

Abstract
Introduction:
The Ca2+release-activated Ca2+ (CRAC) channel, composed of Orai and STIM proteins, represents one of the main routes of Ca2+ entry in most non-excitable cells. There is accumulating evidence to suggest that CRAC channel can influence various processes associated with tumorigenesis. Overexpression of CRAC channel proteins has been observed in several types of cancer tissues and cells, indicating that blocking CRAC channel activated Ca2+ influx can have therapeutic benefits for cancer patients.
Areas covered:
In this review, we have primarily focused on the molecular composition and activation mechanism of CRAC channel as well as the myriad roles this Ca2+ channel play in various cancers. We further describe relevant information about several efforts aimed at developing CRAC channel blockers and their likely implications for cancer therapy. We have extensively utilized the available literature on PubMed to this end.
Expert opinion:
The possibility of targeting CRAC channel mediated Ca2+ entry in cancer cells has generated considerable interest in recent years. Use of CRAC channel blockers in cancer preclinical studies and clinical trials has been relatively limited as compared to other diseases. The future lies in developing and testing more potent and selective drugs that target cancer cell specific CRAC channel proteins, hence opening better avenues for cancer therapeutic development.
Keywords: Calcium, Cancer therapeutics, CRAC channel, CRAC channel inhibitor, Orai1, STIM1, Store operated Ca2+ entry
1. Introduction
As one of the most multifaceted, ubiquitous and dynamic intracellular secondary messengers, calcium (Ca2+) is known to be involved in regulating multiple cellular processes, including cell proliferation, differentiation, apoptosis, migration, as well as cell contraction and secretion [1,2]. It is believed that dysregulation of Ca2+ cellular homeostasis could result in several pathological conditions such as cardiovascular diseases, neurodegenerative ailments, developmental abnormalities, diabetes, immunodeficiency disorders and cancer [3,4]. Maintaining the cellular homeostasis of Ca2+ depends on the synchronized functioning of various calcium channels, exchangers and pumps, which are located either on the plasma membrane of the cell or the membranes of endoplasmic reticulum and mitochondria [5,6].
One of the most important mechanisms of Ca2+ entry into the mammalian cell is known as the store operated Ca2+ entry (SOCE). SOCE, which results in an increase in the cytoplasmic Ca2+ concentration, is triggered by the release of Ca2+ stored in endoplasmic reticulum (intracellular Ca2+ store) and the subsequent entry of extracellular Ca2+ across the cell membrane through Ca2+ channels, that are referred to as calcium release activated calcium (CRAC) channels [7–9]. Ca2+ influx through CRAC channels causes the activation of various downstream signaling pathways which are involved in the regulation of gene expression, cell proliferation, apoptosis, cytokine generation and many other cellular activities [10]. CRAC channels are present widely across various cell types [11–16]. They possess extremely low conductance in comparison to other Ca2+ channels, while being highly selective for Ca2+ [17]. Several studies have linked altered CRAC channel activity with various ailments, such as thrombosis, pancreatitis, inflammatory bowel disease, severe combined immunodeficiency disorder, as well as cancer [18–23].
Cancer is one of the major causes of mortalities across the globe [24] and therefore, there is an ever-increasing need for identifying novel cellular and molecular components that can be targeted for effective therapy. In this regard, Ca2+ activated downstream signal transduction pathways have been considered promising cancer therapeutic targets [1]. Cancer cells, unlike their non-cancerous counterparts, are known to undergo continual Ca2+ signal remodeling due to changes in the activity and expression of calcium pumps and channels [25], as also alterations in the Ca2+ associated intracellular signaling pathways. It is believed that such a strategy is adopted by cancer cells in order to sustain their rapid growth and to circumvent cell death mechanisms [26]. Therefore, a clear understanding of Ca2+ signal remodeling and alterations in calcium channel protein expression in cancer would set the scene for development of new therapeutic drugs targeting cancer cells.
In this article, we review the relevant literature on the molecular identities of CRAC channel in context of the roles they play in cancer development. We also provide a critical overview of the therapeutic potential of various CRAC channel inhibitors for cancer treatment. Although numerous CRAC channel inhibitors have been developed over the years and their effects subsequently investigated on various humans diseases, we have limited ourselves to discussing only those inhibitors that have been reported to show some promising results on cancer models in preclinical studies or those that have reached clinical trials on cancer patients.
2. CRAC channel: molecular basis and mode of activation
CRAC channel comprises of a combination of two protein components - Orai and STIM. Orai, which is a tetra-spanning transmembrane protein, is situated on the cell membrane and is responsible for forming the ion channel pore through which extracellular Ca2+ enters the cell. It has three homologues, namely Orai1, Orai2 and Orai3. On the other hand, STIM (stromal interaction molecule) protein, which functions both as a sensor of endoplasmic reticulum Ca2+ levels as well as an activator of SOCE, is a single-pass transmembrane protein located on the membrane of endoplasmic reticulum. STIM is known to have two isoforms, STIM1 and STIM2. The combination of Orai1 and STIM1 is the prototypical, best characterized CRAC channel and is present in most of the mammalian cells [10,23].
Despite sharing significant structural homology, STIM1 and STIM2 exhibit remarkable differences in their properties and functionalities. Unlike STIM1, STIM2 can be activated by minor alterations in Ca2+ levels owing to a lower affinity towards Ca2+ [27]. Therefore, STIM1 is considered the primary sensor of Ca2+ concentration in the lumen of endoplasmic reticulum which enhances CRAC channel activity as a result of fast and considerable depletion of intracellular Ca2+ stores. In contrast, STIM2 stabilizes the level of Ca2+ following a slow and moderate depletion of Ca2+ stores and thereby acts as a housekeeping Ca2+ sensor in the endoplasmic reticulum [28,29]. Also, STIM1 is reportedly a more efficient activator of CRAC channel activity, possibly due to its rapid aggregation kinetics and stronger ability to associate with Orai [30,31]. In fact, all the three types of Orai proteins can complex with STIM1 to form functional CRAC channels. However, there is variation in their tissue distribution and Ca2+ selectivity [32]. It has also been reported that while Orai2 and Orai3 can be activated by the depletion of Ca2+ stores, the CRAC currents induced by them are weaker than those induced by Orai1 [33].
Ca2+ signaling in the cells gets activated either by the release of Ca2+ stored in the endoplasmic reticulum or the influx of extracellular Ca2+ into the cells. CRAC channel functions to combine and coordinate these two routes through which intracellular Ca2+ signaling may be induced. Figure 1 shows the mechanism of their activation under physiological conditions. The first step in the activation of CRAC channel involves the stimulation of cell surface receptors, such as G-protein coupled receptors (GPCRs) or receptors tyrosine kinases (RTKs), through binding of their respective ligands (agonists), that activates membrane-bound enzyme phospholipase C (PLC), which in turn hydrolyzes membrane phospholipid phosphatidylinositol-4,5-bisphosphate (PIP2) into inositol triphosphate (IP3) and diacylglycerol (DAG). The subsequent binding of IP3 to the Ca2+ permeable IP3 receptors located on the endoplasmic reticulum membrane elicits the release of Ca2+ from the endoplasmic reticulum into the cytosol [34,23]. This results in the depletion of Ca2+ levels in the lumen of endoplasmic reticulum, which is eventually sensed by STIM proteins. Consequently, STIM proteins respond by oligomerizing and translocating to the junction of endoplasmic reticulum and plasma membrane where they form large clusters, termed as STIM puncta. A cytoplasmic CRAC activation domain (CAD) on the C terminus of STIM directly interacts with the intracellular C terminus of Orai [35, 36]. This physical coupling of STIM and Orai proteins induces the opening of CRAC channel, thereby allowing the influx of extracellular Ca2+ into the cytosol. Ultimately, such an influx of Ca2+ facilitates the replenishment of the depleted intracellular Ca2+ stores, in addition to evoking the activation of the Ca2+ associated signal transduction pathways.
Figure 1.
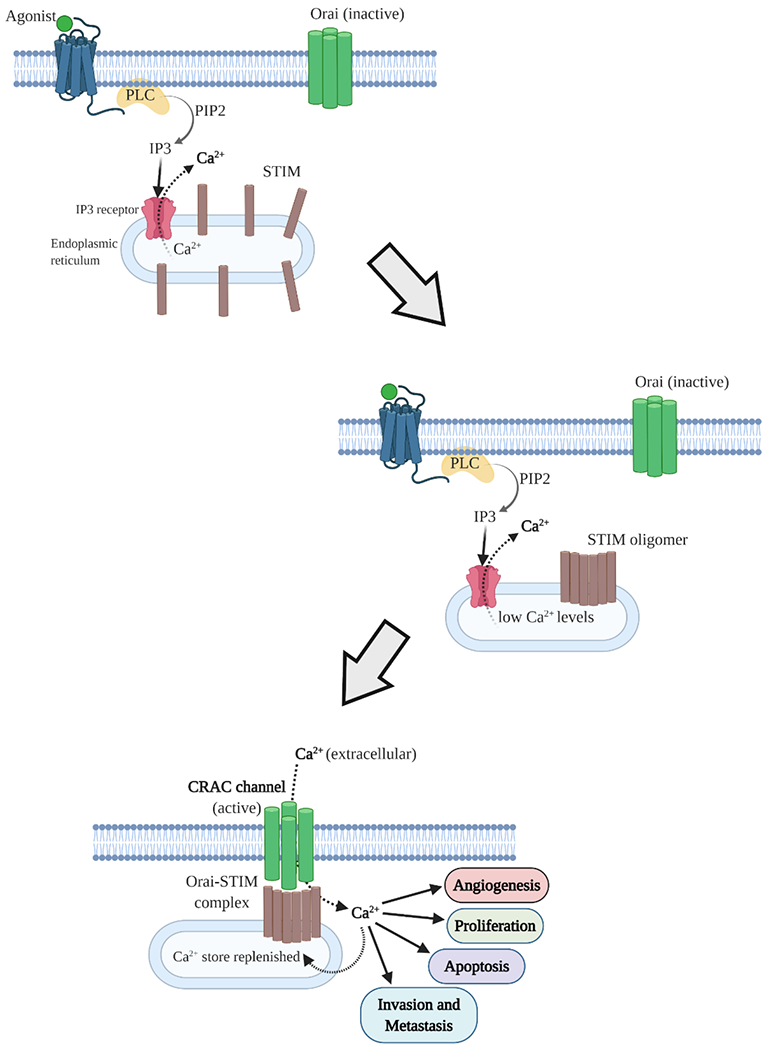
Schematic diagram showing the mechanism of CRAC channel activation. Binding of their corresponding agonists (ligand) stimulates diverse plasma membrane receptors which activate Phospholipase C (PLC). PLC then generates inositol triphosphate (IP3) from phosphatidylinositol-4,5-bisphosphate (PIP2). IP3 binds to the IP3 receptor on the endoplasmic reticulum and triggers Ca2+ efflux. STIM proteins, which are evenly distributed on endoplasmic reticulum membrane at resting state, sense the drop in Ca2+ levels in endoplasmic reticulum lumen and undergo rapid conformational changes that enable them to bind with Orai proteins. This results in the functional activation and opening of CRAC channel through which extracellular Ca2+ enters the cell. Sustained increase in intracellular Ca2+ not just refills the endoplasmic reticulum Ca2+ stores but also activates downstream signal transduction pathways that regulate various cellular processes.
Activation of CRAC channel, as described above, brings about a sustained increase in cytosolic Ca2+ levels that result in the activation of calmodulin (CaM), an intracellular Ca2+ sensor protein. Calmodulin, in turn, activates calcineurin, a Ca2+/CaM dependent phosphatase. Calcineurin further causes the dephosphorylation of several phosphoserines located in the regulatory domain of NFAT (nuclear factor of activated T cells). This results in the translocation of NFAT into the nucleus, where it collaborates with various transcription factors to initiate the expression of several genes that are involved in the regulation of multiple cellular processes such as proliferation, survival, migration and angiogenesis [37]. Ca2+ entry through CRAC channels is also known to activate other transcription factors like NF-kB (nuclear factor-kB) and CREB (cAMP responsive element binding protein). NF-kB is activated through activation of IKK (IkB kinase), whereas CREB gets activated via CaMKII/IV (calmodulin dependent protein kinase II/IV) activation. NF-kB plays a critical role in different cellular phenomena including, carcinogenesis and inflammation [38]. Ca2+ mediated activation of CREB has been reported to enhance the proliferation of vascular smooth muscle cells [39].
3. CRAC channel and cancer
SOCE mediated through the activation of CRAC channels is considered to be the major route of Ca2+ entry in non-excitable cells [40,41]. Interestingly, elevated intracellular Ca2+ levels have been reported in various human and animal cancer cells [42], suggesting the possibility of an augmented Ca2+ entry through CRAC channels in malignant cells. Also, it is now well established that the Ca2+ influx mediated by CRAC channels and its ensuing signal transduction play quite significant and diverse roles in various cellular events associated with cancer development. These include regulation of cell cycle, cell proliferation, cell death, cell migration, invasion and metastasis. In addition, it also regulates tumor neovascularization and antitumor immunity [43].
A concise summary of the aberrant expression of CRAC channel proteins and their consequential effects as reported by several studies on different types of cancers is given in Table 1. Evidence from a considerable body of literature about the role of CRAC channel proteins in regulating some key cancer hallmarks for various types of cancers are enumerated below.
Table 1:
Altered expression of CRAC channel proteins and their reported effects in different types of cancers
| Cancer type | Cancer model | Change in expression of CARC channel protein | Effect | Reference |
|---|---|---|---|---|
| Breast | Microarray profiles of 295 human breast tumors | High STIM1, low STIM2 | Associated with poor prognosis | 45 |
| Breast | MDA-MB-231, MC-7, T47D, BT-483 | Elevated Orai1 | Remodeling of Ca2+ influx associated with invasive stimuli | 45 |
| Breast | MCF-7 cells and cancerous breast tissue samples | High Orai3 expression | Promotes cell proliferation, cell cycle progression and apoptosis resistance | 49 |
| Cervical cancer | primary cervical cancer tissues | Overexpression of STIM1 | Reduces five-year survival rate; Promotes tumor cell growth, cell cycle progression, migration, invasion and angiogenesis | 50 |
| Ovarian cancer | A2780cis (cisplatin resistant) cells | High Orai1 and STIM1 expression | Contributes to therapy resistance | 52 |
| Renal Cancer | ccRCC tissues | High expression of Orai1 | Promotes cell proliferation and migration | 53 |
| Colorectal cancer | Patient tumor tissue specimen | STIM1 overexpression | Increases tumor size, invasion and migration | 54 |
| Colorectal cancer | SW620 and LOVO cells | STIM1 overexpression | Promotes metastasis and EMT | 55 |
| Colorectal cancer | Tissue microarray of 90 CRC patients | STIM1 overexpression | Indicative of poor prognosis | 55 |
| Gastric cancer | Tumor tissues from 327 GC patients | High Orai and STIM1 expression | Correlates with frequent recurrence and high mortality rate | 57 |
| Liver cancer | Hepatoma tissues and HCC cell lines | Higher expression of STIM1 | Correlates with high metastatic potential in patients and higher migration ability in cell lines | 58 |
| Liver cancer | Hepatocarcinoma tissues | Elevated Orai1 expression | Serves as predictor of 5-FU sensitivity | 59 |
| Pancreatic cancer | Panc-1 cells | High expression of Orai1 and STIM1 | Contributes to apoptosis resistance | 60 |
| Esophageal cancer | Tumor tissues from ESCC patients | Elevated expression of Orai1 | Linked to poor overall and recurrence-free survival | 61 |
| Lung cancer | NSCLC tumor tissues | Higher STIM1 expression | Promotes tumorigenesis | 62 |
| Lung cancer | Tumor samples from 60 lung adenocarcinoma patients | Orai3 overexpression | Correlates with high tumor grade | 63 |
| Glioblastoma | U251 cells | Higher expression of STIM1 | Promotes cell proliferation and invasion | 65 |
| Glioblastoma | Primary human cell lines established from glioblastoma surgical samples | Increased Orai1 levels | Promotes cell invasion | 66 |
| Glioblastoma | Tumor tissues from 61 glioma patients | Elevated Orai expression | Correlates with high tumor grade | 67 |
| Glioblastoma | Human glioma cell lines U251, SNB19, U87, and LN229 | Elevated Orai expression | Promotes cell migration and invasion | 67 |
| Leukemia | HL60 cells | High expression of Orai2 | Promotes cell migration | 68 |
| Multiple myeloma | Tumor tissues from 60 Multiple Myeloma patients | Increased expression of STIM1 and Orai1 | Associated with poor prognosis | 69 |
| Multiple myeloma | KM3 and U266 cells | Increased expression of STIM1 and Orai1 | Promotes cell growth and apoptosis resistance | 69 |
3.1. Breast cancer
Breast cancer cells remodel the expression and functional role of the molecular components of CRAC channel [44]. A microarray data analysis has shown that the breast cancer subtype with the poorest prognosis is linked to high STIM1 and low STIM2 tumor expression profile, indicating that alteration in CRAC channel pathway may be associated with poor prognosis in breast cancer patients [45]. A recent study has reported pro-survival and anti-apoptotic effects of Orai1 in T47D and MCF-7 breast cancer cells grown on a collagen coated surface [46]. The essential role of Orai1 and STIM1 in breast cancer cell migration and metastasis has been delineated by Yang et al. [22]. Silencing STIM1 or ORAI1 in highly metastatic human breast cancer cells or treatment with a pharmacological inhibitor of CRAC channel resulted in reduced tumor metastasis in mice. The study further suggested that CRAC channel might act as a potential target for cancer therapeutics.
It has been shown that Orai3 and STIM1/STIM2 are involved in mediating SOCE in estrogen receptor positive breast cancer cells, while estrogen receptor negative breast cancer cells predominantly relied on the canonical Orai1/STIM1 CRAC channel to bring about SOCE [47]. The same group later demonstrated that silencing Orai3 could cause reduction in the anchorage-independent growth and the invasion of MCF-7 breast cancer cells (estrogen receptor positive), while having no such effect on MDA-MB-231 cells (estrogen receptor negative) [48]. Evidence for the role of Orai3 mediated SOCE in breast cancer cell growth and apoptosis resistance was provided by another study [49]. When compared with normal breast tissues and normal breast epithelial cell line (MCF-10A), the expression of Orai3 was found to be elevated in breast cancer tissue samples and breast cancer cell line (MCF-7). Furthermore, Orai3 silencing led to inhibition of MCF-7 cell proliferation and cell cycle arrest at G1 phase. Mechanistically, this was attributed to a decline in the expression of cyclins E/D1 as well as cyclin-dependent kinases (CDKs 4/2) and a concomitant accumulation of cyclin-dependent kinase inhibitor p21(Waf1/Cip1) and tumor suppressor protein p53. Silencing of Orai3, however, did not affect cell proliferation, cell cycle progression and the expression of cyclins D1/E, CDKs 4/2 and p21(Waf1/Cip1) in MCF-10A cells. Taken together, these reports underscore the significance of Orai3 CRAC channel as a selective therapeutic target for estrogen receptor positive breast cancers.
3.2. Gynecologic cancers
In a seminal study, Chen et al. [50] have identified the role of STIM1 in cervical cancer. Their clinical investigation revealed that STIM1 protein was overexpressed in the primary cervical cancer tissue of more than 70% of the patients in comparison to the adjoining non-malignant tissue from the same patient. Moreover, the expression of STIM1 correlated with tumor size and the clinical outcome. The five-year survival rate was considerably lower in cervical cancer patients with high expression of STIM1. This comprehensive study further demonstrated that silencing of STIM1 could inhibit proliferation of cervical cancer cells by inducing cell cycle arrest at the S and G2/M phases. In addition, the overexpression of STIM1 was found to promote migration of cervical cancer cells, while its knockdown reduced such invasive migration. Also, high STIM1 expression correlated with tumor growth, local invasion and angiogenesis in animal models.
It has also been shown that the activity of CRAC channel mediated SOCE fluctuates during cell cycle progression, where SOCE plays an important role in controlling G1/S cell cycle transition in cervical cancer cells [51]. Enhanced SOCE resulting from an increased expression of CRAC channel proteins ORAI1 and STIM1 has been reported in cisplatin-resistant ovary carcinoma cells, suggesting that CRAC channel could be a contributing factor toward therapy resistance in ovarian carcinoma [52].
3.3. Renal cancer
A study on clear cell renal cell carcinoma (ccRCC) has implied that SOCE mediated by CRAC channel may facilitate the development of ccRCC by enhancing cell proliferation and migration. It was observed that ccRCC cell migration and proliferation declined considerably upon knocking down Orai1 or STIM1 [53].
3.4. Colorectal cancer
Wang et al. [54] have observed the overexpression of STIM1 in patients of colorectal cancer (CRC). Interestingly, STIM1 overexpression in CRC was found to be related significantly with size of the tumor, lymph node metastasis, depth of invasion and the levels of carcinoembryonic antigen in serum (p < 0.02). The authors further reported that CRC cell motility gets enhanced by ectopic expression of STIM1, whereas STIM1 silencing via shRNA could prevent migration of CRC cells. In addition, their results indicated that STIM1 mediated CRC cell migration involved enhanced COX-2 (cyclooxygenase-2) expression and production of PGE2 (prostaglandin E2).
Another study which demonstrated that STIM1 induced metastasis in CRC cells, has also established poor prognosis in CRC patients to be associated with STIM1. Overexpression of STMI1 was observed in highly invasive CRC cell lines as well as in CRC tumor tissues compared to surrounding non-malignant tissues obtained from the same patient. Furthermore, STIM1 overexpression in CRC cells was found to promote metastasis and EMT (epithelial to mesenchymal transition), while STIM1 silencing with siRNA reduced both metastasis and EMT [55]. A separate study has implicated that the downregulation of STIM2 may contribute to apoptosis resistance in HT29 CRC cells [56].
3.5. Gastric cancer
In gastric cancer tissues, the expression levels of Orai1 and STIM1 have been reported to be higher than adjacent non-cancerous tissues. Such an increased expression of Orai1 and STIM1 was found to be associated with poor prognosis and high mortality rates in an investigation of 327 patients of gastric cancer. Moreover, this study showed that knockdown of Orai1 and STIM1 reduced the proliferation, migration and invasion of gastric cancer cells in vitro, as well as resulted in significant inhibition of tumor growth and metastasis in vivo. In addition, downregulation of Orai1 and STIM1 altered cell cycle and epithelial-mesenchymal transition (EMT) markers [57].
3.6. Liver cancer
Evidence for the higher expression of STIM1 in hepatocellular carcinoma cell lines than a normal hepatocyte cell line was provided by Yang et al. [58]. The study also reported elevated STIM1 expression in hepatoma tissues as compared to pre-cancerous tissues of the same patients. Another interesting observation was a five-fold elevation in STIM expression in HCC-LM3 cell line, known to possess a higher potential for migration, in comparison to other hepatocellular carcinoma cell lines.
CRAC channel activated SOCE has been implicated in chemoresistance of human hepatocellular carcinoma to 5-fluorouracil therapy. 5-fluorouracil has been shown to induce autophagic cell death in HepG2 hepatoma cells by reducing the expression of Orai1 and consequently preventing SOCE. Moreover, increased expression of Orai1 was also observed in hepatocellular carcinoma tissues. Therefore, Orai1 expression may be considered as a prognostic marker of 5-fluorouracil sensitivity for hepatocellular carcinoma therapy and blocking CRAC channel mediated SOCE may sensitize hepatocellular carcinoma cells to 5-fluorouracil chemotherapy [59].
3.7. Pancreatic cancer
CRAC channel proteins ORAI1 and STIM1 have been demonstrated to play anti-apoptotic role in pancreatic ductal adenocarcinoma cells. It was observed that apoptosis induced by gemcitabine or 5-fluorouracil increased upon RNAi mediated silencing of ORAI1 and STIM1. The authors also reported relatively enhanced expression of Orai1 and STIM1 in several pancreatic ductal adenocarcinoma cell lines as compared to normal pancreatic ductal epithelial cells, suggesting that cancer cells possibly upregulate Orai1 and STIM1 as a mechanism to safeguard themselves from undergoing apoptosis [60].
3.8. Esophageal cancer
Tumor Orai1 expression was found to be considerably increased in comparison to adjacent non-cancerous esophageal tissues obtained from patients of esophageal squamous cell carcinoma (ESCC). This elevated expression of Orai1 correlated with poor overall survival and recurrence-free survival in ESCC patients. Further, downregulating Orai1 activity/expression was accompanied by reduced ESCC cell proliferation and migration in vitro and suppressed growth of tumor xenograft in vivo [61].
3.9. Lung cancer
Cisplatin has been shown to induce apoptosis through modulating STIM1 in non-small cell lung cancer (NSCLC) cell lines A549 and H460. The same study reported significantly higher expression of STIM1 in lung carcinoma tissue than in the adjoining non-cancerous lung tissue. Based on their findings, the authors concluded that STIM1 might play an important role in the development of lung cancer [62].
Ouadid-Ahidouch and co-workers [63] have also investigated the role of CRAC channel in NSCLC. They observed higher Orai3 expression in cancer tissues in comparison to non-cancerous tissues. Further, blocking CRAC channel pharmacologically or silencing Orai3 in NSCLC cell lines, namely NCI-H23 and NCI-H460, resulted in significant suppression of cell proliferation and arrest of cell cycle progression in G0/G1 phase. These phenomena were found to be associated with a reduced expression of cyclins D1/E and CDKs 4/2. In addition, knockdown of Orai3 reduced the phosphorylation levels of Akt, indicating that Orai3 CRAC channel mediated SOCE in NSCLC regulated tumorigenesis through Akt signaling pathway.
3.10. Glioblastoma
Ca2+ entry through CRAC channel has been shown to regulate the proliferation and apoptosis of glioblastoma cells. Use of CRAC channel blockers or siRNA knockdown of ORAI1 and STIM1 proteins, resulted in significant reduction in proliferation and a marked increase in apoptosis of C6 rat glioma cells. Interestingly, a more significant antiproliferative effect was noticed in cells with siRNA knockdown of ORAI1 than those with STIM1 knockdown [64]. In another study, STIM1 silencing by siRNA suppressed the proliferation of U251 glioblastoma cells by triggering cell cycle arrest in G0/G1 phase via regulation of p21Waf1/Cip1, cyclin D1 and CDK4 (cyclin-dependent kinase 4). The antiproliferative effect of STIM1 downregulation was also seen in U251 glioblastoma xenograft tumor model [65].
Motiani et al. [66] reported that glioblastoma cells had elevated Orai1 expression associated with an upregulated SOCE when compared to human primary astrocytes. The authors further examined the functional significance of SOCE in glioblastoma by evaluating the effects of silencing STIM1 and Orai1 on cell proliferation and invasion. It was demonstrated that silencing of STIM1 and Orai1 led to a significant reduction in cell invasion in glioblastoma cells, but not in human primary astrocytes. The effects of STIM1 and Orai1 downregulation on glioblastoma cell proliferation were, however, found to be marginal. Furthermore, Zhu et al. [67] have also found elevated expression of Orai1 in multiple glioma cell lines and glioma tissues as compared to non-malignant brain tissues. They observed reduced glioma cell migration and invasion upon downregulating Orai1 expression or pharmacologically inhibiting CRAC channel. The results further indicated that phosphorylation of Pyk2 (proline-rich tyrosine kinase 2) could be essential in the pathway through which CRAC channel regulated migration and invasion of glioma cells.
3.11. Hematologic cancers
There is evidence to suggest that Orai1 and Orai2 are essential for the proliferation of HL60 leukemia cells and can regulate HL60 cell migration and FAK phosphorylation. Also, Orai2 was found to be overexpressed in HL60 cells [68]. Similarly, overexpression of Orai1 and STIM1 has been reported in multiple myeloma tissue and cell lines. The study also demonstrated that silencing of Orai1 or STIM1 or inhibition of SOCE resulted in a reduction of cell viability and caused apoptosis and cell cycle arrest in multiple myeloma cell lines [69].
3.12. Melanoma
Inhibition of CRAC channel activated SOCE in melanoma cells caused reduction in cell proliferation and migration. Results from the study suggested that CaMKII/Raf-1/ERK signaling pathway controlled the SOCE mediated proliferation and migration in melanoma cells. Moreover, STIM1 and Orai1 were reported to be highly expressed in human melanoma tissues and various melanoma cell lines [70]. Melanoma growth and invasion have also been reported to be controlled by STIM2 and Orai1 mediated Ca2+ entry [71].
3.13. Prostate Cancer
The exact role of CRAC channel is not clearly understood in prostate cancer, perhaps due to its heterogeneous nature. Orai1 mediated SOCE was reported to induce apoptosis in human prostate cancer cells by Flourakis et al., suggesting a tumor suppressive action of CRAC channel. Further, the study correlated low Orai1 levels with apoptosis resistant phenotype in prostate cancer cells [72]. Kappel et al. [73] have reviewed the dysregulation of CRAC channel components in prostate cancer. Interestingly. It has been reported that the expression levels of STIM1 and Orai1 depend on the clinical stage of prostate cancer. Orai1 and STIM1 expression levels have been found to be elevated in early stages of prostate cancer, but they get reduced during the later castration resistant stages of prostate cancer [74].
4. Pharmacologic inhibition of CRAC channel as a cancer treatment strategy
Over the past three decades, our understanding of the molecular components and mechanism of action of CRAC channels has grown significantly. Concomitantly, the critical roles CRAC channels purportedly play in various human pathologies, including cancer, have been explored extensively. Moreover, the fact that CRAC channel proteins have been widely reported to be overexpressed in several types of human tumor tissues in contrast to their normal counterparts, has led to an increased realization that pharmacologically inhibiting CRAC channel mediated Ca2+ entry may possibly have significant implications for the development of cancer therapeutics. In other words, CRAC channels have emerged as potential targets for cancer therapy and this has ignited great interest in developing small molecule inhibitors to block this channel and its associated signaling in tumor cells.
Inhibitors of CRAC channel may act by either directly blocking the Orai pore of the channel or by targeting STIM or by interfering with Orai-STIM interactions, thereby regulating the overall activity of the CRAC channel. Several small molecule inhibitors targeting CRAC channel dependent Ca2+ entry have been developed over the years [10]. Some of them have shown promise as potential candidates for cancer therapy in preclinical studies [75]. Besides, these pharmacological inhibitors of CRAC channel have also proved quite helpful in understanding the roles of CRAC channel in tumorigenesis. A comprehensive description of the various challenges involved in the discovery and development of CRAC channel inhibitors has been provided by Stauderman [76]. Here, we discuss some of the CRAC channel blockers that have shown potential vis a vis cancer therapy. Figure 2 illustrates the chemical structures of these agents.
Figure 2.
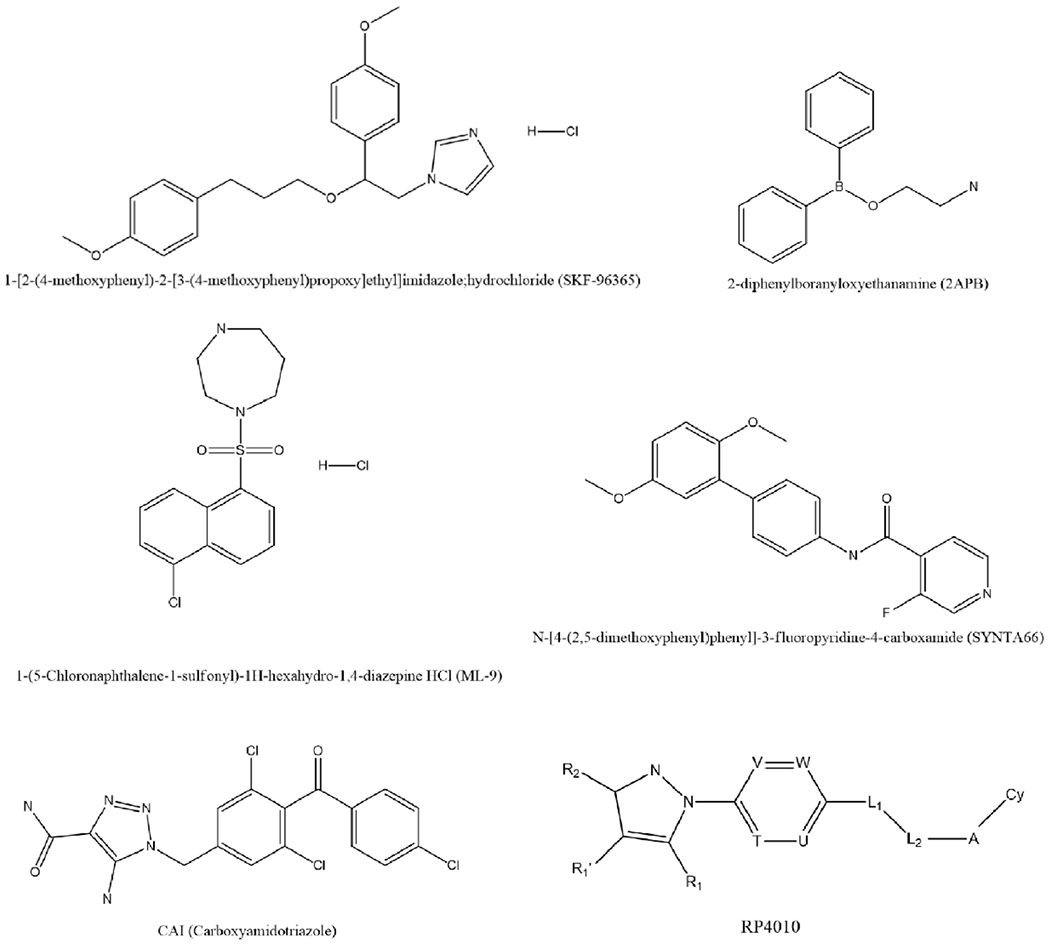
Chemical structures of CRAC channel inhibitors that have shown potential anticancer properties.
4.1. SKF-96365
SKF-96365, an imidazole compound, was shown to block SOCE in Jurkat cells [77] and rat basophilic leukemia cells [78]. It also reportedly blocked SOCE and NFAT nuclear translocation induced by STIM1 overexpression [79]. SKF-96365 was found capable of reducing tumor metastasis in animal models of breast cancer [22]. It also prevented angiogenesis and decreased the growth of cervical cancer xenografts in mice by blocking SOCE [50]. Inhibition of CRAC channel activity by SKF-96365 was shown to suppress the proliferation and migration of esophageal cancer cells in vitro and retard tumor growth in xenografted mice [61]. Interestingly, SKF-96365 was reported to be non-selective for CRAC channels as it could block other calcium channels as well [80]. Nevertheless, it should be noted that silencing or overexpressing Orai1 and STIM1 in the above-mentioned studies gave similar results which corroborate the observation that the effects of SKF96365 in those models stem from its ability to block Orai1-STIM1 CRAC channel.
4.2. 2-APB
2-APB (2-aminoethyldiphenyl borate) has been reported to influence CRAC channel activity in a paradoxical manner, such that it inhibits activity of CRAC channel at high doses while stimulating it at lower doses. It was suggested that 2-APB, at high concentrations, was able to inhibit CRAC channel mediated Ca2+ entry by inhibiting STIM1 redistribution, while stimulating CRAC channel activity by enhancing STIM–ORAI1 interactions at low concentrations [81]. The effects of 2-APB on CRAC channel vary depending on the Orai homologues [82]. While at higher doses 2-APB acts as an inhibitor on Orai1 and Orai2, it stimulates Orai3 and reduces its Ca2+ selectivity independent of STIM1 or store depletion [83,84]. Multiple studies have shown that 2-APB has the potential to inhibit the proliferation of cervical [50], hepatoma [85] and gastric cancer cells [86]. It was also found to reduce the migration of CRC cells [54] and cervical cancer cells [87]. Despite exhibiting effective antitumor activities in multiple studies, 2-APB is considered not suitable for cancer chemotherapy, due to its non-specific and multi-target behavior. In an effort to overcome this problem, more selective 2-APB analogues, namely DPB-162AE and DPB-163AE, were developed, which proved to be 100 times more effective than 2-APB in blocking CRAC channel mediated SOCE [88]. However, the effect of these compounds on in vitro and in vivo cancer models are yet to be explored.
4.3. ML-9
ML-9 is an inhibitor of Akt kinase as well as STIM1, which is believed to block SOCE by interfering with STIM1 translocation [89]. ML-9 was shown to potently induce a concentration and Ca2+ dependent autophagic cell death in prostate cancer cells. It was further observed that ML-9 significantly promoted antitumor effects of docetaxel, which underscored its potential to be used in combination with existing anticancer chemotherapy [90].
4.4. Synta66
Synta66, developed by Synta Pharmaceuticals, is another selective blocker of CRAC channel activated SOCE. It has recently been reported to decrease EGF stimulated migration and proliferation in a breast cancer basal cell line model (MDA-MB-468) [91]. Despite promising initial findings, further investigations on Synta66 and its structural analogues, especially using in vivo models, are still needed.
4.5. CAI
CAI (Carboxyamidotriazole), a synthetic inhibitor of CRAC channel mediated Ca2+ influx, has been shown to decrease cell proliferation, invasion, migration and matrix metalloproteinase production in several head and neck squamous cell carcinoma cell lines, which has helped in establishing CRAC channel activated calcium signaling as a novel target for head and neck cancers [92]. CAI also demonstrated antiproliferative effect on hepatoma cell lines almost ten-fold greater than 2-APB [78]. Anti-angiogenic effects of CAI have also been reported [93]. Further evidence for the anti-angiogenic properties were provided by a study where CAI was shown to inhibit the proliferation and tubulogenesis of endothelial progenitor cells from renal cellular carcinoma patients [94].
Besides preclinical studies, CAI has also been investigated in multiple clinical trials. In one such phase II trial, CAI was found to enhance disease stabilization in patients with relapsed epithelial ovarian carcinoma and was suggested to be useful as a maintenance therapy owing to its limited toxicity profile [95]. However, in another Phase II trial in patients with metastatic renal cell carcinoma, CAI was reported to have little to no effect on response rate and disease progression [96]. Promising activity and effective brain penetration of CAI orotate was observed in a recent multicenter Phase IB study, which led to the conclusion that CAI orotate could be safely used in combination with temozolomide or chemoradiation in hard to treat glioblastoma and anaplastic gliomas [97].
4.6. RP4010
RP4010 is a new and potent CRAC channel inhibitor developed by Rhizen Pharmaceuticals SA. Cui et al. [98] have examined the anti-cancer effects of RP4010 in esophagus squamous cell carcinoma. The authors demonstrated that RP4010 caused reduction in intracellular Ca2+ oscillations, concomitant with cell growth inhibition and cell cycle arrest at G0/G1 phase in cultured esophageal cancer cells as well as a decrease in tumor growth in vivo. Recently our lab has also published a study on the effect of CRAC channel inhibition by RP4010 in pancreatic ductal adenocarcinoma models. RP4010 proved to be effective in suppressing the proliferation of various pancreatic cancer cell lines and was also able to reduce the growth of patient derived tumor xenograft in mice, more potently when combined with gemcitabine and nab-paclitaxel [99]. These findings, therefore, imply that CRAC channel inhibition can have translational relevance as a potential chemotherapy or adjuvant therapy in cancers such as esophagus squamous cell carcinoma and pancreatic ductal adenocarcinoma. However, the exact mode of binding and the mechanism underlying inhibitory action on CRAC channel components needs to be further elucidated. RP4010 continued to be under clinical development until very recently. A multicenter Phase I/Ib clinical trial for evaluation of the safety and efficacy of escalating doses of RP4010 in patients with relapsed or refractory non-Hodgkin’s lymphoma had to be stopped after a review of the pharmacokinetic data (ClinicalTrials.gov Identifier: NCT03119467).
5. Conclusion
CRAC channel regulates intracellular Ca2+ homeostasis in non-excitable cells through mediating SOCE which is triggered in response to depletion of Ca2+ from the endoplasmic reticulum stores. STIM proteins sense the loss of Ca2+ from endoplasmic reticulum and their subsequent coupling with plasma membrane bound Orai proteins forms a functional CRAC channel that allows the influx of Ca2+ into the cell. The critical role of CRAC channel mediated Ca2+ entry in promoting cell proliferation, cell cycle arrest, resistance to apoptosis, invasion and metastasis in several types of cancers has been demonstrated in numerous independent studies. Moreover, there is ample evidence that the expression of CRAC channel proteins, Orai and STIM, is upregulated in various types of cancer cells and patient tumor tissues as compared to normal cells and non-cancerous tissue samples, respectively.
The established role of CRAC channel mediated SOCE in cancer progression also has some strong therapeutic implications, as it gives rise to the possibility of pharmacologically inhibiting CRAC channel to selectively target cancer cells. This anticancer therapeutic potential of targeting CRAC channel gets strengthened by the observation that pharmacological inhibition of CRAC channel or silencing of CRAC channel proteins can effectively suppress tumor growth and metastasis in several cancer models. In addition, CRAC channel inhibition has been shown to enhance the effect of standard of care chemotherapy drugs. A number of CRAC channel blockers have shown anticancer effects in preclinical studies, but a majority of them did not manage to reach the clinical trial stage, mainly because of their toxicity and off-target effects. Nonetheless, there are some selective CRAC channel blockers that hold promise for future drug development.
6. Expert Opinion
The process of SOCE was first described by J. W. Putney in 1986 [100] as the depletion of Ca2+ stores directly resulting in activation of Ca2+ channels in the cell membrane, but the mechanism of SOCE remained largely enigmatic for over two decades until STIM and Orai proteins were identified as the key components for CRAC channel activation. Therefore, our understanding of CRAC channels is relatively recent. Regardless of this, CRAC channel mediated SOCE and its function in various cellular processes has been a subject of considerable interest. Moreover, the implications of CRAC channel activity in various human diseases, including cancer, has garnered a lot of attention. As the field evolved over the last two decades and new findings continued to emerge, our knowledge about this Ca2+ channel and its associated signaling pathways has kept on growing. Despite dissecting much of the molecular basis of CRAC channel activation and the functional association these channels possibly have with tumor development, there are still gaps in our understanding of the molecular mechanisms through which CRAC channels modulate tumor growth and metastasis. Nonetheless, the pleiotropic roles CRAC channel plays in tumorigenesis makes it an attractive target for cancer therapy. Yet, there is a long way to go before the full potential of blocking CRAC channel activity is harnessed as an anticancer therapeutic strategy.
So far, no inhibitor of CRAC channel mediated SOCE has been approved by FDA for clinical use in cancer treatment. But, a few CRAC channel inhibitors have succeeded in entering clinical trials. An increased understanding of CRAC channel activation and mechanisms, together with the development of new techniques to manipulate channel activity and a growing interest shown by several pharmaceutical companies, will hopefully accelerate the development of novel, more potent drug candidates that may specifically target CRAC channels for cancer therapy. More importantly, developing drugs that can specifically target a particular CRAC channel component may prove to be more effective in augmenting the selectivity and minimizing off-target effects. In this regard, structure based rational designing of drugs targeting CRAC channel proteins with greater efficacy and specificity may be expected to come up with promising leads for cancer therapy in the future. It is also worth mentioning that most of the studies on CRAC channels have focused on STIM1-Orai1 complex, which is undoubtedly the best characterized CRAC channel. However, the role of other STIM and Orai isoforms in cancer development and drug resistance remains understudied. Consequently, no CRAC channel inhibitor specific for these underappreciated CRAC components has been developed.
Since CRAC channel proteins are expressed on normal cells as well, another challenge in drug development would be to minimize toxicity to normal cells when CRAC channel blockers are administered systemically in cancer patients. This may be achieved by a CRAC channel inhibiting drug candidate that specifically targets cancer cells by recognizing their overexpressed STIM-Orai signature. Moreover, overexpression of STIM and Orai proteins observed in several types of cancers not only aid in selective targeting of cancer cells by CRAC channel blockers but can also have implications for the adverse prognosis of the cancer. For instance, overexpression of Orai3 in the tissues of lung adenocarcinoma patients [63] can be a prognostic marker for the disease and any prospective CRAC channel blocker which may be specific for Orai3 can be used to selectively target lung cancer cells. Therefore, identifying the specific type of CRAC channel components that contribute to tumor development in an individual cancer patient or a certain patient population can pave the way for possible selective targeting of cancer in a precision medicine approach.
It may be noted that so far only the overexpression of CRAC channel proteins has been widely considered to play the major role in cancer development. However, there is emerging evidence that certain mutations in Orai can result in its constitutive activation and consequent Ca2+ influx. These mutations have been found in large-scale cancer genomics datasets for dysfunctional Orai1 mutants [101]. More such studies in the future may lead to a paradigm shift in our understanding of the functional roles that proteins of the CRAC channel complex play in cancer development and progression. This may also provide new vistas for designing drugs aimed at targeting specific chinks in the armor of CRAC channel machinery.
CRAC channel proteins are also reportedly overexpressed in response to treatment with certain standard of care drugs, thereby contributing to drug resistance in various cancers. The use of CRAC channel inhibitors as adjuvant to chemotherapy can overcome this drug resistance and can even possibly enhance the efficacy of the chemotherapeutic drug. In fact, studies from our lab have recently demonstrated that CRAC channel inhibitor RP4010 was able to synergize with gemcitabine and nab-paclitaxel in pancreatic ductal adenocarcinoma preclinical models [99]. Even though the Phase I/Ib clinical trial of RP4010 as a monotherapy in non-Hodgkin’s lymphoma proved unyielding, the way forward would be to use RP4010 or any comparable CRAC channel blocker in combination with existing chemotherapy or even immunotherapy in the future clinical trials.
Further, recombinant Orai1 monoclonal antibodies have been shown to bind Orai1 strongly and specifically, leading to the internalization of Orai1 proteins, thereby causing loss of SOCE activity [102]. Such potential anti-Orai based therapy has been used for immune disorders. A similar application for cancer therapy holds promise and needs to be explored in future preclinical and clinical studies. Only time will tell if the safety and efficacy of CRAC channel blockers get established by the yet to be initiated clinical trials. Meanwhile, preclinical studies continue to shed more light on the myriad roles that CRAC channels play in carcinogenesis and suggest the potential therapeutic value of targeting these channels in cancer.
Article Highlights:
CRAC channel is responsible for maintaining cellular Ca2+ homeostasis.
CRAC channel is activated by the coupling of channel pore forming Orai protein and endoplasmic reticulum Ca2+ sensing STIM protein.
CRAC channel plays vital roles in cancer cell proliferation, tumor growth, metastasis and tumor neovascularization.
CRAC channel proteins are overexpressed in cancer cells and tissues.
Pharmacological inhibition of CRAC channel represents a promising strategy for anticancer therapy.
Acknowledgments
Funding
Work in the lab of ASA is supported by NIH > National Cancer Institute 5R37CA215427
Declaration of interest
ASA received funding from Rhizen Pharmaceuticals. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Footnotes
Reviewers Disclosure
Peer reviewers on this manuscript have no relevant financial relationships or otherwise to disclose.
References
* = of interest, ** = of considerable interest
- 1.Monteith GR, McAndrew D, Faddy HM, et al. Calcium and cancer: targeting Ca2+ transport. Nat Rev Cancer. 2007;7(7):519–530. [DOI] [PubMed] [Google Scholar]
- 2.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4(7):517–529. [DOI] [PubMed] [Google Scholar]
- 3.Rizzuto R, Pozzan T. When calcium goes wrong: genetic alterations of a ubiquitous signaling route. Nat Genet. 2003;34(2):135–141. [DOI] [PubMed] [Google Scholar]
- 4.Parekh AB. Store-operated CRAC channels: function in health and disease. Nat Rev Drug Discov. 2010;9(5):399–410. [DOI] [PubMed] [Google Scholar]; * Summarizes the progress in the study of CRAC channels in relation to human disease.
- 5.Carafoli E The calcium-signalling saga: tap water and protein crystals. Nat Rev Mol Cell Biol. 2003;4(4):326–332. [DOI] [PubMed] [Google Scholar]
- 6.Carafoli E Calcium signaling: a tale for all seasons. Proc Natl Acad Sci U S A. 2002;99(3):1115–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roos J, DiGregorio PJ, Yeromin AV, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169(3):435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luik RM, Wu MM, Buchanan J, et al. The elementary unit of store-operated Ca2+ entry: local activation of CRAC channels by STIM1 at ER-plasma membrane junctions. J Cell Biol. 2006;174(6):815–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergmeier W, Weidinger C, Zee I, et al. Emerging roles of store-operated Ca2+ entry through STIM and ORAI proteins in immunity, hemostasis and cancer. Channels (Austin). 2013;7(5):379–391. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Summarizes the regulatory roles of CRAC proteins in immunity and cancer.
- 10.Tian C, Du L, Zhou Y, et al. Store-operated CRAC channel inhibitors: opportunities and challenges. Future Med Chem. 2016;8(7):817–832. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** A comprehensive review of CRAC channel inhibitors.
- 11.Lyfenko AD, Dirksen RT. Differential dependence of store-operated and excitation-coupled Ca2+ entry in skeletal muscle on STIM1 and Orai1. J Physiol. 2008;586(20):4815–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vig M, DeHaven WI, Bird GS, et al. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9(1):89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tolhurst G, Carter RN, Amisten S, et al. Expression profiling and electrophysiological studies suggest a major role for Orai1 in the store-operated Ca2+ influx pathway of platelets and megakaryocytes. Platelets. 2008;19(4):308–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaw PJ, Feske S. Regulation of lymphocyte function by ORAI and STIM proteins in infection and autoimmunity. J Physiol. 2012;590(17):4157–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maus M, Medgyesi D, Kiss E, et al. B cell receptor-induced Ca2+ mobilization mediates F-actin rearrangements and is indispensable for adhesion and spreading of B lymphocytes. J Leukoc Biol. 2013;93(4):537–547. [DOI] [PubMed] [Google Scholar]
- 16.Trebak M STIM1/Orai1, ICRAC, and endothelial SOC. Circ Res. 2009;104(9):e56–e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cahalan MD. STIMulating store-operated Ca(2+) entry. Nat Cell Biol. 2009;11(6):669–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Braun A, Varga-Szabo D, Kleinschnitz C, et al. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood. 2009;113(9):2056–2063. [DOI] [PubMed] [Google Scholar]
- 19.Gerasimenko JV, Gryshchenko O, Ferdek PE, et al. Ca2+ release-activated Ca2+ channel blockade as a potential tool in antipancreatitis therapy. Proc Natl Acad Sci U S A. 2013;110(32):13186–13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feske S Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7(9):690–702. [DOI] [PubMed] [Google Scholar]
- 21.Feske S, Gwack Y, Prakriya M, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441(7090):179–185. [DOI] [PubMed] [Google Scholar]
- 22.Yang S, Zhang JJ, Huang XY. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell. 2009;15(2):124–134. [DOI] [PubMed] [Google Scholar]; * First study on the role of CRAC channel in breast cancer migration and metastasis.
- 23.Xie J, Pan H, Yao J, et al. SOCE and cancer: Recent progress and new perspectives. Int J Cancer. 2016;138(9):2067–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Excellent review on the molecular basis of CRAC channel.
- 24.Bray F, Ferlay J, Soerjomataram I, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. [DOI] [PubMed] [Google Scholar]
- 25.Monteith GR, Davis FM, Roberts-Thomson SJ. Calcium channels and pumps in cancer: changes and consequences. J Biol Chem. 2012;287(38):31666–31673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roderick HL, Cook SJ. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer. 2008;8(5):361–375. [DOI] [PubMed] [Google Scholar]
- 27.Thiel M, Lis A, Penner R. STIM2 drives Ca2+ oscillations through store-operated Ca2+ entry caused by mild store depletion. J Physiol. 2013;591(6):1433–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kar P, Bakowski D, Di Capite J, et al. Different agonists recruit different stromal interaction molecule proteins to support cytoplasmic Ca2+ oscillations and gene expression. Proc Natl Acad Sci U S A. 2012;109(18):6969–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bird GS, Hwang SY, Smyth JT, et al. STIM1 is a calcium sensor specialized for digital signaling. Curr Biol. 2009;19(20):1724–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stathopulos PB, Zheng L, Ikura M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J Biol Chem. 2009;284(2):728–732. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Wang Y, Zhou Y, et al. Distinct Orai-coupling domains in STIM1 and STIM2 define the Orai-activating site. Nat Commun. 2014;5:3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shuttleworth TJ. Orai3--the ‘exceptional’ Orai? J Physiol. 2012;590(2):241–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoth M, Niemeyer BA. The neglected CRAC proteins: Orai2, Orai3, and STIM2. Curr Top Membr. 2013;71:237–271. [DOI] [PubMed] [Google Scholar]
- 34.Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446(7133):284–287. [DOI] [PubMed] [Google Scholar]
- 35.Park CY, Hoover PJ, Mullins FM, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136(5):876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Muik M, Frischauf I, Derler I, et al. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283(12):8014–8022. [DOI] [PubMed] [Google Scholar]
- 37.Müller MR, Rao A. NFAT, immunity and cancer: a transcription factor comes of age. Nat Rev Immunol. 2010;10(9):645–656. [DOI] [PubMed] [Google Scholar]
- 38.DiDonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev. 2012;246(1):379–400. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi Y, Watanabe H, Murakami M, et al. Functional role of stromal interaction molecule 1 (STIM1) in vascular smooth muscle cells. Biochem Biophys Res Commun. 2007;361(4):934–940. [DOI] [PubMed] [Google Scholar]
- 40.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soboloff J, Rothberg BS, Madesh M, et al. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. 2012;13(9):549–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anghileri LJ, Miller ES, Robinette J, et al. Calcium metabolism in tumors. Its relationship with chromium complex accumulation. II. Calcium, magnesium and phosphorus in human and animal tumors. Oncology. 1971;25(3):193–209. [DOI] [PubMed] [Google Scholar]
- 43.Chen YF, Lin PC, Yeh YM, et al. Store-Operated Ca2+ Entry in Tumor Progression: From Molecular Mechanisms to Clinical Implications. Cancers (Basel). 2019;11(7):899. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** A good review of the clinical implications of CRAC channel in tumor progression.
- 44.Jardin I, Lopez JJ, Salido GM, et al. Store-Operated Ca2+ Entry in Breast Cancer Cells: Remodeling and Functional Role. Int J Mol Sci. 2018;19(12):4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McAndrew D, Grice DM, Peters AA, et al. ORAI1-mediated calcium influx in lactation and in breast cancer. Mol Cancer Ther. 2011;10(3):448–460. [DOI] [PubMed] [Google Scholar]
- 46.Badaoui M, Mimsy-Julienne C, Saby C, et al. Collagen type 1 promotes survival of human breast cancer cells by overexpressing Kv10.1 potassium and Orai1 calcium channels through DDR1-dependent pathway. Oncotarget. 2017;9(37):24653–24671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Motiani RK, Abdullaev IF, Trebak M. A novel native store-operated calcium channel encoded by Orai3: selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J Biol Chem. 2010;285(25):19173–19183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Motiani RK, Zhang X, Harmon KE, et al. Orai3 is an estrogen receptor α-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 2013;27(1):63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Faouzi M, Hague F, Potier M, et al. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J Cell Physiol. 2011;226(2):542–551. [DOI] [PubMed] [Google Scholar]
- 50.Chen YF, Chiu WT, Chen YT, et al. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc Natl Acad Sci U S A. 2011;108(37):15225–15230. [DOI] [PMC free article] [PubMed] [Google Scholar]; * A comprehensive study exploring the role of STIM1 in the tumorigenesis of cervical cancer.
- 51.Chen YW, Chen YF, Chen YT, et al. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci Rep. 2016;6:22142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmidt S, Liu G, Liu G, et al. Enhanced Orai1 and STIM1 expression as well as store operated Ca2+ entry in therapy resistant ovary carcinoma cells. Oncotarget. 2014;5(13):4799–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim JH, Lkhagvadorj S, Lee MR, et al. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem Biophys Res Commun. 2014;448(1):76–82. [DOI] [PubMed] [Google Scholar]
- 54.Wang JY, Sun J, Huang MY, et al. STIM1 overexpression promotes colorectal cancer progression, cell motility and COX-2 expression. Oncogene. 2015;34(33):4358–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang Z, Liu X, Feng B, et al. STIM1, a direct target of microRNA-185, promotes tumor metastasis and is associated with poor prognosis in colorectal cancer. Oncogene. 2015;34(37):4808–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sobradillo D, Hernández-Morales M, Ubierna D, et al. A reciprocal shift in transient receptor potential channel 1 (TRPC1) and stromal interaction molecule 2 (STIM2) contributes to Ca2+ remodeling and cancer hallmarks in colorectal carcinoma cells. J Biol Chem. 2014;289(42):28765–28782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xia J, Wang H, Huang H, et al. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett. 2016;381(1):31–40. [DOI] [PubMed] [Google Scholar]
- 58.Yang N, Tang Y, Wang F, et al. Blockade of store-operated Ca(2+) entry inhibits hepatocarcinoma cell migration and invasion by regulating focal adhesion turnover. Cancer Lett. 2013;330(2):163–169. [DOI] [PubMed] [Google Scholar]
- 59.Tang BD, Xia X, Lv XF, et al. Inhibition of Orai1-mediated Ca2+ entry enhances chemosensitivity of HepG2 hepatocarcinoma cells to 5-fluorouracil. J Cell Mol Med. 2017;21(5):904–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kondratska K, Kondratskyi A, Yassine M, et al. Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance of pancreatic adenocarcinoma. Biochim Biophys Acta. 2014;1843(10):2263–2269. [DOI] [PubMed] [Google Scholar]
- 61.Zhu H, Zhang H, Jin F, et al. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget. 2014;5(11):3455–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li W, Zhang M, Xu L, et al. The apoptosis of non-small cell lung cancer induced by cisplatin through modulation of STIM1. Exp Toxicol Pathol. 2013;65(7–8):1073–1081. [DOI] [PubMed] [Google Scholar]
- 63.Ay AS, Benzerdjeb N, Sevestre H, et al. Orai3 constitutes a native store-operated calcium entry that regulates non small cell lung adenocarcinoma cell proliferation. PLoS One. 2013;8(9):e72889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu H, Hughes JD, Rollins S, et al. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp Mol Pathol. 2011;91(3):753–760. [DOI] [PubMed] [Google Scholar]
- 65.Li G, Zhang Z, Wang R, et al. Suppression of STIM1 inhibits human glioblastoma cell proliferation and induces G0/G1 phase arrest. J Exp Clin Cancer Res. 2013;32(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Motiani RK, Hyzinski-García MC, Zhang X, et al. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflugers Arch. 2013;465(9):1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhu M, Chen L, Zhao P, et al. Store-operated Ca(2+) entry regulates glioma cell migration and invasion via modulation of Pyk2 phosphorylation. J Exp Clin Cancer Res. 2014;33(1):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Diez-Bello R, Jardin I, Salido GM, et al. Orai1 and Orai2 mediate store-operated calcium entry that regulates HL60 cell migration and FAK phosphorylation. Biochim Biophys Acta Mol Cell Res. 2017;1864(6):1064–1070. [DOI] [PubMed] [Google Scholar]
- 69.Wang W, Ren Y, Wang L, et al. Orai1 and Stim1 Mediate the Majority of Store-Operated Calcium Entry in Multiple Myeloma and Have Strong Implications for Adverse Prognosis. Cell Physiol Biochem. 2018;48(6):2273–2285. [DOI] [PubMed] [Google Scholar]
- 70.Umemura M, Baljinnyam E, Feske S, et al. Store-operated Ca2+ entry (SOCE) regulates melanoma proliferation and cell migration. PLoS One. 2014;9(2):e89292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stanisz H, Saul S, Müller CS, et al. Inverse regulation of melanoma growth and migration by Orai1/STIM2-dependent calcium entry. Pigment Cell Melanoma Res. 2014;27(3):442–453. [DOI] [PubMed] [Google Scholar]
- 72.Flourakis M, Lehen’kyi V, Beck B, et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010;1(9):e75. Published 2010. September 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kappel S, Marques IJ, Zoni E, et al. Store-Operated Ca2+ Entry as a Prostate Cancer Biomarker - a Riddle with Perspectives [published correction appears in Curr Mol Biol Rep. 2018;4(3):142]. Curr Mol Biol Rep. 2017;3(4):208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Perrouin Verbe MA, Bruyere F, Rozet F, Vandier C, Fromont G. Expression of store-operated channel components in prostate cancer: the prognostic paradox. Hum Pathol. 2016;49:77–82. [DOI] [PubMed] [Google Scholar]
- 75.Cui C, Merritt R, Fu L, et al. Targeting calcium signaling in cancer therapy. Acta Pharm Sin B. 2017;7(1):3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Describes various types of calcium channels, pumps and transporters in cancer and the possibility of targeting them.
- 76.Stauderman KA. CRAC channels as targets for drug discovery and development. Cell Calcium. 2018;74:147–159. [DOI] [PubMed] [Google Scholar]; ** Discusses the various challenges associated with the discovery and development of CRAC channel inhibitors.
- 77.Chung SC, McDonald TV, Gardner P. Inhibition by SK&F 96365 of Ca2+ current, IL-2 production and activation in T lymphocytes. Br J Pharmacol. 1994;113(3):861–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kozak JA, Kerschbaum HH, Cahalan MD. Distinct properties of CRAC and MIC channels in RBL cells. J Gen Physiol. 2002;120(2):221–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liou J, Kim ML, Heo WD, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15(13):1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Franzius D, Hoth M, Penner R. Non-specific effects of calcium entry antagonists in mast cells. Pflugers Arch. 1994;428(5–6):433–438. [DOI] [PubMed] [Google Scholar]
- 81.DeHaven WI, Smyth JT, Boyles RR, et al. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J Biol Chem. 2008;283(28):19265–19273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lis A, Peinelt C, Beck A, et al. CRACM1, CRACM2, and CRACM3 are store-operated Ca2+ channels with distinct functional properties. Curr Biol. 2007;17(9):794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Peinelt C, Lis A, Beck A, Fleig A, Penner R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J Physiol. 2008;586(13):3061–3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schindl R, Bergsmann J, Frischauf I, et al. 2-aminoethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. J Biol Chem. 2008;283(29):20261–20267. [DOI] [PubMed] [Google Scholar]
- 85.Enfissi A, Prigent S, Colosetti P, et al. The blocking of capacitative calcium entry by 2-aminoethyl diphenylborate (2-APB) and carboxyamidotriazole (CAI) inhibits proliferation in Hep G2 and Huh-7 human hepatoma cells. Cell Calcium. 2004;36(6):459–467. [DOI] [PubMed] [Google Scholar]
- 86.Sakakura C, Hagiwara A, Fukuda K, et al. Possible involvement of inositol 1,4,5-trisphosphate receptor type 3 (IP3R3) in the peritoneal dissemination of gastric cancers. Anticancer Res. 2003;23(5A):3691–3697. [PubMed] [Google Scholar]
- 87.Chen YT, Chen YF, Chiu WT, et al. The ER Ca2+ sensor STIM1 regulates actomyosin contractility of migratory cells. J Cell Sci. 2013;126(Pt 5):1260–1267. [DOI] [PubMed] [Google Scholar]
- 88.Goto J, Suzuki AZ, Ozaki S, et al. Two novel 2-aminoethyl diphenylborinate (2-APB) analogues differentially activate and inhibit store-operated Ca(2+) entry via STIM proteins. Cell Calcium. 2010;47(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Smyth JT, Dehaven WI, Bird GS, et al. Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J Cell Sci. 2008;121(Pt 6):762–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kondratskyi A, Yassine M, Slomianny C, et al. Identification of ML-9 as a lysosomotropic agent targeting autophagy and cell death. Cell Death Dis. 2014;5(4):e1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Azimi I, Bong AH, Poo GXH, et al. Pharmacological inhibition of store-operated calcium entry in MDA-MB-468 basal A breast cancer cells: consequences on calcium signalling, cell migration and proliferation. Cell Mol Life Sci. 2018;75(24):4525–4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu Y, Palad AJ, Wasilenko WJ, et al. Inhibition of head and neck squamous cell carcinoma growth and invasion by the calcium influx inhibitor carboxyamidotriazole. Clin Cancer Res. 1997;3(11):1915–1921. [PubMed] [Google Scholar]
- 93.Faehling M, Kroll J, Föhr KJ, et al. Essential role of calcium in vascular endothelial growth factor A-induced signaling: mechanism of the antiangiogenic effect of carboxyamidotriazole. FASEB J. 2002;16(13):1805–1807. [DOI] [PubMed] [Google Scholar]
- 94.Lodola F, Laforenza U, Bonetti E, et al. Store-operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS One. 2012;7(9):e42541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hussain MM, Kotz H, Minasian L, et al. Phase II trial of carboxyamidotriazole in patients with relapsed epithelial ovarian cancer. J Clin Oncol. 2003;21(23):4356–4363. [DOI] [PubMed] [Google Scholar]
- 96.Dutcher JP, Leon L, Manola J, et al. Phase II study of carboxyamidotriazole in patients with advanced renal cell carcinoma refractory to immunotherapy: E4896, an Eastern Cooperative Oncology Group Study. Cancer. 2005;104(11):2392–2399. [DOI] [PubMed] [Google Scholar]
- 97.Omuro A, Beal K, McNeill K, et al. Multicenter Phase IB Trial of Carboxyamidotriazole Orotate and Temozolomide for Recurrent and Newly Diagnosed Glioblastoma and Other Anaplastic Gliomas. J Clin Oncol. 2018;36(17):1702–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cui C, Chang Y, Zhang X, et al. Targeting Orai1-mediated store-operated calcium entry by RP4010 for anti-tumor activity in esophagus squamous cell carcinoma.. Cancer Lett. 2018;432:169–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Khan HY, Mpilla GB, Sexton R, et al. Calcium Release-Activated Calcium (CRAC) Channel Inhibition Suppresses Pancreatic Ductal Adenocarcinoma Cell Proliferation and Patient-Derived Tumor Growth. Cancers (Basel). 2020;12(3):750. [DOI] [PMC free article] [PubMed] [Google Scholar]; * First study to demonstrate the effect of a CRAC channel inhibitor in a PDx model of PDAC.
- 100.Putney JW Jr. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7(1):1–12. [DOI] [PubMed] [Google Scholar]
- 101.Frischauf I, Litviňuková M, Schober R, et al. Transmembrane helix connectivity in Orai1 controls two gates for calcium-dependent transcription. Sci Signal. 2017;10(507):eaao0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cox JH, Hussell S, Søndergaard H, et al. Antibody-mediated targeting of the Orai1 calcium channel inhibits T cell function. PLoS One. 2013;8(12):e82944. Published 2013. December 23. [DOI] [PMC free article] [PubMed] [Google Scholar]