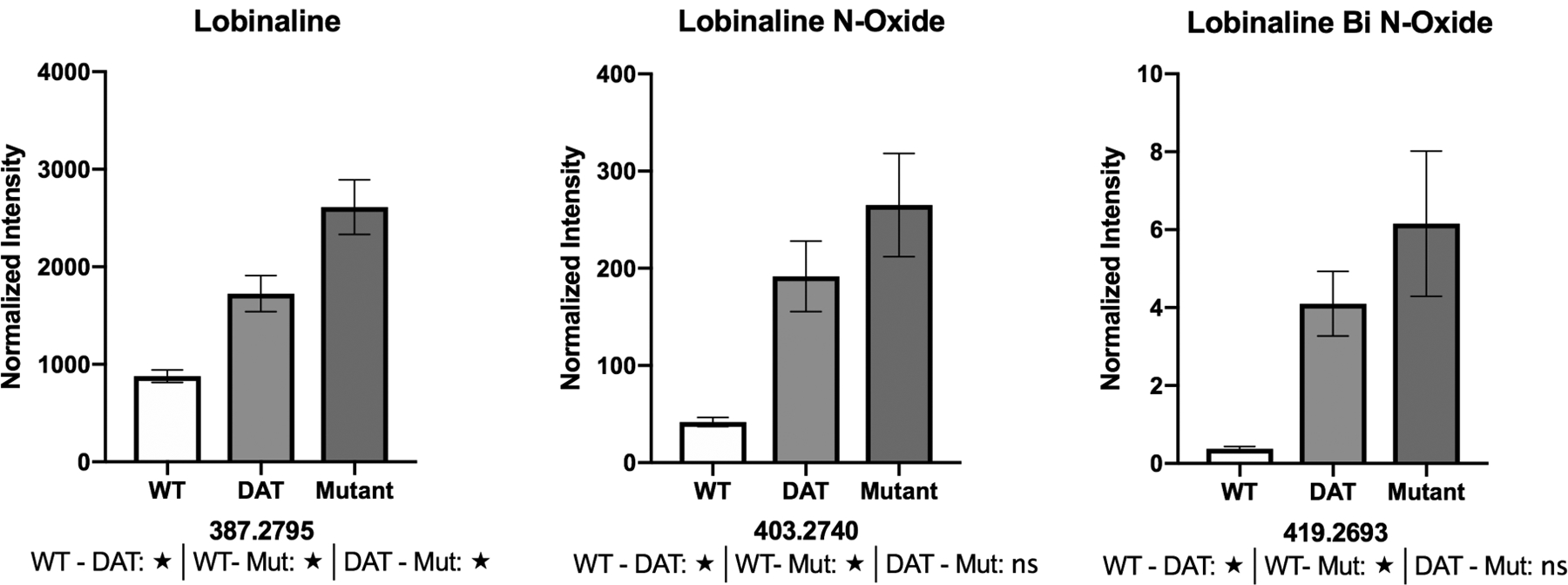
★-statistically significant based on post-hoc Tukey’s multiple comparisons test (ns- not significant).
From the resulting data set and statistical analyses, multiple putative features have been selected as the potential source of the observed hDAT inhibitory activity. Cultures currently being propagated on 15N isotopically labeled media will be extracted and used for future subsequent quantitative assays with this method.
Following the initial screening for ions of interest and evaluation of the microfluidic device, tandem mass spectrometry methods were developed to acquire evidence to support the putative structures. Here, both targeted and untargeted MS/MS was applied for analysis of the mutant plant extract (Mutant). Prior to any MS/MS experiments, adjustments to the separation method were made to increase the time in which ions were present to allow for adequate sampling of each ion. The applied field strength drives the separation event and analyte band compactions in the microfluidic chip therefore lower field strengths were utilized to broaden the analyte peaks. At the standard field strength of 1000 V/cm, for example, the lobinaline peak is 2.4 seconds wide, approximately 21 scans, under the most inclusive MS acquisition parameters. Decreasing the field strength to 300 V/cm increases the peak to over 6 seconds wide consisting of 74 scans. This also changes the retention time, however, increasing it from 0.76 minutes to 2.87. Reducing the field strength to this level also caused the slower moving ions to remain at the end of the separation channel and elute in subsequent analyses, regardless of the application of the pressure assist feature. To address this, a field strength gradient was used. By increasing the field strength linearly from 300 to 1000 V/cm over the 6 minute separation, peak widths were at least doubled and no carry-over was observed. The development of this separation method allowed the DDA and PRM experiments to be performed. For the targeted MS/MS PRM inclusion list development, seven of the ions of interest were selected: lobinaline and the mono- and bi- N-oxide versions and the lobeline-like molecule. Tandem spectra can be found in the supplementary material (S9). Six full scan acquisitions were acquired to record peak durations for each ion of interest. Using the average durations, the peaklist table was constructed to minimize multiplexing (more than four ions at once) while also preventing under-sampling (number of MS/MS scans for each ion: >5). To compare the efficiency of these parameters, the average number of MS/MS scans for each feature under both the PRM and DDA experiments was compared to the average number of scans in which that feature was present in the full scan acquisition. Table 2 displays this comparison.
Table 2:
Comparison of the number of times each feature was selected for both the targeted (PRM) and untargeted (DDA) MS/MS analyses compared to the average number of scans each was detected in during the full scan acquisitions.
| m/z | FS | PRM-1 | Ave | Effciency (Avg/FS) | DDA-1 | Ave | Effciency (Avg/FS) |
|---|---|---|---|---|---|---|---|
| 387 | 30 | 15 | 14 | 47% | 6 | 5 | 18% |
| 403 | 21 | 14 | 11 | 52% | 3 | 2 | 10% |
| 419 | 19 | 11 | 7 | 37% | 0 | 0 | 0% |
| 228 | 91 | 57 | 56 | 61% | 12 | 9 | 10% |
| 242 | 37 | 23 | 16 | 44% | 0 | 4 | 11% |
| 242 | 18 | 15 | 13 | 72% | 0 | 4 | 22% |
| 242 | 30 | 20 | 21 | 69% | 0 | 5 | 17% |
| 258 | 10 6 |
26 | 24 | 23% | 13 | 11 | 11% |
| 272 | 95 | 24 | 22 | 23% | 12 | 10 | 11% |
| 48% | 12% |
The PRM experiment resulted in >40% coverage of the ions compared to their prevalence in the full scan experiments. Those with <25% coverage most likely eluted from the separation channels during multiplexing events, specifically when alternating between three or four ions at once, or outside of the calculated window. The latter is more likely, due to retention time inconsistencies being prevalent in CE mobility experiments. Generally, with the PRM approach, under-sampling did not occur as each ion was analyzed more than five times other than m/z 419 and the first eluting m/z 242 isomer in the third trial. Conversely, the DDA analysis did not yield similar results. Despite the usage of the exclusion list and a relatively short dynamic exclusion time, on average only five of the nine ions of interest were sampled adequately. In some cases, those with low abundance, such as m/z 419, were missed entirely. These results suggest that microfluidic CZE is better suited for the targeted MS/MS approach rather than the untargeted MS/MS approach. While useful for major metabolites with substantial peaks in the sample, the untargeted approach is not recommended for the identification of trace components via MS/MS with the microfluidic device as even broadened bands are missed by the selection algorithm.
Concluding Remarks
The microfluidic CZE-MS device allowed for both a fast and sensitive high-throughput method for screening of plant alkaloids. Previous alkaloid MS screening assays traditionally rely on HPLC or GC as a separation method. These methods can be time consuming and can also lack the capability of isomeric separation. Here, the utilization of the microfluidic chip displayed that more efficient separation can be achieved in approximately half the time or more compared with other methods with a total run time from injection to sample well washing in less than eight minutes and separations being completed in less than three minutes. When coupled with processing software like MZmine2, the process is further streamlined resulting in a fast, comprehensive assay for plant secondary metabolites. These characteristics are particularly valuable for the use described here in which it is essential to rapidly compare alkaloid profiles in many different plant extracts in very small sample volumes. This method enables the identification and separation of novel putatively bioactive metabolites from transgenic mutant plant cells. These metabolites then become a novel source of lead compounds for the pharmaceutical industry.
Supplementary Material
Acknowledgements
The authors gratefully acknowledge support from the National Institute on Alcohol Abuse and Alcoholism of the National Institutes of Health under the award number R44AA025804. Additionally, ZDK and BCL acknowledge the UK Office of Research for funding the acquisition of the ZipChip equipment.
References:
- 1.Jiang M, Severson KA, Love JC, Madden H, Swann P, Zang L, Braatz RD, Biotechnol. Bioeng 2017, 114, 2445–2456. [DOI] [PubMed] [Google Scholar]
- 2.Petruczynik A, Cent. Eur. J. Chem 2012, 10, 802–835. [Google Scholar]
- 3.Nardin T, Piasentier E, Larcher R, Drug Test. Anal 2017, 10, 1–26. [DOI] [PubMed] [Google Scholar]
- 4.Nardin T, Piasentier E, Larcher R, J. Mass Spectrom 2016, 51, 729–741. [DOI] [PubMed] [Google Scholar]
- 5.Beri J, Kirkwood KI, Muddiman DC, Bereman MS, Anal. Bioanal. Chem 2018, 410, 2597–2605. [DOI] [PubMed] [Google Scholar]
- 6.Mellors JS, Gorbounov V, Ramsey RS, Ramsey JM, Anal Chem 2008, 80, 6881– 6887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.908 Devices. Tech Note 1.0 ZipChip: What They Are & How They Work 1–9. [Google Scholar]
- 8.Khatri K, Klein JA, Haserick JR, Leon DR, Costello CE, Mccomb ME, Zaia J, Anal Chem. 2017, 89, 6645–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Feng P, Sosic Z, Zang L, Anal J. Bioanal Tech. 2017, 8, 1–8. [Google Scholar]
- 10.Chen C, Feng H, Guo R, Li P, Laserna AKC, Ji Y, Ng BH, Li SFY, Khan SH, Paulus A, Chen S, Karger AE, Wenz M, Ferrer DL, Huhmer AF, Krupke A Cogent Chem. 2018, 4, 1–13. [Google Scholar]
- 11.Zhang Y, Wang Y, Sosic Z, Zang L, Bergelson S, Zhang W, Anal. Biochem 2018, 555, 22–25. [DOI] [PubMed] [Google Scholar]
- 12.Brown DP, Rogers DT, Gunjan SK, Gerhardt GA, Littleton JM, J. Biotechnol. 2016, 238, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown DP, Rogers DT, Pomerleau F, Siripurapu KB, Gerhardt GA, Littleton JM, Hall K, Fitoterapia. 2016, 111, 109–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gupta RN, Spenser ID, Chem. Commun 1966, 24, 4–5. [Google Scholar]
- 15.Manske R, Can. J. Res 1938, 16, 445–448. [Google Scholar]
- 16.Robinson M, Pierson W, Dorfman L, Lambert B, Lucas R, J. Org. Chem 1966, 31, 3206–3213. [DOI] [PubMed] [Google Scholar]
- 17.Pluskal T, Castillo S, Villar-Briones A, Orešič M, BMC Bioinformatics. 2010, 11, 395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olivon F, Grelier G, Roussi F, Litaudon M, Touboul D, Anal Chem. 2017, 89, 7836–7840. [DOI] [PubMed] [Google Scholar]
- 19.Wong F, Yong A, Sim K, Ong H, Trop J Pharm Res. 2014, 13, 1085–1092. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.